DOI:
10.1039/C7DT00394C
(Paper)
Dalton Trans., 2017,
46, 4929-4942
DFT calculations as a ligand toolbox for the synthesis of active initiators for ROP of cyclic esters†
Received
1st February 2017
, Accepted 6th March 2017
First published on 7th March 2017
Abstract
New heteroleptic zinc dimeric complexes bearing an aminophenolate ligand of a single-site initiator framework were synthesized and characterized by spectroscopic methods, X-ray analysis, and DFT calculations. The theoretical study, verified by the experimental data, explains the catalytic behaviour in the ROP of lactide in the examined zinc complexes. The presented simple DFT protocol constitutes a valuable method for the qualification of the ancillary ligand to rationally design new complexes to improve their catalytic activity.
Introduction
Recently, the synthesis of polymers with programmable by design properties – those suitable for various applications – have attracted significant attention. The most attractive polymers are those dedicated to nanomedicine, molecular engineering and bio-base active packages.1–5 Polylactide (PLA) is one of the advanced green polymers, recently commercialized in worldwide production, that has tremendous application potential in the fields of bio-interface materials for surgery, regenerative medicine, tissue engineering, pharmacy, biodegradable packaging and environmentally friendly polymers.6–10
The ring opening polymerization (ROP) of cyclic esters initiated by metal complexes is the most effective procurement method of PLA among the numerous synthetic procedures studied to date. The heteroleptic metal complexes of the general formula L–M–OR (where, L-ancillary ligand, M-metal centre, and OR-initiating group) are regarded as the best option to access PLA in a controlled and stereo-selective manner. Therefore, the synthesis of well-defined, single-site initiators based on these structural motifs has been the subject of intensively developed studies over the past decade.11–15 Excellent initiators have been obtained that were supported by di-, tri- and recently tetra-valent metal centres and coordinated by numerously ancillary ligand families.16–19 In the context of biomedical applications and environmentally friendly technologies, the complexes that contain biometals (Ca, Mg, and Zn) are still the most searched. However, the kinetic instability of these heteroleptic compounds requires precision in matching appropriate ancillary ligands with a metal centre to prevent a deactivation pathway or ligand redistribution reaction. Excellent initiators have been obtained using biometals coordinated by numerous ancillary ligands such as β-diketiminate,20–24 tris(pirazolyl)borate,25–29 and related N,O-donor ligands.29–45 However, most of them have been generated by in situ alcoholysis of alkyl LMR/(LMR)2 compounds.
In contrast, over the last few years, aminophenol ligands have been used in the construction of new single-site initiators. As a result of the prevailing trend, extensive attention was paid to this type of ancillary ligand, which caused the appearance of a significant and still growing database of alkyl aminophenolate metal complexes. However, for previously studied ligands, the set of well-defined aminophenolate complexes with single-site motifs is very limited. The single-site type zinc complexes with aminophenol ligands are known, but it is worth remembering that although the motif (LZnOR)2 seems simple, its synthesis is elaborate, which has been supported by only four documented (X-ray) compounds to date (Scheme 1). Another alternative, the binary catalytic systems based on the L2M/ROH combination, are also worth noticing because some of them might have the tendency to indicate selectivity towards polymerization or alcoholysis of lactides.41,42 Particularly, this feature is of interest in view of the growing need for PLA and other valuable materials, such as lactyl-lactate/lactate esters, which have applications in cosmetics, fragrances and food additives.
 |
| | Scheme 1 Examples of well-defined (CCDC data base) imino/aminophenolate zinc complexes.29,37,38,40 | |
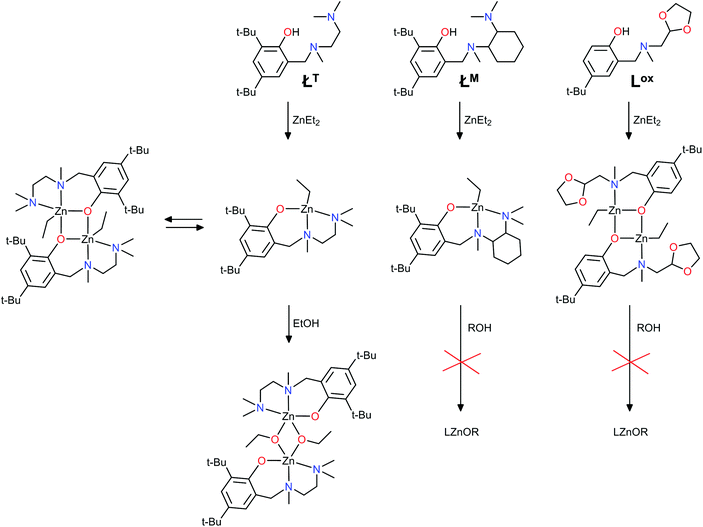
The synthesis of both (LZnEt)2 and L2Zn compounds is strongly connected with the coordination properties of aminophenolate ligands, and one of the most important factors is mutual interaction of substituents introduced to the amine arm and aryl core. Recently, we have explored the combination of functionalized aminophenol ligands with ZnEt2 for targeted hetero/homoleptic (LZnEt)2 complexes synthesis (Scheme 2).43,44 Our study extensively correlated the experimental outcomes with DFT calculations in rationalization of ancillary ligand design for zinc complexes with a programmable structural motif. Likewise, for these complexes, the modulation of the substituents located on the phenyl and amine arm permit the matching of the precise synthetic protocol for exactly one type of complex formation: hetero/homoleptic dimers or homoleptic monomers. Herein, we described the design of ancillary ligands for zinc complexes with the view of predicting their catalytic activity in the ROP of lactides using DFT calculations as a synthesis rating before its experimental verification.
 |
| | Scheme 2 Examples of ethyl zinc homo (left) and hetero (right) dimers. | |
Experimental
Materials and methods
All reactions and operations were performed under an inert atmosphere of N2 while using a glove-box (MBraun) or standard Schlenk apparatus and vacuum line techniques. The solvents were purified by standard methods: methanol, distilled from Mg; toluene, distilled from Na; CH2Cl2, distilled from P2O5; hexanes, distilled from Na; C6D6, distilled from CaH2. L-LA ((3S)-cis-3,6-dimethyl-1,4-dioxane-2,5-dione) (98%; Aldrich) was sublimed and recrystallized from toluene prior to use. Benzyl alcohol (Aldrich) was distilled under dry nitrogen gas and freeze/thaw degassed prior to use. ZnEt2 (1.0 M solution in hexanes), 4-tert-butylphenol, 2-methylaminomethyl-1,3-dioxolane (98%), and formaldehyde (37% solution in H2O) were purchased from Aldrich and used as received. (LRZnEt)2 was obtained according to the literature method.441H and 13C NMR spectra were detected over a temperature range from 233 to 333 K using Bruker ESP 300E or 500 MHz spectrometers. Chemical shifts are reported in parts per million and referenced to residual protons in deuterated solvents. Microanalyses were conducted with an ARL Model 3410 + ICP spectrometer (Fisons Instruments) and a VarioEL III CHNS (in-house).
Syntheses
N-[Methyl(2-hydroxy-5-tert-butylphenyl)]-N-methyl-N-methyl-1,3-dioxolaneamine (Lox-H).
To a stirred solution of 1.32 g (8.78 mmol) of 4-tert-butylphenol and 1.00 mL (8.78 mmol) of 2-methylaminomethyl-1,3-dioxolane in MeOH (50.00 mL), 0.90 mL (11.97 mmol) of formaldehyde (37% solution in H2O) was added at ambient temperature. The mixture was stirred, and the clear solution was heated under reflux for 24 h. The crude product was obtained as a colourless oil after evaporation of the solvent in vacuo. Next, the oil product was dissolved in hexanes, and after addition of cold MeOH, a white solid precipitated. It was collected by filtration, washed with cold MeOH and dried in vacuo to give Lox-H in 89% yield (2.18 g, 7.81 mmol). Anal. Calcd (Found) for C16H25NO3 (%): C, 68.79 (68.68); H, 9.02 (9.24); N, 5.01 (5.07); ESI/MS: 279.2 [M + 1]+; 1H NMR (500 MHz, CDCl3, RT): δ = 7.17 (dd, JHH = 8.4, 2.5 Hz, ArH, 1H), 6.95 (d, JHH = 2.4 Hz, ArH, 1H), 6.76 (d, JHH = 8.4 Hz, ArH, 1H), 5.05 (t, JHH = 4.1 Hz, O2CH–CH2–N, 1H), 4.06–3.81 (m, O–CH2–CH2–O, 4H), 3.78 (s, Ar–CH2–N, 2H), 2.73 (d, JHH = 4.1 Hz, O2CH–CH2–N, 2H), 2.38 (s, N–CH3, 3H), 1.27 (s, C(CH3)3, 9H); 13C{H1}NMR (126 MHz, CDCl3, RT): δ = 155.4 (ArC, 1C), 141.7 (ArC, 1C), 125.5 (ArCH, 1C), 125.4 (ArCH, 1C), 121.3 (ArCH, 1C), 115.6 (ArCH, 1C), 102.6 (O2CH–CH2–N, 1C), 65.0 (O–CH2–CH2–O, 2C), 62.5 (O2CH–CH2–N, 1C), 59.1 (Ar–CH2–N, 1C), 42.7 (N–CH3, 1C), 34.0 (C(CH3)3, 1C), 31.7 (C(CH3)3, 3C).
(LoxZnEt)2
.
The ligand Lox-H (0.56 g, 2.00 mmol) was dissolved in hexanes (30 mL) and stirred until the solution became clear. Next, ZnEt2 (2.00 mL, 2.00 mmol) was added dropwise at ambient temperature, and the solution was stirred until a white solid precipitated. The resulting suspension was filtered off, washed with hexanes (10 mL) and dried in vacuo. The resulting white solid was recrystallized from toluene at −15 °C to give colourless crystals of (LoxZnEt)2. Yield 91% (1.36 g, 1.82 mmol). Anal. Calcd (Found) for C36H58N2O6Zn2 (%): C, 57.99 (58.11); H, 7.84 (7.96); N, 3.76 (3.89); 1H NMR (500 MHz, C6D6, RT): δ = 7.39–7.35 (m, ArH, 2H), 7.32 (s, ArH, 2H), 7.03 (s, ArH, 2H), 5.32 (s, O2CH–CH2–N, 2H), 4.67 (s, Ar–CH2–N, 2H), 3.98 (s, O2CH–CH2–N, 2H), 3.51–3.17 (m, O–CH2–CH2–O, 8H), 2.88 (s, Ar–CH2–N, 2H), 2.53 (s, O2CH–CH2–N, 2H), 2.23 (s, N–CH3, 6H), 1.34 (t, JHH = 8.1 Hz, CH2CH3, 6H), 1.31 (s, C(CH3)3, 18H), 0.36 (q, JHH = 8.1 Hz, CH2CH3, 4H); 13C{H1}NMR (126 MHz, C6D6, RT): δ = 161.6 (ArC–O, 2C), 140.3 (ArC, 2C), 127.6 (ArCH, 4C), 124.4 (ArC, 2C), 120.4 (ArCH, 2C), 102.5 (O2CH–CH2–N, 2C), 65.4 (O–CH2–CH2–O, 2C), 64.3 (O–CH2–CH2–O, 2C), 63.8 (Ar–CH2–N, 2C), 62.6 (O2CH–CH2–N, 2C), 42.3 (N–CH3, 2C), 34.0 (C(CH3)3, 2C), 32.0 (C(CH3)3, 6C), 13.3 (CH2CH3, 2C), −2.3 (CH2CH3, 2C).
LRLoxZn2Et2
.
(LRZnEt)2 (0.78 g, 1.00 mmol) and (LoxZnEt)2 (0.75 g, 1.00 mmol) were dissolved in toluene (20 mL) at room temperature. The solution was stirred and concentrated in vacuo and then placed at −15 °C. After 24 h, LRLoxZn2Et2 was obtained as a white precipitate in an 84% yield (1.28 g, 1.68 mmol). Anal. Calcd (Found) for C40H60N2O4Zn2: C, 62.91 (62.80); H, 7.92 (8.09); N, 3.67 (3.69)%; 1H NMR (500 MHz, C6D6, RT): LR part: δ = 7.33 (dd, JHH = 8.5, 3.0 Hz, ArH, 1H), 7.24–7.11 (m, ArH, 5H), 7.05 (d, JHH = 8.6 Hz, ArH, 1H), 6.82 (d, JHH = 2.3 Hz, ArH, 1H), 5.03 (q, JHH = 6.8 Hz, N–CH–CH3, 1H), 4.79 (d, JHH = 11.9 Hz, Ar–CH2–N, 1H), 3.05 (d, JHH = 11.9 Hz, Ar–CH2–N, 1H), 1.97 (s, N–CH3, 3H), 1.68 (d, JHH = 7.0 Hz, N–CH–CH3, 1H), 1.46 (t, JHH = 8.1 Hz, CH2CH3, 3H), 1.14 (s, 1H), 0.46 (q, JHH = 8.1 Hz, CH2CH3, 2H); Lox part: δ = 7.35–7.31 (m, ArH, 1H), 7.29 (d, JHH = 8.1 Hz, ArH, 1H), 7.06 (d, JHH = 2.9 Hz, ArH, 1H), 5.31 (dd, JHH = 6.2, 2.3 Hz, O2CH–CH2–N, 1H), 4.72 (d, JHH = 12.1 Hz, Ar–CH2–N, 1H), 4.00 (dd, JHH = 12.8, 1.8 Hz, O2CH–CH2–N, 1H), 3.53–3.40 (m, O–CH2–CH2–O, 2H), 3.33–3.18 (m, O–CH2–CH2–O, 2H), 2.91 (d, JHH = 12.1 Hz, Ar–CH2–N, 1H), 2.53 (dd, JHH = 12.7, 6.3 Hz, O2CH–CH2–N, 1H), 2.24 (s, N–CH3, 3H), 1.32 (s, C(CH3)3, 9H), 1.31 (t, JHH = 7.9 Hz, CH2CH3, 3H), 0.32 (q, JHH = 8.1 Hz, CH2CH3, 2H); 13C NMR (126 MHz, C6D6, RT): δ = LR part: 161.5 (ArC–O, 1C), 140.5 (ArC–C, 1C), 136.3 (ArC–CH, 1C), 130.3 (ArCH, 1C), 128.5 (ArCH, 4C), 128.4 (ArCH, 1C), 127.6 (ArCH, 1C), 124.7 (ArC–CH2, 1C), 120.1 (ArCH, 1C), 64.5 (N–CH–Ar, 1C), 61.5 (N–CH2–Ar, 1C), 35.1 (N–CH3, 1C), 33.9 (C(CH3)3, 1C), 31.8 (C(CH3)3, 3C), 19.9 (CH3–CH, 2C), 13.4 (CH2–CH3, 1C), −2.1 (CH2–CH3, 1C); Lox part δ = 161.5 (ArC–O, 1C), 140.4 (ArC, 1C), 128.7 (ArCH, 1C), 127.4 (ArCH, 1C), 124.5 (ArC, 1C), 120.2 (ArCH, 1C), 102.5 (O2CH–CH2–N, 1C), 65.4 (O–CH2–CH2–O, 1C), 64.3 (O–CH2–CH2–O, 1C), 64.0 (Ar–CH2–N, 1C), 62.7 (O2CH–CH2–N, 1C), 42.3 (N–CH3, 1C), 34.0 (C(CH3)3, 1C), 32.0 (C(CH3)3, 3C), 13.6 (CH2CH3, 1C), −1.7 (CH2CH3, 1C).
Representative procedure for solution polymerization
In a typical polymerization experiment, the solution of an initiator (LoxZnEt)2 dissolved in CH2Cl2 was placed in a Schlenk flask, and external alcohol in a stoichiometric amount (LoxZnEt)2/ROH = 0.5/1 (ROH = MeOH, BzOH) was added (0.35 mmol of (LoxZnEt)2 and 0.70 mmol of ROH). Then, after 10 minutes, the monomer L-LA (7.00 mmol) was added at a fixed molar ratio (LoxZnEt)2/L-LA/ROH = 0.5/n/1; n = 20, 50, 100. The resulting solution was stirred at room temperature for a prescribed time until the reaction was completed. At certain time intervals (ca. 10 min), about 1 mL aliquots were removed, precipitated with hexanes and dried in vacuo. The conversion was determined by observing the 1H NMR resonances of the polymer and monomer after dissolving the precipitates in C6D6. When the reaction was completed, an excess of hexanes was added to the reaction mixture. Filtration and vacuum drying yielded a white polymer. The resulting solid was dissolved in CH2Cl2, and the polymer was precipitated with an excess of cold methanol. The polymer was collected by filtration, washed with methanol to remove the non-reacted monomer, and dried under reduced pressure. The reaction mixtures were prepared in a glove-box, and subsequent operations were performed using standard Schlenk techniques.
Crystallography
X-ray diffraction data for (LoxZnEt)2 and (LRŁcyZn2Et2) crystals were collected on a Xcalibur diffractometer with a Sapphire2 detector (MoKα radiation; λ = 0.71073 Å) or a Xcalibur diffractometer with an Onyx detector (see ESI†) with a ω scan technique at 100 K. The data collection and processing utilized the CrysAlis suit of programs.46 Space groups were determined based on systematic absences and intensity statistics. Lorentz, polarization and analytical absorption corrections were applied. The structure was solved by direct methods and refined by full-matrix least-squares on F2. All calculations were performed using the SHELXTL-2013 suite of programs.47 All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were placed at their calculated positions. Before the last cycle of refinement, all H atoms were fixed and were allowed to ride on their parent atoms. Thermal ellipsoid plots were prepared with 50% probability displacements for non-hydrogen atoms using the Mercury 3.1 program.48 All data have been deposited with the Cambridge Crystallographic Data Centre CCDC 1511741 for (LoxZnEt)2 and 1511742 for LRŁcyZn2Et2.
Computational details
All density functional theory (DFT) calculations were performed with the Gaussian 03 program suite.49 The geometries of the complexes and ligands were optimized (Fig. S13–20, Tables S2–9 in ESI†) using B3LYP density functional theory and the 6-31G** basis sets, implemented in Gaussian 03 on all atoms.50,51 The starting geometries of the complexes (LoxZnEt)2 and (Łox)2Zn were generated from their crystal structures. Structures of all isomers of the calculated complexes were first optimized in vacuo.
Results and discussion
The well-defined aminophenolate zinc complexes with a single-site motif determined by X-ray analysis are limited to merely a few examples.29,37,38,40 All of them possess a di-tert-butyl phenol core in the ancillary ligands, and their coordination properties are controlled by backbone variations on nitrogen atoms. The modification of the aryl core or amine substituents with sterically bulky groups has been routinely used to restrict the aggregation of metal aryloxides formed during an alcoholysis. Recently, we published two new zinc examples with aminonaphtholate ancillary ligands, and our results indicated that a sizable group next to the hydroxyl of the aminophenol/aminonaphthol ligands is the reason for the mixture of metal complexes forming.45 The nuances in the synthetic procedure can be used to create a complex of planned structural motif. Thereby, the design of new periphery groups, according to these requirements, constitutes a continuously interesting synthetic strategy.
DFT assisted implications for complex design
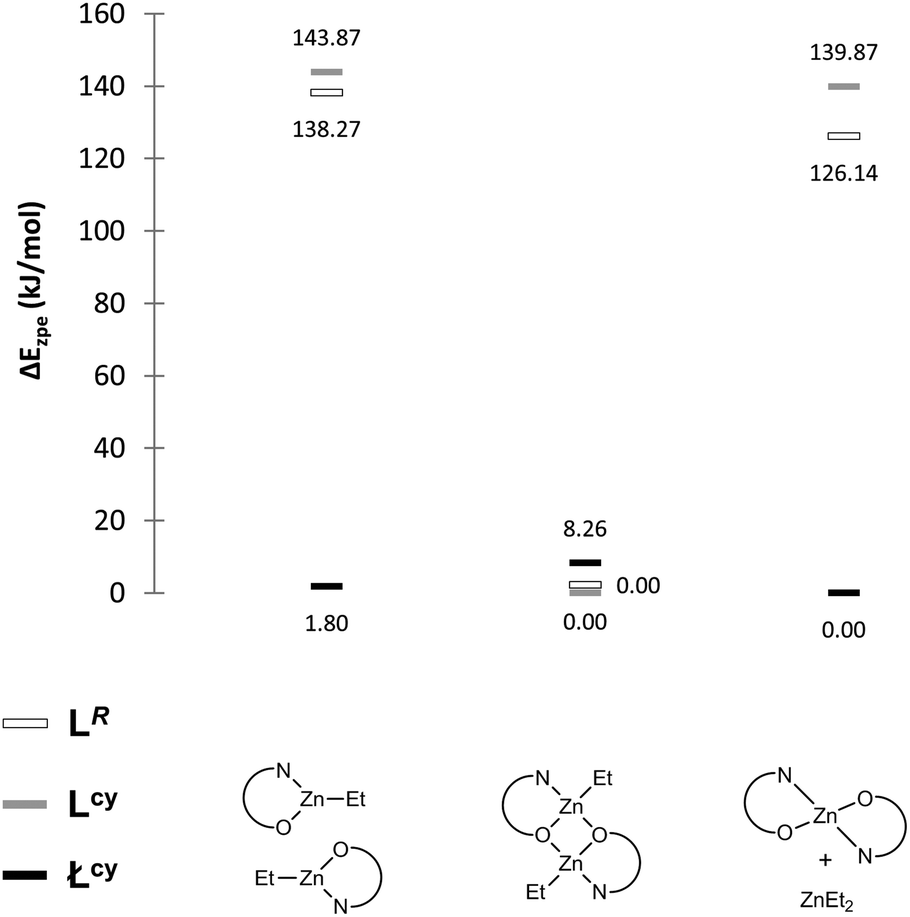
Recently, the family of stable aminophenolate zinc dimers and homoleptic monomers have been synthesized in our group (Scheme 3).33,41,43,44 Both the experimental data and DFT calculations indicate that aminophenol ligands (LR/cy with free ortho-positions of phenol ring) prefer a dimeric alkylzinc compound formation. This dimeric structure is retained in the solution, and this dependence is evidenced by DFT calculations that show that the energy for the monomeric forms (LZnEt or L2Zn) of the zinc complexes with ortho-free ligands Lcy and LR is evidently higher than for the dimers (LZnEt)2 (Scheme 3, left side and Fig. 1, grey and white bars). Instead, for analogous ancillary ligands (Łcy, the same substituent on N-donor atom as for L), the presence of the bulky tert-butyl groups in the ortho-position leads to homoleptic Ł2Zn bis-chelate complex crystallization (Scheme 3, right side). Despite that only the homoleptic monomer precipitated from the reaction mixture, the calculated small differences between the energy of the potential monomers, Ł2Zn, ZnEt2, ŁZnEt compared to the dimers syn/anti(ŁZnEt)2, explain their potential coexistence in the solution (Fig. 1, black bars).
 |
| | Scheme 3 Synthetic procedure for aminophenolate zinc complexes. Blue arrow – favourable synthesis; (A) favourable heteroleptic dimers formation; (B) favourable homoleptic monomers formation. L-para-tert-Butylphenyl core, Ł-ortho,para-di-tert-butylphenyl core, R,cy, ox substituents on amine arm. | |
 |
| | Fig. 1 The calculated relative energies in the gas-phase for the geometrically optimized zinc compounds. | |
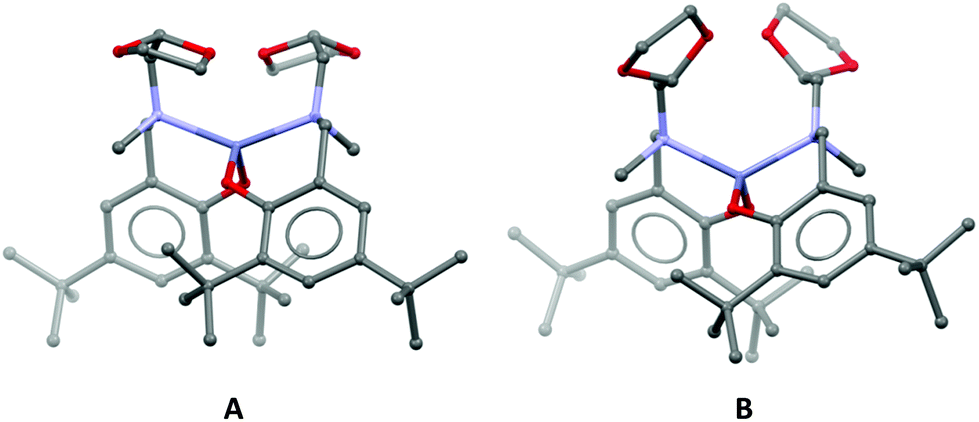
The homoleptic complex (Łox)2Zn containing a hemilable amino-arm with a dioxolane ring has been subjected to our previous structural study by X-ray analysis and DFT calculations.41 This complex is flexible, and during the optimization, we obtained nineteen structures. Moreover, one of them, possessing a “fully open” structure (both oxolane arms pendant), is dominant at higher temperatures, and this isomer forms in the presence of an alcohol an active catalytic system for the ROP of lactides (structure B in Fig. 2). Instead, at room temperature, a mixture of the “closed” and “half-opened” (with one pendant oxolane arm) structures are present.
 |
| | Fig. 2 Isomers of (Łox)2Zn: (A) inactive in ROP and (B) active form. | |
However, at the time, we supposed that the heteroleptic species (ŁoxZnEt)2/ŁoxZnEt were absent in the solution because in the reaction between ZnEt2 and Łox-H, the homoleptic monomer (Łox)2Zn was the only isolated product. Now, we have verified this result, and the potential heteroleptic dimers (ŁoxZnEt)2 and appropriate monomers have been investigated by DFT calculations (see details in the Experimental section and ESI†). The results and the optimized geometrical structures are presented in Fig. 3 (red bars, DFT optimized structures see ESI†). The energies of the generated heteroleptic dimers ΔE(ŁoxZnEt)2, heteroleptic monomers ΔE(ŁoxZnEt), and homoleptic species ΔE[(Łox)2Zn + ZnEt2] are similar, especially for “closed” and “opened” isomers in adequate couples of homomonomers (50.29 and 49.22 kJ mol−1) and heterodimers (8.73 and 0.00 kJ mol−1), as shown in Fig. 3. These additional new theoretical data explain the predictable coexistence of all possible homo/heteroleptic compounds in the reaction mixture due to the low energetic cost of the transformation between them. Then, precipitation/crystallization of e.g. homoleptic (Łox)2Zn may be enough to move the reaction equilibrium towards formation of solely one product of the Łox-H and ZnEt2 reaction. Likely, it may be the reason that the crystallization of “single-site” type zinc complexes is difficult for aminophenolate complexes bearing ligands with bulky substituents in the ortho-position of phenol.
 |
| | Fig. 3 The relative calculated energies in the gas-phase for the geometrically optimized heteroleptic dimers (LZnEt)2, couple of homoleptic monomers (L)2Zn with ZnEt2 and monomers LZnEt; (L = Lox, Łox). Below schematic structures of possible complexes with the ancillary ligand Lox, Łox (open and closed forms). | |
Relying on our previously published results,43 the exclusive formation of a heteroleptic zinc complex should be possible for an analogue ligand with a free ortho-position Lox-H. Therefore, to verify this assumption, we performed gas-phase geometrical optimization of the new dimeric complex (LoxZnEt)2 by means of DFT calculations. The relative energies ΔEZPE calculated for the optimized conformers are presented in Table S1–S5 in the ESI† and in Fig. 3 (green bars). Additionally, the potential monomeric forms of the zinc complex with the Lox ligand have been investigated. The relative energy values for the monomer LoxZnEt and homoleptic species (Lox)2Zn with ZnEt2 are in this case higher than that for the preferable heteroleptic dimer (LoxZnEt)2. These theoretical studies indicated that the heteroleptic dimer (LoxZnEt)2 should be the most stable species in the reaction mixture (both isomers in closed and opened forms), so we decided to verify the theoretical outcome by experimental tests.
Synthesis and characterization of compounds
The new aminophenolate proligand Lox-H was prepared by a standard one-pot Mannich condensation between para-substituted phenol, formaldehyde and 2-methylaminomethyl-1,3-dioxolane in methanol in a similar way as described in previous literature.43 The proligand was obtained in high yields (91%) and was characterized by standard elemental analysis, MS and spectroscopic methods. The 1H and 13C {1H} NMR studies gave well-resolved resonances for all proton and carbon environments, indicating the high purity of the isolated compound (ESI, Fig. S1 and S2†). Next, the Lox-H was used for zinc complex synthesis in the reaction with ZnEt2 in hexanes at room temperature (Scheme 4).
 |
| | Scheme 4 Synthesis of a zinc complex with the aminophenolate ligand Lox-H. | |
In the first attempt, stoichiometric amounts of Lox-H and ZnEt2 (1/1) were used for the isolation of the molecular heteroleptic zinc complex (LoxZnEt)2. Next, we tried to obtain a homoleptic zinc compound (Lox)2Zn, however, it appeared impossible, both with the use of an appropriate molar ratio of reagents (Lox-H/ZnEt2 = 2/1) and when reactions were carried out in an excess of Lox-H proligand. This result correlated strongly with the presented theoretical prediction, which shows that the dimeric complex is the most favourable structural motif.
The obtained dimer (LoxZnEt)2 is well-soluble in toluene, thf, CH2Cl2 and insoluble in aliphatic hydrocarbons. The complex was characterized by 1H and 13C NMR spectroscopy and X-ray crystallography. The distinctive 1H NMR signals of (LoxZnEt)2 include broad singlets (methylene backbone Ar–CH2–N, CH–CH2–N and methine protons –CH at 4.67, 2.88; 3.98, 2.53 and 5.32 ppm, respectively) (Fig. 4 and ESI†). The spectrum includes typical features of (LoxZnEt)2: singlets at 2.23 ppm (N–CH3) and 1.31 (t-Bu), multiplets for oxolane –CH2– protons at 3.51–3.17 ppm and a group of aromatic protons: multiplet at 7.39–7.35 ppm and singlets at 7.32 and 7.03 ppm. The 1H NMR spectrum also shows a triplet at 1.34 and a quartet at 0.36 belonging to Zn–CH2–CH3; the triplet is obscured by the singlet of the tert-butyl protons. Broad singlets form appropriate doublets and multiplets in the 1H NMR spectrum obtained at 223 K (Fig. 4B). All signals are distinguished in a COSY spectrum (see ESI†).
 |
| | Fig. 4 Fragment of 1H NMR spectrum of (LoxZnEt)2 at: (a) 298 K and (b) 223 K. | |
The dimeric structure of (LoxZnEt)2 in solution was confirmed by DOSY NMR. The estimated translation diffusion coefficient Dexp in deuterated benzene was 6.54 × 10−10 m2 s−1 (calculated DSESf = 6.64 × 10−10 m2 s−1 for DFT optimised structure). In order to compare the structure in the solid state and in solution, the adequate diffusion coefficients were estimated from the solid state structure determined by X-ray crystallography and DFT study. The obtained values for the diffusion coefficient of (LoxZnEt)2 estimated by experimental X-ray, NMR and theoretical study were comparable.
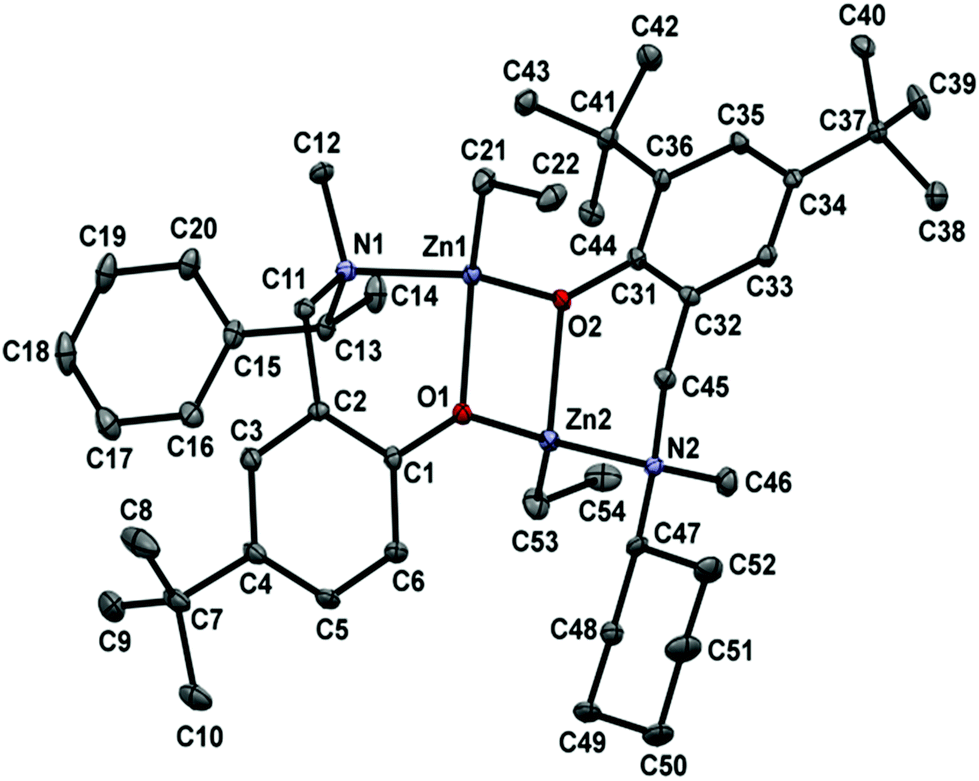
X-ray study of (LoxZnEt)2
The colourless crystals of (LoxZnEt)2, suitable for X-ray analysis, were grown by slow evaporation of a toluene solution. The solid state structure of (LZnEt)2 was determined as outlined in Table 1 and Fig. 5.
 |
| | Fig. 5 The molecular structure of (LoxZnEt)2. The thermal ellipsoids are drawn at the 30% probability level. H atoms are omitted for the sake of clarity. | |
Table 1 Selected bond distances (Å) and angles (°) for (LoxZnEt)2
| Atoms |
(LoxZnEt)2 (X-ray) |
(LoxZnEt)2 (DFT) |
| i = −x + 1, −y + 1, −z + 1. Values in parentheses refer to the non-bonding interactions. |
|
Bond distance [Å]
|
| Zn–C17 |
1.980(2) |
1.9813 |
| Zn1–O1 |
2.009(2) |
2.0234 |
| Zn1–O1′i |
2.093(2) |
2.0843 |
| Zn1–N1 |
2.156(2) |
2.1770 |
| Zn1–Zn1i,a |
3.111(2) |
3.1469 |
| O1–O1i,a |
2.675(3) |
2.6404 |
| Zn1–O2i,a |
2.779(2) |
2.7381 |
|
Angels [°]
|
| C17–Zn1–O1 |
128.92(8) |
130.172 |
| C17–Zn1–O1i |
115.98(9) |
119.636 |
| O1–Zn1–O1i |
81.39(7) |
79.986 |
| C17–Zn1–N1i |
127.25(9) |
122.980 |
| O1–Zn1–N1i |
97.33(7) |
99.382 |
| O1i–Zn1–N1i |
91.85(7) |
91.600 |
| Zn1–O1–Zn1i |
98.61(7) |
100.005 |
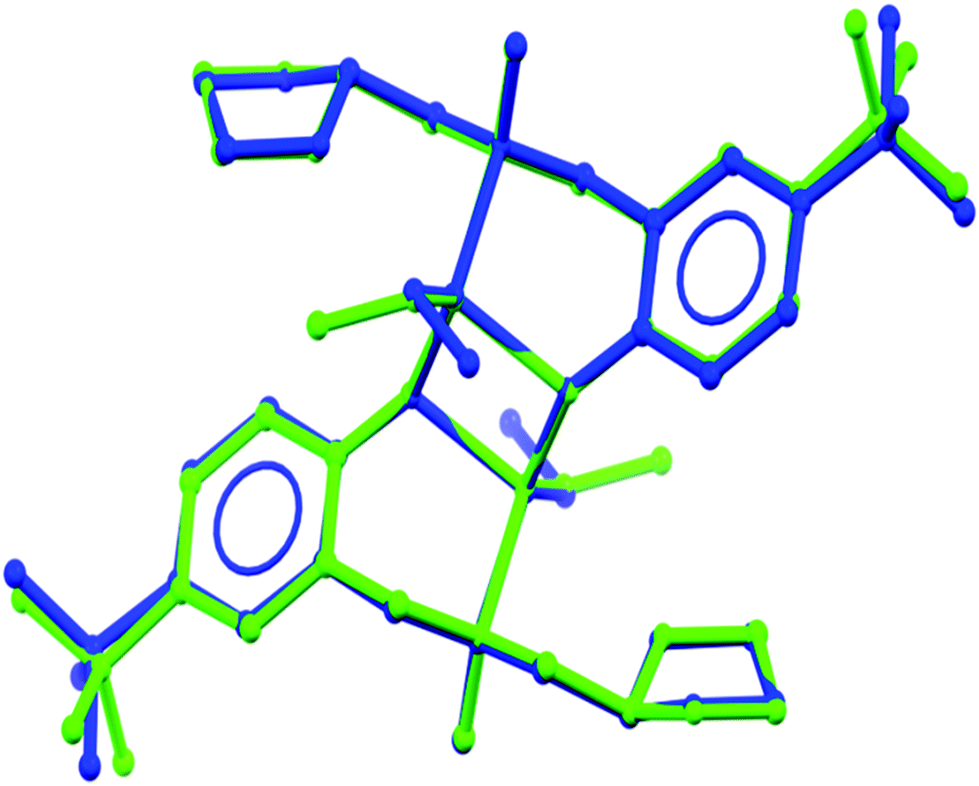
The X-ray single-crystal structure of (LoxZnEt)2 showed dimeric species with tetrahedral zinc centres bridged via phenyl oxygen atoms with ethyl groups located trans to each other. The nitrogen and phenyl oxygen atoms of the aminophenolate ligand form a six-membered metalacycle with a twisted, boat-shaped conformation, but the ether oxygen atoms of the oxolane rings failed to chelate, leaving ethereal functionality free in the crystal. All the bond distances between zinc and O, N, C atoms are typical and range: Zn–O, 2.014(3) and 2.095(3); Zn–N, 2.160(3); Zn–C, 1.976(4) Å. The distances and angles are all comparable with bond lengths and angles observed for similar complexes described in the literature.37,38,43,44 The geometric parameters for (LoxZnEt)2 obtained by DFT calculations were compared with the experimental data (Table 1). The agreement between the optimized geometric structure and the experimental data from the X-ray analysis is fully satisfactory, and the only essential difference may be the position of the ethyl group bonding to the zinc centres (Fig. 6). As it can be seen, the lowest ΔE[(LoxZnEt)2] value found for the optimized isomer (LoxZnEt)2 (see Fig. 3) has been adequate for a solid-state structure, so the experimental data confirm the theoretical studies, indicating that the heteroleptic dimer is the most stable species in the reaction mixture.
 |
| | Fig. 6 Superimposed crystallographic (green) and DFT calculated (blue) structures of (LoxZnEt)2. | |
As already mentioned, the most common single-site initiators for the ROP process are heteroleptic metal alkoxides (sometimes formed in situ by the reaction of an appropriate alcohol and alkylmetal complex, for example LZnEt). The (LoxZnEt)2 dimer possesses all these structural features; therefore, after the in situ alcoholysis it was used as an initiator for the ROP of lactides. Unfortunately, the ROP was slow even at an elevated temperature (60 °C), and (LoxZnEt)2 polymerized 20 equiv. of L-LA in the presence of MeOH or benzyl alcohol (as an external alcohol) in 5 days. The poor polymerization result excluded (LoxZnEt)2 from the cluster of interesting initiators for the ROP of lactides. Therefore, the focus of our attention has now been shifted towards the verification of this unexpectedly low catalytic reactivity.
The well-defined, confirmed by X-ray analysis aminophenolate complexes of the single-site motif are rare, but one of the most active in the ROP is Hillmyer's and Tolman's complex (ŁTZnOEt)2, shown in Scheme 5. It was synthesized via the reaction of the monomer ŁTZnEt with ethanol at room temperature.29 Surprisingly, the chiral ethyl zinc analogue ŁMZnEt, obtained by Mehrkhodavandi,34 was inert towards alcohols with the use of ethanol, methanol, isopropanol, and this complex reacted only with phenol or hydrochloric acid (Scheme 5). The reactivity/inertness of the mentioned complexes may be explained by the lability (dynamic behaviour)/stability of the amine functionality of the ligand, which is crucial during alcoholysis. Hence, we verified this literature data by DFT. The relative calculated energies for the model zinc complexes obtained for Tolman and Mehrkhodavandi initiators are presented in Fig. 7 (for details see, ESI†). Although the heteroleptic monomeric species ŁT/MZnEt are preferred for both examples, the “open structures” facilitating molecular fitting between reagents are energetically disfavoured for ŁMZnEt, which confirms the difficulties during alcoholysis. The large energy difference between “open” and “closed” monomers for ŁTZnEt (0.00 vs. 101.01 kJ mol−1) and the relatively low energy cost for the formation of an open dimer (ŁTZnEt)2 may indicate more advanced but effective transformation pathways between them. In all the considered zinc complexes, the theoretical study significantly confirms the experimental data. This indicates that complexes that are easily transformable coexisted in solution, and homo/heteroleptic species containing a flexible amine arm, may act as a potential precatalysts in ROP. The presented zinc complex with the aminophenolate ligand Łox additionally confirms this phenomenon.
 |
| | Scheme 5 Syntheses of “single-site” type initiators. | |
 |
| | Fig. 7 The relative calculated energies in the gas-phase for the geometrically optimized heteroleptic dimers (LZnEt)2, couple of homoleptic monomers (L)2Zn with ZnEt2 and monomers LZnEt; ŁT – Tolman's ligand – yellow line; ŁM – Mehrkhodavandi ligand – violet line. | |
Nevertheless, the dimer (LoxZnEt)2 is unreactive towards stoichiometric amounts of methanol and alcoholysis according to the procedure published by us recently.42 To clarify the easiness/difficulties in the coordination of alcohols to the discussed zinc dimers, in the next stage, we optimized the structures of these species with methanol (Scheme 6). For dimers with our ancillary ligands (LR, Lcy), two coordination modes are possible to delineate. A similar proposition is clear for Tolman's complex, and all of them are able to easily undergo further intramolecular alcoholysis via a reaction between the coordinated ligands (ROH and Et). However, the coordination of methanol for complexes with Lox or LM is difficult to verify by DFT calculations because generation of appropriate adducts requires decoordination of nitrogen atoms. Whereas, the experimental data indicated that it is impossible. These calculations may serve as direct support for the prediction of the catalytic activity of the related complexes. The energy profile of the intermediates for methoxy-zinc species accessible during alcoholysis is shown in Fig. 8. The calculations for corresponding structural motifs indicated that complexes with flexible ancillary ligands easily undergo alcoholysis and form appropriate alkoxy species.
 |
| | Scheme 6 Structures of LZnEt/(LZnEt)2 species in the presence of an alcohol molecule (schematic on the left, DFT optimized on right), green circle means easy coordination, red circle – difficult/impossible. | |
 |
| | Fig. 8 The relative calculated energies in the gas-phase for the geometrically optimized zinc species potentially formed during methanolysis of LZnEt/(LZnEt)2. ŁT Tolman ligand – yellow line; Lcy – grey line. | |
The simple modification of the ancillary ligand by expurgation of the tert-butyl group gave an excellent structural motif, agreeable with the single-site precursor but insignificantly active towards the ROP of lactides. Here, this conclusion was also verified by theoretical study. Additionally, DFT data gave the leads for modification of the complex (synthesized or calculated) for improved catalytic activity. For example, the last chance for our dimer (LoxZnEt)2 to be an active catalyst could be its dynamic behaviour in solution, but the same stable dimeric structure, as in the solid state, is retained after dissolution. Therefore, “the last bastion of hope” for the synthesis of an “active in ROP” complex based on the Lox ligand was the use of the recently described by us “zebra reaction”, which is a useful procedure for heterodimeric zinc complex formation, as shown in Fig. 9. This procedure is likely supported by the presented DFT calculations indicating the possibility of a potential reaction between two homodimers; one of them is a zinc complex with the chiral ligand LR.44 This complex (LRZnEt)2 well matches the unreactive (LoxZnEt)2 because it is the most dynamic in the solution homodimer from the family of our zinc complexes, which can easily recognize (LoxZnEt)2 by the coordination fitting code. The selective formation of a new heterodimer made by DFT inspiration is presented in Fig. 9.
 |
| | Fig. 9 Synthesis of heterodimeric zinc complex (LRLoxZn2Et2) using the zebra reaction protocol. | |
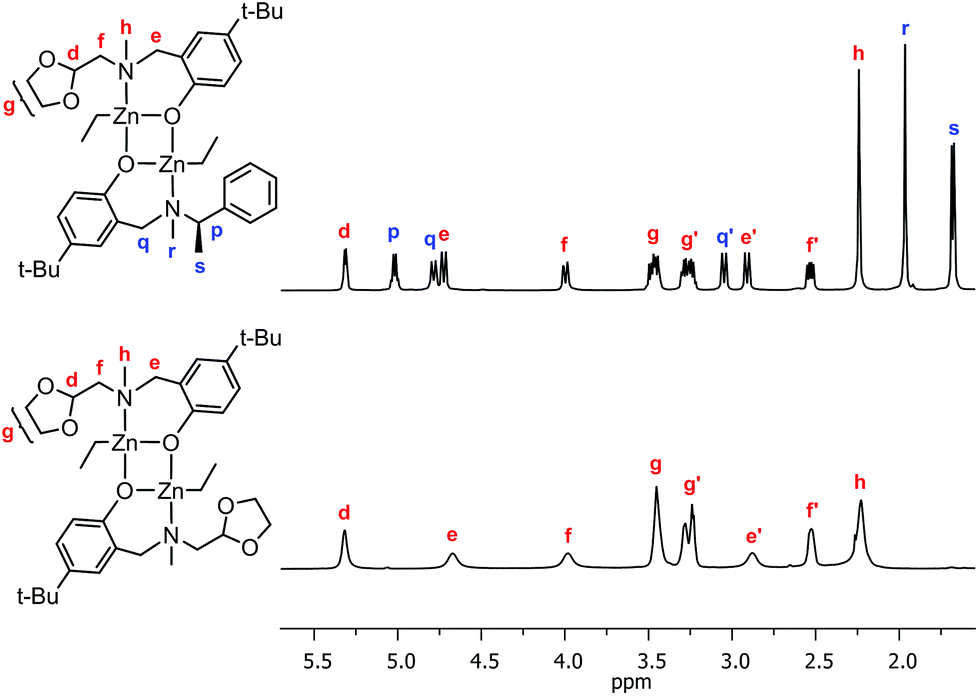
The old (LRZnEt)2 and new (LoxZnEt)2 homodimers reacted readily to form the corresponding zinc heterodimer (LRLoxZn2Et2), and although the X-ray analysis is not available, this complex presents clear NMR spectra confirming the expected structural motif (Fig. 10 and ESI†).
 |
| | Fig. 10 The fragment of 1H NMR spectrum of the substrate and product of the zebra reaction. | |
The polymerization test for the new heterodimer LRLoxZn2Et2 was positive. The initiator formed during an alcoholysis reaction between LRLoxZn2Et2 and methanol polymerized 100 equiv. of L-LA yielding PLA with a narrow PDI = 1.14. The molecular weights of the PLAs were higher than expected for PLA-100 based on the initial monomer-to-catalyst ratio (Table 2, entry 5). The divergence from the anticipated molecular weights (in this example PLA-185 according to Mn determined by GPC) is not accidental, but it may be explained by one effective metal centre that is active during ROP. The fragment LoxZnEt plays the role of the inactive ancillary “metalloligand” stabilizing the working ROP second fragment LRZnEt. What is worth emphasizing is that the homodimer with the ancillary ligand LR is not so precise (PLA-100, PDI = 1.29 and 1.34 for LS respectively; Table 2, entry 1–2).
Table 2 ROP of LA initiated by zinc complexes [I]
| Entry/[I] |
ROH |
[I]/[LA]/ROH |
Time [min] |
C
[%] |
103Mn,cal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
103Mnc |
PDId |
| Reaction conditions: CH2Cl2; T = 25 °C. Obtained from 1H NMR. Calculated from Mn,cal = [L-LA]0/[ROH]0 × C × 144.13 + MROH. Determined by GPC calibrated versus polystyrene standards and corrected by a factor of 0.58 according to literature recommendations.52 Obtained from GPC. rac-LA was used to give atactic PLA (Pi = 0.48 determined by 1H {1H} NMR). |
| 1/(LRZnEt)2 |
MeOH |
0.5/100/1 |
80 |
99.8 |
14.43 |
14.12 |
1.29 |
| 2/(LSZnEt)2 |
MeOH |
0.5/100/1 |
80 |
99.9 |
14.43 |
13.87 |
1.34 |
| 3/(LoxZnEt)2 |
MeOH |
0.5/100/1 |
5 d |
20.2 |
2.92 |
— |
— |
| 4/LRLoxZn2Et2 |
MeOH |
0.5/50/1 |
80 |
99.9 |
7.23 |
11.90 |
1.14 |
| 5/LRLoxZn2Et2 |
MeOH |
0.5/100/1 |
240 |
98.2 |
14.19 |
26.55 |
1.14 |
| 6/LRLoxZn2Et2e |
BnOH |
0.5/100/1 |
220 |
99.2 |
14.41 |
22.06 |
1.18 |
| 7/LRLoxZn2Et2 |
BnOH |
0.5/150/1 |
360 |
99.8 |
21.73 |
33.15 |
1.17 |
It is worth noting that the idea of this reaction and the method for activation of the zinc complex are possible by DFT selection. The optimized structure of the (LRLoxZn2Et2) complex with the methanol molecule confirms the experimental results (Fig. 11 and Scheme 7).
 |
| | Fig. 11 The structures of (LRLoxZn2Et2) complex in the presence of MeOH molecules (schematic on left, DFt optimized on right). | |
 |
| | Scheme 7 Schematic for the first cycle in the ROP of lactide initiated by zinc dimer (LRLoxZn2Et2). I – active species, II – coordination of lactide, III – insertion, IV – ring opening, V – decoordination of lactyl-lactate ester group. | |
Based on the successful synthesis of the (LRLoxZn2Et2) complex, we tried to design another heterodimer from theoretical studies. We would like to freeze by “zebra reaction” the heteroleptic motif ŁcyZnEt, which is impossible to isolate by simple synthetic strategy (Fig. 1 and Scheme 2). The reaction between ZnEt2 and Łcy-H gave a mixture of complexes in the parent toluene solution (all coexisted complexes are well soluble), which clearly transformed to one homoleptic (Łcy)2Zn in hexanes. Therefore, we performed the “zebra reaction” between the chiral homodimer (LRZnEt)2 and in situ prepared a mixture of ZnEt2 and Łcy-H. In this case, the active homodimer was recognized by the well-tailored reagent (ŁZnEt)2, and after several hours, the expected crystals of (LRŁcyZn2Et2) appeared (Fig. 12 and 13, for X-ray structure see ESI†).
 |
| | Fig. 12 Synthesis of heterodimeric zinc complex LRŁcyZn2Et2 using a “zebra reaction” strategy. | |
 |
| | Fig. 13 The molecular structure of (LRŁcyZn2Et2). The thermal ellipsoids are drawn at a 30% probability level. H atoms are excluded for the sake of clarity. | |
The structure of the new unique heterodimer (LRŁcyZn2Et2) has been confirmed by X-ray analysis (Fig. 13); however, dissolution of the crystals in toluene resulted in a mixture of dimer (LRZnEt)2 and monomer (Łcy)2Zn. This data proves that heteroleptic complexes with the Łcy ligand are really present in the reaction mixture. Superimposed solid-state structures of (LRŁcyZn2Et2) and previously published (LRLcyZn2Et2)44 show that the interposition of the sizable tert-butyl hindrance in the ortho-position of the phenol core provoked drift in the methylbenzyl substituent located at the nitrogen atom (Fig. 14, Table 3). This behavior may be the reason for the instability in the solution of the heterodimeric zinc complex (LRŁcyZn2Et2).
 |
| | Fig. 14 Superimposed molecular structures of (LRŁcyZn2Et2) (red) and (LRLcyZn2Et2)44 (blue). H atoms are omitted for the sake of clarity. | |
Table 3 Selected bond distances (Å) and angles (°) for molecular structures of (LRLcyZnEt)2 and (LRŁcyZnEt)2
| Atoms |
LRLcyZn2Et2a |
LRŁcyZn2Et2
|
|
Previously published.44
C53 = C49 for LRLcyZn2Et2.
Values in parentheses refer to the non-bonding interactions.
|
|
Bond distance [Å]
|
| Zn1–C21 |
1.990(6) |
1.977(6) |
| Zn2–C53b |
2.002(6) |
1.993(7) |
| Zn1–O1 |
2.033(3) |
2.017(4) |
| Zn2–O2 |
2.029(3) |
2.044(4) |
| Zn1–O2 |
2.046(4) |
2.025(4) |
| Zn2–O1 |
2.038(4) |
2.055(4) |
| Zn1–N1 |
2.164(4) |
2.194(5) |
| Zn2–N2 |
2.151(4) |
2.147(5) |
| Zn1–Zn2c |
3.0581(12) |
3.0592(10) |
| O1–O2c |
2.690(4) |
2.683(4) |
|
Angels [°]
|
| C21–Zn1–O1 |
126.3(2) |
132.9(2) |
| C53b–Zn2–O2 |
125.57(19) |
119.8(2) |
| C21–Zn1–O2 |
126.04(18) |
122.4(2) |
| C53b–Zn2–O1 |
120.43(19) |
129.3(2) |
| O1–Zn1–O2 |
82.52(14) |
83.18(17) |
| O1–Zn2–O2 |
82.83(14) |
81.79(17) |
| C21–Zn1–N1 |
122.23(18) |
110.4(2) |
| C53b–Zn2–N2 |
123.2(2) |
121.6(2) |
| O1–Zn1–N1 |
94.04(14) |
87.62(15) |
| O2–Zn2–N2 |
93.27(14) |
94.26(14) |
| O2–Zn1–N1 |
95.02(14) |
114.76(15) |
| O1–Zn2–N2 |
102.08(15) |
99.28(15) |
| Zn1–O1–Zn2 |
97.37(13) |
97.40(13) |
| Zn1–O2–Zn2 |
97.24(14) |
97.50(13) |
Conclusions
A successful, based on DFT calculations, synthetic strategy has been developed for the synthesis of different aminophenolate zinc complexes. We have presented here a new heteroleptic (LoxZnEt)2 complex, which showed low catalytic activity in the ROP of lactides due to the stable dimeric structure and flexibility of an oxolane arm efficiently shielding the active metal centre during alcoholysis. The research showed that a greater steric hindrance of the aminophenolate ligand located on the aryloxo fragment results in homoleptic (Łox)2Zn complex formation, which was determined in the solid state as the most stable species among the numerously possible homo- and heteroleptic isomers. The theoretical studies, which were verified by the experimental data, suggested that the lability of the dioxolane fragment is essential to ensure a suitable structure of an active centre for zinc. The subtle changes observed in the calculated structures of the complexes allowed for the rational selection of substituents of the designed ligands in order to improve the active ROP initiators. The detailed analysis of the DFT data has been the inspiration for the synthesis of two heterodimers, (LRLoxZn2Et2) and (LRŁcyZn2Et2), which were impossible to obtain by a classical, planned synthetic way. Additionally, the complex (LRLoxZn2Et2) works in ROP reactions as an initiator with one active metal centre. While, the complex (LRŁcyZn2Et2), proved the existence in the solution of the monomeric species ŁcyZnEt, which is difficult to isolate or even detect during the reaction between Łcy and ZnEt2.
The presented here examples of new zinc complexes indicated that theoretical DFT calculations can be a valuable synthetic toolbox to tailor new complexes that are interesting for coordination chemistry. Additionally, the simple DFT assisted approach constitutes a valuable method for the qualification of an ancillary ligand for the design of new complexes not only as potential initiators for the ROP of lactides. Research showed that the free ortho-position of the aminophenol ligand Lox is one step too far because the aminophenolate zinc complex is inactive in the ROP reactions. Instead, the use of an appropriate ligand, including “exotic one”, for example zinc containing moieties introduced via the “zebra reaction”, is crucial for the improved catalytic activity.
The presented synthetic scenario is not dedicated to zinc complexes exclusively but is useful for the synthesis of other M(II) complexes with an appropriate coordination fitting code. The new examples of heterometal complexes constitute a subject of our intensive research at the moment, and the findings of this research will be soon published in another paper.
Acknowledgements
The author gratefully acknowledges the National Science Centre in Poland (Grant NN 204 200640), KNOW (under the project for Wrocław Biotechnology Center) and the Wrocław Centre for Networking and Supercomputing (http://www.wcss.wroc.pl).
Notes and references
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487 CrossRef CAS PubMed.
- Z. Wang, H. Hu, Y. Wang, Q. Wu, L. Liu and G. Chen, Biomaterials, 2006, 27, 2550 CrossRef CAS PubMed.
- H. Sun, F. Meng, A. A. Dias, M. Hendriks, J. Feijen and Z. Zhong, Biomacromolecules, 2011, 12, 1937 CrossRef CAS PubMed.
- M. P. Arrieta, M. M. Castro-Lopez, E. Rayon, L. F. Barral-Losada, J. M. Lopez-Vilarino, J. Lopez and M. V. Gonzalez-Rodriguez, J. Agric. Food Chem., 2014, 62, 10170 CrossRef CAS PubMed.
- H. K. Lau and K. L. Kiick, Biomacromolecules, 2015, 16, 28 CrossRef CAS PubMed.
- M. Murariu, A. Doumbia, L. Bonnaud, A. L. Dechief, Y. Paint, M. Ferreira, C. Campagne, E. Devaux and P. Dubois, Biomacromolecules, 2011, 12, 1762 CrossRef CAS PubMed.
- H. Kim, H. Park, J. Lee, T. H. Kim, E. S. Lee, K. T. Oh, K. C. Lee and Y. S. Youn, Biomaterials, 2011, 32, 1685 CrossRef CAS PubMed.
- P. Zhang, H. Wu, Z. Lu, C. Deng, Z. Hong, X. Jing and X. Chen, Biomacromolecules, 2011, 12, 2667 CrossRef CAS PubMed.
- A. Bertrand and M. A. Hillmyer, J. Am. Chem. Soc., 2013, 135, 10918 CrossRef CAS PubMed.
- M. Dusselier, P. Van Wouwe, A. Dewaele, P. A. Jacobs and B. F. Sels, Science, 2015, 349, 78 CrossRef CAS PubMed.
- B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215 RSC.
- J. Wu, T.-L. Yu, C.-T. Chen and C.-C. Lin, Coord. Chem. Rev., 2006, 250, 602 CrossRef CAS.
- R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11 CrossRef CAS.
- C. A. Wheaton, P. G. Hayes and B. J. Irealand, Dalton Trans., 2009, 25, 4832 RSC.
- N. Ajellal, J. F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363 RSC.
- S. Dagorne, M. Normand, E. Kirillov and J.-F. Carpentier, Coord. Chem. Rev., 2013, 257, 1869 CrossRef CAS.
- S. Dagorne and C. Fliedel, Top. Organomet. Chem., 2013, 41, 125 CrossRef CAS.
- A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007 RSC.
- S. Dagorne, F. Le Bideau, R. Welter, S. Bellemin-Laponnaz and A. Maisse-François, Chem. – Eur. J., 2007, 13, 3202 CrossRef CAS PubMed.
- M. Cheng, A. B. Attygalle, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 11583 CrossRef CAS.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229 CrossRef CAS PubMed.
- M. H. Chisholm, J. Gallucci and K. Phomphrai, Inorg. Chem., 2002, 41, 2785 CrossRef CAS PubMed.
- M. H. Chisholm and K. Phomphrai, Inorg. Chim. Acta, 2003, 350, 121 CrossRef CAS.
- A. P. Dove, V. C. Gibson, E. L. Marshall, A. J. P. White and D. J. Wiliams, Dalton Trans., 2004, 570 RSC.
- R. Han and G. Parkin, J. Am. Chem. Soc., 1992, 114, 748 CrossRef CAS.
- M. H. Chisholm and N. W. Eilerts, Chem. Commun., 1996, 853 RSC.
- M. H. Chisholm, N. W. Eilerts, J. C. Huffman, S. S. Iyer, M. Pacold and K. Phomphrai, J. Am. Chem. Soc., 2000, 122, 11845 CrossRef CAS.
- M. H. Chisholm, J. Gallucci and K. Phomphrai, Inorg. Chem., 2004, 43, 6717 CrossRef CAS PubMed.
- C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young Jr., M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350 CrossRef CAS PubMed.
- J. Ejfler, M. Kobyłka, L. B. Jerzykiewicz and P. Sobota, Dalton Trans., 2005, 2047 RSC.
- H. Y. Chen, H. Y. Tang and C. C. Lin, Macromolecules, 2006, 39, 3745 CrossRef CAS.
- Z. Zheng, G. Zhao, R. Fablet, M. Bouyahyi, C. M. Thomas, T. Roisnel, O. Casagrande Jr. and J. F. Carpentier, New J. Chem., 2008, 32, 2279 RSC.
- J. Ejfler, S. Szafert, K. Mierzwicki, L. B. Jerzykiewicz and P. Sobota, Dalton Trans., 2008, 6556 RSC.
- G. Labourdette, D. J. Lee, B. O. Patrick, M. B. Ezhova and P. Mehrkhodavandi, Organometallics, 2009, 28, 1309 CrossRef CAS.
- J. Ejfler, K. Krauzy-Dziedzic, S. Szafert, L. B. Jerzykiewicz and P. Sobota, Eur. J. Inorg. Chem., 2010, 3602 CrossRef CAS.
- V. Poirier, T. Roisnel, J.-F. Carpentier and J. Sarazin, Dalton Trans., 2011, 40, 523 RSC.
- H.-J. Chuang, S.-F. Weng, C.-C. Chang, C.-C. Lin and H.-Y. Chen, Dalton Trans., 2011, 40, 9601 RSC.
- N. Ikpo, L. N. Saunders, J. L. Walsh, J. M. B. Smith, L. N. Dawe and F. M. Kerton, Eur. J. Inorg. Chem., 2011, 35, 5347 CrossRef.
- A. Grala, J. Ejfler, L. B. Jerzykiewicz and P. Sobota, Dalton Trans., 2011, 40, 4042 RSC.
- S. Song, X. Zhang, H. Ma and Y. Yang, Dalton Trans., 2012, 41, 3266 RSC.
- J. Wojtaszak, K. Mierzwicki, S. Szafert, N. Gulia and J. Ejfler, Dalton Trans., 2014, 43, 2424 RSC.
- D. Jędrzkiewicz, I. Czeluśniak, M. Wierzejewska, S. Szafert and J. Ejfler, J. Mol. Catal. A: Chem., 2015, 196, 155 CrossRef.
- D. Jędrzkiewicz, J. Ejfler, N. Gulia, Ł. John and S. Szafert, Dalton Trans., 2015, 44, 13700 RSC.
- D. Jędrzkiewicz, J. Ejfler, Ł. John and S. Szafert, Dalton Trans., 2016, 45, 2829 RSC.
- D. Jędrzkiewicz, G. Adamus, M. Kwiecień, Ł. John and J. Ejfler, Inorg. Chem., 2017, 56, 1349 CrossRef PubMed.
-
CrysAlisRED Software, Oxford Diffraction, Wrocław, Poland, 1995–2004 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruna, J. A. Chisholm, P. R. Edgington, P. Mccabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery,Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 1998, 31, 2114 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic CIF files for (LoxZnEt)2, LRŁcyZn2Et2; spectroscopic data for Lox-H, (LoxZnEt)2, LRLoxZn2Et2; computational data: schematic and gas-phase optimized structures of zinc isomers; energies of structures optimized in vacuo; selected bond distances and angles for DFT optimized structures. CCDC 1511741 and 1511742. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt00394c |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  * and
S.
Szafert
* and
S.
Szafert