
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anion exchange dynamics in the capture of perchlorate by a cationic Ag-based MOF†
Ian R.
Colinas
a,
Kenneth K.
Inglis
b,
Frédéric
Blanc
 *b and
Scott R. J.
Oliver
*a
*b and
Scott R. J.
Oliver
*a
aUniversity of California, Santa Cruz, Department of Chemistry and Biochemistry, 1156 High Street, Santa Cruz, California 95064, USA. E-mail: soliver@ucsc.edu
bDepartment of Chemistry, Stephenson Institute for Renewable Energy, University of Liverpool, L69 7ZD, UK. E-mail: frederic.blanc@liverpool.ac.uk
First published on 28th March 2017
Abstract
We report a detailed study of the host–guest interaction for a cationic metal–organic framework that can reversibly capture perchlorate. The structural transformation and flexibility of silver 4,4′-bipyridine nitrate (SBN) upon formation of silver 4,4′-bipyridine perchlorate (SBP) was evaluated by monitoring the anion exchange dynamics using a combination of powder X-ray diffraction (PXRD) with multinuclear 13C, 15N and 109Ag solid-state NMR spectra at different time intervals of the anion exchange. The structural transformation from SBN to SBP is complete within 70 minutes and was determined to take place by a solvent-mediated process. This pathway is confirmed by the morphological changes of the two crystalline materials observed by SEM. This key understanding may lead to application of this material towards perchlorate capture.
1. Introduction
Porous structures possessing weakly bound charge-balancing ions are essential for the development of new separation technologies and the removal of pollutant ions from aqueous environments. The capture of toxic perchlorate (ClO4−) presents unique challenges due its high solubility and non-complexing nature, making it highly mobile and resistant to traditional wastewater treatment technologies.1 Perchlorate is an emerging trace contaminant in groundwater and has gained significant attention, as it has become widespread in many countries.2 This anionic pollutant can block the uptake of iodide by the thyroid due to their equivalent charge and similar ionic radii. This blockage can disrupt the production of thyroid hormones and affect metabolism, possibly leading to hypothyroidism or mental retardation in fetuses and infants.3 Perchlorate salts are used as the conventional solid oxidant in the industrial manufacture of rocket fuel, explosives, flares and fireworks.4 Commercially available ion exchange resins are single use and exhibit limited thermal and chemical stability,5 while other ion exchanging materials such as layered double hydroxides (LDHs) display poor selectivity6 and adsorption capacity7 for perchlorate. Higher performing ion exchangers are therefore sought to reversibly capture perchlorate at greater capacity and selectivity. In particular, metal–organic frameworks (MOFs) have gained significant attention in the selective capture of pollutants via ion exchange.8Recently, MOFs have been under intense investigation due to their ability to exchange their intrapore or interlayer ions with other ionic species in solution. Their structural integrity is not compromised, which make these materials excellent candidates for the separation of pollutants.9 The adsorptive separation of pollutant species in the gas phase, such as carbon dioxide or methane, is driven by the reversible dynamic behavior of the network, often referred to as “breathing”. This action results in a change of interaction strength between the guest molecules and host framework due to framework displacement accompanied by a change in the unit cell volume.10 There has been much less effort into understanding the structural flexibility and dynamic transformations that solid MOFs exhibit during adsorption and/or ion exchange. The mechanism by which such ion exchange processes take place has been proposed to be either by a solid state or a solvent-mediated transformation. In the case of a solid state transition, the ion exchange proceeds through the diffusion of free ions within the channels of the coordination polymer crystals. In contrast, the solvent-mediated process involves the dissolution of the initial coordination polymer followed by the formation and crystallization of a new coordination polymer from the solution phase.11
The structural transformations that coordination polymers undergo during ion exchange processes can be monitored by single crystal or powder X-ray diffraction (PXRD) of the MOF in combination with analytical measurements of the solution towards incoming and outgoing guests. If possible, single crystal X-ray crystallographic methods are preferred for the exact structural determination of the new MOF phase upon adsorption. This methodology, however, provides limited structural information regarding the dynamic behavior of the framework during exchange due to the absence or sufficient size/quality of crystals. In contrast, magic-angle spinning (MAS) nuclear magnetic resonance (NMR) spectroscopy provides structural information about the local environment of various nuclei in solids, including both the metal nodes and the organic linkers of the MOFs.12 Hence, NMR spectroscopy could be used in conjunction with XRD methods as a complementary technique for analyzing the exchange dynamics of MOF materials.13 NMR is well suited to probe the structural flexibility and reversible dynamic behavior of a framework undergoing ion exchange. Jiang et al. reported the structural flexibility of a Cu(bipy)2(H2O)2(BF4) (bipy = 4,4′-bipyridine) framework by chemisorption of probe molecules that could induce reversible structural changes. The transitions could be monitored by 11B MAS NMR as a result of the change in polarization of the probe molecule and its interaction with the Cu(II) paramagnetic sites.14
In our previous work,15 we reported the successful capture of aqueous perchlorate by Silver 4,4′-Bipyridine Nitrate (SBN)16 to yield a Silver 4,4′-Bipyridine Perchlorate (SBP) MOF.17 We showed that SBN effectively exchanged guest NO3− ions for ClO4− ions at a record capacity of 354 mg g−1 within 90 minutes. The material also selectively captured this pollutant in the presence of 50-fold molar SO42− and CO32−, suggesting this methodology could be applied to treat perchlorate pollution at underground plume sites. In addition, SBN displayed excellent reusability: 96% of SBP could be regenerated back to SBN over multiple cycles while maintaining uptake capacity. Anion exchange by metal bipyridine and pyrazine coordination polymers was evaluated by Schröder and coworkers utilizing several anions such as BF4− and PF6−.18 The uptake was determined to be a solvent mediated process driven by the relative solubilities of the complex pairs as well as the relative hydration energy of the anions residing in the pores.
Herein, we report a study of the host–guest interaction of an Ag-bipy coordinated polymer during NO3− and ClO4− anion exchange dynamics. The structural transformation is followed by analysis of the [Ag-bipy]+ material at different time intervals using PXRD to assess the presence of two structural phases corresponding to SBN and SBP. The structural transformation during exchange is further investigated by recording multinuclear 13C, 15N and 109Ag cross-polarization (CP) MAS NMR spectra. 13C [1.13% natural abundance (nat. ab.), nuclear spin I = 1/2] NMR is used to (semi)-quantify the SBN to SBP ratio during the anion exchange. 15N (0.37% nat. ab., I = 1/2) NMR determines whether NO3− is present and probes the bonding between the nitrogen of bipy and silver via the scalar J coupling interaction and is further confirmed by 109Ag (48.18% nat. ab., I = 1/2) NMR. In addition, this exchange is further investigated by inspecting the morphological changes of the crystalline materials by scanning electron microscopy (SEM).
2. Experimental
2.1 Synthesis
In short, a mixture of AgNO3 (AgNO3, Fisher, 99%, 0.1 g, 0.59 mmol), 4,4′-bipyridine (Acros Organics, 98%, 0.1 g, 0.64 mmol) and deionized water (10 mL) was stirred at room temperature for 3 days in a sealed beaker. Pale gray crystals were isolated after filtration and rinsed with water (10 mL) and acetone (5 mL) to yield SBN (0.187 g, 97.6% based on silver nitrate). Batch anion exchange experiments were carried out under ambient conditions by mildly stirring the ground SBN (80 mg, 0.25 mmol) with 50 mL deionized water and NaClO4 (Fluka Analytical, 98%, 35 mg, 0.25 mmol). The post-exchange crystalline SBP product was recovered at the different time intervals by vacuum filtration and rinsed with water (10 mL) and acetone (5 mL).2.2 Regeneration
The regeneration of SBN from SBP was carried out by stirring the ground SBP (80 mg, 0.22 mmol) in a 0.1 M NaNO3 solution (NaNO3, Fisher, 99%) at 70 °C for 24 h. The regeneration of the SBN was studied by recording the PXRD, 13C, 15N, 109Ag CP MAS NMR data and SEM images after treatment of the SBP material with excess nitrate at 70 °C under stirring.2.3 Characterization
PXRD data were measured on a Rigaku Americas Miniflex Plus diffractometer, scanning from 2 to 40° (2θ) at a rate of 2° min−1 with a 0.04° step size under Cu Kα radiation (λ = 1.5418 Å). Ion Chromatography (IC) analysis was performed to assess the perchlorate concentration using a Dionex ICS-3000 with an IonPac AS20 column and a detection limit of 3 μg L−1 (ppb). SEM data were collected with a FEI Quanta 3D Dualbeam microscope. Solid-state NMR experiments were performed at room temperature on a Wide Bore 400 MHz 9.4 T Bruker Avance III HD NMR spectrometer equipped with a 4 mm HXY triple-resonance MAS probe (in double resonance mode) for 13C and 15N and with a 4 mm HX MAS probe for 109Ag or on a Wide Bore 850 MHz 20 T Bruker Avance II NMR spectrometer equipped with a 3.2 mm HXY triple-resonance MAS probe (in double resonance mode) for 13C. Data acquisition was performed with cross polarization (CP) and SPINAL-64 heteronuclear decoupling.19 Recycle delays ranging from 30 to 180 s were used and correspond to 1.3 × 1H T1 (measured by 1H saturation recovery experiments). All other experimental details are given in Table S1.† The 13C, 15N and 109Ag chemical shifts were referenced to the CH carbon of adamantane at 29.45 ppm,20 glycine at 33.4 ppm (corresponding to liquid NH3 at 0 ppm)21 and a saturated solution of AgNO3 in D2O [with 0.24 M of Fe(NO3)3] at 0 ppm,22 respectively. All samples were packed in air. Using the Haeberlen convention,23 the principal components δ11, δ22, δ33 of the chemical shift tensors were defined such that |δ33 − δiso| ≥ |δ11 − δiso| ≥ |δ22 − δiso| where δiso is the isotropic chemical shift and related to the principal components of the shielding tensor by δiso = (δ11 + δ22 + δ33)/3. The chemical shift anisotropy δaniso is defined as δaniso = δ33 − δiso and the deviation from axial symmetry given by the asymmetry parameter η = (δ22 − δ11)/δaniso.3. Results and discussion
The synthesis of SBN can be carried out at room temperature by the initial dissociation of silver nitrate followed by the self-assembly of the cationic polymer chains, charge balanced by nitrate ions. SBN can undergo quantitative anion exchange upon exposure to ClO4− in solution to yield SBP with a 99% conversion as determined by monitoring the concentration of ClO4− in solution from ion chromatography (IC). The PXRD data confirms the identity of the two crystalline phases as they match the theoretical patterns based on the single crystal solution (Fig. S1†).16,17Both SBN and SBP structures consist of Ag(I) chelated by the nitrogens of two different (μ-2)-4,4′-bipyridine units that result in extended polymeric chains. The chains align parallel into a non π-stacked layer where the rings are coplanar with the plane of the page. The two structures differ in the arrangement of their subsequent layers. For SBN, the cationic layers are rotated by 90°, forming a criss-cross or T-shaped pattern in which half the pyridine rings π-stack in a staggered manner with a distance of 3.55 Å from the next layer (Fig. 1). In addition, half of the Ag(I) centers are dimerized and cross linked to the next layer by the formation of Ag–Ag bonds with a distance of 2.97(1) Å. In contrast, the cationic layers of SBP are eclipsed, resulting in π-stacking of all the pyridine rings and a shorter interlayer distance of 3.47 Å (Fig. 1). The Ag(I) centers have a long contact distance of 3.60(1) Å, which is well above the covalent range. These longer Ag–Ag distances allow for the spherical ClO4− anions to be more spread out between the polymeric interlayers. The ClO4− to Ag(I) distance is therefore greater for SBP than for SBN but closer contact of the anions to the bipyridine CHCIV in SBP (Table 1).
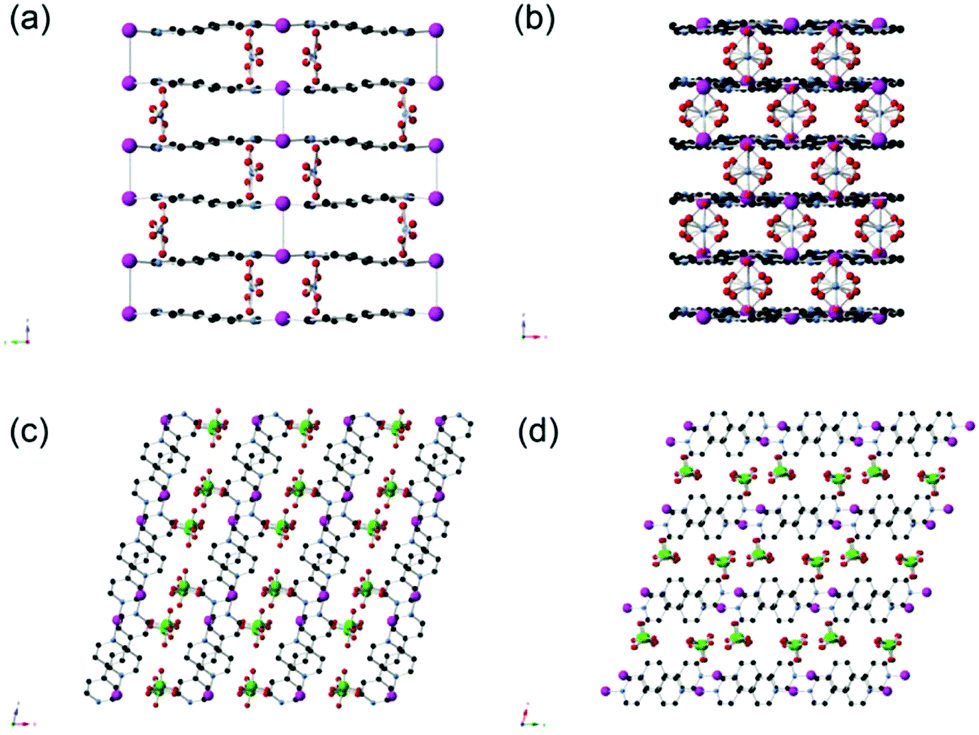 | ||
| Fig. 1 (a) Crystallographic view of SBN in the “a” plane and (c) “b” plane. (b) Crystallographic view of SBP in the “a” plane and (d) “c” plane. The nitrate oxygens are disordered between two sites (Ag, magenta; N, blue; O, red; C, black; Cl, green). | ||
| SBN | SBP | |
|---|---|---|
| Distance | ||
| Ag(I)–Ag(I)/Å | 2.97(1) | 3.60(1) |
| Anion O–Ag(I)/Å | 2.826(17), 2.782(10) | 2.945(15), 2.863(10) |
| Anion O–CHCIV/Å | 3.374(10) | 3.352(10) |
| π-Stacking/Å | 3.55 | 3.47 |
| Angle | ||
| N–Ag(I)–N/° | 173.73 | 174.76 |
Based on the above structural considerations, it is clear that the two coordination polymers are structurally different in the arrangement of the cationic layers as well as the location of the anions. Such structural differences suggest that the anion exchange may proceed by a solvent-mediated process involving the initial dissolution of the SBN T-shaped polymers followed by the crystallization of eclipsed SBP polymers from the perchlorate solution (Fig. S2†).15 In order to support this hypothesis, the anion exchange was monitored versus time by both PXRD (Fig. 2) and multinuclear MAS NMR (Fig. 3). After 10 minutes of exposure to ClO4−, the intensity of the low angle (002) peak decreases and a new set of peaks [e.g. 22° (2θ), corresponding to the (211) plane of the SBP crystalline phase] begin to appear, indicating initiation of the process.
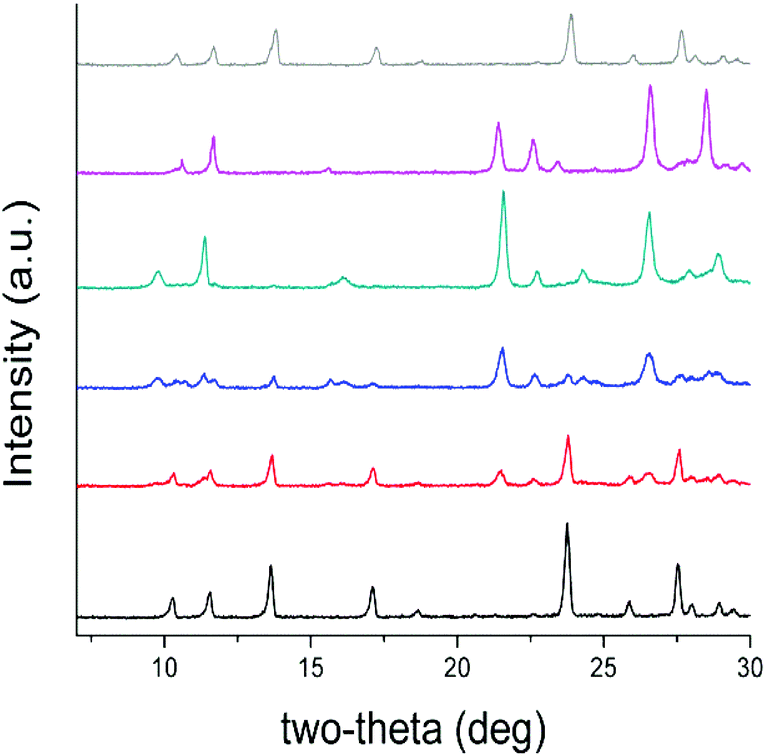 | ||
| Fig. 2 PXRD patterns of the SBN/SBP anion exchange of SBN pre-exchange (black), after 10 min (red), 25 min (blue), 40 min (green), complete exchange to SBP after 70 min (purple), and regeneration to SBN (grey). | ||
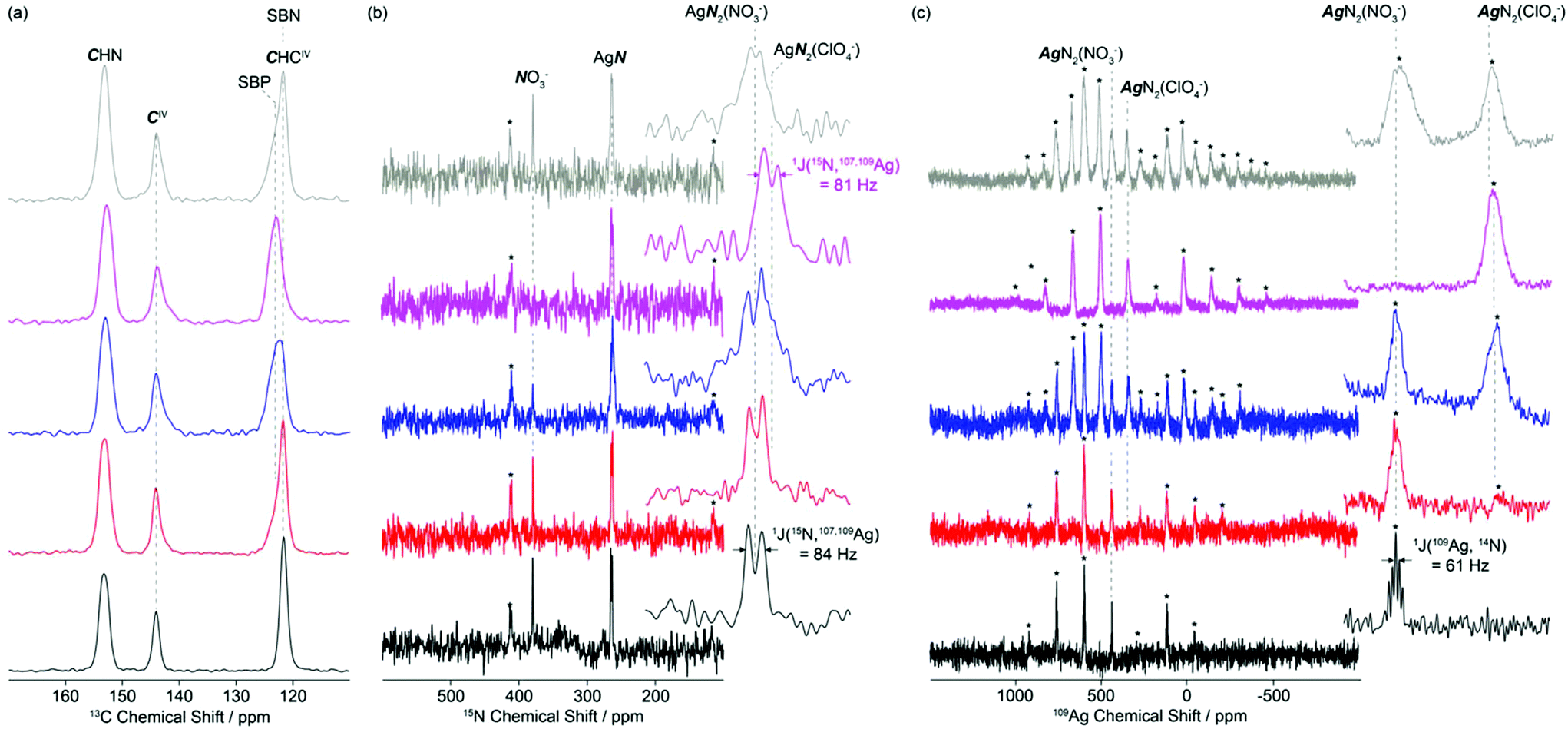 | ||
| Fig. 3 (a) 13C, (b) 15N and (c) 109Ag CP MAS NMR of the SBN/SBP anion exchange of SBN pre-exchange (black), after 10 min (red), 25 min (blue), complete exchange to SBP (purple) and regeneration back to SBN (grey). Spectral assignments are given in the figure. The 13C spectra were obtained at 20 T while the 15N and 109Ag were obtained at 9.4 T. In (b), the insets show magnified views of the 280–250 ppm isotropic region of the AgN resonances. In (c), the insets show magnified views of the first left spinning sidebands in the 650–450 ppm region. Spinning sidebands are marked with asterisks (*). | ||
After 25 minutes, the PXRD pattern is an approximately equal mixture of both the SBN and SBP crystal phases, indicating that the anions have been partially exchanged. The relative amounts of both phases were verified by measuring the residual concentration of perchlorate in the anion exchange solution at different time intervals with ion chromatography (Fig. S3†). The predominance of the SBP phase is observed after 40 minutes, where the low angle peaks corresponding to (001) and (100) of SBP are now higher intensity relative to the peaks of SBN. After 70 minutes, the anion exchange is complete and the PXRD pattern is pure phase SBP (see below for recovery to SBN). In addition, a shift in the (001) and (100) phases to a higher 2-theta indicates that the d-spacing decreased, likely due to a contraction of the structure upon completion of the anion exchange.
The 13C CP MAS NMR spectra of SBN and SBP (black and purple spectra, Fig. 3a) obtained at 20 T show three different isotropic resonances δiso at 153.3–152.7, 144.1–143.8 and 123.3–121.7 ppm for the individual carbons in the asymmetric unit and correspond to the CHN, CIV and CHCIV environments in the 4,4′-bipyridine ligand, respectively. As the NO3−–ClO4− anion exchange progressed by dissolution of SBN and subsequent capture of the ClO4− by the crystallization of SBP, a downfield shift of around 1.6 ppm is clearly observed in the signal of the CHCIV carbon (Fig. 3a, S4 and S5†).
Comparison of the CHCIV signal intensities at 121.7 and 123.3 ppm for SBN and SBP, respectively, enables a semi-quantitative analysis of the rates of exchanges to be monitored (note that while CP MAS NMR experiments are not quantitative, integration is performed on the same CHCIV carbon, therefore a SBN/SBP ratio could be approximately estimated). After 10 and 25 minutes, approximately 20 and 60% of SBN have been transformed into SBP, respectively. Partial degree of exchange at these time intervals agrees well with the PXRD data (red and blue patterns, Fig. 3).
This shift difference between the CHCIV carbon in SBN and SBP is probably due to the higher degree of carbon deshielding caused by the perchlorate anions. As stated above, the cationic layers of SBP have a shorter π-stacking distance relative to SBN due to the fully eclipsed arrangement of the Ag-bipy chains. This shorter distance causes the ClO4− anions to be more tightly packed between the aromatic rings and in turn a longer Ag–O distance. Consequently, the ClO4− oxygens are in closer proximity to CHCIV carbon, resulting in a higher degree of deshielding as reflected by the observed chemical shift.
The 15N CP MAS NMR spectrum of SBN (Fig. 3b, black) at 9.4 T reveals two isotropic resonances δiso at 379.3 and 263.9 ppm and are assigned to the NO3− anions and the nitrogen of the bipy ligand, respectively. A doublet of 84 Hz is observed for the former resonance and results from the indirect scalar 1J(15N, 107,109Ag) coupling arising from coordination of the nitrogen atoms of bipy to the silver atoms, as expected from the crystal structure (Fig. 1). Similar scalar coupling constants were obtained in a series of linear or almost linear two-coordinate [Ag(NH3)2]+ ions.24
The 109Ag CP MAS NMR25 spectrum of SBN (purple, Fig. 3c) shows a series of intense spinning sidebands spanning over approximately 1000 ppm, in agreement with a large chemical shift anisotropy of δaniso = −774 ppm. Note that the relatively low signal to noise ratio of this 109Ag spectrum arises from the very low magnetic moments of this nucleus. A single isotropic resonance appears at δiso = 438.5 ppm and is consistent with a single silver atom environment in SBN. This resonance is a quintet with a spacing of 61 Hz, corresponding to the indirect scalar coupling between 109Ag and 14N (99.63% nat. ab., I = 1). The approximate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3:2:1 relative intensity of the quintet is expected for coupling to two equivalent I = 1 nuclei and the crystal structure of SBN presenting a silver atom nitrogen-bonded by two bipy ligands. Note that the 1J(15N, 107,109Ag):1J(109Ag, 14N) ratio is 1.38, close to the expected 15N:14N magnetic moment ratio of 1.40. Additionally, the spinning sideband manifold yields an axially symmetric chemical shift tensor (i.e. η = 0). This observation agrees well with the almost linear environment23 of the N–Ag(I)–N moieties in the bipy–Ag–bipy polymers (173.73°, Table 1).
:2:3:2:1 relative intensity of the quintet is expected for coupling to two equivalent I = 1 nuclei and the crystal structure of SBN presenting a silver atom nitrogen-bonded by two bipy ligands. Note that the 1J(15N, 107,109Ag):1J(109Ag, 14N) ratio is 1.38, close to the expected 15N:14N magnetic moment ratio of 1.40. Additionally, the spinning sideband manifold yields an axially symmetric chemical shift tensor (i.e. η = 0). This observation agrees well with the almost linear environment23 of the N–Ag(I)–N moieties in the bipy–Ag–bipy polymers (173.73°, Table 1).
As SBN is converted into SBP by addition of ClO4−, the relative ratio of the NO3− anions and the bipy ligand signals at 379.3 and 263.9 ppm in the 15N CP MAS NMR spectra decreases, until a single nitrogen environment is detected upon total exchange to SBP. The corresponding 15N spectrum (purple, Fig. 3b) shows a doublet with 1J(15N, 107,109Ag) = 81 Hz centred at δiso = 261.4 ppm. This signal is due to the nitrogen atoms of bipy that are bonded to Ag. No resonance could be detected at 379.3 ppm in SBP, consistent with the complete NO3−–ClO4− anion exchange as seen by PXRD. The very small difference in chemical shift between the nitrogen atoms of bipy in SBN (263.9 ppm) and SBP (261.4 ppm) likely arises from the greater deshielding of the bipy nitrogen in SBN by the NO3− that is in closer proximity to the silver atom. The 15N CP MAS NMR spectra for SBN and SBP are therefore consistent with the 13C CP MAS NMR spectra in the arrangement of the Ag-bipy cationic layers upon exchange of NO3− for ClO4− perchlorate anions.
Upon SBN/SBP anion exchange, the 109Ag CP MAS NMR spectra (Fig. 3c) reveal the appearance of an additional isotropic resonance at approximately δiso = 341 ppm. Its intensity increases with ClO4− content and is attributed to the [Ag-bipy]+ moiety in SBP. Upon total exchange, the 109Ag signal of SBN completely disappears, in agreement with the PXRD, 13C and 15N data discussed above. Note that no 1J(109Ag, 14N) scalar coupling could be clearly detected in the 109Ag CP MAS NMR spectrum of SBP, probably resulting from slightly broader NMR lines and disorder. An axially symmetric 109Ag chemical shift tensor with δaniso = −776 ppm and η = 0 is also obtained, the later value in agreement with the linear N–Ag(I)–N angle in SBP (Table 1).
The change in morphology of the SBN crystals during anion exchange with ClO4− was investigated by performing batch tests experiments, stopping the reaction at various time intervals to analyze the resulting crystals by SEM (Fig. 4). Initially, the synthesis of SBN yields smooth block-shaped crystals with an average width of 4 to 10 μm (black, top left, Fig. 4). Upon 10 min of exposure of the SBN crystals to the ClO4− aqueous solution, the smooth block shaped crystals began to display indentations at the surface (red, Fig. 4). These indentations show that the new SBP phase is beginning to take form on the SBN crystals, similar to that reported by Schröder and coworkers for [Ag(4,4-bipy)+][BF4−] to [Ag(4,4-bipy)+][NO3−].18 After 25 min, small needles corresponding to the SBP crystal phase are observed in addition to SBN blocks with indentations (blue, Fig. 4). After 40 min, the crystals have lost their block-shaped morphology and consist primarily of bundles of needles that appear to be nucleated from the SBN block crystals (green, Fig. 4). The anion exchange is complete after 70 minutes and only long SBP needles could be identified (purple, Fig. 4), with average length 12 to 15 μm and width 2 to 4 μm.
 | ||
| Fig. 4 SEM images of SBN pre-exchange (black, top left) and the solid after 10 min (red), 25 min (blue), 40 min (green), complete exchange to SBP (purple), and the first regeneration to SBN (grey). | ||
The gradual change in morphology of the SBN block crystals to SBP needles upon exposure to ClO4− solution confirms that the structural transformation between the two coordination polymers occurs by a solvent-mediated process, consistent with the PXRD patterns and MAS NMR spectra described above. The self propagating growth of a new crystal phase from a parent phase has been proposed to facilitate the exchange of anions in the inside core of [Ag(4,4-bipy)+][BF4−] crystals undergoing the structural transformation to [Ag(4,4-bipy)+][NO3].11
The reversibility of the anion exchange was demonstrated in our previous work and seven cycles of perchlorate uptake were monitored by PXRD.15 The metastability of the [Ag-bipy]+ solids allow for the reversible solvent mediated transformation that can be controlled by the concentration of the incoming anions and temperature. The lower solubility of SBP as well as its higher stability due to the greater degree of π-stacking account for the rapid SBN to SBP transition. In contrast, the re-intercalation of nitrate is a less favoured process due to the larger hydration energy of nitrate. Additionally, SBN has a lower stability due to the partial π-stacking.
The regeneration of the SBN was further studied by recording the PXRD, 13C, 15N, 109Ag CP MAS NMR data and SEM images after treatment of the SBP material with excess nitrate at 70 °C under stirring (grey spectra, Fig. 3 and 4). The heat promotes the dissolution of the SBP polymers, while the excess nitrate shifts the dynamic equilibrium of the exchange to return to block shaped crystals with morphology identical to the as-synthesized SBN material (cf. black and grey SEMs, Fig. 4). The corresponding PXRD, 13C and 109Ag NMR data showed that although SBN has been mostly regenerated, approximately 10% of SBP is still present (as evidenced by integration of the NMR spectra and PXRD peaks).
4. Conclusion
This work shows by multiple complementary solid state methods that ClO4− uptake by Silver 4,4′-Bipyridine Nitrate (SBN) [Ag-bipy+][NO3−] occurs via a solvent-mediated process to yield Silver 4,4′-Bipyridine Perchlorate (SBP). The structural features of the silver coordination polymers in terms of π-stacking and electrostatics concur with the dynamics of the anion exchange process. All three characterization methods show that the two crystalline phases are present throughout the anion exchange process. This study provides evidence of the structural flexibility displayed by the interaction of cationic [Ag-bipy+] MOFs with different adsorbates upon anion exchange. Such understanding of structural dynamics is crucial for the rational design of these and related MOFs toward the selective capture of environmental pollutant oxo-anions such as perchlorate, chromate and arsenate. Reusability over many cycles is also crucial for the cost-effective deployment of these materials. The formation of new crystals on each exchange cycle circumvents the degradation issues of current ion exchange solids such as resins and LDHs that force them to be single-use.Acknowledgements
This work was supported by the National Science Foundation under Grant No. 1603754 from the CBET Environmental Engineering Program. I. R. C. was supported by a fellowship from the IMSD program funded by the National Institute of Health under Grant No. 2R25GM058903-17 and thanks Nick J. Brownbill (University of Liverpool, Department of Chemistry) for technical assistance with the NMR spectrometer. The PXRD data in this work were recorded on an instrument supported by the NSF Major Research Instrumentation (MRI) Program under Grant No. 1126845. K. K. I. thanks the EPSRC for a studentship. The UK 850 MHz solid-state NMR Facility used in this research was funded by EPSRC and BBSRC (contract reference PR140003), as well as the University of Warwick including via part funding through Birmingham Science City Advanced Materials Projects 1 and 2 supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF). Collaborative assistance from the 850 MHz Facility Manager (Dr Dinu Iuga, University of Warwick) is acknowledged. We also acknowledge Dr Tom Yuzvinsky and the W. M. Keck Center for Nanoscale Optofluidics at UC Santa Cruz for the use and assistance in image acquisition on the FEI Quanta 3D Dualbeam microscope. Original data are available at the University of Liverpool's DataCat repository at http://datacat.liverpool.ac.uk/id/eprint/283.References
- E. T. Urbansky and M. R. Schock, J. Environ. Manage., 1999, 56, 79–95 CrossRef.
- Y. Yang, N. Gao, W. Chu, Y. Zhang and Y. Ma, J. Hazard. Mater., 2012, 209–210, 318–325 CrossRef CAS PubMed.
- A. Srinivasan and T. Viraraghavan, Int. J. Environ. Res. Public Health, 2009, 6, 1418–1442 CrossRef CAS PubMed.
- P. K. Dasgupta, P. K. Martinelango, W. A. Jackson, T. A. Anderson, K. Tian, R. W. Tock and S. Rajagopalan, Environ. Sci. Technol., 2005, 39, 1569–1575 CrossRef CAS PubMed.
- W. Song, X. Xu, X. Tan, Y. Wang, J. Ling, B. Gao and Q. Yue, Carbohydr. Polym., 2015, 115, 432–438 CrossRef CAS PubMed.
- K.-H. Goh, T.-T. Lim and Z. Dong, Water Res., 2008, 42, 1343–1368 CrossRef CAS PubMed.
- J. Y. Kim, S. Komarneni, R. Parette, F. Cannon and H. Katsuki, Appl. Clay Sci., 2011, 51, 158–164 CrossRef CAS.
- S. R. J. Oliver, Chem. Soc. Rev., 2009, 38, 1868 RSC.
- R. Custelcean, T. J. Haverlock and B. A. Moyer, Inorg. Chem., 2006, 45, 6446–6452 CrossRef CAS PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- C. Thompson, N. R. Champness, A. N. Khlobystov, C. J. Roberts, M. Schroder, S. J. B. Tendler and M. J. Wilkinson, J. Microsc., 2004, 214, 261–271 CrossRef CAS PubMed.
- D. D. Laws, H.-M. L. Bitter and A. Jerschow, Angew. Chem., Int. Ed., 2002, 41, 3096–3129 CrossRef CAS PubMed.
- H. Hoffmann, M. Debowski, P. Müller, S. Paasch, I. Senkovska, S. Kaskel and E. Brunner, Materials, 2012, 5, 2537–2572 CrossRef CAS.
- Y. Jiang, J. Huang, B. Kasumaj, G. Jeschke, M. Hunger, T. Mallat and A. Baiker, J. Am. Chem. Soc., 2009, 131, 2058–2059 CrossRef CAS PubMed.
- I. R. Colinas, R. C. Silva and S. R. J. Oliver, Environ. Sci. Technol., 2016, 50, 1949–1954 CrossRef CAS PubMed.
- O. M. Yaghi and L. Hailian, J. Am. Chem. Soc., 1996, 118, 295–296 CrossRef CAS.
- L.-S. Wang, J.-F. Zhang and S.-P. Yang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, m1484–m1486 CAS.
- X. Cui, A. N. Khlobystov, X. Chen, D. H. Marsh, A. J. Blake, W. Lewis, N. R. Champness, C. J. Roberts and M. Schröder, Chem. – Eur. J., 2009, 15, 8861–8873 CrossRef CAS PubMed.
- B. M. Fung, A. K. Khitrin and K. Ermolaev, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS PubMed.
- C. R. Morcombe and K. W. Zilm, J. Magn. Reson., 2003, 162, 479–486 CrossRef CAS PubMed.
- P. Bertani, J. Raya and B. Bechinger, Solid State Nucl. Magn. Reson., 2014, 61–62, 15–18 CrossRef CAS PubMed.
- J. K. Plischke, A. J. Benesi and M. A. Vannice, J. Phys. Chem., 1992, 96, 3799–3806 CrossRef CAS.
- M. Mehring, Principles of High Resolution NMR in Solids, Springer Verlag, Berlin, 2nd edn, 1983 Search PubMed.
- G. A. Bowmaker, R. K. Harris, B. Assadollahzadeh, D. C. Apperley, P. Hodgkinson and P. Amornsakchai, Magn. Reson. Chem., 2004, 42, 819–826 CrossRef CAS PubMed.
- L. H. Merwin and A. Sebald, J. Magn. Reson., 1992, 97, 628–631 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and NMR acquisition parameters. See DOI: 10.1039/c7dt00475c |
| This journal is © The Royal Society of Chemistry 2017 |