
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flexible organofunctional aerogels†
C. R.
Ehgartner
,
S.
Grandl
,
A.
Feinle
and
N.
Hüsing
 *
*
Materials Chemistry, Paris Lodron University Salzburg, Jakob-Haringer Street 2A, 5020 Salzburg, Austria. E-mail: nicola.huesing@sbg.ac.at
First published on 16th March 2017
Abstract
Flexible inorganic–organic silica aerogels based on methyltrimethoxysilane (MTMS, CH3Si(OCH3)3) can overcome the drawbacks of conventional silica aerogels by introducing high mechanical strength, elastic recovery and hydrophobicity to monolithic materials. In this work, MTMS is co-condensed with organofunctional alkoxysilanes RSi(OMe)3 (R = vinyl, chloropropyl, mercaptopropyl, methacryloxypropyl, etc.) yielding aerogels that are not only flexible but also contain reactive functional groups. Sol–gel parameters, such as the MTMS/RSi(OMe)3 ratio, have been systematically investigated in terms of gelation behavior, complete/incomplete incorporation of the functional organic groups (confirmed by FTIR-ATR and Raman spectroscopy) and flexibility of the resulting gel. Sterically more demanding functional moieties lead to macroscopic phase separation; however, this problem was overcome by the employment of surfactants. Functional aerogels dried by supercritical extraction with carbon dioxide showed promising results in uniaxial compression tests and had an elastic recovery up to 60%. Furthermore, the accessibility of the functional groups was demonstrated by simple reactions, e.g. conversion of the chloro into azido groups via a nucleophilic substitution reaction with NaN3 followed by click reactions.
Introduction
Conventional silica aerogels have a unique combination of properties, including high porosities (80–99.8%), high surface areas (100–1600 m2 g−1), low bulk densities (0.003–0.5 g cm−3), low Young's modulus (0.002–100 MPa) and low dielectric constants (1–1.24).1 However, their commercialized applications are still limited mainly due to their low mechanical strength. The filigree porous structure collapses easily even under small applied forces. In recent years numerous attempts have been made to improve the mechanical behavior of silica aerogels in terms of compression, bendability and the drying process. This can be achieved by aging the said aerogels in specific silane solutions, reinforcing the aerogels with fibers or by employing a polymer crosslinking approach.2–6 Another promising way to improve the mechanical properties is to make aerogels hydrophobic by post-treatment with surface modification agents or by the hybridization of silica aerogels via a co-condensation approach of silicon alkoxides with organosilane precursors that contain hydrophobic groups.7–11Recently, different flexible organic–inorganic hybrid aerogels were made solely from a methyltrimethoxysilane (MTMS) or vinyltrimethoxysilane (VTMS) precursor. Rao et al. produced flexible as well as bendable, superhydrophobic and low-density gels, that were dried supercritically and had a reversible elastic compression of up to 60%.12 Further studies by the group of Xu et al. showed that these gels can even be dried under ambient conditions by choosing the right conditions.13 Kanamori et al. showed that by employing suitable surfactants, MTMS aerogels can be made transparent.14 The group also reported an improved elastic recovery, where these aerogels can endure up to 80% of compression and then spring back to 95% of their original length. Shimizu et al. fabricated transparent polyvinylsilsesquioxane aerogels made from VTMS that show 100% resilience at 50% compression, achieved by a radical polymerization of the vinyl groups.15 “Marshmallow-like” aerogels were produced by Hayase et al., by combining MTMS with dimethyldimethoxysilan (DMDMS) to yield aerogels and xerogels that show unusual flexibility and bendability with a reversible compression up to 80%.16 One comment regarding the term aerogel at this point: a clear definition for an aerogel is somewhat lost with the manifold new compositions and properties. In some cases, the prepared gels show the well-known characteristic aerogel properties, such as an open-celled, mesoporous, solid network of interconnected nanostructures exhibiting porosities of no less than 50%, in others, the gel is meso/macroporous but dried supercritically which is a different definition for an aerogel based on the processing. In the course of this work, the term aerogel will be used even for a structure that exhibits pores larger than 50 nm.
The above mentioned gels all show interesting mechanical properties but lack chemical functionality. The methyl groups only render hydrophobicity to the system. Therefore, Hayase et al. used vinyltrimethoxysilane and vinylmethyldimethoxysilane instead of MTMS/DMDMS to create superamphiphobic “marsh-mallow-like” monoliths which can float on the surface of water and oil alike.17 The group of Xu reported flexible bridged silsesquioxane aerogels produced from a thiol–ene click reaction of mercaptopropyltrimethoxysilane and vinyltrimethoxysilane. The soft thioether segments result in materials that show a reversible compression of up to 50%.18 Yun et al. fabricated other highly flexible bridged silsesquioxane aerogels, resulting from a click reaction of VTMS and 2,2′-(ethylenedioxy)diethanethiol (EDDET), which get their flexibility (they can bend 180°) from the soft C–S and C–O bonds.19 Aravind et al. used a co-condensation reaction of MTMS and (2,3-epoxypropoxy)propyltrimethoxysilane, with the aid of a surfactant, to yield aerogels with impressive mechanical properties (reversible compression up to 70%).20
Nevertheless, there have been few reports of silsesquioxane aerogels that combine flexibility or bendability with active functional groups on their surface after sol–gel processing.21–23 So far and to the best of our knowledge, there is no study published on the variability and accessibility of these functionalities. Herein, we describe the first systematic investigation of the co-condensation approach of MTMS and further organofunctional alkoxysilanes RSi(OMe)3 with reactive functional groups, e.g. R = vinyl, chloropropyl, mercaptopropyl, and methacryloxypropyltrimethoxysilane, aided by a two-step acid–base sol–gel process. The co-condensation approach avoids time and cost intensive multi-step grafting and post-treatment processes. Special emphasis will be placed on the successful gelation behavior of the different organosilanes, the accessibility of the functional organic moieties and the flexibility of the resulting gel.
Experimental
Materials
Methyltrimethoxysilane (98% purity, MTMS), (3-mercaptopropyl)trimethoxysilane (95% purity, MPTMS), hexadecyltrimethylammonium bromide (98% purity, CTAB), methanol (99.8%, MeOH) and tetrakis(acetonitrile)copper(I) hexafluorophosphate were acquired from Sigma Aldrich. Oxalic acid, vinyltrimethoxysilane (97% purity, VTMS), Ellman's reagent (5,5′-dithiobis-(2-nitrobenzoic acid) or DTNB) and sodium azide (NaN3) were from Merck. (3-Chloropropyl)-trimethoxysilane (97% purity; CPTMS) and (3-methacryloxypropyl)trimethoxysilane (98% purity, MAPTMS) were purchased from ABCR. Propiolic acid (98%) was obtained from Acros Organics. Ammonium hydroxide (28% in H2O), sodium carbonate (Na2CO3), potassium permanganate (KMnO4), N,N-dimethylformamide (DMF), acetonitrile (ACN) and sodium azide were procured from VWR.Synthetic procedures
Functional polyorganosilsesquioxane aerogels and xerogels were prepared via a co-condensation approach of methyltrimethoxysilane (MTMS) and an organofunctional trialkoxysilane RSi(OMe)3 with a reactive functional group, e.g. R = vinyl (VTMS), chloropropyl (CPTMS), mercaptopropyl (MPTMS) or methacryloxypropyl (MAPTMS). The number of hydrolysable silicon centers was kept at a constant value of 8.3 mmol, and an increasing molar% of MTMS (10%, 15% and 20%) was substituted by the organofunctional silane, RSi(OMe)3. The amount of hexadecyltrimethylammonium bromide (CTAB) was increased as needed to obtain aerogels without macroscopic phase separation. All other ratios were kept constant with 249.7 mmol (8.0 g) MeOH, 33.3 mmol (0.6 g) aqueous oxalic acid (0.01 M) and 33.3 mmol (0.6 g) ammonium hydroxide solution (10 M). The exact ratios and starting compositions of the employed silanes and CTAB are listed in Table 1. The sols were prepared in a polystyrene container aided by a two-step acid–base approach similar to the one described by Rao et al., where first oxalic acid (0.01 M) is added under stirring to a mixture of methanol (MeOH), CTAB, MTMS and additional functional trialkoxysilane.12 This mixture is then stirred at RT for 24 h, when ammonium hydroxide solution (10 M) is added dropwise over 30 min. The mixture is then placed in closed containers in a ventilated oven at 40 °C for gelation. After gelation the gels are aged in a ventilated oven at 60 °C for 2 days. For the removal of residual surfactants the whole monoliths were completely immersed in water and then soaked three times in methanol (double the volume of the monoliths) with at least 8 h in between the subsequent solvent exchange cycles. The obtained alcogels were dried supercritically with CO2 at 45 °C and in the pressure range between 80–90 bar, to yield aerogels.| Sample | MTMS [g (mmol)] | RSi(OMe)3 [g (mmol)] | CTAB [g (mmol)] | GTa [h] | LSb [%] |
|---|---|---|---|---|---|
| a Gel time. b Linear shrinkage in comparison with the original container. | |||||
| RS | 1.13 (8.3) | — | — | 5 | 6.0 |
| V10 | 1.02 (7.5) | 0.12 (0.8) | 0.6(1.6) | 2 | 6.1 |
| V15 | 0.96 (7.1) | 0.18 (1.2) | 0.6(1.6) | 1.5–2 | 2.1 |
| V20 | 0.91 (6.7) | 0.25 (1.7) | 0.6(1.6) | 1.5 | 6.9 |
| CP10 | 1.02 (7.5) | 0.17 (0.8) | 0.6(1.6) | 1.5–2 | 6.5 |
| CP15 | 0.96 (7.1) | 0.25 (1.2) | 0.6(1.6) | 1–1.5 | 4.7 |
| CP20 | 0.91 (6.7) | 0.33 (1.7) | 0.8(2.2) | 1 | 6.3 |
| MP10 | 1.02 (7.5) | 0.16 (0.8) | 0.6(1.6) | 2.5 | 5.7 |
| MP15 | 0.96 (7.1) | 0.25 (1.2) | 0.8(2.2) | 2 | 7.5 |
| MP20 | 0.91 (6.7) | 0.33 (1.7) | 0.8(2.2) | 1.5 | 7.4 |
| MAP 10 | 1.02 (7.5) | 0.21 (0.8) | 0.6(1.6) | 2.5 | 5.7 |
| MAP15 | 0.96 (7.1) | 0.31 (1.2) | 0.6(1.6) | 2.5 | 6.4 |
| MAP 20 | 0.91 (6.7) | 0.41 (1.7) | 0.6(1.6) | 2 | 6.6 |
The samples are labeled as follows: the letters correspond to the kind of functional group incorporated into the RSiO1.5 matrix. The letter V stands for the vinyl group, CP for the chloropropyl group, MP for the mercaptopropyl group and MAP for the methacryloxypropyl group. The numbers correspond to the molar% of the portion of MTMS replaced by organotrialkoxysilanes (R = vinyl, chloropropyl, mercaptopropyl and methacryloxypropyl). For example, the sample CP10 was prepared with 10 molar% CPTMS and 90 molar% MTMS in which the molar% are related to the constant amount of hydrolysable silicon centers (8.3 mmol). The sample RS is a reference sample made only with MTMS and without CTAB.
Qualitative and quantitative determination of functional groups
Measurements
SEM images were taken with a scanning electron microscope (Zeiss ULTRA Plus) operating at 2–5 kV with an in-lens detector. Nitrogen adsorption–desorption measurements were carried out at 77 K using a Micrometrics ASAP 2420. The specific surface area was calculated with the Brunauer, Emmett and Teller 5-point method in the relative pressure range of 0.05–0.3.26 Prior to the measurement the samples were degassed in a vacuum for 12 h at 300 °C. Thermogravimetric analyses (TGA) were conducted using a Netzsch STA 449C Jupiter. The samples were heated from room temperature to 1000 °C with a heating rate of 10 K min−1 in air. Raman spectra of the samples were acquired on a Thermo DXR Raman microscope with a laser excitation at 532 nm. The measurements were performed in the wavenumber range from 100 cm−1 to 3500 cm−1, with an exposure time of 20 s and up to 10 accumulations. FTIR-ATR spectra were taken using a Bruker Vertex 70 spectrometer with a 4 cm−1 resolution. The spectra were measured in the wavenumber range from 500 cm−1 to 4500 cm−1. The bulk density (ρb) of the cylindrically shaped monoliths was calculated from their mass to volume ratio. The skeletal density (ρs) was measured on a Micromeritics AccuPyc II 1340. The porosity P was calculated using the equation P = [1 − ρb/ρs]/100. A uniaxial compression instrument (Latzke ZDT Universal testing machine) with a 100 N load was used for determining the elasticity and Young's modulus of the cylindrical samples. Compression was performed up to 60% with a compression rate of 1 mm min−1. The initial slope of the stress–strain curve was used to calculate the Young's Modulus (up to 20% of the strain). UV/Vis measurements were performed on a PerkinElmer Lambda 750 device. The absorbance of the Ellman's assay was recorded at 412 nm.Results and discussion
With appropriate synthetic sol–gel parameters, trialkoxysilanes can create flexible open-porous silsesquioxane gel networks. The organic moieties R of the silanes are covalently attached to the silicon atom and do not take part in the hydrolysis and condensation reaction. These moieties are incorporated as dangling bonds into the final network and lead to a lower crosslinking density and higher deformability compared to tetraalkoxysilane-based systems.27 Furthermore, steric and charge effects of trifunctional silanes with larger functional moieties (other than methyl, ethyl) severely hamper the successful condensation to form cross-linked monolithic networks.28 Often condensation of organosilanes results in competitive cyclization reactions that lead to soluble polyhedral oligomers, amorphous oligomers and other cage or ladder like structures.29 Monolithic silsesquioxane gels only form at extreme pH-values or by applying a two-step sol–gel process, with an acid-catalyzed hydrolysis followed by a base-catalyzed condensation.30 Methylsilsesquioxane systems (CH3SiO1.5) are the best investigated organosilane networks forming stable, flexible and superhydrophobic gels. Therefore, co-condensation of methyltrialkoxysilane with other functional organosilanes seems to be the rational approach towards functional and flexible silsesquioxane networks. Nevertheless, for applying a co-condensation approach there are a few things to consider: electronic and steric hindrances, different hydrolysis and condensation rates and control of the homogeneity are key issues when designing an experiment with different precursors.31 Another factor to consider is the hydrophobicity of the sol–gel network that results from the methyl group of the MTMS. In this work, the co-condensation and gelation behavior of MTMS with further organosilanes is carefully monitored. The phase diagram in Fig. 1 illustrates the relationship between the starting compositions of the silanes and the gelation behavior of the MTMS–CPTMS system. The axis on the left hand shows the weight% of MTMS employed, whereas the axis on the right shows the weight% of CPTMS in the starting composition. The filled circles indicate for which compositions successful gelation took place and where stable monolithic systems were obtained. The half circles show unsuccessful gelation attempts, where macroscopic phase separation, or precipitation was encountered. It can be clearly seen that without the employment of CTAB, formation of monolithic systems only occurs for the MTMS system (without CPTMS). This is due to the aforementioned polarity difference of the applied organotrialkoxysilanes. The siloxane polymers become increasingly immiscible with polar solvents in the process of polycondensation, which in turn gives rise to heterogeneous networks, macroscopic phase separation, or precipitation.32 This problem can be overcome by the employment of a suitable surfactant, e.g. the ionic surfactant CTAB.33 CTAB makes the siloxane condensates hydrophilic via the weak interactions between the hydrophobic chains of the CTAB salt. The complex relationship between the silane starting composition and the surfactant can be seen in Fig. 1. The axis at the bottom shows the increasing weight% of CTAB employed. By adjusting the molar ratios (see Table 1) between CTAB and silanes, uniform and flexible monolithic aerogels can be obtained and by increasing the molar percentage of trialkoxysilane (VTMS, CPTMS, MPTMS and MAPTMS) in comparison with MTMS, more surfactant (a minimum of 1.6 mmol) is needed to obtain homogeneous gelation and avoid macroscopic phase separation or precipitation. The compositions have to be adjusted independently for the different functional moieties investigated, but overall all functional groups showed similar trends. The gel times (Table 1) generally decrease with a decreasing amount of MTMS. The reference sample has the highest gelation time (5 h), while the samples with a molar ratio of MTMS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :organosilane 8:2 have the lowest (≤2 h). This is due to the different hydrolysis and condensation rates and different polarities of the organotrialkoxysilanes. Normally, one would expect that the bulky organic group (which increases from methyl to methacryloxypropyl) has an inhibitory effect on the sol–gel polymerization. The group of Loy et al. conducted studies on the gelation times of different organotrialkoxysilanes (e.g. MTMS, VTMS and chloromethyltrimethoxysilane) and found that under certain pH conditions, the vinyl and chloromethyl groups showed faster gelation than the MTMS gels and with other pH values an opposite trend was observed.29 The gelation time is therefore considerably affected by the pH, temperature, water content and the type of organic moiety used and the system becomes increasingly more complex with two different precursors, different polarities, hydrophobic moieties and the inclusion of surfactants. The linear shrinkage (Table 1) of the aerogels is relatively small compared to conventional silica aerogels due to the hydrophobic methyl groups and is similar to the reference sample made only with MTMS. The hydrophobic character reduces the interaction of the solvent (alcohol) to the aerogel surface and decreases interfacial energies and capillary stresses.34
:organosilane 8:2 have the lowest (≤2 h). This is due to the different hydrolysis and condensation rates and different polarities of the organotrialkoxysilanes. Normally, one would expect that the bulky organic group (which increases from methyl to methacryloxypropyl) has an inhibitory effect on the sol–gel polymerization. The group of Loy et al. conducted studies on the gelation times of different organotrialkoxysilanes (e.g. MTMS, VTMS and chloromethyltrimethoxysilane) and found that under certain pH conditions, the vinyl and chloromethyl groups showed faster gelation than the MTMS gels and with other pH values an opposite trend was observed.29 The gelation time is therefore considerably affected by the pH, temperature, water content and the type of organic moiety used and the system becomes increasingly more complex with two different precursors, different polarities, hydrophobic moieties and the inclusion of surfactants. The linear shrinkage (Table 1) of the aerogels is relatively small compared to conventional silica aerogels due to the hydrophobic methyl groups and is similar to the reference sample made only with MTMS. The hydrophobic character reduces the interaction of the solvent (alcohol) to the aerogel surface and decreases interfacial energies and capillary stresses.34
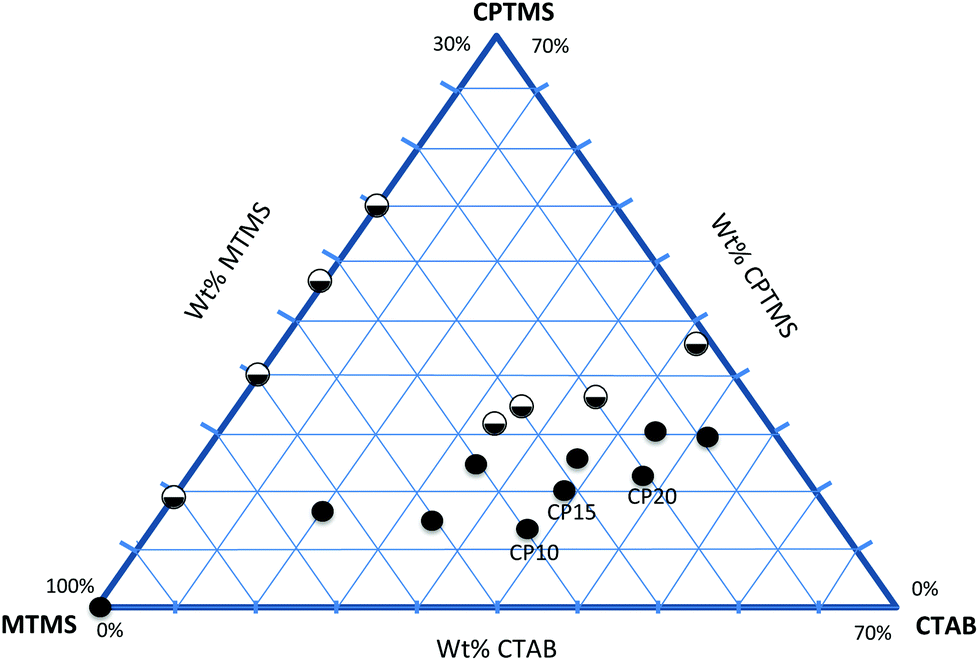 | ||
| Fig. 1 Relationship between starting compositions (in weight%) and successful gelation behavior. The filled circles represent successful gelation and the half circles stand for macroscopic phase separation/precipitation. | ||
Influence of chemical functionalization on micro-, macrostructure and mechanical properties
The properties of the CTAB optimized aerogels (supercritically dried) are listed in Table 2. This list only includes monolithic aerogels that showed the best mechanical properties in terms of flexibility. All displayed gels show low bulk densities between 0.04 g cm−3 and 0.06 g cm−3 and high porosities between 95% and 97%. These values are comparable with the MTMS based monolithic reference sample and of aerogels synthesized by the group of Rao showing densities of 0.037 g cm−3 and porosities of 98%.35| Sample |
ρ
ba [g cm−3] |
P [%] |
S
BETc [m2 g−1] |
E [kPa] | C [%] |
|---|---|---|---|---|---|
| a Bulk density. b Percentage porosity calculated by using the equation: P = [1 − ρb/ρs] × 100, where ρs is the skeletal density determined with He pycnometry. c Specific surface area determined using the BET model. d Youngs’ modulus determined from the initial slope of the stress–strain curve (up to 20% of the strain). e Elastic compressibility in comparison with the original length. | |||||
| CS | 0.067 | 97.0 | 508 | — | 60 |
| V10 | 0.051 | 96.5 | 388 | 12.0 | 60 |
| V15 | 0.041 | 97.2 | 300 | 7.9 | 50 |
| V20 | 0.056 | 96.2 | 376 | 5.9 | 60 |
| CP10 | 0.056 | 95.9 | 317 | 9.6 | 60 |
| CP15 | 0.056 | 95.9 | 263 | 5 | 60 |
| CP20 | 0.061 | 95.6 | 1 | 4.7 | 60 |
| MP10 | 0.045 | 96.4 | 319 | 7.3 | 60 |
| MP15 | 0.060 | 95.4 | 109 | 8.0 | 60 |
| MP20 | 0.051 | 96.0 | 1 | 7.6 | 50 |
| MAP 10 | 0.058 | 96.0 | 338 | 8.6 | 60 |
| MAP15 | 0.062 | 95.5 | 115 | 7.8 | 60 |
| MAP 20 | 0.058 | 96.1 | 21 | 7.2 | 60 |
SEM pictures of the samples display a globular aggregated cluster structure (Fig. 2). All investigated systems show the presence of large empty voids (at the micrometer scale) in the network, which are responsible for the opaque appearance of the monoliths. The large voids result from the strong dilution of the silanes in methanol (silane:MeOH molar ratio of 1:30), since higher precursor concentrations lead to denser aerogels. Another factor to consider is the presence of the organic dangling bonds (methyl, vinyl, chloropropyl, mercaptopropyl, and methacryloxypropyl) that decrease the crosslinking density of the aerogels. These rather large voids act as shock absorbers and are one of the main reasons why the aerogels exhibit flexible behavior.12 Increasing the molar% of trialkoxysilane (VTMS, CPTMS, MPTMS and MAPTMS) and decreasing the molar% of MTMS change the microstructure significantly. The size of the network forming particles and pores increases from nanosized particles to globular micrometer-sized particles, with a more spherical character and smoother surface. On comparing the morphological changes with the BET specific surface areas in Table 2, one can observe that with an increasing amount of functional groups the specific surface areas decrease significantly. Aerogels with a molar ratio of MTMS:organosilane of 9:1 have surface areas >300 m2 g−1. These values are comparable with the surface area of the reference sample and MTMS xerogels synthesized according to Xu et al., that have values between 300 and 400 m2 g−1.36 Aerogels with a molar ratio of MTMS:organosilane of 8:2 have much lower values <25 m2 g−1.
 | ||
| Fig. 2 SEM images of functionalized aerogels prepared with different molar% of the organosilanes VTMS/CPTMS/MPTMS/MAPTMS. | ||
The only exceptions are VTMS-modified aerogels, probably due to the comparable molar mass and polarity of the vinyl moieties and the methyl groups. The adsorption–desorption isotherms (see the ESI†) show that the aerogels have larger pores, but experience a loss of the mesoporous character and of the adsorbed volume in comparison with the reference sample with increasing degree of organic functionalization. All samples generally show features of type IV isotherms, with a narrow H3 hysteresis loop, according to IUPAC classification.37–39 H3 hysteresis loops indicate the presence of slit-shaped mesopores. On the other hand, it was reported in the literature that during BET analysis the network of highly flexible and soft aerogels is significantly compressed. Therefore nitrogen cannot penetrate into the compressed pores leading to lower specific surface areas and pore volumes.39–41 An indicator for this phenomenon is the open hysteresis loops for samples with higher loadings of functional organic moieties (over 10 molar%). If the slit-like pores are compressed during the course of the nitrogen sorption measurement, nitrogen cannot escape from the pores. To gain more insight if the decrease in the surface area and the loss of the mesoporous character are related to the measurement set-up and the softness of the samples further studies have to be conducted.
A photograph of a chloropropyl-modified sample undergoing compression and the corresponding stress and strain curves (see Fig. 3a/b) of three different chloropropyl-modified aerogels demonstrate the high reversible compressibility, flexibility and the elastomeric behavior of the chloropropyl-modified silsesquioxane gel samples. The shown examples (CP10, CP15 and CP20) were compressed to 60% of their original length and recovered to 100% after this treatment. All presented samples (V10–20, CP10–20, MP10–20 and MAP10–20) in Table 2 show this reversible compression behavior between 50 and 60%.
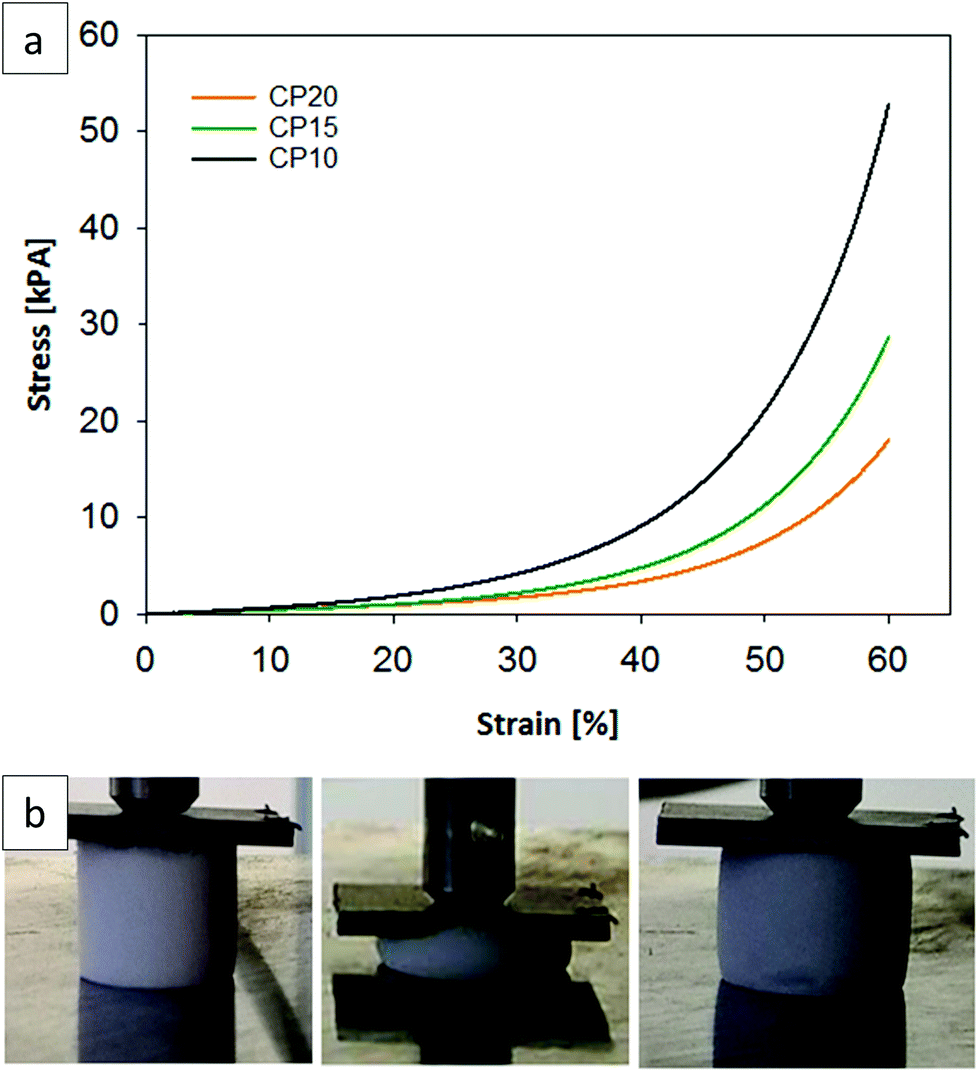 | ||
| Fig. 3 (a) Stress–strain curves of the chloropropyl functionalized gels (CP10, CP15 and CP20), (b) uniaxial compression test (60% of the original height) on a CP10 sample showing reversible compression. | ||
The stress and strain curves and the calculated Young's modulus (Table 2) indicate that the samples become softer with an increasing molar percentage of the organofunctional silanes. Sample CP10 has a Young's Modulus of 9.6 kPa, whereas sample CP20 has a lower value of 4.7 kPa. The highest Young's modulus was reached for the 10% vinyl functionalized sample, with a value of 12 kPa. The original system of Rao et al. had a Young's Modulus of 30 kPa and conventional silica aerogels have even higher values (0.002–100 MPa).35,42 This reveals that functionalization with sterically more demanding moieties creates softer and more open porous networks resulting from hindered condensation processes and weaker particle connectivity.
Determination of the functional groups
The presence of the functional groups was confirmed by Raman spectroscopy (see Fig. 4). The silica fingerprint region of all samples is located in the range between 1250 and 700 cm−1, where the bands between 1000 and 1250 cm−1 can be assigned to Si–O–Si bonds. Raman bands at 791 cm−1 correspond to the symmetric mode of the Si–O–Si vibration and the band at 472 cm−1 is associated with the siloxane ring breathing mode. Moreover, bands attributed to the symmetric and antisymmetric stretching of the methyl group (–CH3) can be identified at 2979 and 2916 cm−1, respectively. The Raman spectrum of aerogels containing vinyl groups is shown in Fig. 4a. For this sample it was possible to identify four bands corresponding to the vinyl group. The band at 3072 cm−1, the stretching vibration at 1603 cm−1 and the bending vibration at 1412 cm−1 and 1278 cm−1 are attributed to a carbon double bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C). The case of the chloropropyl-modified sample is shown in Fig. 4b. Bands at 1445 and 1412 cm−1 are attributed to the angular deformations of the H–C–Cl group and the bands at 646 cm−1 correspond to the C–Cl bond. In Fig. 4c the Raman spectrum of the aerogel containing mercaptopropyl groups is displayed. A well-defined band is detected at 2574 cm−1, which corresponds to the stretching vibration of the S–H bond. Other bands were observed at 1260 cm−1 for the vibration of the S–CH2 group and at 651 cm−1 for the stretching vibration of the C–S bond. Finally Fig. 4d shows the spectrum of a methacryloxypropyl modified sample. The band at 1720 cm−1 is attributed to the CO double bond and the vibration of the CC double bond can be found at 1640 cm−1 and at 3072 cm−1.43
C). The case of the chloropropyl-modified sample is shown in Fig. 4b. Bands at 1445 and 1412 cm−1 are attributed to the angular deformations of the H–C–Cl group and the bands at 646 cm−1 correspond to the C–Cl bond. In Fig. 4c the Raman spectrum of the aerogel containing mercaptopropyl groups is displayed. A well-defined band is detected at 2574 cm−1, which corresponds to the stretching vibration of the S–H bond. Other bands were observed at 1260 cm−1 for the vibration of the S–CH2 group and at 651 cm−1 for the stretching vibration of the C–S bond. Finally Fig. 4d shows the spectrum of a methacryloxypropyl modified sample. The band at 1720 cm−1 is attributed to the CO double bond and the vibration of the CC double bond can be found at 1640 cm−1 and at 3072 cm−1.43
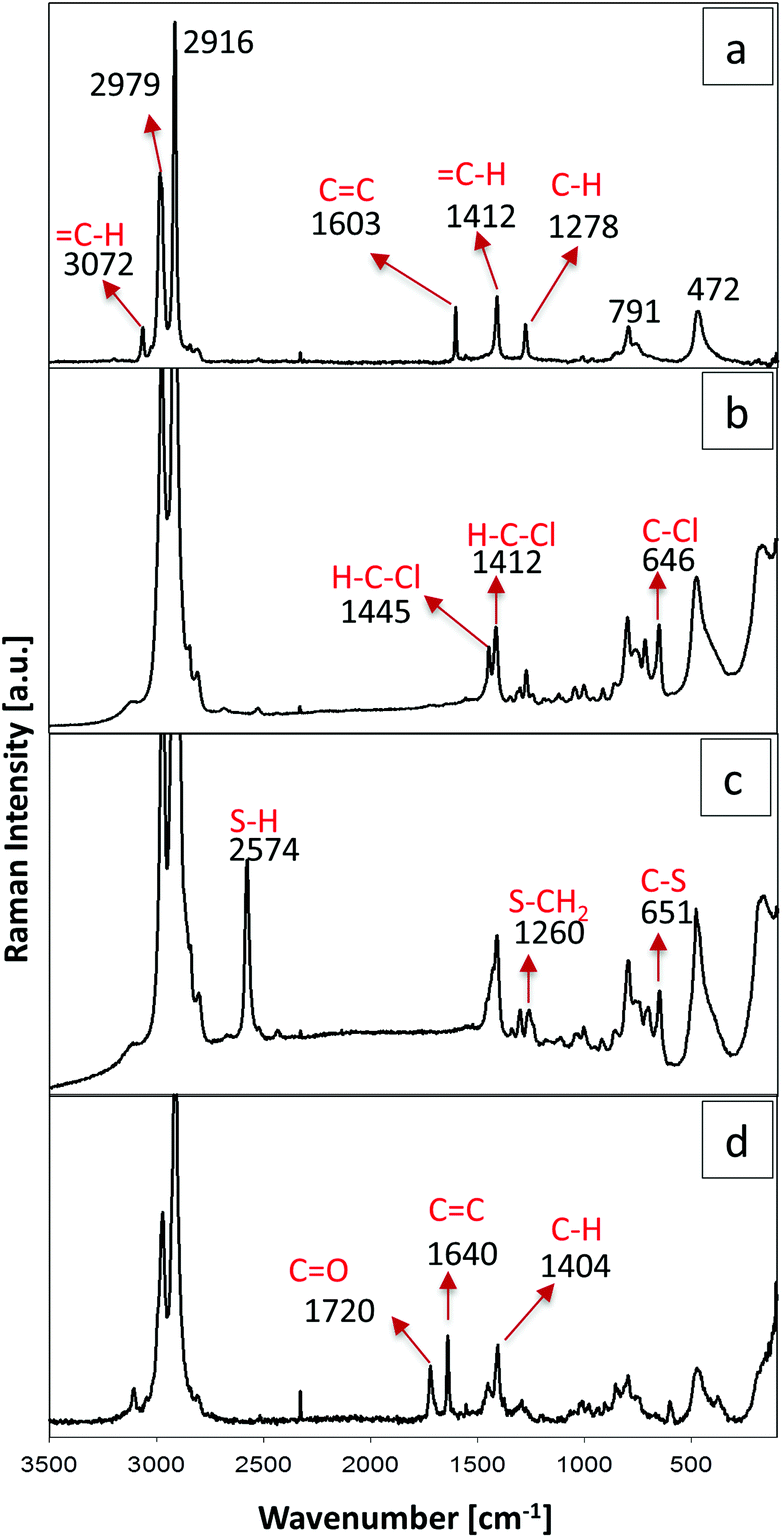 | ||
| Fig. 4 Raman spectra of the samples (a) V20, (b) CP20, (c) MP20 and (d) MAP20. | ||
The thermal stability of the organic units of the organic–inorganic hybrid aerogel samples was analyzed by thermogravimetric analyses (see Fig. 5). There is only a negligible weight loss up to 200 °C, which can be attributed to the hydrophobic character of the samples. The next weight loss step corresponds to the oxidation of the organic groups. The functional moieties (vinyl, chloropropyl, methacryloxypropyl and mercaptopropyl) start to decompose above 250–260 °C. The weight loss between 450–460 °C is associated with the degradation of the methyl groups and condensation reactions of residual silanols and alkoxides. Overall decomposition is complete at 700 °C. The final weight loss correlates very well with the molar ratios of the precursors and the molar mass of the respective organic moiety, which is indicated by the negligible difference between the theoretical and actual weight loss (see Table 3). The actual weight loss is often higher than the calculated one, indicating that the surfactant has not been completely removed by the given washing process. The amount of functional groups (mmol g−1) that are incorporated into the silica network can also be quantitatively determined by thermogravimetric analyses using the following formula (1):
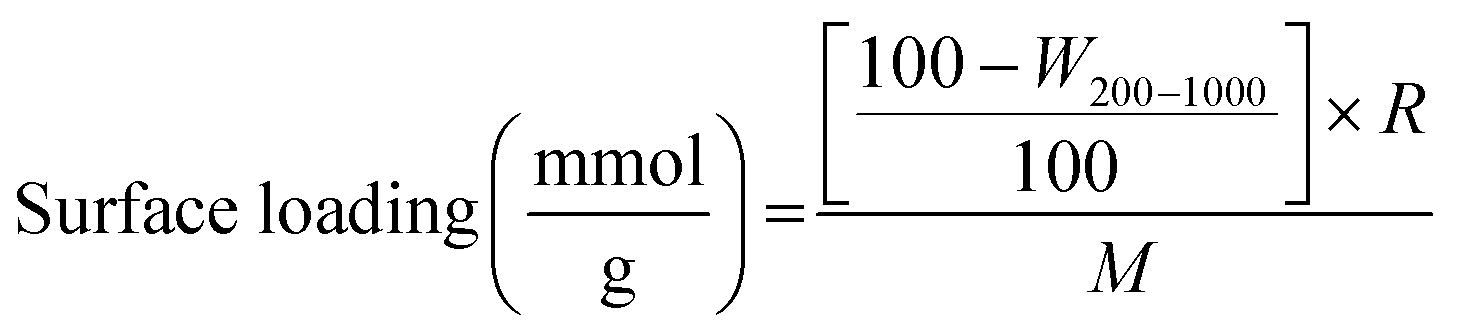 | (1) |
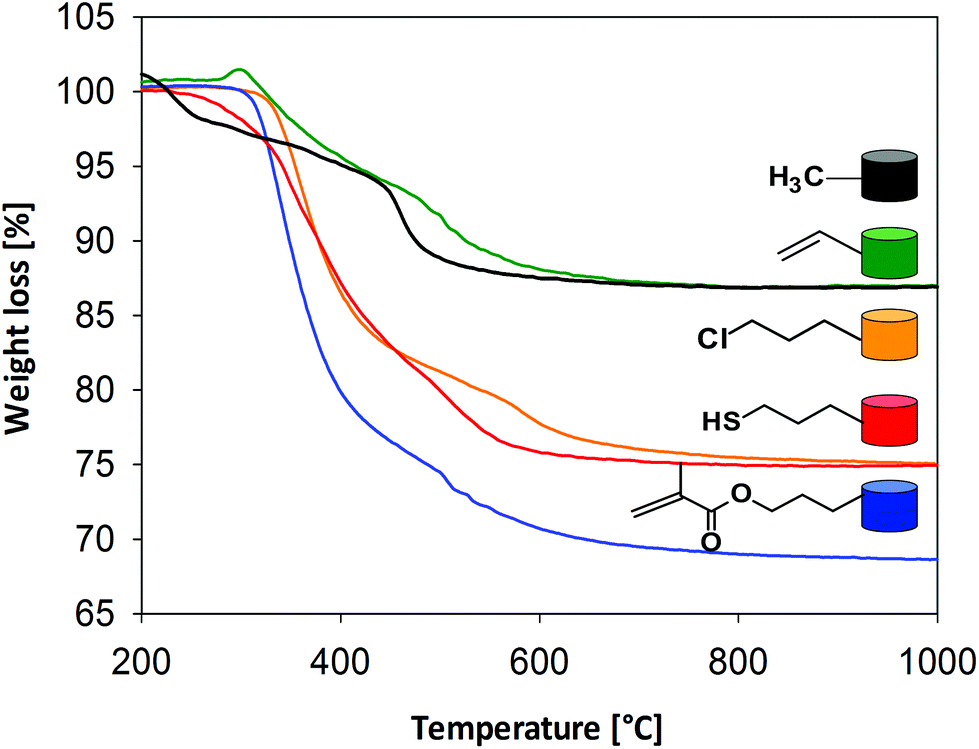 | ||
| Fig. 5 TGA analysis of the samples (a) V20, (b) CP20, (c) MP20 and (d) MAP20 under oxygen flow. | ||
| Sample | Weight loss [%] | Calc. weight loss [%] | Surface loading [mmol g−1] |
|---|---|---|---|
| V10 | 12 | 12 | 0.44 |
| V15 | 14 | 13 | 0.70 |
| V20 | 13 | 13 | 0.99 |
| CP10 | 20 | 17 | 0.24 |
| CP15 | 23 | 21 | 0.42 |
| CP20 | 26 | 23 | 0.65 |
| MP10 | 20 | 18 | 0.24 |
| MP15 | 23 | 21 | 0.42 |
| MP20 | 25 | 24 | 0.65 |
| MAP 10 | 22 | 23 | 0.18 |
| MAP15 | 26 | 28 | 0.33 |
| MAP 20 | 31 | 33 | 0.51 |
Functional group accessibility
Aerogels that are functional and flexible have manifold potential applications, such as an absorbent, sensor or catalytic (support) materials, due to their high surface areas and the possibilities given by the functional moiety, e.g. affinity to bind specific molecules, etc. The mercapto moiety is a prime example of a functional group that can bind heavy metals and such aerogels are therefore of high interest in water purification applications.44 The functional group density as well as their accessibility is a key factor for the applicability. Fig. 6 shows some examples that indicate the successful accessibility of the functional groups within the synthesized aerogels. In Fig. 6a it can be seen that the double bond of the vinyl and methacryl groups (of the V20 and MAP20 aerogels) successfully reacts in redox reactions with potassium permanganate, by turning the purple permanganate solution to a yellow-brownish color forming manganese(IV) oxide and a diol. The solution remained purple for the methyl group containing reference sample. The photographs in Fig. 6b depict a chloropropyl-modified monolith (CP20) before (left) and after (right) nucleophilic substitution followed by a click reaction. First, the CP20 monolith was reacted with sodium azide to yield azido functionalized gels as confirmed by ATR FT-IR spectroscopy (see the ESI†), with a new band appearing at 2100 cm−1 that is attributed to the stretching vibration of the double bond of the azido group.24 Second, the nucleophilic substitution was followed by a copper(I) catalyzed click reaction with propiolic acid. The successful click reaction step was confirmed by the disappearance of the characteristic azido band (see the FT-IR spectrum in the ESI†) and by a color change of the monolith to blue (see Fig. 6b/vi). Mercapto groups are qualitatively determined by using Ellman's reaction which is commonly used for the determination of thiol group contents in protein samples.45Fig. 6c shows a mercapto modified monolith in methanol (left hand side) and in an Ellman's reactant/buffer solution (right hand side), where the conversion to the yellow product (the TNB2− ion) of the Ellman's reactant is clearly evident. This reaction also allows for a quantitative determination of the thiol groups in the system by simple UV/vis spectroscopy, thus, being a direct indicator for the accessibility of the (–SH) group towards other reagents. Results of the Ellman's assay are displayed in Table 4. Interestingly, the accessibility of the functional (–SH) groups decreases with the increasing theoretical mercapto content (from MP10 with 4% to MP20 with 0.2%) of the samples. This can be explained by taking the decreasing specific surface areas into account. The sample MP10 has a much higher surface area than the MP20 sample and has therefore more accessible functional groups available for the reaction with the Ellman's reactant. The generally low accessibility (∼4%) of the MP10 sample results from the hydrophobic character of the gels. The assay is conducted in an aqueous medium and the reactant solution cannot fully penetrate the monoliths. It is suspected that the accessibility is much higher in nonpolar solvents and will be investigated more thoroughly in future research. | ||
| Fig. 6 Demonstration of functional group accessibility: (a) potassium permanganate solution (i) without a monolith, (ii) with V20 alcogel, (iii) with a pure MTMS alcogel and (iv) with VTMS silane; (b) (v) CP20 alcogel and (vi) CP20 alcogel after click reaction; (c) (vii) MP20 alcogel in methanol and (viii) MP20 alcogel immersed in Ellman's reactant/buffer solution. | ||
| Sample | Abs. 412 nm [−] | n NTB2− [mmol L−1] | c MPTMS [mmol g−1] | Accessibility [%] |
|---|---|---|---|---|
| MP10 | 0.36 | 0.032 | 0.008 | 4.2 |
| MP15 | 0.18 | 0.016 | 0.012 | 1.4 |
| MP20 | 0.03 | 0.003 | 0.016 | 0.2 |
Conclusions
Organofunctional monolithic aerogels with relatively large functional moieties (such as vinyl, mercaptopropyl, chloropropyl and methacryloxypropyl) could be synthesized via a one-pot co-condensation approach with the aid of a surfactant. All presented functional gels showed an excellent elastic compression behavior up to 60% and had accessible functional groups. The co-condensation with MTMS proved to be fruitful, since it gave the aerogels additional hydrophobic character that aided the drying and handling processes of the aerogels. The results illustrate that the bulk density, linear shrinkage and the porosity of the aerogels remain similar with increasing size of the functional moieties. On the other hand the specific surface area and the accessibility of the functional group decrease with further modification of the gel system. The ratio of MTMS:organofunctional silane should therefore be kept above 8.5:1.5, otherwise the aerogels will lose a part of their interesting characteristics and will get too soft for further handling. The redox, nucleophilic substitution and click reactions illustrate the suitability of the monolithic systems for further chemical modifications. Future applications including absorbent materials in non-polar solvents or mechanoresponsive materials will be further investigated.
Acknowledgements
The authors thank M. Suljic for nitrogen sorption measurements and Professor M. Musso for Raman measurements performed at the University of Salzburg. We also gratefully acknowledge Maria Schestakow and Barbara Milow for being able to measure He-pycnometry and uniaxial compression test at DLR Cologne.References
- N. Hüsing and U. Schubert, Angew. Chem., Int. Ed., 1998, 37, 22–45 CrossRef.
- H. Maleki, L. Durães and A. Portugal, J. Non-Cryst. Solids, 2014, 385, 55–74 CrossRef CAS.
- L. Li, B. Yalcin, B. N. Nguyen, M. A. B. Meador and M. Cakmak, ACS Appl. Mater. Interfaces, 2009, 1, 2491–2501 CAS.
- M. Shi, C. Tang, X. Yang, J. Zhou, F. Jia, Y. Han and Z. Li, RSC Adv., 2017, 7, 4039–4045 RSC.
- M. A. B. Meador, E. F. Fabrizio, F. Ilhan, A. Dass, G. Zhang, P. Vassilaras, J. C. Johnston and N. Leventis, Chem. Mater., 2005, 17, 1085–1098 CrossRef CAS.
- A. Katti, N. Shimpi, S. Roy, H. Lu, E. F. Fabrizio, A. Dass, L. A. Capadona and N. Leventis, Chem. Mater., 2006, 18, 285–296 CrossRef CAS.
- A. V. Rao, M. M. Kulkarni, D. Amalnerkar and T. Seth, Appl. Surf. Sci., 2003, 206, 262–270 CrossRef CAS.
- Y. Duan, S. C. Jana, B. Lama and M. P. Espe, J. Non-Cryst. Solids, 2016, 437, 26–33 CrossRef CAS.
- S. Cui, Y. Liu, M.-H. Fan, A. T. Cooper, B.-L. Lin, X.-Y. Liu, G.-F. Han and X.-D. Shen, Mater. Lett., 2011, 65, 606–609 CrossRef CAS.
- L. Martin, J. O. Osso, S. Ricart, A. Roig, O. Garcia and R. Sastre, J. Mater. Chem., 2008, 18, 207–213 RSC.
- A. V. Rao, M. M. Kulkarni, G. M. Pajonk, D. P. Amalnerkar and T. Seth, J. Sol-Gel Sci. Technol., 2003, 27, 103–109 CrossRef CAS.
- A. V. Rao, S. D. Bhagat, H. Hirashima and G. Pajonk, J. Colloid Interface Sci., 2006, 300, 279–285 CrossRef CAS PubMed.
- B. Xu, J. Y. Cai, N. Finn and Z. Cai, Microporous Mesoporous Mater., 2012, 148, 145–151 CrossRef CAS.
- K. Kanamori, M. Aizawa, K. Nakanishi and T. Hanada, Adv. Mater., 2007, 19, 1589–1593 CrossRef CAS.
- T. Shimizu, K. Kanamori, A. Maeno, H. Kaji, C. M. Doherty, P. Falcaro and K. Nakanishi, Chem. Mater., 2016, 28, 6860–6868 CrossRef CAS.
- G. Hayase, K. Kanamori and K. Nakanishi, J. Mater. Chem., 2011, 21, 17077–17079 RSC.
- G. Hayase, K. Kanamori, G. Hasegawa, A. Maeno, H. Kaji and K. Nakanishi, Angew. Chem., Int. Ed., 2013, 52, 10788–10791 CrossRef CAS PubMed.
- Z. Wang, Z. Dai, J. Wu, N. Zhao and J. Xu, Adv. Mater., 2013, 25, 4494–4497 CrossRef CAS PubMed.
- S. Yun, H. Luo and Y. Gao, J. Mater. Chem. A, 2015, 3, 3390–3398 CAS.
- P. R. Aravind, P. Niemeyer and L. Ratke, Microporous Mesoporous Mater., 2013, 181, 111–115 CrossRef CAS.
- H. Guo, B. N. Nguyen, L. S. McCorkle, B. Shonkwiler and M. A. B. Meador, J. Mater. Chem., 2009, 19, 9054–9062 RSC.
- G. Hayase, K. Kanamori, M. Fukuchi, H. Kaji and K. Nakanishi, Angew. Chem., Int. Ed., 2013, 52, 1986–1989 CrossRef CAS PubMed.
- T. Matias, C. Varino, H. C. de Sousa, M. E. Braga, A. Portugal, J. F. Coelho and L. Durães, J. Mater. Sci., 2016, 51, 6781–6792 CrossRef CAS.
- M. Keppeler and N. Hüsing, New J. Chem., 2011, 35, 681–690 RSC.
- G. S. Irmukhametova, G. A. Mun and V. V. Khutoryanskiy, Langmuir, 2011, 27, 9551–9556 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- H. Dong and J. D. Brennan, Chem. Mater., 2006, 18, 541–546 CrossRef CAS.
- L. Matějka, O. Dukh, D. Hlavatá, B. Meissner and J. Brus, Macromolecules, 2001, 34, 6904–6914 CrossRef.
- D. A. Loy, B. M. Baugher, C. R. Baugher, D. A. Schneider and K. Rahimian, Chem. Mater., 2000, 12, 3624–3632 CrossRef CAS.
- H. Dong, M. A. Brook and J. D. Brennan, Chem. Mater., 2005, 17, 2807–2816 CrossRef CAS.
- U. Schubert, N. Hüsing and A. Lorenz, Chem. Mater., 1995, 7, 2010–2027 CrossRef CAS.
- K. Kanamori and K. Nakanishi, Chem. Soc. Rev., 2011, 40, 754–770 RSC.
- G. Hayase, K. Kanamori and K. Nakanishi, Microporous Mesoporous Mater., 2012, 158, 247–252 CrossRef CAS.
- H. Xu, Y. Huang, L. Liu, J. Song, C. Wang and L. Zhang, J. Non-Cryst. Solids, 2010, 356, 1837–1841 CrossRef CAS.
- N. D. Hegde and A. V. Rao, J. Mater. Sci., 2007, 42, 6965–6971 CrossRef CAS.
- B. Xu, J. Y. Cai, Z. Xie, L. Wang, I. Burgar, N. Finn, Z. Cai and L. Wong, Microporous Mesoporous Mater., 2012, 148, 152–158 CrossRef CAS.
- K. S. Sing, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- M. Kruk and M. Jaroniec, Chem. Mater., 2001, 13, 3169–3183 CrossRef CAS.
- S. D. Bhagat, C.-S. Oh, Y.-H. Kim, Y.-S. Ahn and J.-G. Yeo, Microporous Mesoporous Mater., 2007, 100, 350–355 CrossRef CAS.
- G. W. Scherer, D. M. Smith, X. Qiu and J. M. Anderson, J. Non-Cryst. Solids, 1995, 186, 316–320 CrossRef CAS.
- P. Aravind and G. Soraru, J. Porous Mater., 2011, 18, 159–165 CrossRef CAS.
- N. Hüsing and U. Schubert, Angew. Chem., Int. Ed., 1998, 110, 22–47 CrossRef.
- L. B. Capeletti, I. M. Baibich, I. S. Butler and J. H. dos Santos, Spectrochim. Acta, Part A, 2014, 133, 619–625 CrossRef CAS PubMed.
- S. Štandeker, A. Veronovski, Z. Novak and Ž. Knez, Desalination, 2011, 269, 223–230 CrossRef.
- I. Bravo-Osuna, D. Teutonico, S. Arpicco, C. Vauthier and G. Ponchel, Int. J. Pharm., 2007, 340, 173–181 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data. See DOI: 10.1039/c7dt00558j |
| This journal is © The Royal Society of Chemistry 2017 |