
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Charge transfer complexes of metal-free phthalocyanine radical anions with decamethylmetallocenium cations: (Cp*2Co+)(H2Pc˙−)·solvent and (Cp*2Cr+)(H2Pc˙−)·4C6H4Cl2†
Dmitri V.
Konarev
 *a,
Salavat S.
Khasanov
b,
Manabu
Ishikawa
cd,
Akihiro
Otsuka
cd,
Hideki
Yamochi
cd,
Gunzi
Saito
ef and
Rimma N.
Lyubovskaya
a
*a,
Salavat S.
Khasanov
b,
Manabu
Ishikawa
cd,
Akihiro
Otsuka
cd,
Hideki
Yamochi
cd,
Gunzi
Saito
ef and
Rimma N.
Lyubovskaya
a
aInstitute of Problems of Chemical Physics RAS, Chernogolovka, Moscow Region, 142432 Russia. E-mail: konarev3@yandex.ru
bInstitute of Solid State Physics RAS, Chernogolovka, Moscow Region, 142432 Russia
cDivision of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan
dResearch Center for Low Temperature and Materials Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan
eFaculty of Agriculture, Meijo University, 1-501 Shiogamaguchi, Tempaku-ku, Nagoya 468- 8502, Japan
fToyota Physical and Chemical Research Institute, 41-1, Yokomichi, Nagakute, Aichi 480-1192, Japan
First published on 14th February 2017
Abstract
Charge transfer complexes (Cp*2Co+)(H2Pc˙−)·0.5C6H4Cl2·0.7C6H5CN·0.3C6H14 (1) and (Cp*2Cr+)(H2Pc˙−)·4C6H4Cl2 (2) have been obtained as single crystals. Both complexes contain metal-free phthalocyanine (Pc) radical anions and decamethylmetallocenium cations. Reduction of the Pc macrocycle leads to the appearance of new bands at 1026–1030 nm in the NIR range and blue shifts of both Soret and Q-bands of H2Pc in the spectra of 1 and 2. The geometry of the Pc macrocycles supports the formation of H2Pc˙− by the alternation of shorter and longer C–N(imine) bonds in the macrocycles in 2. Complex 1 contains pairs of H2Pc˙− having effective π–π interactions with two sandwiched Cp*2Co+ cations, whereas complex 2 contains stacks composed of alternating Cp*2Cr+ and H2Pc˙− ions. The magnetic moment of 1 is 1.64 μB at 300 K due to the contribution of the H2Pc˙− spins with the S = 1/2 state and diamagnetism of Cp*2Co+. This is supported by the observation of a narrow EPR signal of 1 with g = 2.0032–2.0036 characteristic of H2Pc˙−. Strong antiferromagnetic coupling of spins with a Weiss temperature of −23 K is observed between H2Pc˙− in 1. This coupling is probably mediated by the Cp*2Co+ cations. The magnetic moment of 2 is 4.18 μB at 300 K indicating the contribution of both paramagnetic H2Pc˙− (S = 1/2) and Cp*2Cr+ (S = 3/2) species. In spite of the presence of stacks of alternating ions in 2, only weak magnetic coupling is observed with a Weiss temperature of −4 K most probably due to ineffective π–π interactions between Cp*2Cr+ and H2Pc˙−. The EPR spectrum of 2 contains an asymmetric signal attributed to CrIII (g1 = 3.9059–3.9220) and a narrow Lorentzian signal from H2Pc˙− with g2 = 1.9943–1.9961. In addition to these signals, a broad EPR signal grows in intensity below 80 K with g4 = 2.1085–2.2438 which can be attributed to both paramagnetic Cp*2Cr+ and H2Pc˙− species having exchange interactions.
Introduction
Metal phthalocyanines can be promising components in the design of conducting and magnetic compounds.1–6 Chemical or electrochemical oxidation of metal phthalocyanines or the {MIII(CN)2Pc}− anions yields partially oxidized phthalocyanine macrocycles which with appropriate packing can provide highly conducting properties including one-dimensional metal conductivity stable down to liquid helium temperatures.1–4 Since metal phthalocyanines can contain paramagnetic metals, they can also be used as active components in the design of magnetic assemblies. For example, the oxidation of manganese(II) phthalocyanine or substituted naphthalocyanines by tetracyanoethylene yields polymeric compounds with the alternation of the (MnIIIPc)+ and TCNE˙− ions. These compounds show ferrimagnetic ordering of spins.5,6Metal-free and metal-containing phthalocyanines have weak acceptor properties7 and can be reduced by strong donors like alkali metals, LiCp*, or sodium fluorenone ketyl to yield radical anion salts.8–17 Decamethylchromocene is also a strong donor and forms a variety of charge transfer complexes with different planar π-acceptors most of which show promising magnetic properties.18–20 The first reduction potentials of metal-free and metal-containing phthalocyanines are in the −0.4 – −0.8 V range.7 Since decamethylchromocene (Cp*2Cr) has an Eox of −1.04 V vs. SCE21 and decamethylcobaltocene (Cp*2Co) has an Eox of −1.44 V vs. SCE,21 they can reduce phthalocyanines to form charge transfer (CT) complexes. The first compound of such type was obtained with iron(II) phthalocyanine, FeIIPc. Cp*2Cr reduces the FeIIPc forming CT complex (Cp*2Cr+){FeI(Pc2−)}−·4C6H4Cl2, which contains stacks of alternating [FeI(Pc2−)]− and Cp*2Cr+ ions. Alternation of these ions in the stacks having different spin states [FeI(Pc2−)]− (FeI, S = 1/2) and Cp*2Cr+ (CrIII, S = 3/2) results in the ferrimagnetic ordering of spins below 5 K.22 However, until now no more CT complexes of phthalocyanines and decamethylmetallocenes have been obtained.
In this work, we have obtained and studied the first charge transfer (CT) complexes of metal-free phthalocyanine (H2Pc) with decamethylchromocene and decamethylcobaltocene: (Cp*2Co+)(H2Pc˙−)·0.5C6H4Cl2·0.7C6H5CN·0.3C6H14 (1) and (Cp*2Cr+)(H2Pc˙−)·4C6H4Cl2 (2). These complexes were obtained in crystalline forms allowing us to study their crystal structures. The optical and magnetic properties of these complexes were also studied for polycrystalline samples. In contrast to previously studied (Cp*2Cr+)[FeI(Pc2−)]−·4C6H4Cl2 which contains metal-reduced [FeIPc(2−)]− anions with spin density localized on the FeI centers,22 the negative charge and spin of H2Pc˙− are delocalized over the Pc macrocycle. The variation of magnetic properties of the CT complexes will be compared referring to the reduced moieties, FeI or H2Pc˙−.
Results and discussion
Synthesis
The interaction of decamethylcobaltocene (Cp*2Co) and decamethylchromocene (Cp*2Cr) with metal-free phthalocyanine (H2Pc) in o-dichlorobenzene under anaerobic conditions produced deep blue solutions characteristic of the reduced Pc macrocycle. In the case of Cp*2Co, the CT complex partially precipitated after cooling the solution, and in this case 3 mL of benzonitrile was added to dissolve this complex. Synthesis with Cp*2Cr was carried out in pure o-dichlorobenzene. Slow mixing of the obtained solutions with n-hexane for 2 months yielded crystals on the walls of the diffusion cell. The obtained crystals were isolated from the solution, washed with n-hexane and dried to give rhombic plates of 1 with copper luster (in 64% yield) and elongated parallelepipeds of 2 with copper luster (in 46% yield). The compositions of complexes were determined from X-ray diffraction studies on single crystals as (Cp*2Co+)(H2Pc˙−)·0.5C6H4Cl2·0.7C6H5CN·0.3C6H14 (1) and (Cp*2Cr+)(H2Pc˙−)·4C6H4Cl2 (2). Several crystals tested from each synthesis had the same unit cell parameters to show that only one crystal phase was formed for both complexes. Elemental analysis cannot be used to confirm the composition of the complexes due to their high air sensitivity.Optical properties
The IR spectra of starting H2Pc, 1 and 2 are shown in Fig. S1 and S2 and are listed in Table S1.† The IR spectra of 1 and 2 are a superposition of the absorption bands of H2Pc, Cp*2M and solvent molecules. However, some absorption bands of H2Pc strongly decreased in intensity (bands at 1004 and 1501 cm−1) or disappear (1118 cm−1). An absorption band corresponding to the vibrations of the N–H bonds of H2Pc is manifested at 3274 cm−1. This band is shifted to larger wavenumbers during the reduction of the Pc macrocycle appearing at 3299 (1) and 3301 (2) cm−1 (the shift is 25–27 cm−1). Similar shifts were previously found for the H2Pc˙− radical anions in (cryptand[2,2,2][Na+])(H2Pc˙−)·1.5C6H4Cl2 and (TOA+)·(H2Pc˙−)·C6H4Cl2 (TOA+ is a tetraoctylammonium cation).15The spectra of H2Pc, 1 and 2 in the UV-visible-NIR range are shown in Fig. 1. The spectrum of starting H2Pc shows the Soret band at 338 nm and the split Q-band at 640 and 698 nm. Reduction of the Pc macrocycle leads to noticeable changes in the spectrum of H2Pc. The Soret band is blue shifted to 305 (1) and 309 (2) nm in the spectra of both complexes, whereas the Q-band is split and slightly blue shifted to 622 and 672 nm (1) and 592, 632, 672 nm (2) (Fig. 1). New bands observed in the NIR range are positioned at 1026 and 913 nm for 1 and 1030 and 932 nm for 2. These bands are attributed to H2Pc˙−. The appearance of the NIR bands and the blue shift of the Soret and Q-bands are characteristic of radical anion salts with the reduced Pc macrocycle.15–17
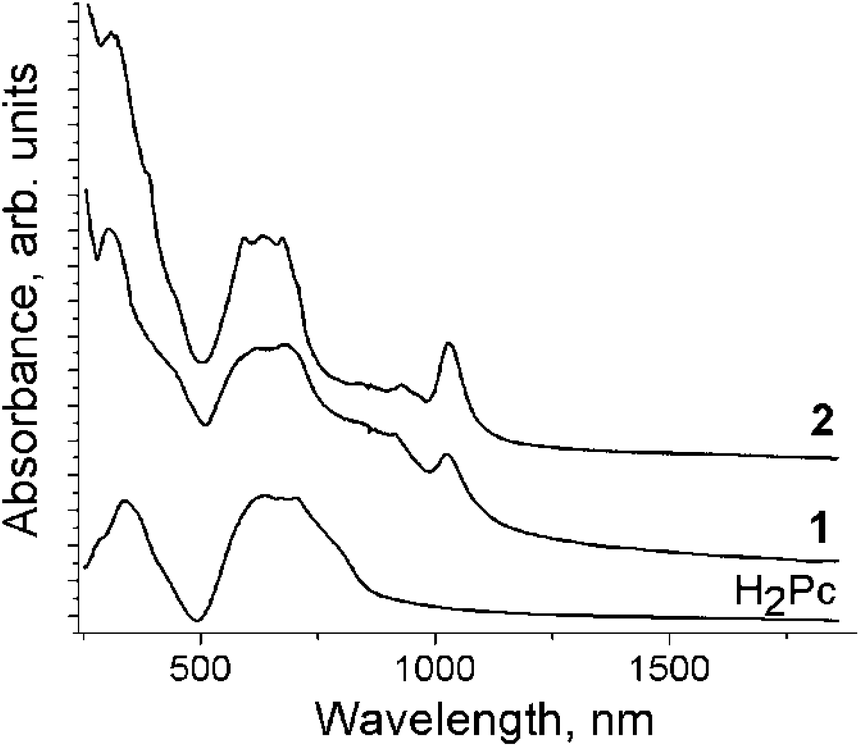 | ||
| Fig. 1 UV-visible-NIR spectra of starting H2Pc and salts 1 and 2 measued in KBr pellets prepared for 1 and 2 in anaerobic conditions. | ||
Crystal structures
There are two independent H2Pc and Cp*2Co units and three sites for solvent molecules in 1. These sites are occupied by disordered C6H5CN, C6H4Cl2 and C6H14 molecules to give the composition of 1 as (Cp*Co+)(H2Pc˙−)·0.5C6H4Cl2·0.7C6H5CN 0.3C6H14. The crystal structure of 1 is shown in Fig. 2. It contains blocks in which two Cp*2Co+ cations are sandwiched between two H2Pc˙− radical anions (Fig. 2). The interplanar distance between the Pc macrocycles in these blocks is 10.373 Å. The block consists of a couple of Cp*2Co and H2Pc˙− units, which are related by the inversion center. The interplanar distances among the Cp* planes of Cp*2Co and the Pc planes of two oppositely located H2Pc˙− are not uniform and are equal to 3.42 and 3.53 Å. This results in the difference of van der Waals C⋯C contacts between Cp*2Co and H2Pc˙−, which are in the 3.42–3.44 and 3.55–3.67 Å ranges, respectively. Projection of two halves of Cp*2Co+ cations on the plane of the Pc macrocycle in 1 is shown in Fig. 2b. Both halves are positioned over oppositely located isoindole units of the Pc macrocycle.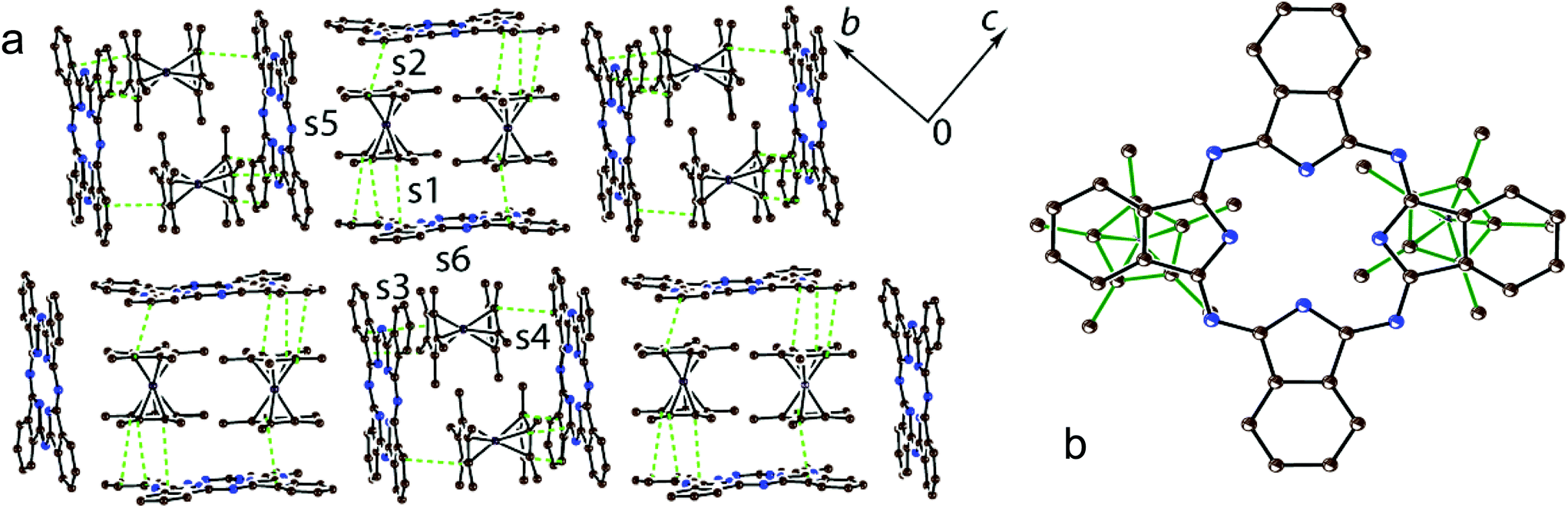 | ||
| Fig. 2 (a) Crystal structure of 1 viewed along the a axis. Solvent molecules are not shown for clarity. The van der Waals C⋯C contacts (<3.56 Å) between H2Pc˙− and Cp*2Co+ are shown by green dashed lines; (b) projection of two halves of Cp*2Co+ (bonds are shown by green solid lines) on the Pc plane. Hydrogen atoms are not shown. The overlap integrals between H2Pc˙− and Cp*2Co+ are shown as s1–s6. | ||
There are halves of independent H2Pc˙− and Cp*2Cr+ ions and two independent disordered C6H4Cl2 molecules in 2 to yield the composition as (Cp*2Cr+)(H2Pc˙−)·4C6H4Cl2. In spite of similar size and shape of the Cp*2M+ cations, complex 2 has a completely different structural motif from that in complex 1 with stacks of alternating H2Pc˙− and Cp*2Cr+ ions directed along the a axis (Fig. 3a and b). This structure is isostructural to that observed previously for the (Cp*2Cr+)[FeI(Pc2−)]−·4C6H4Cl2 CT complex showing ferrimagnetic ordering of spins below 5 K.22 The Cp*2Cr+ cations are positioned closer to the center of the Pc ligand in comparison with 1 but not exactly over its center (Fig. 3c). The interplanar distance between the Pc macrocycles in the stacks of 10.639 Å is longer than those in 1 (10.373 Å) and (Cp*2Cr+)[FeI(Pc2−)]−·4C6H4Cl2 (10.471 Å).22 As a result, the interplanar distances between Cp* and H2Pc˙− are increased in 2 up to 3.54 Å, and there are no C(Cp*)⋯C(H2Pc˙−) atomic contacts shorter than 3.56 Å. This situation is different from that in the [FeI(Pc2−)]− complex which has the interplanar distances between the Cp* and [FeI(Pc2−)]− planes of 3.46 Å and multiple van der Waals contacts between them in the 3.47–3.57 Å range.22 The solvent C6H4Cl2 molecules are incorporated between the Cp*2Cr+ ions belonging to the neighboring stacks of ions (Fig. 3a and b).
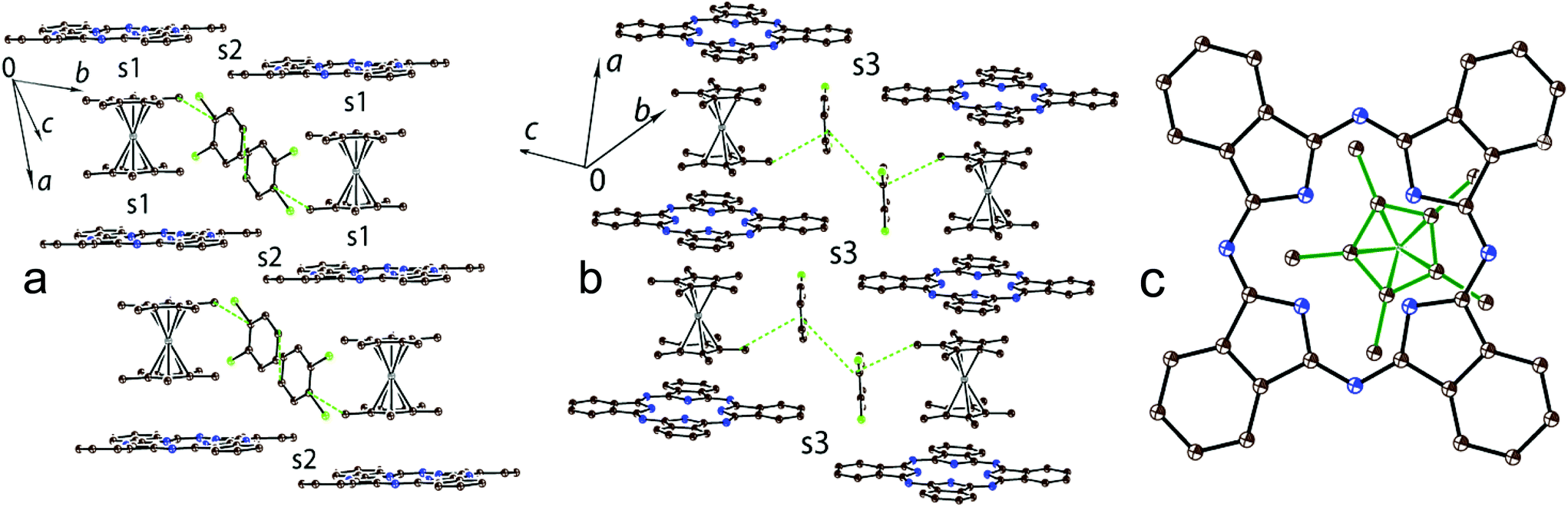 | ||
| Fig. 3 Crystal structure of 2: side view on the stacks of alternating H2Pc˙− and Cp*2Cr+ ions (a) and (b). The van der Waals C⋯C contacts (<3.56 Å) between the components are shown by green dashed lines; (c) projection of one half of Cp*2Cr+ (bonds are shown by green solid lines) on the Pc plane. Hydrogen atoms are not shown. Types of overlap integrals between H2Pc˙− and Cp*2Cr+ (s1) and between H2Pc˙− (s2 and s3) are shown. | ||
The intermolecular overlap integrals in 1 and 2 were examined by the extended Hückel method23,24 based on the results of X-ray single crystal structure analyses. The interactions between Cp*2M+ and H2Pc˙− were evaluated from the overlap integrals among the frontier orbitals {highest occupied molecular orbital (HOMO) for Cp*2M+ and singly occupied molecular orbital (SOMO) for H2Pc˙−}. As we discussed in the Crystal structure section, the Cp*2Co+ and H2Pc˙− ions form blocks in which two Cp*2Co+ cations are sandwiched between two H2Pc˙−. Since there are two independent Cp*2Co+ and each Cp*2Co+ approaches closer only to one of the two H2Pc˙−, four different face-to-face overlap integrals s1–s4 are observed in 1 (Fig. 2a). There are also side-to-face contacts between the Cp* ligand of Cp*2Co+ and two H2Pc˙− (overlap integrals s5 and s6, Fig. 2a). The overlap integrals s1, s2, s3 and s4 were estimated to be 0.0038, 0.0041, 0.0036 and 0.0010, respectively. As was expected, side-to-face overlap integrals s5 and s6 are essentially smaller to be 0.0002 and 0.0004, respectively. These data indicate rather effective face-to-face π–π interactions between the Cp* of Cp*2Co+ and H2Pc˙− in 1. At the same time all overlap integrals between H2Pc˙− in 1 are in the 0.0001–0.0002 range to predict the band insulating state for the H2Pc˙− network.
Chains of alternating Cp*2Cr+ and H2Pc˙− ions are formed in 2 (Fig. 3a and b). Since the Cp*2Cr+ cations are positioned at an equal distance from the planes of both surrounding H2Pc˙−, only one type of face-to-face overlap integral is observed between them (s1). The s1 overlap integral is equal to 0.0005 (several times smaller than those in 1) indicating ineffective π–π interactions between Cp*2Cr+ and H2Pc˙− in the alternating stacks. This can be explained by the positioning of the Cp* ligand of Cp*2Cr+ over an empty central part of the Pc macrocycle (Fig. 3c) and the absence of short van der Waals contacts between them. There are also weak side-by-side van der Waals contacts between H2Pc˙− along the b axis (Fig. 3a, overlap integral s2) and the [0, 1 −1] direction (Fig. 3b, overlap integral s3). In both cases the estimated overlap integrals did not exceed 0.0002 indicating weak π–π interactions between H2Pc˙− in 2.
The structure of (Cp*2Cr+)[FeI(Pc2−)]−·4C6H4Cl2 is isostructural to that of 2 with stacks of alternating Cp*2Cr+ and [FeI(Pc2−)]− ions along the a axis.22 However, in this case the overlap integral between the HOMOs of both [FeI(Pc2−)]− and Cp*2Cr+ was calculated to be 0.0013 showing a larger π–π interaction between the Cp* of Cp*2Cr and [FeI(Pc2−)]− in (Cp*2Cr+)[FeI(Pc2−)]−·4C6H4Cl2 than that in 2.
There are shorter C–N(imine) and longer C–N(pyrrole) bonds in the Pc macrocycle. Previous studies of the radical anions of metal-free and metal-containing phthalocyanines showed that the formation of a 19-π-electron system in Pc˙3− from a more stable aromatic 18-π-electron system of Pc2− generally results in the alternation of the C–N(imine) bonds due to the partial loss of aromaticity in the Pc ligand.11,12,15–17 Changes in the C–N(imine) bonds cannot be analyzed in 1 due to low bond length precision. However, this alternation is well pronounced in 2 to give the shorter and longer C–N(imine) bonds with 1.315(3) and 1.354(3) Å length, respectively, and are positioned in such a way that they belong to two oppositely located isoindole units. The geometry of two independent H2Pc˙− is slightly different in 1. One macrocycle has twisted conformation but another macrocycle is nearly planar with deviation of only one phenylene group from planarity (the dihedral angle between the 24-atom Pc plane and the plane of this phenylene group is 7.59°). The Pc macrocycle in 2 has a slightly non-planar shape with two phenylene groups located above the 24-atom Pc plane and two such groups located below this plane. These effects can be attributed to the packing effects with the Cp*2M+ cations.
Both Cp*2Co+ cations have unusual eclipsed conformation in 1 with methyl substituents of one Cp* ligand positioned nearly facing each other to the methyl substituents of another Cp* ligand. Most probably such unfavorable conformation is stabilized by specific interactions with two Pc macrocycles. The average length of the Co–C(Cp*) bonds for two independent Cp*2Co+ cations in 1 is 2.051(9) and 2.048(9) Å. This length corresponds to the formation of the Cp*2Co+ cations which have the average Co–C(Cp*) bond length of 2.04–2.05 Å,25 whereas neutral Cp*2Co has longer Co–C(Cp*) bonds of 2.101(3) Å.26 The Cp*2Cr+ cations have staggered conformation in 2 which is typical for decamethylmetallocenes. The average Cr–C(Cp*) bond length in 2 is 2.198(3) Å. This value is close to that for the Cp*2Cr+ cations (the average Cr–C(Cp*) bond length is 2.176(3)–2.180(3) Å).27,28 These bonds are longer than those for neutral Cp*2Cr (2.152(4) Å (ref. 18)).
Magnetic properties
The magnetic properties of 1 and 2 were analyzed for polycrystalline samples by SQUID and EPR techniques. The effective magnetic moment of 1 is equal to 1.64 μB at 300 K (Fig. 4a) corresponding to the contribution of one non-interacting S = 1/2 spin per formula unit (the calculated value is 1.73 μB). Since it is well known that the Cp*2Co+ cations containing CoIII are diamagnetic,21 the observed magnetic moment was attributed to the H2Pc˙− spins. Previously it was shown that these radical anions have the S = 1/2 spin state.15 The Weiss temperature determined from the linear temperature dependence of the reciprocal molar magnetic susceptibility of 1 in the 300–50 K range is −23 K (Fig. 4c) showing rather strong antiferromagnetic coupling of spins. The magnetic moment of 1 decreases below 120 K (Fig. 4a) due to the antiferromagnetic coupling of spins. However, long-range antiferromagnetic ordering is not observed down to 1.9 K (Fig. 4a and c). Magnetic exchange between H2Pc˙− can be mediated by diamagnetic Cp*2Co+ cations. We can suppose that this is a rather effective route for magnetic coupling between H2Pc˙− due to effective π–π interactions between Cp*2Co+ and H2Pc˙−.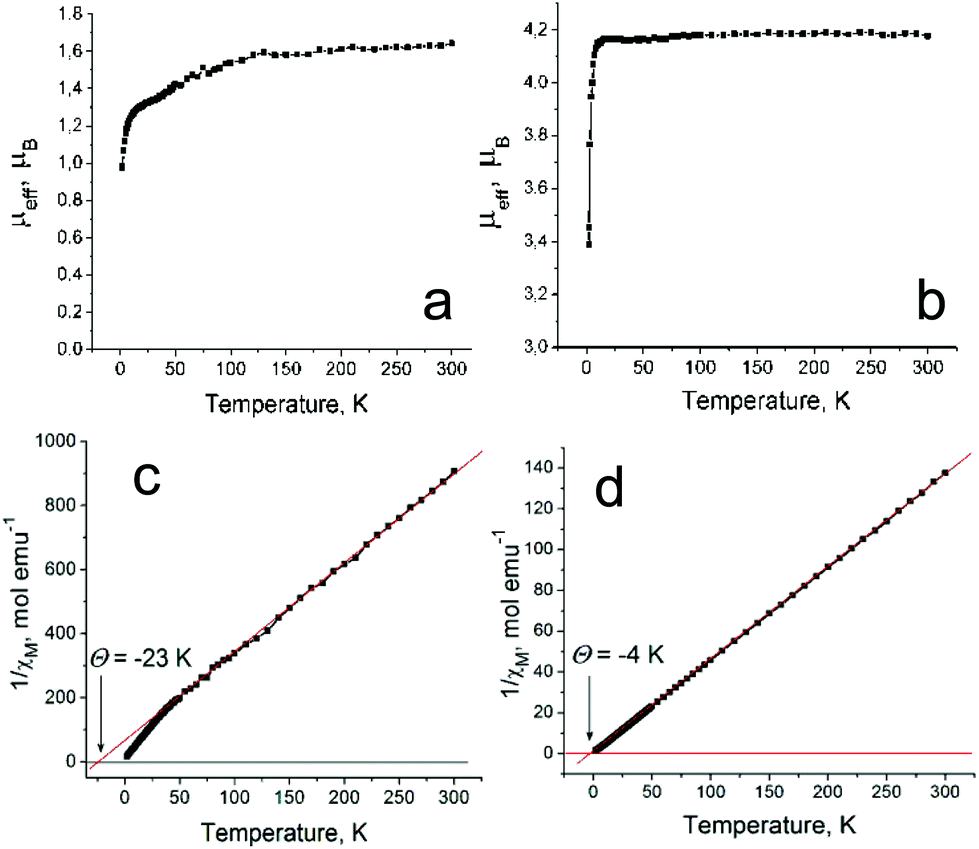 | ||
| Fig. 4 Temperature dependencies of the effective magnetic moment of 1 (a) and 2 (b) and those of the reciprocal molar magnetic susceptibility for 1 (c) and 2 (d). | ||
The presence of spin on H2Pc˙− is supported by the EPR data since only one narrow EPR signal is observed in the spectrum of 1 with a g-factor of 2.0036 and a linewidth (ΔH) of 0.54 mT at 297 K (Fig. 5a). The signal is even more narrowed with the temperature decrease (Fig. 5b) and has g = 2.0032 and ΔH = 0.28 mT at 4.2 K. Narrow EPR signals (ΔH = 0.1–0.2 mT) with similar g-factors were observed previously for the H2Pc˙− radical anions in the salts with organic cations.15,16
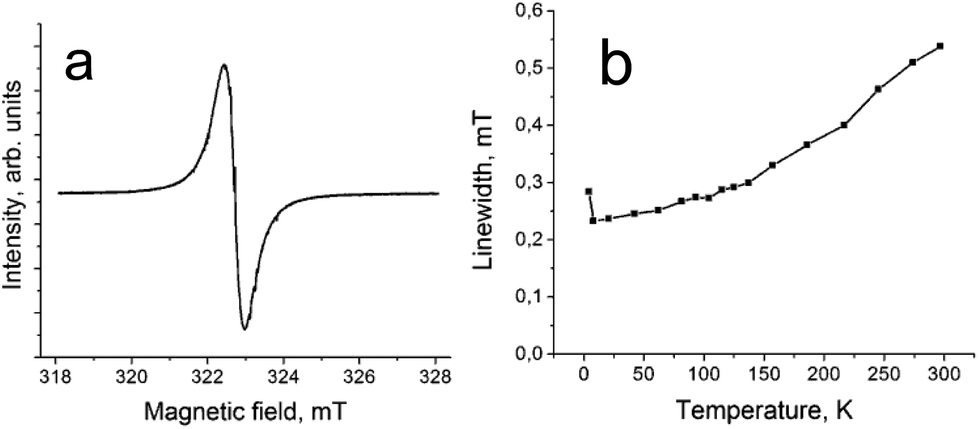 | ||
| Fig. 5 (a) EPR signal in complex 1 measured at 297 K; (b) temperature dependence of linewidth for the EPR signal of 1. | ||
The effective magnetic moment of 2 is equal to 4.18 μB at 300 K (Fig. 4b). It is close to the value of 4.24 μB calculated for the system of two non-interacting S = 3/2 and S = 1/2 spins per formula unit. Therefore, in contrast to 1, complex 2 contains paramagnetic Cp*2Cr+ cations with a high S = 3/2 spin state. This spin state was previously found for different salts of Cp*2Cr+.18,22,28,29 The H2Pc˙− radical anions have the S = 1/2 spin state as in 1. Therefore, uniform stacks of magnetic centers with different spin states {S = 3/2 (Cp*2Cr+) and S = 1/2 (H2Pc˙−)} are formed in 2. Similar stacks of the alternating spins with S = 3/2 (Cp*2Cr+) and S = 1/2 [FeI(Pc2−)]− spin states are formed in (Cp*2Cr+)[FeI(Pc2−)]−·4C6H4Cl2 which shows ferrimagnetic ordering of spins below 5 K.22 However, in contrast to this complex, only weak antiferromagnetic coupling of spins is observed in 2 with a Weiss temperature of −4 K determined in the 10–300 K range (Fig. 4d). This value is even smaller than that for 1 containing diamagnetic Cp*2Co+. The difference in the magnetic behavior of 2 and (Cp*2Cr+)[FeI(Pc2−)]−·4C6H4Cl2 in spite of a similar structure and spin state of the components can be explained by different conditions for π–π interactions between the components. The center of the Cp* ligand of Cp*2Cr+ is positioned exactly over the FeI atom in (Cp*2Cr+)[FeI(Pc2−)]−·4C6H4Cl2 leading to the formation of short Fe⋯C(Cp*) contacts of 3.40–3.56 Å. As a result, the Cp* ligand can effectively mediate magnetic coupling between the paramagnetic FeI and CrIII centers. On the contrary, the Cp* ligand of Cp*2Cr+ is positioned over the empty central part of the H2Pc macrocycle in 2 (Fig. 3c). Short van der Waals contacts between Cp*2Cr+ and H2Pc˙− are absent and, hence, the π–π interaction is relatively weak between them (due to small overlap integrals), while the additional electron of H2Pc˙− is delocalized over the Pc macrocycle. In this case the Cp* ligand cannot effectively mediate magnetic coupling between CrIII and H2Pc˙− resulting in only weak antiferromagnetic interactions. Therefore, compounds with stacks of alternating paramagnetic metal atoms like FeI and CrIII between which magnetic coupling is mediated through the Cp* ligands are more promising in the design of magnetic assemblies. In the case of compounds with stacks of alternating organic paramagnetic radical anions like H2Pc˙− and CrIII, magnetic coupling depends strongly on the conditions for the π–π interaction between the components.
Complex 2 manifests a complicated EPR spectrum due to the presence of different paramagnetic species. An asymmetric signal with g1 = 3.922 at 20 K can be attributed unambiguously to Cp*2Cr+ with the S = 3/2 spin state (Fig. 6, spectrum at 20 K). Asymmetry of this signal can be due to polycrystallinity of the sample. A narrower intense signal containing two lines at 20 K with g2 = 1.9943 and a linewidth (ΔH) of 1.8 mT (Fig. 6, green curve) and g3 = 1.9831 and ΔH = 4.2 mT at 20 K (Fig. 6, red curve) can be ascribed to H2Pc˙−. These signals are observed from 4.1 K up to room temperature. They have the following parameters at 4.1 K: g1 = 3.9059, g2 = 1.9961 (ΔH = 1.78 mT) and g3 = 1.9832 (ΔH = 3.24 mT) (Fig. 6). A new signal appears below 80 K (Fig. 6, blue curve). It grows in intensity and broadens with the temperature decrease. The parameters of this signal are g4 = 2.1085 and ΔH = 46.8 mT at 20 K and g4 = 2.2438 and ΔH = 80 mT at 4.1 K. Most probably the broad signal originates from both paramagnetic H2Pc˙− and Cp*2Cr+ species having exchange interactions since the g-factor of this signal is approximately intermediate between those characteristic of H2Pc˙− and Cp*2Cr+. The g⊥-value of the EPR signal from Cp*2Cr+ is also close to 2.000.28,30 However, generally this component has weaker intensity in the spectrum of Cp*2Cr+ and it is not so broad.28,30
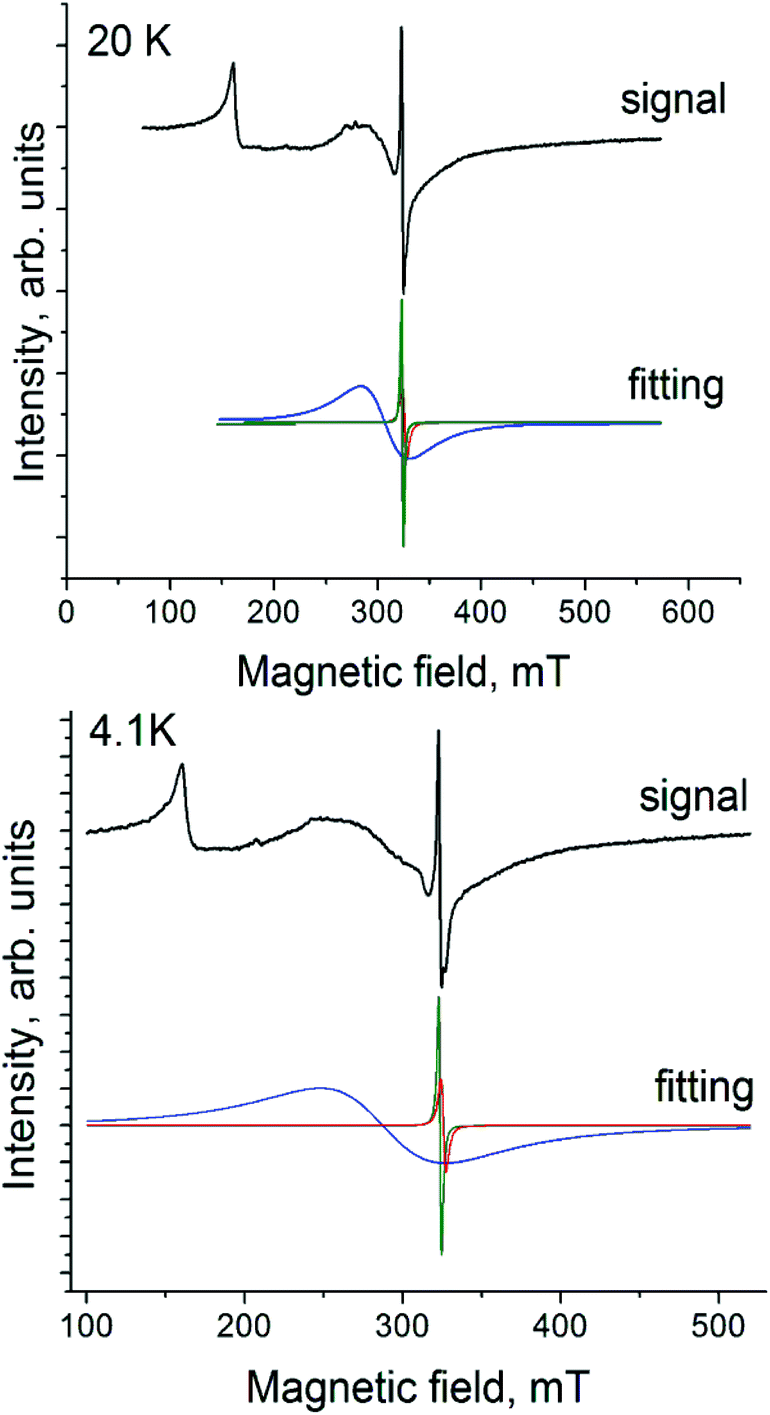 | ||
| Fig. 6 EPR spectrum of polycrystalline 2 at 20 and 4.1 K. | ||
Experimental
Materials
Decamethylcobaltocene (Cp*2Co) and metal-free phthalocyanine (H2Pc, 98%) were purchased from Aldrich. Decamethylchromocene (Cp*2Cr, >95%) was purchased from Strem. o-Dichlorobenzene (C6H4Cl2, Acros) was distilled over CaH2 under reduced pressure, n-hexane was distilled over Na/benzophenone, and benzonitrile (C6H5CN, Aldrich) was distilled over sodium under reduced pressure. Solvents were degassed and stored in an MBraun 150B-G glove box. Complexes 1 and 2 were synthesized and stored in the glove box under a controlled atmosphere containing less than 1 ppm of water and oxygen. KBr pellets used for the IR and UV-visible-NIR analyses were prepared in the glove box. EPR and SQUID measurements were performed on the polycrystalline samples of 1 and 2 sealed in 2 mm quartz tubes in the glove box under ambient pressure.General
UV-visible-NIR spectra were recorded in KBr pellets on a PerkinElmer Lambda 1050 spectrometer in the 250–2500 nm range. FT-IR spectra were obtained in KBr pellets with a PerkinElmer spectrum 400 spectrometer (400–7800 cm−1). EPR spectra were recorded for the polycrystalline samples of 1 and 2 with a JEOL JES-TE 200 X-band ESR spectrometer equipped with a JEOL ES-CT470 cryostat in the temperature range from 293 down to 4 K. A Quantum Design MPMS-XL SQUID magnetometer was used to measure the static magnetic susceptibility of 1 and 2 at 100 mT magnetic field under cooling and heating conditions in the 300–1.9 K range. A sample holder contribution and core temperature independent diamagnetic susceptibility (χd) were subtracted from the experimental values. The χd values were estimated by the extrapolation of the data in the high-temperature range by fitting the data with the expression: χM = C/(T − Θ) + χd, where C is the Curie constant and Θ is the Weiss temperature. The effective magnetic moment (μeff) was calculated with the following formula: μeff = (8·χM·T)1/2.Synthesis
Crystals of 1 and 2 were obtained by a diffusion technique. A reaction mixture was filtered into a 1.8 cm-diameter, 50 mL glass tube with a ground glass plug, and then 30 mL of n-hexane was layered over the solution. Slow mixing of the solutions resulted in the precipitation of crystals over 2 months. The solvent was then decanted from the crystals, and they were washed with n-hexane. The compositions of the obtained compounds were determined from X-ray diffraction analysis on a single crystal. Several crystals from one synthesis were found to consist of single crystalline phase. Due to the high air sensitivity of 1 and 2, elemental analysis could not be used to determine the composition because they reacted with oxygen in the air before the quantitative oxidation procedure could be performed.(Cp*2Co+)(H2Pc˙−)·0.5C6H4Cl2·0.7C6H5CN·0.3C6H14 (1) was obtained via the reduction of H2Pc (21.5 mg, 0.042 mmol) by slight excess of Cp*2Co (14 mg, 0.0425 mmol) in 15 mL of o-dichlorobenzene by stirring at 100 °C for 4 hours. After cooling down to room temperature, the complex partially precipitated from the solution and 3 mL of benzonitrile was added to dissolve the precipitate by stirring the mixture at 100 °C for 2 hours. This resulted in complete dissolution of the precipitate and the formation of deep blue solution characteristic of the reduced Pc macrocycle. The solution was cooled down to room temperature and filtered into the tube for diffusion. Rhombic plates of 1 with copper luster were obtained in 64% yield.
(Cp*2Cr+)(H2Pc˙−)·4C6H4Cl2 (2) was obtained via the reduction of H2Pc (21.5 mg, 0.042 mmol) by slight excess of Cp*2Cr (14 mg, 0.0435 mmol) in 16 mL of o-dichlorobenzene by stirring at 100 °C for 4 hours. This resulted in complete dissolution of H2Pc and the formation of deep blue solution which was cooled down to room temperature and filtered into the tube for diffusion. Elongated parallelepipeds of 2 with copper luster were obtained in 46% yield.
X-ray crystal structure determination
X-ray diffraction data‡ for 1 were collected on an Oxford diffraction “Gemini-R” CCD diffractometer with graphite monochromated MoKα radiation using an Oxford Instrument Cryojet system. Raw data reduction to F2 was carried out using CrysAlisPro, Oxford Diffraction Ltd. X-ray diffraction data for 2 were collected on a Bruker Smart Apex II CCD diffractometer with graphite monochromated MoKα radiation using a Japan Thermal Engineering Co. cooling system DX-CS190LD. Raw data reduction to F2 was carried out using Bruker SAINT.32 The structures were solved by direct methods and refined by the full-matrix least-squares method against F2 using SHELX-2013.33 Non-hydrogen atoms were refined anisotropically. The positions of hydrogen atoms were calculated geometrically. The tiny crystal sample of 1 showed very low diffraction resulting in a low number of intense reflections. As a result, the observed/unique reflection ratio is low. Nevertheless the crystal structure solution and refinement were stable. The structure of 1 contains three positions of solvent molecules. In all these positions solvent molecules are disordered and the positions are shared by several types of molecules. The occupancies for the C6H4Cl2 molecules are 0.221(3)/0.605(3)/0.114(5)/0.061(2), for the C6H5CN molecules are 0.886(5)/0.350(3)/0.173(3) and the C6H14 molecules have only one orientation with the 0.589(3) occupancy. The structure of 2 contains two positions of strongly disordered solvent C6H4Cl2 molecules with the 0.510(4)/0.203(4)/0.121(3)/0.108(3)/0.058(3) and 0.355(13)/0.229(11)/0.227(4)/0.189(6) occupancies. To keep the anisotropic thermal parameters of the disordered atoms within the reasonable limits the displacement components were restrained using ISOR, SIMU and DELU SHELXL instructions. This resulted in 1173 and 1393 restraints used for the refinement of the crystal structures of 1 and 2, respectively.Conclusions
Summarizing the studies here, new charge transfer complexes of metal-free phthalocyanine (H2Pc) and decamethylmetallocenes (Cp*2Co, Cp*2Cr) were obtained. In both complexes, the nearly full transfer of an electron from decamethylmetallocenes to H2Pc is realized forming the H2Pc˙− radical anions and the Cp*2M+ cations. This can be explained by essentially more negative oxidation potentials of Cp*2M (Eox = −1.04 − −1.47 V vs. SCE in acetonitrile)21 in comparison with the first reduction potential of H2Pc (Ered = −0.66 V vs. SCE in DMF).31 The formation of these ions is supported by their geometric parameters, the appearance of new bands of H2Pc˙− in the NIR range and blue shifts of both Soret and Q-bands of H2Pc. Magnetic data also support the formation of diamagnetic Cp*2Co+ (1), high-spin paramagnetic Cp*2Cr+ with the S = 3/2 spin state (2), and paramagnetic H2Pc˙− with the S = 1/2 spin state. An effective antiferromagnetic coupling between the H2Pc˙− spins is attained in 1 which is most probably mediated through the diamagnetic Cp*2Co+ cations. Complex 2 has stacks of alternating Cp*2Cr+ and H2Pc˙− ions and is isostructural to the previously studied complex (Cp*2Cr+)[FeI(Pc2−)]−·4C6H4Cl2 which manifested ferrimagnetic ordering of spins below 5 K.22 However, complex 2 shows only weak antiferromagnetic coupling of spins. Since the additional electron in H2Pc˙− is delocalized over the Pc macrocycle, we suppose that the additional factor to modulate the relative orientation between the Cp* ligand of Cp*2Cr+ and H2Pc˙− will improve the magnetic interactions, e.g. the incorporation of solvent molecules with different sizes and shapes than those in 2.Acknowledgements
The work was supported by RSF Grant No. 17-13-01215, and by JSPS KAKENHI Grant Numbers JP23225005 and JP26288035.Notes and references
- J. L. Petersen, C. S. Schramm, D. R. Stojakovic, B. M. Hoffman and T. J. Marks, J. Am. Chem. Soc., 1977, 99, 286 CrossRef CAS.
- H. Hasegawa, T. Naito, T. Inabe, T. Akutagawa and T. Nakamura, J. Mater. Chem., 1998, 8, 1567 RSC.
- M. Matsuda, T. Naito, T. Inabe, N. Hanasaki, H. Tajima, T. Otsuka, K. Awaga, B. Narymbetov and H. Kobayashi, J. Mater. Chem., 2000, 10, 631 RSC.
- T. Inabe and H. Tajima, Chem. Rev., 2004, 104, 5503 CrossRef CAS PubMed.
- J. S. Miller, C. Vazquez, J. C. Calabrese, M. L. McLean and A. J. Epstein, Adv. Mater., 1994, 6, 217 CrossRef CAS.
- D. K. Rittenberg, L. Baars-Hibbe, A. B. Böhm and J. S. Miller, J. Mater. Chem., 2000, 10, 241 RSC.
- A. B. P. Lever, E. R. Milaeva and G. Speier, in The phthalocyanines, properties and applications, ed. C. C. Leznoff and A. B. P. Lever, VCH Publishing, Weinheim, 1993, vol. 3, pp. 1–69 Search PubMed.
- M. Tahiri, P. Doppelt, J. Fischer and R. Weiss, Inorg. Chim. Acta, 1987, 127, L1 CrossRef CAS.
- H. Hückstädt and H. Homborg, Z. Anorg. Allg. Chem., 1998, 624, 715 CrossRef.
- E. W. Y. Wong and D. B. Leznoff, J. Porphyrins Phthalocyanines, 2012, 16, 154 CrossRef CAS.
- E. W. Y. Wong, C. J. Walsby, T. Storr and D. B. Leznoff, Inorg. Chem., 2010, 49, 3343 CrossRef CAS PubMed.
- W. Zhou, R. H. Platel, T. T. Tasso, T. Furuyama, N. Kobayashi and D. B. Leznoff, Dalton Trans., 2015, 44, 13955 RSC.
- D. V. Konarev, S. S. Khasanov, M. Ishikawa, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Inorg. Chem., 2013, 52, 3851 CrossRef CAS PubMed.
- D. V. Konarev, A. V. Kuzmin, S. S. Khasanov and R. N. Lyubovskaya, Dalton Trans., 2013, 42, 9870 RSC.
- D. V. Konarev, L. V. Zorina, S. S. Khasanov, A. L. Litvinov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2013, 42, 6810 RSC.
- D. V. Konarev, A. V. Kuzmin, M. A. Faraonov, M. Ishikawa, Y. Nakano, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Chem. – Eur. J., 2015, 21, 1014 CrossRef CAS PubMed.
- D. V. Konarev, S. I. Troyanov, M. Ishikawa, M. A. Faraonov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, J. Porphyrins Phthalocyanines, 2014, 18, 1157 CrossRef CAS.
- F. Zuo, A. J. Epstein, C. Vazquez, R. S. McLean and J. S. Miller, J. Mater. Chem., 1993, 3, 215 RSC.
- W. E. Broderick and B. M. Hoffman, J. Am. Chem. Soc., 1991, 113, 6334 CrossRef CAS.
- Metallocene-based magnets. G. T. Yee and J. S. Miller. in Magnetism: Molecules to Materials V, ed. J. S. Miller and M. Drillon, Wiley-Vch Verlag GmbH & Co, Weinheim, 2005, p. 223 Search PubMed.
- J. L. Robbins, N. Edelstein, B. Spencer and J. C. Smart, J. Am. Chem. Soc., 1982, 104, 1882 CrossRef CAS.
- D. V. Konarev, L. V. Zorina, S. S. Khasanov, E. U. Hakimova and R. N. Lyubovskaya, New J. Chem., 2012, 36, 48 RSC.
- M.-H. Whangbo and R. Hoffmann, J. Am. Chem. Soc., 1978, 100, 6093 CrossRef CAS.
- J. Ren, W. Liang and M.-H. Whangbo, Crystal and Electronic Structure Analysis Using CAESAR, Prime Color Software, Inc., 1998, (this book can be downloaded free of charge from the website: http://www.PrimeC.com/). Default parameters were used Search PubMed.
- D. V. Konarev, S. S. Khasanov, G. Saito, I. I. Vorontsov, A. Otsuka, R. N. Lyubovskaya and Yu. M. Antipin, Inorg. Chem., 2003, 42, 3706 CrossRef CAS PubMed.
- H. Heise, F. H. Kohler, M. Herker and W. Hillez, J. Am. Chem. Soc., 2002, 124, 10823 CrossRef CAS PubMed.
- M. M. Clark, W. W. Brennessel and P. L. Holland, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m391 CAS.
- D. V. Konarev, S. S. Khasanov, A. Otsuka and G. Saito, J. Am. Chem. Soc., 2002, 124, 8520 CrossRef CAS PubMed.
- D. Belo, J. Mendonca, I. C. Santos, L. C. J. Pereira, M. Almeida, J. I. Novoa, C. Rovira, J. Veciana and V. Gama, Eur. J. Inorg. Chem., 2008, 5327 CrossRef CAS.
- D. V. Konarev, S. S. Khasanov, G. Saito, A. Otsuka, Y. Yoshida and R. N. Lyubovskaya, J. Am. Chem. Soc., 2003, 125, 10074 CrossRef CAS PubMed.
- D. W. Clack, N. S. Hush and I. S. Woolsey, Inorg. Chim. Acta, 1976, 19, 129 CrossRef.
- SAINT Software Users Guide, Bruker Analytical X ray Systems, Inc., Madison, Wisconsin, U.S.A, 1999.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: IR spectra of starting compounds and complexes 1 and 2. CCDC 1529825 and 1529823. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt00336f |
‡ Crystal data of 1 at 150(1) K: C61.70H57.65ClCoN8.70, Mr = 1015.51 g mol−1, black plate, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 14.339(2), b = 18.376(3), c = 20.764(3) Å, α = 90.130(13), β = 101.362(14), γ = 103.949(13)°, V = 5198.5(15) Å3, Z = 4, dcalc = 1.298 g cm−3, μ = 0.431 mm−1, F(000) = 2134, 2θmax = 59.538°, reflections measured 46 , a = 14.339(2), b = 18.376(3), c = 20.764(3) Å, α = 90.130(13), β = 101.362(14), γ = 103.949(13)°, V = 5198.5(15) Å3, Z = 4, dcalc = 1.298 g cm−3, μ = 0.431 mm−1, F(000) = 2134, 2θmax = 59.538°, reflections measured 46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 575, unique reflections 23594, reflections with I > 2σ(I) = 5358, parameters refined 1555, restraints 1173, R1 = 0.1182, wR2 = 0.2503, G.O.F. = 0.976, CCDC 1529825. 575, unique reflections 23594, reflections with I > 2σ(I) = 5358, parameters refined 1555, restraints 1173, R1 = 0.1182, wR2 = 0.2503, G.O.F. = 0.976, CCDC 1529825.Crystal data of 2 at 150(1) K: C76H64Cl8CrN8, Mr = 1424.95 g mol−1, black parallelepiped, triclinic, P |
| This journal is © The Royal Society of Chemistry 2017 |