
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of coenzyme Q0 through divanadium-catalyzed oxidation of 3,4,5-trimethoxytoluene with hydrogen peroxide†
Olga V.
Zalomaeva
 a,
Vasilii Yu.
Evtushok
ab,
Gennadii M.
Maksimov
a,
Raisa I.
Maksimovskaya
a and
Oxana A.
Kholdeeva
*ab
a,
Vasilii Yu.
Evtushok
ab,
Gennadii M.
Maksimov
a,
Raisa I.
Maksimovskaya
a and
Oxana A.
Kholdeeva
*ab
aBoreskov Institute of Catalysis, Pr. Ac. Lavrentieva 5, Novosibirsk 630090, Russia. E-mail: khold@catalysis.ru; Fax: +7-383-330-9573; Tel: +7-383-326-9433
bNovosibirsk State University, Pirogova str. 2, Novosibirsk 630090, Russia
First published on 20th March 2017
Abstract
The selective oxidation of methoxy/methyl-substituted arenes to the corresponding benzoquinones has been first realized using aqueous hydrogen peroxide as a green oxidant, acid tetrabutylammonium salts of the γ-Keggin divanadium-substituted phosphotungstate [γ-PW10O38V2(μ-O)2]5− (I) as a catalyst, and MeCN as a solvent. The presence of the dioxovanadium core in the catalyst is crucial for the catalytic performance. The reaction requires an acid co-catalyst or, alternatively, a highly protonated form of I can be prepared and employed. The industrially relevant oxidation of 3,4,5-trimethoxytoluene gives 2,3-dimethoxy-5-methyl-1,4-benzoquinone (ubiquinone 0 or coenzyme Q0, the key intermediate for coenzyme Q10 and other essential biologically active compounds) with 73% selectivity at 76% arene conversion. The catalyst retains its structure under turnover conditions and can be easily recycled and reused without significant loss of activity and selectivity.
Introduction
The selective aromatic oxidation of arenes bearing methoxy and methyl substituents offers an efficient access to a range of benzoquinones, which play an important role in biological systems and are useful intermediates in the synthesis of fine chemicals, nutraceuticals and pharmaceuticals.1 Ubiquinones (also called coenzymes Qn, among which Q10 is the most known coenzyme) act as biochemical oxidizing agents that mediate electron-transfer processes involved in energy production.2,3 Their synthetic analogue, idebenone, has been developed as a drug for the treatment of Alzheimer's disease and other cognitive defects.4 2,3-Dimethoxy-5-methyl-1,4-benzoquinone, known as ubiquinone 0 or coenzyme Q0 (CoQ0), can serve as a key intermediate in the synthesis of coenzyme Q10 and other ubiquinones (Scheme 1).5,6 | ||
| Scheme 1 Bioactive compounds derived from CoQ0. | ||
The selective oxidation of methoxytoluenes to the corresponding p-benzoquinones can be accomplished using dimethyldioxirane as an oxidant, mineral acid as a catalyst and acetone as a solvent.7 Several synthetic methods were reported for the production of CoQ0 through the oxidation of commercially available 3,4,5-trimethoxytoluene (TMT) with the green oxidant – hydrogen peroxide.8–16 Among catalysts applied were potassium hexacyanoferrate(III),8 methyltrioxorhenium(VII) (MTORe),11,12 mineral acids (H2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10,16 or HNO315), and heteropolyacids of the general formula HnXM12O40 (where X = P or Si, n = 3 (P) or 4 (Si), and M = Mo or W).13,14 So far, the best yields of CoQ0 have been claimed for systems that employed formic acid as a solvent and phosphomolybdic heteropolyacid as a catalyst14 or a mixture of acetic and formic acids without any catalysts.16 However, the use of hydrogen peroxide in combination with carboxylic acids imposes practical problems related to reactor corrosion and safety risks associated with the in situ formation of explosive peroxy acids. Growing environmental concerns stimulated the development of more safe and sustainable catalytic methods for the production of methoxy-substituted p-benzoquinones.1d,17,18
10,16 or HNO315), and heteropolyacids of the general formula HnXM12O40 (where X = P or Si, n = 3 (P) or 4 (Si), and M = Mo or W).13,14 So far, the best yields of CoQ0 have been claimed for systems that employed formic acid as a solvent and phosphomolybdic heteropolyacid as a catalyst14 or a mixture of acetic and formic acids without any catalysts.16 However, the use of hydrogen peroxide in combination with carboxylic acids imposes practical problems related to reactor corrosion and safety risks associated with the in situ formation of explosive peroxy acids. Growing environmental concerns stimulated the development of more safe and sustainable catalytic methods for the production of methoxy-substituted p-benzoquinones.1d,17,18
In 2012, Mizuno and co-workers discovered an efficient system for hydroxylation of arenes that involved the divanadium-substituted γ-Keggin phosphotungstate (Bu4N)4[γ-PW10O38V2(μ-O)(μ-OH)] (hereinafter, TBA4H-I) as a catalyst, mineral acid as a co-catalyst, H2O2 as an oxidant, and MeCN/t-BuOH (1:1) as a solvent.19 At a substrate to oxidant ratio of 50, mono- and dialkyl(alkoxy)arenes afforded the corresponding phenols with excellent chemoselectivity and unusual regioselectivity. More recently, we investigated the catalytic performance of TBA4H-I in some industrially important reactions, such as oxidation of 2,3,6-trimethylphenol (TMP)20 and pseudocumene21 to 2,3,5-trimethyl-p-benzoquinone (TMBQ, Vitamin E precursor) using H2O2, and found that TMP can be converted to TMBQ with a nearly quantitative yield and 80–90% oxidant utilization efficiency.20 In contrast to the oxidation of alkylarenes, this reaction did not require an acid co-catalyst.
In the present work, we explored further the catalytic properties of TBA5−nHn-I (n = 1–2) in H2O2-based aromatic oxidation and first employed this catalyst system to accomplish the challenging oxidative transformation of TMT into coenzyme Q0 (Scheme 2). The extension of this method toward other methoxyarenes is also reported. We carefully investigated the role of the acid co-catalyst and a possibility to replace it by using a highly protonated form of I. The catalyst stability and recyclability issues have also been addressed.
 | ||
| Scheme 2 Oxidation of TMT in the presence of TBA5−nHn-I. | ||
Results
In most procedures reported in the literature for TMT oxidation with H2O2, the reaction proceeds in acidic medium, such as acetic or formic acid.9–11,13,15,16 Some of them require addition of mineral acid in catalytic amounts to accelerate the formation of peroxy acid, which is the real oxidizing species in these systems.10,15,16 In view of our interest in the development of a more safe and environmentally friendly approach to the synthesis of CoQ0, we have checked whether a mineral acid can play a role of the sole catalyst in a less corrosive and harmful solvent, for example, MeCN. While TMT conversion was negligible without any catalyst (Table 1, entry 1), in the presence of HClO4, it attained 22% after 40 min at 60 °C. However, selectivity toward the target product was low (Table 1, entry 2) and the yield of CoQ0 did not exceed 4%.Previously, the group of Mizuno demonstrated that efficient hydroxylation of arenes can be realized in the presence of the divanadium-substituted polyoxometalate (POM) TBA4H-I and 1 equiv. of HClO4 as the co-catalyst.19,21 They rationalized the effect of the co-catalyst in terms of the in situ generation of a catalytically active diprotonated form of I, TBA3[γ-PW10O38V2(μ-OH)2] (TBA3H2-I).19 To minimize the use of the acid co-catalyst, we attempted to develop a simple and affordable procedure for the preparation of a TBA salt of polyanion I that would contain an increased amount of protons as counter cations. Some modifications of the previously reported synthetic procedure,22viz. additional acidification of the reaction mixture (pH 0.8 versus pH 2.0 used for the preparation of TBA4H-I) before final precipitation with TBABr (see Experimental for details), allowed us to obtain POM with an increased amount of protons (1.5–1.7 H+ per POM molecule). IR spectroscopy corroborated the retention of the polyanion structure (Fig. S1†). In dry dilute MeCN solution, such POM revealed two separate 31P NMR signals at −13.7 and −14.1 ppm, which according to the literature,22 can be assigned to di- and monoprotonated forms, TBA3H2-I and TBA4H-I, respectively. Fig. 1 shows a typical 31P NMR spectrum where the ratio of the two signals is ca. 2:1, which implies the formulation of TBA3.3H1.7-I. The corresponding 51V NMR spectrum revealed two poorly resolved signals at −579 (TBA3H2-I) and −581 (TBA4H-I) ppm (Fig. S2 in ESI†). In more concentrated or wet solutions only one averaged NMR signal is observed at −13.9 (31P) and −579 (51V) due to fast exchange on the NMR time scale. Potentiometric titration with aqueous TBAOH confirmed the presence of two types of acid protons in the POM and made possible accurate determination of the ratio between the two forms of I. Fig. S3† depicts a representative potentiometric titration curve for the sample of TBA3.5H1.5-I. The results of the potentiometric titration are in good agreement with CNH analysis (see Experimental).
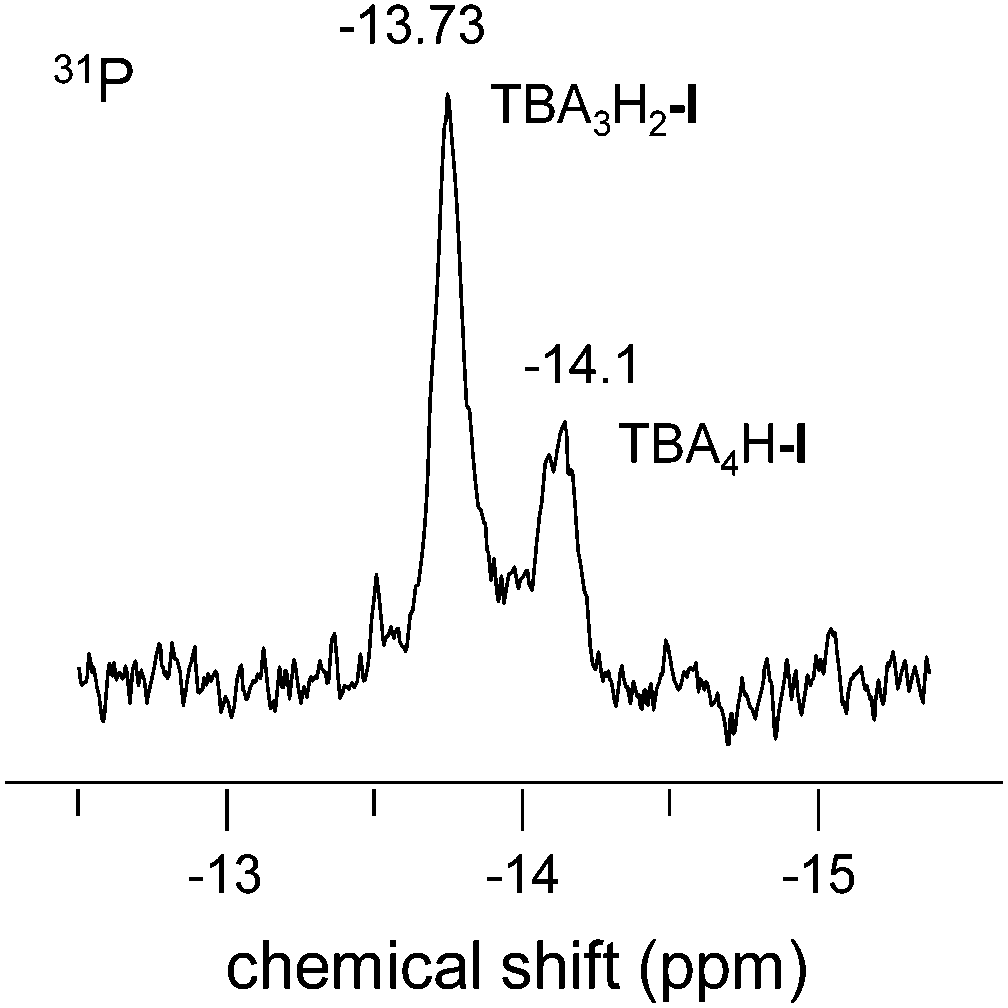 | ||
| Fig. 1 31P NMR spectrum of TBA3.3H1.7-I (0.0015 M) in dry CH3CN. | ||
It is noteworthy that TBA3.5H1.5-I itself is able to convert TMT to CoQ0 with selectivity as high as 83% (Table 1, entry 3). However, arene conversion was only 36% and could not be increased by increasing the reaction time. The addition of HClO4 led to improvement in both substrate conversion and CoQ0 yield. The optimal amount of acid turned out to be close to 0.5 equiv. (Table 1, compare entries 4–6), corroborating the above mentioned hypothesis that the role of an acid co-catalyst is to generate TBA3H2-I in the diprotonated form.
Fig. 2 demonstrates how the protonation state of I affects its catalytic performance. Both TMT conversion and CoQ0 yield increase with increasing amounts of protons in the catalyst. In the presence of TBA3H2-I, 73% selectivity could be attained at 68% TMT conversion. Importantly, a similar result was acquired using either TBA3.5H1.5-I and 0.5 equiv. of HClO4 or TBA3.3H1.7-I and 0.3 equiv. of HClO4, indicating that it does not matter what is the source of protons, POM or mineral acid. Given that the synthesis of TBA3.5H1.5-I is more simple and reproducible than the synthesis of TBA3H2-I (see Experimental for details), we performed further studies and optimization of the catalyst system using TBA3.5H1.5-I.
 | ||
| Fig. 2 TMT oxidation in the presence of TBA5−nHn-I. Reaction conditions: TMT (0.1 M), H2O2 (0.2 M), I (2.5 mM), MeCN 1 mL, 60 °C, 30 min. | ||
To verify the uniqueness of polyanion I in the oxidation of TMT, we compared the catalytic performance of TBA3.5H1.5-I with some other representative POMs, including a Si-containing analog, TBA4H2[γ-SiW10V2O40], and conventional α-Keggin heteropoly acids, such as vanadium-free H3PW12O40 and divanadium-substituted H5PMo10V2O40 (the latter has two vanadium atoms statistically distributed over 12 positions of the α-Keggin structure). A comparison of their catalytic properties is shown in Fig. 3. One can see that both the Si-analogue of I and the heteropoly acids are poor catalysts for the production of CoQ0. Although 35–60% conversion of TMT was attained after 1 h, selectivity towards ubiquinone 0 did not exceed 27%. On the other hand, 73% selectivity at 70% TMT conversion was reached after 0.5 h in the presence of 2.5 mol% of TBA3.5H1.5-I combined with 0.5 equiv. of mineral acid. These results indicate that the presence of both the dimeric vanadium core and the P central atom in the specific γ-Keggin structure are imperative for efficient catalysis of the title reaction. A simple vanadium complex, VO(acac)2, revealed 42% TMT conversion in 5 min, but the yield of CoQ0 was below 2%.
 | ||
| Fig. 3 TMT oxidation in the presence of different POMs. Reaction conditions: TMT (0.1 M), H2O2 (0.2 M), POM (2.5 mM), MeCN 1 mL, 60 °C. | ||
To figure out optimal reaction conditions, we studied the influence of all reaction parameters on TMT conversion and CoQ0 selectivity. The results are presented in Table 2. Previously, it was shown that I-based oxidation of alkylarenes gave better yields of oxygenated products in a solvent mixture of MeCN and t-BuOH (1:1, v/v).19,21 However, in the case of TMT, the addition of t-BuOH resulted in worsening of both arene conversion and quinone selectivity relative to the reaction in MeCN (Table 2, compare entries 1 and 2).
| Entry | Cat:TMT:H2O2 |
TMT (M) | Time (min) | TMT conv. (%) | CoQ0 select.b (%) |
|---|---|---|---|---|---|
|
a Reaction conditions: HClO4 (1.25 mM), MeCN 1 mL, 60 °C.
b GC yield based on converted TMT.
c Isolated yield.
d MeCN/t-BuOH (1:1, v/v) instead of MeCN.
e 30 °C.
f 80 °C.
g H2O2 added in three portions.
h H2O2 added in two portions.
i 0.1 M of MeOH was added.
|
|||||
| 1 | 0.025:1:2 |
0.1 | 30 | 70 | 73 (67)c |
| 2d | 0.025:1:2 |
0.1 | 15 | 43 | 56 |
| 3e | 0.025:1:2 |
0.1 | 240 | 39 | 77 |
| 4f | 0.025:1:2 |
0.1 | 5 | 71 | 66 |
| 5 | 0.013:1:2 |
0.1 | 30 | 69 | 70 |
| 6 | 0.05:1:2 |
0.1 | 5 | 66 | 64 |
| 7 | 0.025:1:2 |
0.05 | 45 | 75 | 67 |
| 8 | 0.025:1:2 |
0.2 | 15 | 51 | 75 |
| 9 | 0.025:1:3 |
0.1 | 15 | 65 | 63 |
| 10g | 0.025:1:3 |
0.1 | 45 | 85 | 64 |
| 11h | 0.025:1:2 |
0.1 | 30 | 76 | 73 |
| 12i | 0.025:1:2 |
0.1 | 30 | 64 | 65 |
The oxidation rate was reduced significantly by decreasing the reaction temperature: after 4 h at 30 °C, TMT conversion reached only 39%. However, some enhancement in the CoQ0 selectivity was observed (Table 2, compare entries 1 and 3). At 80 °C, the attainable level of conversion was the same as at 60 °C, but selectivity to CoQ0 decreased to 66% (Table 2, entry 4). A 2-fold reduction of the catalyst concentration with simultaneous alteration of the co-catalyst amount did not strongly affect the catalytic performance (Table 2, compare entries 1 and 5). On the other hand, a 2-fold augmentation of the catalyst concentration led to some decrease of TMT conversion and CoQ0 selectivity (Table 2, entry 6). A proportional diminution of concentrations of all the reactants relative to the standard conditions resulted in some increase of TMT conversion but led to a reduction in CoQ0 selectivity (Table 2, entry 7). Oppositely, only 51% substrate conversion was reached in a more concentrated reaction mixture, showing a similar level of selectivity (Table 2, compare entries 1 and 8).
According to the reaction stoichiometry (see Scheme 2), two equivalents of H2O2 are required to convert TMT to CoQ0. However, under the standard reaction conditions of entry 1 (Table 2), the reaction stopped after 30 min, reaching a substrate conversion of 70%. Semiquantitative evaluation with Quantofix peroxide test sticks revealed that practically no oxidant was present in the final reaction mixture. Therefore, incomplete TMT conversion with 2 equiv. of H2O2 may be an indication of some unproductive decomposition of the oxidant. When the concentration of H2O2 was reduced twice relative to the required stoichiometric amount, the oxidant utilization efficiency was improved considerably (70% versus 52%) along with selectivity to the target quinone (82% versus 73%). Fig. 4 shows that H2O2 efficiency tends to decrease with increasing H2O2 concentration. The reaction selectivity follows a similar trend. A stepwise addition of the oxidant to the reaction mixture allowed higher TMT conversions to be achieved, keeping quinone selectivity at the same level (Table 2, compare entries 9 and 10, 1 and 11).
 | ||
| Fig. 4 The effect of H2O2 concentration on TMT oxidation in the presence of TBA3.5H1.5-I. Reaction conditions: TMT (0.1 M), I (2.5 mM), HClO4 (1.25 mM), MeCN 1 mL, 60 °C. | ||
During the reaction course, we revealed the formation of a product that reached a maximum yield of ca. 15% at the initial stage and then disappeared (Fig. 5). In the final reaction mixture, only 1–2% of this compound was detected. GC-MS analysis identified it as 2,3,4-trimethoxy-6-methylphenol (TMMP). The bell-shaped accumulation curve depicted in Fig. 5 suggests that TMMP is, most likely, an intermediate product formed during the conversion of TMT to CoQ0. GC-MS also detected trace amounts of a compound that could be assigned to 3,4-dimethoxy-6-methylpyrocatechol or 2,3-dimethoxy-6-methyl-hydroquinone. These facts strongly support a mechanism that involves electrophilic hydroxylation of TMT to form TMMP at the first step of the oxidation process (Scheme 3).
 | ||
| Fig. 5 TMT consumption and product formation versus time. Reaction conditions: TMT (0.1 M), H2O2 (0.05 M), TBA3.5H1.5-I (2.5 mM), HClO4 (1.25 mM), MeCN 1 mL, 40 °C. | ||
 | ||
| Scheme 3 Plausible route of TMT oxidative transformation to CoQ0. | ||
At high conversions, CoQ0 was the only product detected by GC, GC-MS, and 1H NMR. However, CoQ0 yields determined by means of GC or 1H NMR using the internal standard suggested the presence of some by-products, most likely, tars.
To examine reusability of TBA3.5H1.5-I in the oxidation of TMT, we performed 3-fold scaled experiments, in which the catalyst was separated from the reaction mixture by precipitation with diethyl ether and used repeatedly under the conditions of entry 1, Table 2. The recycling performance is shown in Fig. 6. Only minor reduction of arene conversion and product selectivity was observed during at least four recycles. Importantly, the reaction time did not increase, indicating stable catalytic activity. The FTIR spectrum of the recovered catalyst confirmed the retention of the POM structure (see Fig. S1†).
 | ||
| Fig. 6 Reuse of TBA3.5H1.5-I in TMT oxidation. Reaction conditions: TMT (0.1 M), H2O2 (0.2 M), TBA3.5H1.5-I (2.5 mM), HClO4 (1.25 mM), MeCN (3 mL), 60 °C, 15 min. | ||
Some decrease of TMT conversion and product yield might be caused by a partial transformation of the POM catalyst to a methoxy derivative, (Bu4N)3[γ-PW10O38V2(μ-OH)(μ-OMe)], that could be formed during the reaction course upon interaction of polyanion I with methanol, which is one of the reaction products (see Scheme 2). Earlier, Nakagawa et al. studied the interaction of methanol and other alcohols with the Si-containing analogue of I and proved the formation of such methoxy derivatives.23 Indeed, 51V NMR detected the appearance of a signal at −562 ppm during the reaction progress, which could be assigned to the methoxy derivative of I (Fig. 7b). To verify this hypothesis, we performed an experiment where 1 equiv. of MeOH was added to a solution of TBA3.5H1.5-I in MeCN. Indeed, the appearance of the 51V NMR signal at −562 ppm was detected (Fig. 7c). The addition of methanol to the initial reaction mixture produced a rate-retarding effect (Fig. 8) and decreased TMT conversion and selectivity to CoQ0 (Table 2, entry 12). It should be noted, however, that most of the methoxy derivative was hydrolyzed back to I upon the recycling workup since the recovered POM was present in its initial state (δ −579 ppm). Therefore, 31P and 51V NMR along with FTIR spectroscopic technique confirmed the retention of the γ-Keggin structure of I after the catalysis.
 | ||
| Fig. 7 51V NMR spectra in CH3CN: (a) initial TBA3.5H1.5-I (0.0025 M), (b) reaction mixture after 10 min, and (c) TBA3.5H1.5-I (0.0025 M) + 1 equiv. of MeOH. | ||
 | ||
| Fig. 8 The effect of MeOH on TMT conversion. Reaction conditions: TMT (0.1 M), H2O2 (0.2 M), TBA3.5H1.5-I (2.5 mM), HClO4 (1.25 mM), [MeOH] 0.1 M, MeCN 1 mL, 60 °C. | ||
To estimate the scope of the TBA3.5H1.5-I/H2O2 catalytic system, we studied the oxidation of some other representative methoxyarenes (Scheme 4). The results are summarized in Table 3. For all substrates, the formation of the corresponding quinones occurred with moderate to good yields (22–70%) at conversions ≥74%. It is noteworthy that the oxidation of 1,2,3-trimethoxybenzene (Scheme 4, d) gave only 2,3-dimethoxy-1,4-benzoquinone while, for K3[Fe(CN)6]-catalyzed8 and non-catalytic9 oxidations, the formation of two isomeric quinones, 2,3- and 2,6-dimethoxy-1,4-benzoquinone, was observed. This fact implies that steric factors, most likely, control the oxidation process in the case of the bulky divanadium-POM catalyst.
 | ||
| Scheme 4 Oxidation of methoxyarenes in the presence of TBA3.5H1.5-I. | ||
For the sake of comparison, we also performed TMT oxidation following two protocols reported in the literature14,16 but in a lower scale (1 mL). The comparison is shown in Fig. 9. In a mixture of formic and acetic acids, CoQ0 yield achieved 58%, which is a bit higher than the yield obtained in the presence of TBA3.5H1.5-I (52%). On the contrary, the quinone yield was significantly lower (ca. 30%) in the presence of phosphomolybdic acid in AcOH. An evident advantage of the TBA3.5H1.5-I-based catalyst system is greater oxidant utilization efficiency (see Fig. 9), which allows one to attain a good product yield using just the stoichiometric amount of the oxidant. In addition, we avoid the use of carboxylic acids as solvents and, therefore, preclude corrosion and formation of explosive peroxy acids upon interaction with H2O2.
 | ||
| Fig. 9 Comparison of I-catalyzed TMT oxidation with reported systems. Reaction conditions: (a) TMT (1.4 M), H2O2 (5.5 M) added dropwise for 10 min, HCOOH 0.35 mL, AcOH 0.18 mL, 35 °C; (b) TMT (2.2 M), 50% H2O2 (7 M), H3PMo12O40 (0.7 M), HCOOH 0.5 mL, 25 °C; and (c) as in Table 2, entry 1. | ||
Conclusions
In summary, we have developed a new method for the synthesis of the important fine chemical coenzyme Q0via oxidation of commercially available 3,4,5-trimethoxytoluene with 30% H2O2 in MeCN employing acid tetrabutylammonium salts of the divanadium-substituted polyoxometalate [γ-PW10V2O40]5− as a catalyst. Coenzyme Q0 was obtained with selectivity as high as 73% at 76% arene conversion. The procedure is affordable, safe and sustainable. The catalyst system is applicable to oxidation of a variety of di- and trimethoxyarenes, producing the corresponding benzoquinones in moderate to good yields. Since the presence of an acid is crucial for the catalytic performance, a simple procedure for the preparation of a highly protonated derivative TBA3.5H1.5-I has been developed. The use of this derivative enables significant reduction of the amount of acid co-catalyst without deterioration of the catalytic performance. The catalyst could be easily separated from the reaction mixture and recycled without appreciable loss of activity and selectivity.Experimental
Materials and instrumentation
3,4,5-Trimethoxytoluene (97%) and 2,3-dimethoxy-5-methyl-1,4-benzoquinone (99%) were obtained from Aldrich, 2,3-dimethoxytoluene (98%) and 2,4,6-trimethoxytoluene (97%) were purchased from Alfa, 3,5-dimethoxytoluene (99%), 1,2,3-trimethoxybenzene (98%) and 1,3,5-trimethoxybenzene (99%) were obtained from Acros. Acetonitrile (Panreac, HPLC grade) was dried and stored over activated 4 Å molecular sieves. All the other compounds were the best available reagent grade and used without further purification. The concentration of H2O2 (ca. 35 wt% aqueous solution) was determined iodometrically prior to use. GC analyses were performed using a gas chromatograph Chromos GC-1000 equipped with a flame ionization detector and a quartz capillary column BPX5 (30 m × 0.25 mm). GC-MS analyses were carried out using an Agilent 7000B system with a triple-quadrupole mass-selective detector Agilent 7000 (HP-5 ms quartz capillary column 30 m × 0.25 mm). 1H, 31P and 51V NMR spectra were recorded on a Brüker AVANCE-400 spectrometer at 400.130, 161.67 and 105.24 MHz, respectively. Chemical shifts for P and V, δ, were determined relative to 85% H3PO4 and VOCl3, respectively. Infrared spectra were recorded with KBr pellets on an Agilent 660 FTIR spectrometer.General catalyst preparation
:P = 5 mol/mol), and then 3 M HNO3 were added to the mother liquor until pH reached 7. During the adjustment of the pH a white precipitate was formed. The mixture was heated to boiling and then filtered. The white solid was separated, washed with hot water and dried. Then another portion of 3 M HNO3 was added to the filtrate until pH reached 7, and a new portion of the white solid was treated as described above. Total yield of γ-Cs7PW10O36·5H2O was ca. 80% (17.5 g). The IR spectrum of the obtained solid corresponded to that of γ-Cs7PW10O36·5H2O prepared by the conventional procedure.24 IR (KBr, cm−1): 1080, 1050, 1025, 937, 892, 829, 755.
General methods for catalytic oxidation and product analysis
Product characterization
Acknowledgements
The authors thank Dr I. E. Soshnikov and Dr M. V. Shashkov for their help with product identification by 1H NMR and GC-MS, respectively. This work was carried out in the framework of budget project no. 0303-2016-0005 for Boreskov Institute of Catalysis and partially supported by the Russian Foundation for Basic Research (RFBR grant 13-03-12042).References
- (a) The Chemistry of the Quinonoid Compounds, ed. S. Patai and Z. Rappoport, Wiley, New York, 1988 Search PubMed; (b) K. C. Nicolaou, J. S. Chen, D. J. Edmonds and A. A. Estrada, Angew. Chem., Int. Ed., 2009, 48, 660 CrossRef CAS PubMed; (c) O. A. Kholdeeva, in Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds, ed. J. Mortier, Wiley, 2016, ch. 14, pp. 365–398 Search PubMed; (d) O. A. Kholdeeva and O. V. Zalomaeva, Coord. Chem. Rev., 2016, 306, 302 CrossRef CAS.
- L. Ernster and G. Dallner, Biochim. Biophys. Acta, Mol. Basis Dis., 1995, 1271, 195 CrossRef.
- Coenzyme Q: Molecular Mechanisms in Health and Disease, ed. V. E. Kagan and P. J. Quinn, CRC Press, Boca Raton, FL, 2000 Search PubMed.
- J. C. Gillis, P. Benfield and D. McTavish, Drugs Aging, 1994, 5, 133 CAS.
- (a) V. Berl, F. Wetterich, H. Ernst and K. Schein, WO patent2006032425, 2006 Search PubMed; (b) E. Negishi, S.-Y. Liou, C. Xu and S. Huo, Org. Lett., 2002, 4, 261 CrossRef CAS PubMed; (c) B. H. Lipshutz, A. Lower, V. Berl, K. Schein and F. Wetterich, Org. Lett., 2005, 7, 4095 CrossRef CAS PubMed.
- J. Wang, S. Li, T. Yang and J. Yang, Tetrahedron, 2014, 70, 9029 CrossRef CAS.
- W. Adam and M. Shimizu, Synthesis, 1994, 560 CrossRef CAS.
- M. Matsumoto, H. Kobayashi and Y. Hotta, J. Org. Chem., 1985, 50, 1766 CrossRef CAS.
- H. Orita, M. Shimizu, T. Hayakawa and K. Takehira, Bull. Chem. Soc. Jpn., 1989, 62, 1652 CrossRef CAS.
- H. Sugihara, M. Watanabe, Y. Kawamatsu and H. Morimoto, Justus Liebigs Ann. Chem., 1972, 763, 109 CrossRef CAS PubMed.
- W. Adam, W. A. Herrmann, C. R. Saha-Möller and M. Shimizu, J. Mol. Catal. A: Chem., 1995, 97, 15 CrossRef CAS.
- R. Bernini, E. Mincione, M. Barontini, G. Fabrizi, M. Pasqualetti and S. Tempesta, Tetrahedron, 2006, 62, 7733 CrossRef CAS.
- (a) H. Orita, M. Shimizu, T. Hayakawa and K. Takehira, React. Kinet. Catal. Lett., 1991, 44, 209 CrossRef CAS; (b) H. Orita, M. Shimizu, T. Hayakawa and K. Takehira, EP0347021, 1989 Search PubMed.
- A. S. Chida, P. V. S. N. Vani, M. Chandrasekharam, R. Srinivasan and A. K. Singh, Synth. Commun., 2001, 31, 657 CrossRef CAS.
- G. Occhipinti, L. Liguori, A. Tsoukala and H.-R. Bjørsvik, Org. Process Res. Dev., 2010, 14, 1379 CrossRef CAS.
- J. Wang, X. Hu and J. Yang, Synthesis, 2014, 2371 CrossRef CAS.
- S. Iwasa, A. Fakhruddin, H. S. Widagdo and H. Nishiyama, Adv. Synth. Catal., 2005, 347, 517 CrossRef CAS.
- M. Crucianelli, R. Saladino and F. De Angelis, ChemSusChem, 2010, 3, 524 CrossRef CAS PubMed.
- K. Kamata, T. Yamaura and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 7275 CrossRef CAS PubMed.
- I. D. Ivanchikova, N. V. Maksimchuk, R. I. Maksimovskaya, G. M. Maksimov and O. A. Kholdeeva, ACS Catal., 2014, 4, 2706 CrossRef CAS.
- O. V. Zalomaeva, V. Yu. Evtushok, G. M. Maksimov and O. A. Kholdeeva, J. Organomet. Chem., 2015, 793, 210 CrossRef CAS.
- K. Kamata, K. Yonehara, Y. Nakagawa, K. Uehara and N. Mizuno, Nat. Chem., 2010, 2, 478 CrossRef CAS PubMed.
- Y. Nakagawa, K. Uehara and N. Mizuno, Inorg. Chem., 2005, 44, 14 CrossRef CAS PubMed.
- P. J. Domaille, G. Hervé and A. Tézé, in Inorganic Syntheses, ed. A. P. Ginsberg, John Wiley & Sons, Inc., 1990, vol. 27, pp. 96–104 Search PubMed.
- Y. Nakagawa, K. Kamata, M. Kotani, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2005, 44, 5136 CrossRef CAS PubMed.
- G. A. Tsigdinos and C. J. Hallada, Inorg. Chem., 1968, 7, 437 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR, NMR and potentiometric titration data. See DOI: 10.1039/c7dt00552k |
| This journal is © The Royal Society of Chemistry 2017 |