
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Variable coordination of tris(2-pyridyl)phosphine and its oxide toward M(hfac)2: a metal-specifiable switching between the formation of mono- and bis-scorpionate complexes†
Alexander V.
Artem'ev
 *a,
Alexey V.
Kashevskii
b,
Artem S.
Bogomyakov
c,
Alexander Yu.
Safronov
b,
Anastasiya O.
Sutyrina
d,
Anton A.
Telezhkin
d and
Irina V.
Sterkhova
d
*a,
Alexey V.
Kashevskii
b,
Artem S.
Bogomyakov
c,
Alexander Yu.
Safronov
b,
Anastasiya O.
Sutyrina
d,
Anton A.
Telezhkin
d and
Irina V.
Sterkhova
d
aNikolaev Institute of Inorganic Chemistry, Siberian Branch of Russian Academy of Sciences, 3, Akad. Lavrentiev Ave., Novosibirsk 630090, Russian Federation. E-mail: chemisufarm@yandex.ru
bIrkutsk State University, Karl Marx str., 1, 664003 Irkutsk, Russian Federation
cInternational Tomography Center, SB RAS, Institutskaya Str. 3A, 630090 Novosibirsk, Russian Federation
dA. E. Favorsky Irkutsk Institute of Chemistry, Siberian Branch of the Russian Academy of Sciences, 1 Favorsky Str., 664033 Irkutsk, Russian Federation
First published on 3rd April 2017
Abstract
An unexpected substitution of the anionic chelating ligands at the MII centre by a neutral tripodal ligand has been observed in the reaction of MnII, CoII, NiII and CuII hexafluoroacetylacetonates (hfac) with tris(2-pyridyl)phosphine (Py3P) or its oxide (Py3P = O). The nature of the metal ion in M(hfac)2 and the M/L ratio determine the degree of substitution of hfac-anions (partial vs. total) and therefore, the structure of the complex formed (scorpionate vs. bis-scorpionate ones, respectively). Hence, the reaction of the ligands with [Cu(hfac)2(H2O)2] in an equimolar ratio affords scorpionate [Cu(N,N′,N′′-Py3P = X)(O,O′-hfac)(O-hfac)], wherein one hfac-ligand chelates metal, while the other hfac acts as an O-monodentate one. Using the two equivalents of Py3P in this reaction leads to [Cu(N,N′,N′′-Py3P)2](hfac)2, which contains a bis-scorpionate cation [Cu(Py3P)2]2+ and two noncoordinated hfac-anions. [Co(hfac)2(H2O)2] and [Ni(hfac)2(H2O)2], regardless of the M/L molar ratio, react with Py3P = O to give cationic scorpionates [M(N,N′,N′′-Py3P = O)(O,O′-hfac)(H2O)](hfac), in which one hfac-anion is noncoordinated. In contrast, [Mn(hfac)2(H2O)2], on interaction with Py3P, results in the cationic complex [Mn(N,N′,N′′-Py3P)2][Mn(hfac)3]2 bearing a bis-scorpionate cation [Mn(Py3P)2]2+ and two [Mn(hfac)3]2− counterions. The synthesized scorpionates have been characterized by X-ray diffractometry, cyclic voltammetry, SQUID magnetometry, FT-IR and UV-Vis techniques.
1. Introduction
During the past decades, pyridylphosphines and their chalcogenides have attracted increasing attention in coordination chemistry, catalysis and material science.1–3 The combination of “soft” (P-atom) and “hard” (N-atoms) donor sites in pyridylphosphines makes them very important and versatile ligands for the design of unique catalysts,4 luminescent materials5 and prospective drugs.6 Among pyridylphosphines, tris(2-pyridyl)phosphine (Py3P) is gaining a special interest owing to the useful properties of its metal complexes. For instance, Pd,7,8 Cr,9 Mo,9 W,9,10 Ru11 and Rh,11 Fe,12 Co,12,13 Ni13 and Mn13 complexes with Py3P have been explored as catalysts for methoxycarbonylation of alkynes,7,8 Diels–Alder cycloaddition,9 Friedel–Crafts/aldehyde cyclotrimerization,10 hydroformylation of alkenes,11 ethylene polymerization,12 and O2-oxidation of tetraline.13 Most significantly, Py3P has recently become readily available due to the development of its synthesis directly from elemental phosphorus and 2-bromopyridine.14Tris(2-pyridyl)phosphine, owing to its tripodal structure and heminal disposition of the N atoms towards the P atom, exhibits numerous coordination patterns, e.g. N-15 and P-monodentate,14,16P,N-bridging,17N,N′-chelating,18,19N,P,N′-pincer,20N,N′/P-bridging21 and N,N′,N′′-tripodal9,22–24 ones. A while ago, N,N′,N′′-/P-coordination of substituted tris(2-pyridyl)phosphine was found in the dinuclear complex [(MeCN)3Cu{P(6-Me-2-Py)3}Cu(MeCN)](PF6)2, wherein this ligand acts as a Janus head ligand.25
In contrast to the plethora of studies related to tris(2-pyridyl)phosphine based complexes, the data on those with tris(2-pyridyl)phosphine chalcogenides, Py3P = X (X = O, S or Se), are scarce, although the latter are equally important ligands. In recent years, these complexes have been employed for the synthesis of the scorpionate complexes [Cu(N,N′,N′′-Py3P = X)]Hal,26,27 showing yellow to red TADF emission with good quantum yields.27 Later, a series of CuI thiocyanate complexes with Py3P = O was designed.28 The RuII scorpionate supported by the Py3P = O, viz. [Ru(N,N′,N′′-Py3P = O)(bpy)(OH2)](OTf)2, has proven to be an excellent electrocatalyst for water oxidation, thus surpassing any known Ru catalyst.29
Therefore, the further development of the coordination chemistry of tris(2-pyridyl)phosphine and its chalcogenides appears to be particularly appealing to access novel functional compounds. Herein, as a part of our ongoing interest in this area, we report on the variable coordination of tris(2-pyridyl)phosphine (1) and its oxide (2) towards M(hfac)2 (M = Cu, Ni, Co, Mn) leading to either scorpionate or bis-scorpionate complexes. In the course of this study, we have observed an unexpected replacement of the anionic chelating ligand at the metal center by the neutral tripodal ligand. The degree of such substitution (partial vs. complete) and, hence, the structure of the resulting complexes (scorpionate vs. bis-scorpionate) depend on the nature of the metal ion in M(hfac)2 and, to a lesser extent, on a metal-to-ligand ratio.
2. Results and discussion
2.1. Synthetic aspects
As our experiments have shown, the reaction of [Cu(hfac)2(H2O)2] with Py3P (1) at the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio occurs immediately upon mixing of the reactants (CHCl3, r.t.) to give scorpionate 3 in 88% yield (Scheme 1). The reaction is strictly selective: a possible formation of the coordination polymers does not take place. In the structure of 3, Py3P acts as N,N′,N′′-tripodal ligand toward the octahedral CuII ion. Importantly, during the reaction, one of the hfac-anions retains an O,O′-chelating pattern, whereas the second hfac-anion adopts an O-monodentate mode. The reaction of Py3P = O (2) with [Cu(hfac)2(H2O)2] under similar conditions (1:1 ratio, r.t., CHCl3) instantly leads to structurally related scorpionate 4 in 80% yield (Scheme 1). Therefore, the charge of the P atom in 1 and 2 (δ− vs. δ+, respectively) has no influence on the result of the reaction with Cu(hfac)2. This can be explained by weak or a complete lack of conjugation between the phosphorus-centered orbitals and the π-electronic system of the pyridine rings.
:1 molar ratio occurs immediately upon mixing of the reactants (CHCl3, r.t.) to give scorpionate 3 in 88% yield (Scheme 1). The reaction is strictly selective: a possible formation of the coordination polymers does not take place. In the structure of 3, Py3P acts as N,N′,N′′-tripodal ligand toward the octahedral CuII ion. Importantly, during the reaction, one of the hfac-anions retains an O,O′-chelating pattern, whereas the second hfac-anion adopts an O-monodentate mode. The reaction of Py3P = O (2) with [Cu(hfac)2(H2O)2] under similar conditions (1:1 ratio, r.t., CHCl3) instantly leads to structurally related scorpionate 4 in 80% yield (Scheme 1). Therefore, the charge of the P atom in 1 and 2 (δ− vs. δ+, respectively) has no influence on the result of the reaction with Cu(hfac)2. This can be explained by weak or a complete lack of conjugation between the phosphorus-centered orbitals and the π-electronic system of the pyridine rings.
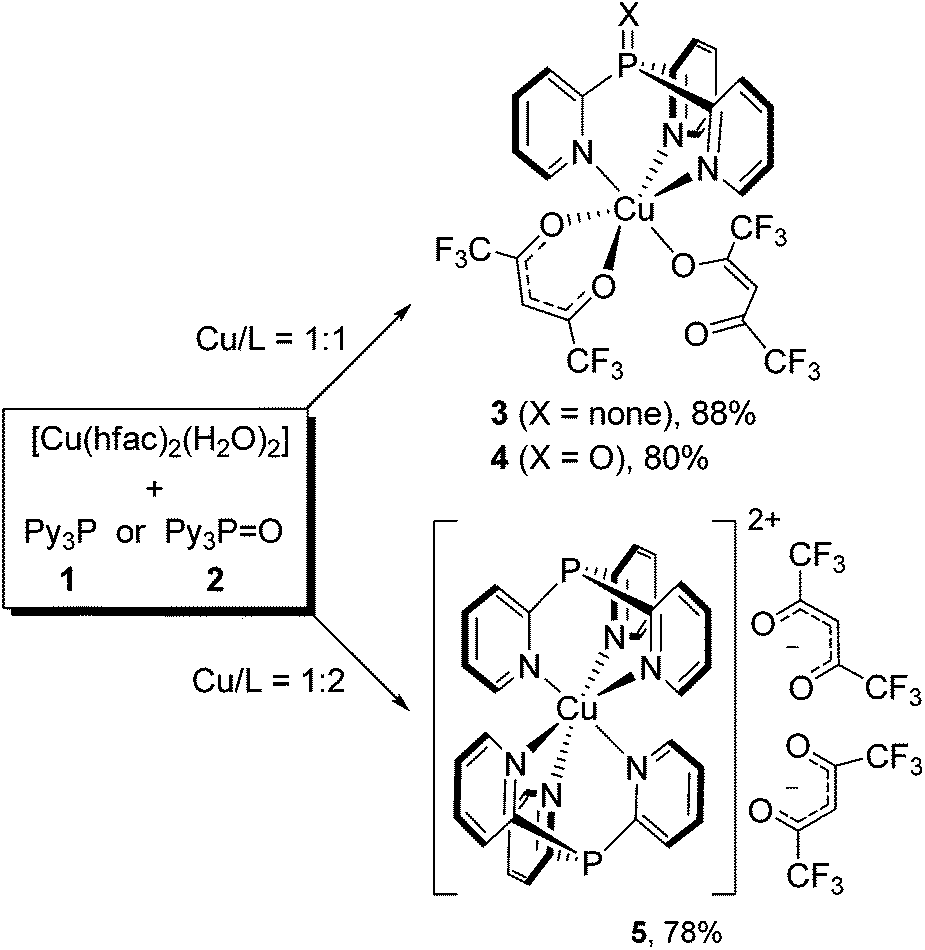 | ||
| Scheme 1 Synthesis of mono- and bis-scorpionate complexes 3–5. | ||
Moreover, the interaction of [Cu(hfac)2(H2O)2] with Py3P in a 1:2 ratio at ambient temperature (CHCl3 or MeCN) produces complex 5 in 78% yield (Scheme 1). Thus, both chelating hfac-anions within the coordination sphere of CuII are replaced by the Py3P ligands. In the complex obtained (5), the CuII ion octahedrally coordinates six N atoms of two Py3P molecules to form a bis-scorpionate cation [Cu(Py3P)2]2+. The two hfac-anions are in the second coordination sphere.
Notably, Cu(acac)2 does not react with Py3P or Py3P = O; after removing the solvent from the reaction mixture, the starting reagents remain unchanged. The observed discrepancy is likely due to a higher Lewis acidity of CuII in Cu(hfac)2 caused by the presence of two strong electron-acceptor hfac-ligands.
To our surprise, the reaction between Py3P = O and [Co(hfac)2(H2O)2] or [Ni(hfac)2(H2O)2], regardless of the M/L molar ratio, always furnishes scorpionates 6 or 7 in 85% and 81% yields, respectively (Scheme 2). Importantly, unlike 3 and 4, in the scorpionates 6 and 7, the MII ion of the {M(N,N′,N′′-Py3P = O)} unit is coordinated by one chelating hfac-anion and H2O molecule, thus adopting an octahedral geometry, while another hfac-ligand is noncoordinated. Our attempts to synthesize bis-scorpionate CoII and NiII complexes by the reaction of [M(hfac)2(H2O)2] with excess 2 were unsuccessful; only complexes 6 and 7 were obtained along with unreacted ligand. In contrast, when Co(NO3)2·7H2O,23 CoCl2·6H2O30 and NiCl2·6H2O,31 react with Py3P in 1:2 molar ratio, bis-scorpionate complexes of [M(Py3P)2]2+ type are readily formed.
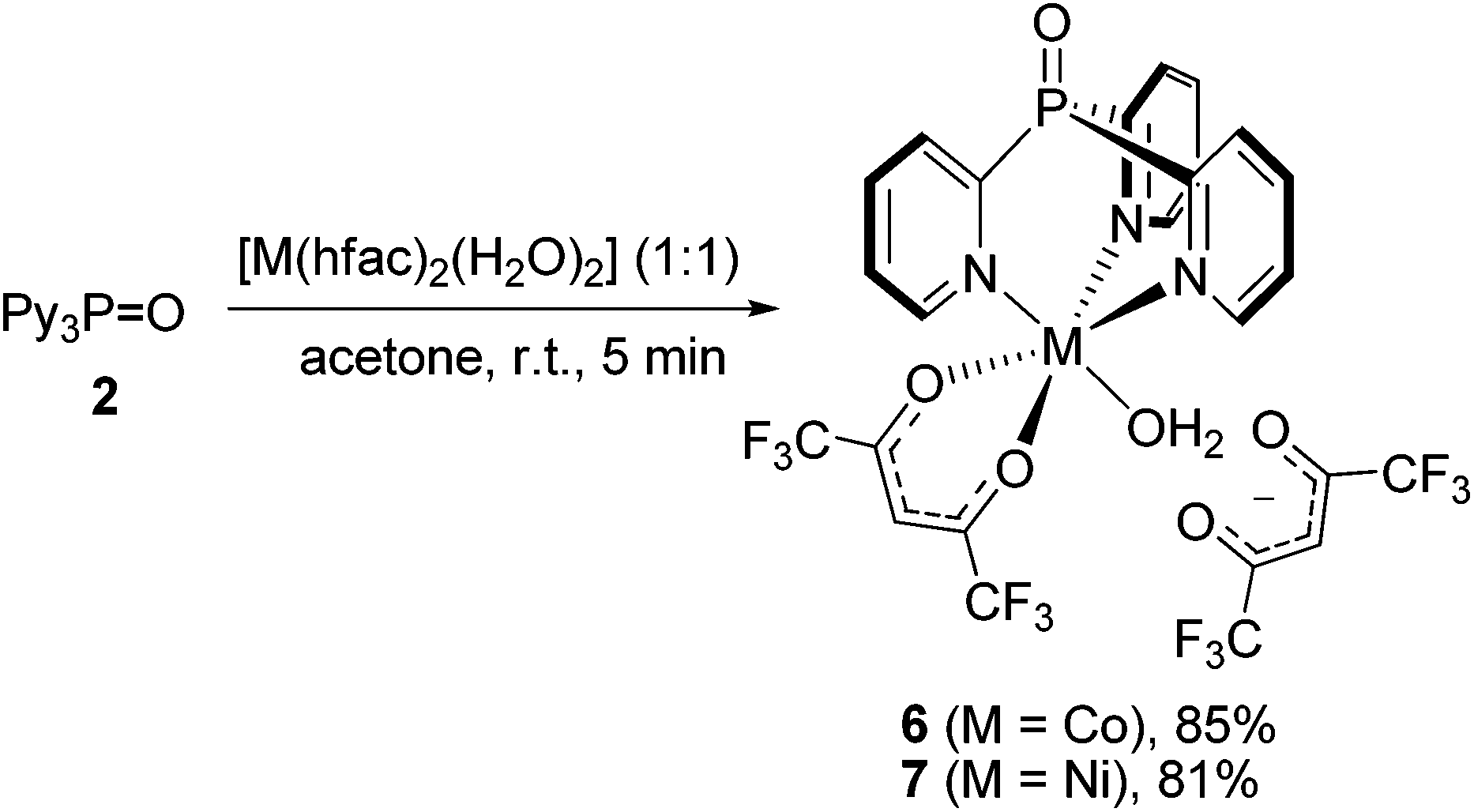 | ||
| Scheme 2 Synthesis of scorpionate complexes 6 and 7. | ||
Surprisingly, the treatment of [Mn(hfac)2(H2O)2] with Py3P in 1:1 or 1:2 molar ratio (r.t., MeCN) immediately produces complex [Mn(N,N′,N′′-Py3P)2][Mn(hfac)3]2 (8), as shown in Scheme 3. Using a stoichiometric ratio of the reactants (3:2) affords 8 in 69% yield. In the bis-scorpionate [Mn(Py3P)2]2+ cation, the metal has an octahedral environment arranged from six N atoms of two Py3P ligands. In the crystal of 8, there are two stereochemically different [Mn(hfac)3]− anions (Δ- and Λ-isomers), in which the MnII coordinates three hfac-anions to form a MnO6 twisted prism.
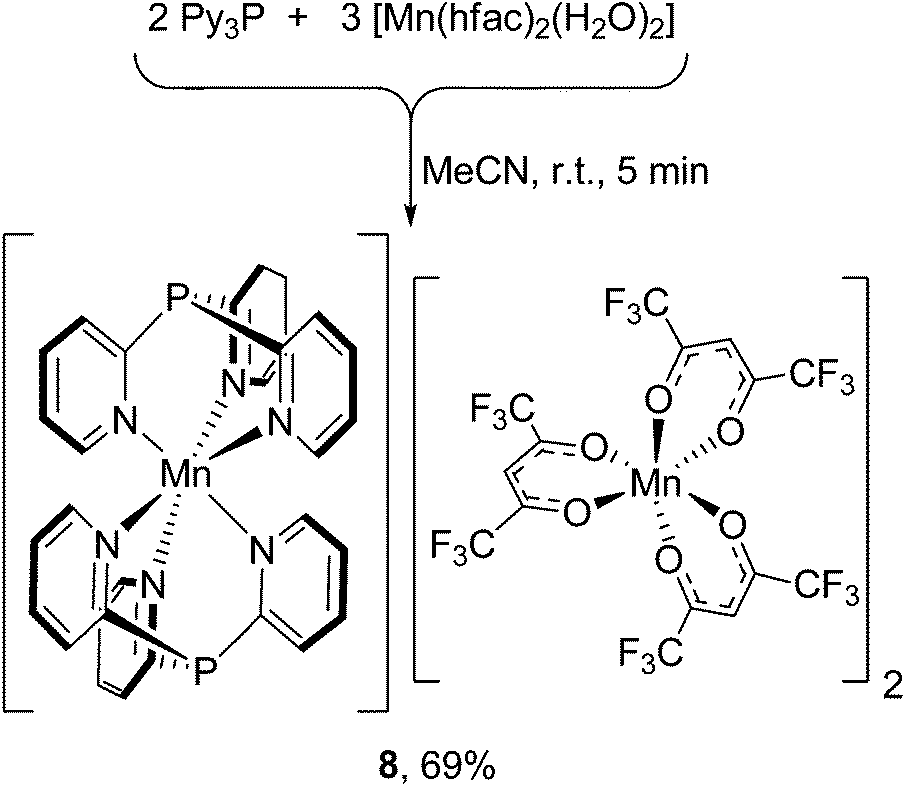 | ||
| Scheme 3 Synthesis of bis-scorpionate complex 8. | ||
These results suggest that the outcome of the reaction between M(hfac)2 and Py3P or Py3P = O is strongly influenced by the nature of MII ions and, in the case of Cu(hfac)2, by the M/L ratio specified. In the reaction with Cu(hfac)2, the final product may be the scorpionate (3 or 4) or bis-scorpionate (5) complex, whilst Co(hfac)2 and Ni(hfac)2 give only mono-scorpionates (6 and 7). Unlike these, Mn(hfac)2 interacts with 1, furnishing the unexpected complex [Mn(N,N′,N′′-Py3P)2][Mn(hfac)3]2 (8). Therefore, in the reactions investigated, the replacement of the charged chelating ligands by the neutral tripodal ligands is observed. When M(acac)2 (exemplified by CuII acetylacetonate), is used as the starting reagent, no reaction with the Py3P or Py3P = O is observed. The different behavior of M(acac)2 and their polyfluorinated analogues is probably due to the better leaving group ability of the hfac-anion compared with that of the acac-anion.
The observed that the difference in reactivity of M(hfac)2 toward Py3P or Py3P = O can be rationalized by the interference of two factors. First, it is well known32 that within a series of octahedral complexes, the lability toward ligand substitution generally increases in the sequence: Cu2+ [t2g(6)eg(3)] > Mn2+ [t2g(3)eg(2)] > Co2+ [t2g(5)eg(2)] > Ni2+ [t2g(6)eg(2)] due to the filling of the t2g and eg orbitals. Therefore, [Cu(hfac)2(H2O)2] and [Mn(hfac)2(H2O)2] are expected to be more substitutionally labile compared to [Co(hfac)2(H2O)2] and [Ni(hfac)2(H2O)2], which agrees with the complete replacement of both hfac-ligands in CuII and MnII precursors. On the other hand, the interaction of lesser labile [Co(hfac)2(H2O)2] and [Ni(hfac)2(H2O)2] with Py3P = O, regardless of their ratio, always gives scorpionates 6 and 7, wherein the parent hfac-anion and water molecule remain coordinated to the metal. In contrast, [Cu(hfac)2(H2O)2], when reacted with Py3P or Py3P = O in a 1:1 ratio, forms scorpionates 3 and 4, in which the metal is coordinated by chelating and monodentate hfac-anions. In other words, the total substitution of the two H2O molecules and partial substitution of a hfac-ligand within [Cu(hfac)2(H2O)2] takes place in this reaction. The possible explanation of this fact is that the [Cu(hfac)2(H2O)2], being subjected to a Jahn–Teller distortion,33 features a faster substitution of axially located H2O molecules compared to the equatorial hfac-anions. Furthermore, the lower reactivity of [Co(hfac)2(H2O)2] and [Ni(hfac)2(H2O)2], for which the partial substitution of hfac-anions is observed, is consistent with maximum charge density (Z2/r) at the Co2+ or Ni2+ ion (5.37 and 5.79)34 compared with the value for Mn2+ (4.82).34 As a consequence, the breaking of the metal–Ohfac and metal–OH2O bonds in [Co(hfac)2(H2O)2] and [Ni(hfac)2(H2O)2] as well as in the resulting complexes 6 and 7 becomes more difficult.
The synthesized complexes are air-stable crystals, which are well soluble in MeCN, Me2CO and CHCl3. Moreover, complex 8 is soluble in Et2O and, partially, in hexane. In the solid state, complexes 3–8 have been characterized by X-ray diffraction analysis (the crystallographic data are given in Table S1†), SQUID magnetometry and ATR-IR spectroscopy, while their redox behaviour has been studied using cyclic voltammetry (CV).
2.2. Crystal structure description
In scorpionate 3 (Fig. 1), the Cu atom has a 4 + 2 pseudooctahedral environment, where the equatorial plane is defined by two O atoms of the chelating hfac-ligand (Cu–O 1.9798 ± 0.0069 Å) and two N atoms of Py3P. The axial positions are occupied by the third N atom of Py3P and the O atom of second hfac-anion, the O3 atom of which remains noncoordinated. The Cu–Neq distance is about 2.01 Å, whereas the Cu–Nax bond length is significantly longer [2.2575(14) Å], indicating a Jahn–Teller axial elongation.35 On the whole, these values are typical for the CuII–NPy lengths.36,37 The geometry of the [Cu(Py3P)] cage of 3 deviates from the perfect C3-symmetry; the dihedral angles between the pyridine plane are 55.66, 61.78 and 62.82°. The peculiarity of the packing of 3 is that the neighboring molecules in 3 are linked by F⋯F [dF3⋯F3′ = 2.759, dF4⋯F9 = 2.887, dF2⋯F8 = 2.923 Å], P⋯F [dP1⋯F1 = 3.267 Å], Chfac⋯F [2.894, 3.071 Å], CPy⋯F [2.928 Å], CPy–H⋯F [2.483–2.620 Å] and CPy–H⋯Ohfac [2.344, 2.640 Å] interactions to generate a 3D supramolecular network.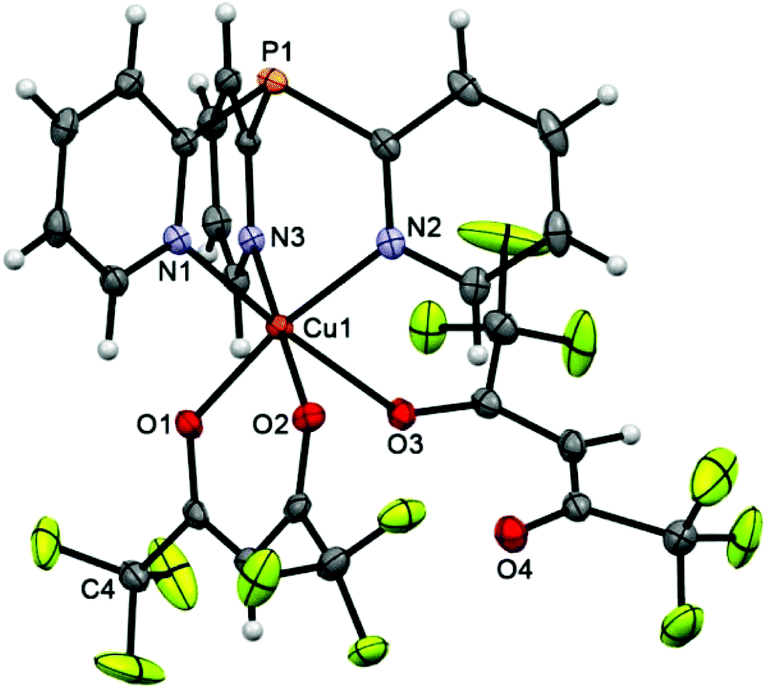 | ||
| Fig. 1 Molecular structure of 3 (50% thermal ellipsoid). The minor disorder of the CF3 (C4) group is omitted for clarity. Selected bond lengths [Å] and angles [°]: Cu(1)–O(1) 1.9729(11), Cu(1)–O(2) 1.9867(12), Cu(1)–O(3) 2.474(10), Cu(1)–N(2) 2.0101(13), Cu(1)–N(3) 2.0152(13), Cu(1)–N(1) 2.2575(14), N(1)–Cu(1)–O(3) 175.87, O(2)–Cu(1)–N(3) 177.78(5), O(1)–Cu(1)–N(2) 169.51(5). | ||
The structure of scorpionate 4 (Fig. 2) is almost perfectly consistent with that of 3 (overlay of these molecules is depicted in Fig. S1†). The major difference between them is the presence of an O atom of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in 4, the length of which [1.468(2) Å] is comparable with that in the free Py3P = O [1.4792(11) Å].38 Similarly, the Cu ion of 4 shows a typical Jahn–Teller axial elongation in its distorted octahedral geometry.
O bond in 4, the length of which [1.468(2) Å] is comparable with that in the free Py3P = O [1.4792(11) Å].38 Similarly, the Cu ion of 4 shows a typical Jahn–Teller axial elongation in its distorted octahedral geometry.
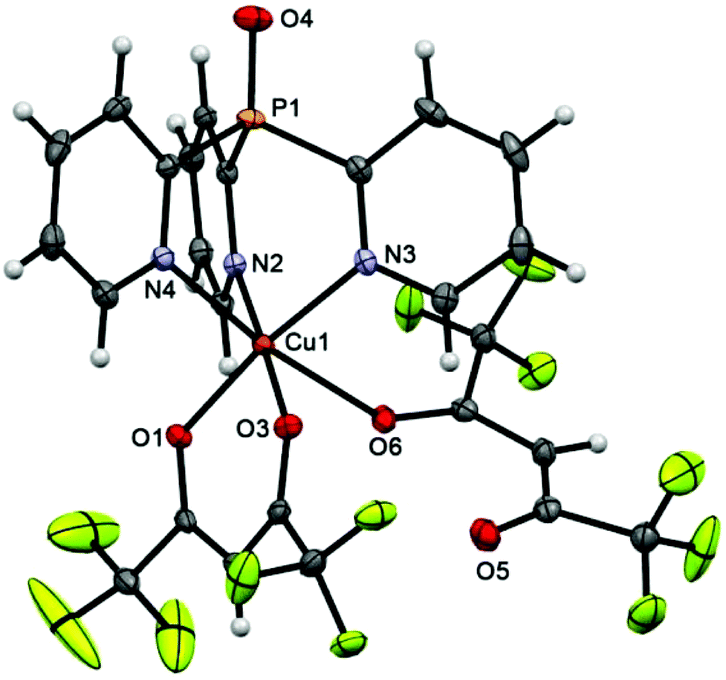 | ||
| Fig. 2 Molecular structure of 4 (50% thermal ellipsoid). Selected bond lengths [Å] and angles [°]: Cu(1)–O(1) 1.9694(19), Cu(1)–O(2) 1.971(2), Cu(1)–O(6) 2.3988(19), Cu(1)–N(2) 2.023(2), Cu(1)–N(3) 2.029(2), Cu(1)–N(4) 2.345(2), P(1)–O(4) 1.468(2), O(1)–Cu(1)–N(3) 171.42(9), O(2)–Cu(1)–N(2) 177.79(9), N(4)–Cu(1)–O(6) 171.62(7). | ||
The asymmetric unit of 5 contains one noncoordinated hfac-anion and half of the cation [Cu(Py3P)2]2+, where the Cu atom is located in an inversion center. The two Py3P molecules are ligated to the Cu atom through six N atoms to form the bis-scorpionate structure that looks like a paddle-wheel (Fig. 3). Thus, the metal atom adopts a distorted octahedral geometry. It should be noted that in the CuN6 unit of 5, there are three pairs of unique Cu–N bonds with Cu–N distances of 2.2883(17) [Cu–Nax], 2.1360(17) and 2.0192(14) Å [Cu–Neq] that suggest a Jahn–Teller elongation along the z-axis. In contrast, in the related complexes, e.g. [Cu(Py3P)2]Br2,39 [Cu(MeCPy3)2]I2,40 and [Cu(MeOCPy3)2](OTf)2,41 two short (ca. 2.0 Å) and four long (ca. 2.2 Å) Cu–N distances are observed. The O1–C19–C18–C17–O2 chain of the noncoordinated hfac-anions in 5 has a syn–syn conformation.
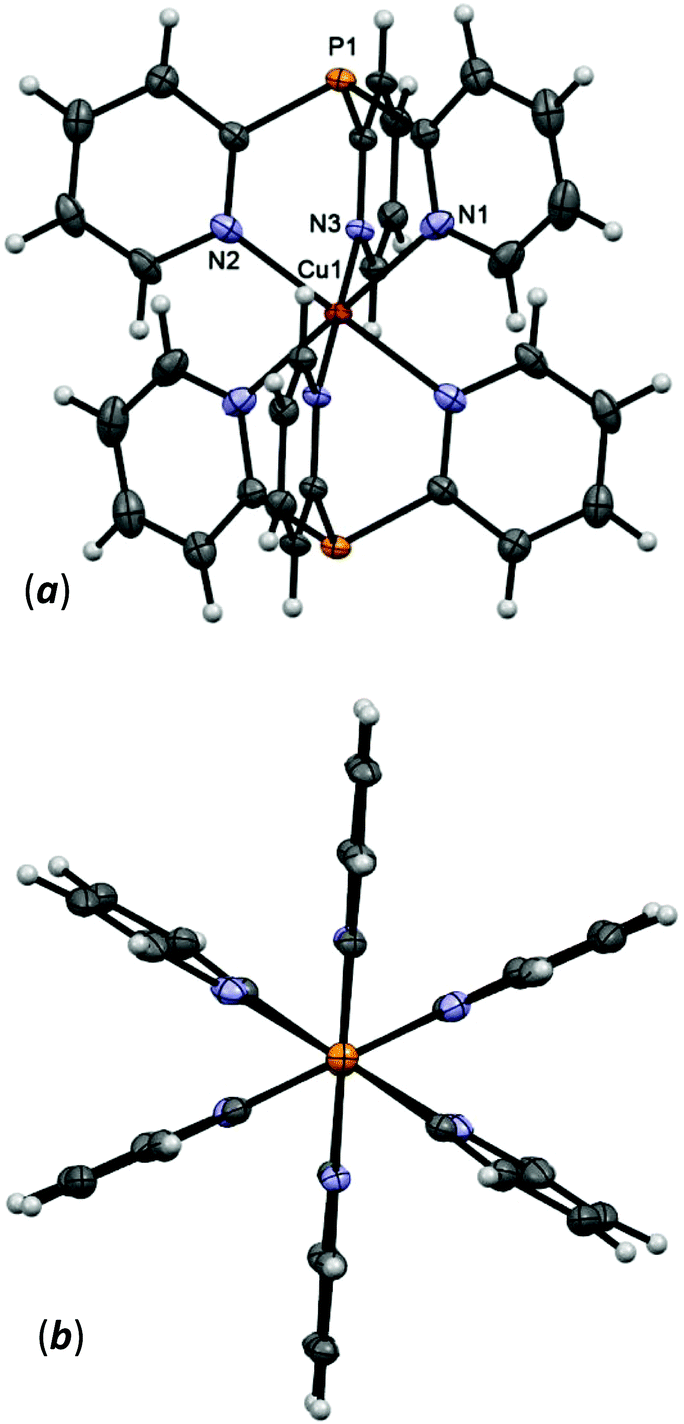 | ||
| Fig. 3 Side-on (a) and top-down (b) views of structure of the [Cu(Py3P)2]2+ cation of 5 (50% thermal ellipsoid). Selected bond lengths [Å] and angles [°]: Cu(1)–N(1) 2.2883(17), Cu(1)–N(2) 2.1360(17), Cu(1)–N(3) 2.0192(14), N(1)′–Cu(1)–N(1) 180.00(5), N(2)–Cu(1)–N(2)′ 180.00(5), N(3)–Cu(1)–N(3)′ 180.0, N(2)–Cu(1)–N(1) 90.75(6), N(3)–Cu(1)–N(2) 90.77(6). | ||
The cationic parts of scorpionates 6 (CoII) and 7 (NiII), [M(Py3P = O)(hfac)(H2O)]+, are isostructural and have almost superimposable structures (see overlay of these molecules in Fig. S2†). As seen from Fig. 4 and 5, in both cations, Py3P = O is bonded to a metal atom in a N,N′,N′′-tripodal manner with three M–N bonds being nearly equal (average 2.126 Å). The O,O′-chelating hfac-anion and the oxygen atom of water completes the coordination sphere of the metal, representing a slightly distorted octahedron. The [M(N–C)3P = O] cage has pseudo-C3-symmetry with the dihedral angles between three mean Py cycles of 117.64–123.02° (for 6) and 117.65–121.79° (for 7). The M–Ohfac distances (ca. 2.052–2.114 Å) are longer than the M–OH2O bond (ca. 2.030 Å). The noncoordinated hfac-anion in 6 and 7 adopts a syn–syn conformation; it is linked with the coordinated water molecule through OH2O–H⋯Ohfac hydrogen bonds (their parameters are given in the captions of Fig. 4 and 5).
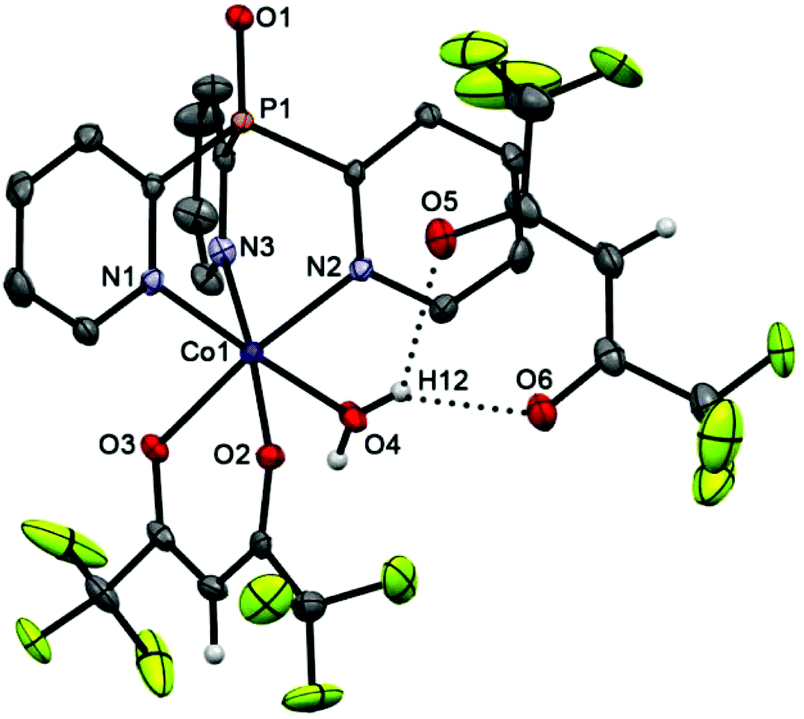 | ||
| Fig. 4 Molecular structure of 6 (50% thermal ellipsoid). The pyridine hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Co(1)–O(4) 2.033(3), Co(1)–O(2) 2.070(3), Co(1)–O(3) 2.114(3), Co(1)–N(3) 2.116(3), Co(1)–N(2) 2.128(3), Co(1)–N(1) 2.133(3), P(1)–O(1) 1.479(3), H(12)–O(6) 2.0091(14), H(12)–O(5) 2.3020(12), O(4)–Co(1)–N(1) 176.86(13), O(3)–Co(1)–N(2) 173.44(12), O(2)–Co(1)–N(3) 175.47(13). | ||
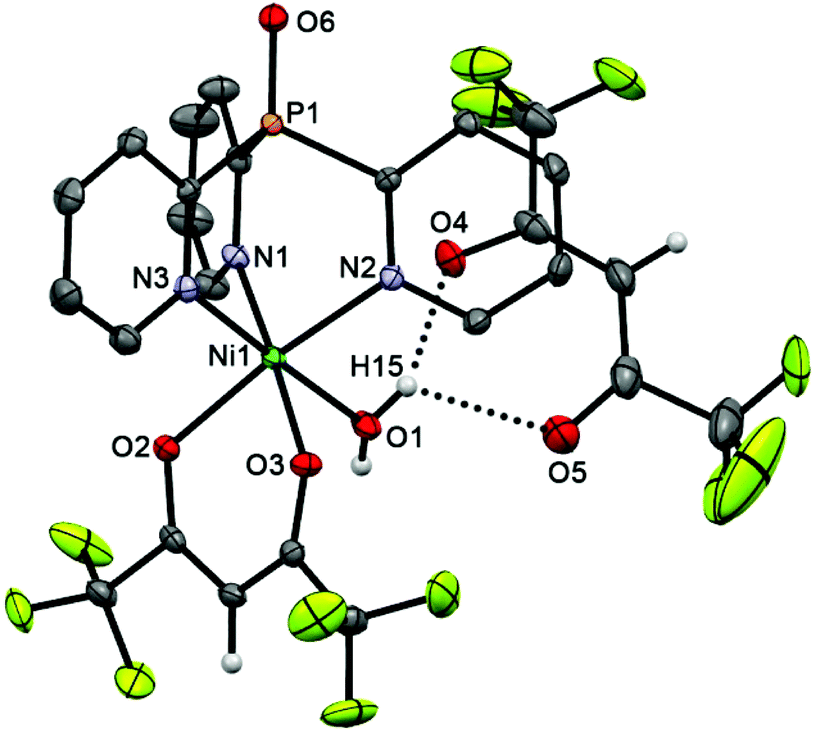 | ||
| Fig. 5 Molecular structure of 7 (50% thermal ellipsoid). The pyridine hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: ]: Ni(1)–O(1) 2.028(2), Ni(1)–O(3) 2.052(2), Ni(1)–O(2) 2.061(2), Ni(1)–N(3) 2.068(3), Ni(1)–N(1) 2.069(3), Ni(1)–N(2) 2.081(2), P(1)–O(6) 1.478(2), H(15)–O(4) 2.0102(13), H(15)–O(5) 2.2952(13), O(1)–Ni(1)–N(3) 176.03(10), O(3)–Ni(1)–N(1) 176.32(9), O(2)–Ni(1)–N(2) 173.83(9). | ||
Complex 8 crystallizes from acetone as the solvate 8·2Me2CO, which consists of a bis-scorpionate cation [Mn(Py3P)2]2+ and two [Mn(hfac)3]2− anions (Δ- and Λ-enantiomers) per formula unit. In the cation, metal lies on a crystallographic inversion center (Fig. 6); it is coordinated in a tripodal manner by the three pyridine nitrogens of each ligand. The Mn(1) atom has a nearly perfect octahedral geometry with N–Mn–N angles of 180.00°. The Mn(1)–N distances (mean 2.255 Å) are comparable with those in [MnCl2(Py3P)(OH2)].13 The [Mn(hfac)3]2− anions have a tris-chelate motif, with the Mn(2)–O distances varying from 2.1372(14) to 2.1714(13) Å. Analysis of bond angles within the MnO6 polyhedron reveals that it can be best described as a twisted prism. On the whole, structures of the [Mn(hfac)3]2− anions in 8 are consistent with those in PyH[Mn(hfac)3]42 and K[Mn(hfac)3].43
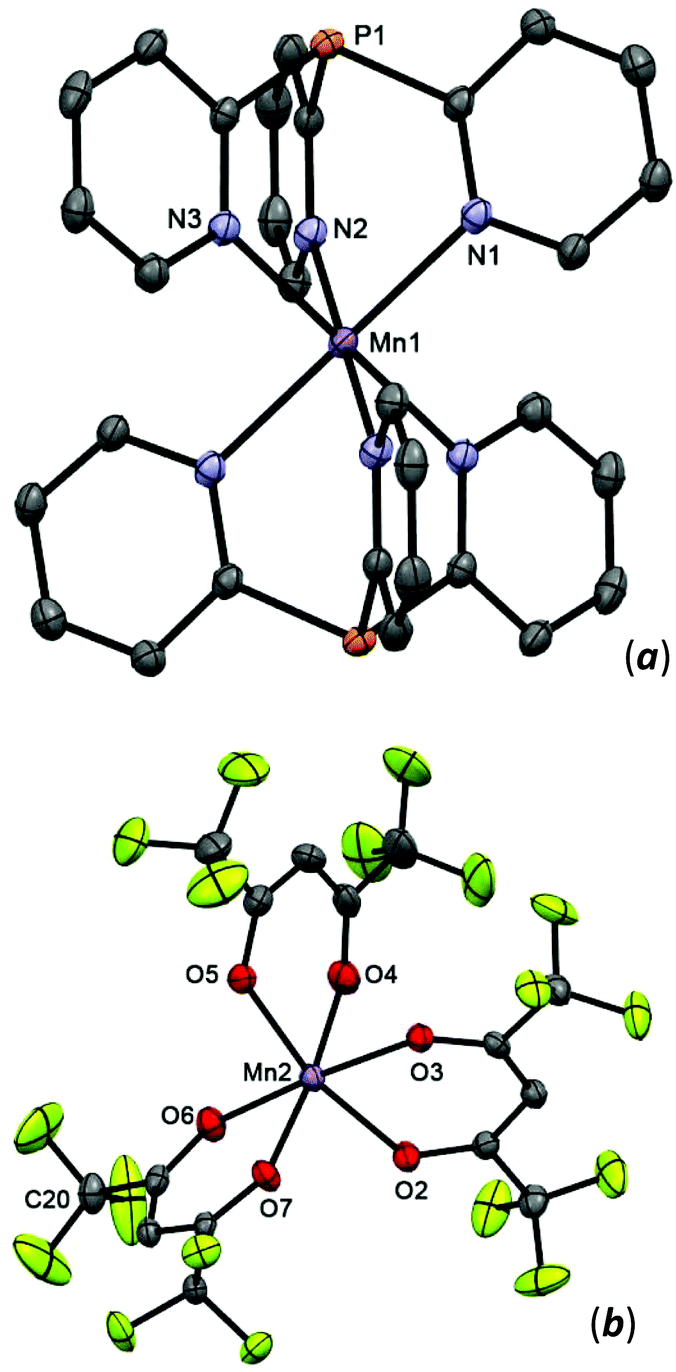 | ||
| Fig. 6 The structure of cationic [Mn(Py3P)2]2+ (a) and anionic [Mn(hfac)3]− (b) of 8 (50% thermal ellipsoid). The pyridine hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Mn(1)–N(1) 2.2647(15), Mn(1)–N(2) 2.2453(15), Mn(1)–N(3) 2.2558(15), Mn(2)–O (3) 2.1714(13), Mn(2)–O(6) 2.1643(14), Mn(2)–O(5) 2.1488(14), Mn(2)–O(7) 2.1469(14), Mn(2)–O(4) 2.1372(14), Mn(2)–O(2) 2.1513(14), N(1)–Mn(1)–N(1)′ 180.0, N(2)′–Mn(1)–N(2) 180.0, N3–Mn(1)–N(3)′ 180.00(4), N(2)–Mn(1)–N(1) 86.79(5), O(6)–Mn(2)–O(3) 175.35(5), O(5)–Mn(2)–O(2) 166.70(5), O(4)–Mn(2)–O(7) 173.90(6). | ||
2.3. Electrochemical properties
To elucidate the electrochemical features of the prepared complexes and possible products of their redox transformations, typically two successive cycles were measured. For the initial scan starting at 0.0 V (Ag/AgI), a potential was swept up to the positive direction. After two cycles were recorded, the experiment was repeated with the negative direction of the initial potential sweep. In all cases, voltammetry results at both Pt and glassy carbon (GC) electrodes were similar. The data for the first oxidation and first reduction peak potentials of the studied complexes have been summarized in the Table 1.| Complex | GC electrode | Pt electrode | ||
|---|---|---|---|---|
| E Ia, V (Ag/Ag+) | E Ic, V (Ag/Ag+) | E Ia, V (Ag/Ag+) | E Ic, V (Ag/Ag+) | |
| a Shoulder of the complex peak measured at the Nafion modified electrode (for details, see below). b Shoulder of the complex peak measured at the bare electrode. | ||||
| 3 | 0.987 | −0.415 | 1.032 | −0.483 |
| 4 | 0.980 | −0.349 | 1.045 | −0.371 |
| 5 | 0.958 | −0.436 | 0.968 | −0.378 |
| 6 | 0.969 | — | 1.057 | — |
| 7 | 0.942 | — | 0.972 | — |
| 8 | 0.94a | — | 1.17b | −0.906 |
Fig. 7 shows characteristic voltammograms of Py3P = O based scorpionates 4, 6 and 7. The anodic current peaks at 0.95–1.00 V correspond to the first oxidation of the complexes. CV measured in the Py3P = O solution of the same concentration demonstrates a weak quasi-reversible couple at the mentioned potentials, whereas the oxidation peaks of the studied complexes are electrochemically irreversible. It is worthwhile to note that neither the nature of the metal nor the type of ligand significantly influences the potential of the first oxidation peak. The peculiarity of CoII complex (6) is the appearance of the second anodic oxidation peak at E = 1.414 V and the cathodic peak at E = 0.552 V on the reverse branch of CV. In our opinion, these peaks might be associated with the CoII/CoIII redox transformations.
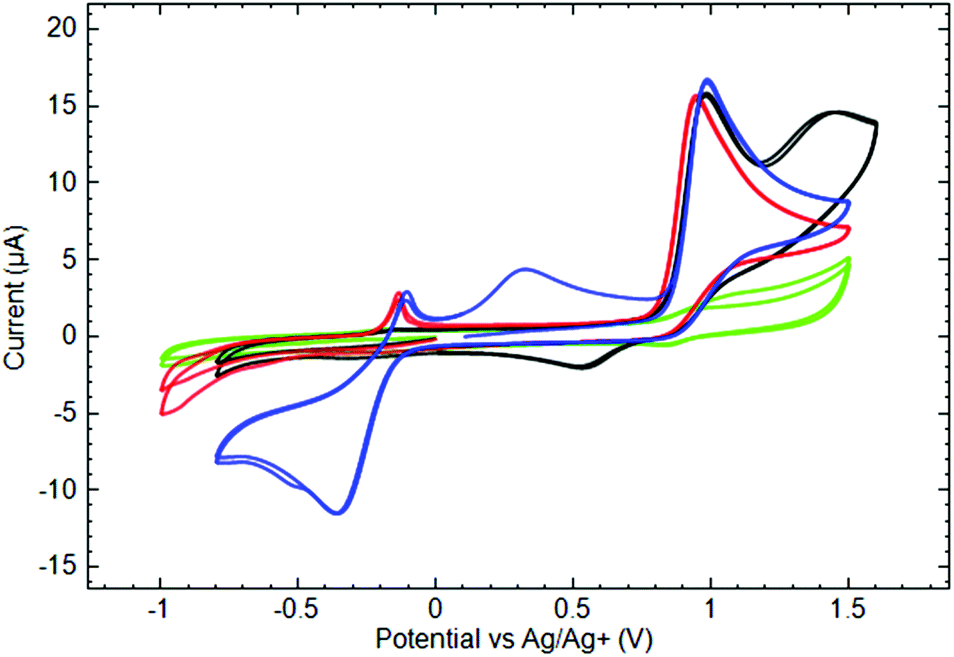 | ||
Fig. 7 Cyclic voltammograms of 10−3 M ( ) Py3P = O, ( ) Py3P = O, ( ) 4, (—) 6, and ( ) 4, (—) 6, and ( ) 7 in 0.1 M TBAP acetonitrile solutions measured at the GC electrode (ν = 50 mV s−1). ) 7 in 0.1 M TBAP acetonitrile solutions measured at the GC electrode (ν = 50 mV s−1). | ||
On the cathodic potential sweep in the solutions of CoII (6) and NiII (7) complexes at potentials more negative than −0.5 V, the growth of a cathodic current was observed, although there were no cathodic peaks in the studied potential window. Even so, the products of cathodic reduction appeared in the solution and might be responsible for the anodic peak at −0.150/−0.140 V on the anodic branch of the CV.
The CuII scorpionate 4 demonstrated different cathodic behavior. Thus for its solution, a clear cathodic current peak was measured at −0.349 V. Some parts of the shape of this peak were quite complicated and probably corresponded to several overlapping redox reactions: one part was the same as for the above-mentioned reduction process of the CoII and NiII complexes and was associated with the anodic peak at −0.140 V on the anodic branch of the CV, and another might have been associated to CuII/CuI reduction. This process is electrochemically irreversible, although the appearance of the broad anodic current peak at 0.317 V indicates an oxidation of a reduced complex on the reverse potential scan. Noteworthy, in this case, the cathodic scan was limited by potentials less negative than that needed for the first reduction, with the only anodic feature being an irreversible oxidation peak at the potential close to that of CoII and NiII complexes.
The data shown in Fig. 8 allow one to estimate the influence of the ligands’ nature and their number on the CuII complexes’ voltammetry. The first oxidation current peaks appear at ca. 0.95 V for all of the studied substances, again evidencing that the nature of the ligand has a minor effect on the oxidation peak position. In contrast, the number of ligands coordinated to the metal strongly affects the value of the anodic current measured at the peak. Indeed, comparing the CV measured in the phosphine complexes’ (3 and 5) solution of the same concentration, one could notice that for complex 5, bearing two ligands, the oxidation current is two times higher than that for mono-scorpionate 3.
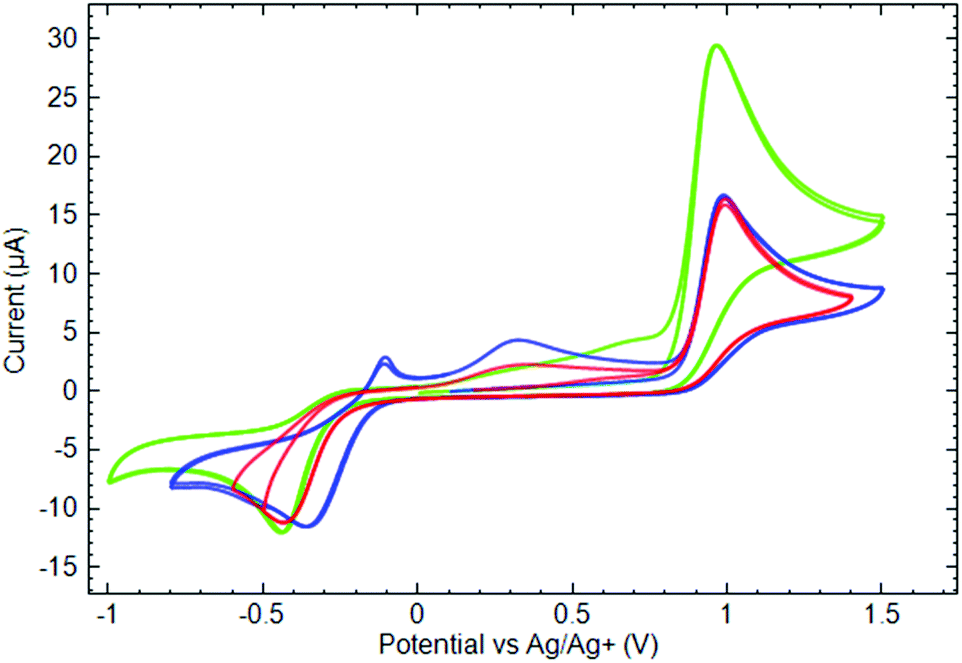 | ||
Fig. 8 Cyclic voltammograms of 10−3 M ( ) 3, ( ) 3, ( ) 4, and ( ) 4, and ( ) 5 in 0.1 M TBAP acetonitrile solutions measured at the GC electrode (ν = 50 mV s−1). ) 5 in 0.1 M TBAP acetonitrile solutions measured at the GC electrode (ν = 50 mV s−1). | ||
In the series of additional experiments by varying the potential sweep rate, it has been found that the oxidation current peak value linearly depends on the square root of the sweep rate (see ESI† for corresponding data). Such a result suggests a diffusion control on the oxidation process and the absence of the hindrances from adsorption.44 Taking into account the above-mentioned oxidation current ratio for mono- and bis-phosphine complexes, two assumptions can be deduced. First, the oxidation of complexes occurs presumably through the ligands. Second, for bis-scorpionate complex 5, both ligands are equally accessible for oxidation in the same potential scan.
On the cathodic branches of the CV (Fig. 8), there are irreversible peaks of cathodic current at −0.35/−0.45 V corresponding to the first reduction of the studied compounds. In contrast to the oxidation, the potentials of the reduction peak depend significantly on the type of ligand. In fact, for both phosphine complexes 3 and 5, their reductions occur at potentials about 100 mV more negative than that for phosphine oxide 4. Considering the oxygen atom in the phosphine oxide ligand as an electron withdrawing substituent, one could speculate on the role of such a center facilitating the reduction of the complex at less negative potentials. Despite the irreversibility of the cathodic peaks, the reduction products of all copper complexes undergo oxidation on the second anodic potential sweep, contributing to the growth of the anodic current in a potential region of 0.0/0.8 V.
As complex [Mn(Py3P)2][Mn(hfac)3]2 (8) contains Mn2+ ions both in the cationic and in the anionic parts, we attempted to separate their voltammetric responses. Because the MnII scorpionate composes the cationic part of the complex, CVs were measured at the Nafion coated GC electrode. According to Zook et al.,45 Nafion behaves as a swelling membrane in acetonitrile in a similar way as in water solutions. The application of Nafion as a cation exchange membrane in non-aqueous solutions containing TBA+ as a background cation is also known.46 Voltammograms measured at the Nafion coated GC electrode remarkably differ from those measured at a bare GC (see ESI† for corresponding data). Fig. 9 shows CVs measured at the Nafion modified GC electrode in the 8 solution. At first glance, the anodic behavior of 8 differs significantly from all other studied complexes and demonstrates quasi-reversible characteristics of the first oxidation. However, in fact, the first oxidation current wave should be considered as a result of at least two overlapping processes. By varying the value limiting the anodic potential sweep, it was possible to confirm this suggestion. Indeed, until the anodic potential limit does not exceed 0.95 V, the oxidation remains irreversible and resembles other studied complexes. An inflection appears on the anodic branch of the voltammogram at the mentioned potential value and indicates the transition to the next redox process. Expanding the anodic potential limit above 0.95 V, leads to a registration of the cathodic current peak at 0.8 V on the reverse potential scan. Finally, at potentials more positive than 1.0 V, there is one more irreversible anodic current wave. Comparing the anodic behavior of 8 with other studied complexes, one can conclude that, in all cases, oxidation first involves the ligand moiety and demonstrates an irreversible characteristic. At the more positive potentials, the Mn site might be considered as an apparent target for oxidation.
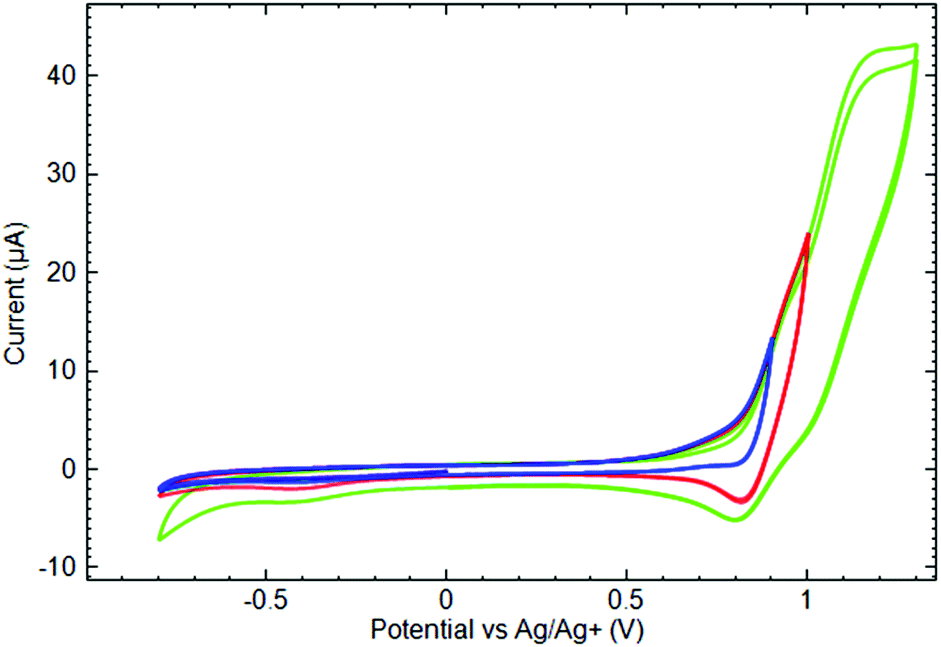 | ||
| Fig. 9 Cyclic voltammograms of 10−3 M [Mn(Py3P)2][Mn(hfac)3]2 (8) in 0.1 M TBAP acetonitrile solution measured at a Nafion modified GC electrode by varying the positive sweep potential limit (ν = 50 mV s−1). | ||
CV measured at the bare Pt electrode in the 8 solution shows the irreversible cathodic current peak at −0.93 V and the anodic peak at 0.09 V on the reverse potential sweep. However, none of them are reproduced at the modified GC electrode. Indeed, in the studied potential region, there were no reduction peaks on the cathodic branch of the CV measured in the solution of 8 on the Nafion coated GC electrode, except for those assigned to the reduction of the products obtained in the anodic potential sweep. Such a difference can be attributed to the presence of MnII complexes both in cationic and in anionic parts of 8.
2.4. Magnetic properties
Magnetic susceptibility data for complexes 3–8 were collected as a function of temperature from 2 to 300 K. Fig. 10 shows a representative plot of CuII complexes 3–5. For 3, the μeff value at 300 K is 1.91, which is in good agreement with the theoretical value (1.86μB) for CuII ion, i.e. one paramagnetic centre with spin S = 1/2 and g = 2.15. The μeff value virtually does not change at lower temperatures down to 2 K. Thus, the characteristic of the μeff(T) dependence reveals the absence of significant exchange interactions and isolation of the paramagnetic centres that is consistent with X-ray analysis data. As seen from Fig. 10, complexes 4 and 5 exhibit nearly the same magnetic behavior; their μeff values at 300 K are 1.84 and 1.83μB, respectively.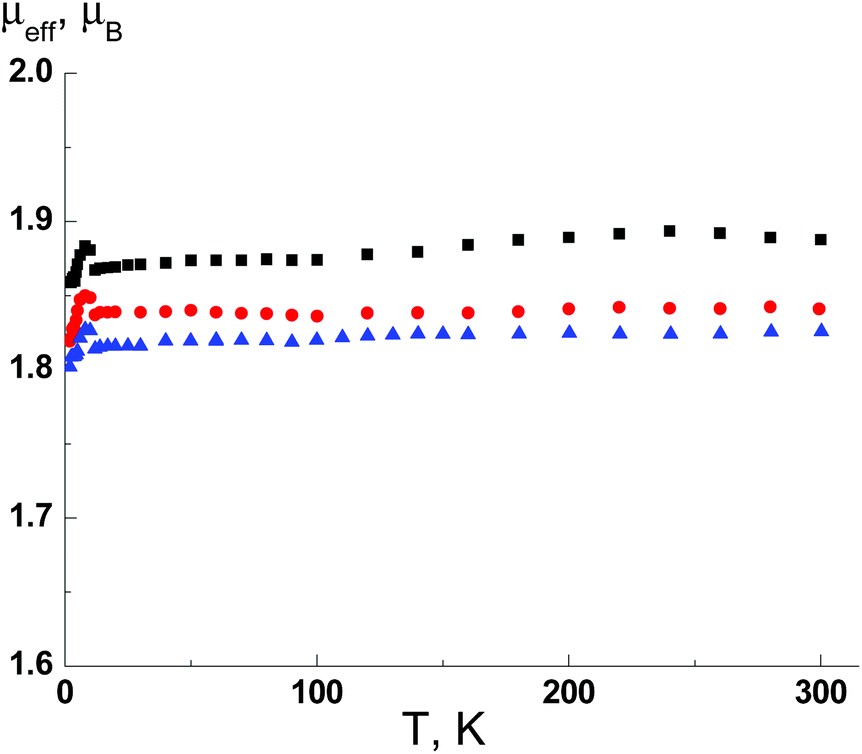 | ||
| Fig. 10 The temperature dependence of the effective magnetic moment μeff(T) for 3 (■), 4 (●) and 5 (▲). | ||
The temperature dependence μeff for CoII complex (6) is presented in Fig. 11. At a lower temperature, μeff smoothly decreases from 4.67μB (300 K) to 3.46μB (2 K). The high temperature value of μeff is higher than the theoretical spin-only value for CoII (S = 3/2, g = 2), which is consistent with the orbital contribution to the magnetic susceptibility, typical for CoII ions in the octahedral environment and g-factors larger than 2. The μeff value at 300 K agrees well with typical values of 4.3–5.2μB for the CoII ion. The decrease of μeff at lower temperatures is due to the spin–orbit interaction. The μeff value at low temperature is close to the theoretical spin-only value for the CoII ion and indicates the absence of significant exchange interactions between the paramagnetic centres.
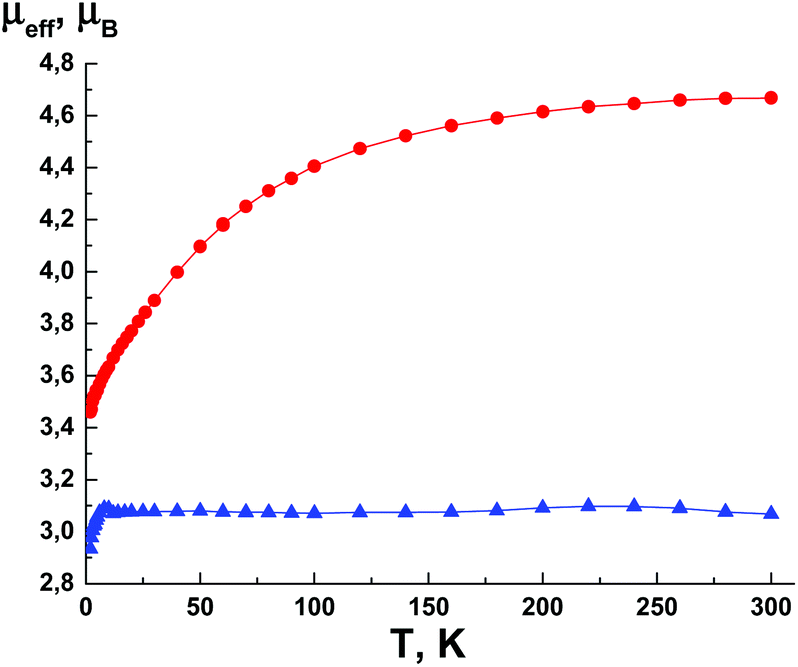 | ||
| Fig. 11 The temperature dependence of the effective magnetic moment μeff(T) for 6 (●)and 7 (▲). | ||
For NiII complex (7), the μeff value at 300 K is 3.07μB (Fig. 11), which agrees well with the theoretical value of 3.11μB for NiII with spin S = 1 and the g-factor of 2.2. Therefore, the dependence μeff(T) for 7 is quite similar to that for CuII complexes 3–5 (Fig. 10), showing the absence of significant exchange interactions between the paramagnetic centres.
For MnII complex (8), the μeff value at 300 K is 10.17μB and practically does not change when the temperature is lowered to 25 K but decreases to 9.77μB at 2 K. The μeff value for the temperature range of 300–25 K is in good agreement with the theoretical spin-only value, 10.25μB, for three non-interacting paramagnetic centers with S = 5/2 and g = 2 (MnII ions). The decreasing of μeff value below 25 K is due to weak antiferromagnetic exchange interactions.
Thus, magnetic measurements show that the paramagnetic nature of complexes 3–8 is defined by the MII ions only; any exchange interactions between the paramagnetic centres are negligibly small. A magnetic saturation plays a role at low temperatures, where μH ∼ kT causes a small decrease of μeff. For comparison, the theoretical dependence of μeff(T), that takes into account the magnetic saturation, is shown in Fig. 12 by the solid line.
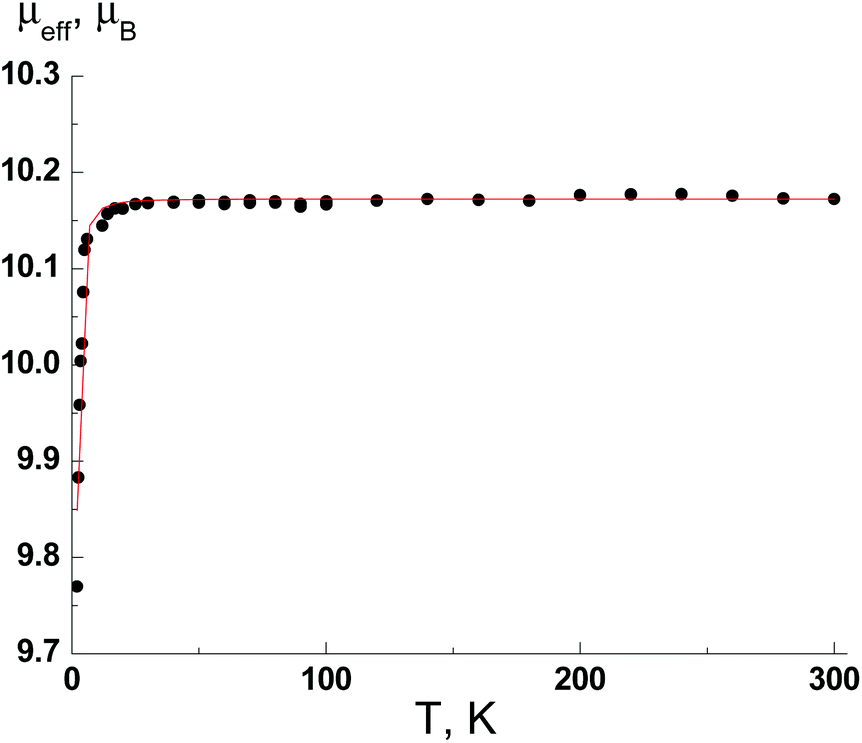 | ||
| Fig. 12 The temperature dependence of the effective magnetic moment μeff(T) for 8. | ||
3. Conclusions
In summary, the coordination of tris(2-pyridyl)phosphine or its oxide toward M(hfac)2 is accompanied by an unexpected replacement of the hfac-ligands by one or two tripodal ligands to afford mono- or bis-scorpionate complexes, respectively. The switching between the formation of one or another structure strongly depends on the nature of MII and, in the case of Cu(hfac)2, on the M/L ratio used. In the reaction with Cu(hfac)2, the alteration of the Cu/L ratio from 1:1 to 1:2 alters the selectivity of the product formation from a mono-scorpionate to a bis-scorpionate complex, correspondingly. Moreover, NiII and CoII hexafluoroacetylacetonates yield exceptional mono-scorpionates, even if the M/L ratio is 1:2. In contrast, Mn(hfac)2, upon reacting with Py3P, gives a complex composed of a bis-scorpionate cation [Mn(Py3P)2]2+ and two [Mn(hfac)3]− counterions.
CV measurements have shown that all synthesized compounds undergo irreversible oxidation at 0.95–1.00 V. The first electrochemical oxidation can be considered as an apparently ligand-centered process. Both the metal nature and the ligand type significantly influence the reduction of the complexes. The irreversibility of the voltammetric peaks suggests that an oxidation as well as a reduction of the studied complexes leads to significant structural changes or can be followed by some chemical transformations.
The magnetic measurements display that the coordination of tris(2-pyridyl)phosphine or its oxide to the MII ion leads to the disappearance of the exchange interactions. As a result, paramagnetic MII ions are well isolated magnetically from each other. Therefore, the tris(2-pyridyl)phosphine and its oxide are promising ligands in the design of single molecular (SMM) or single ion (SIM) magnets.
Keeping in mind the known data,12,13 the synthesized complexes can be regarded as potential catalysts for organic transformations. Moreover, they are useful building blocks for the assembly of new heterometallic complexes, e.g. through coordination of the P atoms of 3, 5 or 8 to the “soft” metal ions.
All of the results obtained contribute to the coordination chemistry of tripodal ligands and scorpionate complexes. The disclosed substitution of the anionic chelating ligand in the coordination sphere by the neutral tripodal ligand opens up new opportunities for the structure-oriented design of coordination compounds.
4. Experimental
4.1 General details
Solvents were purified following standard procedures prior to use. Tris(2-pyridyl)phosphine (1) was synthesized from red phosphorus and 2-bromopyridine according to the published method.14 Phosphine oxide 2 was prepared by oxidation of 1 with H2O2 in water/acetone media. The M(hfac)2 were prepared by the method of Bertrand and Kaplan.47Elemental analyses were performed using a Flash EA 1112 CHNS analyzer. Melting points were determined with a Kofler micro hot stage. The IR spectra were recorded on a Varian 3100 FT-IR spectrometer with an ATR sample setting. XPRD analysis of samples 3–8 (see Fig. S20–S24†) was performed on a Shimadzu XRD-7000 diffractometer (Cu Kα radiation, Ni-filter, 5–35° 2θ range, 0.03° 2θ step, 5 s per point).
4.2 Electrochemistry
CVs have been recorded using Autolab PGSTAT 128N (The Netherlands) in MeCN solutions containing 0.1 M TBAP as a supporting electrolyte. Measurements were performed at room temperature in a convenient three electrode cell with either Pt (area 2.01 mm2; BAS) or glassy carbon (GC, area 7.07 mm2) working electrodes, Pt mesh counter electrode and a Ag/AgI reference electrode prepared with a BAS reference electrode kit filled with a CH3CN solution containing 0.1 M TBAP and 0.01 M AgNO3. The Ag/AgI reference electrode has been characterized in 0.1 M TBAP CH3CN solution containing 1 mM ferrocene (FeCp2). Both on Pt and on GC working electrodes, the FeCp2+/FeCp2 couple shows E1/2 = 0.120 V [estimated as (Eap − Ecp)/2] vs. a thus-prepared Ag/Ag+ reference electrode. All potentials are given against this reference electrode. The concentrations of studied complexes were 1 mM, and a potential sweep rate (ν) was 50 mV s−1. Prior to CV measurements, argon gas (99.99%) was passed through the solution in the cell for 10 min. For several CV experiments, the GC electrode modified by Nafion was prepared. Nafion solution (Aldrich) was coated onto the GC surface in a manner described earlier.48 In order to equilibrate the Nafion film against the background electrolyte before CV measurement, the electrode was kept overnight in the MeCN 0.1 M TBAP solution.4.3 Magnetochemistry
The magnetic susceptibility of the polycrystalline samples 3–8 was measured with a Quantum Design MPMSXL SQUID magnetometer in the temperature range of 2–300 K with a magnetic field of up to 5 kOe. None of the complexes exhibited any field dependence of molar magnetization at low temperatures. Diamagnetic corrections were made using the Pascal constants. The effective magnetic moment was calculated as μeff(T) = [(3k/NAμB2)χT]1/2 ≈ (8χT)1/2.4.4 X-Ray crystallography
The single crystals of 3–7 and 8·2Me2CO were grown by slow liquid diffusion of hexane (ca. 7 mL) into an acetone or chloroform solution of these complexes (about 50 mg in 5 mL of the solvent). The mixture was allowed to stay at room temperature for a day. The precipitated crystals were collected, washed with a hexane/acetone (or chloroform) mixture and dried in air. Data were collected on a Bruker D8 Venture diffractometer with Mo Kα (λ = 0.71073 Å) radiation using the φ and ω scans. An empirical absorption correction was applied using the SADABS program.49 The structures were solved and refined by direct methods using SHELX.50 All non-hydrogen atoms were refined anisotropically using SHELX.50 The coordinates of the hydrogen atoms were calculated from geometrical positions. The CF3 groups within 3 (C4) and 8 (C20) were rotationally disordered, and two conformers were thus observed in the solid state.CCDC 1431158 (3), 1431160 (4), 1431159 (5), 1033904 (6), 1036445 (7) and 1035512 (8) contain the supplementary crystallographic data for this article.
4.5 Synthetic procedures
:1) mixture and dried in air. Yield: 197 mg (88%). Green crystals, mp 198–199 °C. IR (ATR): ν = 574 (m), 588 (m), 658 (s), 673 (m), 753 (m), 772 (s), 787 (s), 793 (s), 799 (m), 908 (w), 935 (w), 1004 (m), 1019 (m), 1059 (m), 1070 (m), 1127–1251 (vs, νC–F in CF3), 1354 (w), 1428 and 1452 (m, νCC in Py), 1530 and 1546 (m, νCN in Py), 1587 (w), 1644 (s, νCO) 1669 (m, νCO), 3014 (w), 3066 (w) cm−1. Anal. Calcd for C25H14CuF12N3O4P (742.90): C, 40.42; H, 1.90; N, 5.66. Found: C, 40.59; H, 1.89; N, 5.46.
[Cu(N,N′,N′′-Py
3
P = O)(O,O’-hfac)(O-hfac)] (4) was prepared from [Cu(hfac)2(H2O)2] (146 mg, 0.28 mmol) and tris(2-pyridyl)phosphine oxide (80 mg, 0.28 mmol) by a procedure similar to the one used for complex 3. Yield: 172 mg (80%). Green crystals, mp 177–182 °C. IR (ATR): ν = 577 (m), 659 (s), 665 (s), 736 (m), 748 (s), 771 (m), 794 (m), 938 (w), 1007 (m), 1019 (m), 1048 (m), 1059 (m), 1088 (s), 1122–1253 (vs, νC–F in CF3), 1433, 1439 and 1455 (m, νCC in Py), 1487 (m), 1526 and 1548 (m, νCN in Py), 1589 (w), 1646 (m, νCO), 2925–3264 br (w) cm−1. Anal. Calcd for C25H14CuF12N3O5P (758.90): C, 39.57; H, 1.86; N, 5.54. Found: C, 39.42; H, 2.01; N, 5.47.
[Cu(N,N′,N′′-Py
3
P)
2
](hfac)
2
(5) was prepared from [Cu(hfac)2(H2O)2] (73 mg, 0.14 mmol) and tris(2-pyridyl)phosphine oxide (80 mg, 0.28 mmol) by a procedure similar to the one used for complex 3 using MeCN as solvent. Yield: 112 mg (78%). Turquois crystals, mp 158–165 °C. IR (ATR): ν = 569 (m), 631 (w), 657 (s), 713 (m), 745 (m), 770 (s), 780 (s), 883 (w), 935 (m), 1004 (m), 1017 (m), 1061 (m), 1113–1242 (vs, νC–F in CF3), 1278 (w), 1422 and 1455 (m, νCC in Py), 1459 (m), 1524 and 1555 (m, νCN in Py), 1570 (m), 1582 (m), 1669 (s, νCO), 3053 (w) cm−1. Anal. Calcd for C40H26CuF12N6O4P2 (1008.15): C, 47.65; H, 2.60; N, 8.34. Found: C, 47.45; H, 2.51; N, 8.27.
[Co(N,N′,N′′-Py
3
P = O)(O,O′-hfac)(O-H2O)](hfac) (6) was prepared from [Co(hfac)2(H2O)2]·H2O (150 mg, 0.28 mmol) and tris(2-pyridyl)phosphine oxide (80 mg, 0.28 mmol) using acetone as solvent. Yield: 187 mg (85%). Orange-red crystals, mp 127–130 °C. IR (ATR): ν = 570 (m), 574 (m), 583 (m), 594 (w), 618 (w), 659 (m), 665 (m), 674 (m), 737 (m), 750 (m), 772 (m), 796 (m), 937 (w), 1006 (w), 1018 (w), 1058 (w), 1072 (m), 1094 (m), 1147–1254 (vs, νC–F in CF3), 1432 and 1455 (m, νCN in Py), 1529 and 1547 (m, νCN in Py), 1589 (w), 1646 and 1669 (s, νCO), 3018–3295 br (w) cm−1. Anal. Calcd for C25H16CoF12N3O6P (772.30): C, 38.88; H, 2.09; N, 5.44. Found: C, 38.71; H, 2.15; N, 5.61.
[Ni(N,N′,N′′-Py
3
P = O)(O,O′-hfac)(O-H2O)](hfac) (7) was prepared from [Ni(hfac)2(H2O)2] (145 mg, 0.28 mmol) and tris(2-pyridyl)phosphine oxide (80 mg, 0.28 mmol) by a procedure similar to the one used for complex 3. Yield: 178 mg (81%). Pale green crystals, mp 151–153 °C. IR (ATR): ν = 576 (m), 587 (m), 659 (m), 670 (m), 738 (m), 750 (m), 772 (m), 795 (m), 938 (w), 1006 (w), 1018 (m), 1058 (m), 1072 (m), 1094 (m), 1099 (m), 1147–1254 (vs, νC–F in CF3), 1433, 1439 and 1455 (m, νCN in Py), 1486 (m), 1528 and 1547 (m, νCN in Py), 1590 (w), 1647 and 1669 (s, νCO), 2952–3396 br (w) cm−1. Anal. Calcd for C25H16NiF12N3O6P (772.06): C, 38.89; H, 2.09; N, 5.44. Found: C, 38.74; H, 2.18; N, 5.35.
[Mn(N,N′,N′′-Py
3
P)
2
][Mn(hfac)
3
]
2
(8) was prepared from [Mn(hfac)2(H2O)2] (172 mg, 0.34 mmol) and tris(2-pyridyl)phosphine (60 mg, 0.23 mmol) in MeCN (3 mL). Yield: 152 mg (69%). Yellow crystals, ca. 220 °C dec. IR (ATR): ν = 579 (s), 642 (m), 662 (s), 711 (w), 720 (w), 740 (m), 748 (m), 762 (m), 773 (s), 792 (s), 806 (m), 900 (w), 946 (m), 976 (w), 1010 (s), 1057 (s), 1091 (s), 1126–1246 (vs, νC–F in CF3), 1341 (w), 1427 and 1459 (m, νCC in Py), 1503 (s), 1524 and 1551 (m, νCN in Py), 1584 (m), 1647 (s, νCO) cm−1. Calcd for C60H30F36Mn3N6O12P2 (1937.62): C, 37.19; H, 1.56; N, 4.34. Found: C, 37.04; H, 2.01; N, 4.10. The crystals of 8 were redissolved in acetone, and the yellow solution was layered with hexane. After several days, X-ray quality crystals of 8·2Me2CO (found: C, 38.41; H, 2.17; N, 4.17. Calcd: C, 38.60; H, 2.06; N, 4.09) were precipitated.
Acknowledgements
The authors acknowledge the financial support from the Russian Foundation for Basic Research (Grant 15-03-05591).Notes and references
- G. R. Newkome, Chem. Rev., 1993, 93, 2067–2089 CrossRef CAS.
- P. Espinet and K. Soulantica, Coord. Chem. Rev., 1999, 193–195, 499–556 CrossRef CAS.
- A. P. Sadimenko, Adv. Heterocycl. Chem., 2011, 104, 391–475 CrossRef CAS.
- R. J. Somerville, Pyridyl phosphine complexes in the design of hydration catalysts, PhD thesis, Victoria University of Wellington, 2015 Search PubMed.
- M. Wallesch, D. Volz, D. M. Zink, U. Schepers, M. Nieger, T. Baumann and S. Bräse, Chem. – Eur. J., 2014, 20, 6578–6590 CrossRef CAS PubMed.
- J. J. Liu, P. Galettis, A. Farr, L. Maharaj, H. Samarasinha, A. C. McGechan, B. C. Baguley, R. J. Bowen, S. J. Berners-Price and M. J. McKeage, J. Inorg. Biochem., 2008, 102, 303–310 CrossRef CAS PubMed.
- E. Drent, P. Arnoldy and P. H. M. Budzelaar, J. Organomet. Chem., 1993, 455, 247–253 CrossRef CAS.
- E. Drent, P. Arnoldy and P. H. M. Budzelaar, J. Organomet. Chem., 1994, 475, 57–63 CrossRef CAS.
- C.-Y. Kuo, Y.-S. Fuh, J.-Y. Shiue, S. J. Yu, G.-H. Lee and S.-M. Peng, J. Organomet. Chem., 1999, 588, 260–267 CrossRef CAS.
- H.-S. Wang and S. J. Yu, Tetrahedron Lett., 2002, 43, 1051–1055 CrossRef CAS.
- K. Kurtev, D. Ribola, R. A. Jones, D. J. Cole-Hamilton and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1980, 55–58 RSC.
- A. Karam, R. Tenia, M. Martinez, F. López-Linares, C. Albano, A. Diaz-Barrios, Y. Sánchez, E. Catarí, E. Casas, S. Pekerar and A. Albornoz, J. Mol. Catal. A: Chem., 2007, 265, 127–132 CrossRef CAS.
- A. N. Kharat, B. T. Jahromi and A. Bakhoda, Transition Met. Chem., 2012, 37, 63–69 CrossRef CAS.
- B. A. Trofimov, A. V. Artem'ev, S. F. Malysheva, N. K. Gusarova, N. A. Belogorlova, A. O. Korocheva, Yu. V. Gatilov and V. I. Mamatyuk, Tetrahedron Lett., 2012, 53, 2424–2427 CrossRef CAS.
- A. Steiner and D. Stalke, Organometallics, 1995, 14, 2422–2429 CrossRef CAS.
- C. J. L. Lock and M. A. Turner, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 2096–2099 CrossRef.
- G. Zhang, J. Zhao, G. Raudaschl-Sieber, E. Herdtweck and F. E. Kuhn, Polyhedron, 2002, 21, 1737–1746 CrossRef CAS.
- S. A. S. Anaya, A. Hagenbach and U. Abram, Polyhedron, 2008, 27, 3587–3592 CrossRef.
- F.-W. Lee, M. C.-W. Chan, K.-K. Cheung and C.-M. Che, J. Organomet. Chem., 1998, 563, 191–200 CrossRef CAS.
- R. P. Shutte, S. J. Rettig, A. M. Joshi and B. R. James, Inorg. Chem., 1997, 36, 5809–5817 CrossRef.
- A. Bakhoda, N. Safari, V. Amani, H. R. Khavasi and M. Gheidi, Polyhedron, 2011, 30, 2950–2956 CrossRef CAS.
- A. V. Artem'ev, N. K. Gusarova, S. F. Malysheva, O. N. Kazheva, G. G. Alexandrov, O. A. Dyachenko and B. A. Trofimov, Mendeleev Commun., 2012, 22, 294–296 CrossRef.
- K. R. Adam, P. A. Anderson, T. Astley, I. M. Atkinson, J. M. Charnock, C. D. Garner, J. M. Gulbis, T. W. Hambley, M. A. Hitchman, F. R. Keene and E. R. T. Tiekink, J. Chem. Soc., Dalton Trans., 1997, 519–530 RSC.
- T. Astley, M. A. Hitchman, F. R. Keene and E. R. Tiekink, J. Chem. Soc., Dalton Trans., 1996, 1845–1851 RSC.
- S. Hanf, R. Garcia-Rodriguez, A. D. Bond, E. Hey-Hawkins and D. S. Wright, Dalton Trans., 2016, 45, 276–283 RSC.
- A. V. Artem'ev, N. K. Gusarova, V. A. Shagun, S. F. Malysheva, V. I. Smirnov, T. N. Borodina and B. A. Trofimov, Polyhedron, 2015, 90, 1–6 CrossRef.
- T. Gneuß, M. J. Leitl, L. H. Finger, N. Rau, H. Yersin and J. Sundermeyer, Dalton Trans., 2015, 44, 8506–8520 RSC.
- A. V. Artem'ev, E. P. Doronina, M. I. Rakhmanova, A. O. Sutyrina, I. Yu. Bagryanskaya, P. M. Tolstoy, A. L. Gushchin, A. S. Mazur, N. K. Gusarova and B. A. Trofimov, New J. Chem., 2016, 40, 10028–10040 RSC.
- A. G. Walden and A. J. M. Miller, Chem. Sci., 2015, 6, 2405–2410 RSC.
- B. A. Trofimov, N. K. Gusarova, A. V. Artem'ev, S. F. Malysheva, N. A. Belogorlova, A. O. Korocheva, O. N. Kazheva, G. G. Alexandrov and O. A. Dyachenko, Mendeleev Commun., 2012, 22, 187–188 CrossRef CAS.
- M. D. Le Page, B. O. Patrick, S. J. Rettig and B. R. James, Inorg. Chim. Acta, 2015, 425, 198–210 CrossRef CAS.
- D. T. Richens, Chem. Rev., 2005, 105, 1961–2002 CrossRef CAS PubMed.
- A. W. Maverick, F. R. Fronczek, E. F. Maverick, D. R. Billodeaux, Z. T. Cygan and R. A. Isovitsch, Inorg. Chem., 2002, 41, 6488–6492 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751–767 CrossRef.
- M. A. Halcrow, Chem. Soc. Rev., 2013, 42, 1784–1795 RSC.
- Md. B. Zaman, K. A. Udachin and J. A. Ripmeester, CrystEngComm, 2002, 4, 613–617 RSC.
- S. Delgado, A. Barrilero, A. Molina-Ontoria, M. E. Medina, C. J. Pastor, R. Jiménez-Aparicio and J. L. Priego, Eur. J. Inorg. Chem., 2006, 2746–2759 CrossRef CAS.
- R. J. Bowen, M. A. Fernandes, P. W. Gitari and M. Layh, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, C60, o258–o260 CAS.
- T. Astley, H. Headlam, M. A. Hitchman, F. R. Keene, J. Pilbrow, H. Stratemeier, E. R. T. Tiekink and Y. C. Zhong, J. Chem. Soc., Dalton Trans., 1995, 3809–3818 RSC.
- A. Santoro, C. Sambiagio, P. C. McGowan and M. A. Halcrow, Dalton Trans., 2015, 44, 1060–1069 RSC.
- R. T. Jonas and T. D. P. Stack, Inorg. Chem., 1998, 37, 6615–6629 CrossRef CAS PubMed.
- E. Bouwman, K. G. Caulton, G. Christou, K. Folting, C. Gasser, D. N. Hendrickson, J. C. Huffman, E. B. Lobkovsky and J. D. Martin, Inorg. Chem., 1993, 32, 3463–3470 CrossRef CAS.
- S. I. Troyanov, O. Yu. Gorbenko and A. A. Bosak, Polyhedron, 1999, 18, 3505–3509 CrossRef CAS.
- R. G. Compton and G. E. Banks, Understanding Voltammetry, Imperial College Press, London, 2nd edn, 2011, 429 p Search PubMed.
- L. A. Zook and J. Leddy, Anal. Chem., 1996, 68, 3793–3796 CrossRef CAS PubMed.
- I. L. Escalante-Garcia, J. S. Wainright, L. T. Thompson and R. F. Savinell, J. Electrochem. Soc., 2015, 162, A363–A372 CrossRef CAS.
- J. A. Bertrand and R. I. Kaplan, Inorg. Chem., 1966, 5, 489–491 CrossRef CAS.
- A. V. Kashevskii, A. Y. Safronov and O. Ikeda, J. Electroanal. Chem., 2001, 510, 86–95 CrossRef CAS.
- SADABS, v. 2008-1, Bruker AXS, Madison, WI, USA, 2008 Search PubMed.
- G. M. Sheldrick, SHELX-97, Programs for Crystal Structure Analysis (Release 97-2), University of Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of FT-IR, excitation and emission spectra; computation details. CCDC 1431158, 1431160, 1431159, 1033904, 1036445 and 1035512. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt00339k |
| This journal is © The Royal Society of Chemistry 2017 |