
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLarge, weakly basic bis(carboranyl)phosphines: an experimental and computational study†‡
Laura E.
Riley
a,
Tobias
Krämer
 *a,
Claire L.
McMullin
b,
David
Ellis
a,
Georgina M.
Rosair
a,
Igor B.
Sivaev
c and
Alan J.
Welch
*a
*a,
Claire L.
McMullin
b,
David
Ellis
a,
Georgina M.
Rosair
a,
Igor B.
Sivaev
c and
Alan J.
Welch
*a
aInstitute of Chemical Sciences, Heriot-Watt University, Edinburgh, UK EH14 4AS. E-mail: t.kraemer@hw.ac.uk; a.j.welch@hw.ac.uk; Tel: +44 (0)131 451 3259 Tel: +44 (0)131 451 3217
bDepartment of Chemistry, University of Bath, Bath, UK BA2 7AY
cA. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, 28 Vavilov Str., 119991 Moscow, Russia
First published on 7th March 2017
Abstract
The bis(carboranyl)phosphines [μ-2,2′-PPh-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (I) and [μ-2,2′-PEt-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (1) have been prepared and spectroscopically and structurally characterised. Crystallographic and DFT computational studies of 1 suggest that the orientation of the ethyl group, relative to the bis(carborane), is the result of intramolecular dihydrogen bonding. This orientation is such that the magnitudes of the 2JPH coupling constants are approximately equal but of opposite sign, and fast exchange between the methylene protons in solution leads to an observed 2JPH close to zero. The steric properties of I, 1 and their derivatives [μ-2,2′-P(Ph)AuCl-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (2) and [μ-2,2′-P(Et)AuCl-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (3) have been assessed by Tolman cone angle and percent buried volume calculations, from which it is concluded that the bis(carboranyl)phosphines I and 1 are comparable to PCy3 in their steric demands. The selenides [μ-2,2′-P(Ph)Se-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (4) and [μ-2,2′-P(Et)Se-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (5) have also been prepared and characterised. The 1JPSe coupling constants for 4 and 5 are the largest reported so far for carboranylphosphine selenides and indicate that I and 1 are very weakly basic.
Introduction
Currently, one of the most active areas of carborane chemistry concerns the compound [1-(1′-1′,2′-closo-C2B10H11)-1,2-closo-C2B10H11], two ortho-carborane units connected by a C–C bond1 and commonly referred to as 1,1′-bis(o-carborane) (Fig. 1). Although it was first reported >50 years ago,2 the chemistry of 1,1′-bis(o-carborane) was not fully explored for long periods because of the lack of a reliable, high-yielding synthesis. However, this has now been achieved3 and consequently a significant amount of new chemistry of 1,1′-bis(o-carborane) has recently appeared.4–20 | ||
| Fig. 1 1,1′-Bis(o-carborane). | ||
Several authors have taken advantage of the functionality of its CcageH units to use 1,1′-bis(o-carborane) as a κ2 ligand, binding transition-metals to the cage through two M–C σ-bonds, with recent studies7,8,13,19 building on the pioneering work of Hawthorne and co-workers.21 In marked contrast, very little has been reported concerning main group elements bound to 1,1′-bis(o-carborane). Zakharkin reported [μ-2,2′-AsMe-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] and [μ-2,2′-PPh-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (I), although the latter was only poorly characterised,22,23 and Johnson and Knobler described [μ-2,2′-PX-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}], X = Cl and F.24 Very recently, Peryshkov and co-workers isolated an interesting 12-vertex closo/12-vertex nido species with both bridging P(i-Pr)2 and non-bridging PH(i-Pr)2 units when attempting to add two P(i-Pr)2 groups onto 1,1′-bis(o-carborane), one on each cage.17
In this contribution we expand on Zakharkin's early work, resynthesising I (in higher yield) and preparing the related μ-2,2′-PEt compound (1), with full characterisation of both bis(carboranyl)phosphines. All phosphine have potential as ligands in homogeneous catalysis, and consequently we have explored the steric and electronic properties of I and 1via the synthesis and study of derivatives in which the P lone pair is bound to {–AuCl} and {![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Se} fragments.
Se} fragments.
Results and discussion
Bis(carboranyl)phosphines
Double deprotonation of 1,1′-bis(o-carborane) in Et2O with n-BuLi in hexanes followed by treatment with PPhCl2 affords [μ-2,2′-PPh-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (I) as a white solid in 72% isolated yield, following work-up involving flash chromatography on silica. Although this bis(carboranyl)phosphine has been reported before it was previously characterised only by elemental analysis.22 We have studied the compound by mass spectrometry, multinuclear NMR spectroscopy and single-crystal X-ray diffraction.The mass spectrum of I is dominated by a typical carborane envelope centred at m/z 392 corresponding to the molecular ion. The 11B{1H} NMR spectrum contains resonances from δ ca. 0 to −13 ppm but is relatively uninformative in terms of the detailed structure because of the overlap of the lower-frequency resonances. The 1H NMR spectrum confirms that no CcageH resonances are present and reveals the expected multiplets for ortho, meta and para H atoms which collapse to a doublet, apparent triplet and triplet, respectively, on 31P decoupling. In the 31P{1H} spectrum there is a simple singlet at δ 40.35 ppm.
The molecular structure of I is shown in Fig. 2. As anticipated the {PPh} fragment binds symmetrically to the bis(carborane) (the P1–C2 and P1–C2′ distances are identical within experimental error), and only a small rotation of the Ph substituent about the P1–C11 bond would afford the molecule Cs symmetry, the likely time-averaged symmetry in solution. The formation of the P1C2C1C1′C2′ ring results in a slight bending of the spine of the bis(carborane) with angles B12⋯C1–C1′ 169.14(16)° and B12′⋯C1′–C1 170.17(15)°, cf. 175.14(5)° in 1,1′-bis(o-carborane) itself.1
 | ||
| Fig. 2 Perspective view of I with the atomic numbering scheme. Displacement ellipsoids are drawn at the 50% probability level except for H atoms. Selected interatomic distances (Å): P1–C2 1.893(3), P1–C2′ 1.887(3), P1–C11 1.819(3), and C1–C1′ 1.534(3). | ||
An analogous compound, [μ-2,2′-PEt-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (1), was similarly prepared. The 11B{1H} NMR spectrum of 1 reveals a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:6:2:2:6 pattern (high frequency to low frequency) between δ ca. 0 and −11 ppm, consistent with time-averaged Cs molecular symmetry. The 1H{31P} NMR spectrum affords the expected quartet and triplet for the PCH2CH3 and PCH2CH3 resonances, respectively, and a singlet at δ 42.75 ppm is observed in the 31P{1H} spectrum.
:2:6:2:2:6 pattern (high frequency to low frequency) between δ ca. 0 and −11 ppm, consistent with time-averaged Cs molecular symmetry. The 1H{31P} NMR spectrum affords the expected quartet and triplet for the PCH2CH3 and PCH2CH3 resonances, respectively, and a singlet at δ 42.75 ppm is observed in the 31P{1H} spectrum.
There are two crystallographically-independent molecules of compound 1 in the asymmetric fraction of the unit cell, and Fig. 3 shows a perspective view of one of them, 1AB (the other is 1CD). Using the Structure Overlay tool in Mercury25 the overall root-mean-square (rms) misfit between the {P(C2B10)2} fragments of the two independent molecules (25 atoms) is 0.024 Å, with the greatest individual misfit between B4B and B4D, 0.049 Å. Including the C atoms of the Et groups results in only a marginal increase in the rms misfit to 0.029 Å (the greatest individual misfit 0.073 Å between C12A and C12C) since the orientation of the ethyl group relative to the bis(carborane) in the two independent molecules is effectively the same.
 | ||
| Fig. 3 Perspective view of one of the two crystallographically-independent molecules of compound 1 (1AB) with the atomic numbering scheme. Molecule 1CD is practically superimposable. Displacement ellipsoids are as in Fig. 2. Selected interatomic distances (Å): P1A–C2A 1.870(5), P1A–C2B 1.890(5), P1A–C11A 1.844(5), C1A–C1B 1.533(7); P1C–C2C 1.886(5), P1C–C2D 1.890(5), P1C–C11C 1.818(5), and C1C–C1D 1.530(6). | ||
This ethyl orientation is significant because of its influence on the 1H NMR spectroscopic properties of 1 (vide infra). We believe that the origin of the orientation is that it maximises intramolecular dihydrogen bonding between the weakly protonic CH atoms and the weakly hydridic BH atoms of the carborane cage, with the key CH⋯HB contacts shown in Fig. 4, an alternative view of 1AB.
 | ||
| Fig. 4 Alternative view of 1AB showing dihydrogen bonds as red lines. Interatomic distances (Å): H11P⋯H3A 2.45(4), 2.39(4), 2.28; H11P⋯H6B 2.28(5), 2.29(5), 2.10; H11Q⋯H7A 2.46(5), 2.47(4), 2.36; H12R⋯H11B 2.51(5), 2.40(5), 2.31. Values in italics are the equivalent distances in molecule 1CD, and values in bold are those from the DFT study of 1. | ||
To investigate this further we have undertaken DFT calculations on compound 1. The optimised (BP86-D3/def2-TZVP/def2-SVP) geometry, 1DFT (Fig. 5), is in excellent agreement with the structure determined crystallographically (see the ESI‡ for key optimised bond parameters), including the orientation of the ethyl group. Thus the overall rms misfit between the computed and experimental {P(C2B10)2} fragments (using molecule 1AB) is only 0.019 Å (the greatest individual misfit 0.031 Å for P1), rising to only 0.029 Å if the ethyl C atoms are included (the greatest individual misfit 0.081 Å for C12A). In terms of the ethyl group orientation, the experimental lp–P1–C11–C12 torsion angles are 38.8(4)° and 42.5(4)° for 1AB and 1CD, respectively, and the computed torsion angle is 38.4°. Moreover, the DFT study provides strong support for the presence of dihydrogen bonding. A topological analysis of the electron density in 1DFT using the QTAIM methodology reveals bond critical points (BCP) between H11PX and the cage hydrogen atoms H3 and H6′, located roughly at the centre of each bond vector (see Fig. S1‡ for a molecular graph). Additional, albeit somewhat weaker interactions, are present for H11QY/H7 and H12R/H11 (see Fig. S1‡). Both the electron density ρ(r) (0.007–0.011 a.u.) and its Laplacian ∇2ρ(r) (0.022–0.038 a.u.) at the relevant BCPs are diagnostic of typical closed-shell interactions and fall within the ranges proposed for dihydrogen bonds.26
 | ||
| Fig. 5 DFT-optimised structure of 1. Note the excellent agreement with the experimentally-determined structure (Fig. 3), including the orientation of the Et group. Selected interatomic distances (Å): P1–C2 1.889, P1–C2′ 1.892, P1–C11 1.852, and C1–C1′ 1.528. | ||
An interesting feature of the 1H NMR spectrum of compound 1 is the lack of observable coupling between the PCH2CH3 H atoms and the P atom. Thus, whilst the PCH2CH3 H atoms appear as a doublet (3JPH = 23.0 Hz) of triplets (3JHH = 7.9 Hz) the PCH2CH3 H atoms appear as only a simple quartet (3JHH = 7.9 Hz). A 1H–31P HMBC experiment shows the presence of the expected coupling between the P atom and the CH2CH3 H atoms but, additionally, a weak correlation between signals due to P and CH2CH3. The lack of any observable splitting in the 1D 1H spectrum suggests that the value of 2J (1H–31P) is of the order of the 1H resonance linewidth (estimated to be ca. 1 Hz).
An understanding of the origin of this small 2-bond P–H coupling comes from the DFT calculations on compound 1. The computed energy profile for ethyl rotation about the P1–C11 vector (Fig. 6) establishes that 1DFT and its isoenergetic rotational conformer 1′DFT dominate the equilibrium distribution, whilst the Boltzmann population of the symmetrical isomer 1′′DFT (+4 kcal mol−1) is near zero at room temperature. Importantly, due to the low energetic barrier associated with TS(1DFT–1′DFT) separating 1DFT and 1′DFT (ΔG‡ = +1.8 kcal mol−1) the methylene protons can undergo rapid exchange in solution. The calculated averaged 3JPH (calc. 26.2 Hz; exp. 23.0 Hz) and 3JHH (calc. 9.7 Hz; exp. 7.9 Hz) coupling constants are in good agreement with the experimental values. The values of 2JPH for coupling to the individual methylene hydrogen atoms are of the same order of magnitude but, importantly, they are of opposite sign (for H11PX 2JPH = −5.4 Hz whilst for H11QY 2JPH = +5.6 Hz). Hence, fast exchange between these hydrogen atoms reduces the observed 2JPH to approximately zero, in line with the experimental upper limit of ∼±1 Hz. The predominant contribution to the total coupling constant is associated with the Fermi contact term.
 | ||
| Fig. 6 Calculated Gibbs free energy profile (kcal mol−1) for ethyl rotation in 1DFT (BP86-D3/def2-TZVP/def2-SVP). Boltzmann populations: 1DFT 50%, 1′DFT 50%, and 1′′DFT 0%. | ||
With the characterisation of I and 1 now complete, we turn to their derivatives. Phosphines are ligands of great importance in homogeneous catalysis by transition-metal complexes,27 and carboranylphosphines have been used extensively in a variety of catalytic applications.28 The two key characteristics of phosphines as ligands in catalysis are their size and basicity, and we have targeted and studied derivatives of I and 1 specifically to assess these features.
Steric properties
The steric demand of a phosphine is classically assessed using its Tolman cone angle (θ),29 and we have used the crystallographically-determined structures of I and 1 to measure these. In addition, a recent alternative to θ is Cavallo and Nolan's percent buried volume parameter, %Vbur.30 Although originally developed for NHCs, %Vbur has been shown to scale linearly with θ for a range of phosphines and, furthermore, there is a strong linear relationship between θ of PR3 and %Vbur of the adduct R3PAuCl.31 For this reason we have prepared and studied the gold–chloride adducts of compounds I and 1.Treatment of a solution of I or 1 in DCM with an equimolar amount of (tht)AuCl affords the new species [μ-2,2′-P(Ph)AuCl-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (2) and [μ-2,2′-P(Et)AuCl-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (3) as white solids in 81 and 32% yields, respectively.
Compounds 2 and 3 were initially characterised by mass spectrometry and 1H, 11B and 31P NMR spectroscopies. The 11B{1H} spectrum of 2 is largely uninformative because of multiple overlapping resonances, but that of 3 is sufficiently well-resolved to allow integration, which is consistent with time-averaged Cs molecular symmetry in solution at room temperature. In both 2 and 3 the 31P{1H} spectrum reveals a simple singlet, shifted ca. 28 and 33 ppm, respectively, to a high frequency relative to those in I and 1. The 1H NMR spectrum of 2 shows the anticipated multiplets for the phenyl protons, whilst for 3 the H atoms of the ethyl group appear as a doublet of quartets (PCH2CH3) and a doublet of triplets (PCH2CH3).
The molecular structures of 2 and 3, as determined crystallographically, are shown in Fig. 7 and 8, respectively. In both cases co-ordination to the {AuCl} fragments causes minimal change in the structures of the bis(carboranyl)phosphines, with the same orientations of the Ph groups or Et groups being effectively maintained between I and 2 and between 1 and 3. Moreover, a DFT-optimised study of 3 was fully consistent with its crystallographic counterpart; a structure overlay of 3 and 3DFT yielded an overall rms misfit of 0.018 Å for {P(C2B10)2} fragments (the greatest individual misfit 0.027 Å for B5′) rising to only 0.020 Å if the ethyl carbon atoms were included (the greatest misfit 0.033 Å for C11). The overlay is somewhat poorer if the {AuCl} fragment is included, with the overall misfit increasing to 0.077 Å and the Cl atoms misfitting by 0.345 Å.
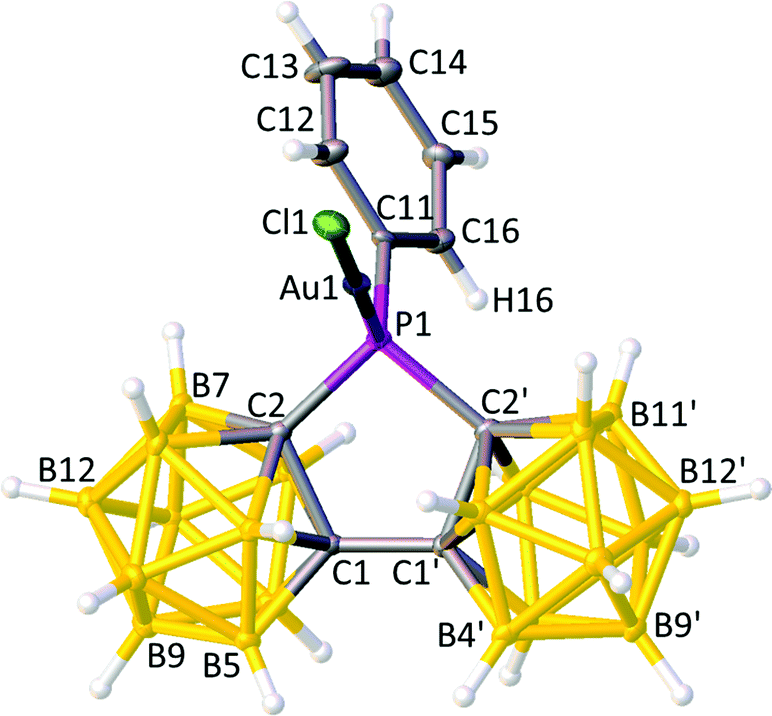 | ||
| Fig. 7 Perspective view of compound 2 with the atomic numbering scheme. Displacement ellipsoids are as in Fig. 2. Selected interatomic distances (Å): P1–Au1 2.2217(8), Au1–Cl1 2.2698(8), P1–C2 1.878(3), P1–C2′ 1.879(3), P1–C11 1.797(3), and C1–C1′ 1.523(4). | ||
 | ||
| Fig. 8 Perspective view of compound 3 with the atomic numbering scheme. Displacement ellipsoids are as in Fig. 2. Selected interatomic distances (Å): P1–Au1 2.2181(11), Au1–Cl1 2.2910(11), P1–C2 1.860(4), P1–C2′ 1.883(4), P1–C11 1.816(4), and C1–C1′ 1.538(5). | ||
The Tolman cone angle for I was calculated to be 172.5°, whilst that for 1 is 171.6° (Table 1). Although Ph is a larger substituent than Et, the similarity of the cone angles for I and 1 reflects the fact that the vast majority of the steric bulk of I and 1 comes from the common bis(carborane) fragment. Percent buried volumes for I and 1 were calculated to be 32.0 and 30.8 (average), respectively. Co-ordination to {AuCl} to afford 2 and 3, respectively, results in small increases in both θ and %Vbur. This arises because the stereochemical influence of the P lone pair of electrons on the bis(carborane) and Ph/Et substituents is reduced on co-ordination, allowing small increases in the C–P–C angles and producing a slightly bulkier ligand.
| θ | %Vbur | |
|---|---|---|
| I | 172.5 | 32.0 |
| 1AB | 171.6 | 30.9 |
| 1CD | 171.6 | 30.7 |
| 2 | 176.2 | 33.2 |
| 3 | 176.5 | 32.0 |
Comparing the θ and %Vbur values for I, 1, 2 and 3 with analogous species in the literature, we conclude that the bis(carboranyl)phosphines I and 1 are, in terms of their size, the most comparable to (but slightly larger than) tricyclohexylphosphine, PCy3.31
Electronic properties
The basicity of phosphines PR3 is conveniently assessed by the measurement of the 1-bond P–Se coupling constant, 1JPSe, of the corresponding selenide R3PSe; the more electron-withdrawing the substituents (i.e. the less basic the phosphine), the greater the degree of 3s character in the phosphorus lone pair and the greater the magnitude of 1JPSe.32 Accordingly, we have synthesised [μ-2,2′-P(Ph)Se-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (4) and [μ-2,2′-P(Et)Se-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (5), the selenides of I and 1 respectively, by the simple process of heating the bis(carboranyl)phosphine with an excess of elemental Se in toluene. Scheme 1 summarises all the syntheses reported in this paper.
 | ||
| Scheme 1 Generalised reaction scheme for compounds I and 1–5. | ||
Compounds 4 and 5 are afforded as pale-pink solids in reasonable isolated yields. Both show the anticipated molecular ion peaks in their mass spectra. Their 11B{1H} NMR spectra are relatively uninformative due to considerable overlap of resonances. In the 31P{1H} NMR spectra are singlets at δ ca. 50 and 58 ppm, respectively, at higher frequency than in the corresponding bis(carboranyl)phosphine but not as deshielded as in the AuCl compounds 2 and 3. The 1H NMR spectra of 4 and 5 show the expected resonances for the Ph or Et substituents, including a doublet of quartets for the PCH2CH3 resonance of 5 with 2JPH = 11.6 Hz, and the re-emergence of this 2-bond P–H coupling can be understood in terms of the rehybridisation of the phosphorus orbitals induced by coordination of the P lone pair to the {Se} fragment.33 These changes closely follow the general trends observed for P(III) and P(V) compounds.34 The computed averaged geminal 2JPH coupling constant in 5DFT increases to −14.1 Hz, a value in good agreement with experiment (2JPH = 11.6 Hz). We note that the phase information of the coupling constant is invisible in first-order NMR-spectra, and thus all coupling constants appear to be positive. The observed decrease of 2JPH (or increase of the modulus |2JPH|) can be correlated with the admixture of a higher degree of 3s character into the P bonds, rendering the hybridisation around P close to sp3, as borne out in the NBO analysis of the associated P–C bonding orbitals (21–26% s, 73–78% p). This is paralleled by a notable reduction of P 3s character in the P–Se σ-bond (32% s, 67% p), compared to 52% 3s character of the phosphorus lone pair in 1DFT. This change of the electron distribution around P as the lone pair is replaced by Se in 5DFT in turn reinforces the direct Fermi contact contribution to the 1- and 2-bond coupling pathways (see the ESI‡ for more details).
Of relevance to the basicities of the bis(carboranyl)phosphine parent compounds I and 1, the 31P{1H} spectra of 4 and 5 show clear Se satellites with 1JPSe = 891 and 894 Hz, respectively.35 These coupling constants are considerably larger than those of a range of common phosphines [typically 670–750 Hz, Table 1 of ref. 32d], suggesting that I and 1 are very weakly basic. There are few carboranylphosphine selenides in the literature, but those that are known all have relatively large values of 1JPSe. In [1-P(Se)Ph2-2-PPh2-1,2-closo-C2B10H10] 1JPSe = 807 Hz, whilst in the 2-Me and 2-Ph analogues 1JPSe = 804 and 812 Hz, respectively.36 Clearly, these large couplings are the result of the strong electron-withdrawing property of carboranes relative to alkyl or aryl substituents, and we attribute the even larger 1JPSe values in 4 and 5 to the fact that here the {PSe} fragment is directly bonded to two carborane cages. Consistent with this, Viñas and co-workers have described species [μ-1,1′-P(R)Se-3,3′-Co(1,2-C2B9H10)2]− in which the {P(R)Se} fragment is also attached to two heteroborane cages and reported 1JPSe values of 833 Hz (R = Ph) and 679 Hz (R = t-Bu).37 In the related species [8,8′-μ-(1′′,2′′-C6H4)-μ-1,1′-P(R)Se-3,3′-Co(1,2-C2B9H9)2]−, 1JPSe is 848 Hz (R = Ph) and 692 Hz (R = t-Bu), respectively.37
Overall, the magnitudes of 1JPSe recorded for 4 and 5 are currently the largest reported for carboranylphosphine selenides, implying that the parent phosphines I and 1 are very weakly basic. Note that, in principle, the degree of s character in the phosphorus lone pair in PR3 can be influenced not only by the electron-withdrawing properties but also by the size of the substituents. In this respect we recall that I and 1 are comparable in their steric demands to PCy3. They are, however, much less basic, since 1JPSe in Cy3PSe is only 673 Hz.38
Compounds 4 and 5 were also studied crystallographically, and perspective views of single molecules are shown in Fig. 9 and 10, respectively. As noted above, compound 5 was also studied computationally, again affording excellent agreement with the crystallographic structure; for the {P(C2B10)2} fragments of 5 and 5DFT the rms misfit is 0.020 Å (the worst individual misfit 0.034 Å for B11′), rising to only 0.034 Å (the poorest individual fit 0.100 Å for C11) when the ethyl C atoms are included and 0.036 Å (the worst fit 0.098 Å, C12) when the Se atom is added. Once again, the general orientation of the Ph or Et substituent relative to the bis(carborane) moiety is maintained, with torsion angles lp/Au/Se–P–C11–C12 broadly consistent for I, 2 and 4 (Ph substituents) and for 1, 1DFT, 3, 3DFT, 5 and 5DFT (Et substituents) albeit with a ca. 10° increase for the selenides 4 and 5 (Table 2). As previously noted, for the ethyl species 1, 3 and 5 this orientation is traced to intramolecular dihydrogen bonding involving H11A (H11P and H11X in 1) with H6′ and with H3, whilst for the phenyl compounds I, 2 and 4 H16 of the phenyl ring is seen to interact primarily with H6′ and to a lesser extent with H3.
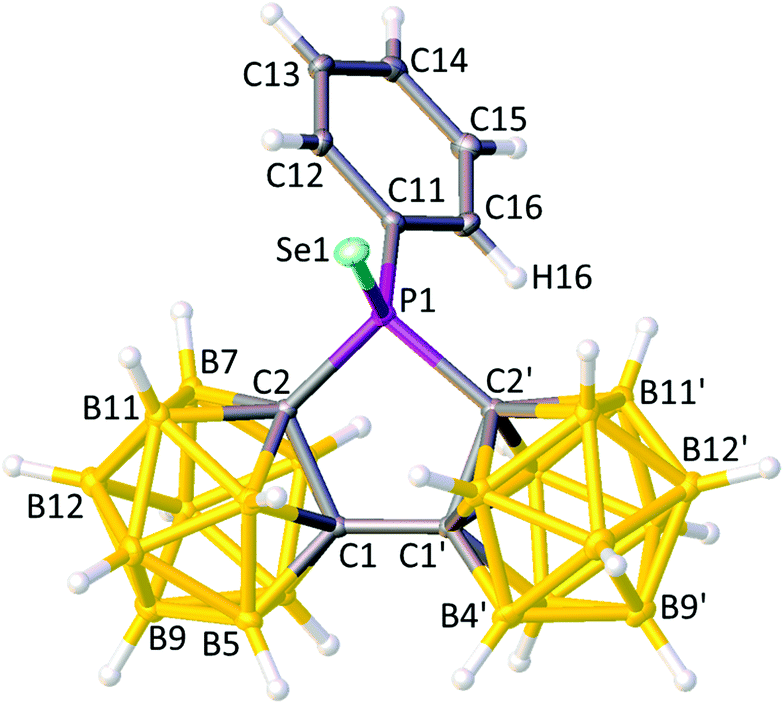 | ||
| Fig. 9 Perspective view of compound 4 with the atomic numbering scheme. Displacement ellipsoids are as in Fig. 2. Selected interatomic distances (Å): P1–Se1 2.0798(6), P1–C2 1.8860(19), P1–C2′ 1.896(2), P1–C11 1.810(2), and C1–C1′ 1.536(3). | ||
 | ||
| Fig. 10 Perspective view of compound 5 with the atomic numbering scheme. Displacement ellipsoids are as in Fig. 2. Selected interatomic distances (Å): P1–Se1 2.0801(7), P1–C2 1.879(2), P1–C2′ 1.893(2), P1–C11 1.822(2), and C1–C1′ 1.543(3). | ||
| Compound | X | X–P–C11–C12 | Dihydrogen bond | Distance | Dihydrogen bond | Distance |
|---|---|---|---|---|---|---|
| (i) Phenyl substituent | ||||||
| I | lp | −29.4(2) | H16⋯H6′ | 2.15(3) | H16⋯H3 | 2.48(2) |
| 2 | Au | −28.8(3) | H16⋯H6′ | 2.20(3) | H16⋯H3 | 2.49(3) |
| 4 | Se | −40.0(2) | H16⋯H6′ | 2.19(2) | H16⋯H3 | 2.84(2) |
| (ii) Ethyl substituent | ||||||
| 1AB | lp | 38.8(4) | H11P⋯H6B | 2.28(5) | H11P⋯H3A | 2.45(5) |
| 1CD | lp | 42.5(4) | H11X⋯H6D | 2.29(5) | H11X⋯H3C | 2.39(4) |
| 1DFT | lp | 38.4 | H11PX⋯H6′ | 2.10 | H11PX⋯H3 | 2.28 |
| 3 | Au | 37.7(3) | H11A⋯H6′ | 2.32(3) | H11A⋯H3 | 2.52(4) |
| 3DFT | Au | 39.8 | H11A⋯H6′ | 2.17 | H11A⋯H3 | 2.34 |
| 5 | Se | 48.6(3) | H11A⋯H6′ | 2.40(3) | H11A⋯H3 | 2.46(2) |
| 5DFT | Se | 39.9 | H11A⋯H6′ | 2.14 | H11A⋯H3 | 2.29 |
The PSe distances in 4 and 5 are 2.0798(6) and 2.0801(7) Å, respectively. These are significantly shorter than that in Cy3PSe, 2.108(1) Å,39 and those in recent compilations of PSe distances.38,40 In fact, the PSe distances in 4 and 5 are amongst the shortest yet reported; of 378 structures containing the fragment {R1R2R3PSe} in the Cambridge Structural Database41 only seven have PSe distances <2.08 Å.42 Short PSe distances in 4 and 5 are fully consistent with a significant degree of s character in the phosphorus lone pair of I and 1, as implied from the 1JPSe values.
Conclusions
The bis(carboranyl)phosphines I and 1 are comparable in their steric demands to PCy3 but are much less basic. In fact, in terms of their 1JPSe values, I and 1 are the least basic of any carboranylphosphines so far reported. As large, weakly basic phosphines I and 1 have potential as ligands in homogeneous catalysis, and future contributions will develop this theme.Experimental
Synthesis
Experiments were performed under dry, oxygen free N2 using standard Schlenk techniques, although subsequent manipulations were sometimes performed in an open laboratory. Solvents were either freshly distilled under nitrogen from the appropriate drying agent [THF, Et2O and 40–60 °C petroleum ether (petrol); sodium wire: CH2Cl2 (DCM); calcium hydride] or were purified by using an MBRAUN SPS-800 and stored over 4 Å molecular sieves, and all were degassed (3× freeze–pump–thaw cycles) before use. Preparative TLC employed 20 × 20 cm Kieselgel F254 glass plates and for column chromatography we used 60 Å silica as the stationary phase. NMR spectra at 400.1 MHz (1H), 128.4 MHz (11B) or 162.0 MHz (31P) were recorded on a Bruker DPX-400 spectrometer from CDCl3 solutions at 298 K, using CDCl3 stored over 4 Å molecular sieves. Electron impact mass spectrometry (EIMS) was carried out using a Finnigan (Thermo) LCQ Classic ion trap mass spectrometer (University of Edinburgh). The starting materials 1,1′-bis(o-carborane)3 and (tht)AuCl43 were prepared by literature methods or slight variations thereof. All other reagents were supplied commercially.:petrol, 1:19) yielded a white solid (Rf = 0.97), subsequently identified as [μ-2,2′-PEt-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (1) (0.154 g, 0.447 mmol, 64%). 11B{1H} NMR, δ −0.3 (2B), −3.8 (2B), −6.6 (6B), −7.6 (2B), −8.5 (2B), −10.6 (6B). 1H NMR, δ 1.98 (q, 2H, PCH2CH3, 3JHH = 7.9 Hz), 1.31 (dt, 3H, PCH2CH3, 3JHH = 7.9 Hz, 3JPH = 23.0 Hz). 1H{31P} NMR, δ 1.99 (q, 2H, PCH2CH3, 3JHH = 7.9 Hz), 1.32 (t, 3H, PCH2CH3, 3JHH = 7.9 Hz). 31P{1H} NMR, δ 42.75 (s). EIMS, envelope centred on m/z 344.3 (M+).
:petrol, 1:9) yielded a colourless band at Rf = 0.59 from which the product, [μ-2,2′-P(Ph)Se-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (4), was isolated as a pale-pink solid (0.057 g, 0.120 mmol, 47%). 11B{1H} NMR, δ 1.2 (2B), −4.1 to −11.2 [overlapping resonances with maxima at −4.1, −6.0, −6.4, −8.5, −9.9, −11.2 (18B)]. 1H NMR, δ 8.31–8.25 (m, 2H, C6H5), 7.73–7.68 (m, 1H, C6H5), 7.63–7.57 (m. 2H, C6H5). 31P{1H} NMR, δ 50.52 (s + Se satellites, 1JPSe = 891 Hz). EIMS, envelope centred on m/z 471.3 (M+).
Crystallography
Diffraction-quality crystals of all compounds were obtained by slow evaporation of a solution of the appropriate compound: I, 1 and 4; petrol: 2 and 3; DCM: 5; CDCl3. Intensity data were collected on a Bruker X8 APEXII diffractometer using Mo-Kα X-radiation, with crystals mounted in inert oil on a cryoloop and cooled to 100 K by using an Oxford Cryosystems Cryostream. Indexing, data collection and absorption correction were performed using the APEXII suite of programs.44 Using OLEX2,45 structures were solved by direct methods using the SHELXS46 or SHELXT47 programme and refined by full-matrix least-squares (SHELXL).46All crystals were single except those of 5 which was treated as a two-component twin. All crystals were also fully ordered and solvate-free, except those of compound 3. In 3 the gold carboranylphosphine complex is fully ordered but there is a disordered solvent in the lattice that was impossible to satisfactorily model. Hence for this structure the intensity contribution of the disordered solvent was removed using the BYPASS procedure48 implemented in OLEX2. The total electron count of the solvent per cell was 420e, which corresponds to 10 DCM molecules. These disordered solvent molecules predominantly occupy four voids of ca. 270 Å3 each.
For all structures H atoms bound to cage B atoms were allowed to refine positionally whilst H atoms bound to C atoms were constrained to idealised geometries; Cphenyl–H = 0.95 Å, Cmethyl–H = 0.98 Å, Cmethylene–H = 0.99 Å. All H displacement parameters, Uiso, were constrained to be 1.2 × Ueq (bound B or C) except for Me H atoms [Uiso(H) = 1.5 × Ueq C(Me)]. Table 3 contains further experimental details.
| I | 1 | 2 | 3·1.25CH2Cl2 | 4 | 5 | |
|---|---|---|---|---|---|---|
| CCDC | 1527028 | 1527029 | 1527030 | 1527031 | 1527032 | 1527033 |
| Formula | C10H25B20P | C6H25B20P | C10H25AuB20ClP | C6H25AuB20ClP | C10H25B20PSe | C6H25B20PSe |
| M | 392.47 | 344.43 | 624.89 | 683.00 | 471.43 | 423.39 |
| Crystal system | Triclinic | Monoclinic | Monoclinic | Orthorhombic | Monoclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n | C2/c | Pbca | P21/n |
P |
| a/Å | 7.1139(10) | 23.441(2) | 17.0588(8) | 10.4380(13) | 10.3457(12) | 7.1663(7) |
| b/Å | 11.4378(14) | 7.2690(6) | 14.2499(7) | 17.711(2) | 16.9431(17) | 11.0308(15) |
| c/Å | 13.3912(15) | 24.083(2) | 20.0698(9) | 27.475(3) | 13.6941(14) | 14.573(2) |
| α/° | 81.884(5) | 90 | 90 | 90 | 90 | 111.478(9) |
| β/° | 84.954(5) | 108.290(5) | 102.497(3) | 90 | 105.612(6) | 92.125(9) |
| γ/° | 81.231(6) | 90 | 90 | 90 | 90 | 104.679(6) |
| U/Å3 | 1063.6(2) | 3896.2(6) | 4763.1(4) | 5079.3(10) | 2311.9(4) | 1026.2(2) |
| Z, Z′ | 2, 1 | 8, 2 | 8, 1 | 8, 1 | 4, 1 | 2, 1 |
| F(000)/e | 400 | 1408 | 2368 | 2596 | 936 | 420 |
| D calc./Mg m−3 | 1.225 | 1.174 | 1.743 | 1.786 | 1.354 | 1.370 |
| μ(Mo-Kα)/mm−1 | 0.127 | 0.128 | 6.357 | 6.224 | 1.693 | 1.898 |
| θ max/° | 26.46 | 23.77 | 32.01 | 27.24 | 28.95 | 30.14 |
| Data measured | 15395 |
44057 |
58934 |
33633 |
43105 |
40190 |
| Unique data, n | 4334 | 5928 | 8266 | 5651 | 6088 | 5691 |
| R int | 0.0636 | 0.1415 | 0.0796 | 0.0526 | 0.0740 | 0.0763 |
| R, wR2 (obs. data) | 0.0685, 0.1260 | 0.0795, 0.1841 | 0.0342, 0.0602 | 0.0297, 0.0585 | 0.0365, 0.0767 | 0.0410, 0.0861 |
| S (all data) | 1.135 | 1.053 | 1.015 | 1.020 | 1.034 | 1.036 |
| Variables | 340 | 609 | 358 | 323 | 349 | 315 |
| E max, Emin/e Å−3 | 0.31, −0.35 | 0.48, −0.41 | 0.93, −1.81 | 0.69, −0.65 | 0.35, −0.42 | 0.61, −0.58 |
Cone angle and percent buried volume calculations
Bis(carborane)phosphine cone angles were calculated from the crystallographically-determined structures using both the free bis(carborane)phosphines (compounds I and 1) and their AuCl complexes (compounds 2 and 3), using the method of Müller and Mingos.49 The P–M distance was set at 2.28 Å. %Vbur calculations were performed (again on all compounds I, 1–3) using the SambVca software of Cavallo and co-workers,50 with a sphere radius of 3.5 Å, a P–M distance of 2.28 Å and scaled Bondi radii.Computational methods
All electronic structure calculations were carried out using the Gaussian 09 (Revision D.01)51 program suite at the DFT level of theory. Geometries of all compounds were fully optimised without imposing symmetry constraints (C1 symmetry), employing the BP86 functional.52 Ahlrich's def2-TZVP basis of triple-ζ quality was used on P, Au, Cl, Se, and C and all hydrogen atoms of the ethyl group, while all B–H units of the carborane cage were described with the def2-SVP basis set.53 The core electrons in Au were replaced by the Stuttgart–Dresden scalar relativistic effective core potential (SDD, ECP60MWB).54 Optimised stationary points were characterised by analysis of their analytical second derivatives, with minima having only positive eigenvalues and transition states having exactly one imaginary eigenvalue. Subsequent geometry optimisations in both directions of the reaction coordinate were performed to confirm the minima linked by each transition state. The frequency calculations also provided thermal and entropic corrections to the total energy in the gas phase at T = 298.15 K and p = 1 atm within the rigid-rotor/harmonic oscillator (RRHO) approximation. Dispersion effects were accounted for by applying Grimme's van der Waals correction (D3 parameterization) protocol including Becke–Johnson damping during the optimisations.55 The topology of the electron density was analysed by means of QTAIM (quantum theory of atoms in molecules),56 as implemented in the AIMAll package.57 For 3-DFT, inner shell electrons on Au modelled by the ECP were fitted by core density functions. Isotropic NMR spin–spin coupling constants were calculated using the coupled-perturbed SCF58 method with the BHandHLYP59 functional, which was found to yield the best agreement with experimental couplings. The J-couplings were obtained as the sum of all four Ramsey terms, i.e. Fermi contact (FC), spin–dipolar (SD), paramagnetic spin–orbit (PSO), and diamagnetic spin–orbit (DSO). The reported coupling constants have been averaged assuming free internal molecular rotation. The basis sets stated above were replaced by aug-cc-pVTZ-J60 on P, and ethyl (C, H), as well as the cc-pVDZ61 basis set (and the associated ECP for Au) on all other atoms. Effects due to the presence of a solvent were treated implicitly with a polarisable dielectric model, using the IEFPCM formalism in conjunction with Truhlar's SMD model.62 The chosen dielectric constant (ε = 4.71) corresponds to that of chloroform. The compositions of molecular orbitals were analysed within the framework of localised natural bond orbitals, using the NBO 6.0 software.63Acknowledgements
We thank the EPSRC for support of LER through grant no. EP/I031545/1. TK wishes to thank Prof. Stuart A. Macgregor for providing access to high-performance computing facilities and helpful discussions.References
- W. Y. Man, G. M. Rosair and A. J. Welch, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70, 462 CAS.
- J. A. Dupont and M. F. Hawthorne, J. Am. Chem. Soc., 1964, 86, 1643 CrossRef CAS.
- S. Ren and Z. Xie, Organometallics, 2008, 27, 5167 CrossRef CAS.
- D. Ellis, G. M. Rosair and A. J. Welch, Chem. Commun., 2010, 46, 7394 RSC.
- D. Ellis, D. McKay, S. A. Macgregor, G. M. Rosair and A. J. Welch, Angew. Chem., Int. Ed., 2010, 49, 4943 CrossRef CAS PubMed.
- W. Y. Man, S. Zlatogorsky, H. Tricas, D. Ellis, G. M. Rosair and A. J. Welch, Angew. Chem., Int. Ed., 2014, 53, 12222 CrossRef CAS PubMed.
- M. J. Martin, W. Y. Man, G. M. Rosair and A. J. Welch, J. Organomet. Chem., 2015, 798, 36 CrossRef CAS.
- Z.-J. Yao, Y.-Y. Zhang and G.-X. Jin, J. Organomet. Chem., 2015, 798, 274 CrossRef CAS.
- D. Mandal, W. Y. Man, G. M. Rosair and A. J. Welch, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 793 CAS.
- G. Thiripuranathar, W. Y. Man, C. Palmero, A. P. Y. Chan, B. T. Leube, D. Ellis, D. McKay, S. A. Macgregor, L. Jourdan, G. M. Rosair and A. J. Welch, Dalton Trans., 2015, 44, 5628 RSC.
- G. S. Kazakov, I. B. Sivaev, K. Yu. Suponitsky, D. D. Kirilin, V. I. Bregadze and A. J. Welch, J. Organomet. Chem., 2016, 805, 1 CrossRef CAS.
- D. Mandal, W. Y. Man, G. M. Rosair and A. J. Welch, Dalton Trans., 2016, 45, 15013 RSC.
- L. E. Riley, A. P. Y. Chan, J. Taylor, W. Y. Man, D. Ellis, G. M. Rosair, A. J. Welch and I. B. Sivaev, Dalton Trans., 2016, 45, 1127 RSC.
- W. Y. Man, D. Ellis, G. M. Rosair and A. J. Welch, Angew. Chem., Int. Ed., 2016, 55, 4596 CrossRef CAS PubMed.
- S. L. Powley, L. Schaefer, W. Y. Man, D. Ellis, G. M. Rosair and A. J. Welch, Dalton Trans., 2016, 45, 3635 RSC.
- D. Zhao, J. Zhang, Z. Lin and Z. Xie, Chem. Commun., 2016, 52, 9992 RSC.
- Y. O. Wong, M. D. Smith and D. V. Peryshkov, Chem. – Eur. J., 2016, 22, 6764 CrossRef CAS PubMed.
- Y. O. Wong, M. D. Smith and D. V. Peryshkov, Chem. Commun., 2016, 52, 12710 RSC.
- K. O. Kirlikovali, J. C. Axtell, A. Gonzalez, A. C. Phung, S. I. Khan and A. M. Spokoyny, Chem. Sci., 2016, 7, 5132 RSC.
- I. B. Sivaev, Commun. Inorg. Synth., 2016, 4, 21 Search PubMed.
- (a) D. A. Owen and M. F. Hawthorne, J. Am. Chem. Soc., 1970, 92, 3194 CrossRef CAS; D. A. Owen and M. F. Hawthorne, J. Am. Chem. Soc., 1971, 93, 873 Search PubMed; (b) R. A. Love and R. Bau, J. Am. Chem. Soc., 1972, 94, 8274 CrossRef CAS; (c) D. E. Harwell, J. McMillan, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 1997, 36, 5951 CrossRef CAS PubMed.
- L. I. Zakharkin and N. F. Shemyakin, Izv. Akad. Nauk SSSR, Ser. Khim., 1978, 1450 CAS.
- A. I. Yanovsky, N. G. Furmanova, Yu. T. Struchkov, N. F. Shemyakin and L. I. Zakharkin, Izv. Akad. Nauk SSSR, Ser. Khim., 1979, 1523 Search PubMed.
- S. E. Johnson and C. B. Knobler, Phosphorus, Sulfur Silicon Relat. Elem., 1996, 115, 227 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- P. L. A. Popelier, J. Phys. Chem. A, 1998, 102, 1873 CrossRef CAS.
- P. W. N. M. van Leeuwen, Homogeneous Catalysis: Understanding the Art, Kluwer, Dordrecht, The Netherlands, 2004 Search PubMed.
- Representative examples: (a) F. A. Hart and D. W. Owen, Inorg. Chim. Acta, 1985, 103, L1 CrossRef CAS; (b) C. Viñas, M. A. Flores, R. Núñez, F. Teixidor, R. Kivekäs and R. Sillanpää, Organometallics, 1998, 17, 2278 CrossRef; (c) F. Teixidor, R. Núñez, M. A. Flores, A. Demonceau and C. Viñas, J. Organomet. Chem., 2000, 614–615, 48 CrossRef CAS; (d) H.-S. Lee, J.-Y. Bae, J. Ko, Y. S. Kang, H. S. Kim, S.-J. Kim, J.-H. Chung and S. O. Kang, J. Organomet. Chem., 2000, 614–615, 83 CrossRef CAS; (e) H.-S. Lee, J.-Y. Bae, J. Ko, Y. S. Kang, H. S. Kim and S. O. Kang, Chem. Lett., 2000, 29, 602 CrossRef; (f) H. Brunner, A. Apfelbacher and M. Zabel, Eur. J. Inorg. Chem., 2001, 917 CrossRef CAS; (g) H.-S. Lee, J.-Y. Bae, D.-H. Kim, H. S. Kim, S.-J. Kim, S. Cho, J. Ko and S. O. Kang, Organometallics, 2002, 21, 210 CrossRef CAS; (h) H. Wang, H.-S. Chan, J. Okuda and Z. Xie, Organometallics, 2005, 24, 3118 CrossRef CAS; (i) V. Lavallo, J. H. Wright, F. S. Tham and S. Quinlivan, Angew. Chem., Int. Ed., 2013, 52, 3172 CrossRef CAS PubMed.
- C. A. Tolman, J. Am. Chem. Soc., 1970, 92, 2956 CrossRef CAS; C. A. Tolman, Chem. Rev., 1977, 77, 313 CrossRef.
- A. C. Hillier, W. J. Sommer, B. S. Yong, J. L. Petersen, L. Cavallo and S. P. Nolan, Organometallics, 2003, 22, 4322 CrossRef CAS.
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841 RSC.
- (a) D. W. Allen and B. F. Taylor, J. Chem. Soc., Dalton Trans., 1982, 51 RSC; (b) D. W. Allen, I. W. Nowell and B. F. Taylor, J. Chem. Soc., Dalton Trans., 1985, 2505 RSC; (c) N. Anderson and B. Keay, Chem. Rev., 2001, 101, 997 CrossRef; (d) U. Beckmann, D. Süslüyan and P. C. Kunz, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 2061 CrossRef CAS.
- (a) C. J. Jameson and H. S. Gutowsky, J. Chem. Phys., 1969, 51, 2790 CrossRef CAS; (b) C. J. Jameson, J. Am. Chem. Soc., 1969, 91, 6232 CrossRef CAS; (c) W. McFarlane, Proc. R. Soc. London, Ser. A, 1968, 306, 185 CrossRef CAS.
- (a) J. P. Alabrand, D. Gagnaire and J. B. Robert, J. Mol. Spectrosc., 1968, 27, 428 CrossRef; (b) A. R. Cullingworth, A. Pidcock and J. D. Smith, Chem. Commun., 1966, 89 RSC.
- Although the sign of 1JPSe has been determined to be negative (e.g. W. McFarlane and D. S. Rycroft, J. Chem. Soc., Dalton Trans., 1973, 2162 RSC ) it is conventionally reported as positive and we will follow this convention here.
- A. R. Popescu, A. Laromaine, F. Teixidor, R. Sillanpää, R. Kivekäs, J. I. Llambias and C. Viñas, Chem. – Eur. J., 2011, 17, 4429 CrossRef CAS PubMed.
- P. Farràs, F. Teixidor, I. Rojo, R. Kivekäs, R. Sillanpää, P. González-Cardoso and C. Viñas, J. Am. Chem. Soc., 2011, 133, 16537 CrossRef PubMed.
- A. Muller, S. Otto and A. Roodt, Dalton Trans., 2008, 650 RSC.
- J. A. Davies, S. Dutremez and A. A. Pinkerton, Inorg. Chem., 1991, 30, 2380 CrossRef CAS.
- S. Otto, A. Ionescu and A. Roodt, J. Organomet. Chem., 2005, 690, 4337 CrossRef CAS.
- C. R. Groom and F. H. Allen, Angew. Chem., Int. Ed., 2014, 53, 662 CrossRef CAS PubMed . For this search we used ConQuest and version 5.37 of the CSD (November 2015).
-
(a) SUNKIX, PSe 2.073(9), 2.079(3) Å: Y. Sun, M.-Q. Yan, Y. Liu, Z.-Y. Lian, T. Meng, S.-H. Liu, J. Chen and G.-A. Yu, RSC Adv., 2015, 5, 71437 RSC;
(b) BAZWEF, PSe 2.073(3) Å: T. Stampfl, R. Gutmann, G. Czermak, C. Langes, A. Dumfort, H. Kopacka, K.-H. Ongania and P. Bruggeller, Dalton Trans., 2003, 3425 RSC;
(c) DAHROV, PSe 2.0588(9), 2.0770(9) Å: K. D. Reichl, C. L. Mandell, O. D. Henn, W. G. Dougherty, W. S. Kassel and C. Nataro, J. Organomet. Chem., 2011, 696, 3882 CrossRef CAS;
(d) NIKBIS, PSe 2.075(15) Å: Q. Zhang, M. Hong and H. Liu, Transition Met. Chem., 1997, 22, 156 CrossRef CAS;
(e) PPHSEA, PSe 2.0697(17) Å: O. Orama, K. Nieminen, M. Karhu and R. Uggla, Cryst. Struct. Commun., 1979, 8, 909 CAS;
(f) UXIWUU, PSe 2.079(3) Å: R. Kabe, V. M. Lynch and P. Anzenbacher Jr., CrystEngComm, 2011, 13, 5423 RSC;
(g) VUHJIR, PSe 2.055(3), 2.056(4), 2.065(3) Å: M. Birkel, J. Schulz, U. Bergstrasser and M. Regitz, Angew. Chem., Int. Ed., 1992, 31, 879 CrossRef.
- R. Uson, A. Laguna and M. Laguna, Inorg. Synth., 1989, 26, 85 CrossRef CAS.
- Bruker AXS APEX2, version 2009-5, Bruker AXS Inc., Madison, Wisconsin, USA, 2009 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 3 CrossRef PubMed.
- (a) P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 194 CrossRef; (b) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- (a) T. E. Müller and D. M. P. Mingos, Transition Met. Chem., 1995, 20, 533 CrossRef; (b) D. M. P. Mingos in Modern Coordination Chemistry: The Legacy of Joseph Chatt, RSC, Cambridge, 2002, p. 69 Search PubMed.
- (a) A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 2009, 1759 CrossRef; (b) http://www.molnac.unisa.it/OMtools/sambvca.php .
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS; (b) J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- D. Andrae, U. Haussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 122, 154101 CrossRef PubMed; (c) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, 1990 Search PubMed.
- T. A. Keith, AIMAll (Version 13.02.26), TK Gristmill Software, Overland Park KS, USA, 2014 Search PubMed (aim.tkgristmill.com), 1997–2013.
- (a) V. Sychrovský, J. Gräfenstein and D. Cremer, J. Chem. Phys., 2000, 113, 3530 CrossRef; (b) T. Helgaker, M. Watson and N. C. Handy, J. Chem. Phys., 2000, 113, 9402 CrossRef CAS; (c) V. Barone, J. E. Peralta, R. H. Contreras and J. P. Snyder, J. Phys. Chem. A, 2002, 106, 5607 CrossRef CAS.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS; (c) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS; (d) A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS.
- (a) P. F. Provasi, G. A. Aucar and S. P. A. Sauer, J. Chem. Phys., 2001, 115, 1324 CrossRef CAS; (b) J. E. Peralta, G. E. Scuseria, J. R. Cheeseman and M. J. Frisch, Chem. Phys. Lett., 2003, 375, 452 CrossRef CAS.
- (a) T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007 CrossRef; (b) A. K. Wilson, D. E. Woon, K. A. Peterson and T. H. Dunning Jr., J. Chem. Phys., 1999, 110, 7667 CrossRef CAS; (c) K. A. Peterson and C. Puzzarini, Theor. Chem. Acc., 2005, 114, 283 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2013; http://nbo6.chem.wisc.edu/ Search PubMed.
Footnotes |
| † Dedicated to Professor Evamarie Hey-Hawkins on the occasion of her 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: NMR spectra of all new compounds reported. Full details of computational studies. CCDC 1527028–1527033. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt00485k |
| This journal is © The Royal Society of Chemistry 2017 |