
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligands and complexes based on piperidine and their exploitation of the ring opening polymerisation of rac-lactide†
Paul
McKeown
a,
James
Brown-Humes
b,
Matthew G.
Davidson
ab,
Mary F.
Mahon
b,
Timothy J.
Woodman
c and
Matthew D.
Jones
 *b
*b
aDoctoral Training Centre in Sustainable Chemical Technologies, University of Bath, Bath BA2 7AY, UK
bDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: mj205@bath.ac.uk; Fax: +44 (0)1225 386231; Tel: +44 (0)1225 384908
cDepartment of Pharmacy and Pharmacology, University of Bath, Claverton Down, Bath BA2 7AY, UK
First published on 23rd March 2017
Abstract
A range of ligands based upon 2-(aminomethyl)piperidine have been successfully complexed to Mg(II), Zn(II) and group IV metal centres. These complexes have been characterised both in solution and solid state with different coordination geometries realised dependant on the nature of the ligand. For the Mg(II) and Zn(II) complexes, M(1–2)2, were isolated and analysed by DOSY NMR spectroscopy. These ligands also furnished diastereomeric group IV complexes, M(1–2)2(OiPr)2. Group IV salalen and salan complexes, M(4–5)(OR)2 were also found to be diastereomeric in nature, with either β-cis or α-cis geometriesrespectively. The tridentate ligand, 6H2, yielded five coordinate complexes with both Ti(IV) and Zr(IV). All complexes were screened for the ring opening polymerisation of rac-lactide under both solvent and melt conditions. For the Mg(II) and Zn(II) complexes, good activity was observed with Zn(1–2)2 demonstrating immortal polymerisation characteristics.
Introduction
There are approximately 300 Mt's of plastics produced annually, of which only 1% are from renewable resources.1 As well as being bio-based, such renewable materials need to have comparable material properties to conventional plastics to fulfill the same range of applications. Poly(lactic acid) (PLA) has received much interest due to its renewable credentials as well as promising mechanical and biodegradation properties.2–7 The challenges for PLA are related to the ability to control polymer tacticity, a property which greatly impacts the thermal characteristics of the final material.8 To achieve this, a wide range of complexes for the ring opening polymerisation (ROP) of lactide (LA) have been detailed in the literature. Success has been achieved with Al(III) complexes, with exquisite stereocontrol often being reported.9–22 However, a common drawback to Al(III) complexes is low activity, with several days being required to achieve high conversions in extreme cases.12,19,20 Although this is not always the case, for example we have shown that Al-salan complexes based on the piperidine back bone can show excellent rates.15 The use of Ga(III) and In(III) complexes has been shown to increase activity relative to Al(III), but the level of stereocontrol is often much reduced.23–32 Group I metals have shown to be highly active for the ROP and in some instances, Na(I) and K(I) complexes have demonstrated strong isotactic tendencies.33–38 Lanthanide based complexes have also received much attention, demonstrating high activities with both heterotactic and isotactic PLA being reported.39–48Relevant to this study is the application of group II, Zn(II) and group IV metal complexes for the ROP of rac-LA. Mg(II) and Zn(II) based complexes have been widely reported and represent popular metal choices due to their high abundance and biocompatibility. Such complexes often demonstrate high activity and selectivity. The application of β-diketimate (BDI) ligands with Mg(II)/Zn(II) typically affords highly active alkoxide bridged species. Coates et al. have demonstrated a [(BDI)Zn(OiPr)]2 complex which facilitates rapid polymerisation at ambient temperatures as well as furnishing highly heterotactic PLA (Pr = 0.94, 0 °C).49 A similar result is achievable with (BDI)Mg(OtBu) with the crucial factor being the choice of solvent; Chisholm et al. have demonstrated high heterotactic preference (Pr = 0.90) for this system but only for polymerisations carried out in THF.50 Ma et al. have reported a series of chiral amino-monophenol ligands with both Mg(II) and Zn(II).51–54 Due to pro-chirality of a coordinating nitrogen, diastereomers are generally observed for these complexes. For the Mg(II) family of complexes, both heterotactic (Pr = 0.78) and isotactic (Pm = 0.67) PLA are achievable in toluene within an hour.51,53 For the corresponding Zn(II) complexes, an increased isotactic bias (Pm = 0.84, Tm = 166 °C) was realised with bulkier ligand substituents.52,54 Abbina and Du have prepared a series of chiral amido-oxazilinate Zn(II) complexes.55 These structures contain a three coordinate metal centre and achieve high isotacticity (Pm = 0.91, Tm = 212 °C) at room temperature. Redshaw et al. have trialled a Mg(II) calixarene complex for the ROP of rac-LA.56 High conversion was achieved in minutes and both heterotactic PLA (THF, Pr = 0.85) and isotactic PLA (toluene, Pm = 0.70) were realised. Almost perfectly heterotactic PLA has been prepared by Cui,57 using Mg(II) phosphinimino–amine complexes. High conversion and selectivity was observed at room temperature (THF, Pr = 0.93) and this control could be enhanced at 0 °C (Pr = 0.98).
A series of group IV imino monophenolate complexes have been prepared by Davidson.58 The Zr(IV) complexes demonstrated a heterotactic bias under both solution and melt conditions (Pr < 0.78) and the system was found to be robust, being resistant to the addition of water. Our work has also focussed on group IV salalen structures.59 For this family of complexes, optimal results were achieved with Hf(IV) which furnished isotactic PLA (Pm < 0.75). Related salan group IV structures were found to demonstrate higher activity and maintained an isotactic bias (Pm < 0.75).60 Use of chiral bipyrrolidine salan complexes increased the degree of stereocontrol and activity (Pm < 0.86, Tm = 176 °C), with successful polymerisations being carried out in both solution and solvent free ROP.21,61 Recently, we have reported a range of complexes based upon 2-(aminomethyl)piperidine (2-AMP) frameworks.15,22 Imino monophenolate Al(III) complexes were shown to be active for the ROP of rac-LA with selectivity and polymerisation rate being greatly influenced by the ortho aryl substituents.22 The related salalen Al(III) complexes had much reduced activity with marginal stereocontrol.22 Remarkably, the corresponding Al(III) salan complexes demonstrated a dramatic increase in activity and selectivity (Pm = 0.83, Tm = 177 °C).15
In this study, a range of ligand types (based on 2-AMP) are prepared and complexed to a variety of metal centres. The initial monophenol ligand set is readily converted to a salalen ligand which is reducible to yield a salan. For the monophenol ligands, Mg(II) and Zn(II) complexes are targeted. The bisphenol systems are amenable to coordination to group IV metal centres. The resultant complexes were then tested for their activity in the ROP of rac-LA.
Results and discussion
Ligand synthesis
The procedure for ligand synthesis (except 2H) has been previously reported (Fig. 1 and 2).15,22 For the monophenol ligands, 1H and 2H, a ring-chain tautomer is present in solution leading to a further bicyclic product (Fig. 1).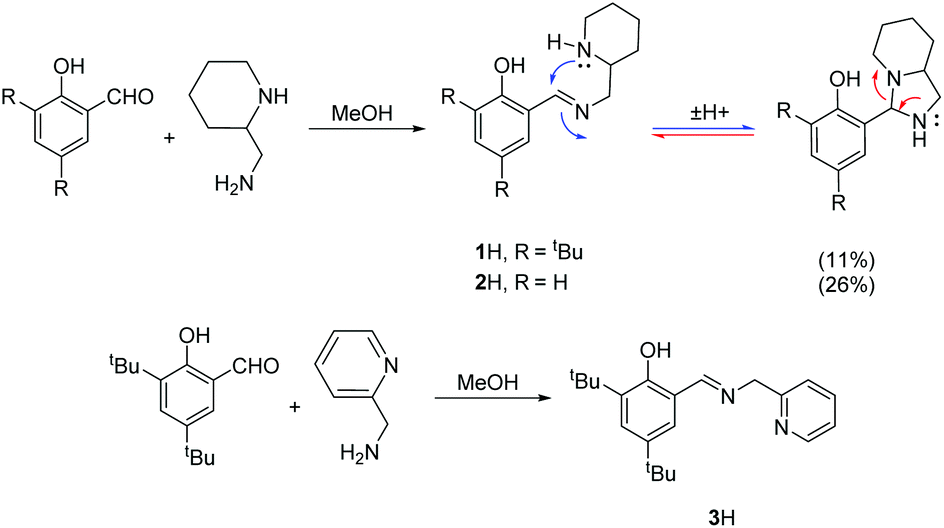 | ||
| Fig. 1 Synthesis and tautomerism of monophenolate ligands, 1–2H and synthesis of 3H. | ||
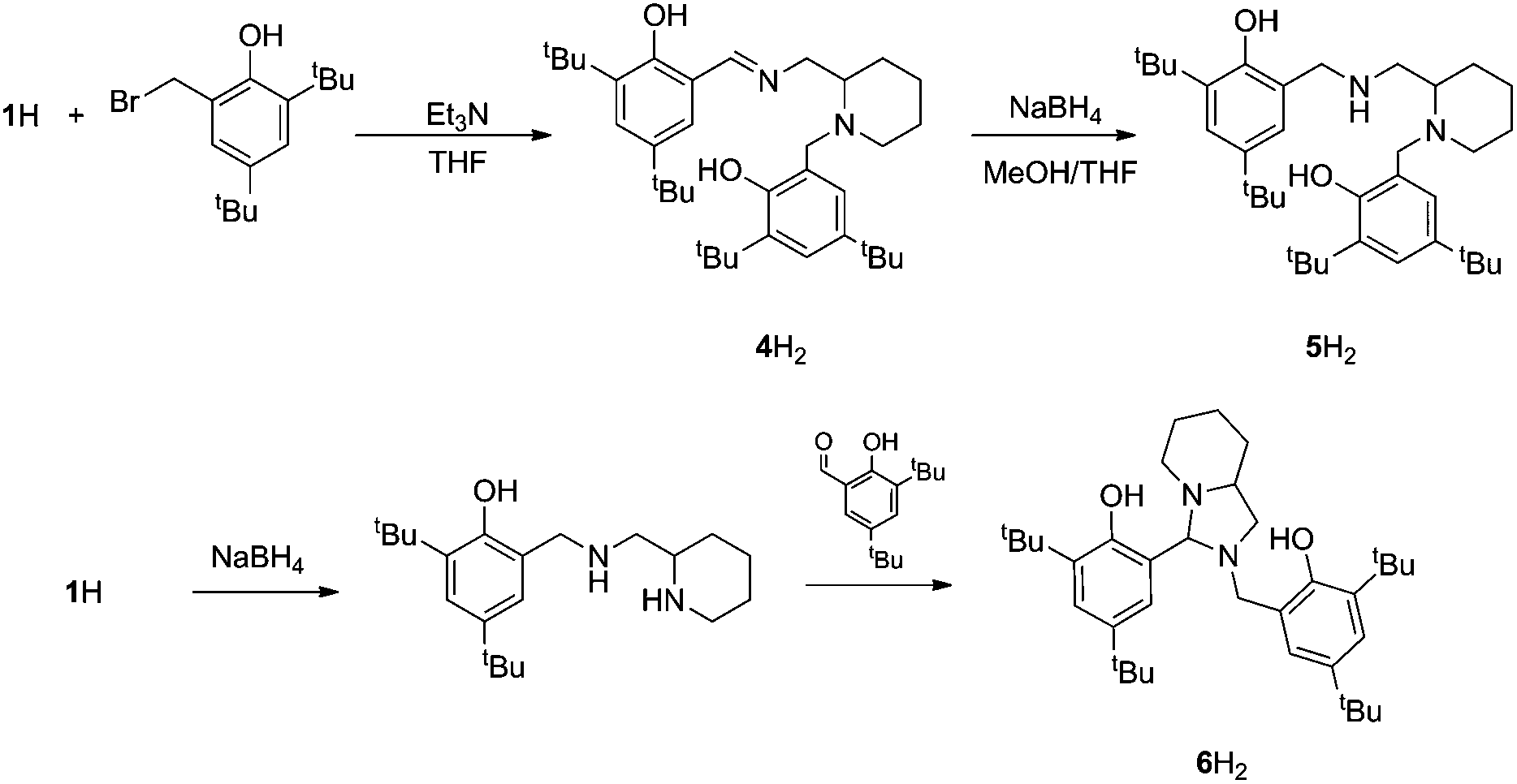 | ||
| Fig. 2 Synthesis of bisphenol ligands, 4–6H2. | ||
For the tBu substituted aryl ring (1H), 89% of the product is observed to be the desired imino form and for the unsubstituted aryl ring (2H), 74% of the product is represented by the imino form (CDCl3, 298 K). For comparison, a pyridine based ligand, 3H, was prepared according to literature methods.62 From 1H, both salalen and salan ligands were also realised (Fig. 2). A small amount (<10%) of bicyclic bisphenol, 6H, is typically isolated with the salalen, 4H2, and this structure is resistant to reduction and is also present in the salan, 5H2. To determine the activity of resultant complex impurities, a route was devised to produce 6H2 in high yield and purity (Fig. 2).
Monophenolate complexes
Ligands, 1–2H were complexed to both Mg(II) and Zn(II) (Fig. 3). The pyridine based ligand, 3H, has previously been complexed with Zn(II) and therefore was only complexed to Mg(II).63,64 Despite a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand-to-metal stoichiometry, only the homoleptic complexes, M(1–3)2 were isolable from reaction with the metal alkyl species. X-ray crystallographic analysis revealed a pseudo octahedral complex for M(1)2 {M = Mg(II) or Zn(II)} (Fig. 4).
:1 ligand-to-metal stoichiometry, only the homoleptic complexes, M(1–3)2 were isolable from reaction with the metal alkyl species. X-ray crystallographic analysis revealed a pseudo octahedral complex for M(1)2 {M = Mg(II) or Zn(II)} (Fig. 4).
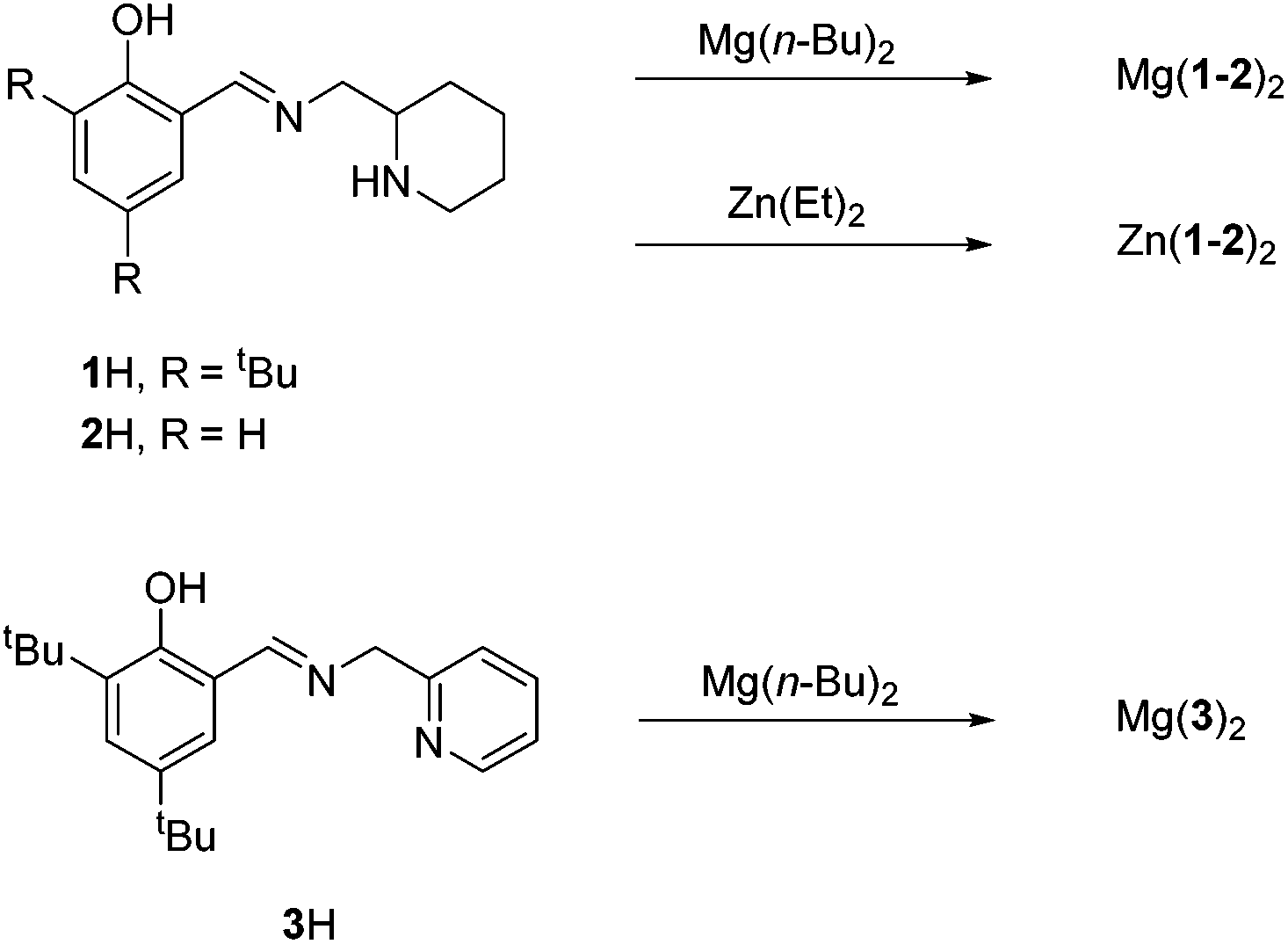 | ||
| Fig. 3 Synthesis of Mg/Zn(II) monophenolate complexes. | ||
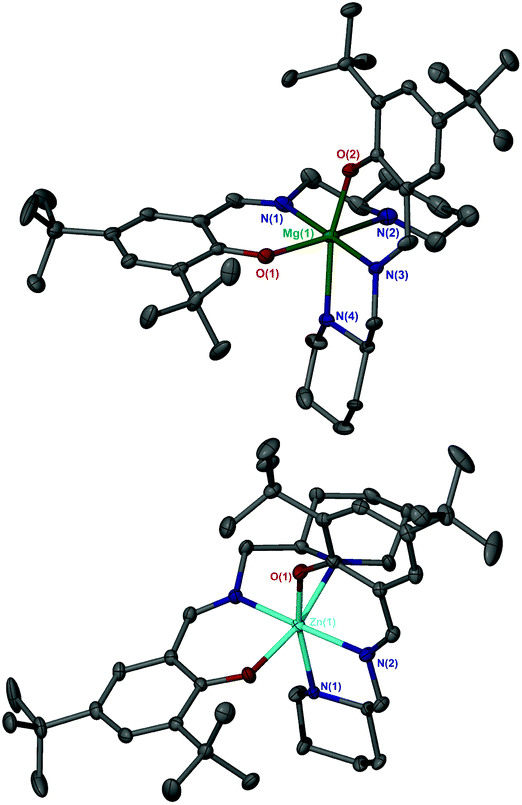 | ||
| Fig. 4 Solid-state structure of Mg(1)2 (Top) and Zn(1)2 (Bottom). Ellipsoids are shown at the 30% probability level and all hydrogen atoms have been removed for clarity. Selected bond length (Å) and angles (°): Mg–O(1) = 1.9963(16), Mg–O(2) = 1.9751(15), Mg–N(1) = 2.1359(18), Mg–Np(2) = 2.326(2), Mg–N(3) = 2.105(9), Mg–N(4) = 2.257(7); O(1)–Mg–Np(2) = 158.95(7), N(1)–Mg–N(3) = 178.62. Zn–O(1) = 2.0139(12), Zn–Np(1) = 2.256(6), Zn–N(2) = 2.1182(14); O(1)–Zn–N(1) = 165.48(16), N(1)–Zn–N(3) = 171.56(8). | ||
The three trans groups show deviation between the anticipated 180° bond angle with the more rigid trans imino groups being closer to ideality {O(1/2)–Mg–Np(2/4) = 158.95(7)°/163.2(2)°, N(1)–Zn–N(3) = 178.62(18)°, O(1)–Zn–Np(2) = 165.48(16)°, N(1)–Zn–N(3) = 171.56(8)°}. Within these geometries, a mer–mer arrangement of ligands was observed.
For both Mg(II) and Zn(II) complexes of 1H disorder is present in the solid-structure, being centred around the aminopiperidine chiral centre. These secondary positions clearly show an exchange in groups at the chiral centre with the ring “flipped” to maintain the imino methyl group in the equatorial position (Fig. 5). These two structures are represented in the form of diastereomeric pairs in the form of (RR/RS) and (SS/SR), for both Mg(1)2 and Zn(1)2. A similar solid-state geometry is anticipated for Mg/Zn(2)2.
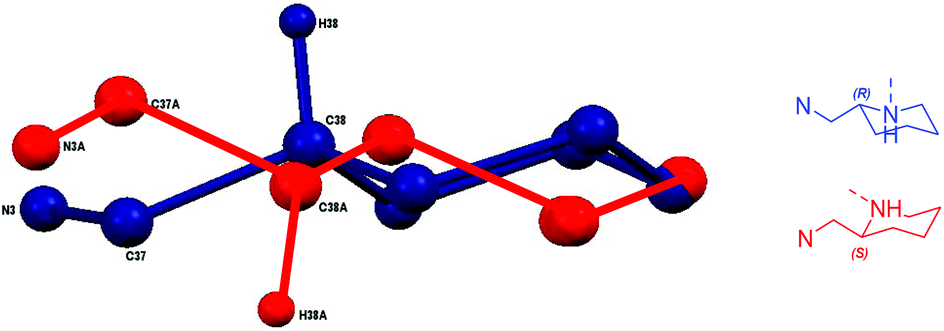 | ||
| Fig. 5 Disordered positions of piperidine ring in Mg(1)2. | ||
The presence of diastereomers for Mg/Zn(1/2)2 is supported by 1H NMR spectroscopy, which shows multiple species in solution. These species typically occur within narrow chemical shift ranges, suggesting similar environments. DOSY NMR spectroscopy of both Mg(1)2 and Zn(1)2 revealed two distinct sets of diffusion coefficients with a small difference be 4.67 × 10−10 m2 s−1 and 4.78 × 10−10 m2 s−1 (C6D6, 298 K) (Table 1).
| Mg(1)2 | Zn(1)2 | 1H | |
|---|---|---|---|
| Major | 4.78 | 5.56 | 6.87 |
| Minor | 4.67 | 5.47 |
Larger values are found for Zn(1)2, equal to 5.47 × 10−10 m2 s−1 and 5.56 × 10−10 m2 s−1 (C6D6, 298 K). The close agreement of these values for each complex series is further evidence that the samples are isomeric. For comparison, the diffusion coefficient of the free ligand is also given; uncoordinated 1H diffuses at a faster rate relative to the complexes {6.87 × 10−10 m2 s−1 (C6D6, 298 K)}.
For 3H, without any potential point of chirality, the 1H and 13C{1H} spectra of the resultant Mg(II) complex reveal one species in solution. Hence, an octahedral complex is likely present in solution in an analogous fashion to Mg(1)2. Unlike the free ligand, the –CH2– groups connected to the pyridine moiety are now split into a pair of diastereotopic doublets, indicating their inequivalence once coordinated.
Group IV complexes
Complexation of the imino monophenol, 1H, to group IV metals yielded M(1)2(OiPr)2, where M = Ti(IV) or Zr(IV) (Fig. 6). Complexes based upon 1H, M(1)2(OiPr)2, were characterised by X-ray crystallography revealing a pseudo octahedral geometry in each case (Fig. 7). The arrangement of ligands around the metal centre conforms to an α-cis conformation with trans phenoxy groups {O(3)–Ti–O(4) = 167.12(5)°/O(3)–Zr–O(4) = 163.02(5)°} and cis isopropoxide moieties {O(1)–Ti–O(2) = 104.60(6)°/O(1)–Zr–O(2) = 101.64(6)°}. The solid-state structure is a racemate with a Δ-SS/Λ-RR configuration. Comparison of the bond lengths and angles for both Ti(1)2(OiPr)2 and Zr(1)2(OiPr)2 show both complexes to be similar and compare well to related literature complexes in terms of their metric data.58 | ||
| Fig. 6 Synthesis of group IV monophenolate complexes. | ||
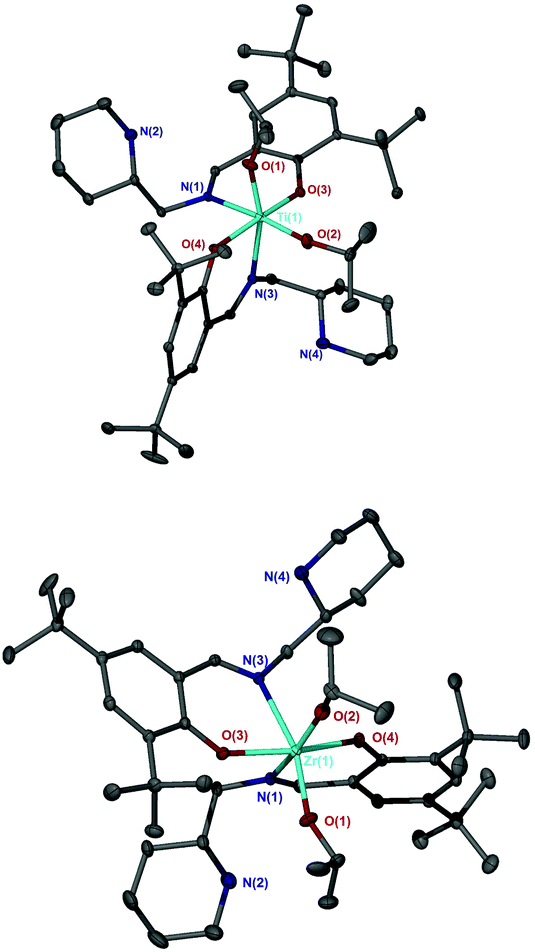 | ||
| Fig. 7 Solid-state structure of Δ-Ti(SS-1)2(OiPr)2 (Top) and Λ-Zr(RR-1)2(OiPr)2 (Bottom). Ellipsoids are shown at the 30% probability level and all hydrogen atoms have been removed for clarity. Selected bond length (Å) and angles (°): Ti–O(1) = 1.8037(12), Ti–O(2) = 1.7852(12), Ti–O(3) = 1.9473(10), Ti–O(4) = 1.9316(10), Ti–N(1) = 2.2628(13), Ti–N(3) = 2.2825(12); O(1)–Ti–O(2) = 104.60(6), O(1)–Ti–N(3) = 162.39(5), O(3)–Ti–O(4) = 167.12(5). Zr–O(1) = 1.9245(13), Zr–O(2) = 1.9289(13), Zr–O(3) = 2.0587(11), Zr–O(4) = 2.0596(12), Zr–N(1) = 2.3979(14), Zr–N(3) = 2.3979(14); O(1)–Zr–O(2) = 104.64(6), O(1)–Zr–N(3) = 161.42(6), O(3)–Zr–O(4) = 163.02(5). | ||
Analysis of the 1H NMR spectra for M(1–2)2(OiPr)2 revealed multiple species in solution (Fig. 8). This is to be expected due to the potential for diastereomeric relationships, with three points of chirality in the structure. It is assumed that the major isomer is that observed for the crystal structure (Δ-SS/Λ-RR) and the remaining species are due to the remaining diastereomers (e.g., Δ-RS/Λ-SR or Λ-RS/Δ-SR). Similar observations and assignments have been previously reported.58 The 13C{1H} NMR spectra provides further evidence for the species to be related diastereomers, having four distinct resonances for the imino functionality in agreement with the number of inequivalent species (Fig. 8). Further analysis via DOSY NMR spectroscopy suggests the species in solution are diffusing at a similar rate {CDCl3, 298 K, Ti(1)2(OiPr)2, D = 5.47 × 10−10 m2 s−1/Zr(1)2(OiPr)2, D = 4.35 × 10−10 m2 s−1}.
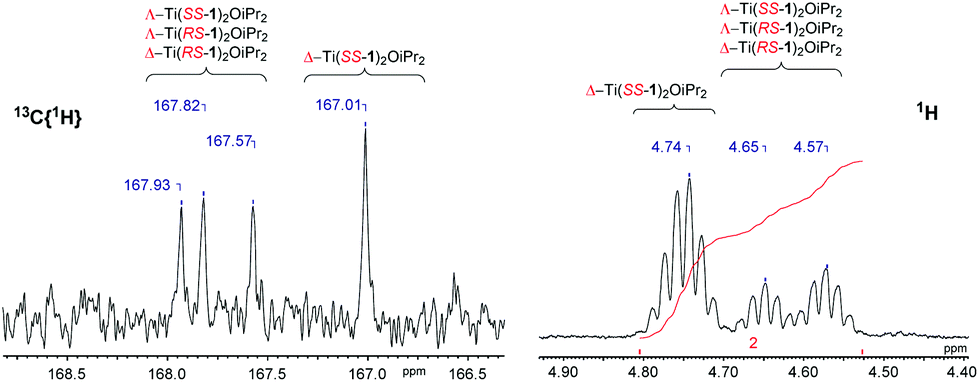 | ||
| Fig. 8 13C{1H} NMR spectrum showing imino region and 1H NMR spectrum showing methine region of Ti(1)2(OiPr)2. Diastereomers have only been indicated but are present as enantiomeric pairs. | ||
The complexation of 3H to Ti(OiPr)4 afforded an unexpected product. Instead of the anticipated bis-ligated species, Ti(3)2(OiPr)2, the isolated complex is the product of a ligand dimerisation (Fig. 9, and ESI†). The resultant Ti(IV) salalen complex is shown to adopt an octahedral geometry in the solid-state with a β-cis arrangement of coordinating groups. The new carbon chiral centres are observed to have opposing stereochemical configurations and the titanium centre also possesses chirality (Λ-RS/Δ-SR).
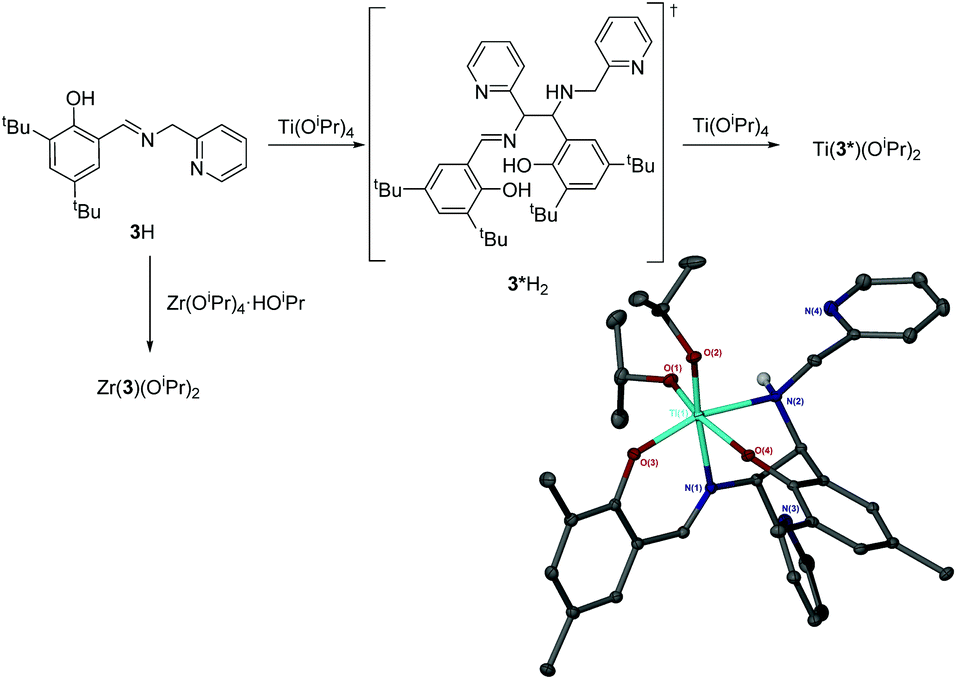 | ||
| Fig. 9 Group IV complexes of 3H. Solid-state structure of Ti(3*)(OiPr)2. Ellipsoids are shown at the 30% probability level and all hydrogen atoms have been removed for clarity. | ||
Analysis of Ti(3*)(OiPr)2 by 1H/13C{1H} NMR spectroscopy reveals an increased number of –CH2– and –CH– resonances due to the ligand dimerisation. A NH resonance is also identifiable in the 1H spectra. Resonances related to inequivalent isopropoxide groups are also observed suggesting the solid-state structure is maintained in solution. There are also several species observed for this complex.
From the complex filtrate, a second precipitate was isolated. Subsequent analysis of which revealed a different NMR spectrum which was also consistent with the structure of Ti(3*)(OiPr)2 with increased purity. It is suggested that the second product is a diastereomer, for which the chirality at the two carbon centres is the same (RR/SS).
The complexation of 3H with Zr(IV) afforded the expected bis-ligated product as shown by NMR spectroscopy. The number of resonances indicates an equivalence of both ligands and isopropoxide groups suggesting an α-cis conformation. There is a broadness associated with the isopropoxide and a –CH2– benzylic resonances which is resolved on cooling (CDCl3, 273 K).
The salalen, 4H2, was also successfully coordinated to Ti(IV) and Zr(IV). For the Zr(IV) complexation, best results were achieved by employing Zr(OtBu)4, which furnished a complex amenable to recrystallisation. The use of Zr(OiPr)4·(HOiPr) afforded the desired product, as suggested by 1H NMR analysis, but the complex was not successfully purified. Solid-state analysis of both metal complexes revealed a pseudo octahedral structure with a β-cis geometry (Fig. 10 and 11). The alkoxide groups have a cis relationship {O(1)–Ti–O(2) = 92.57(5)°/O(1)–Zr–O(2) = 96.75(4)°} and the phenoxy groups of both complexes are also mutually cis for {O(3)–Ti–O(4) = 90.30(6)°/O(3)–Zr–O(4) = 91.45(5)°}. The imino bond-to-metal is observed to be shorter relative to the amino bond {for Zr(4)(OtBu)2, Zr–N(1) = 2.3417(16) Å vs. Zr–N(2) = 2.3417(16) Å}. The stereochemical configuration of both complexes is observed to be Λ-RS/Δ-SR. The ligand wrapping and bond lengths/angles are consistent with group IV complexes reported in the literature.65–67
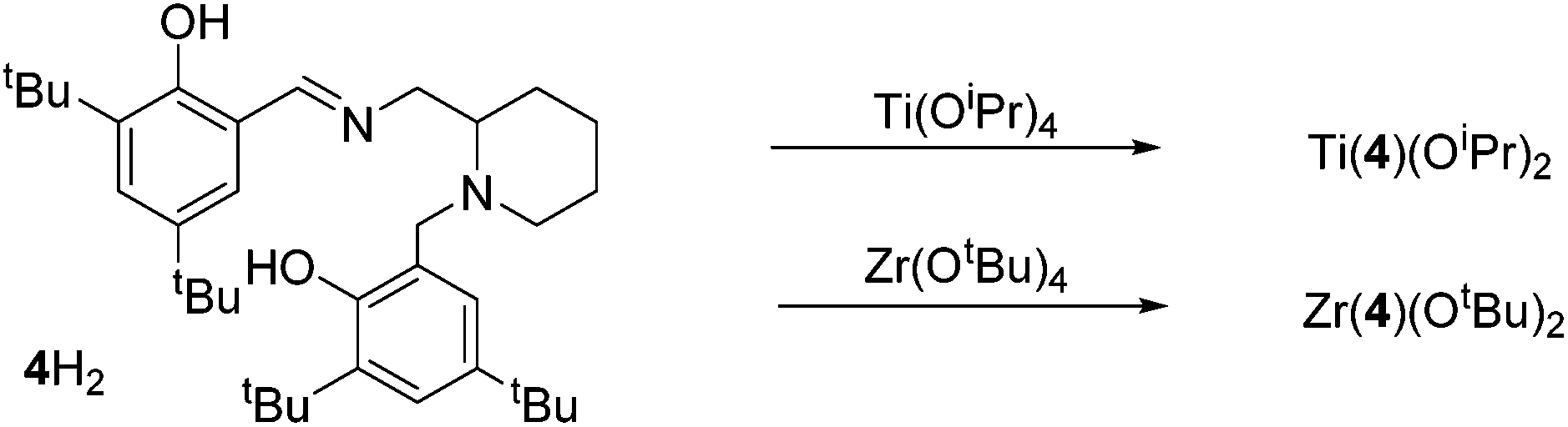 | ||
| Fig. 10 Synthesis of group IV salalen complexes. | ||
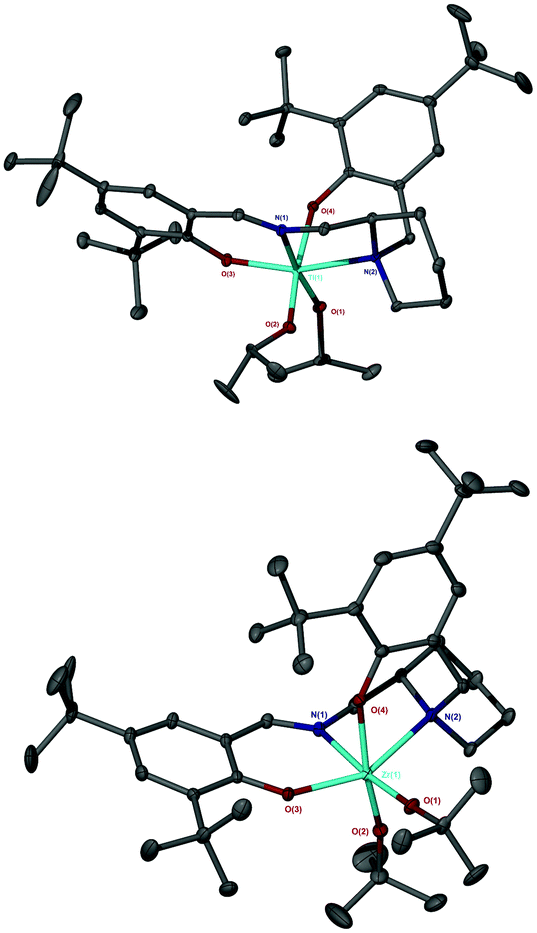 | ||
| Fig. 11 Solid-state structure of Λ-Ti(RS-4)(OiPr)2 (Top) and Δ-Zr(SR-4)(OtBu)2 (Bottom). Ellipsoids are shown at the 30% probability level and all hydrogen atoms have been removed for clarity. Selected bond length (Å) and angles (°): Ti–O(1) = 1.8351(11), Ti–O(2) = 1.8537(12), Ti–O(3) = 1.8890(12), Ti–O(4) = 1.9257(11), Ti–N(1) = 2.2038(13), Ti–N(2) = 2.3101(13); O(1)–Ti–O(2) = 92.57(5), O(1)–Ti–N(1) = 169.83(5), O(3)–Ti–O(4) = 91.79(5). Zr–O(1) = 1.9398(14), Zr–O(2) = 1.9535(14), Zr–O(3) = 2.0209(12), Zr–O(4) = 2.0731(13), Zr–N(1) = 2.3417(16), Zr–N(2) = 2.4371(15); O(1)–Zr–O(2) = 96.75(4), O(1)–Zr–N(1) = 174.48(6), O(3)–Zr–O(4) = 163.02(5). | ||
Analysis via1H NMR spectroscopy reveals two species in solution at a ratio of approximately 5:1 for both salalen Ti(IV) and Zr(IV) complexes (Fig. 12). It is proposed that the minor series of resonances is due to the presence of diastereomers in solution. Application of achiral salalens typically lead to the formation of single diastereomers in solution despite the chirality at both metal and nitrogen centres.65–67 It is therefore tentatively suggested that the extra resonances are due to Λ-SS/Δ-RR species, with the metal and nitrogen chirality remaining unchanged.
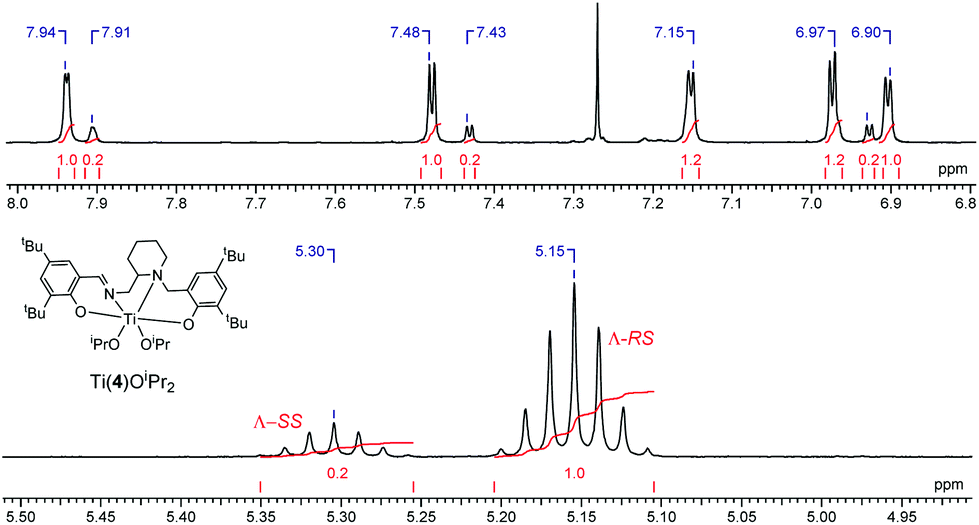 | ||
| Fig. 12 1H NMR (CDCl3, 298 K) spectra of Ti(4)(OiPr)2 showing imino-aromatic region and methine region with Λ-RS and Λ-SS assignment. Diastereomers have only been indicated but are present as enantiomeric pairs. | ||
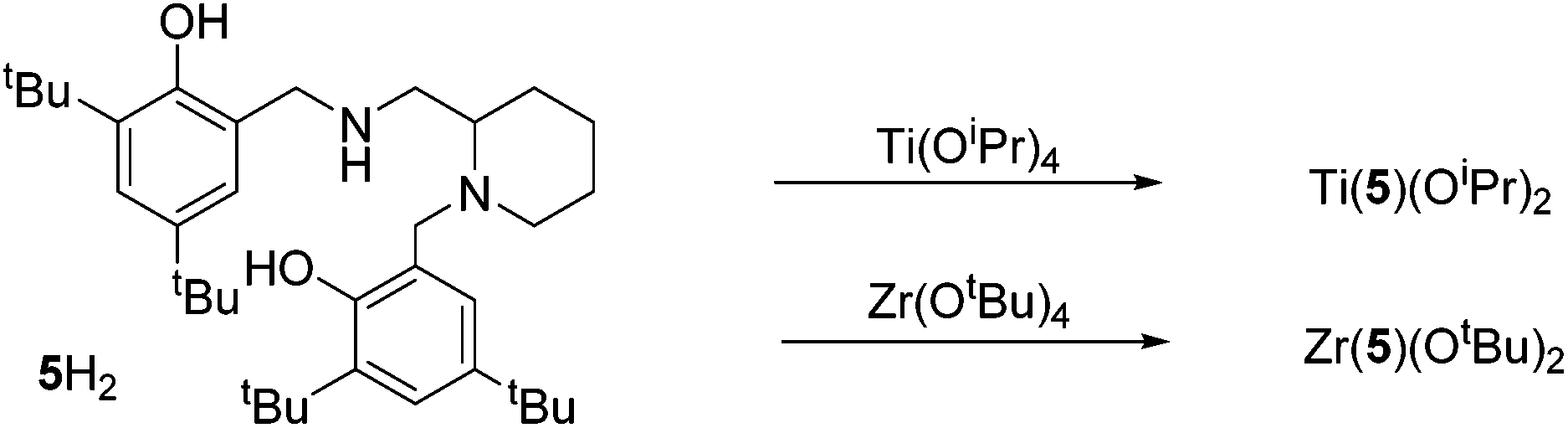
Coordination of the related salan, 5H2, with group IV metals was also realised (Fig. 13). For Ti(5)(OiPr)2 due to the reduction of the imino functionality, an α-cis geometry is adopted having a trans relationship between phenoxy groups {O(3)–Ti–O(4) = 161.33(5)°}. The two isopropoxide groups are observed to be mutually cis {O(1)–Ti–O(2) = 105.58(6)°} in the structure of Ti(5)(OiPr)2. The nitrogen centre within the piperidine ring has a lengthened bond relative to that of the secondary amine {Ti–Np(1) = 2.3876(13) Å/Ti–N(2) = 2.2797(13) Å}. The stereochemical configuration of the ligand is observed to be SRS/RSR relating to the nitrogen, carbon and nitrogen centres respectively. The metric data for Ti(5)(OiPr) is consistent with that of literature Ti(IV) salan complexes.60,68–70 The related Zr(IV) complex, Zr(5)(OtBu)2 is assumed to adopt a similar geometry to the Ti(IV) analogue. Investigation of the solution state structure of Ti(5)(OiPr)2 by 1H NMR spectroscopy revealed a minor series which corresponds to ∼10%. Nevertheless, the resonances of the major product correlate to the solid-state structure of Ti(5)(OiPr)2. While likely diastereotopic in nature, the exact assignment of the extra resonances is increasingly complex due to four points of chirality for Ti(5)(OiPr)2. Previous reports concerning the coordination of chiral ligands to Ti(IV) have demonstrated a change in chirality at the metal centre is common for these systems.68,70,71 A switch to Λ chirality at the metal could be the origin of the diastereomers for this system, with the Δ chirality being preferred during synthesis or recrystallisation (Fig. 14). In contrast, the 1H NMR spectrum for Zr(5)(OtBu)2 revealed the presence of a pure complex in solution.
 | ||
| Fig. 13 Synthesis of group IV salan complexes. | ||
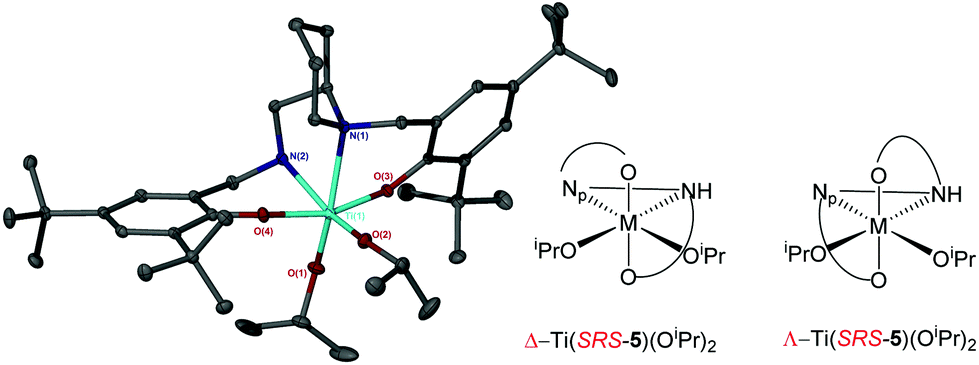 | ||
| Fig. 14 Solid-state structure of Δ-Ti(SRS-5)(OiPr)2 and potential diastereomeric configuration. Ellipsoids are shown at the 30% probability level and all hydrogen atoms have been removed for clarity. Selected bond length (Å) and angles (°): Ti–O(1) = 1.8079(12), Ti–O(2) = 1.7906(12), Ti–O(3) = 1.9138(11), Ti–O(4) = 1.9121(11), Ti–N(1) = 2.3876(13), Ti–N(2) = 2.2797(13); O(1)–Ti–O(2) = 105.58(6), O(1)–Ti–N(1) = 163.11(5), O(3)–Ti–O(4) = 161.33(5). | ||
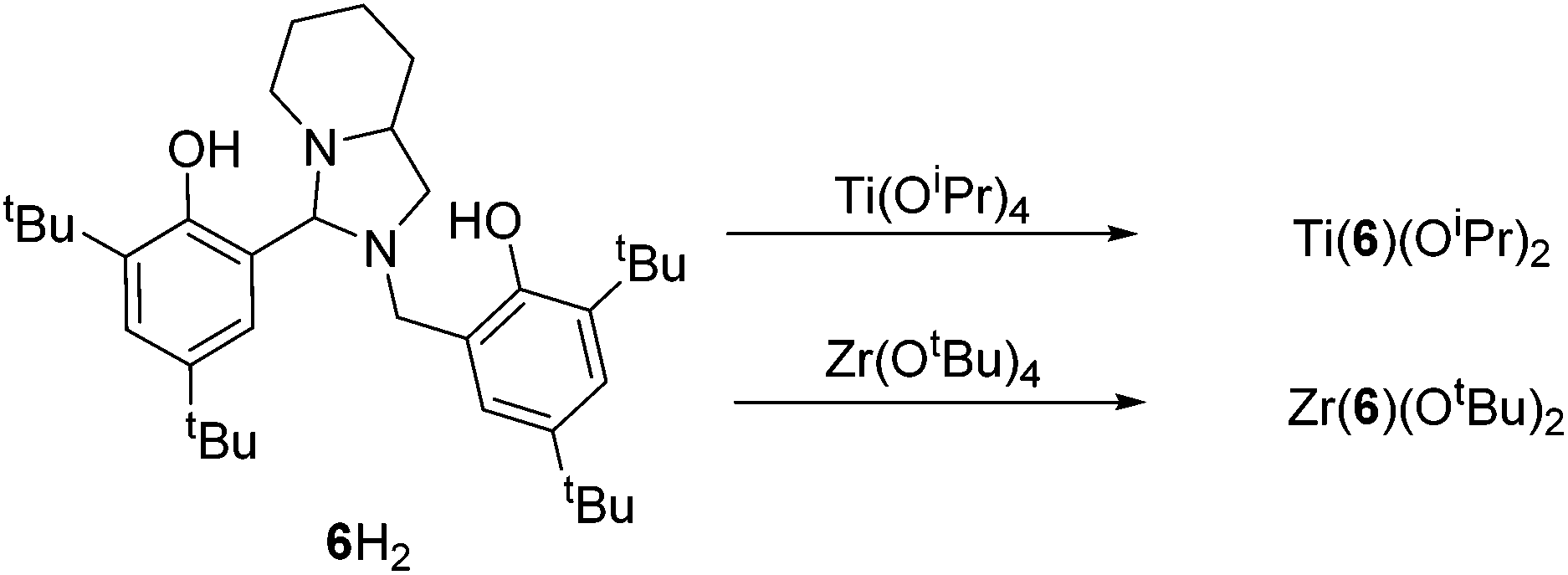
The bicyclic ligand, 6H2, was also coordinated to group IV metals (Fig. 15). Complexation with Zr(OtBu)4 afforded the crystalline product, Zr(6)(OtBu)2. The complex geometry is observed to be trigonal bipyramidal with a tert-butoxide and amine group occupying the pseudo axial positions {N(1)–Zr–O(1) = 168.83(7)°} (Fig. 16). Use of a bulkier Zr(IV) source as well as sterically demanding aryl groups furnishes exclusively the alkoxide complex; we have previously reported a related series of Zr(IV) complexes that demonstrated a strong tendency in forming the bis-ligated species, Zr(L)2.22
 | ||
| Fig. 15 Synthesis of group IV salan complexes. | ||
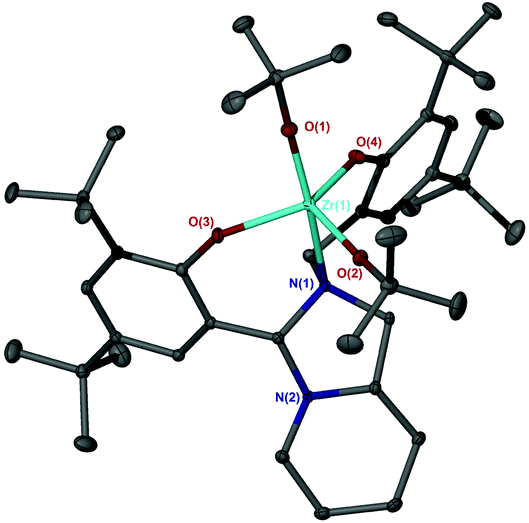 | ||
| Fig. 16 Solid-state structure of Zr(6)(OtBu)2. Ellipsoids are shown at the 30% probability level and all hydrogen atoms have been removed for clarity. Selected bond length (Å) and angles (°): Zr–O(1) = 1.9229(18), Zr–O(2) = 1.9300(17), Zr–O(3) = 2.0181(18), Zr–O(4) = 1.9890(18), Zr–N(1) = 2.420(2); O(1)–Zr–O(2) = 104.28(8), O(1)–Zr–N(1) = 168.83(7), O(3)–Zr–O(4) = 122.23(8). | ||
The metric data is similar to that of a related Ti(IV) complex published previously, albeit with a reduced ideality towards the trigonal bipyramidal geometry (τ = 0.78).22 Analysis of both Zr(IV) and Ti(IV) complexes of 6H2via NMR spectroscopy demonstrates the maintaining of the solid-state structure in solution and the presence of a single species.
Polymerisation studies
Polymerisations were carried out with rac-LA which was singly recrystallised before use. Polymerisations with Mg(1–3)2 and Zn(1–2)2 were performed in solution (toluene) at 80 °C. In the first instance, an equivalence of benzyl alcohol was added to aid initiation (Table 2). For the majority of complexes, high conversion was achievable within one hour, with the Zn(II) complexes generally displaying greater activity. In exception to this, Mg(2)2 was shown to be less active in this time frame. The PLA produced by these complexes was essentially atactic. There was very little difference between the activity and stereocontrol exerted by Mg(1)2 and Mg(3)2.| Complex | Time/h | Conv.g, % |
P
rh |
M
n,theoi |
M
nj |
Đ |
|---|---|---|---|---|---|---|
| Conditions:a [LA]:[I]:[BnOH] = 100:1:1, 80 °C, toluene.b [LA]:[I] = 1000:1:10, 80 °C, toluene.c [LA]:[I]:[BnOH] = 100:1, 80 °C, toluene.d [LA]:[I] = 300:1, 130 °C.e [LA]:[I]:[BnOH] = 300:1:1, 130 °C.f [LA]:[I]:[BnOH] = 3000:1:10, 130 °C.g Determined via1H NMR spectroscopy.h Pr is the probability of heterotactic enchainment, determined via homonuclear decoupled 1H NMR spectroscopy.i Theoretical molecular weight calculated from conversion {[LA]/[BnOH] × (conv. × 144.13) + 108.14} (rounded to the nearest 50).j Determined from GPC (in THF) referenced against polystyrene standards with a correction factor of 0.58 applied. |
||||||
| Mg(1)2a |
1 | 72 | 0.51 | 10500 |
8300 | 1.10 |
| Mg(2)2a |
1 | 40 | 0.47 | 6450 | 2100 | 1.10 |
| Mg(3)2a |
1 | 81 | 0.53 | 11800 |
10000 |
1.10 |
| Zn(1)2a |
0.08 | 90 | 0.57 | 13050 |
10550 |
1.05 |
| Zn(1)2b |
0.5 | 95 | 0.57 | 13800 |
12200 |
1.10 |
| Zn(2)2a |
0.25 | 74 | 0.56 | 9300 | 6650 | 1.08 |
| Zn(2)2b |
0.5 | 58 | 0.59 | 8300 | 8050 | 1.05 |
| Mg(1)2c |
1 | 17 | 0.53 | 2600 | 28800 |
1.14 |
| Mg(3)2c |
1 | 25 | 0.52 | 3700 | 48450 |
1.19 |
| Zn(1)2c |
0.5 | 41 | 0.58 | 6000 | 82300 |
1.28 |
| Zn(2)2c |
0.5 | 9 | 0.59 | 1400 | 28300 |
1.07 |
| Mg(1)2d |
0.5 | 61 | 0.61 | 26450 |
23250 |
1.81 |
| Mg(1)2e |
0.5 | 74 | 0.60 | 32050 |
17400 |
1.49 |
| Mg(2)2e |
1.5 | 67 | 0.57 | 29050 |
5550 | 1.57 |
| Mg(3)2d |
0.07 | 53 | 0.66 | 23000 |
28100 |
1.43 |
| Mg(3)2e |
0.07 | 63 | 0.66 | 27300 |
22000 |
1.41 |
| Zn(1)2d |
0.15 | 49 | 0.59 | 21250 |
42300 |
1.37 |
| Zn(1)2e |
0.1 | 69 | 0.57 | 29900 |
32250 |
1.18 |
| Zn(1)2f |
0.3 | 69 | 0.57 | 29900 |
24750 |
1.12 |
| Zn(2)2e |
1.5 | 84 | 0.51 | 36700 |
7650 | 1.51 |
For this series of complexes, it was found that the addition of excess alcohol, during the polymerisation quenching process, facilitated depolymerisation reactions. This is illustrated by GPC traces of such polymer before and after MeOH wash which show a broadening of the polymer chain distribution {for Mg(1)2, see ESI†}. As a consequence of this, further polymerisations were not quenched. Such observations have previously been noted in the literature.72,73 This depolymerisation was found to be more pronounced for Mg(1)2 which lead to the rapid formation of oligomers and lactates, whereas for Zn(1)2 treatment with methanol resulted in a broadening of the GPC trace and a tail towards low molecular weight. It is also noted that Mg(3)2 initially afforded a pale yellow polymer which became red on prolonged exposure to air. This has previously been observed and attributed to a ligand oxidation process.63 The Zn(II) complexes were also active with under immortal polymerisation conditions ([LA]:[Zn]:[BnOH] = 1000:1:10), achieving reasonable conversion with molecular weight being dependent on the [LA]:[BnOH] ratio. An NMR scale polymerisation was carried out for Zn(1)2 (CDCl3, 298 K, [LA]:[Zn]:[BnOH] = 20:1:1). The initial combination of complexes and alcohol demonstrated the Zn(II) complex to be inert towards alcoholysis. On addition of rac-LA, polymer was observed to be produced without any change in the complex resonances suggesting the operation of an activated monomer mechanism (see ESI†). Further to this, DOSY analysis of the system suggested there was no major interaction between polymer and Zn(II) centre. Kinetic analysis of the polymerisation with Zn(2)2 (toluene, 353 K, [LA]:[Zn]:[BnOH] = 100:1:1) yielded a linear plot of ln([LA]0/[LA]t) against time (kapp = 5.21 ± 0.51 h−1) indicating the polymerisation proceeds with a first order dependence on monomer (see ESI†).
The resultant molecular weight of solution polymerisations with BnOH co-initiator demonstrates a reasonable correlation with the predicted value. However, the molecular weight achieved by Mg(2)2, is much lower than anticipated. In each case, the distribution of molecular weights is narrow again implying a controlled polymerisation (Đ ≤ 1.10). Further analysis via MALDI-ToF mass spectrometry gives comparable values of molecular weight and generally shows the presence of polymer chains with a BnO- and -H end groups (see ESI, Table S1†). For each Mg(1/3) complex, there is a minor series due to transesterification side reactions. For Mg(3)2 there is also evidence of intramolecular transesterification with low molecular weight cyclic oligomers being observed For Mg(2)2, only cyclic species are observed.
Analysis of polymer derived from Zn(1/2)2via MALDI-ToF mass spectrometry indicates a minimal degree of undesirable transesterification reactions. The literature complex, Zn(3)2, was shown to be active in the absence of co-initiator under solution conditions.64 Under similar conditions, the activity of the complexes Mg(1/3)2 and Zn(1–2)2 is observed to be much reduced within the same time frame, with Zn(1)2 being the most active. Despite the low conversion, relatively high molecular weights are afforded with the distribution of polymer chain remaining relatively narrow. It is unclear if initiation is due to a ligand phenoxy moiety or monomer impurities (see ESI†).
The complexes are also tested under solvent free conditions (130 °C, [LA]:[I] = 300:1 or [LA]:[I] = 300:1:1). The polymerisations with Mg(1)2 was hampered by the insolubility of this complex in the molten LA. As a consequence, the reaction time was lengthened. The molecular weight control afforded by these complexes is poor, with lower than theoretical values being realised, even in the absence of BnOH. However, slight heterotacticity is demonstrated by these Mg(II) systems (Pr ≈ 0.60). A faster reaction, with slightly more of a heterotactic preference, is achieved with Mg(3)2, which was fully soluble in the LA melt. A more controlled polymerisation is achieved by employing Zn(1)2, which is also fully soluble in the LA melt. At this higher temperature, the reaction in the absence of benzyl alcohol achieves higher conversion, indicating the initiation step may have a thermal barrier, perhaps due to insertion of a ligand phenoxy moiety. Under these conditions, a higher molecular weight than predicted is afforded, indicating fewer chains are initiated relative to the amount of metal centres. The addition of BnOH yields PLA with a good agreement between experimental molecular weight and predicted. Zn(1)2 can also perform under immortal melt conditions (3000:1:10, 130 °C). While there is a slight increase in polymerisation time, the molecular weight achieved is comparable to the LA to co-initiator ratio, with a relative improvement of polymer chain distribution. The solvent-free polymerisation of Mg/Zn(2)2 was slow and poorly controlled. It is tentatively suggested that the less hindered metal centres are susceptible to decomposition under these conditions. The titanium based complexes were tested under melt conditions (130 °C, 300:1), having isopropoxide groups to initiate polymerisation directly (Table 3). The bis-ligated complex, Ti(1)2(OiPr)2, was found to be a poor complexes for the ROP of rac-LA achieving 58% under melt conditions after 24 hours. The observed molecular weight is much lower than anticipated suggesting the formation of oligomers. The salalen complexes, Ti(3*/4)(OiPr)2, were observed to only reach 15–17% conversion after 24 hours under these conditions. In this instance, coordination of LA may be restricted due to high metal coordination and bulky aryl substituents. The titanium salan complex, Ti(5)(OiPr)2 is more active reaching high conversion within 3 hours. The Ti(IV) salan complex affords a narrow distribution of polymer chains with a molecular weight consistent with two chain per metal centre. Geometrically, the structures are different with the salalen adopting a fac–mer geometry and the salan, a fac–fac arrangement. This increased activity on going from salalen to salan also correlates with the observations made for the respective aluminium complexes.15,22
| Complex | Time/h | Conv.a, % |
P
rb |
M
n,theoc |
M
nd |
Đ |
|---|---|---|---|---|---|---|
| Conditions: [LA]:[I] = 300:1, 130 °C, solvent free.a Determined via1H NMR spectroscopy.b Pr is the probability of heterotactic enchainment, determined via homonuclear decoupled 1H NMR spectroscopy.c Theoretical molecular weight calculated from conversion {300 × (conv. × 144.13) + Mn(end group)}/2 (rounded to the nearest 50).d Determined from GPC (in THF) referenced against polystyrene standards with a correction factor of 0.58 applied. |
||||||
| Ti(1)2(OiPr)2 | 24 | 58 | 0.49 | 12550 |
2850 | 1.36 |
| Ti(3*)(OiPr)2 | 24 | 15 | — | — | — | — |
| Ti(4)(OiPr)2 | 24 | 17 | — | — | — | — |
| Ti(5)(OiPr)2 | 3 | 75 | 0.52 | 16250 |
13900 |
1.04 |
| Ti(6)(OiPr)2 | 18 | 72 | 0.51 | 15550 |
10350 |
1.31 |
The bicyclic complex Ti(6)(OiPr)2 achieved reasonable conversion within 18 hours, however, molecular weight control was poor (Đ = 1.31). The activity of Ti(6)(OiPr)2 is much reduced compared to a previously reported bicyclic analogue.22 In no instance is stereocontrol exerted by this series of complexes which is analogous to the majority of Ti(IV) complexes in the literature.58,66,74–77
The bis-ligated species, Zr(1)2(OiPr)2 was more active than the related Ti(IV) complex, Ti(1)2(OiPr)2. In solution (toluene, 80 °C), reasonable conversion was achieved after 2 hours and under solvent free conditions, less than 1 hour was required. However, similar to the Ti(IV) complex, only lower molecular weight was achievable. In comparison, Zr(3)2(OiPr)2 achieves a higher molecular weight with improved control albeit with a longer polymerisation time. The Zr(IV) salalen complex was also assessed for activity in the ROP of rac-LA. For Zr(4)(OtBu)2, improved activity is observed relative to the Ti(IV) salalen.
However, there is a large difference between observed and theoretical molecular weight values for the solution polymerisation, with a relatively broad distribution of chain lengths (Đ = 1.34). Both of these facts may indicate a slow initiation process, due to the disfavoured insertion of tBuO- into the monomer.42 To test this, polymerisation was carried out in the presence of one equivalence of BnOH. Under these conditions, the both molecular weight and distribution are reduced (Đ = 1.21) indicating an increase in control, which is mirrored by better agreement between theoretical and actual Mn. Furthermore, the polymerisation is observed to be more rapid (71%, 3 h). The solvent-free polymerisation achieves reasonable conversion within 30 minutes, however, a broad molecular weight distribution is observed under these conditions (Đ = 1.42). Under solution or melt conditions no stereocontrol is achieved by Zr(4)(OtBu)2 (Table 4).
| Complex | Time/h | Conv.d, % |
P
re |
M
n,theof |
M
ng |
Đ |
|---|---|---|---|---|---|---|
| Conditions:a [LA]:[I] = 100:1, 80 °C, toluene.b [LA]:[I] = 300:1, 130 °C, solvent free.c [LA]:[I]:[BnOH]: = 100:1:1, 80 °C, toluene.d Determined via1H NMR spectroscopy.e Pr is the probability of heterotactic enchainment, determined via homonuclear decoupled 1H NMR spectroscopy.f Theoretical molecular weight calculated from conversion {[LA]/[I] × (conv. × 144.13)/2 + Mn(end group)} (rounded to the nearest 50).g Determined from GPC (in THF) referenced against polystyrene standards with a correction factor of 0.58 applied. |
||||||
| Zr(1)2(OiPr)2a |
2 | 62 | 0.53 | 4500 | 2950 | 1.12 |
| Zr(1)2(OiPr)2b |
0.8 | 80 | 0.60 | 17350 |
6950 | 1.17 |
| Zr(3)2(OiPr)2a |
6 | 63 | 0.45 | 4500 | 4000 | 1.07 |
| Zr(4)(OtBu)2a |
4 | 72 | 0.53 | 5250 | 16350 |
1.34 |
| Zr(4)(OtBu)2c |
3 | 71 | 0.56 | 5200 | 5900 | 1.21 |
| Zr(4)(OtBu)2b |
0.4 | 69 | 0.52 | 15000 |
13400 |
1.42 |
| Zr(6)(OtBu)2a |
4 | 77 | 0.60 | 5600 | 6500 | 1.10 |
| Zr(6)(OtBu)2b |
0.25 | 75 | 0.60 | 16300 |
15600 |
1.19 |
Unusually, attempts to polymerise rac-LA with the related Zr(IV) salan, Zr(5)(OtBu)2, were unsuccessful in solution and for solvent-free reactions. For the five coordinate Zr(6)(OtBu)2 complex, similar molecular weight control and activity is demonstrated in both solution and under melt conditions. However, closer examination of the homonuclear decoupled 1H spectrum for PLA derived from Zr(6)(OtBu)2 shows an unusual enhancement of the isi resonance (see ESI†). This is due to stereorandom transesterification giving rise to polymer linkages not typically observed for rac-LA.60,78,79 These linkages are determined to be related to iss, sss, and ssi tetrads from analysis of the methine region in the 13C{1H} NMR spectrum (see ESI†) and should not be present for the ROP of rac-LA.80 These tetrads coincide with the isi resonance in the 1H NMR spectrum, hence the overall dominance of this resonance.
MALDI-ToF mass spectrometry analysis was performed on polymer derived from Zr(IV) alkoxides (see ESI†). For Zr(1)2(OiPr)2, in solution, the observed molecular weight series is higher than GPC and more consistent with the theoretical value (Mn,MALDI = 4200 g mol−1). There is one major series with 72 g mol−1 spacing indicating transesterification reactions are occurring under these conditions, with iPrO- and H- end-groups. There is also evidence of cyclic PLA oligomers from application the of Zr(1)2(OiPr)2 as well as TFA (trifluoroacetic acid) capped polymer (the TFA is the counterion for the Na(I) source in MALDI).
In another instance, a series related to methoxy capped PLA was observable due to washing with MeOH. The polymer derived from the solvent-free polymerisation was also amenable to MALDI-ToF analysis. Similar observations are made as for the solution polymerisation, with iPrO-, H- and TFA- end-groups present as well as cyclic structures. For the solvent-free MALDI-ToF spectrum, the distribution is not symmetrical with a substantial tail to low molecular weight. Zr(3)2(OiPr)2 affords one main polymeric series with a spacing of 72 g mol−1 indicating the presence of undesirable transesterification reactions, however, the correct end groups are evident. For the salalen complex, Zr(5)(OtBu)2, the polymer prepared in the absence of co-initiator is too large to be analysed by MALDI-ToF. However, the addition of BnOH afforded lower molecular weights amenable to characterisation. The spectrum is dominated by polymer containing the expected BnO- end group, with a symmetrical distribution and a 72 g mol−1 peak spacing. Trace amounts of tBuO- capped polymer is observed at lower molecular weight. For polymer derived from Zr(6)(OtBu)2, there are no peaks associated with the expected tBuO- end group via MALDI-ToF analysis. Instead, the main series, which has a spacing of 72 g mol−1, correlates well with methoxy end groups. Evidently, transesterification or cyclic oligomer ring opening occurs during polymer work-up. A minor series is also present which is shown to possess no end-groups.
Conclusions
Racemic ligands based upon 2-(aminomethyl)piperidine have been prepared and complexed to a range of metals. The majority of complexes reported are found to be diastereomeric in solution and characterised by a combination of X-ray crystallography and NMR spectroscopic methods. These complexes were assessed for their ability to polymerise lactide. For the Mg(II) and Zn(II) complexes, good activity and molecular weight control is generally achievable in solution. For Zn(1–2)2, immortal polymerisation was shown to be feasible. These complexes demonstrated activity under solvent free conditions. The Ti(IV) complexes were found to be relatively poor for the ROP of rac-LA with only Ti(5)(OiPr)2 demonstrating a controlled polymerisation. Increased activity and control was achievable with the Zr(IV) complexes, which is in agreement with literature precedent.Acknowledgements
We wish to thank the EPSRC for funding the CDT at Bath (EP/G03768X/1) and Purac for the donation of lactide. X-ray diffraction, NMR, MALDI-ToF and GPC facilities were provided through the Chemical Characterisation and Analysis Facility (CCAF) at the University of Bath.References
- M. R. Thomsett, T. E. Storr, O. R. Monaghan, R. A. Stockman and S. M. Howdle, Green Mater., 2016, 4, 115 CrossRef.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835 CrossRef CAS PubMed.
- R. Auras, S. Singh and J. Singh, J. Test. Eval., 2006, 34, 1 Search PubMed.
- R. A. Auras, S. P. Singh and J. J. Singh, Packag. Technol. Sci., 2005, 18, 207 CrossRef CAS.
- R. G. Sinclair, J. Macromol. Sci., Pure Appl. Chem., 1996, A33, 585 CrossRef CAS.
- E. T. H. Vink, D. A. Glassner, J. J. Kolstad, R. J. Wooley and R. P. O′Connor, Ind. Biotechnol., 2007, 3, 58 CrossRef CAS.
- M. R. Yates and C. Y. Barlow, Resour., Conserv. Recycl., 2013, 78, 54 CrossRef.
- H. Tsuji, Macromol. Biosci., 2005, 5, 569 CrossRef CAS PubMed.
- H.-L. Chen, S. Dutta, P.-Y. Huang and C.-C. Lin, Organometallics, 2012, 31, 2016 CrossRef CAS.
- E. D. Cross, L. E. N. Allan, A. Decken and M. P. Shaver, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1137 CrossRef CAS.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, R. I. Pugh and A. J. P. White, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15343 CrossRef CAS PubMed.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688 CrossRef CAS PubMed.
- S. M. Kirk, H. C. Quilter, A. Buchard, L. H. Thomas, G. Kociok-Kohn and M. D. Jones, Dalton Trans., 2016, 45, 13846 RSC.
- K. Majerska and A. Duda, J. Am. Chem. Soc., 2004, 126, 1026 CrossRef CAS PubMed.
- P. McKeown, M. G. Davidson, G. Kociok-Kohn and M. D. Jones, Chem. Commun., 2016, 52, 10431 RSC.
- N. Nomura, R. Ishii, Y. Yamamoto and T. Kondo, Chem. – Eur. J., 2007, 13, 4433 CrossRef CAS PubMed.
- A. Pilone, K. Press, I. Goldberg, M. Kol, M. Mazzeo and M. Lamberti, J. Am. Chem. Soc., 2014, 136, 2940 CrossRef CAS PubMed.
- N. Spassky, M. Wisniewski, C. Pluta and A. LeBorgne, Macromol. Chem. Phys., 1996, 197, 2627 CrossRef CAS.
- Z. Zhong, P. J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291 CrossRef CAS PubMed.
- Z. Y. Zhong, P. J. Dijkstra and J. Feijen, Angew. Chem., Int. Ed., 2002, 41, 4510 CrossRef CAS PubMed.
- M. D. Jones, L. Brady, P. McKeown, A. Buchard, P. M. Schafer, L. H. Thomas, M. F. Mahon, T. J. Woodman and J. P. Lowe, Chem. Sci., 2015, 6, 5034 RSC.
- P. McKeown, M. G. Davidson, J. P. Lowe, M. F. Mahon, L. H. Thomas, T. J. Woodman and M. D. Jones, Dalton Trans., 2016, 45, 5374 RSC.
- D. C. Aluthge, J. M. Ahn and P. Mehrkhodavandi, Chem. Sci., 2015, 6, 5284 RSC.
- D. C. Aluthge, B. O. Patrick and P. Mehrkhodavandi, Chem. Commun., 2013, 49, 4295 RSC.
- D. C. Aluthge, E. X. Yan, J. M. Ahn and P. Mehrkhodavandi, Inorg. Chem., 2014, 53, 6628 CrossRef PubMed.
- J.-C. Buffet, J. Okuda and P. L. Arnold, Inorg. Chem., 2010, 49, 419 CrossRef CAS PubMed.
- M. Cybularczyk, M. Dranka, J. Zachara and P. Horeglad, Organometallics, 2016, 35, 3311 CrossRef CAS.
- A. F. Douglas, B. O. Patrick and P. Mehrkhodavandi, Angew. Chem., Int. Ed., 2008, 120, 2322 CrossRef.
- S. Ghosh, R. R. Gowda, R. Jagan and D. Chakraborty, Dalton Trans., 2015, 44, 10410 RSC.
- K. M. Osten, D. C. Aluthge and P. Mehrkhodavandi, Dalton Trans., 2015, 44, 6126 RSC.
- A. Pietrangelo, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2009, 2736 RSC.
- I. Yu, A. Acosta-Ramírez and P. Mehrkhodavandi, J. Am. Chem. Soc., 2012, 134, 12758 CrossRef CAS PubMed.
- Z. Dai, Y. Sun, J. Xiong, X. Pan, N. Tang and J. Wu, Catal. Sci. Technol., 2016, 6, 515 CAS.
- Z. Dai, Y. Sun, J. Xiong, X. Pan and J. Wu, ACS Macro Lett., 2015, 556 CrossRef CAS.
- Y. Sun, J. Xiong, Z. Dai, X. Pan, N. Tang and J. Wu, Inorg. Chem., 2016, 55, 136 CrossRef CAS PubMed.
- J. Xiong, J. Zhang, Y. Sun, Z. Dai, X. Pan and J. Wu, Inorg. Chem., 2015, 54, 1737 CrossRef CAS PubMed.
- J. Zhang, C. Jian, Y. Gao, L. Wang, N. Tang and J. Wu, Inorg. Chem., 2012, 51, 13380 CrossRef CAS PubMed.
- J. Zhang, J. Xiong, Y. Sun, N. Tang and J. Wu, Macromolecules, 2014, 47, 7789 CrossRef CAS.
- A. Amgoune, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Chem. – Eur. J., 2006, 12, 169 CrossRef CAS PubMed.
- P. L. Arnold, J.-C. Buffet, R. P. Blaudeck, S. Sujecki, A. J. Blake and C. Wilson, Angew. Chem., Int. Ed., 2008, 47, 6033 CrossRef CAS PubMed.
- C. Bakewell, T.-P.-A. Cao, X. F. Le Goff, N. J. Long, A. Auffrant and C. K. Williams, Organometallics, 2013, 32, 1475 CrossRef CAS.
- C. Bakewell, T.-P.-A. Cao, N. Long, X. F. Le Goff, A. Auffrant and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 20577 CrossRef CAS PubMed.
- C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 9226 CrossRef CAS PubMed.
- C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Inorg. Chem., 2015, 54, 2204 CrossRef CAS PubMed.
- T.-P.-A. Cao, A. Buchard, X. F. Le Goff, A. Auffrant and C. K. Williams, Inorg. Chem., 2012, 51, 2157 CrossRef CAS PubMed.
- Z. Mou, B. Liu, X. Liu, H. Xie, W. Rong, L. Li, S. Li and D. Cui, Macromolecules, 2014, 47, 2233 CrossRef CAS.
- K. Nie, L. Fang, Y. Yao, Y. Zhang, Q. Shen and Y. Wang, Inorg. Chem., 2012, 51, 11133 CrossRef CAS PubMed.
- S. Yang, K. Nie, Y. Zhang, M. Xue, Y. Yao and Q. Shen, Inorg. Chem., 2014, 53, 105 CrossRef CAS PubMed.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229 CrossRef CAS PubMed.
- M. H. Chisholm, J. Gallucci and K. Phomphrai, Inorg. Chem., 2002, 41, 2785 CrossRef CAS PubMed.
- H. Wang, J. Guo, Y. Yang and H. Ma, Dalton Trans., 2016, 45, 10942 RSC.
- H. Wang and H. Ma, Chem. Commun., 2013, 49, 8686 RSC.
- H. Wang, Y. Yang and H. Ma, Macromolecules, 2014, 47, 7750 CrossRef CAS.
- H. Wang, Y. Yang and H. Ma, Inorg. Chem., 2016, 55, 7356 CrossRef CAS PubMed.
- S. Abbina and G. Du, ACS Macro Lett., 2014, 3, 689 CrossRef CAS PubMed.
- M. J. Walton, S. J. Lancaster and C. Redshaw, ChemCatChem, 2014, 6, 1892 CrossRef CAS.
- H. Xie, Z. Mou, B. Liu, P. Li, W. Rong, S. Li and D. Cui, Organometallics, 2014, 33, 722 CrossRef CAS.
- A. J. Chmura, D. M. Cousins, M. G. Davidson, M. D. Jones, M. D. Lunn and M. F. Mahon, Dalton Trans., 2008, 1437 RSC.
- E. L. Whitelaw, G. Loraine, M. F. Mahon and M. D. Jones, Dalton Trans., 2011, 40, 11469 RSC.
- A. J. Chmura, M. G. Davidson, M. D. Jones, M. D. Lunn, M. F. Mahon, A. F. Johnson, P. Khunkamchoo, S. L. Roberts and S. S. F. Wong, Macromolecules, 2006, 39, 7250 CrossRef CAS.
- M. D. Jones, S. L. Hancock, P. McKeown, P. M. Schafer, A. Buchard, L. H. Thomas, M. F. Mahon and J. P. Lowe, Chem. Commun., 2014, 50, 15967 RSC.
- P. A. Cameron, V. C. Gibson, C. Redshaw, J. A. Segal, A. J. P. White and D. J. Williams, J. Chem. Soc., Dalton Trans., 2002, 415 RSC.
- L. Cuesta-Aluja, A. Campos-Carrasco, J. Castilla, M. Reguero, A. M. Masdeu-Bultó and A. Aghmiz, J. CO2 Util., 2016, 14, 10 CrossRef CAS.
- J. B. L. Gallaway, J. R. K. McRae, A. Decken and M. P. Shaver, Can. J. Chem., 2012, 90, 419 CrossRef CAS.
- K. Press, A. Cohen, I. Goldberg, V. Venditto, M. Mazzeo and M. Kol, Angew. Chem., Int. Ed., 2011, 50, 3529 CrossRef CAS PubMed.
- E. L. Whitelaw, M. D. Jones and M. F. Mahon, Inorg. Chem., 2010, 49, 7176 CrossRef CAS PubMed.
- A. Yeori, S. Gendler, S. Groysman, I. Goldberg and M. Kol, Inorg. Chem. Commun., 2004, 7, 280 CrossRef CAS.
- E. Sergeeva, J. Kopilov, I. Goldberg and M. Kol, Chem. Commun., 2009, 3053 RSC.
- H. Glasner and E. Y. Tshuva, Inorg. Chem., 2014, 53, 3170 CrossRef CAS PubMed.
- J. Balsells, P. J. Carroll and P. J. Walsh, Inorg. Chem., 2001, 40, 5568 CrossRef CAS PubMed.
- A. Yeori, S. Groysman, I. Goldberg and M. Kol, Inorg. Chem., 2005, 44, 4466 CrossRef CAS PubMed.
- C. Fliedel, D. Vila-Viçosa, M. J. Calhorda, S. Dagorne and T. Avilés, ChemCatChem, 2014, 6, 1357 CrossRef CAS.
- E. L. Whitelaw, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 10004 RSC.
- A. J. Chmura, M. G. Davidson, M. D. Jones, M. D. Lunn and M. F. Mahon, Dalton Trans., 2006, 887 RSC.
- S. Gendler, S. Segal, I. Goldberg, Z. Goldschmidt and M. Kol, Inorg. Chem., 2006, 45, 4783 CrossRef CAS PubMed.
- M. Kol, M. Shamis, I. Goldberg, Z. Goldschmidt, S. Alfi and E. Hayut-Salant, Inorg. Chem. Commun., 2001, 4, 177 CrossRef CAS.
- A. L. Zelikoff, J. Kopilov, I. Goldberg, G. W. Coates and M. Kol, Chem. Commun., 2009, 6804 RSC.
- M. Bero, J. Kasperczyk and Z. J. Jedlinski, Makromol. Chem., 1990, 191, 2287 CrossRef CAS.
- B. Calvo, M. G. Davidson and D. García-Vivó, Inorg. Chem., 2011, 50, 3589 CrossRef CAS PubMed.
- M. T. Zell, B. E. Padden, A. J. Paterick, K. A. M. Thakur, R. T. Kean, M. A. Hillmyer and E. J. Munson, Macromolecules, 2002, 35, 7700 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full analysis of 1H and 13C{1H} NMR spectra and data are provided as well as examples of polymer characterisation. CCDC 1534471–1534479. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt00751e |
| This journal is © The Royal Society of Chemistry 2017 |