
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hexahalorhenate(IV) salts of metal oxazolidine nitroxides†
Anders H.
Pedersen
 a,
Blaise L.
Geoghegan
b,
Gary S.
Nichol
a,
David W.
Lupton
c,
Keith. S.
Murray
c,
José
Martínez-Lillo
*d,
Ian A.
Gass
*b and
Euan K.
Brechin
*a
a,
Blaise L.
Geoghegan
b,
Gary S.
Nichol
a,
David W.
Lupton
c,
Keith. S.
Murray
c,
José
Martínez-Lillo
*d,
Ian A.
Gass
*b and
Euan K.
Brechin
*a
aEaStCHEM School of Chemistry, The University of Edinburgh, David Brewster Road, EH9 3FJ Edinburgh, Scotland, UK. E-mail: E.Brechin@ed.ac.uk
bSchool of Pharmacy and Biomolecular Sciences, University of Brighton, Brighton BN2 4GJ, UK. E-mail: I.Gass@brighton.ac.uk
cSchool of Chemistry, Monash University, Clayton, Victoria 3800, Australia
dDepartament de Química Inorgànica/Instituto de Ciencia Molecular (ICMol), Universitat de València, C/Catedrático José Beltrán 2, 46980, Paterna (València), Spain. E-mail: F.Jose.Martinez@uv.es
First published on 20th March 2017
Abstract
Eight coordination compounds of formulae [FeII(L˙)2][ReIVCl6] (1a), [FeII(L˙)2][ReIVBr6] (1b), [CoII(L˙)2][ReIVCl6]·CH3CN (2a), [CoII(L˙)2][ReIVBr6] (2b), [NiII(L˙)(CH3CN)3][ReIVCl6]·CH3CN (3a), [NiII(L˙)(CH3CN)3][ReIVBr6]·3CH3CN (3b), [CuII(L˙)2][ReIVCl6] (4a) and [CuII(L˙)2][ReIVBr6] (4b), where L˙ is the aminoxyl radical chelating ligand, 4,4′-dimethyl-2,2′-di(2-pyridyl)oxazolidine-N-oxide, have been synthesised. Structural and magnetic studies reveal metal–radical intramolecular antiferromagnetic interactions in the [MII(L˙)2]2+ cations in the iron, cobalt and copper based compounds (1a, 1b, 2a, 2b, 4a and 4b) with the central metal ion low-spin in the case of iron (1a and 1b) and a gradual, cobalt based, spin-crossover transition present in 2a and 2b. The nickel based compounds, 3a and 3b, were analysed in the dried form (3a(dried) and 3b(dried)) and directly in acetonitrile (3a(solvated) and 3b(solvated)). Microanalysis and IR spectroscopy on 3a(dried) and 3b(dried) suggest that the dried samples are best formulated as [NiII(L˙)(H2O)3][ReIVX6], where X = Cl (3a(dried)) and Br (3b(dried)). All forms of 3a and 3b exhibit cationic metal–radical ferromagnetic interactions resulting in S = 3/2 ground states. In addition, 3a(dried) exhibits spin-canting behaviour with an ordering temperature of 2.7 K, an open hysteresis loop with a coercive field Hc = 580 Oe, and a remanent magnetisation Mr = 0.21μB, resulting in a canting angle of ∼1.8°. In contrast, 3b(dried) shows no spin-canting behaviour; a maximum in χMvs. T at T = 3 K suggesting long-range antiferromagnetic ordering. 3a(solvated) and 3b(solvated) show no indication of long-range magnetic ordering, unlike 4a and 4b where anomalies are evident in the low-temperature magnetic susceptibility measurements.
Introduction
Since the initial discovery of a dodecanuclear Mn molecule exhibiting slow relaxation of its magnetisation in zero field at low temperature, magneto-structural studies of discrete paramagnetic systems have grown exponentially.1–3 This research encompasses a wide range of magnetic materials including organic radicals, mono- and multinuclear homometallic transition metal or lanthanide based complexes, as well as heterometallic 3d/4f systems.3–6 The energy barrier for magnetisation reversal in such systems depends on several factors, but in all cases magnetic anisotropy is a key parameter. This has led to increased interest in the giant magnetic anisotropy offered by certain 4f/5f ions with unquenched orbital angular momenta, and selected 4d/5d metal ions possessing considerable spin–orbit coupling.5,7–11The 5d3 ReIV ion is characterised by large magnetic anisotropy originating from second order spin–orbit coupling, with λ ≈ 1000 cm−1 for the free ion, often resulting in large values of the axial zero field splitting parameter, D.12,13 In addition, the diffuse nature of the 5d orbitals gives rise to significant spin delocalisation onto the ligand atoms directly bonded to it, leading to non-negligible intermolecular magnetic exchange interactions, commonly mediated by Re–X⋯X–Re![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14–19 or Re–X⋯H2O⋯X–Re contacts.20 Extensive research on the hexahalorhenate moiety [ReIVX6]2− (X = F, Cl, Br or I) has shown that the magnetic behaviour of the anion in the solid state depends greatly on the nature of the cation employed. The latter include organic radicals,21 alkali metals,22 ferrocenium,23 and the Single-Molecule Magnet (SMM) ‘Mn6’.24,25 For example, research conducted on the [ReIVI6]2− anion with alkali metal cations, Li+ to Cs+, illustrated the effect of cation size on the intermolecular Re–I⋯I–Re interaction, where it was found that the magnetic ordering temperature increased with decreasing cation size,22 whilst replacing the perchlorate counter ions with [ReIVCl6]2− in the Mn626–34 SMM led to the energy barrier for magnetisation relaxation increasing by 30%.24
14–19 or Re–X⋯H2O⋯X–Re contacts.20 Extensive research on the hexahalorhenate moiety [ReIVX6]2− (X = F, Cl, Br or I) has shown that the magnetic behaviour of the anion in the solid state depends greatly on the nature of the cation employed. The latter include organic radicals,21 alkali metals,22 ferrocenium,23 and the Single-Molecule Magnet (SMM) ‘Mn6’.24,25 For example, research conducted on the [ReIVI6]2− anion with alkali metal cations, Li+ to Cs+, illustrated the effect of cation size on the intermolecular Re–I⋯I–Re interaction, where it was found that the magnetic ordering temperature increased with decreasing cation size,22 whilst replacing the perchlorate counter ions with [ReIVCl6]2− in the Mn626–34 SMM led to the energy barrier for magnetisation relaxation increasing by 30%.24
Investigations into the exchange interactions and magnetic properties present in molecule-based magnets containing a coordinated radical, through the ‘metal–radical approach’, were instigated in the late 1980s and early 1990s.35,36 This focused initially on the use of nitroxide-based radicals, resulting in the discovery of the first Single-Chain Magnet (SCM), [CoII(hfac)2(NITPhOMe)], and the ferrimagnetically ordered system {MnII(hfac)2(L)}n37,38 (hfac = hexafluoro-acetylacetonate, NITPhOMe = 4′-methoxy-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide and L = 1,3,5-tris{p-(N-oxy-N-tert-butylamino)phenyl}benzene). Here, the hfac co-ligand was used to increase the Lewis acidity of the central metal ion, enabling the nitroxide N–O moiety, a weak Lewis base, to coordinate directly to the metal.
A second approach for coordinating a radical based group directly to the metal ion is to create ligands combining a radical species in close proximity to a conventional ligating group such as bipyridine, imidazole or pyridine.39–41 The [MII(L˙)2]2+ unit, MII = Mn, Fe, Co, Cu and Zn, with L˙ = 4,4′-dimethyl-2,2′-di(2-pyridyl)oxazolidine-N-oxide (Scheme 1), has been studied over the past decade leading to the discovery of interesting magnetic phenomena such as spin-crossover, ferromagnetic exchange and reductively induced oxidation.41–46 Herein we report an extension to these studies with the synthesis and magnetic characterisation of a series of coordination compounds containing both the [MII(L˙)2]2+ cation and the [ReIVX6]2− anion. When the transition metal in the [MII(L˙)2]2+ cation is Fe, Co or Cu and used in combination with [ReIVCl6]2− and [ReIVBr6]2− (1a, 1b, 2a, 2b, 4a and 4b) we observe predominantly antiferromagnetic metal–radical exchange interactions and typical [ReIVX6]2− anion behaviour, with a gradual spin-crossover transition present in 2a and 2b. Using Ni yields the crystalline products [NiII(L˙)(CH3CN)3][ReIVCl6]·CH3CN (3a) and [NiII(L˙)(CH3CN)3][ReIVBr6]·3CH3CN (3b), which exhibit cationic metal–radical ferromagnetic interactions, and intermolecular antiferromagnetic interactions (3b(dried)) or spin-canting (3a(dried)).
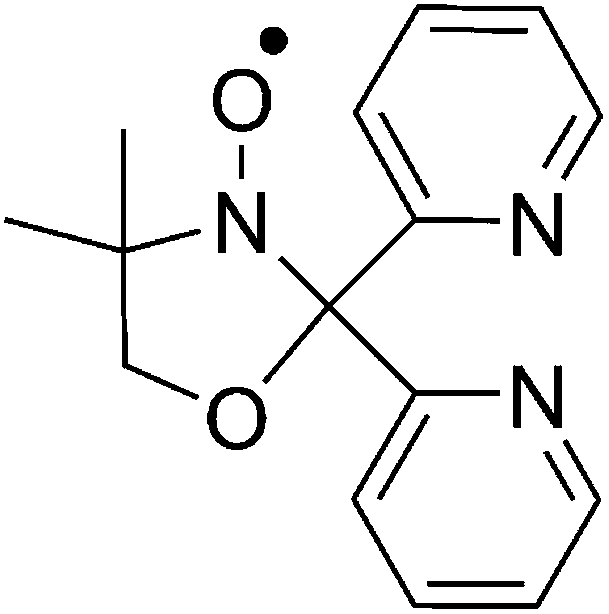 | ||
| Scheme 1 Molecular structure of 4,4′-dimethyl-2,2′-di(2-pyridyl)oxazolidine-N-oxide, L˙. | ||
Experimental
Materials and methods
All chemicals were used as received. Syntheses were carried out under aerobic conditions using CH3CN dried over 3 Å molecular sieves. (NBu4)2[ReIVCl6] and (NBu4)2[ReIVBr6] were prepared as described previously.19,47 The neutral radical ligand, 4,4′-dimethyl-2,2′-di(2-pyridyl)oxazolidine-N-oxide (L˙), was prepared as per literature methods.42 Crystals of all eight compounds were collected and left open to air for use in further analysis. Elemental analyses (C, H, N) were performed by MEDAC Ltd. Direct current (dc) magnetic susceptibility measurements on all eight compounds were collected on a Quantum Design MPMS-XL SQUID magnetometer equipped with a 7 T dc magnet in the temperature range of 2–300 K, under an applied field of 0.1 T. Crystalline samples were powdered, dried, and restrained in gelatine capsules for measurements. Magnetic susceptibility measurements were also carried out on crystalline samples of 3a and 3b immersed directly in acetonitrile from 200–2 K under an applied field of 0.1 T (3a(solvated) and 3b(solvated)). Diamagnetic corrections were applied using Pascal's constants.48 The formula and subsequent molecular weight used in the magnetic measurements were determined from the microanalysis of dried samples. Infrared (IR) spectra from 3800 to 600 cm−1 were recorded on a PerkinElmer Spectrum 65 ATR-IR spectrometer. Powder X-ray diffraction measurements were carried out on a Rigaku Oxford Diffraction SuperNova X-ray diffractometer at 298 K using a scan step size of 0.086° at 1° s−1. Calculated patterns were obtained using Mercury 3.7.Crystallography
Data were measured on a Rigaku Oxford Diffraction SuperNova (1a, 1b, 2a, 2a·150 K, 2a·200 K, 2a·250 K, 2b, 3b, 4a, and 4b) and Rigaku Oxford Diffraction XCalibur (3a) X-ray diffractometers using Mo-Kα (1a, 1b, 2a, 2a·150 K, 3a, 3b and 4b) or Cu-Kα (2b and 4a) radiation. The crystal temperature was maintained at 120 K using an Oxford Cryosystems Cryostream 700+ low temperature device for all eight complexes. CrysAlisPro was used for diffractometer control and data processing. Structures were solved with olex2.solve (1a, 1b, 2a, 2a·150 K, 2a·200 K, 2a·200 K, 2b, 3b, 4a and 4b)49 or ShelXS (3a)50 and refined by full-matrix least-squares on F-squared using ShelXL, interfaced through Olex2.51 In 1a C12 is disordered over two positions with partial occupancies of 0.56 and 0.44. In 2a the nitrogen atom associated with N4 on the acetonitrile solvate is disordered over two positions with partial occupancies of 0.45 and 0.55. In 4a C12 and C14 are disordered over two positions with partial occupancies of 0.49 and 0.51, and 0.53 and 0.47. All non-hydrogen atoms were refined anisotropically and hydrogens placed at calculated positions. Crystallographic data, selected bond lengths and angles can be found in Tables S1–S7 and Fig. S1 in the ESI.†CCDC 1534665–1534675.
Synthesis
Results and discussion
Structural studies
Complex 1a (Fig. 1) crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with inversion centres on both the ReIV and FeII ions. The asymmetric unit contains one half of the [FeII(L˙)2]2+ cation and one half of the [ReIVCl6]2− anion. The ReIV ion is in a regular octahedral environment coordinated to six chloride ions, with Re–Cl bond lengths between 2.3547(8) and 2.3748(9) Å, in accordance with previously published compounds containing the [ReIVCl6]2− anion (Table S4†).21,52 Two neutral radical ligands are coordinated facially to the FeII ion, bonding in an η3-fashion through the pyridyl nitrogen atoms and oxygen atoms of the nitroxide (Fig. 1a). This results in a slightly distorted octahedral trans-N4O2 coordination sphere around the central FeII ion. The Fe–N bond distances are 1.984(3) and 1.967(3) Å, and the Fe–O bond is 1.872(2) Å. The cis angles range between 87.60(12) and 92.40(12)°, while the trans angles are generated by the inversion centre resulting in angles of 180°. The nitroxide N–O bond length of 1.323 (3) Å shows the ligand to be in its neutral radical form and not the reduced hydroxylamino anionic form (L−).42 The structural features of the cation are in accordance with previously published results on the [FeII(L˙)2]2+ cation containing a central low-spin FeII ion with both ligands in the neutral radical form.42 Examination of the crystal packing reveals alternating layers of [FeII(L˙)2]2+ cations and [ReIVCl6]2− anions in the crystallographic ac-plane, as seen in Fig. 1b. In the plane of the cations, short CH⋯π interactions force the [FeII(L˙)2]2+ moieties to pack in chains along the crystallographic a-axis. The CH⋯π distances range from 3.5 to 3.8 Å (Fig. S4 and Table S8†) between the methyl group on the oxazolidine ring and the centroid of the pyridyl groups. The [ReIVCl6]2− ions are surrounded by four cations and are thus well isolated from each other, with the shortest Cl⋯Cl distance being 4.7 Å. The [ReIVCl6]2− anion interacts with two cations from opposite layers via Cl⋯π interactions of 3.7 Å (Fig. S4†), creating a 1-D network with alternating [ReIVCl6]2− and [FeII(L˙)2]2+ ions running diagonally in the unit cell (Fig. 1b, red lines).
with inversion centres on both the ReIV and FeII ions. The asymmetric unit contains one half of the [FeII(L˙)2]2+ cation and one half of the [ReIVCl6]2− anion. The ReIV ion is in a regular octahedral environment coordinated to six chloride ions, with Re–Cl bond lengths between 2.3547(8) and 2.3748(9) Å, in accordance with previously published compounds containing the [ReIVCl6]2− anion (Table S4†).21,52 Two neutral radical ligands are coordinated facially to the FeII ion, bonding in an η3-fashion through the pyridyl nitrogen atoms and oxygen atoms of the nitroxide (Fig. 1a). This results in a slightly distorted octahedral trans-N4O2 coordination sphere around the central FeII ion. The Fe–N bond distances are 1.984(3) and 1.967(3) Å, and the Fe–O bond is 1.872(2) Å. The cis angles range between 87.60(12) and 92.40(12)°, while the trans angles are generated by the inversion centre resulting in angles of 180°. The nitroxide N–O bond length of 1.323 (3) Å shows the ligand to be in its neutral radical form and not the reduced hydroxylamino anionic form (L−).42 The structural features of the cation are in accordance with previously published results on the [FeII(L˙)2]2+ cation containing a central low-spin FeII ion with both ligands in the neutral radical form.42 Examination of the crystal packing reveals alternating layers of [FeII(L˙)2]2+ cations and [ReIVCl6]2− anions in the crystallographic ac-plane, as seen in Fig. 1b. In the plane of the cations, short CH⋯π interactions force the [FeII(L˙)2]2+ moieties to pack in chains along the crystallographic a-axis. The CH⋯π distances range from 3.5 to 3.8 Å (Fig. S4 and Table S8†) between the methyl group on the oxazolidine ring and the centroid of the pyridyl groups. The [ReIVCl6]2− ions are surrounded by four cations and are thus well isolated from each other, with the shortest Cl⋯Cl distance being 4.7 Å. The [ReIVCl6]2− anion interacts with two cations from opposite layers via Cl⋯π interactions of 3.7 Å (Fig. S4†), creating a 1-D network with alternating [ReIVCl6]2− and [FeII(L˙)2]2+ ions running diagonally in the unit cell (Fig. 1b, red lines).
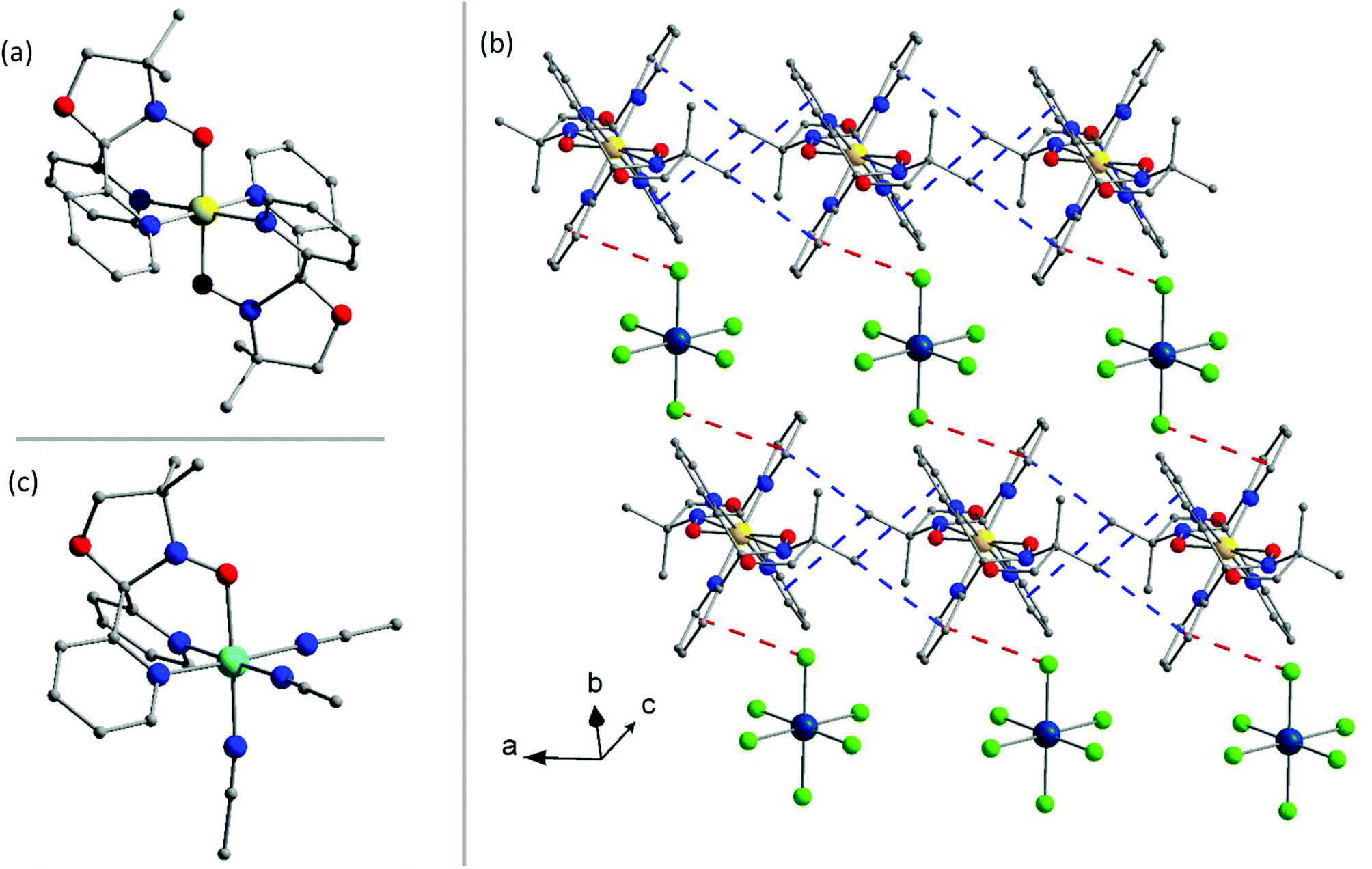 | ||
| Fig. 1 (a) The structure of the [FeII(L˙)2]2+ cation in 1a. (b) The crystal packing of 1a. (c) The [NiII(L˙)(CH3CN)3]2+ cation present in 3a and 3b. Colour code: Re, dark blue; Ni, cyan; Fe, yellow; Cl, green; O, red; N, blue; C, grey. Hydrogen atoms are omitted for clarity. CH⋯π and Cl⋯π interactions are indicated by blue and red lines, respectively. | ||
Complex 1b crystallises in the triclinic space group P and possesses a structure that resembles 1a (Fig. S5†); the most obvious and significant difference being the presence of the hexabromorhenate(IV) anion instead of the hexachlororhenate(IV) ion. Inversion centres are located on the FeII and ReIV ions with the asymmetric unit containing one half of the [FeII(L˙)2]2+ cation and one half of the [ReIVBr6]2− anion. The ReIV ion is coordinated to six bromide ions in an octahedral geometry with Re–Br bond lengths of 2.5029(6), 2.5249(6) and 2.5132(6) Å, similar to that seen in previously published reports on the [ReIVBr6]2− anion.25,53 The bond lengths and angles of the [FeII(L˙)2]2+ unit are similar to the cation in 1a (Table S4†) suggesting the presence of a central low-spin FeII ion with both ligands in their neutral radical (L˙) form. The packing of 1b in the crystal is analogous to 1a, with minor deviations originating from the larger [ReIVBr6]2− ion. The anions are well isolated from each other, as in 1a, with the shortest Br⋯Br distance being 4.6 Å along the crystallographic a-axis. The shortest CH⋯π interaction between the cations is approximately 3.6 Å, and the Br⋯π interactions between the [FeII(L˙)2]2+ cations and the [ReIVBr6]2− anions are of the order of 3.8 Å (Fig. S5 and Table S8†).
Complex 2a has similar structural features to 1a in that it crystallises in the space group P with inversion centres located on both the CoII and ReIV ions, and the asymmetric unit contains one half of the [CoII(L˙)2]2+ cation and one half of the [ReIVCl6]2− anion along with a disordered acetonitrile solvate molecule. This results in an overall formula of [CoII(L˙)2][ReIVCl6]·2CH3CN (2a). The bond lengths and bond angles of the [ReIVCl6]2− ion are similar to the anion described in 1a (Table S4†). The [CoII(L˙)2]2+ is structurally similar to the cation described in 1a (Fig. 1a). The CoII ion is in a regular, trans-N4O2 octahedral geometry, coordinated to two L˙ ligands. The Co–N bond lengths are 1.9251(11) and 1.9556(10) Å, and the Co–O bond length is 1.9103(10) Å. The cis bond angles are 85.76(4)–94.24(4)° and the trans angles all 180°. The nitroxide N–O bond on the ligand is 1.3137(16) Å consistent with the ligand in the neutral radical form (L˙).42 The packing of 2a in the crystal is only slightly different from that observed in 1a. The acetonitrile solvent molecules lie in the plane of the anions leading to a different orientation of the [ReIVCl6]2− unit, with the shortest Cl⋯Cl interaction being 3.8 Å. No Cl⋯π interactions are observed between the [CoII(L˙)2]2+ and [ReIVCl6]2− ions. The cations pack as described in 1a, with the shortest CH⋯π interactions approximately 3.4 Å in length (Fig. S6 and Table S8†).
The bond lengths in the cation in 2a are shorter than that expected for a low-spin CoII ion with an axially elongated pseudo-Jahn–Teller distortion and clearly do not correspond to that of a high-spin CoII ion. To clarify the unusual bond lengths contained in the [CoII(L˙)2]2+ cation in 2a, a new set of crystals were grown and X-ray crystallography measurements undertaken on the same single crystal at 150 (2a·150 K), 200 (2a·200 K) and 250 K (2a·250 K) (Tables S1, S2 and S4, Fig. S7†). These, however, all crystallised in the monoclinic space group, P21/c, with an overall formula of [CoII(L˙)2][ReIVCl6]·4CH3CN in contrast to the initial crystal structure which crystallised in the triclinic space group, P, and formulated as [CoII(L˙)2][ReIVCl6]·2CH3CN (2a). An identical synthetic route and crystallising conditions were used for both solvates so we can only conclude that it is very sensitive to external perturbations such as temperature and humidity. Both solvates contain the [CoII(L˙)2]2+ cation and the [ReIVCl6]2− anion and differ only in their intermolecular arrangement in the solid state driven mainly by the differing degrees of solvation.
The variable temperature study on 2a·150 K, 2a·200 K and 2a·250 K showed evidence of spin-crossover behaviour: an axially elongated pseudo-Jahn–Teller distortion of the low-spin CoII ion was observed at 150 K which diminished as the temperature was increased (Table S4 and Fig. S7†). This suggests a gradual, thermally induced, spin transition between the low-spin and high-spin states of the central CoII ion. A comparison of the bond lengths in 2a, 2a·150 K, 2a·200 K and 2a·250 K with similar species can be found in Table S9.†
The bond lengths and angles of the [ReIVCl6]2− anion in 2a·150 K, 2a·200 K, 2a·250 K are similar to those described in 1a and 2a. The crystal packing is similar in 2a·150 K, 2a·200 K and 2a·250 K with alternate layers of the [CoII(L˙)2]2+ cations and the [ReIVCl6]2− anions (Fig. S8†). There is no indication of any significant CH⋯π or Cl⋯Cl intermolecular interactions and the shortest Cl⋯Cl distance is 6.455 Å in 2a·150 K, 6.572 Å in 2a·200 K and 6.715 Å in 2a·250 K (Table S8†). A comparison of the intermolecular packing in both solvates and their relation to any magnetic properties (vide infra) is rendered moot by the fact that the microanalysis of both solvates suggests that they coalesce upon drying into the same material, namely, [CoII(L˙)2][ReIVCl6].
Complex 2b (Fig. S9†) crystallises in the triclinic space group P with inversion centres located on both the CoII and ReIV ions; subsequently the asymmetric unit contains one half of the [CoII(L˙)2]2+ cation and one half of the [ReIVBr6]2− anion. The geometrical parameters of the [ReIVBr6]2− anion are analogous to that described in 1b (Table S5†). The CoII ion is in a distorted, trans-N4O2 octahedral geometry, coordinated to two L˙ ligands. The Co–N bond lengths are 1.963(4) and 1.976(4) Å, and the Co–O bond length is 2.092(4) Å. The cis bond angles range from 87.98(17)–92.02(17)° and the trans angles all 180°. The N–O bond on the ligand is 1.269(6) Å which confirms the neutral radical (L˙) form of the ligand. Bond lengths and angles suggest a central low-spin CoII ion showing an axially elongated pseudo Jahn–Teller distortion, consistent with previously reported studies on the [CoII(L˙)2]2+ cation (Table S9†).43 The packing of [CoII(L˙)2][ReIVBr6] in the crystal is identical to that described for 1b. The shortest CH⋯π interactions between the cations are ca. 3.6 and 3.7 Å, and the Br⋯Br interactions approximately 4.6 Å. The Br⋯π interactions between the [CoII(L˙)2]2+ cations and the [ReIVBr6]2− ions are 3.8 Å (Fig. S9 and Table S8†).
Complex 3a (Fig. 2) crystallises in the orthorhombic space group Pbca with the asymmetric unit containing a single [NiII(L˙)(CH3CN)3]2+ cation, the [ReIVCl6]2− anion and one acetonitrile solvate molecule. The geometrical parameters of the [ReIVCl6]2− ion are similar to those described for 1a and 2a (Tables S4 and S6†). The NiII ion is atypically coordinated to one tridentate, facially capping L˙ ligand, not two as in the other complexes described in this paper, and all other previously published results (Fig. 1c).41–43,45 The coordination sphere of the NiII ion is completed by three acetonitrile molecules, creating a slightly distorted octahedral environment around the metal ion with cis angles varying in the range 86.1(17)–93.9(17)°, the most acute trans angle being 176.19(18)°. The Ni–N bond lengths are 2.068(4) and 2.058(4) Å, with the Ni–O distance being 2.073(4) Å. The N–O bond length of 1.271 (6) Å shows the ligand again to be in the neutral radical state L˙.42 The acetonitrile molecules are coordinated to the nickel(II) ion with Ni–N bond lengths of 2.071(5), 2.058(5) and 2.037(5) Å, which correlates with the previously published values.54 In the crystal the [ReIVCl6]2− anions are well isolated from each other, being ‘encapsulated’ by four cations, resulting in a short Cl⋯Cl distance of 4.55 Å along the crystallographic a-axis (Fig. 2a). Short CH⋯π interactions of 3.6 and 3.8 Å between the methyl group on the radical ligand and pyridyl rings of the neighbouring cation create a 1-D network travelling along the crystallographic a-axis (Fig. S10 and Table S8†). In the crystallographic ab plane, chains of cations order in layers separated by [ReIVCl6]2− ions (Fig. 2a). The acetonitrile molecule of crystallisation is sandwiched between two [NiII(L˙)(CH3CN)3]2+ cations in the ab plane.
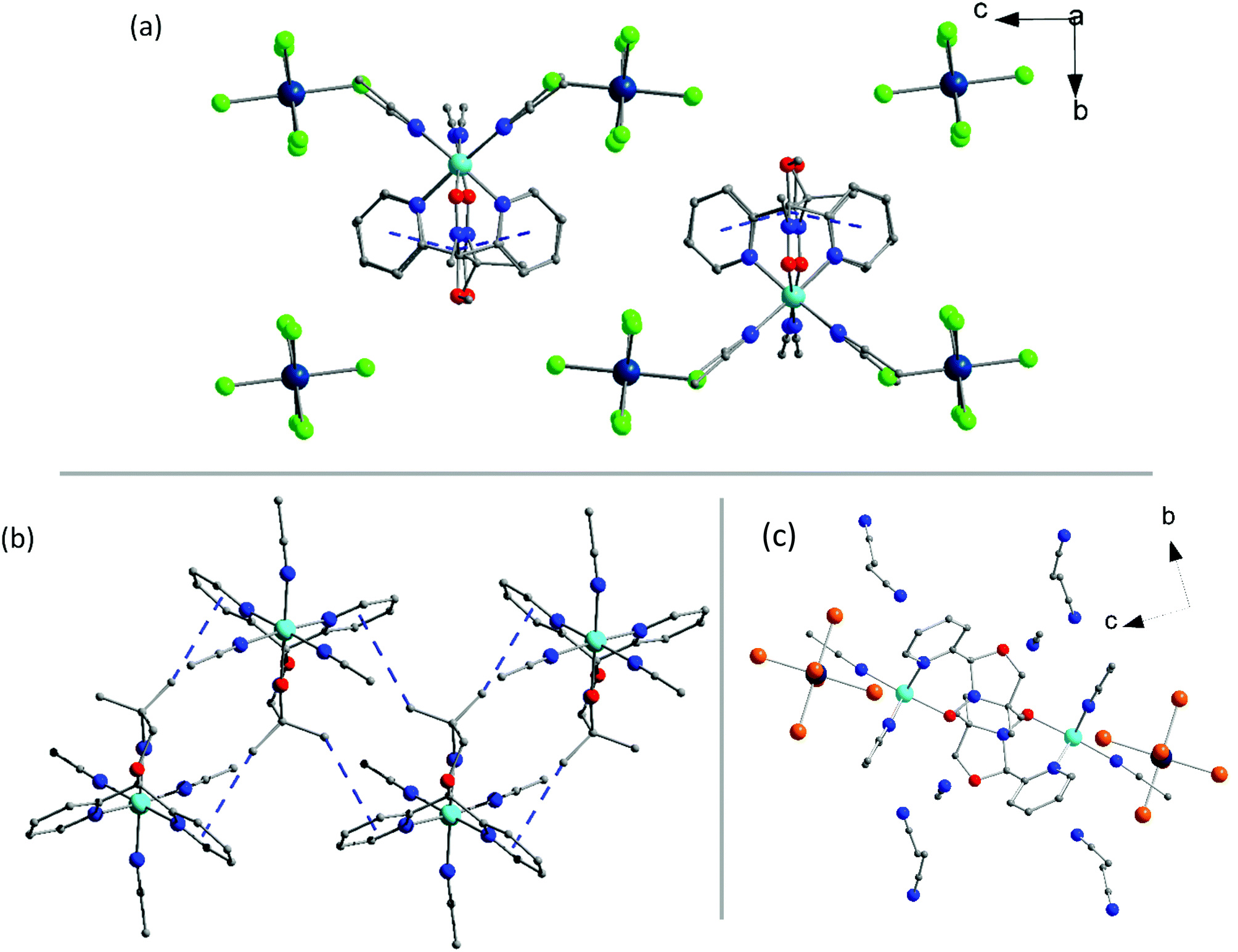 | ||
| Fig. 2 The packing of 3a viewed down the crystallographic a-axis. (b) The intermolecular interactions between the cations in 3b along the crystallographic a-axis. (c) The packing in 3b viewed down the crystallographic a-axis. Colour code: Re, dark blue; Ni, cyan; Br, orange; Cl, green; O, red; N, blue; C, grey. Hydrogen atoms are omitted for clarity. CH⋯π interactions are indicated by blue lines. In (a) and (b) solvent molecules are removed for clarity. | ||
Complex 3b crystallises in the monoclinic space group P21/c. The asymmetric unit contains a [NiII(L˙)(CH3CN)3]2+ cation, one [ReIVBr6]2− anion and three solvent acetonitrile molecules. The structure of the [NiII(L˙)(CH3CN)3]2+ cation is identical to the one presented in 3a, and the [ReIVBr6]2− anion is as described in 1b (Fig. 1c). The sole L˙ ligand on the cation has close contacts through the methyl groups to two other ligands’ pyridyl moieties, creating a 1-D zigzag structure along the crystallographic a-axis, as illustrated in Fig. 2b and S11.† The CH⋯π interactions between the methyl groups and the two nearest cations are 3.5 and 3.8 Å, which imposes an alternating chain motif. In each chain the ligands are facing ‘inwards’ and the coordinated acetonitrile molecules are facing ‘outwards’. The [ReIVBr6]2− ions sit in the plane of the NiII ions between two neighbouring cations. The anions are isolated from each other with a short Br⋯Br distance of 3.9 Å along the direction of the crystallographic c-axis, creating a 1D network (Table S8 and Fig. S11†). The acetonitrile solvent molecules are positioned above and below the plane of the cations (Fig. 2c). Although we have presented structural information on 3b in the monoclinic space group P21/c above, it must be noted that an alternative solvate is also possible containing a single solvate acetonitrile per formula unit. This crystallises in the monoclinic space group C2/c and is formulated as [NiII(L˙)(CH3CN)3][ReIVBr6]·CH3CN. The crystal structures, their intermolecular interactions and subsequent magnetic measurements (vide infra) of both solvates are similar so only that of the P21/c solvate has been reported here.
Complex 4a (Fig. S12†) crystallises in the triclinic space group P with inversion centres located on the CuII and ReIV ions with the asymmetric unit containing one half of the [CuII(L˙)2]2+ cation and one half of the [ReIVCl6]2− anion. The [ReIVCl6]2− anion in 4a is isostructural to the anions described in 1a, 2a and 3a (Tables S4, S6 and S7†) and the [CuII(L˙)2]2+ cation is structurally similar to the cation described in 1a and 2a (Fig. 1a). The CuII ion is six coordinate and in a pseudo-Jahn–Teller distorted octahedral environment, with elongation along the O–Cu–O vector. The Cu–N bond lengths are 2.0135(18) and 2.0116(18) Å, with the Cu–O bond length of 2.3021(16) Å (Table S7†). These values are similar to those observed in a previously published complex.41 The cis angles vary between 86.31(7) and 93.69(7)°, and the trans angles are constrained to 180°. The nitroxide N–O bond length is 1.270(3) Å consistent with that of the ligand in the neutral radical form. The packing in the crystal is identical to that observed in 1a. The shortest CH⋯π interactions between the cations are between 3.5–3.8 Å, the shortest Cl⋯Cl distance is 4.8 Å, and the Cl⋯π interactions between the [CuII(L˙)2]2+ and [ReIVCl6]2− ions are 3.7 Å (Fig. S12 and Table S8†).
Complex 4b crystallises in the triclinic space group P, with a structure similar to that of 4a. The [CuII(L˙)2]2+ moiety is isostructural with the cation described in 4a, with the geometrical parameters of the [ReIVBr6]2− unit being the same as those present in the anion of 1b (Tables S4 and S7†). In the extended structure the packing of [CuII(L˙)2][ReIVBr6] is analogous to that described in 1b. The anions display the shortest Br⋯Br distance of 4.6 Å, with CH⋯π interactions between cations of approximately 3.6 Å. The Br⋯π interactions between the [CuII(L˙)2]2+ cations and [ReIVBr6]2− anions are of the order of 3.7 Å (Fig. S13 and Table S8†).
Magnetic studies
The χMT value for complex 1a (Fig. 3) at T = 300 K is 1.96 cm3 mol−1 K, in the range expected with contributions of ∼1.52–1.69 cm3 mol−1 K from a magnetically isolated [ReIVCl6]2− anion (assuming g = 1.8–1.9 and S = 3/2) and the previously published value of ca. 0.16 cm3 mol−1 K for the [FeII(L˙)2]2+ unit.42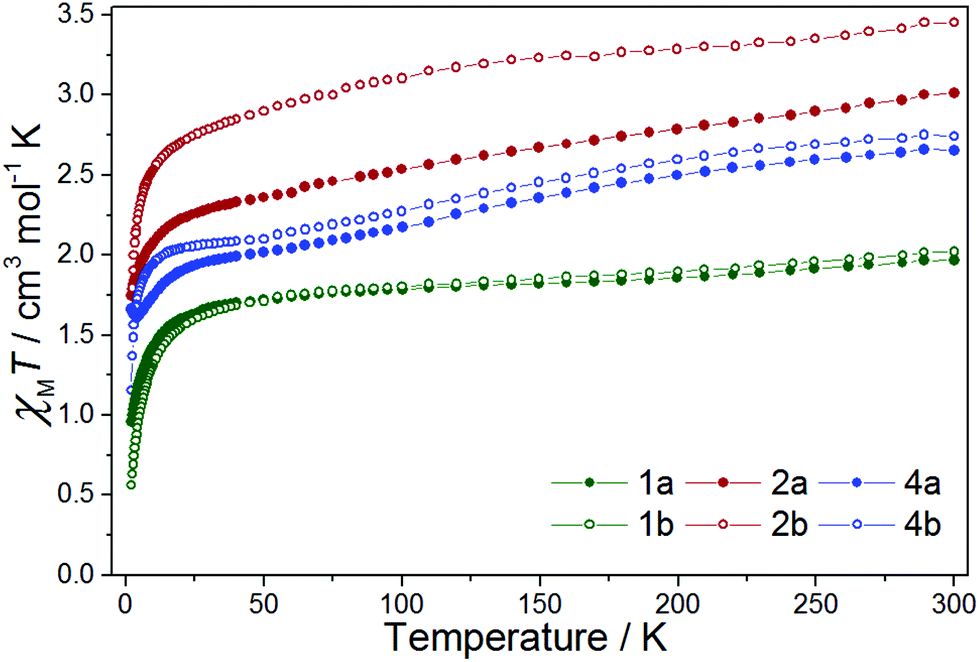 | ||
| Fig. 3 χ M T vs. T for 1a, 1b, 2a, 2b, 4a and 4b in the indicated temperature range, with H = 0.1 T. | ||
The χMT value decreases with decreasing temperature, first to a value of 1.82 cm3 mol−1 K at T = 150 K, then more slowly to a value of 1.70 cm3 mol−1 K at T = 40 K, before plunging rapidly to 0.96 cm3 mol−1 K at T = 2 K. The high temperature decrease in χMT is likely due to a large antiferromagnetic radical–radical coupling in the [FeII(L˙)2]2+ cation, as previously reported for the species [FeII(L˙)2](BF4)2, where Jrad–rad = −315 cm−1 (Ĥ = −2JŜ1Ŝ2).42 The rapid low temperature decrease is attributed to the zero-field splitting (zfs) of the Re(IV) ion, with a value of 0.96 cm3 mol−1 K corresponding to that expected from an isolated [ReIVCl6]2− ion.21 Compound 1b behaves in a largely similar manner, albeit with a smaller value of χMT (0.56 cm3 mol−1 K) at T = 2 K.12χMversus T data for 1a and 1b, shown in Fig. S14,† show no evidence of any significant intermolecular interactions.
For complex 2a the χMT value at T = 300 K (3.01 cm3 mol−1 K) correlates with that expected from the sum of the previously published results for the [CoII(L˙)2]2+ and [ReIVCl6]2− ions (Fig. 3).21,43,45 In the temperature range from T = 300 to 30 K, the χMT value decreases linearly, after which there is a sharp decline to a minimum value of 1.75 cm3 mol−1 K at T = 2 K. Variable temperature structural studies, carried out on solvated samples, reveal the CoII ion to be undergoing a gradual spin-crossover transition (Fig. S7†). It is likely, therefore, that the decrease in the χMT product from T = 300 to 30 K is due to a combination of a spin-crossover transition and antiferromagnetic exchange interactions between the radical ligands and the central CoII ion, as seen previously.43 The sharp low temperature downturn below T = 30 K is again assigned to zero-field splitting – but here from both ReIV and CoII. There is no evidence of any significant intermolecular contributions to the susceptibility data at low temperatures (Fig. S15†). Complex 2b behaves in a similar fashion.
Crystals of complex 3a and 3b were prone to solvent loss and therefore measured in both their dried (3a(dried), 3b(dried)) and fully solvated (3a(solvated), 3b(solvated)) forms (Fig. 4). The former consisted of crystals that had been left to air dry in an open vial for up to five days, during which time they changed colour from brown to green (3a(dried)), and from brown to yellow (3b(dried)). The fully solvated forms were measured on freshly prepared samples suspended in acetonitrile. X-ray powder diffraction data obtained on the fully solvated samples agree with the calculated patterns obtained from the relevant crystal structures, confirming phase purity (Fig. S16†). PRXD patterns of the dried samples indicate that these are in a different crystallographic phase to that present in the crystal structures (Fig. S17†). Microanalyses and IR spectra (Fig. S2†) of air dried crystals of complex 3a(dried) suggest the formulation as [NiII(L˙)(H2O)3][ReIVCl6] and not [NiII(L˙)(CH3CN)3][ReIVCl6]·CH3CN. This is perhaps unsurprising given the labile nature of coordinated acetonitrile and the fact that the samples were left open to air before analysis. The loss of coordinated acetonitrile has been seen previously in the octahedral nickel complexes [TpmMe,MeNi(CH3CN)3](BF4)255 and [TpR2Ni(CH3CN)3]OTf,56 where TpmMe,Me = tris(3,5-dimethylpyrazol-1-yl)methane and TpR2 = hydrotrispyrazolyl borato with R = 3,5-iPr2. This is in contrast to previously published members of this family,42–46 and the majority of previously published complexes of octahedral NiII containing three coordinated acetonitrile molecules which are stable to desolvation (Table S10†).
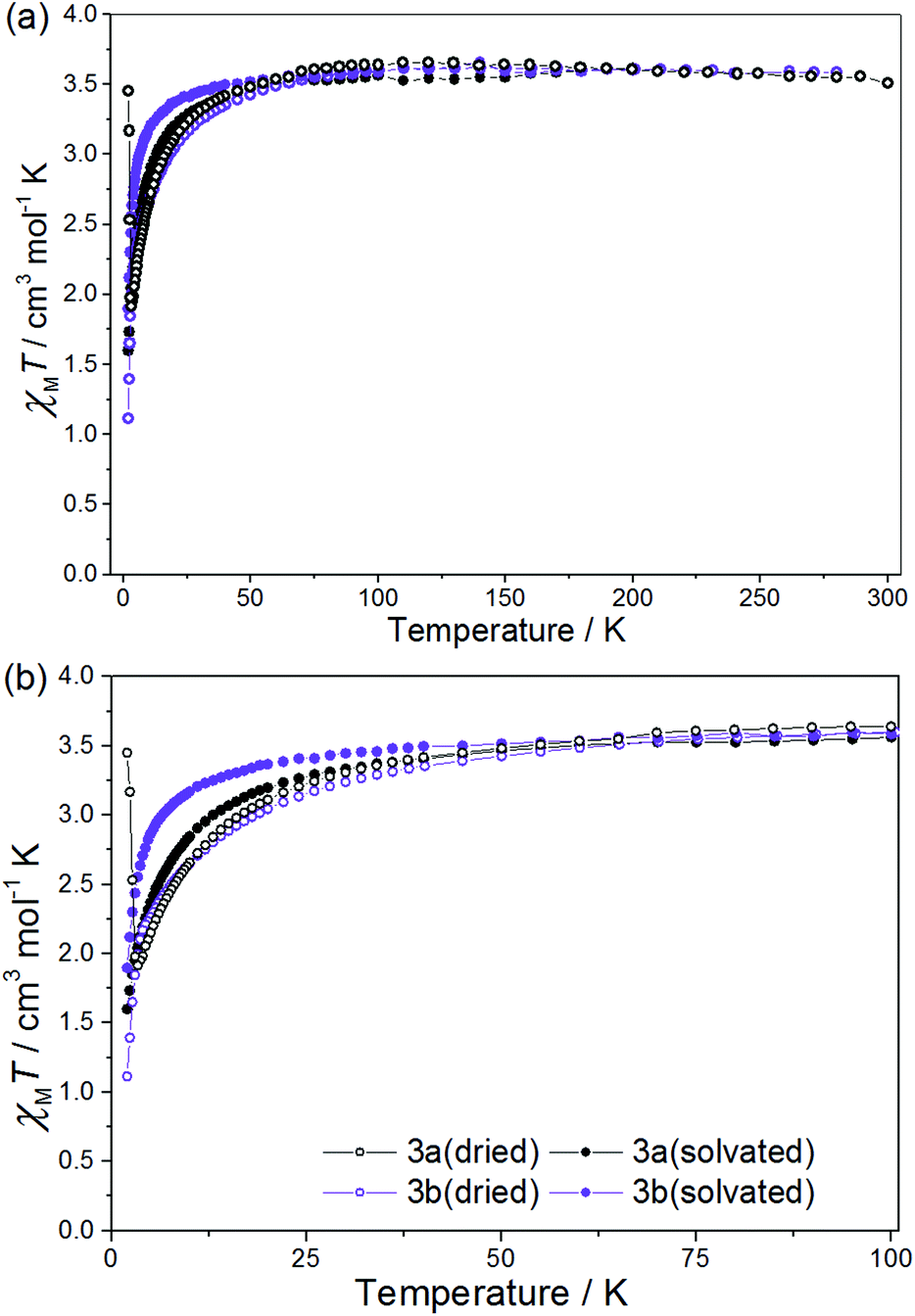 | ||
| Fig. 4 χ M T vs. T for 3a(dried), 3a(solvated), 3b(dried) and 3b(solvated) between T = 300–2 K (a) and 100–2 K (b), in a field of H = 0.1 T. | ||
For 3a(dried) the χMT value at T = 300 K is 3.51 cm3 mol−1 K, greater than the expected value for the sum of an isolated [ReIVCl6]2− anion (1.52–1.69 cm3 mol−1 K), an octahedral NiII ion (1.0–1.2 cm3 mol−1 K) and a neutral radical ligand (0.375 cm3 mol−1 K; Fig. 4). Upon cooling the χMT value gradually increases to a maximum value of 3.66 cm3 mol−1 K at T = 110 K, before decreasing gradually to 50 K and then more abruptly to a minimum value of 1.92 cm3 mol−1 K at T = 3.30 K. The high temperature value of χMT and the initial increase in its magnitude with decreasing temperature is suggestive of the presence of a ferromagnetic exchange interaction between the central NiII ion and the coordinated radical ligand. A ferromagnetic CoII–radical exchange interaction (J = 63.8 cm−1) was reported previously in the species [CoII(L˙)2](NO3)2, but this is the first reported instance of any interaction between a NiII ion and the neutral radical form of the ligand used here.43 The abrupt decrease at lower temperatures is again apportioned to the zfs of the NiII/ReIV ions. At the very lowest temperatures measured (T = 3.30–2.00 K) the χMT value increases once more, reaching a value of 3.45 cm3 mol−1 K (Fig. 4 and 5). Further measurements reveal magnetic ordering with a peak in the zero-field cooled–field-cooled (ZFC–FC) data at T = 2.4 K (Fig. 5b). Susceptibility measurements at T = 1.8 K reveal a field-dependent increase in χM (Fig. 5a), and a frequency-independent peak at T = 2.7 K in out-of-phase ac susceptibility (χ′′) measurements. Magnetisation versus field data (inset of Fig. 5a) taken at T = 1.8 K and H = +3 ↔ −3 kOe show an open hysteresis loop with a coercive field Hc = 580 Oe, and a remanent magnetisation Mr = 0.21μB. This behaviour is indicative of spin-canting. The canting angle can be deduced from sin(α) = Mc/Ms, where α is the canting angle, Mc is the canting magnetisation induced by a weak field, and Ms is the magnetic saturation value.57,58 From the FC measurement Mc = 0.181μB and Ms = 5.8μB, resulting in a canting angle α = 1.8°, similar to the previously published values on systems containing the ReIV ion.22,57,58 Complexes 3a(solvated), 3b(dried) (which were analysed as the hydrated analogue [NiII(L˙)(H2O)3][ReIVBr6]) and 3b(solvated) all behave in a similar manner to 3a(dried), but with no evidence of any significant intermolecular interactions/spin canting at low temperature. The origin of these differences is clearly the different packing effects/intermolecular interactions in the extended structures.
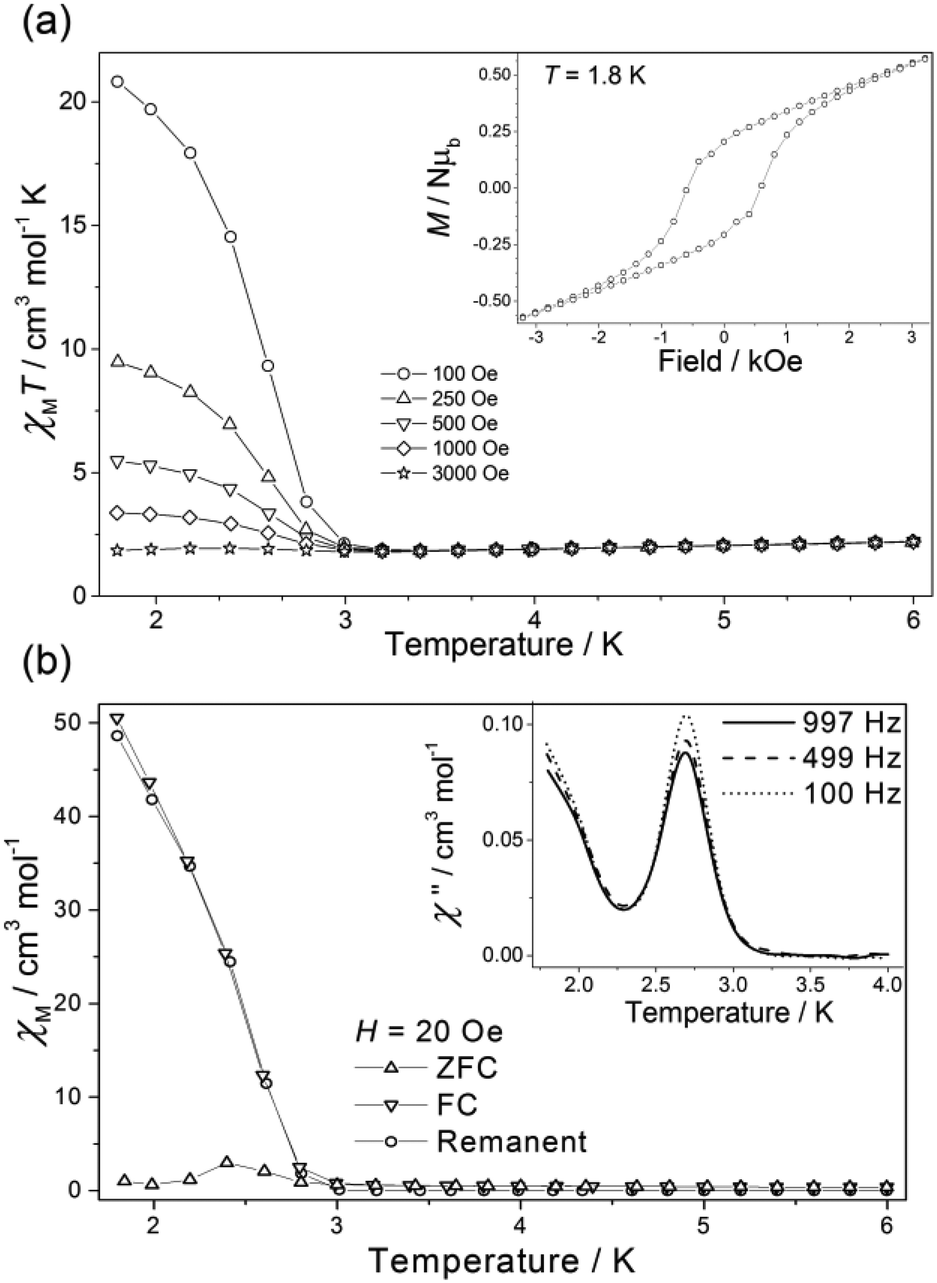 | ||
| Fig. 5 (a) Susceptibility (as χMT vs. T) of 3a(dried) measured in the indicated field and temperature ranges. The inset illustrates the open hysteresis loop at T = 1.8 K. (b) Zero-field-cooled (ZFC), field-cooled (FC) and remanent susceptibility measurement of 3a(dried). The inset shows the out-of-phase ac signal at three frequencies. | ||
The χMT value for complex 4a at 300 K is 2.66 cm3 mol−1 K (Fig. 3) in agreement with the previously published values for the sum of the [CuII(L˙)2]2+ cation and the [ReIVCl6]2− ion (vide supra).21,23,41 As the sample is cooled, the χMT value decreases gradually to ca. T = 50 K where it plateaus at a value of 2.01 cm3 mol−1 K. Upon further cooling the χMT value decreases rapidly, reaching a minimum of 1.62 cm3 mol−1 K at T = 4 K, before increasing slightly between T = 4–2 K. The high temperature decrease stems from a substantial antiferromagnetic coupling between the radical ligands and the CuII ion in the [CuII(L˙)2]2+ cation; a previously reported value of JCu–rad = −81.6 cm−1 (Ĥ = −2JŜ1Ŝ2) resulting in a fully populated S = 1/2 state at T = 50 K.41 This is corroborated by the χMT value of 2.01 cm3 mol−1 K at T = 50 K for 4a which matches the χMT value expected for an isolated [ReIVCl6]2− anion (1.52–1.69 cm3 mol−1 K) and an isolated S = 1/2 [CuII(L˙)2]2+ cation (0.375 cm3 mol−1 K). The behaviour of 4b is analogous to that of 4a, with the only difference occurring in the very low temperature region (T < 4 K) where the χMT product of 4b continues to decrease with decreasing temperature. The origin of this difference may be the presence of very weak ferro/antiferromagnetic intermolecular interactions in 4a/4b, respectively. Further studies would require collection of magnetic data at T < 2 K, which is beyond the scope of this paper.
Conclusions
We have reported the synthesis and magnetic characterisation of a series of coordination compounds containing both the [MII(L˙)2]2+ cation and the [ReIVX6]2− anion, where L˙ is the aminoxyl radical chelating ligand, 4,4′-dimethyl-2,2′-di(2-pyridyl)oxazolidine-N-oxide, the first examples of salts of this cation with any 5d complex anion. Complexes 1a, 1b, 2a and 2b show antiferromagnetic metal–radical exchange interactions, with the data for 2a and 2b suggesting a gradual, thermally induced spin-crossover transition at the CoII centres. The NiII containing cations in compounds 3a and 3b contain one tridentate neutral radical ligand and three coordinated acetonitrile molecules. Upon drying, the coordinated acetonitrile molecules dissociate and are replaced by atmospheric water. Subsequent magnetic susceptibility measurements on both dried (3a(dried), 3b(dried)) and solvated samples (3a(solvated), 3b(solvated)) revealed nickel–radical ferromagnetic exchange coupling. 3a(dried) exhibits spin-canting behaviour with an ordering temperature of T = 2.7 K and an open hysteresis loop with a coercive field Hc = 580 Oe, remanent magnetisation Mr = 0.21μB and a spin-canting angle, α = 1.8°. 4a and 4b show antiferromagnetic metal–radical exchange interactions, with no significant interactions with the [ReIVX6]2− ions in the extended structure. The synthesis of compounds 1–4 hints at the possibility of creating a very large family of magnetic salts of the type [MII(L˙)2][M′X6] whereby the anionic moiety can be changed at will for a variety of species whose magnetic behaviour will depend on the structural relationship between the cation and anion.Acknowledgements
EKB thanks the EPSRC for funding. JML thanks the Spanish Ministry of Economy and Competitiveness (MINECO) for projects CTQ2016-75068P and MDM-2015-0538 (Excellence Unit “María de Maeztu”).References
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- G. A. Craig and M. Murrie, Chem. Soc. Rev., 2015, 44, 2135–2147 RSC.
- S. Demir, I.-R. Jeon, J. R. Long and T. D. Harris, Coord. Chem. Rev., 2015, 289–290, 149–176 CrossRef CAS.
- K. S. Pedersen, J. Bendix and R. Clérac, Chem. Commun., 2014, 50, 4396–4415 RSC.
- X.-Y. Wang, C. Avendano and K. R. Dunbar, Chem. Soc. Rev., 2011, 40, 3213–3238 RSC.
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC.
- R. J. Blagg, L. Ungur, F. Tuna, J. Speak, P. Comar, D. Collison, W. Wernsdorfer, E. J. L. McInnes, L. F. Chibotaru and R. E. P. Winpenny, Nat. Chem., 2013, 5, 673–678 CrossRef CAS PubMed.
- M. A. Antunes, L. C. J. Pereira, I. C. Santos, M. Mazzanti, J. Marçalo and M. Almeida, Inorg. Chem., 2011, 50, 9915–9917 CrossRef CAS PubMed.
- K. R. Dunbar, E. J. Schelter, A. V. Palii, S. M. Ostrovsky, V. Y. Mirovitskii, J. M. Hudson, M. A. Omary, S. I. Klokishner and B. S. Tsukerblat, J. Phys. Chem. A, 2003, 107, 11102–11111 CrossRef CAS.
- J. Martínez-Lillo, D. Armentano, G. De Munno, W. Wernsdorfer, M. Julve, F. Lloret and J. Faus, J. Am. Chem. Soc., 2006, 128, 14218–14219 CrossRef PubMed.
- D. Pinkowicz, H. I. Southerland, C. Avendaño, A. Prosvirin, C. Sanders, W. Wernsdorfer, K. S. Pedersen, J. Dreiser, R. Clérac, J. Nehrkorn, G. G. Simeoni, A. Schnegg, K. Holldack and K. R. Dunbar, J. Am. Chem. Soc., 2015, 137, 14406–14422 CrossRef CAS PubMed.
- J. Martinez-Lillo, J. Faus, F. Lloret and M. Julve, Coord. Chem. Rev., 2015, 289–290, 215–237 CrossRef CAS.
- S. K. Singh and G. Rajaraman, Nat. Commun., 2016, 7, 10669 CrossRef CAS PubMed.
- P. A. Reynolds, B. Moubaraki, K. S. Murray, J. W. Cable, L. M. Engelhardt and B. N. Figgis, Dalton Trans., 1997, 263–268 RSC.
- C. M. Nelson, G. E. Boyd and W. T. Smith Jr., J. Am. Chem. Soc., 1954, 76, 348–352 CrossRef CAS.
- B. N. Figgis, J. Lewis and F. E. Mabbs, J. Chem. Soc., 1961, 3138–3145 RSC.
- R. H. Busey and E. Sonder, J. Chem. Phys., 1962, 36, 93–97 CrossRef CAS.
- J. Martinez-Lillo, D. Armentano, G. De Munno, F. Lloret, M. Julve and J. Faus, Cryst. Growth Des., 2006, 6, 2204–2206 CAS.
- R. Chiozzone, R. González, C. Kremer, G. De Munno, J. Cano, F. Lloret, M. Julve and J. Faus, Inorg. Chem., 1999, 38, 4745–4752 CrossRef CAS PubMed.
- J. Martínez-Lillo, D. Armentano, G. De Munno, N. Marino, F. Lloret, M. Julve and J. Faus, CrystEngComm, 2008, 10, 1284–1287 RSC.
- R. Gonzalez, F. Romero, D. Luneau, D. Armentano, G. De Munno, C. Kremer, F. Lloret, M. Julve and J. Faus, Inorg. Chim. Acta, 2005, 358, 3995–4002 CrossRef CAS.
- R. Gonzalez, R. Chiozzone, C. Kremer, G. De Munno, F. Nicolo, F. Lloret, M. Julve and J. Faus, Inorg. Chem., 2003, 42, 2512–2518 CrossRef CAS PubMed.
- R. Gonzalez, R. Chiozzone, C. Kremer, F. Guerra, G. De Munno, F. Lloret, M. Julve and J. Faus, Inorg. Chem., 2004, 43, 3013–3019 CrossRef CAS PubMed.
- J. Martinez-Lillo, J. Cano, W. Wernsdorfer and E. K. Brechin, Chem. – Eur. J., 2015, 21, 8790–8798 CrossRef CAS PubMed.
- J. Martinez-Lillo, A. H. Pedersen, J. Faus, M. Julve and E. K. Brechin, Cryst. Growth Des., 2015, 15, 2598–2601 CAS.
- C. J. Milios, C. P. Raptopoulou, A. Terzis, F. Lloret, R. Vicente, S. P. Perlepes and A. Escuer, Angew. Chem., Int. Ed., 2004, 43, 210–212 CrossRef CAS PubMed.
- C. J. Milios, A. Vinslava, W. Wernsdorfer, S. Moggach, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2007, 129, 2754–2755 CrossRef CAS PubMed.
- C. J. Milios, A. Vinslava, P. A. Wood, S. Parsons, W. Wernsdorfer, G. Christou, S. P. Perlepes and E. K. Brechin, J. Am. Chem. Soc., 2007, 129, 8–9 CrossRef CAS PubMed.
- C. J. Milios, A. Vinslava, W. Wernsdorfer, A. Prescimone, P. A. Wood, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2007, 129, 6547–6561 CrossRef CAS PubMed.
- C. J. Milios, R. Inglis, A. Vinslava, R. Bagai, W. Wernsdorfer, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2007, 129, 12505–12511 CrossRef CAS PubMed.
- C. J. Milios, R. Inglis, R. Bagai, W. Wernsdorfer, A. Collins, S. Moggach, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, Chem. Commun., 2007, 3476–3478 RSC.
- C. J. Milios, S. Piligkos and E. K. Brechin, Dalton Trans., 2008, 1809–1817 RSC.
- A.-R. Tomsa, J. Martinez-Lillo, Y. Li, L.-M. Chamoreau, K. Boubekeur, F. Farias, M. A. Novak, E. Cremades, E. Ruiz, A. Proust, M. Verdaguer and P. Gouzerh, Chem. Commun., 2010, 46, 5106–5108 RSC.
- R. Inglis, C. J. Milios, L. F. Jones, S. Piligkos and E. K. Brechin, Chem. Commun., 2012, 48, 181–190 RSC.
- A. Caneschi, D. Gatteschi and P. Rey, in Prog. Inorg. Chem, John Wiley & Sons, Inc., 1991, pp. 331–429 Search PubMed.
- A. Caneschi, D. Gatteschi, R. Sessoli and P. Rey, Acc. Chem. Res., 1989, 22, 392–398 CrossRef CAS.
- A. Caneschi, D. Gatteschi, N. Lalioti, C. Sangregorio, R. Sessoli, G. Venturi, A. Vindigni, A. Rettori, M. G. Pini and M. A. Novak, Angew. Chem., Int. Ed., 2001, 40, 1760–1763 CrossRef CAS PubMed.
- K. Inoue and H. Iwamura, J. Am. Chem. Soc., 1994, 116, 3173–3174 CrossRef CAS.
- D. Luneau, F. M. Romero and R. Ziessel, Inorg. Chem., 1998, 37, 5078–5087 CrossRef CAS.
- K. Fegy, D. Luneau, T. Ohm, C. Paulsen and P. Rey, Angew. Chem., Int. Ed., 1998, 37, 1270–1273 CrossRef CAS.
- A. Ito, Y. Nakano, M. Urabe, K. Tanaka and M. Shiro, Eur. J. Inorg. Chem., 2006, 3359–3368 CrossRef CAS.
- I. A. Gass, C. J. Gartshore, D. W. Lupton, B. Moubaraki, A. Nafady, A. M. Bond, J. F. Boas, J. D. Cashion, C. Milsmann, K. Wieghardt and K. S. Murray, Inorg. Chem., 2011, 50, 3052–3064 CrossRef CAS PubMed.
- I. A. Gass, S. Tewary, A. Nafady, N. F. Chilton, C. J. Gartshore, M. Asadi, D. W. Lupton, B. Moubaraki, A. M. Bond, J. F. Boas, S.-X. Guo, G. Rajaraman and K. S. Murray, Inorg. Chem., 2013, 52, 7557–7572 CrossRef CAS PubMed.
- S. Tewary, I. A. Gass, K. S. Murray and G. Rajaraman, Eur. J. Inorg. Chem., 2013, 2013, 1024–1032 CrossRef CAS.
- I. A. Gass, S. Tewary, G. Rajaraman, M. Asadi, D. W. Lupton, B. Moubaraki, G. Chastanet, J.-F. Letard and K. S. Murray, Inorg. Chem., 2014, 53, 5055–5066 CrossRef CAS PubMed.
- I. A. Gass, M. Asadi, D. W. Lupton, B. Moubaraki, A. M. Bond, S.-X. Guo and K. S. Murray, Aust. J. Chem., 2014, 67, 1618–1624 CrossRef CAS.
- J. Kleinberg, Inorg. Synth, McGraw-Hill, 1963 Search PubMed.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 59–75 CrossRef CAS PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- D. Armentano and J. Martinez-Lillo, Inorg. Chim. Acta, 2012, 380, 118–124 CrossRef CAS.
- J. Martinez-Lillo, Polyhedron, 2014, 67, 213–217 CrossRef CAS.
- W.-Z. Lee, H.-S. Tseng and T.-S. Kuo, Dalton Trans., 2007, 2563–2570 RSC.
- S. Liang, H. Wang, T. Deb, J. L. Petersen, G. T. Yee and M. P. Jensen, Inorg. Chem., 2012, 51, 12707–12719 CrossRef CAS PubMed.
- K. Uehara, S. Hikichi and M. Akita, J. Chem. Soc., Dalton Trans., 2002, 3529–3538 RSC.
- J. Martinez-Lillo, D. Armentano, G. De Munno, M. Julve, F. Lloret and J. Faus, Dalton Trans., 2013, 42, 1687–1695 RSC.
- J. Martínez-Lillo, F. Lloret, M. Julve and J. Faus, J. Coord. Chem., 2009, 62, 92–99 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional structural and magnetic data/figures. CCDC 1534665–1534675. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt00752c |
| This journal is © The Royal Society of Chemistry 2017 |