
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dibenzothiophene-platinated complexes: probing the effect of ancillary ligands on the photophysical performance†
Anastasia I.
Solomatina‡
 a,
Irina O.
Aleksandrova‡
bc,
Antti J.
Karttunen
*c,
Sergey P.
Tunik
a and
Igor O.
Koshevoy
*b
a,
Irina O.
Aleksandrova‡
bc,
Antti J.
Karttunen
*c,
Sergey P.
Tunik
a and
Igor O.
Koshevoy
*b
aSt.-Petersburg State University, 7/9 Universitetskaya nab., 199034 St. Petersburg, Russia
bUniversity of Eastern Finland, Department of Chemistry, 80101, Joensuu, Finland. E-mail: igor.koshevoy@uef.fi
cAalto University, Department of Chemistry, FI-00076 Aalto, Finland. E-mail: antti.karttunen@aalto.fi
First published on 7th March 2017
Abstract
Cyclometalation of dibenzothienyl-pyridine (HPyDBT) afforded a series of platinum(II) complexes Pt(PyDBT)(L)Cl (L = DMSO, 1; P(p-C6H4-X)3 (X = H, 2; CF3, 3; OMe, 4; NPh2, 5); 1,3,5-triaza-7-phosphaadamantane, 6; 2,6-dimethylphenyl isocyanide, 7). Chelating bidentate LL ligands formed cationic compounds [Pt(PyDBT)(LL)]+ (LL = 1,2-bis(diphenylphosphino)benzene, 8; 2,2′-bipyridine, 9; 1,10-phenanthroline, 10). Oxidation of a thienyl sulfur atom allowed for the isolation of the sulfone derivative Pt(PyDBT)(PPh3)Cl (11). The title complexes were characterized crystallographically (except 7). Investigation of their photophysical behavior revealed solid state phosphorescence with quantum yields up to 0.45 for neat powders. The ancillary ligands L show a minor influence on the emission energies of the neutral compounds, but affect dramatically the intensity of luminescence. In contrast, the cationic species with diimine ligands demonstrate a significant contribution of the LL fragments into the emissive T1 states that leads to a certain mixing of 3IL and 3LL′CT transitions and causes a substantial bathochromic shift of emission.
Introduction
Cyclometalated aromatic ligands play an essential role in the coordination chemistry of transition metals. The organometallic compounds, formed upon complexation of these anionic heterodentate moieties (e.g.−C^N, −N^N, −C^C, −C^P metallates of the LX type, and more sophisticated LLX/LXL/XLX species), demonstrate remarkable stability due to particularly strong chelating metal–ligand bonding. Also, the incorporation of the heavy elements into the conjugated motifs dramatically perturbs their electronic properties and offers intriguing means of control over the photophysical behaviour, which substantially differs from that of the parent organic species. These features make the cyclometalated complexes notably appealing candidates for the development of novel optically functional materials.1Large spin–orbit coupling constants of late d-elements (especially those of iridium and platinum) induce fast intersystem crossing S1 → T1 to attain the lowest lying triplet excited state, the radiative relaxation of which (T1 → S0) results in efficient room temperature phosphorescence.2 The photoemission of the triplet origin, typically not observable for pure organic luminophores under ambient conditions, often occurs from platinated intraligand π–π* electronic transitions. This intrinsic attribute allows for a convenient modulation of luminescence parameters (wavelength, lifetime, and quantum yield) of Pt complexes, varying the π–π* energy gap via rational design of the metalated ligand. The success of this approach resulted in extensive studies of the photoactive cycloplatinated compounds,2 accessible functionalization of which led to impressively wide applications in the fields of sensing,3,4 imaging,5 molecular switches6,7 and optoelectronic devices.4,8,9
A general strategy to influence the energies of the frontier molecular orbitals and therefore to tune the photophysical characteristics of Pt(II) cyclometalated complexes implies the alteration of the aromatic system of the platinated LX ligand (X = C−, N−; L = neutral N-heterocyclic or carbene donor). Modification of the HOMO and LUMO levels by the systematic use of electron withdrawing or donating substituents on LX chelates provides a way to govern the change of emission energy.10 Another synthetic alternative involves an extension of the degree of π-conjugation, which is capable of considerably increasing the quantum efficiency11 and is usually accompanied by a red shift of emission.9,12–14 Furthermore, introducing the O,S-containing heterocyclic fragments into the metalated motifs has proven to be a facile method to improve the luminescence intensity and to affect delicately the intraligand emission. Thus, the utilization of dibenzofuranyl and dibenzothienyl groups as the constituents of −C^Ccarbene ligands allowed one to reach blue-green phosphorescence with quantum yields up to 90%.15–17 On the other side, platinated −C^N moieties having thienyl/thiazolyl functionalities can exhibit red luminescence7,9,18–20 or dual triplet/singlet emission,19 suitable for ratiometric oxygen sensing.21 The ancillary ligands coordinated to {Pt(LX)}+ moieties have also been used to tune the photoluminescence performance of the target complexes. Due to the pronounced domination of π–π* LX intraligand transitions there has been a little effect of auxiliary mono- and bidentate groups on the energy of the observed emission, but a substantial modulation of the luminescence intensity and excited state lifetime was reported.20 For example, a prominent 10-fold increase of quantum yield from 5 to 53% was noticed for [Pt(C^N)(LL)]+ (HC^N = phenylpyridine, LL = dicarbene) compounds upon changing the dicarbene stoichiometry.22 The electronic characteristics of the non-chelating mono- and dicarbenes also show an important impact on the quantum efficiency of Pt(C^N)(L)Hal species.23,24 In the case of the diphosphine ligands the origin of photoluminescence of [Pt(C^N)(PP)]+ and {Pt(C^N)Cl}2(PP) was ascribed to have mainly the 3IL and 3MLCT character, respectively, due to the difference in PP ligand coordination (chelate or bridging) and therefore change of the ligand field strength.25 Moreover, binding of two phosphinine ligands to the Pt(II) center results in a prevailing localization of the excited state mostly on one of the P-donor ligands in the [Pt(C^N)(phosphinine)2]+ complex26 that is in drastic contrast to the majority of the congener {Pt(C^N)}-based species. Similarly, the neutral and cationic [Pt(C^N)(NN)]n+ (NN = diimine, n = 0, 1) species display phosphorescence mainly associated with the 3IL and 3MLCT excited states involving the cyclometalated −C^N ligand.13,27 However, depending on the properties of the constituting ligands, a substantial contribution of 3ML′CT and 3LL′CT (L′ = NN ligand) electronic transitions into the photoemission processes can be observed. This confirms the non-innocent role of the ancillary groups bound to the metal ion.13,14
To further develop the studies of the effect of ancillary ligands on the behaviour of emitting platinated motifs, we have chosen 2-(4-dibenzothienyl)pyridine (HPyDBT) as an HC^N precursor. The utilization of accessible dibenzothienyl-derived carbanionic ligands affords Pt(II) complexes with considerably higher quantum efficiencies16 compared to their phenyl relatives.15 In addition, the presence of sulfur functionality in the π-conjugated chromophore system might introduce complementary ways to alter the optical behaviour of these organometallic compounds. Moreover, the PyDBT moiety has not been employed yet for the preparation of Pt(II) luminophores, except few derivatives recently mentioned in the patent.28 Herein we report on a series of novel platinum(II) complexes with a variety of auxiliary ligands (phosphines, isocyanide, diimines) in an attempt to reveal their influence on the photophysical performance of the materials, ultimately aiming at the possibility of mixing the 3IL excited states with 3LL′ charge transfer transitions. The investigation of their photoluminescence properties is supported by the TD-DFT analysis of the electronic structures to elucidate the subtle influence of the ligand sphere on the optical properties of the chromophoric {Pt(C^N)} fragment.
Results and discussion
Synthesis and structural characterization
The cycloplatination of HPyDBT was carried according to a conventional protocol (Scheme 1), which involves the reaction of K2PtCl4 with 2-fold excess of the HC^N precursor in an ethoxyethanol/water mixture at 85 °C. Further treatment of a crude product with DMSO gave the complex Pt(PyDBT)(DMSO)Cl (1) in good yield. Facile substitution of the DMSO ligand with a series of monophosphines P(p-C6H4-X)3 (X = H, 2; CF3, 3; OMe, 4; NPh2, 5), 1,3,5-triaza-7-phosphaadamantane (PTA, 6) and 2,6-dimethylphenyl isocyanide (7) provided a family of the neutral complexes Pt(PyDBT)(L)Cl, which are in general rather poorly soluble in common organic solvents. The cationic compounds [Pt(PyDBT)(LL)]+ (LL = 1,2-bis(diphenylphosphino)benzene (dppb), 8; 2,2′-bipyridine, 9; 1,10-phenanthroline, 10) were initially generated similarly to the published procedures, reacting 1 with a suitable bidentate ligand at elevated temperatures.25,27 The products with chloride counterions demonstrated sparing solubility in the majority of the solvents except DMSO. However, the NMR spectroscopic data indicate that the diimine derivatives 9 and 10 dissociate completely in the latter solvent to generate the mixtures of [Pt(PyDBT)(DMSO)2]+ and the free N,N ligands. The room temperature NMR spectrum of the diphosphine complex 8 in DMSO-d6 similarly showed the spectral pattern, where all the signals are substantially broadened that is indicative of a dynamic process evidently due to the equilibrium between dppb and DMSO substituted products. Carrying out the reactions with LL ligands in the presence of a stoichiometric amount of sodium tetrakis(3,5-bis(trifluoromethyl)phenyl)borate (NaBARF) under ambient conditions efficiently produced complexes 8–10 as BARF− salts (Scheme 1), which are readily soluble in common non-coordinating solvents.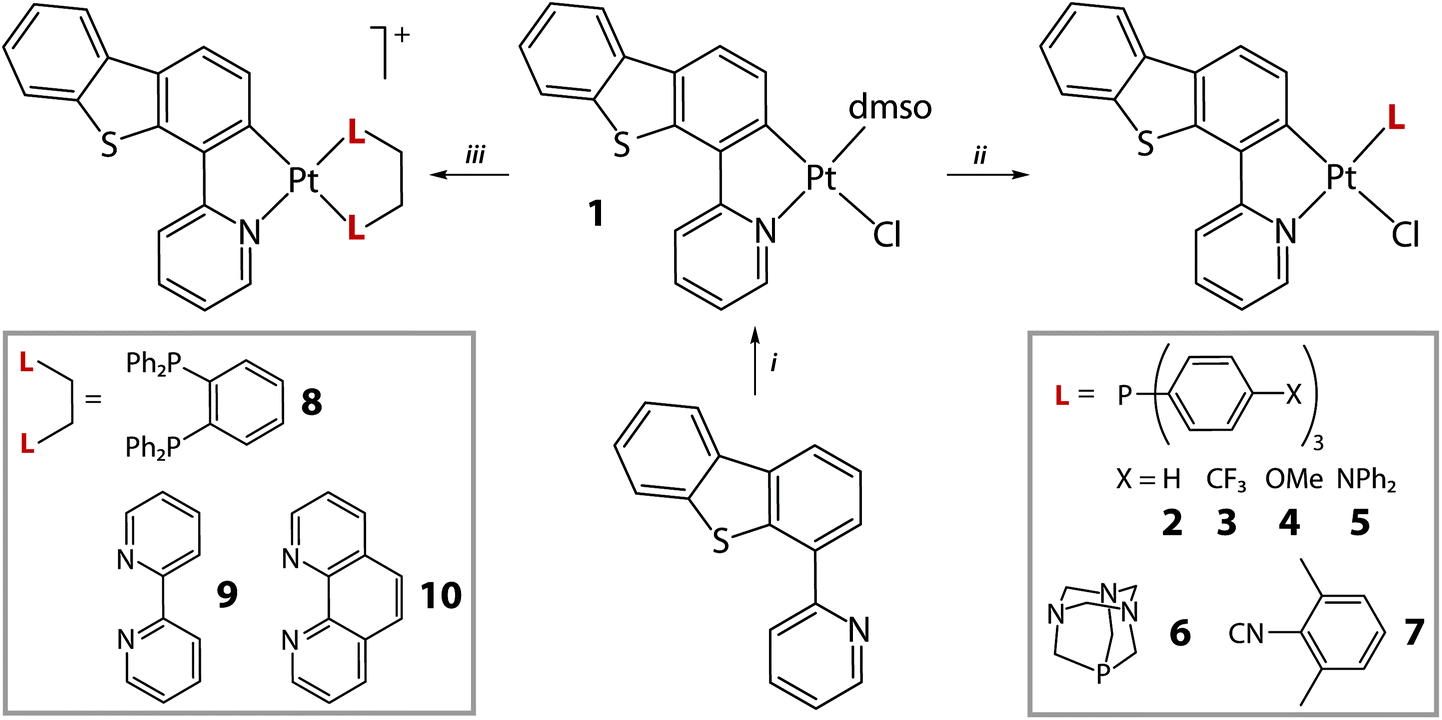 | ||
| Scheme 1 Synthesis of complexes 1–10 (i: K2PtCl4, ethoxyethanol/H2O, 85 °C, 24 h, N2, then DMSO, 74%; ii: CH2Cl2, 25 °C, 1 h, 58–91%; iii: CH2Cl2/Et2O, NaBARF, 25 °C, 1 h, 72–90%). | ||
The structures of 1–6 and 8–10 were determined by X-ray diffraction analysis and are shown in Fig. 1, 2 and S1, S2 (ESI†); selected structural parameters are listed in Table S2.† The neutral DSMO (1) and phosphine (2–6) complexes have a very close stereochemical arrangement, in which the platinum center adopts a square-planar geometry with five-membered metallacycles formed by the chelating PyDBT ligand. Analogously to the congener C^N Pt(II) complexes, the chloride is found in the trans-position to the carbanion, while the P-donor occupies the trans-coordination site relative to the pyridyl nitrogen atom.18,24,29,30
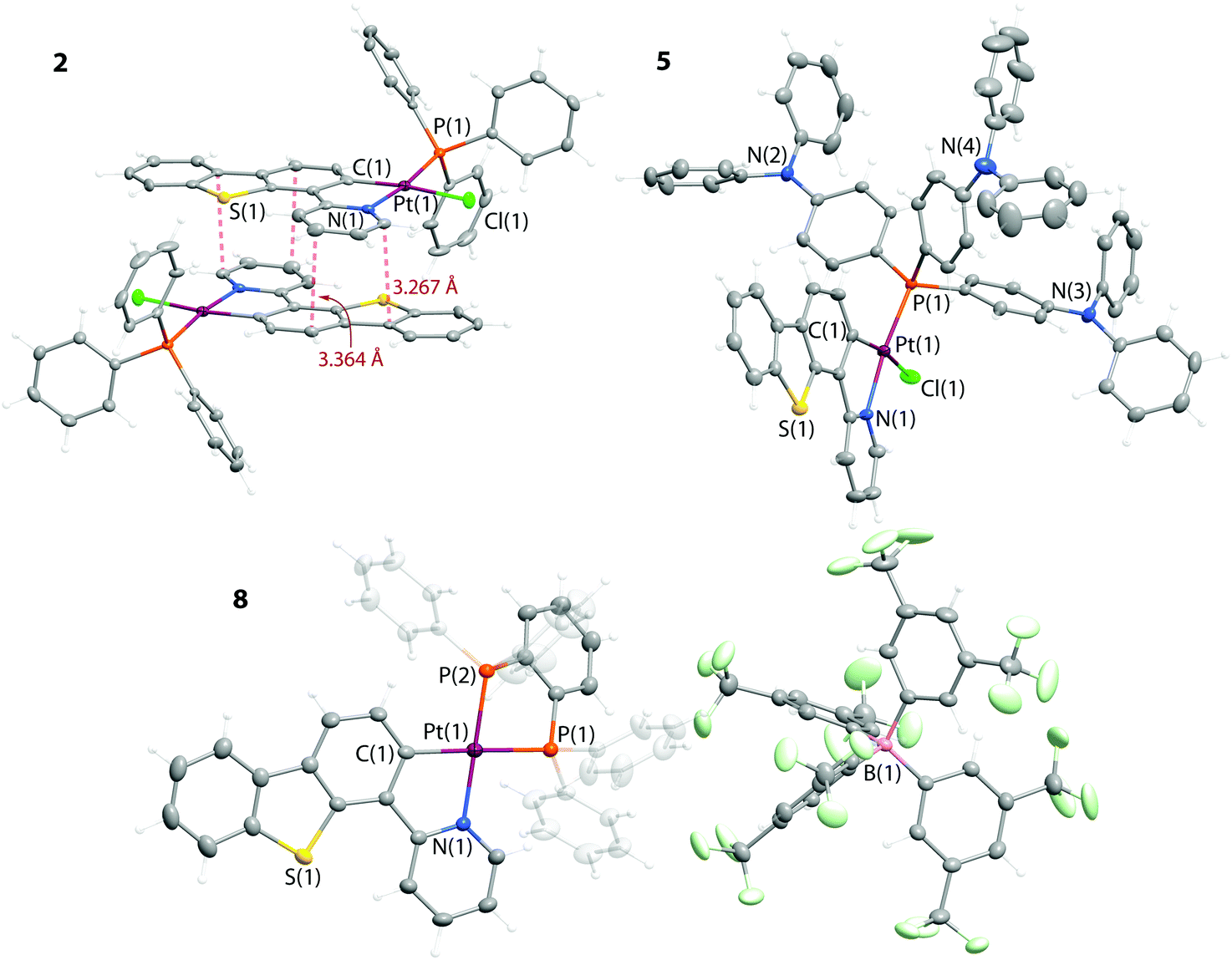 | ||
| Fig. 1 Molecular views of complexes 2, 5 and 8. Thermal ellipsoids are shown at the 50% probability level. | ||
 | ||
| Fig. 2 Molecular view of the cation 9. BARF− counterions are omitted for clarity. Thermal ellipsoids are shown at the 50% probability level. | ||
The bond distances and angles around the metal centers in 1–6 are very much alike and do not demonstrate a systematic dependence on the electronic properties of the phosphines (Table S2†). All the complexes Pt(PyDBT)(L)Cl in the solid state exhibit π–π interactions between the adjacent metalated ligands (the intermolecular separations are in the range of 3.3–3.4 Å) that results in pairwise or infinite stacking with head to head (1–5) and head to tail (6) configurations, respectively. The large Pt⋯Pt distances, exceeding the value of 5.9 Å, exclude any substantial metallophilic interactions in these systems.
The cationic complex 8 contains chelating diphosphine dppb (Fig. 1), which evidently introduces an additional strain as the C(1)–Pt(1)–N(1) angle (79.5°) is visibly smaller than those in 1–6 (92.0–93.1°). The Pt–P bond lengths in 8 are inequivalent (2.329 and 2.253 Å) due to the different trans influence of C− and N donor functions. These structural characteristics agree with those observed for the other recently characterized [Pt(C^N)(PP)]+ species.25 The angle between the planes of the phenylene backbone of the diphosphine and the metalated PyDBT ligand is 39° that deviates considerably from the idealized planar geometry. However, such stereochemistry keeps the optimal tetrahedral geometry of the phosphorus atoms and simultaneously reduces the possible repulsion between the H atoms of the phenyl rings and the PyDBT ligand.
The major features of the platinum(II) coordination environment in the diimine complexes 9 and 10 are in line with the trends observed for the compounds described above (Fig. 2 and S2†). The observed distortions are associated with the intramolecular H⋯H repulsion of the −C^N and NN ligands, that leads to considerable bending of both chelating motifs. This relatively unfavorable configuration may account for the instability of 9 and 10 with respect to the diimine substitution in coordinating solvents like DMSO. Similarly to the neutral complexes 1–6, the cationic species 8–10 in the solid state also demonstrate extensive π-stacking which is not accompanied by metal–metal interactions.
In addition to the variation of the ancillary ligands, we also investigated the effects of the electronic characteristics of the metalated fragment onto the structural and photophysical properties of the products. Thus, the thiophene sulphur in 1 has been oxidized by H2O2 in acidic medium to afford a sulfone derivative (Scheme 2), which appeared to be relatively unstable and could not be isolated in a pure form. Therefore, the oxidized DMSO intermediate was directly converted into the triphenylphosphine complex Pt(PyDBTO2)(PPh3)Cl (11) to give a stable product in a moderate overall yield.
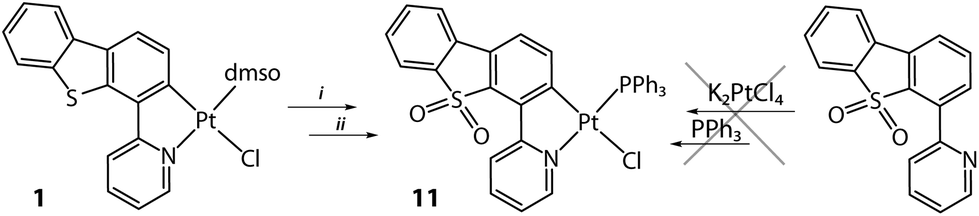 | ||
| Scheme 2 Synthesis of complex 11 (i: aq. H2O2/CH3COOH, 25 °C, 12 h; ii: PPh3, CH2Cl2, 25 °C, 12 h, N2, 46%). | ||
It has to be noted that the use of the HPyDBTO2 ligand (its preparation is described in the ESI†) in the cyclometalation reaction was unsuccessful and resulted in a complete degradation of the reaction mixture.
The analysis of the solid-state structure of 11 (Fig. 3) did not reveal any noticeable changes of the metal coordination environment in comparison to that of 2 (see Table S2†). Oxidation of the sulphur atom mainly affected the intermolecular packing due to the participation of the polar sulfone functionality in the formation of hydrogen bonding.
 | ||
| Fig. 3 Molecular view of complex 11. Thermal ellipsoids are shown at the 50% probability level. | ||
In solution, all the studied complexes retain their stoichiometry according to the 1H and 31P NMR spectroscopic data (see the Experimental section). The monophosphine-containing compounds 3, 4, and 6 demonstrate the 31P resonances with typical 1JPtP splitting in the range 3854–4332 Hz (the isotopomeric satellites due to the 1JPPt coupling was not observed for 2 and 5 due to their very poor solubility), the value of which indicates the trans-position of the phosphorus atom with respect to the nitrogen atom.18,30 The diphosphine complex 8 expectedly shows two 31P NMR signals with significantly different coupling constants (1JPPt = 1819 and 3747 Hz, Fig. S3†) that correlates well with the chemical inequivalence of the PPh2 groups, induced by the C^N ligand.25,26
The 1H NMR spectra of compounds 9 and 10 point to the absence of detectable dissociation of the NN ligands in a non-coordinating solvent (CD2Cl2). This conclusion is evidenced by clearly visible 3JHPt coupling constants (33 and 34 Hz, respectively) in the signals at δ 8.85 (9) and 8.98 (10) ppm, which are assigned to the ortho-H atoms of the diimines. Additionally, the composition of the cationic species 8–10 in the fluid medium was confirmed by ESI-MS, which display the signals at m/z 901.15 (8), 611.09 (9) and 635.09 (10), corresponding to the singly charged molecular ions (Fig. S4†).
Photophysical properties and computational studies
The absorption spectra of complexes 1–11 in solution are shown in Fig. S5† and are summarized in Table S4.† The excitation and emission spectra are presented in Fig. 4, 5, S6 and S7,† and the relevant data are listed in Tables 1 and 2. The photophysical properties of complexes 1–11 were also investigated using density functional theory and time-dependent density functional approach (DFT-PBE0 and TDDFT-PBE0, see Experimental for full computational details). The wavelengths predicted for the S0 → S1 and T1 → S0 electronic transitions are listed in Table 3; Fig. 6 illustrates the electron density difference plots for representative complexes 1 and 10 (see the ESI† for the corresponding plots for the other complexes).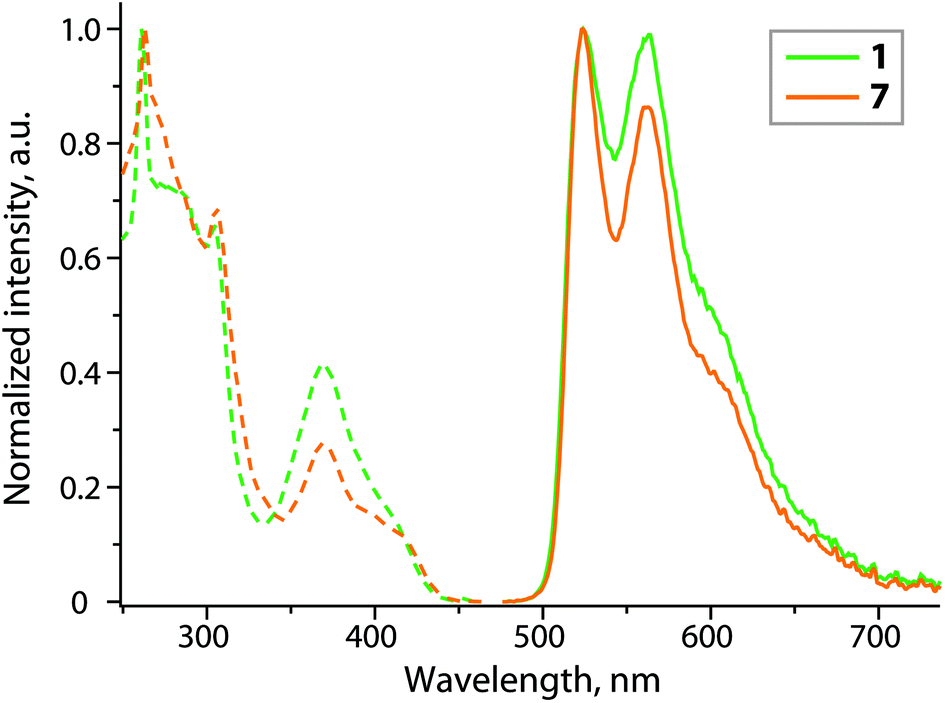 | ||
| Fig. 4 Normalized excitation (dashed lines; λem = 523 nm) and emission (solid lines; λex = 380 nm) spectra of complexes 1 and 7 (CH2Cl2, 298 K). | ||
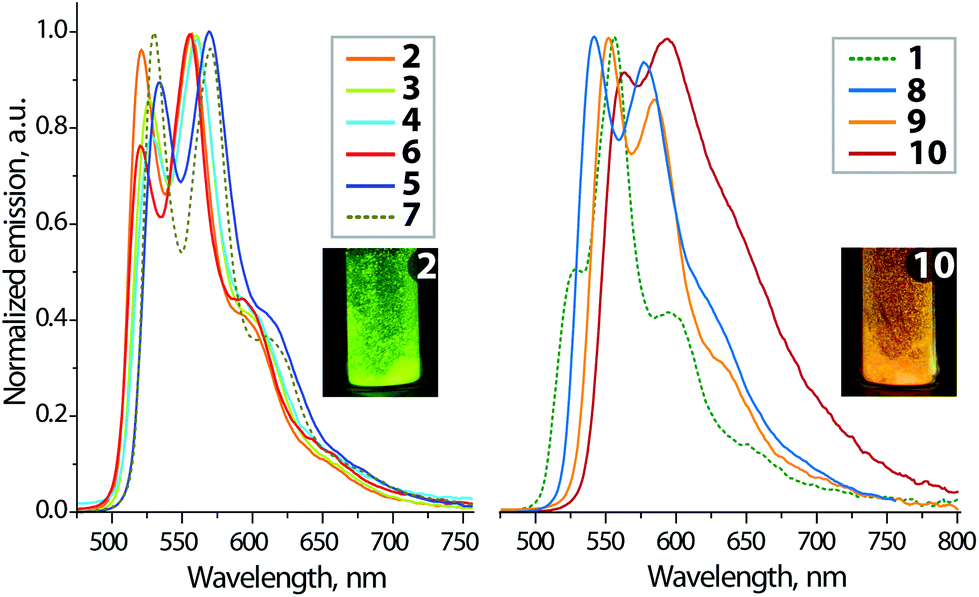 | ||
| Fig. 5 Normalized solid-state emission spectra of complexes 1–10 (298 K). The inset photos show the visual appearance of 2 and 10 under UV excitation (λex = 365 nm). | ||
 | ||
| Fig. 6 Electron density difference plots for the lowest energy singlet excitation (S0 → S1) and the lowest energy triplet emission (T1 → S0) of complexes 1 and 10 (isovalue 0.002 a.u.). During the electronic transition, the electron density increases in the blue areas and decreases in the red areas. Hydrogen atoms are omitted for clarity. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| λ abs, nm (ε × 10−3, M−1 cm−1) | λ ex, nm | λ em, nm |
τ
obs, μsb |
Φ
emb |
|
|---|---|---|---|---|---|
| a CH2Cl2, 298 K. b Aerated/degassed solution. | |||||
| 1 | 250sh (24), 266 (33), 285sh (24), 293sh (18), 305 (19), 370 (9.4), 405sh (3.5) | 261, 274sh, 286sh, 305, 370, 405sh | 523, 563, 600sh | 0.22/0.28 | 0.002/0.004 |
| 7 | 265 (54), 285sh (34), 295sh (29), 307 (33), 370 (9.8), 395sh (5.1), 414sh (3.9) | 263, 270sh, 285sh, 305, 370, 395sh, 414sh | 523, 563, 600sh | 0.73/2.52 | 0.016/0.088 |
| λ ex, nm | λ em, nm |
τ
obsb, μs |
Φ em |
k
rc, 104 s−1 |
k
nrd, 104 s−1 |
|
|---|---|---|---|---|---|---|
| a λ exc = 440 nm; the uncertainty of the quantum yield measurement was in the range of ±5% (an average of three replicas). b Average emission lifetimes for the two-exponential decay determined using the equation τav = (A1τ12 + A2τ22)/(A1τ1 + A2τ2), where Ai is the weight of the i-exponent. c k r were estimated using Φ/τobs. d k nr were estimated using (1 − Φ)/τobs. | ||||||
| 1 | 438, 481, 511 | 528, 556, 593, 648sh | 2.2 | 0.05 | 2.3 | 43 |
| 2 | 432, 480, 511 | 521, 557, 590sh, 650sh | 5.2 | 0.14 | 2.7 | 17 |
| 3 | 444, 479, 513 | 526, 560, 598sh, 650sh | 17.0 | 0.14 | 0.8 | 5.0 |
| 4 | 446, 480, 511 | 529, 560, 598sh, 650sh | 2.1 | 0.10 | 4.8 | 43 |
| 5 | 441, 484, 520 | 534, 569, 610sh, 668sh | 4.4 | 0.33 | 7.6 | 15 |
| 6 | 431, 479, 510 | 520, 555, 594, 644sh | 2.0 | 0.08 | 4.0 | 46 |
| 7 | 443, 486, 522 | 530, 570, 610, 663sh | 3.0 | 0.29 | 9.6 | 23 |
| 8 | 424, 491, 526 | 542, 577, 624sh, 685sh | 29.5 | 0.45 | 1.5 | 1.9 |
| 9 | 492, 533 | 552, 584, 630sh, 685sh | 3.1 | 0.11 | 3.6 | 29 |
| 10 | 499, 540 | 562, 593, 640sh, 700sh | 1.4 | 0.10 | 7.3 | 65 |
| 11 | 477, 508 | 523, 553, 592sh, 645sh | 1.2 | 0.07 | 5.7 | 76 |
The neutral species 1–7 and 11 except 5 show akin spectroscopic profiles with high energy (HE) bands (λabs < 310 nm), assigned mainly to the 1IL(π → π*) electronic transitions localized on the metalated PyDBT ligand, with a possible small contribution from the aromatic systems of the phosphine/isocyanide moieties. Complex 5, which contains the phosphine functionalized with electron-donating NPh2 groups, shows a rather different absorption pattern. The HE bands in the range ca. 290–350 nm with significantly larger extinction coefficients are dominated by the P(p-Ph2NC6H4)3 ligand, as similar absorption spectra were observed for its chalcogen and Au derivatives.31 The calculated absorption spectrum of 5 (Fig. S5†) also clearly shows significant participation of the phosphine in the electronic transitions with the wavelength below 350 nm.
The cationic complexes 8–10 display somewhat more intense HE absorption bands, which correspond to the intraligand transitions involving both chelating motifs. The red shifted bands of lower intensity extend to ca. 430 nm (1–7 and 11) and 480 nm (9 and 10), suggesting metal-to-ligand charge transfer (1MLCT) mixed with ligand-to-ligand charge transfer (1LLCT), particularly in the case of the diimine cations 9 and 10.14,25 The TDDFT-PBE0 calculations support this assignment, showing clear contributions from the Pt metal orbitals (Fig. 6, S8, and S9†). The predicted S0 → S1 wavelengths for the neutral complexes 1–7 and 11 vary between 405 and 417 nm (Table 3) and are in agreement with the corresponding experimental absorption wavelengths. For the diimine cationic complexes 9 and 10, the predicted S0 → S1 wavelengths are correctly red-shifted in comparison to the neutral complexes, but the predicted wavelengths are clearly too large in comparison to the experiment. The S0 → S1 excitation is clearly delocalized and TDDFT is known to predict substantially smaller excitation energies for such systems. For the cationic complex 8, the wavelength is only slightly overestimated.
Among the studied complexes only 1 and 7 exhibit detectable luminescence in solution at room temperature (Table 1 and Fig. 4). Such weak or even negligible emission of Pt(II) cyclometalated complexes in fluid medium is not exceptional24,25 and can be explained in terms of efficient nonradiative pathways of the excited state deactivation, which may arise from the collisional interaction with solvent molecules.1 Moreover, compounds 2–6 containing sterically unhindered phosphine ligands are likely to have additional degrees of freedom, which cause motional and vibrational quenching of luminescence in solution. Despite a simple configuration Pt(C^N)(phosphine)Hal, there is very limited photophysical data on these types of complexes.18,25,32 The available reports are in accordance with our observation and confirm low quantum yields in fluid media, which substantially grow in a rigid environment. Structured luminescence with vibronic progression (ν = ca. 1360 cm−1) indicates the prevailing contribution of the cyclometalated ligand into the frontier molecular orbitals, associated with emissive states. Additionally, the absence of a concentration dependence of the photophysical characteristics and relatively long radiation decay times (τobs = 0.28 and 2.52 μs for 1 and 7 in degassed solutions) suggest that the observed photoemission in solution occurs from the lowest excited triplet state T1 of the monomeric molecules, without aggregation neither in the ground nor in the excited states. The ancillary ligands show virtually no influence on the emission energy, but dramatically vary the quantum efficiency, which demonstrates an over 20-fold increase in degassed dichloromethane solution upon substitution of DMSO (1) for isocyanide (7).
In the solid state, all the title complexes show moderate to intense room temperature photoluminescence (Table 2, Fig. 5, 6, S6 and S7†). The spectra of the neutral compounds 1–7 display essentially common profiles, which reveal vibronic progression reminiscent to those of 1 and 7, observed in solution. Comparable emission energies found for the solid and fluid media evidently point to the same electronic nature of the ground and excited states participating in the radiative processes. This indicates only a slight effect of solid state π-stacking interactions on the photophysical behaviour, which in both phases is determined by the IL transitions centered at the {Pt(PyDBT)} chromophore. The excited state lifetimes, found for the crystalline samples 1–7, range from 2 to 17 μs that correlate well with the congener phosphorescent complexes of the Pt(C^N)(L)Hal type.18,24,25 The intraligand π → π* (PyDBT) origin of the 1–7 luminescence is also supported by a small influence of the ancillary L ligands on the emission wavelength. The latter demonstrates a limited bathochromic shift for 5 and 7, introduced by the increase in the electron-donating ability of L, that possibly points to a certain contribution of the dπ → π* 3MLCT character to the lowest triplet excited states. The simulated T1 → S0 emission wavelengths (Table 3) for the neutral complexes 1–7 and 11 are in line with the experimental data. The Pt metal contributes to the T1 → S0 emission, but the contribution is clearly smaller than for the S0 → S1 excitation. More importantly, the ligands L show a significant ability to vary the quantum yield obtained for neat powder, which ranges from 0.05 (1, L = DMSO) and 0.08 (6, L = PTA) to moderately high values of 0.29 (7, L = CN-C6H3(CH3)2) and 0.33 (5, L = P(C6H4-p-NPh2)3). The obtained efficiencies are systematically higher than those reported for [Pt(C^N)Cl]2(diphosphine) compounds (C^N = phenylpyridine, ppy),25 whose solid state Φem do not exceed the magnitude of 0.08. The improved intensities are probably determined by the extended π-system of PyDBT compared to ppy that could facilitate radiative relaxation of the excited state.15,16 A substantial growth of the emission intensity has been achieved for the electron-rich ligands L (5 and 7). The radiative rate constants kr (Table 2) for the series of Pt(PyDBT)(L)Cl complexes show a steady growth for the series with increasing σ-donation of L ligands (P(p-C6H4-X)3, isocyanide): 0.8 (3, X = CF3) < 2.7 (2, X = H) < 4.8 (4, X = OMe) < 7.6 (5, X = NPh2) < 9.6 (7, CNR) × 104 s−1. This trend reflects a non-negligible role of the electronic influence of the ligand L onto the excited state dynamics, which in the case of phosphines is induced by peripheral groups in phenyl substituents. However, solid state quantum yield is a parameter determined by a few independent effects, like electronic and structural features, crystal packing, etc. This makes it difficult to correlate it only with the contribution of the ancillary ligands and/or metal d orbitals into the frontier MOs of 5 and 7 on the basis of computational analysis. Therefore, the observed improvement of the quantum efficiency for 5 and 7 may be tentatively assigned to the subtle cooperative effects of substituents’ electron donation and crystal packing, which increase the radiative rates kr and provide higher local structural rigidity to suppress radiationless decays (knr), see Table 2.
A comparison of complexes 2 and 11, which contain PyDBT and oxidized PyDBTO2 metalated ligands, respectively, manifests a nearly negligible deviation of the emission profiles (Fig. S7†). However, the presence of the sulfone moiety apparently has a detrimental effect on the luminescence intensity, as it apparently provides efficient nonradiative pathways of the excited state relaxation, indicated by the largest knr value (11) among the studied compounds (Table 2). Consequently, the quantum yield of the non-oxidized complex 2 (Φem = 0.14) is two times larger than that of its sulfone derivative 11 (Φem = 0.07).
The cationic species 8–10 of the [Pt(PyDBT)(LL)]+ type display a gradual red shift of the emission bands as a result of LL ligand contribution into frontier MOs and due to the variations in the ligand field effect, confirmed by the TDDFT calculations, as well. However, while the calculated results for complex 8 are rather close to the experimental data, complexes 9 and 10 with substantially delocalized excited states show clear overestimation of the emission wavelength. The diphosphine complex 8 demonstrates the highest quantum efficiency (Φem = 0.45) and the longest lifetime (τobs = 29.5 μs) within the studied series. Such a high intensity, which radically contrasts with virtually no emission in solution, may be attributed to a favourable crystal packing containing extensive hydrogen bonding between the complex and the fluorinated BARF− counterion along with some π–π stacking of the PyDBT chromophores. These intermolecular interactions evidently lead to a significant increase in the structural rigidity of the emissive centers and therefore severely decrease the radiationless decay rate (Table 2). The diimine complexes 9 and 10 display the lowest energy luminescence, particularly visible for the phenanthroline-containing 10 (see the inset in Fig. 5). The broadened poorly structured band profile suggests a considerable mixing of the 3IL (L = PyDBT) excited state with the 3MLCT and 3LL′CT (L′ = phen), the latter is determined by the close proximity of the orbital energy for both the coordinated aromatic systems. This assignment is supported by TDDFT calculations, which reveal a highly delocalized T1 state for complexes 9 and 10.
Conclusions
We have synthesised a family of cycloplatinated complexes exploiting a virtually unexplored 2-(4-dibenzothienyl)pyridine ligand (PyDBT). Variation of the ancillary donors afforded the neutral and cationic compounds of Pt(PyDBT)L(Cl) (1–7) and [Pt(PyDBT)(LL)]+ (8–10) types, respectively (L, LL – neutral 2- and 4-electron donors). The title species display weak to moderately strong room temperature photoluminescence in the solid state, which is dominated by the triplet intraligand (PyDBT) emission for complexes 1–7. The electronic and steric properties of the ligands L (DMSO for 1, phosphines for 2–6, isocyanide for 7) show a minor influence on the emission energy. The experimental observations correlate well with the results of TD-DFT analysis, which reveals a negligible contribution of L motifs into the emissive T1 → S0 transitions. However, the nature of L has a pronounced effect on the quantum efficiency of 1–7, which changes from a low (Φem = 0.05 for 1 and 0.08 for 6, L = PTA) to a reasonably high intensity of 0.33 (5, L = P(p-Ph2N-C6H4)3). The oxidation of the thiophene fragment of the PyDBT chromophore to form the sulfone derivative 11 (L = PPh3) causes a minor deviation of the emission profile along with a 2-fold drop of the quantum yield.The bidentate LL ligands induce a visible red shift and broadening of luminescence signals for complexes 8 and 9 due to a substantial delocalization of the T1 state between the aromatic systems of the metalated PyDBT and diimine moieties. The latter suggests a considerable mixing of 3IL (L = PyDBT) and 3LL′CT excited states and offers a convenient way of tuning the photophysical parameters of this class of organometallic luminophores without stereochemical alteration of the emissive center.
Experimental
General comments
1,3,5-Triaza-7-phosphaadamantane (PTA),33 tris-(4-diphenylaminophenyl)phosphine31 and 2-dibenzothienylpyridine (HPyDBT)34 were prepared according to the published procedures. Other reagents were used as received. The solution 1H, 31P{1H} NMR and 1H–1H COSY spectra were recorded on Bruker AMX 400, Avance 400 and Avance 500 spectrometers. Mass spectra were determined with a maXis II ESI-QTOF instrument (Bruker) in the ESI+ mode. Microanalyses were carried out at the analytical laboratory of the University of Eastern Finland.:1 v/v mixture) at +5 °C gave a yellow crystalline material (305 mg, 74%). 1H NMR (DMSO-d6, 298 K, δ): 9.72 (dd with unresolved 3JHPt, 3JHH 5.8 Hz, 4JHH 0.9 Hz, 1H, py), 8.56 (d with unresolved 3JHPt, 3JH–H 8.4 Hz, 1H, dbt), 8.41–8.37 (m, 2H, dbt and py), 8.33 (d, 3JHH 7.8 Hz, 1H, py), 8.22 (d, 3JHH 8.4 Hz, 1H, dbt), 8.08 (m, 1H, dbt), 7.65 (td, 3JHH 6.0, 5.8 Hz 4JHH 1.2 Hz, 1H, py), 7.56 (m, 2H, dbt). Anal. calc. for C19H16PtClOS2N (%): C 40.11; H 2.83; N 2.46; S 11.27. Found: C 40.08; H 2.88; N 2.44; S 11.16.
:1 v/v mixture) at +5 °C. 1H NMR (CD2Cl2, 298 K, δ): 10.05 (m, 1H, py), 8.45 (d, 3JHH 8.3 Hz, 1H, py), 8.18 (dd, 3JHH 8.3 and 8.1 Hz, 1H, py), 7.96 (d, 3JHH 8.3 Hz, 1H, dbt), 7.92 (d, 3JHH 7.4 Hz, 1H, dbt), 7.86 (dd, 3JHP 11.5 Hz, 3JHH 7.2 Hz, 6H, ortho-H Ph), 7.48 (m, 1H, py), 7.46 (m, 2H, dbt), 7.39 (d, 3JHH 8.3 Hz, 1H, dbt), 6.97 (ddd, 3JHPtca. 50 Hz, 3JHH 8.3 Hz, 4JHP 3.5 Hz, 1H, dbt), 7.52–7.41 (m, 9H, Ph). The 31P{1H} NMR spectrum could not be measured due to low solubility of the sample. Anal. calc. for C35H25PtPClSN·0.5CH2Cl2 (%): C 53.59; H 3.29; N 1.76; S 4.03. Found: C 53.47; H 3.27; N 1.73; S 3.70.
:1 v/v mixture) at +5 °C to give a yellow crystalline material (49 mg, 73%). 1H NMR (DMSO-d6, 298 K, δ): 9.87 (dd, 3JHH 5.2 Hz, 4JHP 4.7 Hz, 1H, py), 8.48–8.43 (m, 2H, py), 8.19 (d, 3JHH 7.6 Hz, 1H, dbt), 8.06 (d, 3JHH 7.7 Hz, 1H, dbt), 8.04 (dm, 3JHH 7.1 Hz, 6H, ortho-H C6H4), 7.93 (d, 3JHH 7.1 Hz, 6H, meta-H C6H4), 7.75 (ddd, 3JHH 5.6 and 5.2 Hz, 4JHH 1.7 Hz, 1H, py), 7.66 (d, 3JHH 8.2 Hz, 1H, dbt), 7.53 (ddd, 3JHH 7.7 and 7.2 Hz, 4JHH 1.0 Hz, 1H, dbt), 7.46 (ddd, 3JHH 7.6 and 7.2 Hz, 4JHH 0.8 Hz, 1H, dbt), 6.75 (ddd, 3JHPtca. 45 Hz, 3JHH 8.2 Hz, 4JHP 3.6 Hz, 1H, dbt) ppm. 31P{1H} NMR (DMSO-d6, 298 K, δ): 25.5 (d, 1JPPt 4332 Hz, 1P). Anal. calc. for C38H22ClF9NPPtS (%): C 47.68; H 2.32; N 1.46; S 3.35. Found: C 47.31; H 2.45; N 1.41; S 3.33.
:1 v/v mixture) at +5 °C to give a green-yellow crystalline material (19 mg, 58%). 1H NMR (DMSO-d6, 298 K, δ): 9.61 (d with unresolved 3JHPt, 3JHH 5.2 Hz, 1H, py), 8.46–8.38 (m, 2H, py + dbt), 8.35 (d, 3JHH 7.7 Hz, 1H, py), 8.23 (d, 3JHH 7.9 Hz, 1H, dbt), 8.11 (d, 3JHH 6.9 Hz, 1H, dbt), 7.91 (dd, 3JHPtca. 60 Hz, 3JHH 7.9 Hz, 1H, dbt), 7.75 (dd, 3JHH 6.5 and 5.2 Hz, 1H, py), 7.58 (m, 2H, dbt), 7.45 (t, 3JHH 7.4 Hz, 1H, isocyanide), 7.37 (d, 3JHH 7.4 Hz, 2H, isocyanide), 2.59 (s, 6H13). Anal. calc. for C26H19ClN2PtS (%): C 50.20; H 3.08; N 4.50; S 5.16. Found: C 50.12; H 2.83; N 4.50; S 4.98.
X-ray structure determination
The crystals of 1–6 and 8–11 were immersed in cryo-oil, mounted in a Nylon loop, and measured at a temperature of 150 K or 120 K (6, 9, and 10). The X-ray diffraction data were collected with Bruker Kappa Apex II, Bruker SMART APEX II, and Bruker Kappa Apex II Duo diffractometers using Mo Kα radiation (λ = 0.71073 Å). The APEX235 program package was used for cell refinements and data reductions. The structures were solved by direct methods using the SHELXS-201436 program with the WinGX37 graphical user interface. A semiempirical absorption correction (SADABS)38 was applied to all data. Structural refinements were carried out using SHELXL-2014.36Some of the CF3 groups of the borate counterions (in 8–10) and of the phosphine ligand (in 3) were disordered and modelled to have two orientations each; the geometry and displacement constraints and restraints were applied to these moieties. The crystallization solvent in 3 was partially lost from the crystal and could not be resolved unambiguously. The contribution of the missing solvent to the calculated structure factors was taken into account by using a SQUEEZE routine of PLATON.39 The missing solvent was not taken into account in the unit cell content. The structure of 6 was refined as a 2-component inversion twin.
All H atoms in 1–6 and 8–11 were positioned geometrically and constrained to ride on their parent atoms, with C–H = 0.95–0.99 Å and Uiso = 1.2–1.5Ueq (parent atom). The crystallographic details are summarized in Table S1, ESI.†
Photophysical measurements
The photophysical measurements in solution were carried out in CH2Cl2 and DMSO, which were distilled prior to use. UV/Vis spectra were recorded with a Shimadzu UV-1800 spectrophotometer at concentrations of ca. 1–5 × 10−5 M (1 cm cuvettes). Emission spectra in solution were recorded on a FluoMax-4 (JY Horiba Inc., Japan) spectrofluorimeter using a concentration of ca. 3 × 10−5 M. The emission quantum yield in solutions was determined by the comparative method40 using coumarin 102 in ethanol (Φr = 0.764) as ref. 41 with the refraction indexes of dichloromethane and ethanol equal to 1.42 and 1.36 respectively. The steady-state emission and excitation spectra of complexes 1–11 in the solid state at room temperature and at 77 K were recorded on a FluoroLog 3 Horiba spectrofluorimeter. Lifetimes in the solid state and in solution were determined by the Time-Correlated Single Photon Counting (TCSPC) method. The lifetime data were fit using the Jobin-Yvon software package and the Origin 9.0 program. Direct quantum yield measurements of the crystalline samples were performed at room temperature with an integrating sphere from Quanta-phi.Computational details
Complexes 1–11 were studied using the hybrid PBE0 density functional method (DFT-PBE0).42 Pt atoms were described by a triple-zeta-valence quality basis set with polarization functions (def2-TZVP).43 Scalar relativistic effects were taken into account by employing a 60-electron relativistic effective core potential for Pt.44 A split-valence basis set with polarization functions on non-hydrogen atoms was used for the other atoms (def2-SV(P)).43 Multipole-accelerated resolution-of-the-identity technique was used to speed up the calculations.45 The geometries of all complexes were first fully optimized without any symmetry constraints using the DFT-PBE0 method. The optimized geometries of complexes 1–11 are in agreement with the available X-ray structures (the coordinates of the optimized structures are included as the ESI†). The excited states were investigated using the Time-Dependent DFT formalism.46 The singlet excitations were determined at the optimized ground state S0 geometries, while the lowest energy triplet emissions were determined at the optimized T1 geometry. All electronic structure calculations were carried out with the TURBOMOLE program package (version 7.1).47Acknowledgements
Financial support from the Academy of Finland (grant 268993, I. O. K., structural characterization) and the Russian Science Foundation (grant 16-43-03003, synthesis and photophysical studies) is gratefully acknowledged. Computational resources were provided by CSC, the Finnish IT Center for Science (A. J. K.). The NMR, photophysical and mass-spectrometry measurements were performed using the following core facilities at St. Petersburg State University Research Park: the Centre for Magnetic Resonance, the Centre for Optical and Laser Materials Research, and the Centre for Chemical Analysis and Materials Research.Notes and references
- Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC
.
- K. Li, G. S. M. Tong, Q. Wan, G. Cheng, W.-Y. Tong, W.-H. Ang, W.-L. Kwong and C.-M. Che, Chem. Sci., 2016, 7, 1653–1673 RSC
- V. Guerchais and J.-L. Fillaut, Coord. Chem. Rev., 2011, 255, 2448–2457 CrossRef CAS
- K. M.-C. Wong, M. M.-Y. Chan and V. W.-W. Yam, Adv. Mater., 2014, 26, 5558–5568 CrossRef CAS PubMed
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762–1785 CrossRef CAS
- Z. M. Hudson, S.-B. Zhao, R.-Y. Wang and S. Wang, Chem. – Eur. J., 2009, 15, 6131–6137 CrossRef CAS PubMed
- J. C.-H. Chan, W. H. Lam, H.-L. Wong, N. Zhu, W.-T. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 12690–12705 CrossRef CAS PubMed
- J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Coord. Chem. Rev., 2011, 255, 2401–2425 CrossRef CAS
- Y.-M. Zhang, F. Meng, J.-H. Tang, Y. Wang, C. You, H. Tan, Y. Liu, Y.-W. Zhong, S. Su and W. Zhu, Dalton Trans., 2016, 45, 5071–5080 RSC
- J. A. G. Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596–2611 CrossRef
- S. C. F. Kui, I. H. T. Sham, C. C. C. Cheung, C.-W. Ma, B. Yan, N. Zhu, C.-M. Che and W.-F. Fu, Chem. – Eur. J., 2007, 13, 417–435 CrossRef CAS PubMed
- B. Yin, F. Niemeyer, J. A. G. Williams, J. Jiang, A. Boucekkine, L. Toupet, H. Le Bozec and V. Guerchais, Inorg. Chem., 2006, 45, 8584–8596 CrossRef CAS PubMed
- J. R. Berenguer, A. Díez, E. Lalinde, M. T. Moreno, S. Ruiz and S. Sanchez, Organometallics, 2011, 30, 5776–5792 CrossRef CAS
- O. J. Stacey, J. A. Platts, S. J. Coles, P. N. Horton and S. J. A. Pope, Inorg. Chem., 2015, 54, 6528–6536 CrossRef CAS PubMed
- Y. Unger, D. Meyer, O. Molt, C. Schildknecht, I. Munster, G. Wagenblast and T. Strassner, Angew. Chem., Int. Ed., 2010, 49, 10214–10216 CrossRef CAS PubMed
- A. Tronnier, A. Risler, N. Langer, G. Wagenblast, I. Münster and T. Strassner, Organometallics, 2012, 31, 7447–7452 CrossRef CAS
- M. Tenne, S. Metz, G. Wagenblast, I. Münster and T. Strassner, Organometallics, 2015, 34, 4433–4440 CrossRef CAS
- S.-W. Lai, Q. K.-W. Chan, J. Han, Y.-G. Zhi, N. Zhu and C.-M. Che, Organometallics, 2009, 28, 34–37 CrossRef CAS
- D. N. Kozhevnikov, V. N. Kozhevnikov, M. Z. Shafikov, A. M. Prokhorov, D. W. Bruce and J. A. G. Williams, Inorg. Chem., 2011, 50, 3804–3815 CrossRef CAS PubMed
- G. R. Kumar and P. Thilagar, Inorg. Chem., 2016, 55, 12220–12229 CrossRef PubMed
- Y. Liu, H. Guo and J. Zhao, Chem. Commun., 2011, 47, 11471–11473 RSC
- H. Uesugi, T. Tsukuda, K. Takao and T. Tsubomura, Dalton Trans., 2013, 42, 7396–7403 RSC
- J. J. Hu, S.-Q. Bai, H. H. Yeh, D. J. Young, Y. Chi and T. S. A. Hor, Dalton Trans., 2011, 40, 4402–4406 RSC
- A. I. Solomatina, D. V. Krupenya, V. V. Gurzhiy, I. Zlatkin, A. P. Pushkarev, M. N. Bochkarev, N. A. Besley, E. Bichoutskaia and S. P. Tunik, Dalton Trans., 2015, 44, 7152–7162 RSC
- R. B. Aghakhanpour, S. M. Nabavizadeh, M. Rashidi and M. Kubicki, Dalton Trans., 2015, 44, 15829–15842 RSC
- J. Moussa, T. Cheminel, G. R. Freeman, L.-M. Chamoreau, J. A. G. Williams and H. Amouri, Dalton Trans., 2014, 43, 8162–8165 RSC
- D. M. Jenkins, J. F. J. Senn and S. Bernhard, Dalton Trans., 2012, 41, 8077–8085 RSC
-
G. Kottas, Z. Elshenawy, G. Szigethy and C. Xia, US20160285014 A1, 2016 Search PubMed
- A. Diez, J. Fornies, C. Larraz, E. Lalinde, J. A. Lopez, A. Martin, M. T. Moreno and V. Sicilia, Inorg. Chem., 2010, 49, 3239–3251 CrossRef CAS PubMed
- V. V. Sivchik, A. I. Solomatina, Y.-T. Chen, A. J. Karttunen, S. P. Tunik, P.-T. Chou and I. O. Koshevoy, Angew. Chem., Int. Ed., 2015, 54, 14057–14060 CrossRef CAS PubMed
- I. Kondrasenko, Z.-H. Tsai, K.-y. Chung, Y.-T. Chen, Y. Y. Ershova, A. Doménech-Carbó, W.-Y. Hung, P.-T. Chou, A. J. Karttunen and I. O. Koshevoy, ACS Appl. Mater. Interfaces, 2016, 8, 10968–10976 CAS
- S. Jamali, R. Czerwieniec, R. Kia, Z. Jamshidi and M. Zabel, Dalton Trans., 2011, 40, 9123–9130 RSC
- D. J. Daigle, Inorg. Synth., 1998, 32, 40–45 CrossRef CAS
- M. Shibue, M. Hirotsu, T. Nishioka and I. Kinoshita, Organometallics, 2008, 27, 4475–4483 CrossRef CAS
-
APEX2 - Software Suite for Crystallographic Programs, Bruker AXS, Inc., Madison, WI, USA, 2010 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS
-
G. M. Sheldrick, SADABS-2008/1 - Bruker AXS Area Detector Scaling and Absorption Correction, Bruker AXS, Madison, Wisconsin, USA, 2008 Search PubMed
-
A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 1.17th edn, 2013 Search PubMed
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CrossRef CAS
- K. Rurack and M. Spieles, Anal. Chem., 2011, 83, 1232–1242 CrossRef CAS PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chem. Acc., 1990, 77, 123–141 CrossRef CAS
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–290 CrossRef CAS
-
F. Furche and D. Rappoport, in Computational Photochemistry, ed. M. Olivucci, Elsevier, Amsterdam, 2005, pp. 93–128 CrossRef CAS
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Cartesian coordinates of the optimized geometries, additional NMR, ESI-MS, computational. CCDC 1529875–1529884 for 1–6 and 8–11. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt00349h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |