
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparative solution equilibrium studies of antitumor ruthenium(η6-p-cymene) and rhodium(η5-C5Me5) complexes of 8-hydroxyquinolines†
Orsolya
Dömötör
ab,
Veronika F. S.
Pape
c,
Nóra V.
May
d,
Gergely
Szakács
ce and
Éva A.
Enyedy
 *a
*a
aDepartment of Inorganic and Analytical Chemistry, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary. E-mail: enyedy@chem.u-szeged.hu
bMTA-SZTE Bioinorganic Chemistry Research Group, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary
cInstitute of Enzymology, Research Centre for Natural Sciences, Hungarian Academy of Sciences, Magyar Tudósok körútja 2, H-1117 Budapest, Hungary
dResearch Centre for Natural Sciences Hungarian Academy of Sciences, Magyar tudósok körútja 2, H-1117 Budapest, Hungary
eInstitute of Cancer Research, Medical University of Vienna, Borschkegasse 8a, A-1090 Vienna, Austria
First published on 3rd March 2017
Abstract
Complex formation processes of [Ru(η6-p-cymene)(H2O)3]+ and [Rh(η5-C5Me5)(H2O)3]+ organometallic cations with 8-hydroxyquinoline (HQ) ligands were studied in aqueous solution by the combined use of 1H NMR spectroscopy, UV-visible spectrophotometry and pH-potentiometry. Solution stability, chloride ion affinity and lipophilicity of the complexes were characterized together with the in vitro cytotoxicity against a pair of cancer cell lines, responsive and resistant to classic chemotherapy. The solid phase structure of the [Rh(η5-C5Me5)(8-quinolinolato)(Cl)] complex was characterized by single-crystal X-ray diffraction analysis. In addition to the unsubstituted HQ its 7-(1-piperidinylmethyl) (PHQ) and 5-sulfonate (HQS) derivatives were involved. PHQ has a significant preference for targeting multidrug resistant cancer cell lines, while HQS served as a water soluble model compound. The equilibrium studies revealed the formation of mono[M(L)(H2O)] complexes with prominently high solution stability, which predominate at physiological pH even in the micromolar concentration range, and the formation of mixed hydroxido [M(L)(OH)] complexes was characterized by relatively high pKa values (8.5–10.3). In comparison to the Rh(η5-C5Me5) species the complexation process with Ru(η6-p-cymene) is much slower, and both the pKa values and the H2O/Cl− co-ligand exchange constants are lower by 1–1.5 orders of magnitude. The stability order obtained for these organometallic complexes is as follows: HQS > HQ > PHQ. The cytotoxicity of the ligands and their Ru(η6-p-cymene) and Rh(η5-C5Me5) complexes was investigated against MES-SA (human uterine sarcoma) cell line and its multidrug resistant counterpart (MES-SA/Dx5). HQ and its complexes show similar cytotoxicity in both cell lines. In contrast, PHQ and its Rh(η5-C5Me5) complex are more potent against MES-SA/Dx5 cells, while this selectivity could not be observed for the Ru(η6-p-cymene) complex.
Introduction
Resistance and the serious side effects associated with the use of anticancer platinum drugs used in chemotherapy are still driving for the design and development of novel metal-based compounds that combine good efficacy, selectivity and low systemic toxicity due to their different modes of action and pharmacokinetics. Ruthenium complexes have been the subject of extensive drug discovery efforts, yielding e.g. the sodium trans-[tetrachloridobis(1H-indazole)ruthenate(III)] (NKP-1339, IT-139) and imidazolium trans-[tetrachlorido(DMSO)(imidazole)ruthenate(III)] (NAMI-A) as the most promising compounds reaching clinical trials.1–4 These ruthenium(III) complexes are considered as prodrugs activated by reduction. Organoruthenium(II) compounds have gained increasing attention recently and numerous [Ru(η6-arene)(X)(Y)(Z)] complexes were found to be active as antitumor compounds.5 One of the most well-known ruthenium(II) arene complexes [Ru(η6-p-cymene)Cl2(PTA)] (PTA = 1,3,5-triaza-7-phosphatricyclo-[3.3.1.1]decane) shows anti-metastatic properties and is ready for translation to clinical evaluation.6 In most of the half-sandwich organoruthenium(II) compounds a bidentate ligand with an (O,O), (O,S), (O,N), (N,N) or (N,S) binding mode is coordinated and a chloride ion acts as the leaving group.7–11 The replacement of the chlorido ligand by a water molecule facilitates the reaction with biological macromolecules such as proteins or DNA,12 while the chelating ligand allows modifications of the chemical properties, ligand exchange rate, lipophilicity, 3D shape and ultimately influences the pharmacological effect. Rhodium(III) is isoelectric with ruthenium(II) and the coordination of e.g. the anionic pentamethylcyclopentadienyl (C5Me5–) ligand results in faster ligand exchange kinetics.13 Promising in vitro antitumor activity has been reported for Rh(η5-C5Me5) complexes of (N,N) donating polypyridyl ligands by Sheldrick and his co-workers.14,15 Recently we reported Rh(η5-C5Me5) complexes formed with (O,O) donor hydroxypyr(id)ones (maltol, deferiprone) showing moderate cytotoxicity on various cancer cell lines,16,17 while the complexes of (N,O) donor picolinates exhibited only poor anticancer activity.17,18 Both Rh(η5-C5Me5) and Ru(η6-p-cymene) half-sandwich complexes of the (N,O) donor 8-hydroxyquinoline (HQ, Chart 1) are reported to possess antitumor activity with IC50 values in the low micromolar concentration range.19–21 These complexes were characterized by standard analytical methods19–24 and in the case of the [Ru(η6-p-cymene)(L)(Cl)] and [Ru(η6-p-cymene)(L)(H2O)]+ (L: 8-quinolinolato) in the solid state by single-crystal X-ray diffraction analysis.23,24 Notably, in a set of Ru(η6-p-cymene) half-sandwich complexes with 8-HQ derivatives it was found that the introduction of halogens in positions R5 and R7 of the scaffold increased both anticancer cytotoxicity and intracellular accumulation, suggesting that these two moieties might be relevant for the fine tuning of the biological activity of these complexes.21 Excellent cytotoxic effect in tumor cell lines was found for water soluble mixed-ligand [Ru(η6-p-cymene)(8-quinolinolato)(H-azole heterocycle)]+ complexes.20 In addition, Ru(η6-p-cymene) complexes of various HQ derivatives have been found to catalyze the hydrogenation of CO2 to formate in aqueous solution and the catalytic activity showed strong pH-dependence.24 Although these 8-quinolinolato complexes have been extensively studied, their solution speciation and stability constants are not available in the literature. For the better understanding of the pharmacokinetic profile and mechanisms of action of these metal complexes in addition to their pH-dependent catalytic activity, the knowledge of the speciation and the most plausible chemical forms in aqueous solution, especially at physiological pH, is a mandatory prerequisite. Therefore, one of the aims of the present study was to characterize the solution speciation of Rh(η5-C5Me5) and Ru(η6-p-cymene) complexes of HQ in aqueous solution involving studies on their chloride ion affinity and lipophilicity.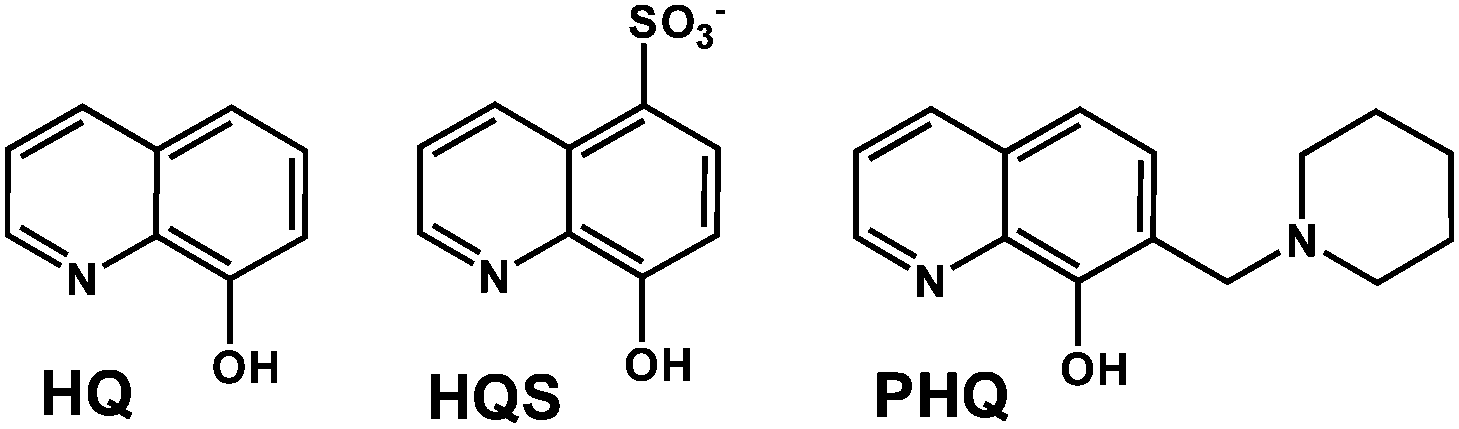 | ||
| Chart 1 Chemical structures of the ligands: 8-hydroxyquinoline (HQ), 8-hydroxyquinoline-5-sulfonate (HQS) and 7-(1-piperidinylmethyl)-8-hydroxyquinoline (PHQ). | ||
HQ is a privileged structure, which appears frequently in drugs, natural compounds, or bioactive molecules and is used as a ligand in the orally active tris(8-quinolinolato)gallium(III) complex (KP46), currently undergoing clinical trials.25,26 The HQ derived Mannich base 7-(1-piperidinylmethyl)-HQ (NSC57969, PHQ, Chart 1) has recently been identified to overcome multidrug resistance (MDR) in cancer, a phenomenon conferring resistance to a wide range of structurally and mechanistically unrelated anticancer agents.27–29 Several related derivatives have been identified to show paradoxically enhanced cytotoxicity against MDR cell lines overexpressing P-glycoprotein (P-gp), a transport protein mediating resistance by effluxing chemotherapeutic agents from cancer cells, thereby keeping their intracellular concentrations below a cell-killing threshold.27–30
In this work, our additional aim was to investigate the complex formation processes of PHQ in comparison to the HQ scaffold and the R5 substituted HQS with [Ru(η6-p-cymene)(H2O)3]+ and [Rh(η5-C5Me5)(H2O)3]+ cations and to reveal their cytotoxic effectiveness.
Results and discussion
Proton dissociation processes of the ligands (HQ, HQS, PHQ) and hydrolysis of the organometallic cations
HQ and 8-hydroxyquinoline-5-sulfonate (HQS) (Chart 1) are well-known compounds and their proton dissociation processes have already been described in the literature.31–34 Due to the insufficient water solubility of HQ, HQS was involved in the studies and considered as a model compound possessing the same coordination mode as HQ. Notably, proton dissociation constants of HQ and HQS (Table 1) were determined herein by UV-visible (UV-vis) spectrophotometry and 1H NMR spectroscopy (pH-dependent NMR spectra for HQS are shown in Fig. S1†) at 1 mM concentration. Data obtained by the different methods under the chloride-free conditions are in good agreement with each other and with the previously published data obtained by pH-potentiometry.31 pK1 is attributed to the deprotonation of the quinolinium (NH+) group and pK2 belongs to the hydroxyl moiety. The sulfonic acid group of HQS is deprotonated in the whole pH range studied due to its strong acidic character. Notably, the deconvolution of the UV-vis spectra recorded at various pH values using ten times more diluted conditions (cL ∼ 0.1 mM) gave lower pK1, and somewhat higher pK2 constants for both ligands (see the dissimilar positions of the inflection points in Fig. S2† for the pH-dependent absorbance values obtained for HQS at two kinds of concentrations).| Method | c L | HQ | HQS | PHQ | |||
|---|---|---|---|---|---|---|---|
| pK1 | pK2 | pK1 | pK2 | pK1 | pK2 | ||
| a Uncertainties (SD) of the last digits are shown in parentheses. b Data are taken from ref. 31. | |||||||
| pH-Metry | 1 mM | 4.99b | 9.51b | 3.90b | 8.37b | — | — |
| 1H NMR | 1 mM | — | — | 3.92(1) | 8.38(1) | — | — |
| UV-Vis | 1 mM | 5.03(1) | 9.66(1) | 3.83(1) | 8.39(1) | — | — |
| UV-Vis | ∼0.1 mM | 4.78(1) | 9.74(1) | 3.63(1) | 8.48(1) | 2.80(1) | 6.93(1) |
The concentration dependent pKa values of these compounds might be the result of a slightly altered ratio of the α and β (or cis/trans) HL forms35 under the two kinds of conditions.
The deprotonation of the piperidine derivative PHQ has also two pKa values attributed to the same moieties as in the case of HQ, but PHQ has 3 protonation sites, and the methylpiperidinium nitrogen is most probably protonated in the pH range 2–11.5. The corresponding pKa values of PHQ and HQS are significantly lower than those of HQ due to the electron withdrawing effect of the protonated piperidinium moiety and the sulfonate substituent, respectively.
The hydrolytic behavior of the aquated organometallic cations [Ru(η6-p-cymene)(H2O)3]2+ and [Rh(η5-C5Me5)(H2O)3]2+ has been studied previously.16,36–38 The structure of the major hydrolysis products of [Rh(η5-C5Me5)(H2O)3]2+, the dimeric [(Rh(η5-C5Me5))2(μ-OH)3]+ species was characterized by single-crystal X-ray analysis39 and the similar structure is assumed for the [(Ru(η6-p-cymene))2(μ-OH)3]+ complex based on 1H NMR studies.40 Overall stability constants were reported for the μ-hydroxido-bridged dinuclear ruthenium(II) species ([(Ru(η6-p-cymene))2(μ-OH)3]+) by Buglyó et al.,36 and for the rhodium(III) species [(Rh(η5-C5Me5))2(μ-OH)3]+, [(Rh(η5-C5Me5))2(μ-OH)2]2+) by some of us,16 and were used in this work for the calculations.
Complex formation equilibria of [Rh(η5-C5Me5)(H2O)3]2+ with HQ, HQS and PHQ
The complex formation equilibrium processes in the case of the [Rh(η5-C5Me5)(H2O)3]2+–HQS system were found to be fast and the species involved in the equilibria have good water solubility. These features allowed us the combined use of pH-potentiometry, 1H NMR spectroscopy and UV-vis spectrophotometry in a chloride-free medium. Notably, HQS was used as a model compound for HQ and PHQ, the biologically more interesting compounds, as its good solubility in water allowed us the simultaneous use of the various techniques. The complexation between [Rh(η5-C5Me5)(H2O)3]2+ and HQS follows a fairly simple scheme (Chart S1†), since a mono-ligand [Rh(η5-C5Me5)(L)(H2O)] (=![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) [ML]) complex is formed with this bidentate ligand, and a mixed hydroxido [ML(OH)]− species appears upon the deprotonation of the coordinated water molecule in the basic pH range, similarly to the behavior of numerous half-sandwich organorhodium complexes studied previously.16–18,41 The pH-potentiometric titration data reveal almost complete complex formation already at the starting pH (∼2), therefore the stability constant of this [ML] type complex (Table 2) was determined by deconvolution of UV-vis spectra measured between pH 0.7 and 3.0 (Fig. 1a) (M always denotes the metal ion moiety: [Rh(η5-C5Me5)(H2O)3]2+ or [Ru(η6-p-cymene)(H2O)3]2+).
[ML]) complex is formed with this bidentate ligand, and a mixed hydroxido [ML(OH)]− species appears upon the deprotonation of the coordinated water molecule in the basic pH range, similarly to the behavior of numerous half-sandwich organorhodium complexes studied previously.16–18,41 The pH-potentiometric titration data reveal almost complete complex formation already at the starting pH (∼2), therefore the stability constant of this [ML] type complex (Table 2) was determined by deconvolution of UV-vis spectra measured between pH 0.7 and 3.0 (Fig. 1a) (M always denotes the metal ion moiety: [Rh(η5-C5Me5)(H2O)3]2+ or [Ru(η6-p-cymene)(H2O)3]2+).
 | ||
| Fig. 1 UV–Vis spectra recorded for the [Rh(η5-C5Me5)(H2O)3]2+–HQS (1:1) system at pH 0.7–1.9 (a) and pH 2.0–11.7 (b). Dashed spectrum shows the sum of those of HQS and [Rh(η5-C5Me5)(H2O)3]2+.{cL = cRh = 160 μM; T = 25 °C; I = 0.20 M (KNO3)}. | ||
K [ML]), pKa [ML] values of the Rh(η5-C5Me5) and Ru(η6-p-cymene) complexes formed with HQS, HQ and PHQ in chloride-free aqueous solutions determined by various methods; H2O/Cl− exchange constants (logK′) for the [Rh(η5-C5Me5)(L)(H2O)] and [Ru(η6-p-cymene)(L)(H2O)] complexes {T = 25 °C; I = 0.2 M (KNO3)}a
| Method | Constants | HQS | HQ | PHQ | |
|---|---|---|---|---|---|
|
a Uncertainties (SD) of the last digits are shown in parentheses. M denotes Rh(η5-C5Me5) and Ru(η6-p-cymene), respectively and aqua ligands and the charges of the complexes are not shown for clarity. Hydrolysis products of the organometallic cations: logβ [(Rh(η5-C5Me5))2H−2(H2O)2]2+ = −8.53, logβ [(Rh(η5-C5Me5))2H−3]+ = −14.26 and logβ [(Ru(η6-p-cymene))2H−3]+ = −9.36 at I = 0.20 M (KNO3) taken from ref. 16 and 37.
b Determined by UV-Vis spectrophotometry at pH 0.7–3.0.
c For the [Rh(η5-C5Me5)(L)(H2O)]+ + Cl− ⇌ [Rh(η5-C5Me5)(L)Cl] + H2O and [Ru(η6-p-cymene)(L)(H2O)]+ + Cl− ⇌ [Ru(η6-p-cymene)(L)Cl] + H2O respectively, equilibria determined at various total chloride ion concentrations by UV-Vis.
d Estimated from the 1H NMR peak integrals of the ligand protons in the bound and unbound forms at pH 0.7.
|
|||||
| Rh(η5-C5Me5) | UV-Vis | logK [ML] |
14.52(2)b | 15.02(3) | 12.38(6)b |
| UV-Vis | pKa [ML] | 10.10(1) | 10.27(5) | 10.08(2) | |
| 1H NMR | pKa [ML] | 10.12(1) | — | — | |
| pH-Metry | pKa [ML] | 9.90(7) | — | — | |
| UV-Vis | logK′ (H2O/Cl−)c |
1.54(1) | 1.81(1) | 1.61(2) | |
| Ru(η6-p-cymene) | UV-Vis | logK [ML] |
— | 16.53(2)b | 13.31(4)b |
| UV-Vis | pKa [ML] | 8.46(2) | 9.19(4) | 9.37(6) | |
| 1H NMR | logK [ML] |
≥16d | — | — | |
| 1H NMR | pKa [ML] | 8.52(3) | — | — | |
| UV-Vis | logK′ (H2O/Cl−)c |
0.64(4) | 0.89(2) | 1.19(2) | |
These spectra were recorded for individual samples, in which the KNO3 was partially or completely replaced by HNO3 keeping the ionic strength constant and the actual pH values were calculated based on the strong acid content. The recorded UV-vis spectra were the same at pH between 2.9 and ∼8, while significant changes of the charge-transfer band are seen at pH > 8.5, and λmax is shifted from 376 nm up to 384 nm. In addition, a well-isolated isosbestic point is observed at 432 nm showing a clean transformation of the [ML] complex to another species, most probably [ML(OH)]−. The appearance of the isosbestic point suggests that the metal complex does not decompose under these conditions, merely it is deprotonated.
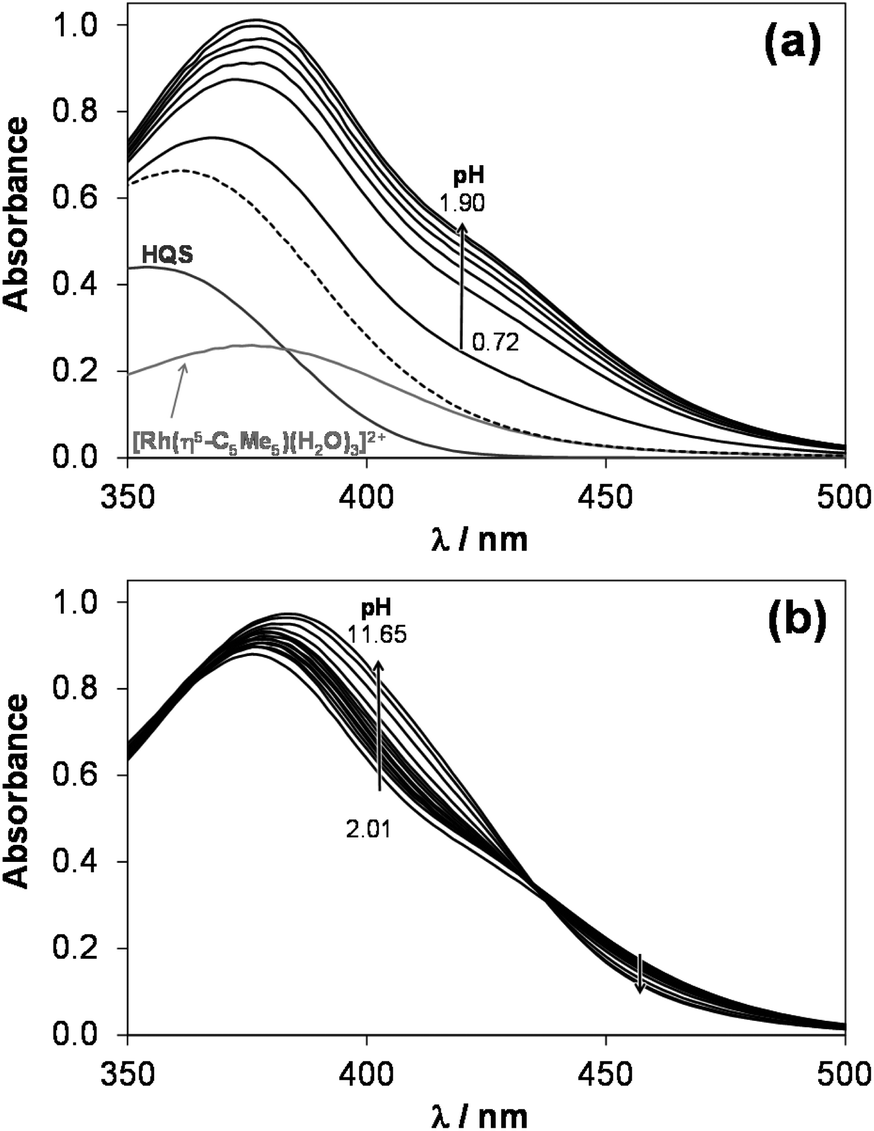
1H NMR spectra recorded for the [Rh(η5-C5Me5)(H2O)3]2+–HQS system at a 1:1 metal-to-ligand ratio at various pH values (Fig. 2) undoubtedly reveal that neither a free metal ion nor a ligand is present at pH > 2.9, which means that the complex does not suffer from decomposition due to its outstanding high stability at 1 mM concentration. An upfield shift of all peaks belonging to the [ML] complex is observed in the basic pH range due to the fast exchange process between the aquated and the mixed hydroxido species. Therefore, pKa of the aqua complex could be determined on the basis of the pH-dependent chemical shift (δ) values, which is in an excellent agreement with the data obtained spectrophotometrically, while a somewhat lower pKa [ML] could be calculated based on the pH-potentiometric titrations (Table 2).
 | ||
| Fig. 2
1H NMR spectra recorded for the [Rh(η5-C5Me5)(H2O)3]2+–HQS (1:1) system at the indicated pH values (peak assignation: CH(2) (■), CH(4) (●), C5Me5 (▼, ∇), empty symbol = unbound organometallic cation). {cHQS = 1 mM; T = 25 °C; I = 0.20 M (KNO3); 10% D2O}. | ||
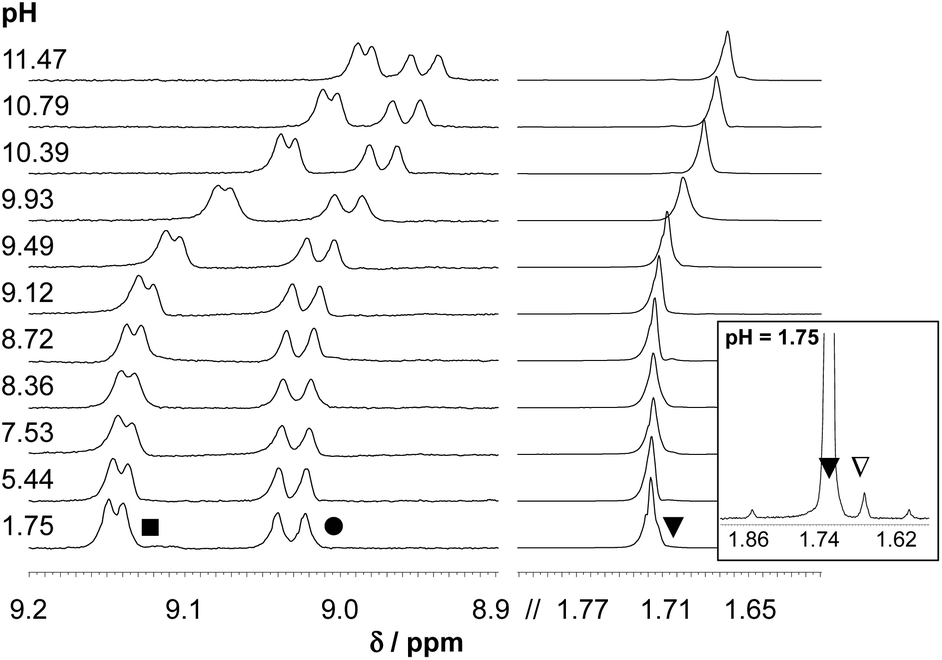
As HQ, PHQ and their metal complexes have much lower solubility in water compared to that of HQS, their complexation processes could only be studied by UV-vis spectrophotometry using much lower concentrations (cL ∼ 50–160 μM). The behavior of these ligands was found to be quite similar to that of the reference compound HQS, however the complex formation with HQ starts at higher pH due to the higher pKa values, thus stronger basicity of this ligand (Fig. 3). The equilibrium constants providing the best fits to the experimental data are listed in Table 2.
 | ||
| Fig. 3 UV–Vis spectra recorded for the [Rh(η5-C5Me5)(H2O)3]2+–HQ (1:1) system at pH 2.0– 11.5. Inset shows the absorbance values at 400 nm plotted against the pH {cL = cRh = 150 μM; T = 25 °C; I = 0.20 M (KNO3)}. | ||
Concentration distribution curves for the [Rh(η5-C5Me5)(H2O)3]2+–HQ/PHQ systems were computed on the basis of the stability constants (Fig. 4), which represent the predominant formation of the [Rh(η5-C5Me5)(L)(H2O)] complexes in the pH range from 4 to 8 in both cases at the biologically more relevant 50 μM concentration.
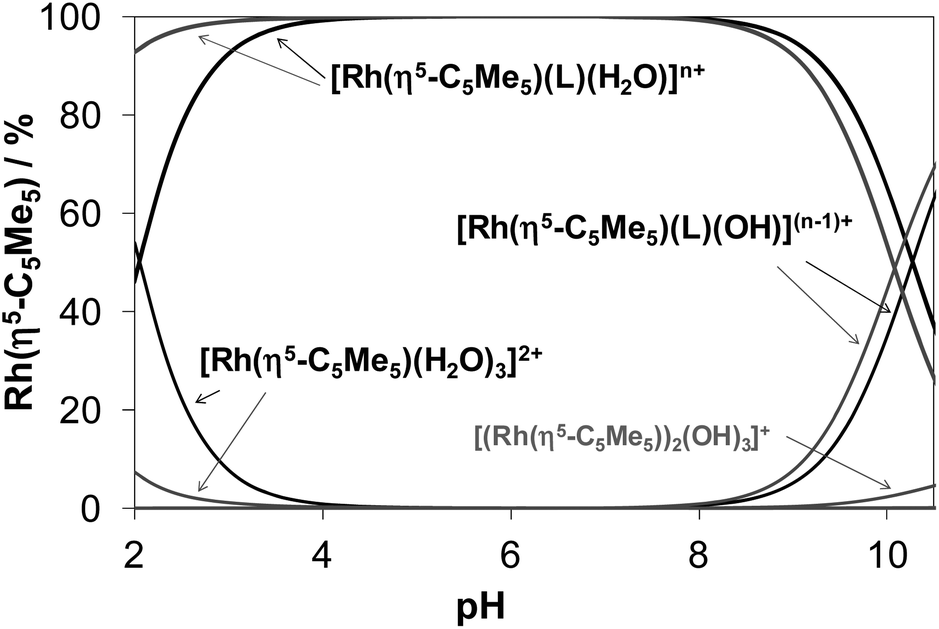 | ||
| Fig. 4 Concentration distribution curves for the [Rh(η5-C5Me5)(H2O)3]2+–HQ (black lines) and [Rh(η5-C5Me5)(H2O)3]2+–PHQ (grey lines) (1:1) systems calculated on the basis of the stability constants determined {cRh = cL = 50 μM; T = 25 °C; I = 0.20 M (KNO3); n = 1 (HQ), 2 (PHQ)}. | ||
Notably, the pKa [ML] values of these Rh(η5-C5Me5) complexes are rather high (∼10) and consequently the formation of mixed hydroxido species at pH 7.4 is negligible in the absence of chloride ions. The presence of the chloride ions generally results in even higher pKa [ML] values,16,17,41 therefore the deprotonation of the [Rh(η5-C5Me5)(L)(H2O)] complexes of 8-hydroxyquinolines is not likely under physiological conditions.
In the studied Rh(η5-C5Me5) complexes the bidentate 8-hydroxyquinoline ligands coordinate most probably via the (N,O−) donor set, which was confirmed by X-ray crystallography in the case of HQ (see the next section).
Crystallographic structure determination of complex [Rh(η5-C5Me5)(8-quinolinolato)(Cl)] (1)
Single crystals of [Rh(η5-C5Me5)(8-quinolinolato)(Cl)] (1) were obtained by the slow diffusion method from an ethanol/water mixture at neutral pH and at room temperature. The crystal structure has been determined by single crystal X-ray diffraction, crystal data and structure refinement parameters are seen in Table S1.† The ORTEP representation of the complex is depicted in Fig. 5a, while the packing arrangement is shown in Fig. S3.† Selected bond distances and angles are collected and given in comparison to the analogous iridium(III) complex22 in Table 3. The complex [Rh(η5-C5Me5)(8-quinolinolato)(Cl)] crystallized in the monoclinic crystal system, in the space group Cc with three water molecules in the asymmetric unit.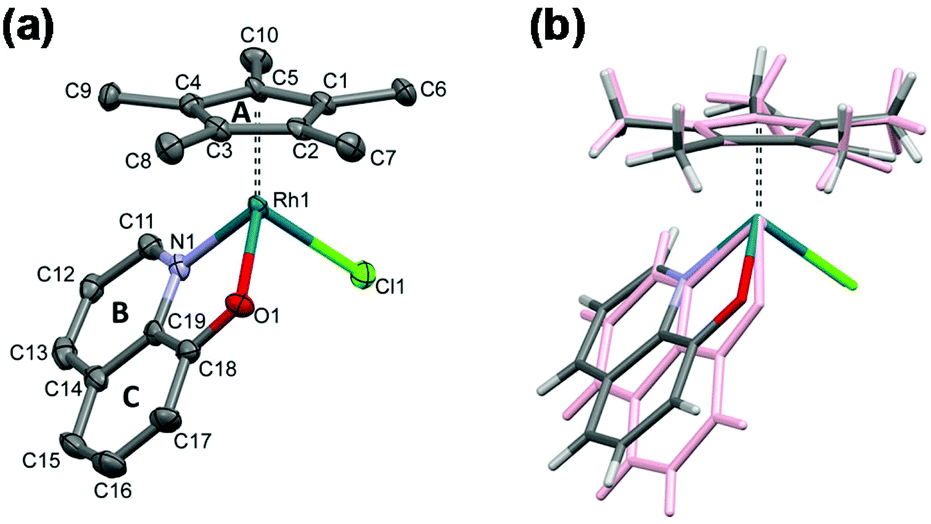 | ||
| Fig. 5 Molecular structure of the metal complex [Rh(η5-C5Me5)(8-quinolinolato)(Cl)] (1) with the indication of rings (A–C). Displacement parameters are drawn at 50% probability level and solvent molecules and hydrogens are omitted for clarity (a). Comparison of the molecular structure of complex 1 (colored by element) with [Ir(η5-C5Me5)(8-quinolinolato)(Cl)] (CSD ref. code VUMQAW) (rose)22 (b). | ||
| M = Rh (1) | M = Ir (VUMQAW)22 | |
|---|---|---|
| a Cg is the center of gravity calculated for rings A or BC. b Angles between planes calculated for the rings A and BC. | ||
| Bond length (Å) | ||
| M–Cl1 | 2.417(2) | 2.386(2) |
| M–O1 | 2.099(4) | 2.091(6) |
| M–N1 | 2.116(5) | 2.088(7) |
| M–C1 | 2.165(5) | 2.163(12) |
| M–C2 | 2.144(5) | 2.135(13) |
| M–C3 | 2.140(6) | 2.155(9) |
| M–C4 | 2.139(6) | 2.177(9) |
| M–C5 | 2.160(6) | 2.164(10) |
| M–Cg(A)a | 1.768(3) | 1.789(5) |
| M–Cg(BC)a | 2.139(6) | 2.177(9) |
| Bond angles (°) | ||
| O1–M–N1 | 78.4(2) | 77.8(3) |
| O1–M–Cl1 | 86.5(1) | 84.6(2) |
| N1–M–Cl1 | 90.7(1) | 85.2(2) |
| Cg(A)–M–O1a | 127.85(12) | 131.2(2) |
| Cg(A)–M–N1a | 130.73(15) | 133.2(2) |
| Cg(A)–M–Cl1a | 126.74(9) | 126.8(2) |
| Cg(A)–Cg(BC)b | 49.0(3) | 60.9(5) |
As it is expected, the rhodium(III) center exhibits a pseudooctahedral (“piano-stool”) geometry, and the C5Me5 moiety occupies facially three coordination sites, while the deprotonated ligand is bidentate via its (N,O) donors and the coordination sphere is completed with a chlorido ligand. The binding of the different donor groups resulted in a pseudochiral center for Rh, notwithstanding the complex crystallized in a racemic form. The measured Rh–N and Rh–Cl bond lengths were found to be very similar to the reported values for this complex based on DFT calculations (Rh–N: 2.115 Å, Rh–Cl: 2.416(2) Å).19 On the other hand the measured Rh–O bond length is somewhat longer, while the Rh-ring centroid distance is significantly shorter than the calculated values (Rh–O: 2.065 Å, Rh–ring centroid: 1.887 Å).19 The molecular structure of the complex was compared directly with that of [Ir(η5-C5Me5)(quinolin-8-olate)(Cl)] (Fig. 5b), which crystallized without solvate inclusion in the orthorhombic Pna21 space group (unit cell dimensions are a = 15.285(3), b = 8.335(2), c = 13.626(3) Å).22
The metal ion–C5Me5 ring centroid distance is very similar for the two complexes (1.768(3) Å in rhodium(III) and 1.789(5) Å in iridium(III) species, respectively); however the angles between the pentamethylcyclopentadienyl (A) and the 8-quinolinolato rings (B, C) differ significantly (Table 3 and Fig. 5b). The difference between the structures of these organorhodium and organoiridium complexes can be due to dissimilar secondary interactions with neighbouring molecules as different molecular arrangements and solvate inclusion realized in the two kinds of crystal structures. In the crystal lattice of the rhodium(III) complex the molecules are packed in a way that channels are formed (Fig. 6). The volume of the solvent accessible voids are 157 Å3 calculated by using program PLATON.42 Selected secondary interactions are shown in Fig. S4,† and the collection of the main intermolecular interactions is listed in Table S2.† It is worth mentioning that the isolated [Rh(η5-C5Me5)(L)(Cl)] complex was also characterized in this work by 1H NMR spectroscopy and electrospray ionization mass spectrometry, confirming the coordination of the ligand to the metal center.
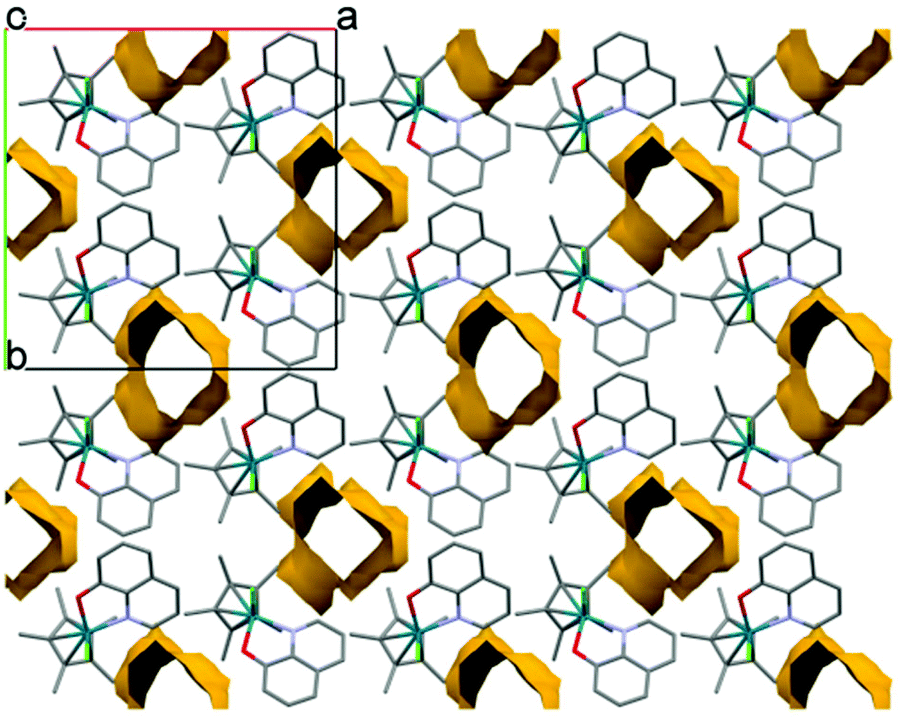 | ||
| Fig. 6 Packing arrangement showing water channels viewing along the c crystallographic axis in crystal [Rh(η5-C5Me5)(8-quinolinolato)(Cl)] (1). | ||
Complex formation equilibria of [Ru(η6-p-cymene)(H2O)3]2+ with HQ, HQS and PHQ
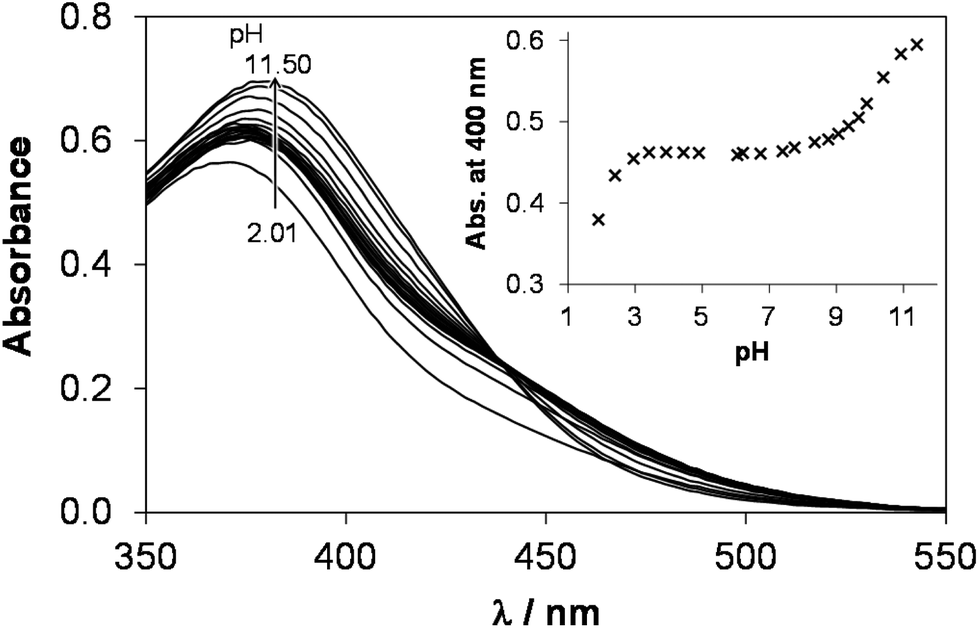
The complex formation of [Ru(η6-p-cymene)(H2O)3]2+ with the chosen 8-hydroxyquinoline ligands is a rather slow process compared to the case of the Rh(η5-C5Me5) species due to the markedly increased trans effect of the anionic pentamethylcyclopentadienyl ligand in comparison to the neutral arene ligand. For example the equilibrium could be reached after more than 120 min in the [Ru(η6-p-cymene)(H2O)3]2+–HQS system at pH 3 as the time-dependence of the UV-vis spectra indicates (Fig. S5†), which hindered the application of conventional pH-potentiometric titrations. To overcome this problem, individual samples were prepared by the addition of different amounts of strong base under Ar, and the actual pH and the UV-vis and/or 1H NMR spectra were measured after 24 h. Besides the altered complexation kinetics of the organorhodium and organoruthenium compounds with the 8-hydroxyquinolines, the other most conspicuous difference is that the organoruthenium cation starts to form complexes at much lower pH values. As a consequence logK [ML] constants were determined from the UV-vis spectral changes in the pH range from 0.7 to 3.0 for the HQ and PHQ complexes (Table 2). Although, the spectra recorded at pH 0.7 and 3 for the [Ru(η6-p-cymene)(H2O)3]2+–HQS were almost identical due to the negligible decomposition of the complex under the strongly acidic conditions. Therefore the stability constant could not be obtained based on these UV-vis spectra. Only a lower limit for the logK [ML] stability constant (Table 2) could be estimated from the 1H NMR spectrum recorded at pH 0.7 at a 1:1 metal-to-ligand ratio based on the integrated peak areas of the isopropyl methyl protons of the p-cymene ring belonging to the bound and unbound fractions of the organometallic fragment.
In the [Ru(η6-p-cymene)(L)(H2O)] complexes in solution the ligand coordinates most probably in the same bidentate manner via the (N,O−) donors as in the case of the analogous rhodium(III) species (vide supra) and as the crystal structures reported for both [Ru(η6-p-cymene)(8-quinolinolato)(Cl)]23 and [Ru(η6-p-cymene)(8-quinolinolato)(H2O)]+ (ref. 24) complexes also show. Deprotonation of the coordinated water molecule in the [Ru(η6-p-cymene)(L)(H2O)] species was characterized by the pKa [ML] values determined by the deconvolution of the 1H NMR (only in the case of HQS) and UV-vis spectra (Table 2). pH-Dependent 1H NMR spectra in a 10% DMSO/D2O mixture were also reported for the [Ru(η6-p-cymene)(L)Cl] complex of HQ, however no time-dependent measurements were performed and no pKa [ML] was provided.21 It is noteworthy that the logK [ML] values are higher, while pKa [ML] constants are lower by ca. 1–1.5 orders of magnitude obtained for the Ru(η6-p-cymene) complexes compared to the those of the Rh(η5-C5Me5) counterparts. Based on these values it can be predicted that the deprotonation of the Ru(η6-p-cymene) complex formed with HQS, where pKa [ML] is the lowest among the studied complexes, takes place still to a low extent at physiological pH. (Formation of ca. 7% [ML(OH)] is estimated in the chloride-free medium at 50 μM concentration of the monocomplex in this particular case.)
In the presence of ligand excess novel bands appeared unexpectedly in the UV-vis spectra recorded for the [Ru(η6-p-cymene)(H2O)3]2+–HQS (1:2) system at pH 7.4 (Fig. 7). As the complex formation is slow, it was expected that the final spectrum is the sum of those of the [Ru(η6-p-cymene)(L)(H2O)] monocomplex and one equivalent unbound ligand (see the red dashed lines in Fig. 7). In addition, the development of these new bands depends on the conditions, namely different spectral changes were observed under aerobic conditions or under an Ar atmosphere. The solution turned green with time in the presence of O2 (λmax ∼ 406 nm), on the contrary when Ar was bubbled though the sample it became reddish (λmax ∼ 530 and 406 nm). It should be noted that in the case of the Ar atmosphere the presence of minor O2 was possible. The absorbance values at both wavelength maxima become much higher with time than it is expected (see the inset of Fig. 7a). The band at 406 nm most probably appears as a consequence of O2, as the absorbance increased significantly when O2 gas was purged through the samples kept under Ar previously, while the band at 530 nm was decreased (Fig. S6†).
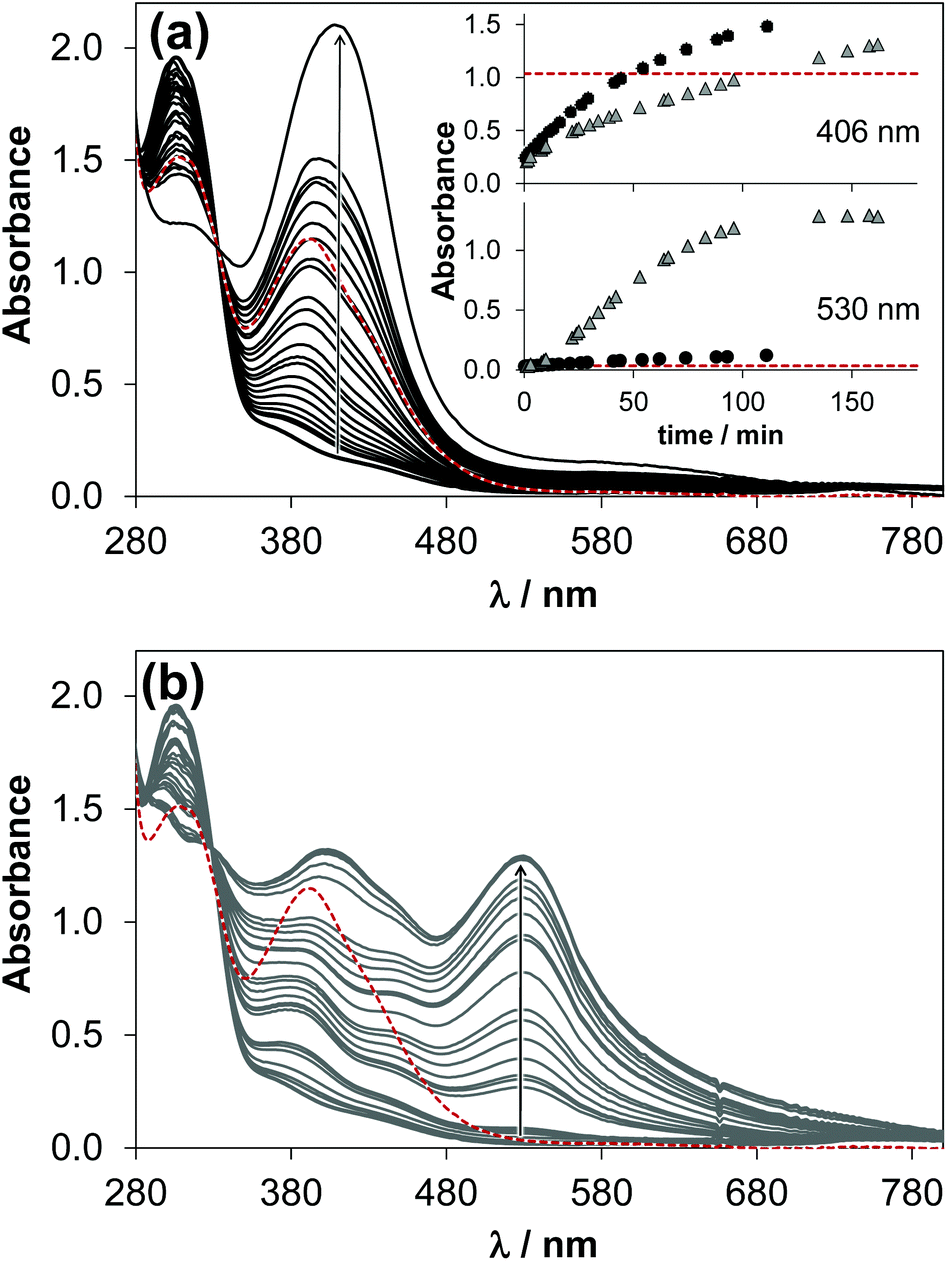 | ||
| Fig. 7 Time-dependent UV–vis absorbance spectra of the [Ru(η6-p-cymene)(H2O)3]2+–HQS (1:2) system at pH 7.4 under aerobic conditions (a) and under an Ar atmosphere (b); red dashed spectrum is calculated as sum of those of [Ru(η6-p-cymene)(HQS)(H2O)] and 1 eq. HQS. Inset (in a) shows the time-dependent changes of absorbance at 406 nm and 530 nm, the symbol colors correspond to the spectrum coloring {cRu = 223 μM; cHQS = 444 μM; pH = 7.4 (20 mM phosphate buffer); T = 25 °C}. | ||
Additionally, the 1H NMR spectrum recorded at a 1:2 metal-to-ligand ratio (in air) at pH 7.4 showed intense broadening of the signals. All these findings strongly suggest that HQS is able to replace the arene ring at least partially, and this process sensitizes ruthenium(II) to oxidation. In order to confirm the formation of ruthenium(III) in the [Ru(η6-p-cymene)(H2O)3]2+–HQS system, EPR spectra were recorded at pH 7.4 and at 11.1 as well at ligand excess (Fig. S7†). The EPR spectra undoubtedly show that the oxidation of the ruthenium center took place indeed at both pH values. The appearance of ruthenium(III) was also seen in the case of HQ (Fig. S8†). The partial loss of the arene ligand was reported for [Ru(η6-biphenyl)(L)Cl] complexes where L = 2,2′-bipyridine or 3,3′-hydroxy-2,2′-bipyridine during aquation, however the report does not indicate the pH-range for the process.43 On the other hand, the co-incubation of the [Ru(η6-p-cymene)(8-quinolinolato)Cl] complex with cysteine, that has a strong binding affinity towards Ru(η6-p-cymene), led to the quick release of the arene moiety and degradation of the complex.21
Comparison of the solution stability of the studied organometallic complexes
In order to compare the solution stability of the Ru(η6-p-cymene) and Rh(η5-C5Me5) complexes formed with the 8-hydroxyquinoline ligands HQ, PHQ and HQS there are several possibilities. However, the direct comparison of the determined logK [ML] values (Table 2) is not adequate, since the complex formation equilibrium is superimposed by other accompanying equilibria, such as (de)protonation of the ligands and hydrolysis of the organometallic cations. Conditional stability constants (logK′ [ML]) taking into consideration the different basicities of the ligands can be computed at a fixed pH value or as a function of pH.44 Thus logK′ [ML] values were calculated at pH 7.4 (Fig. 8a) for the complexes of the studied 8-hydroxyquinolines, which give the following order: HQS > HQ > PHQ in the case of both organometallic cations.
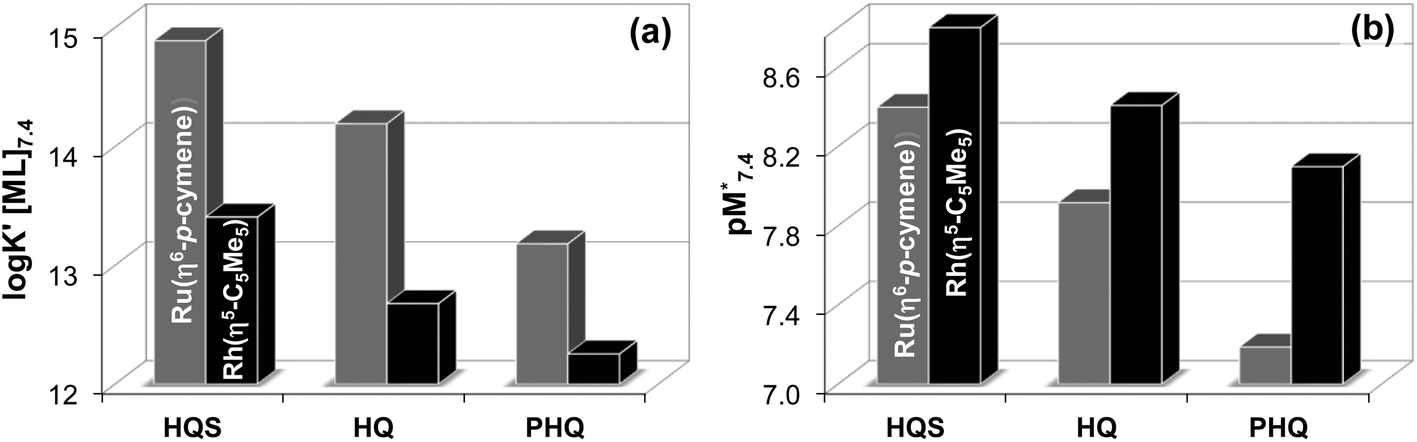 | ||
Fig. 8 Conditional stability constants (logK′ [ML]) of [Rh(η5-C5Me5)(L)(H2O)] (black bars) and [Ru(η6-p-cymene)(L)(H2O)] (grey bars) complexes of HQS, HQ and PHQ at pH 7.4. K′ [ML] = K [ML]/αH, where 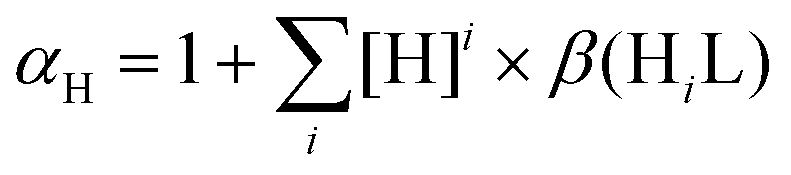 . {T = 25 °C; I = 0.20 M (KNO3)} (a). pM* values at pH 7.4, where pM* = −log([M] + [M2(OH)3] + [M2(OH)2]) {cM = 50 μM; M:L = 1:1; T = 25 °C; I = 0.20 M (KNO3)} (b). . {T = 25 °C; I = 0.20 M (KNO3)} (a). pM* values at pH 7.4, where pM* = −log([M] + [M2(OH)3] + [M2(OH)2]) {cM = 50 μM; M:L = 1:1; T = 25 °C; I = 0.20 M (KNO3)} (b). | ||
Another option is the calculation of pM values for a particular ligand. Basically pM is the negative logarithm of the equilibrium concentration of the unbound metal ion, and a higher pM value indicates a stronger metal ion binding ability of the ligand under given circumstances. The tendency of these organometallic fragments to hydrolyze shows remarkable differences, which has to be taken into account for a more adequate comparison of the complex stabilities. Namely, the hydrolysis of the [Ru(η6-p-cymene)(H2O)3]2+ cation is stronger and occurs at lower pH values compared to [Rh(η5-C5Me5)(H2O)3]2+,16,36 thus the extent of competition between a given ligand and the hydroxide ion for the metal is different as well. Therefore the formation of the various hydroxido species ([(Rh(η5-C5Me5))2(μ-OH)3]+, [(Rh(η5-C5Me5))2(μ-OH)2]2+ and [(Ru(η6-p-cymene)2(μ-OH)3]+), which are all unbound forms of the metal ions besides the triaqua species, should be considered and pM* values (pM* = −log([M] + [M2(OH)3] + [M2(OH)2])) were computed at pH 7.4 using the experimentally determined stability constants instead of the simple pM (Fig. 8b). pM* values are shown in the pH range from 2 to 10.5 for complexes of HQ in Fig. S9.†
The 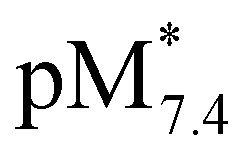 values show the same trend of the metal binding effectiveness of the investigated ligands as the conditional stability constants (Fig. 8a), although they are always higher for the Rh(η5-C5Me5) complexes due to the higher affinity of Ru(η6-p-cymene) towards the hydroxide ions diminishing the ligand-bound fractions. It is worth mentioning that these
values show the same trend of the metal binding effectiveness of the investigated ligands as the conditional stability constants (Fig. 8a), although they are always higher for the Rh(η5-C5Me5) complexes due to the higher affinity of Ru(η6-p-cymene) towards the hydroxide ions diminishing the ligand-bound fractions. It is worth mentioning that these 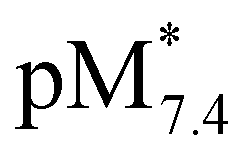 values indicate the formation of very high stability complexes, even in the case of the lowest value (Ru(η6-p-cymene) complex of PHQ) decomposition of less than 1% is estimated at 50 μM concentration. The predominant species at pH 7.4 is the [ML] type complex in all cases. These findings suggest that the studied organometallic complexes are able to retain their bidentate 8-hydroxyquinoline ligand in the coordination sphere under physiological conditions (at pH 7.4, biologically relevant low concentrations) and merely substitution of the aqua ligand by chloride (or vice versa) or by donor atoms of bioligands such as proteins in the biofluids is probable.
values indicate the formation of very high stability complexes, even in the case of the lowest value (Ru(η6-p-cymene) complex of PHQ) decomposition of less than 1% is estimated at 50 μM concentration. The predominant species at pH 7.4 is the [ML] type complex in all cases. These findings suggest that the studied organometallic complexes are able to retain their bidentate 8-hydroxyquinoline ligand in the coordination sphere under physiological conditions (at pH 7.4, biologically relevant low concentrations) and merely substitution of the aqua ligand by chloride (or vice versa) or by donor atoms of bioligands such as proteins in the biofluids is probable.
Chloride ion affinity and lipophilicity of the studied organometallic complexes
In most of the half-sandwich Ru(η6-p-cymene) and Rh(η5-C5Me5) complexes of bidentate ligands a chloride ion is coordinated as a leaving group in the solid forms. Aquation (Cl−/H2O exchange) followed by dissolution in aqueous solution is known to be an important step of mechanism of activation for many anticancer drugs such as cisplatin,45 and has a key role in the DNA/protein interactions. In the case of Ru(η6-arene) complexes it is also assumed that the aqua complex [Ru(η6-arene)(L)H2O] is responsible for the bioactivity, therefore the exchange of the chlorido ligand to water should occur with adequate rate and extent.46 The hydrolysis of the M–Cl bond was found to be fairly fast for the studied 8-hydroxyquinoline complexes, the equilibrium could be reached within some minutes. The immediate formation of the aqua species from the [Ru(η6-p-cymene)(8-quinolinolato)Cl] complex was reported by Kubanik et al.21 in conjunction with our findings. In our experimental setup the following equilibrium process was studied spectrophotometrically: [M(L)(H2O)] + Cl− ⇌ [M(L)(Cl)] + H2O. The displacement of water by the chlorido ligand results in characteristic spectral changes in the UV-vis spectra as Fig. 9 shows for the Rh(η5-C5Me5) complex of HQS. Namely, λmax is increased with increasing absorbance upon higher chloride ion concentrations at pH 7.4.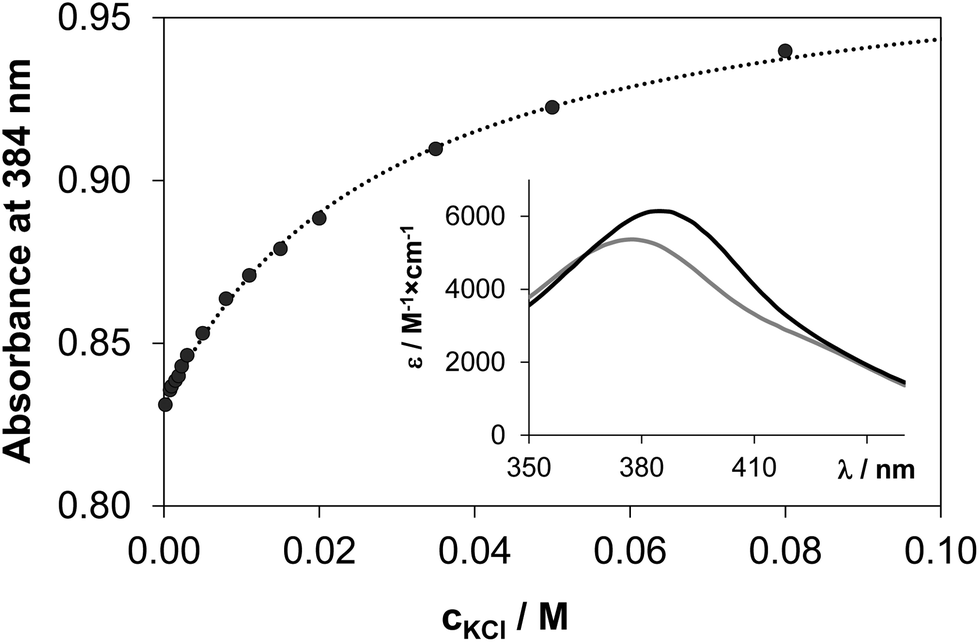 | ||
| Fig. 9 Measured (●) and fitted (dotted line) absorbance values at 384 nm at various chloride ion concentrations in the [Rh(η5-C5Me5)(H2O)3]2+–HQS (1:1) system at pH 7.4. Inset shows the individual calculated molar absorbance spectra of [Rh(η5-C5Me5)(L)(H2O)] (grey spectrum) and [Rh(η5-C5Me5)(L)(Cl)]− (black spectrum) {cL = cRh = 160 μM; cCl− = 0–0.08 M; T = 25 °C}. | ||
Equilibrium constants (see logK′ (H2O/Cl−) values in Table 2) and the individual spectra of the aquated and chlorinated complexes could be estimated by the deconvolution of the measured spectra (see the inset in Fig. 9). The equilibrium constants of the Ru(η6-p-cymene) complexes are more than one order of magnitude lower compared to those of the Rh(η5-C5Me5), reflecting a significantly lower affinity of these ruthenium(II) species towards the chloride ion and an easier replacement by water or by donor atoms of biomolecules. The ratio of the aquated and chlorinated species depends on the actual concentration of the chloride ions. The distribution of these species was estimated for the HQ complexes based on the determined logK′ (H2O/Cl−) constants at 100, 24 and 4 mM chloride content in accordance with the blood serum, cell plasma and cell nucleus,45 respectively (Fig. 10). It can be concluded that the extent of aquation is higher for the Ru(η6-p-cymene) complexes, and it is assumed that 97% and 80% of the organoruthenium and the organorhodium complexes respectively are present in solution as the more reactive aqua species at 4 mM chloride ion concentration.
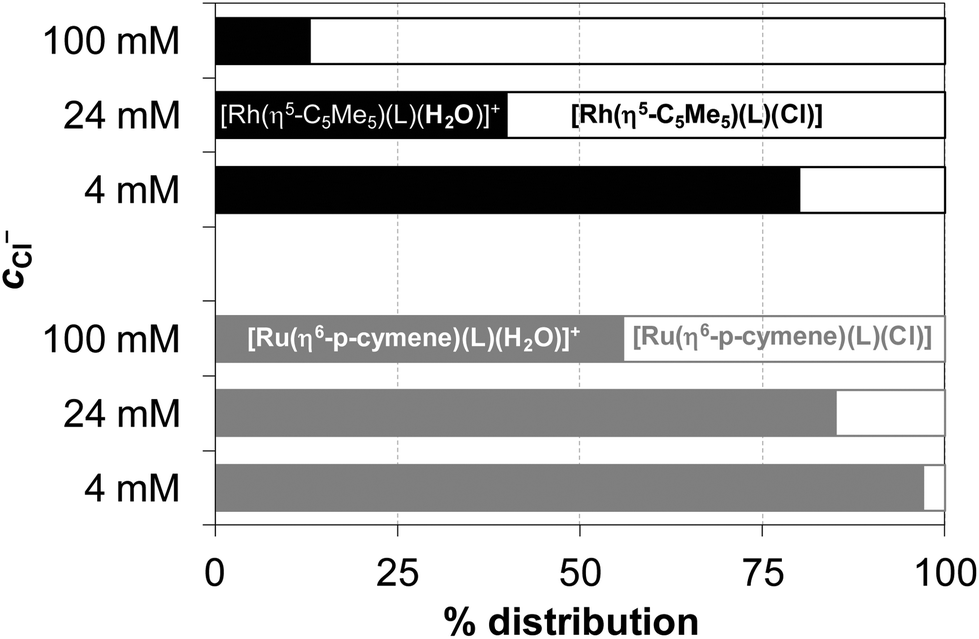 | ||
| Fig. 10 Estimated distribution (%) of the aqua (filled bars) and chlorido (empty bars) complexes of HQ formed with Rh(η5-C5Me5) (black) and Ru(η6-p-cymene) (grey) at 100, 24 and 4 mM concentration of chloride ions calculated on the basis of the exchange constants (logK′ (H2O/Cl−)) {cL = cM = 100 μM; pH = 7.40; T = 25 °C}. | ||
The dependence of cytotoxicity on chloride ion affinity has been reported for several Ru(η6-arene) complexes47 as well as for a series of Rh(η5-C5Me5) compounds.41 However, besides the chloride affinity the lipophilicity is another crucial factor determining the antiproliferative activity as it influences the solubility and the passage through the cell membrane.
Lipophilicity of the half-sandwich organometallic complexes is not only governed by the lipophilic character of the coordinated ligand and the arene/arenyl moiety, but the chloride/water exchange also has an impact on the lipophilic character as it alters the net charge of the complexes. Therefore, distribution coefficients at pH 7.4 (logD7.4) were determined for the complexes of HQ and PHQ at various chloride ion concentrations (Table 4). logD7.4 values for the ligands and the organometallic cations are also shown for comparison, but the data were not determined for the HQS complexes as they were found to be non-cytotoxic (IC50 > 100 μM in the cell lines studied, see the next section) most likely due to their much stronger hydrophilic character. A calculated logP value of +0.46 was reported for the complex [Ru(η6-p-cymene)(8-quinolinolato)Cl],21 but the possible Cl−/H2O exchange was not taken into consideration. Based on the data in Table 4 it can be concluded that the HQ complexes are more lipophilic than the PHQ species similarly to the case of the metal-free ligands. The lower logD7.4 value of PHQ is a consequence of the protonated piperidinium moiety. On the other hand the general trend for the increasing lipophilicity of the metal complexes is observed with increasing chloride ion concentration as the compounds become more chlorinated, thus the net charge turns to be lower (e.g. [M(L)(H2O)]+ → [M(L)(Cl)] in the case of HQ complexes). In the absence of chloride ions both the triaqua and the mono-ligand aqua complexes are more lipophilic in the case of the Ru(η6-p-cymene), however the logD7.4 values are higher for the Rh(η5-C5Me5) complexes of HQ, PHQ when chloride ions are present in the solution, thus in the coordination sphere.
D7.4) for the [Rh(η5-C5Me5)(L)(Z)] and [Ru(η6-p-cymene)(L)(Z)] (Z = H2O/Cl−; charges are omitted for clarity) complexes formed with HQ and PHQ as well as the corresponding free ligands and organometallic precursors for comparison at various chloride ion concentrations {T = 25 °C, pH = 7.4 (20 mM phosphate buffer)a
| logD7.4 |
Rh(η5-C5Me5) | Ru(η6-p-cymene) | ligand alone | ||||
|---|---|---|---|---|---|---|---|
| c Cl− = | 0.0 M | 0.1 M | 0.5 M | 0.0 M | 0.1 M | 0.5 M | 0.1 M |
|
a Uncertainties (SD) of the last digits are shown in parentheses.
b logD7.4 = +1.78.48
c Data are taken from ref. 18.
|
|||||||
| HQ | −0.63(2) | +0.75(3) | +0.80(1) | +0.10(1) | +0.54(1) | +0.78(8) | +1.81(2)b |
| PHQ | −1.22(4) | −0.55(1) | −0.31(1) | −1.05(1) | −0.78(1) | −0.59(1) | +0.93(4) |
| No ligand | <−2.00c | −0.61c | −0.46c | −1.71(2) | −0.46(2) | +0.33(8) | — |
Consequently, the lipophilicity of these organometallic complexes shows a strong dependence on the actual chloride ion concentration.
Cytotoxic activity and MDR-selective activity in cancer cell lines
The cytotoxic effect of ligand HQ and its Ru(η6-p-cymene) and Rh(η5-C5Me5) complexes measured in various cancer cell lines has been already reported. A recent study of Kubanik el al. provides IC50 values of 1.97–5.96 μM for HQ and 11.4–19.3 μM for [Ru(η6-p-cymene)(8-quinolinolato)Cl] in human colorectal (HCT116), non-small cell lung (NCI-H460), and cervical carcinoma (SiHa) cells.21 The metal complex showed good activity that is clearly associated with the cytotoxic activity of the ligand. Similar IC50 values were obtained for this ruthenium(II) complex in ovarian (CH1) and colon carcinoma (SW480) cell lines.20 The [Rh(η5-C5Me5)(8-quinolinolato)Cl] complex was found to be active in human melanoma and glioblastoma cells (IC50 ∼ 0.8–100 μM) and showed good activity against Gram-positive bacteria as well.19 The piperidine derivative of HQ (PHQ) is also a cytotoxic compound and has a strong preference for targeting MDR cell lines, while HQ does not exert MDR-selectivity.29Here, our aim was to reveal whether the complexation of PHQ with the studied organometallic cations resulting in the formation of very high stability complexes can modify the intrinsic cytotoxic effectiveness and the MDR-selectivity of the ligand.
The cytotoxic activity of PHQ, HQ and HQS was investigated in the absence and in the presence of one equivalent of [Ru(η6-p-cymene)(H2O)3]2+ or [Rh(η5-C5Me5)(H2O)3]2+ cations in MES-SA (human uterine sarcoma) and in its multidrug-resistant counterpart (MES-SA/Dx5) cell lines by means of the colorimetric 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay, as detailed in the Experimental section. The resistance of MES-SA/Dx5 cells is primarily mediated by the overexpression of P-gp, a member of the ABC transporter family, which pumps out xenobiotics from the cells. P-gp expression is significantly increased in multidrug-resistant tumor cells resulting in decreased intracellular drug accumulation. In order to show the effect of the active pump, experiments were also performed in the presence of the P-gp-inhibitor Tariquidar (TQ), which binds with high affinity to the P-gp transporter. The clinical drug and the P-gp substrate doxorubicin was used as a positive control. As a further control, the cytotoxicity of the organometallic cations was measured as well.
The IC50 values are collected in Table 5. Notably, the toxicity of the organometallic cations and the HQS containing samples is negligible. The cytotoxicity of the ligands HQ and PHQ in MES-SA cell lines is very similar, and both are more active in MES-SA/Dx5 cells, however the selectivity ratio, i.e. IC50 (MES-SA)/IC50 (MES-SA/Dx5), is much higher for PHQ that is considered as a MDR selective compound.29 For a better comparison of the biological data the pIC50 values obtained in the MES-SA/Dx5 cells are plotted against those of the MES-SA cells (Fig. 11a and b) and the selectivity ratio is also shown for the ligands and the metal complexes (Fig. 11c).
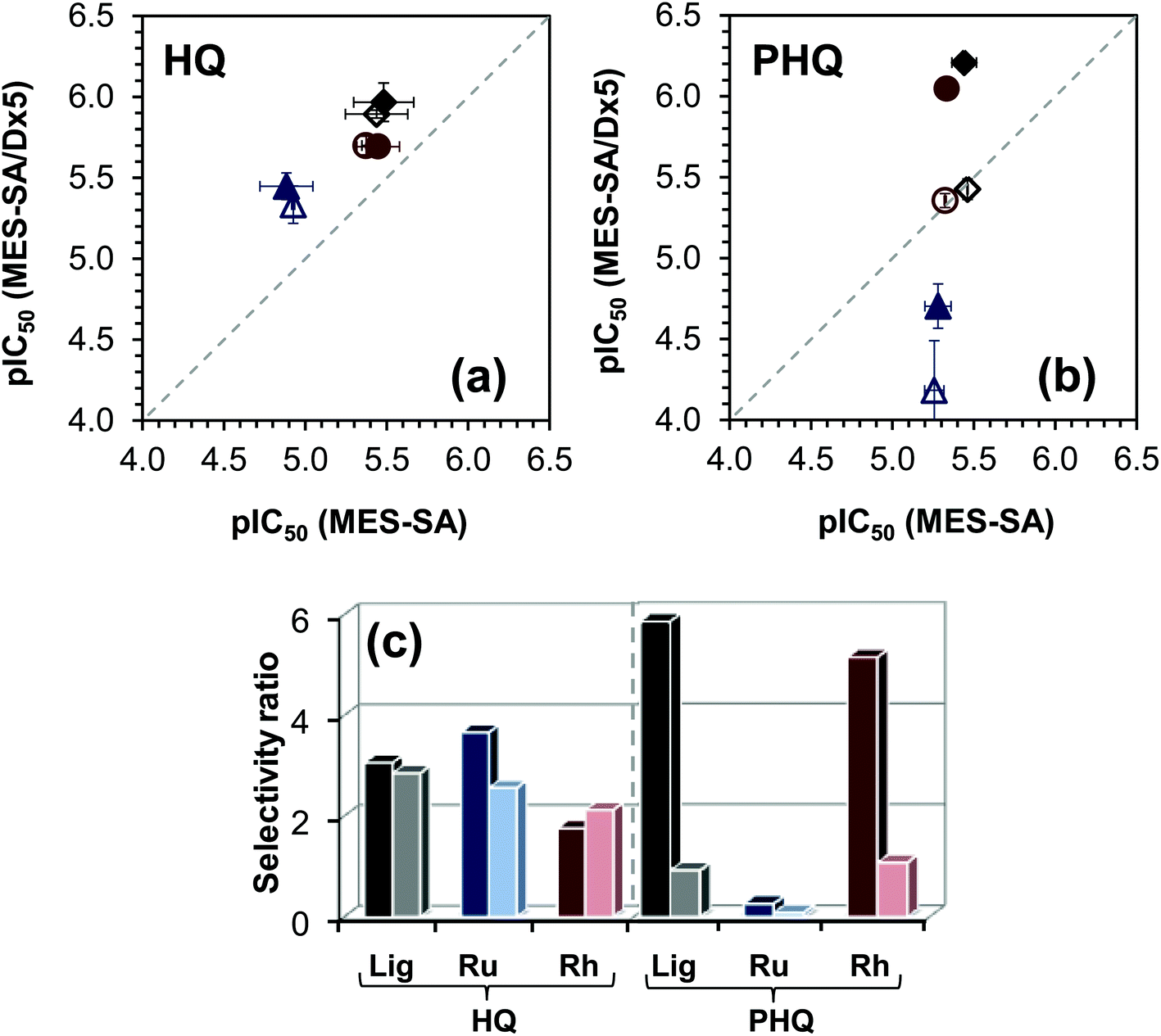 | ||
| Fig. 11 pIC50 (= −log(IC50 expressed in M)) values measured in MES-SA/Dx5 cells of the ligand HQ (a) and PHQ (b) with their Rh(η5-C5Me5) and Ru(η6-p-cymene) complexes plotted against those obtained in MES-SA cells in the absence (filled symbols) or in the presence (empty symbols) of TQ. Symbols: ligand alone (◆), Rh(η5-C5Me5) complexes (●), Ru(η6-p-cymene) complexes (▲). The selectivity ratio (IC50 (MES-SA)/IC50 (MES-SA/Dx5)) for ligands (Lig: HQ, PHQ) and their Ru(η6-p-cymene) (Ru) and Rh(η5-C5Me5) (Rh) complexes in the absence (left darker bars) or in the presence (right lighter bars) of TQ (c). | ||
| IC50/μM | MES-SA | MES-SA/Dx5 | MES-SA + 1 μM TQ | MES-SA/Dx5 + 1 μM TQ | |
|---|---|---|---|---|---|
| Doxorubicin | 0.087 ± 0.022 | 3.1 ± 1.1 | 0.064 ± 0.013 | 0.036 ± 0.008 | |
| [Ru(η6-p-cymene)(Z)3] | >100 | >100 | >100 | >100 | |
| [Rh(η5-C5Me5)(Z)3] | >100 | >100 | >100 | >100 | |
| HQ | Ligand | 3.3 ± 1.1 | 1.08 ± 0.26 | 3.7 ± 1.3 | 1.28 ± 0.07 |
| [Ru(η6-p-cymene)(L)(Z)] | 13.1 ± 4.1 | 3.57 ± 0.61 | 11.87 ± 0.27 | 4.6 ± 1.1 | |
| [Rh(η5-C5Me5)(L)(Z)] | 3.57 ± 0.93 | 2.03 ± 0.27 | 4.24 ± 0.24 | 2.00 ± 0.25 | |
| PHQ | Ligand | 3.63 ± 0.58 | 0.62 ± 0.03 | 3.46 ± 0.02 | 3.75 ± 0.51 |
| [Ru(η6-p-cymene)(L)(Z)] | 5.25 ± 0.88 | 19.8 ± 5.4 | 5.54 ± 0.70 | 66 ± 33 | |
| [Rh(η5-C5Me5)(L)(Z)] | 4.63 ± 0.35 | 0.90 ± 0.12 | 4.75 ± 0.07 | 4.41 ± 0.41 | |
| HQS | Ligand | >100 | >100 | >100 | >100 |
| [Ru(η6-p-cymene)(L)(Z)] | >100 | >100 | >100 | >100 | |
| [Rh(η5-C5Me5)(L)(Z)] | >100 | >100 | >100 | >100 | |
The selectivity ratio is an indicator of the increased sensitivity of the MDR MES-SA/Dx5 cell line as compared to the parental MES-SA cell line. A selectivity ratio >2 indicates that MES-SA/Dx5 cells are paradoxically more sensitive, rather than more resistant, to a given compound. It is important to note that collateral sensitivity may be limited to a particular cell line.30,49 To establish the requirement of functional P-gp, we repeated the cytotoxicity experiments in the presence of the P-gp-inhibitor TQ. As expected, TQ has no impact on the IC50 values of all the studied compounds in the case of the P-gp negative parental MES-SA cells. In contrast, by inhibition of the transporter, MES-SA/Dx5 cells become more sensitive to the P-gp substrate doxorubicin. The cytotoxicity of the ligand HQ and its complexes is independent of the presence of TQ in the MES-SA/Dx5 cell lines. On the contrary, higher IC50 values were observed for the ligand PHQ and its Rh(η5-C5Me5) complex in MES-SA/Dx5 cells when TQ was added to the samples, proving that the cytotoxicity is indeed potentiated by P-gp. The IC50 values obtained for Ru(η6-p-cymene) and Rh(η5-C5Me5) complexes of HQ and PHQ are similar to those of the free ligands, although the values are somewhat higher in the case of the Ru(η6-p-cymene) complexes in MES-SA and MES-SA/Dx5 cells. As a consequence, the selectivity ratio remained low and unaffected by the complexation in the case of the non-MDR selective compound HQ (Fig. 11c). The selectivity ratio calculated for PHQ and its Rh(η5-C5Me5) complex is fairly high, while the complex formation with Ru(η6-p-cymene) seems to abolish MDR selective activity. The altered behavior of the Ru(η6-p-cymene) and Rh(η5-C5Me5) complexes of PHQ most likely cannot be explained simply on the basis of the solution speciation data and their lipophilic character, which shows dependence on the chloride ion concentration. Both complexes possess undoubtedly high stability, whereas their decomposition is assumed to be negligible in solution under physiological conditions. On the other hand, the Ru(η6-p-cymene) complex is kinetically more inert, it has somewhat lower chloride-affinity, thus more double positively charged mono-aqua [Ru(η6-p-cymene)(L)(H2O)]2+ species are present in the solution, and the complex has slightly more hydrophilic character in the medium containing chloride ions at concentration ≤100 mM. These features can affect the biological activity; however other factors such as the 3D structure, or the interaction with bioligands might also be relevant.
Experimental
Chemicals
All solvents were of analytical grade and used without further purification. HQ, HQS, [Rh(η5-C5Me5)(μ-Cl)Cl]2, [Ru(η6-p-cymene)(μ-Cl)Cl]2, KCl, KNO3, AgNO3, HCl, HNO3, KOH, 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS), dimethyl sulfoxide (DMSO), NaH2PO4 and Na2HPO4 were purchased from Sigma-Aldrich in puriss quality. The ligand PHQ (NSC57969) was acquired from the drug repository of the Developmental Therapeutics Program of the National Cancer Institute. Doubly distilled Milli-Q water was used for sample preparation. The exact concentration of the ligand stock solutions together with the proton dissociation constants were determined by pH-potentiometric titrations with the use of the computer program HYPERQUAD.50 The aqueous [Rh(η5-C5Me5)(H2O)3](NO3)2 and [Ru(η6-p-cymene)(H2O)3](NO3)2 stock solutions were obtained by dissolving the exact amounts of [Rh(η5-C5Me5)(μ-Cl)Cl]2 or [Ru(η6-p-cymene)(μ-Cl)Cl]2 respectively in water followed by the removal of chloride ions by the addition of equivalent amounts of AgNO3. The exact concentrations of [Rh(η5-C5Me5)(H2O)3]2+ and [Ru(η6-p-cymene)(H2O)3]2+ were determined by pH-potentiometric titrations employing stability constants for [(Rh(η5-C5Me5))2(μ-OH)i](4−i)+ (i = 2 or 3)16 and [(Ru(η6-p-cymene))2(μ-OH)3]+ (ref. 37) complexes, respectively.pH-Potentiometric measurements
pH-Potentiometric measurements determining proton dissociation constants of ligands HQ and HQS and overall stability constants for tested Rh(η5-C5Me5)–HQS complexes were carried out at 25.0 ± 0.1 °C in water and at a constant ionic strength of 0.20 M KNO3. The titrations were performed in a carbonate-free KOH solution (0.20 M). The exact concentrations of HNO3 and KOH solutions were determined by pH-potentiometric titrations. An Orion 710A pH-meter equipped with a Metrohm “double junction” combined electrode (type 6.0255.100) and a Metrohm 665 Dosimat burette were used for the pH-potentiometric measurements. The electrode system was calibrated to the pH = −log[H+] scale by means of blank titrations (strong acid vs. strong base: HNO3vs. KOH), as suggested by Irving et al.51 The average water ionization constant, pKw, was determined as 13.76 ± 0.01 at 25.0 °C, I = 0.20 M (KNO3), which is in accordance with the literature.52 The reproducibility of the titration points included in the calculations was within 0.005 pH units. The pH-potentiometric titrations were performed in the pH range between 2.0 and 11.5. The initial volume of the samples was 10.0 mL. The ligand concentration was 1.0 mM and was investigated at metal ion-to-ligand ratios of 1:1, 1:1.5, and 1:2. The accepted fitting between the measured and calculated titration data points regarding the volume of the titrant was <10 μL. The samples were degassed by bubbling purified argon through them for about 10 minutes prior to the measurements and the inert gas was also passed over the solutions during the titrations.
The computer program PSEQUAD53 was utilized to establish the stoichiometry of the complexes and to calculate the overall stability constants. β(MpLqHr) is defined for the general equilibrium: pM + qL + rH ⇌ MpLqHr as β(MpLqHr) = [MpLqHr]/[M]p[L]q[H]r; where M denotes the metal moiety [Rh(η5-C5Me5)(H2O)3]2+ or [Ru(η6-p-cymene)(H2O)3]2+ and L the completely deprotonated ligand. β values for the various hydroxido complexes [(Rh(η5-C5Me5))2(μ-OH)i](4−i)+ (i = 2 or i = 3) or [(Ru(η6-p-cymene))2(μ-OH)3]+ were calculated based on the pH-potentiometric titration data in the absence of chloride ions and were found to be in good agreement with our previously published data.16,37
UV–vis spectrophotometric, 1H NMR EPR and ESI-MS measurements
A Hewlett Packard 8452A diode array spectrophotometer was used to record the UV-vis spectra in the interval 200–800 nm. The path length was 1 or 0.2 cm. Equilibrium constants (proton dissociation, stability constants and H2O/Cl− exchange constants) and the individual spectra of the species were calculated with the computer program PSEQUAD.53 The spectrophotometric titrations were performed in pure water on the samples containing the ligands with or without the organometallic cations and the concentration of the ligands was 150–200 μM or 1 mM (HQ and HQS) and 75 μM (PHQ). The organometallic cations were also titrated (200 μM). The metal-to-ligand ratios were 1:1 and 1:2 in the pH range from 2 to 11.5 at 25.0 ± 0.1 °C at an ionic strength of 0.20 M (KNO3). Measurements for 1:1 metal-to-ligand systems were also carried out by preparing individual samples in which KNO3 was partially or completely replaced by HNO3 and pH values, varying in the range ca. 0.7–2.0, were calculated from the strong acid content. In the case of the Ru(η6-p-cymene) complexes the absorbance data were always recorded after 24 h of incubation. UV-Vis spectra were used to investigate the H2O/Cl− exchange processes of complexes [Rh(η5-C5Me5)(L)(H2O)] and [Ru(η6-p-cymene)(L)(H2O)] at 120–160 μM (HQ, HQS) or 50 μM (PHQ) concentration and at pH 7.40 (using 20 mM phosphate buffer) as a function of chloride concentrations (0–330 mM).
1H NMR studies were carried out on a Bruker Ultrashield 500 Plus instrument. All 1H NMR spectra were recorded with the WATERGATE water suppression pulse scheme using a DSS internal standard. The ligand HQS was dissolved in a 10% (v/v) D2O/H2O mixture to yield a concentration of 1 mM and were titrated at 25 °C, at I = 0.20 M (KNO3) in the absence or presence of [Rh(η5-C5Me5)(H2O)3]2+ at a 1:1 metal-to-ligand ratio in the pH range between 2.0 and 11.5. 1H NMR spectra were recorded on samples containing [Ru(η6-p-cymene)(H2O)3]2+ and HQS at a 1:2 ratio after 24 h of incubation. Stability constants for the complexes were calculated by the computer program PSEQUAD.53
All continuous-wave (CW) EPR spectra were recorded with a BRUKER EleXsys E500 spectrometer (microwave frequency 9.81 GHz, microwave power 10 MW, modulation amplitude 5 G, modulation frequency 100 kHz). EPR spectra were recorded at 10.0 mM of [Ru(η6-p-cymene)(H2O)3]2+ and the metal-to-HQS ratios were 1:1 and 1:2 at pH 7.4 and 11.1.
Electrospray ionization mass spectrometric (ESI-MS) measurements were performed using a Micromass Q-TOF Premier (Waters MS Technologies) mass spectrometer equipped with an electrospray ion source.
Determination of the distribution coefficients
Distribution coefficient (D7.4) values of the [Ru(η6-p-cymene)(L)(Z)] and [Rh(η5-C5Me5)(L)(Z)] complexes (where L = deprotonated HQ or PHQ; Z = H2O/Cl−, charges are omitted for simplicity) and the ligands as well as the organoruthenium Ru(η6-p-cymene) fragment were determined by the traditional shake-flask method in n-octanol/buffered aqueous solution at pH 7.40 (20 mM phosphate buffer) at various KCl concentrations (cKCl = 0.0, 0.10, 0.50 M) at 25.0 ± 0.2 °C as described previously.54 The data for the organorhodium Rh(η5-C5Me5) fragment were already published under similar conditions.18 Two parallel experiments were performed for each sample. The complexes or ligands were dissolved in n-octanol pre-saturated aqueous solution of the buffer at 200 μM (HQ and HQS complexes) or 75 μM (PHQ complexes) concentrations. The aqueous solutions and n-octanol (1:1 ratio) were gently mixed with 360° vertical rotation (∼20 rpm) for 3 h to avoid emulsion formation, and the mixtures were centrifuged at 5000 rpm for 3 min by a temperature controlled centrifuge (Sanyo) at 25 °C. After separation, UV−vis spectra of the compounds in the aqueous phase were compared to those of the original aqueous solutions and D7.4 values of the complexes or ligands were calculated as follows: [absorbance (original solution)/absorbance (aqueous phase after separation) − 1]. Absorbances were recorded in the region of λmax (250–600 nm).
Synthesis of complex [chlorido(8-hydroxyquinolinato-κN1,κO8)(η5-1,2,3,4,5-pentamethylcyclopentadienyl)rhodium(III)] (1) and crystallographic structure determination
Single crystals suitable for X-ray diffraction experiment of compound [Rh(η5-C5Me5)(8-quinolinolato)Cl] (1) were grown from a water/ethanol solution mixture (2.0 mL) containing the HQ ligand (2.9 mg, 0.02 mmol) and rhodium dimer [Rh(η5-C5Me5)(μ-Cl)Cl]2 (6.2 mg, 0.01 mmol) at neutral pH (adjusted by NaOH solution). The mixture was kept in the dark at room temperature and after 4 h dark orange crystals were formed.A single crystal was mounted on a loop and transferred to the goniometer. X-ray diffraction data were collected at −170 °C on a Rigaku RAXIS-RAPID II diffractometer using Mo-Kα radiation. A multi-scan absorption correction was carried out using the program CrystalClear.55 Sir201456 and SHELXL57 under the WinGX58 software were used for structure solution and refinement, respectively. The structures were solved by direct methods. The models were refined by full-matrix least squares on F2. Refinement of non-hydrogen atoms was carried out with anisotropic temperature factors. Hydrogen atom positions were located in difference electron density maps (for water hydrogens) or placed into geometric positions. They were included in structure factor calculations but they were not refined. The isotropic displacement parameters of the hydrogen atoms were approximated from the U(eq.) value of the atom they were bonded to. The summary of data collection and refinement parameters for complex [Rh(η5-C5Me5)(8-quinolinolato)Cl]·3H2O are collected in Table S1.† Selected bond lengths and angles of compounds were calculated by using the PLATON software.42 The graphical representation and the edition of CIF files were done by using the Mercury59 and PublCif60 software. The crystallographic data files for the complexes have been deposited with the Cambridge Crystallographic Database as CCDC 1530884.
The orange crystals were filtered and analyzed by 1H NMR (500.13 MHz, CD3OD): δ (ppm) 8.82 (m, 1H, CH2); 8.28 (d, 3J (H,H) = 8 Hz, 1H, CH4); 7.56 (m 1H, CH6); 7.36 (t, 3J (H,H) = 8 Hz, 1H, CH3); 6.97 (d, 3J (H,H) = 8 Hz, 1H, CH5); 6.94 (d, 3J (H,H) = 8 Hz, 1H, CH7); 1.72 (s, 15H, CH3, C5Me5) and by ESI-MS: m/z = 382 [complex − Cl−]+. NMR and ESI-MS data obtained are similar to those published by Sliwinska et al.19 and Thai et al.22
Cell lines, culture conditions and cytotoxicity tests in cancer cell lines
000 cells per well and allowed to settle and resume exponential growth in drug-free complete culture medium for 12 h to 24 h. Ligands HQ, PHQ and HQS were dissolved in an 80% (v/v) ethanol/water mixture first, diluted in complete culture medium and added to the plates where the final ethanol content did not exceed 0.5%. Whereas the Rh(η5-C5Me5) and Ru(η6-p-cymene) complexes of the ligands were prepared in an 80% (v/v) ethanol/water mixture in situ by mixing the ligand with one equimolar concentration of the organometallic cations using their stock solutions containing known amounts of [Rh(η5-C5Me5)(H2O)3]2+ and [Ru(η6-p-cymene)(H2O)3]2+. Following the addition of the serial dilutions of ligands and complexes and an incubation period of 72 h, the supernatant was removed and fresh medium supplemented with the MTT reagent (0.83 mg mL−1) was added. Incubation with MTT at 37 °C was terminated after 1 h by removing the supernatants and lysing the cells with 100 μL DMSO per well. Viability of the cells was measured spectrophotometrically by absorbance at 540 nm using an EnSpire microplate reader. The data were background corrected by subtraction of the signal obtained from unstained cell lysates and normalized to untreated cells. Curves were fitted by using the Prism software62 using the sigmoidal dose–response model (comparing variable and fixed slopes). Curve fit statistics were used to determine the concentration of the test compound that resulted in 50% toxicity (IC50). Evaluation is based on means from three independent experiments, each comprising three replicates per concentration level. Co-incubation experiments were also performed in the presence of the P-gp inhibitor TQ (kind gift of Dr S. Bates, NCI NIH). Doxorubicin (Sigma-Aldrich) was used as a positive control.
Conclusions
The rational design and optimization of the bioactivity of metallodrugs require detailed information about the solution behavior, stability and speciation characteristics, especially under physiological conditions. Solution equilibrium studies provide information about the chemical species present in aqueous solution and are of utmost importance for the understanding of the mechanism of action of biologically active compounds. The main objective of this work was to characterize and compare the solution speciation of Ru(η6-p-cymene) and Rh(η5-C5Me5) complexes formed with various bidentate 8-hydroxyquinoline compounds (HQ, PHQ and HQS). Stoichiometry and stability of these organometallic complexes were determined in aqueous solution via a combined approach using 1H NMR spectroscopy, UV-visible spectro-photometry and pH-potentiometry. X-ray diffraction study of [Rh(η5-C5Me5)(8-quinolinolato)(Cl)] showed a pseudo-octahedral “piano-stool” geometry, and the deprotonated ligand coordinates in a bidentate mode via (N,O−) donor atoms. Based on the comparative equilibrium studies we concluded that mono-ligand complexes with a general formula of [M(L)(H2O)] are formed with significantly high solution stability, and their decomposition cannot occur even at low micromolar concentrations at physiological pH. The trend for the stability of the studied organometallic complexes is the following: HQS > HQ > PHQ. The relative affinities of the 8-hydroxyquinolines towards the organometallic cations are somewhat higher with Rh(η5-C5Me5) at pH 7.4. However, 1–1.5 orders of magnitude lower pKa values were obtained for the [Ru(η6-p-cymene)(L)(H2O)] complexes, mixed hydroxido species [M(L)(OH)] are formed only in the basic pH range in all cases due to the relatively high pKa values (8.5–10.3). Notably, the complex formation rate with [Ru(η6-p-cymene)(H2O)3]2+ is much lower compared to the organorhodium triaqua cation. Additionally, H2O/Cl− co-ligand exchange constants show the stronger affinity of the Rh(η5-C5Me5) complexes towards chloride ions. As a consequence of the aquation of the chlorinated compounds ([M(L)(Cl)]) the lipophilic character of the studied organometallic complexes is decreasing with decreasing chloride ion concentrations. A somewhat stronger hydrophilic character of the Ru(η6-p-cymene) complexes was found at chloride ion concentrations which are biologically relevant.In vitro cytotoxicity of the unsubstituted HQ, as well as of the substituted derivatives PHQ (which is preferentially toxic to multidrug resistant cell lines) and HQS was measured in the absence and in the presence of the [Ru(η6-p-cymene)(H2O)3]2+ and [Rh(η5-C5Me5)(H2O)3]2+ cations. A cell line pair, namely MES-SA (human uterine sarcoma) and its P-gp-expressing multidrug resistant counterpart (MES-SA/Dx5), was used. IC50 values in the low μM range were observed except for the HQS containing samples. The effect of Tariquidar, a high-affinity P-gp inhibitor, on the selectivity ratio was also investigated. Ligand HQ and its organometallic complexes exhibit a similar cytotoxicity in both cell lines, with a low selectivity ratio that is unaffected by the complexation and P-gp activity. At the same time PHQ and its Rh(η5-C5Me5) complex have an increased P-gp mediated MDR-selective ratio, however the selectivity is abrogated in the case of the Ru(η6-p-cymene) complex, which might be a consequence of its more inert feature and slightly more hydrophilic character.
Acknowledgements
We thank Ms Nikolett Jabronka and Mr Ádám Szitás for expert technical assistance. This work was supported by the National Research, Development and Innovation Office-NKFIH through project GINOP-2.3.2-15-2016-00038, OTKA PD103905, K115762, and the J. Bolyai Research Scholarship of the Hungarian Academy of Sciences (E. A. E.). G. S. was supported by a Momentum Grant of the Hungarian Academy of Sciences and ERC (StG-260572).References
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CAS.
- E. Alessio, G. Mestroni, A. Bergamo and G. Sava, Curr. Top. Med. Chem., 2004, 4, 1525–1535 CrossRef CAS PubMed.
- E. Alessio, Eur. J. Inorg. Chem. DOI:10.1002/ejic.201600986.
- A. K. Singh, D. S. Pandey, Q. Xua and P. Braunstein, Coord. Chem. Rev., 2014, 270, 31–56 CrossRef.
- A. Weiss, R. H. Berndsen, M. Dubois, C. Müller, R. Schibli, A. W. Griffioen, P. J. Dyson and P. Nowak-Sliwinska, Chem. Sci., 2014, 5, 4742–4748 RSC.
- W. Kandioller, C. G. Hartinger, A. A. Nazarov, C. Bartel, M. Skocic, M. A. Jakupec, V. B. Arion and B. K. Keppler, Chem. – Eur. J., 2009, 15, 12283–12291 CrossRef CAS PubMed.
- W. H. Ang, A. Casini, G. Sava and P. J. Dyson, J. Org. Chem., 2011, 696, 989–998 CrossRef CAS.
- B. Therrien, Coord. Chem. Rev., 2009, 253, 493–519 CrossRef CAS.
- S. H. van Rijt and P. J. Sadler, Drug Discovery Today, 2009, 14, 1089–1097 CrossRef CAS PubMed.
- É. A. Enyedy, É. Sija, T. Jakusch, C. G. Hartinger, W. Kandioller, B. K. Keppler and T. Kiss, J. Inorg. Biochem., 2013, 127, 161–168 CrossRef PubMed.
- A. Merlino, Coord. Chem. Rev., 2016, 326, 111–134 CrossRef CAS.
- Y. Geldmacher, M. Oleszak and W. S. Sheldrick, Inorg. Chim. Acta, 2012, 393, 84–102 CrossRef CAS.
- Y. Geldmacher, K. Splith, I. Kitanovic, H. Alborzinia, S. Can, R. Rubbiani, M. A. Nazif, P. Wefelmeier, A. Prokop, I. Ott, S. Wölfl, I. Neundorf and W. S. Sheldrick, J. Biol. Inorg. Chem., 2012, 17, 631–646 CrossRef CAS PubMed.
- M. A. Scharwitz, I. Ott, Y. Geldmacher, R. Gust and W. S. Sheldrick, J. Organomet. Chem., 2008, 693, 2299–2309 CrossRef CAS.
- O. Dömötör, S. Aicher, M. Schmidlehner, M. S. Novak, A. Roller, M. A. Jakupec, W. Kandioller, C. G. Hartinger, B. K. Keppler and É. A. Enyedy, J. Inorg. Biochem., 2014, 134, 57–65 CrossRef PubMed.
- É. A. Enyedy, O. Dömötör, C. M. Hackl, A. Roller, M. S. Novak, M. A. Jakupec, B. K. Keppler and W. Kandioller, J. Coord. Chem., 2015, 68, 1583–1601 CrossRef.
- O. Dömötör, C. M. Hackl, K. Bali, A. Roller, M. Hejl, M. A. Jakupec, B. K. Keppler, W. Kandioller and É. A. Enyedy, unpublished results.
- U. Sliwinska, F. P. Pruchnik, S. Ułaszewski, M. Latocha and D. Nawrocka-Musiał, Polyhedron, 2010, 29, 1653–1659 CrossRef CAS.
- R. Schuecker, R. O. John, M. A. Jakupec, V. B. Arion and B. K. Keppler, Organometallics, 2008, 27, 6587–6595 CrossRef CAS.
- M. Kubanik, H. Holtkamp, T. Söhnel, S. M. F. Jamieson and C. G. Hartinger, Organometallics, 2015, 34, 5658–5668 CrossRef CAS.
- T.-T. Thai, B. Therrien and G. Süss-Fink, Inorg. Chem. Commun., 2009, 12, 806–807 CrossRef CAS.
- C. Gemel, R. John, C. Slugovc, K. Mereiter, R. Schmid and K. Kirchner, J. Chem. Soc., Dalton Trans., 2000, 2607–2612 RSC.
- T.-T. Thai, B. Therrien and G. Süss-Fink, J. Organomet. Chem., 2009, 694, 3973–3981 CrossRef CAS.
- M. A. Jakupec and B. K. Keppler, Curr. Top. Med. Chem., 2004, 4, 1575–1583 CrossRef CAS PubMed.
- R. D. Hofheinz, C. Dittrich, M. A. Jakupec, A. Drescher, U. Jaehde, M. Gneist, N. Graf von Keyserlingk, B. K. Keppler and A. Hochhaus, Int. J. Clin. Pharmacol. Ther., 2005, 43, 590–591 CrossRef CAS PubMed.
- G. Szakacs, J. K. Paterson, J. A. Ludwig, C. Booth-Genthe and M. M. Gottesman, Nat. Rev. Drug Discovery, 2006, 5, 219–234 CrossRef CAS PubMed.
- D. Türk, M. D. Hall, B. F. Chu, J. A. Ludwig, H. M. Fales, M. M. Gottesman and G. Szakacs, Cancer Res., 2009, 69, 8293–8301 CrossRef PubMed.
- A. Füredi, S. Tóth, K. Szebényi, V. F. S. Pape, D. Türk, N. Kucsma, L. Cervenak, J. Tóvári and G. Szakács, Mol. Cancer Ther., 2017, 16, 45–56 CrossRef PubMed.
- G. Szakács, M. D. Hall, M. M. Gottesman, A. Boumendjel, R. Kachadourian, B. J. Day, H. Baubichon-Cortay and A. Di Pietro, Chem. Rev., 2014, 114, 5753–5774 CrossRef PubMed.
- É. A. Enyedy, O. Dömötör, E. Varga, T. Kiss, R. Trondl, C. G. Hartinger and B. K. Keppler, J. Inorg. Biochem., 2012, 117, 189–197 CrossRef PubMed.
- S. Tsakovski, K. Benkhedda, E. Ivanova and F. C. Adams, Anal. Chim. Acta, 2002, 453, 143–154 CrossRef CAS.
- P. Letkeman, A. E. Martell and R. J. Motekaitis, J. Coord. Chem., 1980, 10, 47–53 CrossRef CAS.
- O. Jarjayes, S. Hamman, F. Sarrazin, T. Benaissa and C. G. Beguin, New J. Chem., 1998, 22, 361–366 RSC.
- M. Amati, S. Belviso, P. L. Cristinziano, C. Minichino, F. Lelj, I. Aiello, M. La Deda and M. Ghedini, J. Phys. Chem. A, 2007, 111, 13403–13414 CrossRef CAS PubMed.
- P. Buglyó and E. Farkas, Dalton Trans., 2009, 8063–8070 RSC.
- L. Bíró, E. Farkas and P. Buglyó, Dalton Trans., 2012, 41, 285–291 RSC.
- M. S. Eisen, A. Haskel, H. Chen, M. M. Olmstead, D. P. Smith, M. F. Maestre and R. H. Fish, Organometallics, 1995, 14, 2806–2812 CrossRef CAS.
- A. Nutton, P. M. Baily and P. M. Maitlis, J. Chem. Soc., Dalton Trans., 1981, 1997–2002 RSC.
- A. F. A. Peacock, M. Melchart, R. J. Deeth, A. Habtemariam, S. Parsons and P. J. Sadler, Chem. – Eur. J., 2007, 13, 2601–2613 CrossRef CAS PubMed.
- É. A. Enyedy, J. P. Mészáros, O. Dömötör, C. M. Hackl, A. Roller, B. K. Keppler and W. Kandioller, J. Inorg. Biochem., 2015, 152, 93–103 CrossRef PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- T. Bugarcic, A. Habtemariam, J. Stepankova, P. Heringova, J. Kasparkova, R. J. Deeth, R. D. L. Johnstone, A. Prescimone, A. Parkin, S. Parsons, V. Brabec and P. J. Sadler, Inorg. Chem., 2008, 47, 11470–11486 CrossRef CAS PubMed.
- G. Schwarzenbach and G. Anderegg, Helv. Chim. Acta, 1957, 40, 1773–1792 CrossRef CAS.
- R. B. Martin, in Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug, ed. B. Lippert, VHCA & Wiley-VCH, Zürich, Switzerland, 1999, pp. 181–205 Search PubMed.
- P. C. A. Bruijnincx and P. J. Sadler, Adv. Inorg. Chem., 2009, 61, 1–62 CrossRef CAS PubMed.
- Y. K. Yan, M. Melchart, A. Habtemariam and P. J. Sadler, Chem. Commun., 2005, 4764–4776 RSC.
- S. Tsakovski, K. Benkhedda, E. Ivanova and F. C. Adams, Anal. Chim. Acta, 2002, 453, 143–154 CrossRef CAS.
- V. F. S. Pape, S. Tóth, A. Füredi, K. Szebényi, A. Lovrics, P. Szabó, M. Wiese and G. Szakács, Eur. J. Med. Chem., 2016, 117, 335–354 CrossRef CAS PubMed.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS PubMed.
- H. M. Irving, M. G. Miles and L. D. Pettit, Anal. Chim. Acta, 1967, 38, 475–488 CrossRef CAS.
- SCQuery, The IUPAC Stability Constants Database, Academic Software (Version 5.5), Royal Society of Chemistry, 1993–2005 Search PubMed.
- L. Zékány and I. Nagypál, in Computational Methods for the Determination of Stability Constants, ed. D. L. Leggett, Plenum Press, New York, 1985, pp. 291–353 Search PubMed.
- É. A. Enyedy, D. Hollender and T. Kiss, J. Pharm. Biomed. Anal., 2011, 54, 1073–1081 CrossRef PubMed.
- CrystalClear SM 1.4.0, Rigaku/MSC Inc., 2008 Search PubMed.
- M. C. Burla, R. Caliandro, B. Carrozzini, G. L. Cascarano, C. Cuocci, C. Giacovazzo, M. Mallamo, A. Mazzone and G. Polidori, J. Appl. Crystallogr., 2015, 48, 306–309 CrossRef CAS.
- SHELXL-2013 Program for Crystal Structure Solution, University of Göttingen, Germany, 2013 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van De Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef.
- S. P. Westrip, J. Appl. Crystallogr., 2010, 43, 920–925 CrossRef CAS.
- K. Juvale, V. F. S. Pape and M. Wiese, Bioorg. Med. Chem., 2012, 20, 346–355 CrossRef CAS PubMed.
- GraphPad Software I, GraphPad Prism, GraphPad Software, Inc., 2007, http://www.graphpad.com, (accessed February 2017) Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectroscopy, UV-Vis, EPR and X-ray diffraction data. CCDC 1530884. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt00439g |
| This journal is © The Royal Society of Chemistry 2017 |