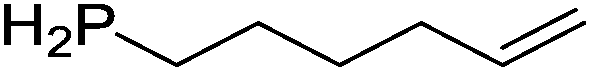DOI:
10.1039/C7DT00319F
(Perspective)
Dalton Trans., 2017,
46, 4483-4498
β-Diketiminate complexes of the first row transition metals: applications in catalysis
Received
26th January 2017
, Accepted 27th February 2017
First published on 27th February 2017
Abstract
Although β-diketiminate complexes have been widely explored in stoichiometric studies, their use as catalysts is largely underdeveloped. With growing interest in the catalytic activity of complexes of the first row transition metals, primarily due to the untapped potential of such metal centers, along with the growing global focus on sustainable chemistry with earth abundant metals, this Perspective focuses on the use of β-diketiminate complexes of the first row transition metals as catalysts for the synthesis of small organic molecules.
Introduction
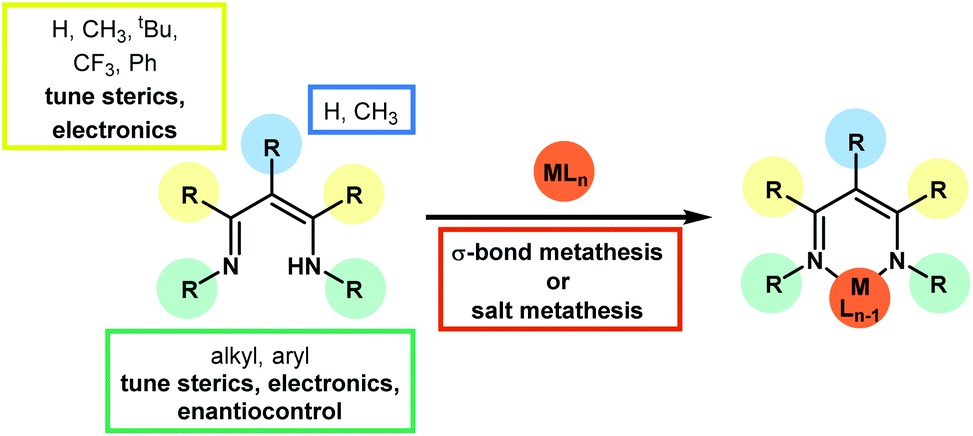
Since the earliest development of the coordination chemistry of β-diketiminates by Bradley1 and Holm2 and the extensive work from Lappert3 there has been a great deal of interest in this ligand in a host of complexes,4 ranging from alkaline earth metals,5 main group elements,6 lanthanides and actinides7 and transition metals.8 Much work has focused on stoichiometric transformations that are supported by these ligands, along with ligand design9 through modification of the functional groups on the β-diketiminate itself (where the ligand is often referred to as nacnac or BDK). This review primarily focuses on catalytic transformations using first row transition metal pre-catalysts which contain the β-diketiminate motif, with a particular focus on catalysis that involves the transformations of small organic motifs. Numerous examples of ring-opening polymerization,10 polycarbonate formation,11 and olefin polymerization12 exist in the literature and these transformations are covered by several comprehensive, specialized reviews elsewhere. Likewise, this review focuses on the classic β-diketiminate motif, where the metal or main group element is bonded in a six-membered ring through a delocalized imine–amine motif. This is typically derived from the condensation of 2,4-pentanedione and two equivalents of a substituted aniline, although many more highly functionalized variants exist (Scheme 1). These ligand syntheses can be carried out on tens of grams scale using a condensation with Dean–Stark apparatus and a catalytic amount of strong acid (for example p-toluene sulfonic acid). β-Diketiminate analogues are known, for example the anilido-aldimine13 and bis-oxazoline (and enamine-oxazoline)14 bonding motifs, which have also been covered elsewhere.
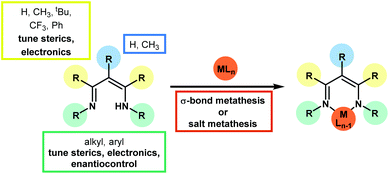 |
| | Scheme 1 The utility of the β-diketiminate ligand stems from the steric and electronic parameters that can be readily tuned, whilst the varied methods available to introduce the metal centre and co-ligands is synthetically advantageous. | |
The growing importance of first row transition metal catalyzed processes is highlighted by recent literature which has seen an increase in the number of publications on, for example, complex organic transformations catalyzed by salts or complexes of even the more ‘unusual’ metals such as manganese15 and cobalt.16 The 3d transition metals often offer complementary reactivity compared to their heavier d-block counterparts, not least due to their small size, ability to readily exist in low coordinate environments and to undertake single electron or radical reactions. This opens up new reaction pathways and the opportunity to undertake completely new transformations. The β-diketiminate ligand is anionic and forms strong metal–ligand bonds, where the ligand acts as a σ-donor, stabilizing the metal center. In the case of late transition metals, there are also opportunities for π-bonding. These bonding modes, along with the steric and electronic effects of the β-diketiminate motif, have been comprehensively reviewed.3,9 As a general overview for selection of a specific β-diketiminate motif, increasing the steric bulk of the N–R groups (often by using 2,6-disubstituted aryl groups) leads to a smaller N–M–N bite angle. Overall this is useful in supporting mononuclear complexes (dimerization is avoided and so high levels of reactivity can be achieved) and can prevent the coordination of neutral donors (such as solvent) thus maintaining a low coordinate and highly reactive environment. However, as sterics at the metal centre increase this can lead to a higher activation barrier and thus a lower rate of reaction. This can be beneficial for mechanistic study in that it can make it possible to observe or even isolate reactive intermediates. The orientation of the aryl groups around the metal center are such that employing electron-donating or electron-withdrawing substituents is not as influential as the substituents on the ligand backbone; rather than using 2,4-pentanedione to construct the ligand, the electron withdrawing ability of 1,1,1,5,5,5-hexafluoro-2,4-pentanedione can have a profound effect on reactivity by increasing the positive redox potential at the metal and reducing the back-bonding between the metal and co-ligands. In short, the β-diketiminate ligand is ideal for supporting the 3d metals: steric protection from the aryl groups flanking the metal centre can give access to low, often three, coordinate environments whilst the non-innocence17 of the ligand can help to promote single electron chemistry.18 Even with all of these benefits associated with both the ligand and the metal, as will be demonstrated in the following pages, there are potential areas in catalysis that are largely undiscovered.
Titanium
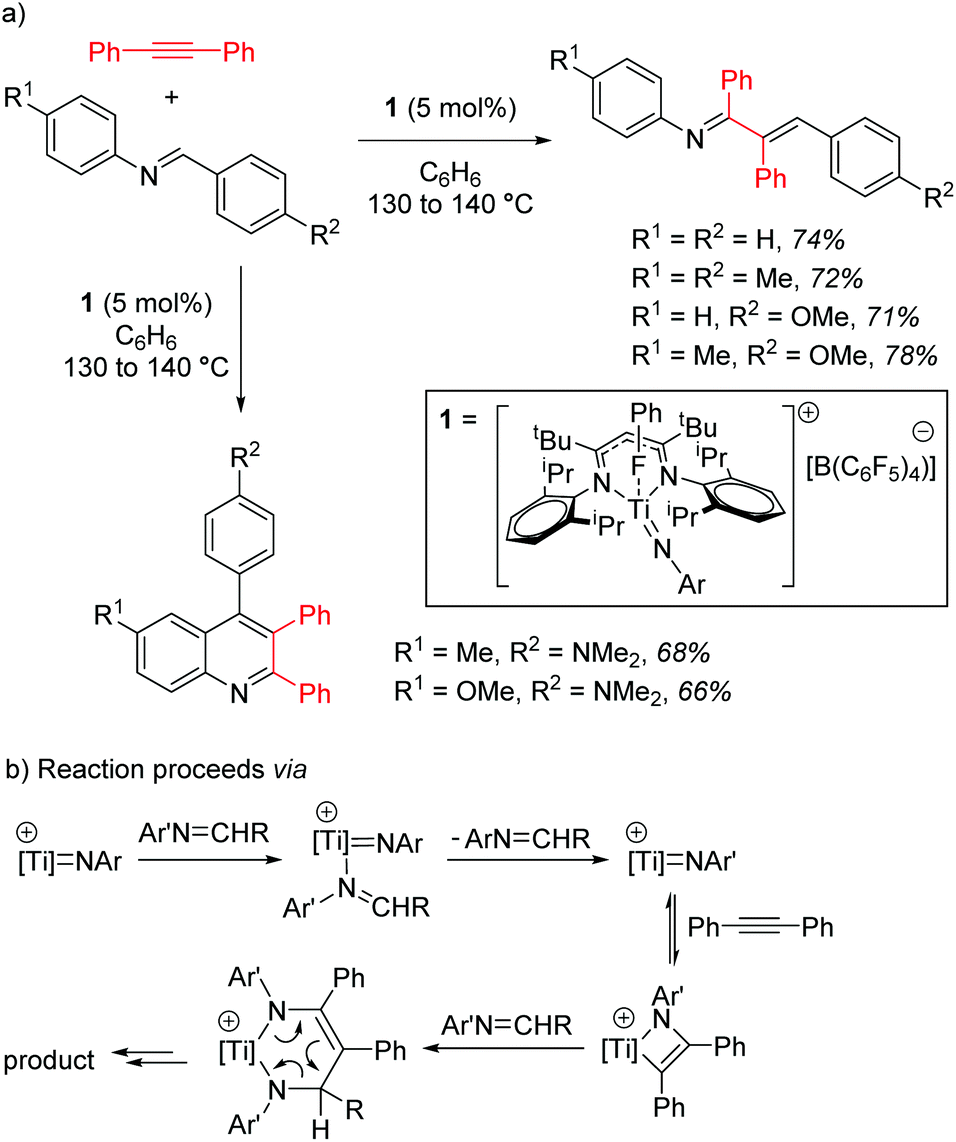
Mindiola and co-workers first demonstrated the catalytic activity of a titanium β-diketiminate complex by using a highly active fluorobenzene adduct to undertake carboamination reactions of diphenylacetylene with aldimines.19 Note that the reaction does not yield product when electron poor aldimines are employed, but good isolated yield of the enimine is obtained when aldimines with electron donating substituents, such as methoxy groups, are employed (Scheme 2a). When even more electron rich substrates are used, for example with a diethylamine group on the aromatic ring, quinolones are obtained. The reaction is postulated to proceed via a Ti-imido intermediate, which then undergoes [2 + 2] cyclization followed by insertion and finally a [4 + 2] retrocyclization releases the product (Scheme 2b).
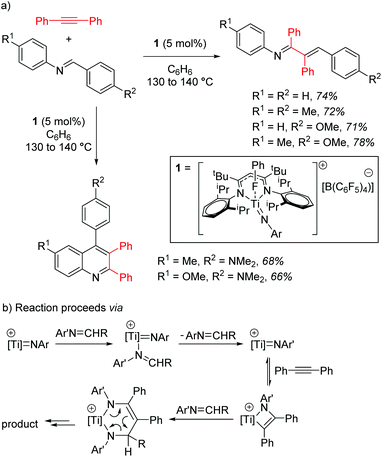 |
| | Scheme 2 (a) Mindiola's carboamination operates for a range of substrates, but highest yields are observed with electron rich aldimine starting materials. (b) The reaction is believed to proceed via a Ti-imido generated from the aldimine starting material, this then undergoes a [2 + 2] cyclization followed by retrocyclization to generate the product. | |
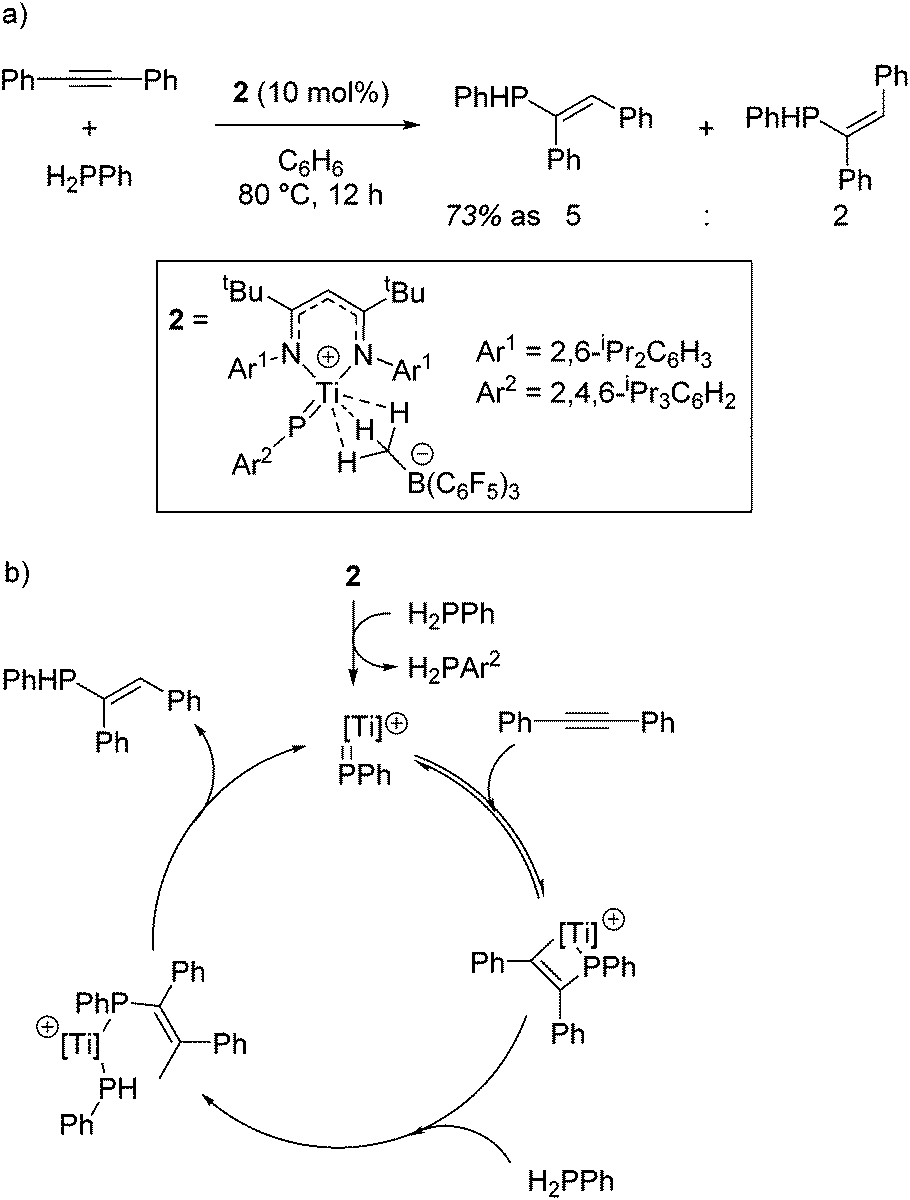
The ability of the titanium imido complex to undergo [2 + 2] cyclizations was then extrapolated to investigate highly unusual titanium phosphinidene complexes which catalyze the controlled hydrophosphination of alkynes, using phenyl phosphine as the phosphorus source (Scheme 3a).20 The difficulty in carrying out hydrophosphination with phenylphosphine is the propensity to undertake double functionalization of the phosphine with two equivalents of alkene, whereas by employing a phosphinidene intermediate, Mindiola avoids this (Scheme 3b). No reaction is observed when diphenylphosphine is used in the reaction, providing more evidence that the reaction is likely to proceed via a (phenyl)phosphinidene intermediate generated from the phosphinidene pre-catalyst, 2.
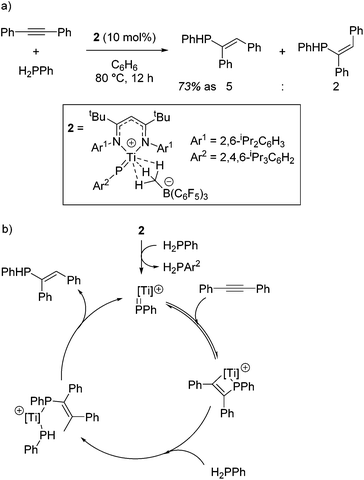 |
| | Scheme 3 (a) By a similar principal to the carboamination chemistry, a sterically hindered Ti-phosphinidene is used to prepare ethenyl phosphines from phenylphosphine. (b) The proposed catalytic cycle. | |
In the context of titanium d0 chemistry, this ligand environment is clearly well-placed to support metal–heteroatom multiple bonds. However, it could be argued that the titanium–imido reactivity is not unique to the β-diketiminate ligand and that other d0 metal–ligand combinations are equally well-positioned to support such chemistry.21 However, the formation of the phosphinidene22 and its use for controlled transfer of a primary phosphine is highly unusual and, in terms of synthetic methodology, is an area in need of further research.
Vanadium
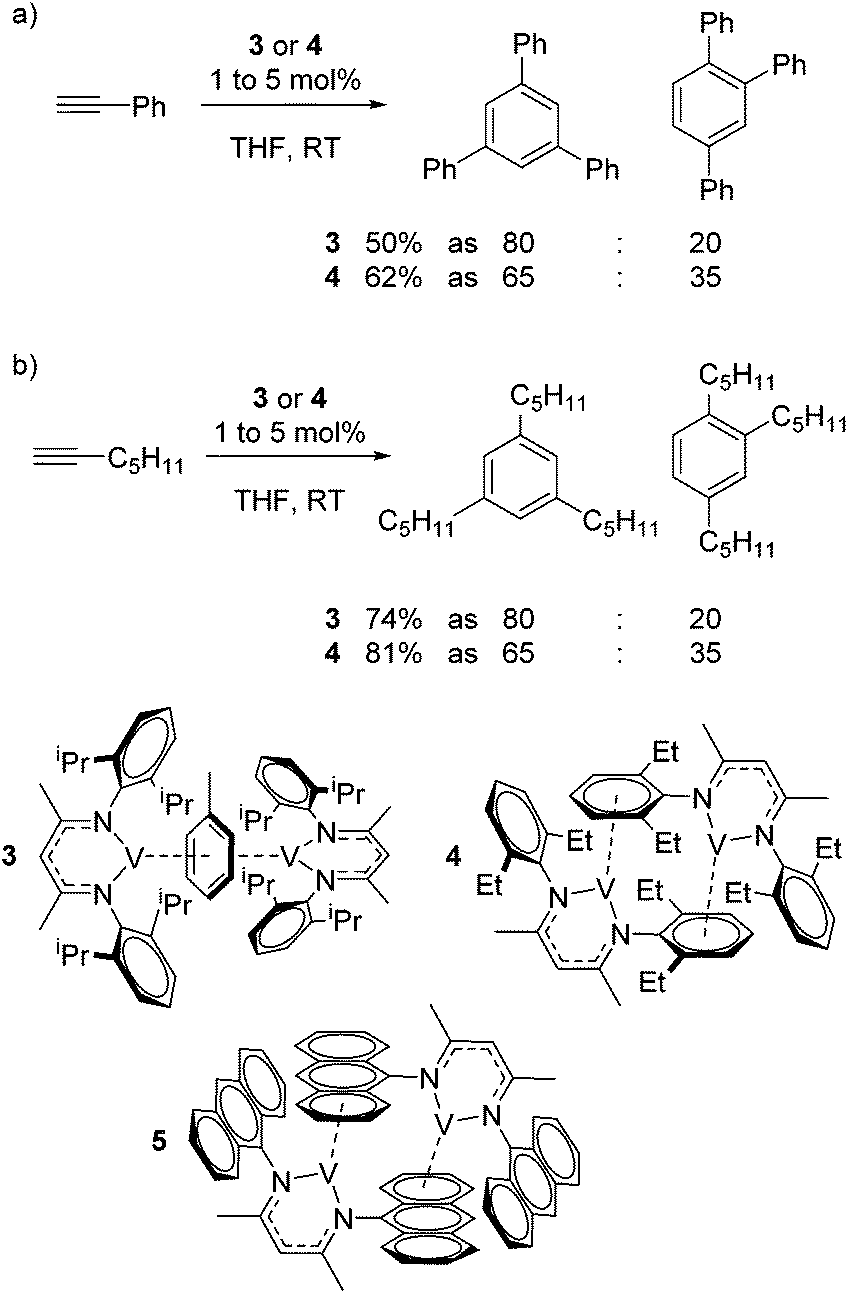
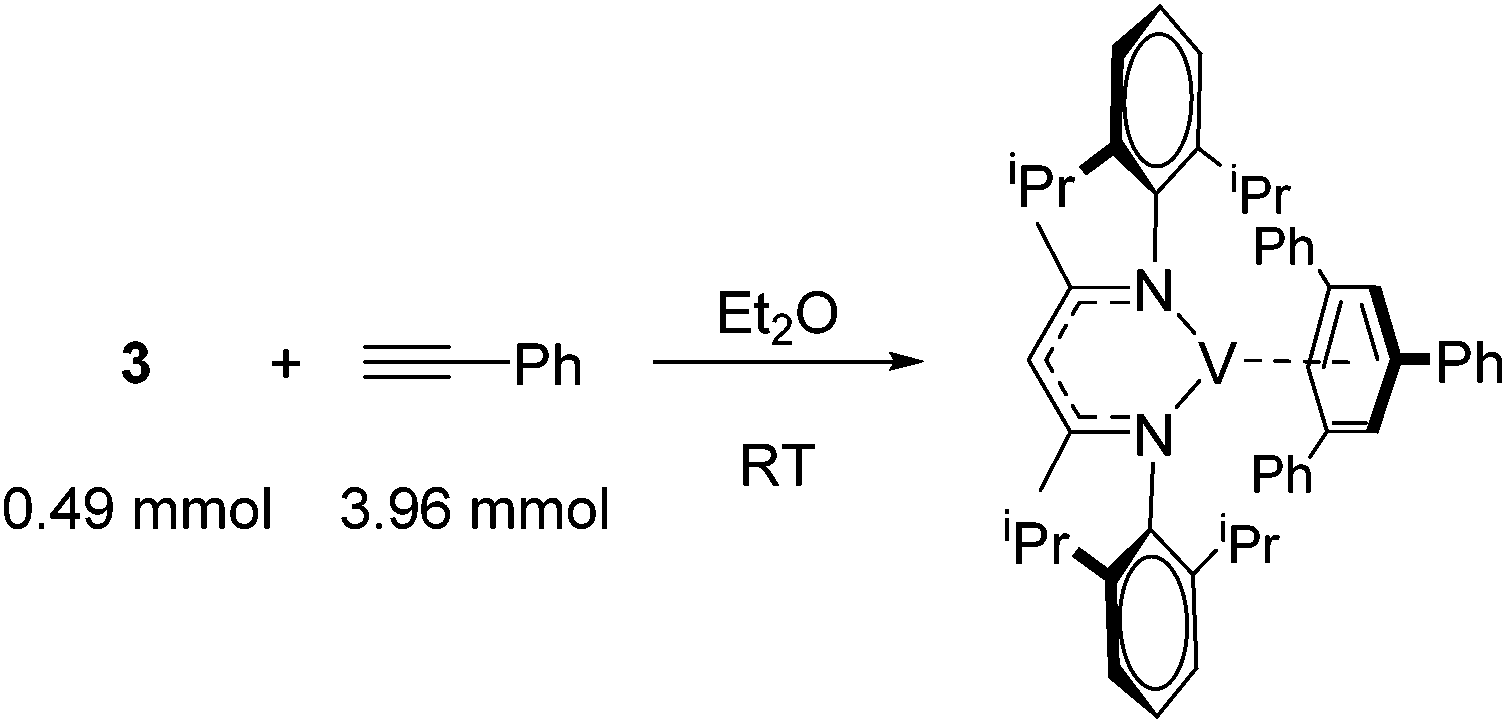
Catalytic reactions using vanadium β-diketiminate complexes are scarce, although the stoichiometric reactivity of group 5 complexes is covered in detail by a recent Perspective from Arnold and co-workers.23 In what is a rare example of catalysis, the η6-coordinating ability of a series of V(I) complexes is exploited by Tsai for the catalytic cyclotrimerization of alkynes.24 Complex 3 gives a modest yield of cyclotrimerized phenylacetylene in a ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 of 1,3,5-substituted:1,2,4-substituted product (Scheme 4a). Interestingly, when the less activated substrate 1-heptyne is used, the yield increases to 74% (Scheme 4b). In contrast complex 4 gives a greater yield of the products (62% using phenyl acetylene and 81% using 1-heptyne), but this is to the detriment of selectivity, where the ratio of 1,3,5-product to 1,2,4-product drops to 65:35. This is postulated to be due to reduced steric hindrance from the ethyl substituents on the ligand. Complex 5 was synthesized during this study but not employed in catalysis; it would be interesting to compare complex 4 to complex extremely hindered complex 5 in order to gain more insight into the effects of sterics.
:20 of 1,3,5-substituted:1,2,4-substituted product (Scheme 4a). Interestingly, when the less activated substrate 1-heptyne is used, the yield increases to 74% (Scheme 4b). In contrast complex 4 gives a greater yield of the products (62% using phenyl acetylene and 81% using 1-heptyne), but this is to the detriment of selectivity, where the ratio of 1,3,5-product to 1,2,4-product drops to 65:35. This is postulated to be due to reduced steric hindrance from the ethyl substituents on the ligand. Complex 5 was synthesized during this study but not employed in catalysis; it would be interesting to compare complex 4 to complex extremely hindered complex 5 in order to gain more insight into the effects of sterics.
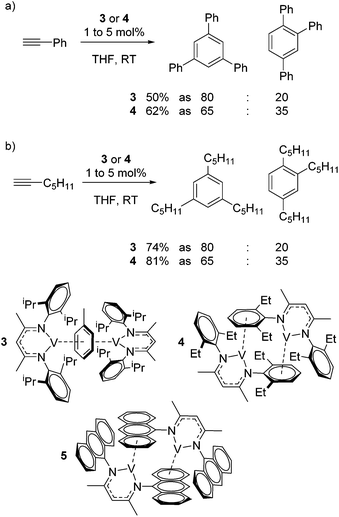 |
| | Scheme 4 Tsai used η6-bound vanadium β-diketiminate complexes to afford the cyclotrimerization of terminal alkynes. | |
Elegantly, the group isolate and characterize a potential reactive intermediate from a reaction of 3 with five equivalents of phenyl acetylene, which would suggest displacement of the η6-coordination mode of the ligand (toluene or β-diketiminate) to allow η6-product coordination during catalysis (Scheme 5).
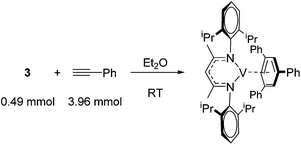 |
| | Scheme 5 A vanadium intermediate isolated on the stoichiometric reaction of pre-catalyst 3 with phenyl acetylene. | |
Chromium
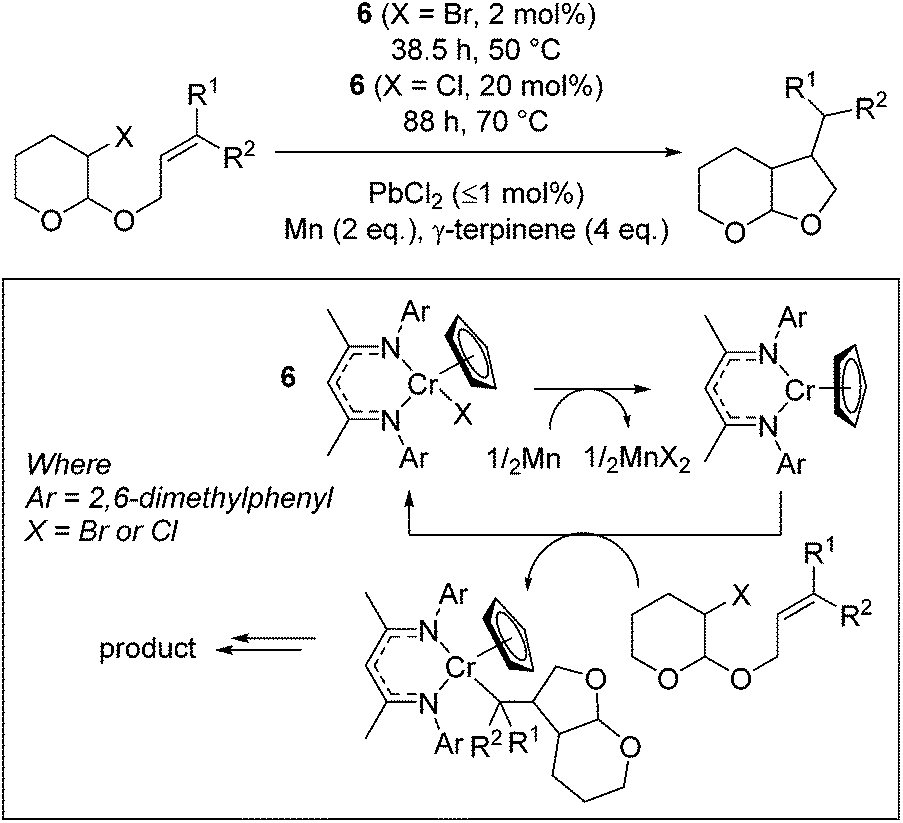
Smith and co-workers used a Cr(III) complex, 6, for catalytic bromo-acetal cyclization.25 The reaction proceeds by reduction of the Cr(III)Cp precursor to form the Cr(II)Cp complex by a stoichiometric loading of elemental Mn (activated by a sub-catalytic amount of PbBr2, Scheme 6). The Cr(II) complex then undergoes oxidation to reform the Cr(III)–halide starting catalyst and Cr(III)–alkyl complex. γ-Terpinene is an inexpensive proton source, which allows release of the organic product from the Cr–alkyl intermediate.
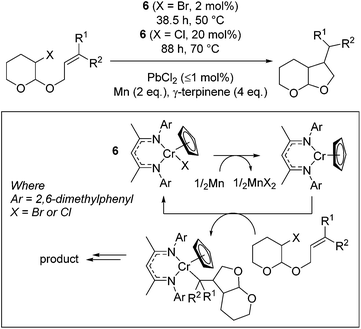 |
| | Scheme 6 Halo-acetals form furo-pyran structures using Cr–Cp catalyst 6. The chemistry can be applied to both bromo- and chloro substrates, with the bromo-acetal undergoing conversion to product more rapidly and at lower temperature. | |
The reaction proceeds most efficiently, in terms of yield and rate, when a tertiary or secondary radical is formed (Table 1, entries 2 and 3). This is in contrast to the yield obtained when sterically undemanding substrates are used (entry 1) or when a kinetically stabilized radical forms (entry 4). The yield of a challenging substrate such as that in entry 1 can be increased under photolytic conditions (entry 1, parentheses). A change to slightly more forcing reaction conditions allows the cyclization of chloro-acetals (entries 5 to 7), which again result in lower yield when a benzylic radical is formed (entry 7).
Table 1 Selected results from Smith's halo-acetal cyclizations
| Entry |
X |
R1 |
R2 |
Yield |
Diastereomeric ratio |
| Conditions: 1 mmol substrate, 2 mol% 6 (X = Br) or 20 mol% 6 (X = Cl), ≤1 mol% PbX2, 38.5 h at 50 °C (X = Br) or 88 h at 70 °C (X = Cl). Isolated yields shown. n.i. = no product isolated: 73% unreacted starting material isolated. |
| 1 |
Br |
H |
H |
n.i. (48) |
−(83:17) |
| 2 |
Br |
H |
C3H7 |
93 |
82:18 |
| 3 |
Br |
CH3 |
CH3 |
85 |
65:35 |
| 4 |
Br |
H |
C6H5 |
28 |
79:21 |
| 5 |
Cl |
H |
C3H7 |
56 |
93:7 |
| 6 |
Cl |
CH3 |
CH3 |
57 |
61:39 |
| 7 |
Cl |
H |
C6H5 |
29 |
83:17 |
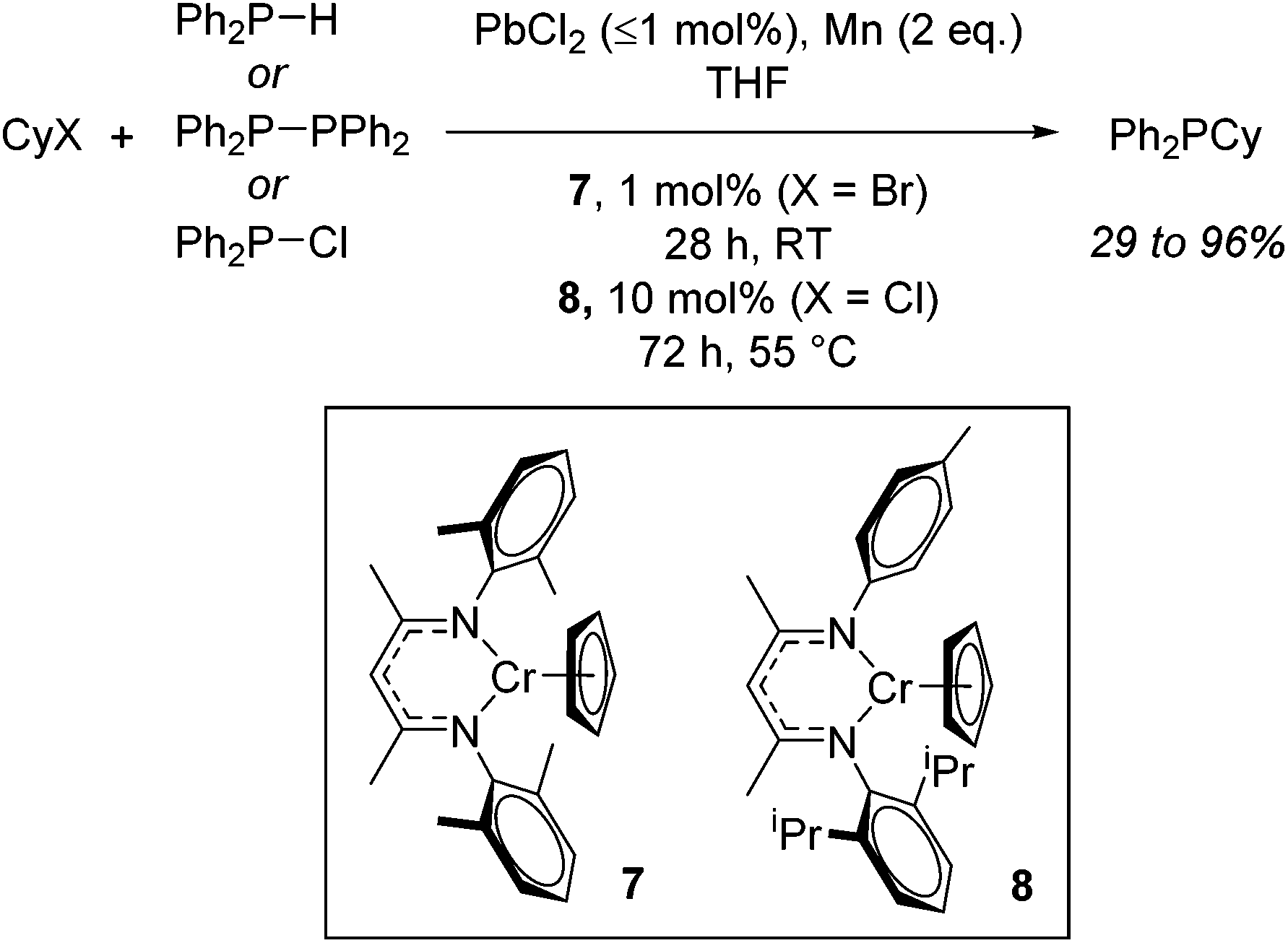
With detailed stoichiometric studies and kinetic understanding in place,26 Smith then extended the reactivity to include P–C bond forming reactions.27 The catalysis exploits the ability of these chromium β-diketiminate complexes to undergo homolytic cleavage of the alkyl halide and thus release organic and phosphinyl radicals which can then react. For example, both cyclohexyl bromide and cyclohexyl chloride can be used to generate cyclohexyl radicals which can then react with P-centred radicals generated from diphenylphosphine, tetraphenyldiphosphane or chlorodiphenylphosphine. Once again the reactions require the addition of PbX2 (or chlorotrimethylsilane) and Mn powder, with reactions performed at room temperature in THF using 1 mol% 7 for cyclohexyl bromide and 10 mol% 8 when cyclohexyl chloride is employed (Scheme 7). Ligand sterics were used to attenuate reactivity for the different halide substrates: the increased steric bulk of 7 results in faster reduction (and generation of the product Ph2PCy from CyBr) whereas 8, with its reduced steric hindrance allows for faster oxidation and formation of the Cr(III) alkyl intermediate from CyCl.
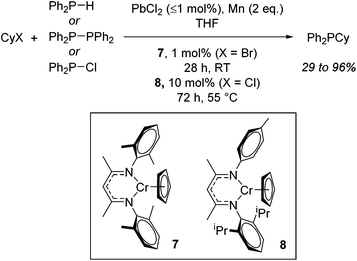 |
| | Scheme 7 P–C bond formation using Cr(II) complexes and involving phosphinyl radicals which can be generated from several sources. | |
Iron
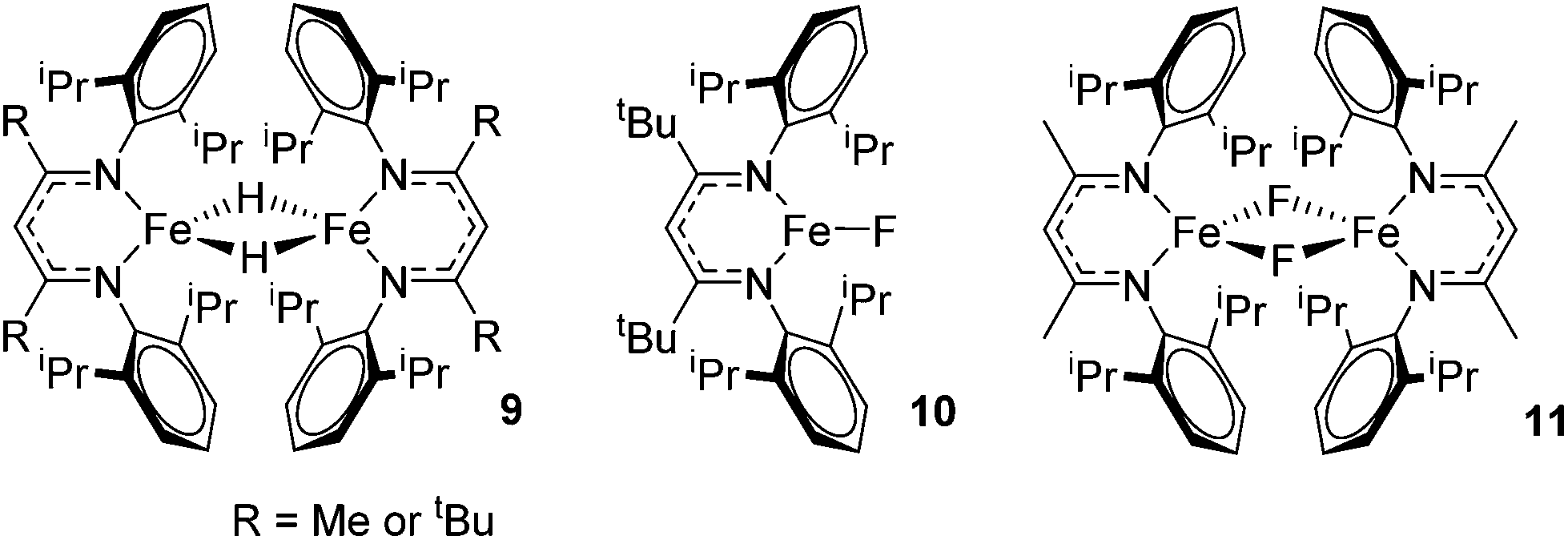
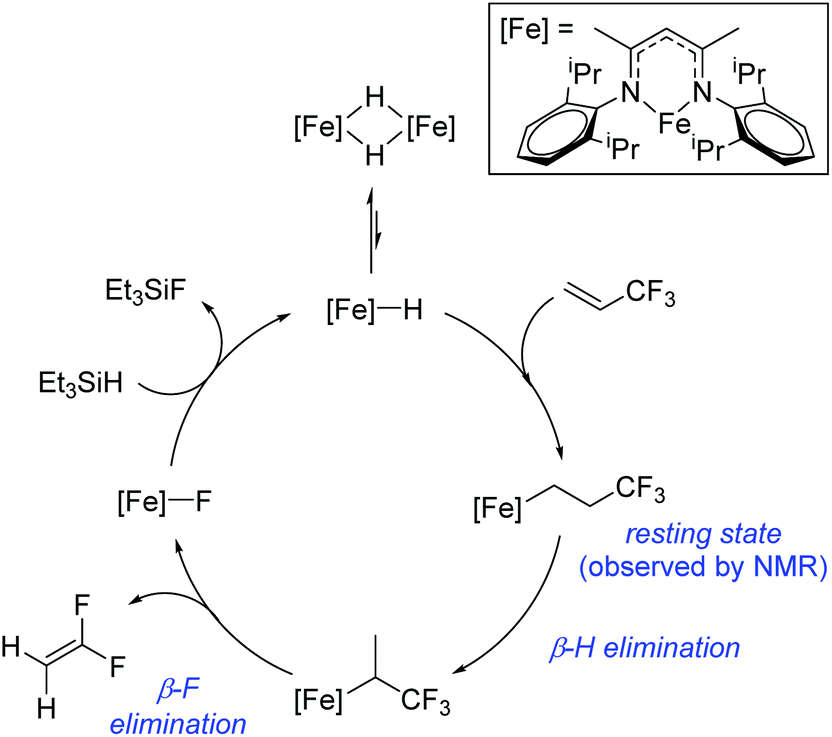
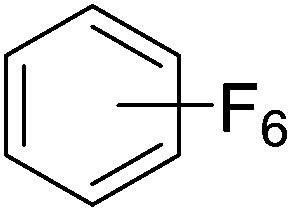
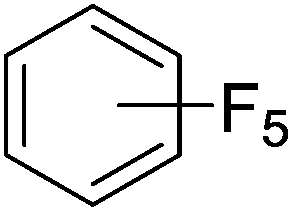
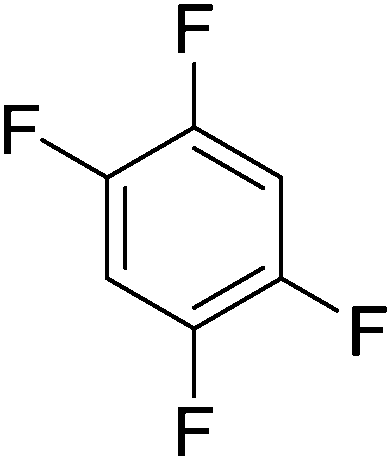
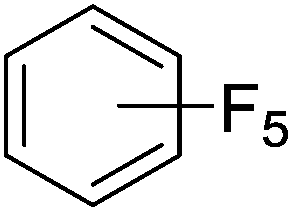
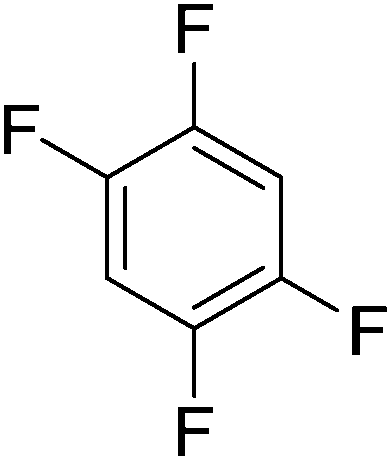
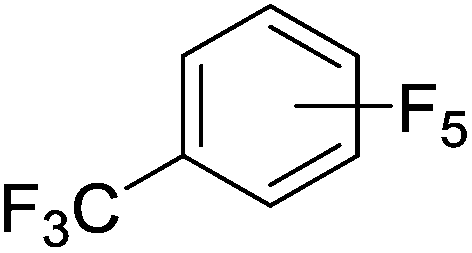
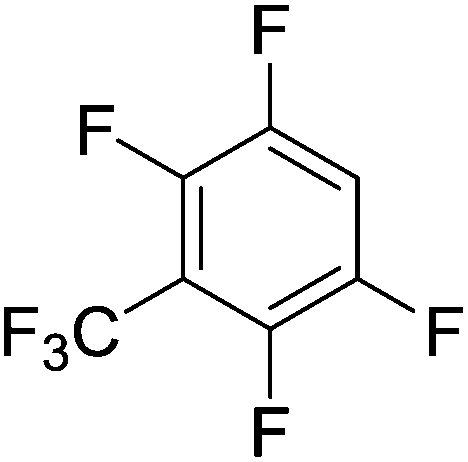
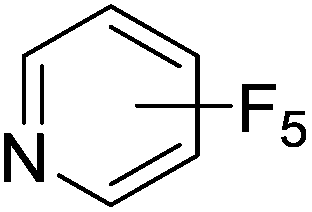
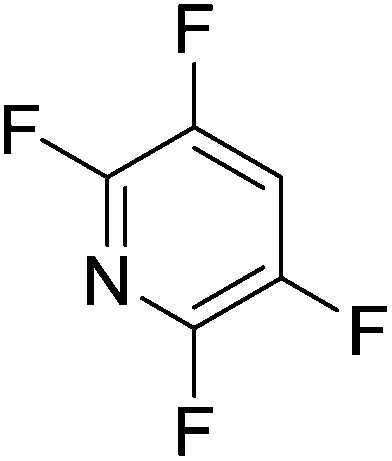
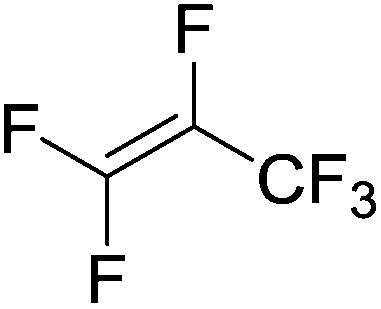
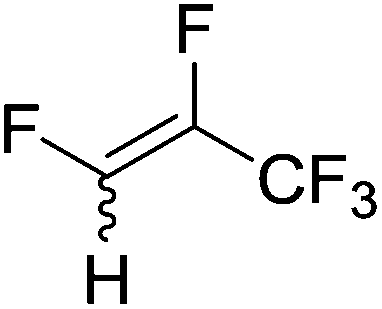
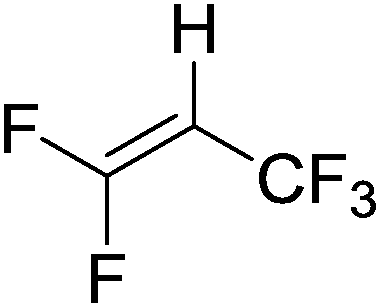
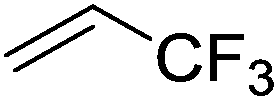
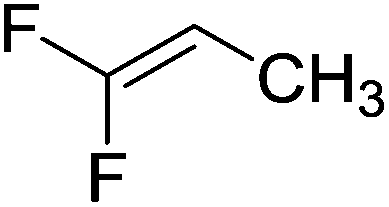
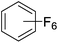
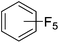
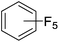
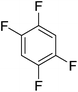
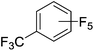
Beyond the earliest report in lactide polymerization from Gibson28 and Liu's work on ethylene polymerization,29 the next report of a catalytic transformation with an iron β-diketiminate complex came from Holland. In this manuscript the hydrodefluorination of aryl and vinyl fluorides is reported in the presence of an Fe(II) fluoride β-diketiminate pre-catalyst and triethylsilane as the hydride source.30 A range of fluorocarbons undergo selective mono-hydrodefluorination (Table 2). However, mechanistic investigations of aryl hydrodefluorination, although detailed, demonstrated that a complex catalytic cycle must be at play, with no reaction observed between the postulated intermediate, iron hydride 9 (Fig. 1) and fluorocarbon. Interestingly, the bulky β-diketiminate complex 10 is most active for the hydrodefluorination of aryl fluorides (Table 2, compare entries 1 and 2), whereas the less bulky analogue 11 is active for hydrodefluorination of fluoroolefins, albeit with lower regioselectivity (entries 7 and 8). In this latter case the data obtained allowed a mechanistic proposal to be made (Scheme 8). Similar to many of the examples presented throughout, this catalytic cycle involves a redox-neutral, σ-bond metathesis-type, series of bond transformations. It should be noted that KHBEt can be used in place of iron pre-catalyst and Et3SiH, giving 79% C6HF5 and p-C6H2F4 in five minutes at room temperature; a more favourable reaction time but at the expensive of selectivity. Increasing the temperature of reaction leads to complex decomposition and a substantial reduction in yield (entry 3).
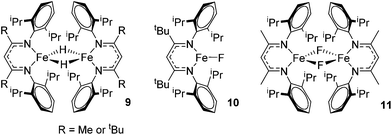 |
| | Fig. 1 Iron complexes involved in catalytic hydrodefluorination. | |
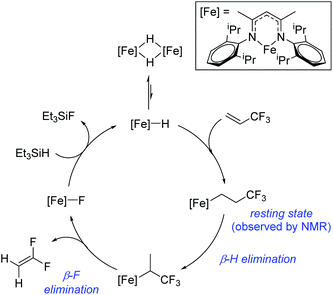 |
| | Scheme 8 Postulated catalytic cycle for hydrodefluorination of fluoroolefins. | |
Table 2 Iron catalyzed hydrodefluorination substrate scope, selected examples
| Entry |
Substrate |
Product (s) |
[Fe] Time temp. (°C) |
TON |
Conversion (%) |
| Conditions: 19 mol% 10 or 10 mol% 11, 0.11 M THF solutions of fluorocarbon and Et3SiH. TON = turnover number. Conversion obtained by NMR and/or GC-MS analysis. |
| 1 |
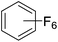
|
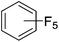
|
10
|
2.5 |
50 |
|
|
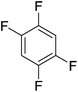
|
4 days |
|
0.8 |
| 45 °C |
|
|
| 2 |
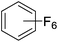
|
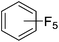
|
11
|
1.0 |
20 |
|
|
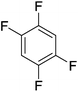
|
4 days |
|
|
| 45 °C |
|
0.2 |
| 3 |
|
|
10
|
1.2 |
24 |
| 4 days |
|
|
| 80 °C |
|
|
| 4 |
|
|
10
|
0.2 |
4 |
| 4 days |
|
|
| 45 °C |
|
|
| 5 |
|
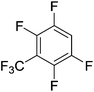
|
10
|
4.5 |
90 |
| 12 h |
|
|
| 45 °C |
|
|
| 6 |
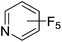
|
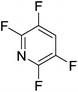
|
10
|
3.6 |
71 |
| 4 days |
|
|
| 45 °C |
|
|
| 7 |
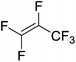
|
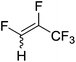
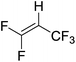
|
11
|
9.8 |
60 E |
|
|
|
3 h |
|
27 Z |
| 100 °C |
|
2 |
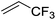
| 8 |
|
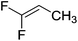
|
11 4 days 100 °C |
1.2 |
11 |
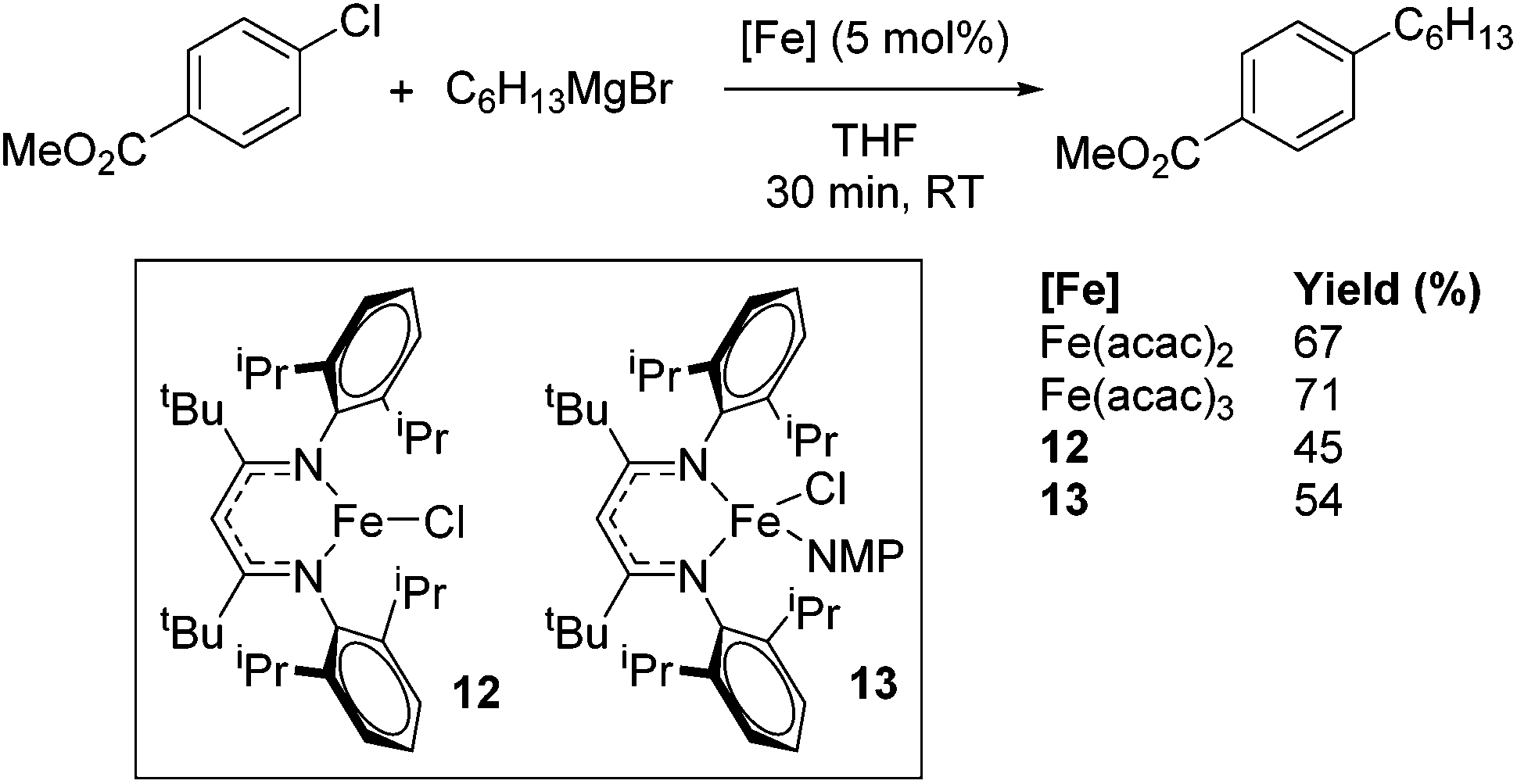
Holland also used Fe(II) chloride β-diketiminate complexes, with moderate success, for Kumada cross-coupling of an aryl halide and alkyl Grignard reagent, although the β-diketiminate pre-catalyst did not perform as well as the simple acetylacetonate (acac) salts, highlighting that the β-diketiminate ligand is not the best choice in every application (Scheme 9).31
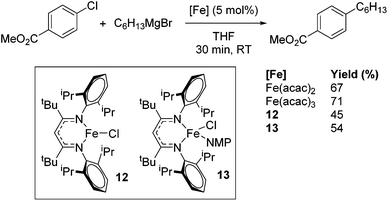 |
| | Scheme 9 Kumada coupling can be performed with several different iron pre-catalysts. Yield determined by GCMS analysis. | |
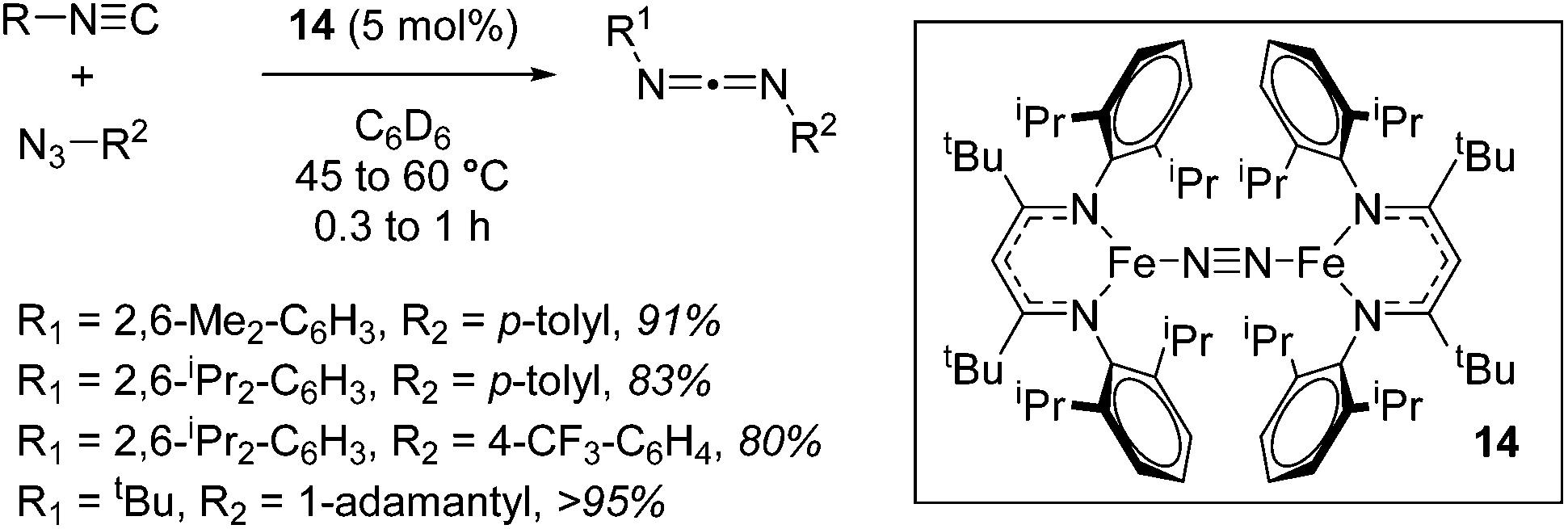
A move to Fe(I) dinitrogen complex 14 allowed Holland to undertake catalytic carbodiimide formation (Scheme 10).32
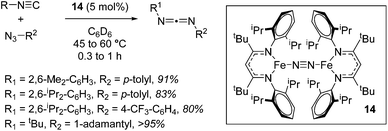 |
| | Scheme 10 Selected examples of Holland's catalytic carbodiimide formation catalyzed by 14. | |
Hannedouche then reported intramolecular hydroamination chemistry in the presence of the iron alkyl complex 15 and cyclopentylamine as an additive.33 The reaction is proposed to proceed via an initial σ-bond metathesis step to generate the on-cycle iron amido complex, this then undergoes insertion to form the iron alkyl intermediate followed by a rate-limiting protonation of alkyl functionality to release the product and regenerate the amido intermediate. The amido intermediate is postulated to be in equilibrium with an amido formed from the cyclopentylamine additive (Scheme 11). The cyclopentylamine appears to be crucial in preventing side-products forming (the dehydrogenated hydroamination product and the hydrogenated aminoalkene starting material).
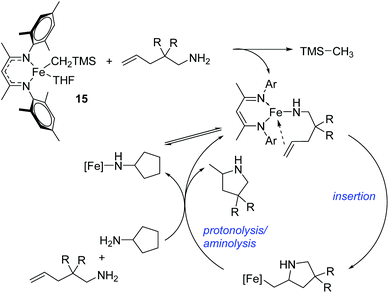 |
| | Scheme 11 Hannedouche's postulated mechanism for intramolecular hydroamination. | |
A range of amino alkenes are tolerated in the reaction mixture, including a trans-substituted internal alkene and an allene (Table 3).
Table 3 Substrate scope for intramolecular hydroamination catalyzed by 15
| Entry |
Substrate |
Product |
Conversion (%) |
| Conditions: 10 mol% 15, 10 mol% cyclopentylamine, C7D8, 90 °C, 48 h. Ar = 4-MeO-C6H4. Yield determined by GCMS or 1H NMR. |
| 1 |
|
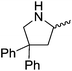
|
100 |
| 2 |
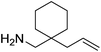
|
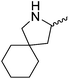
|
91 |
| 3 |
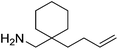
|
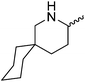
|
90 |
| 4 |
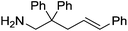
|
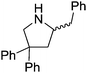
|
74 |
| 5 |
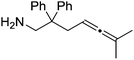
|
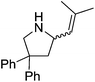
|
20 |
| 6 |
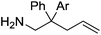
|
|
100 |
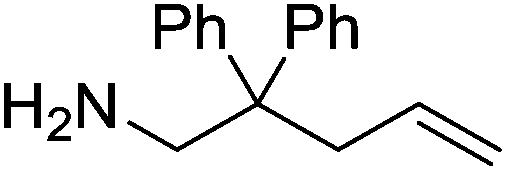
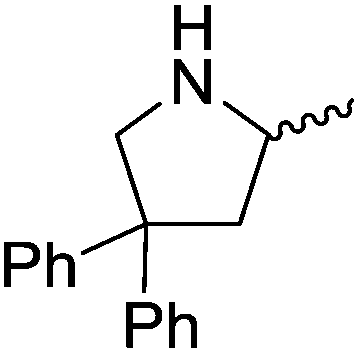
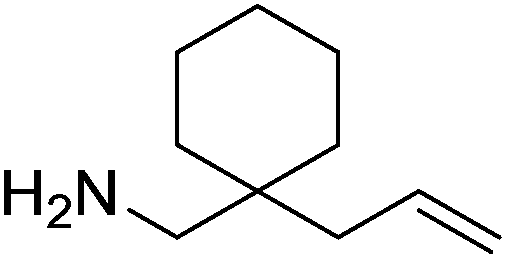
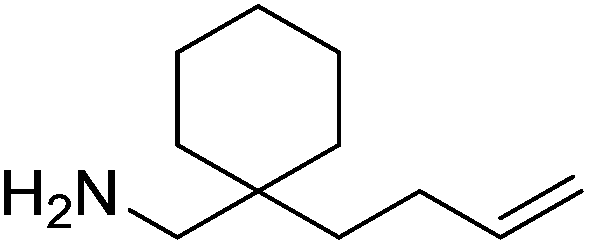
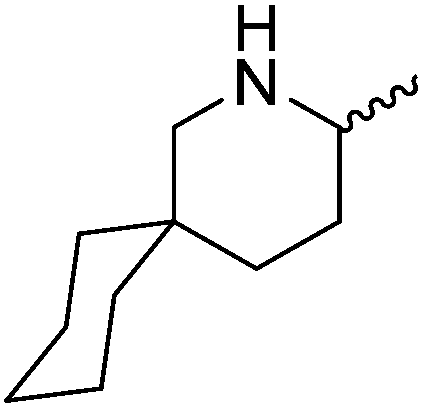
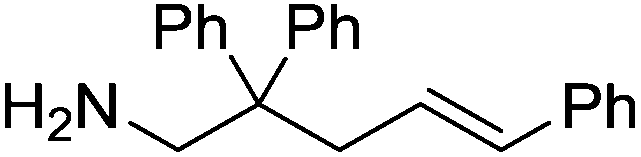
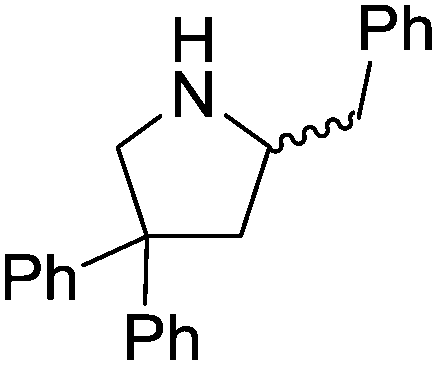
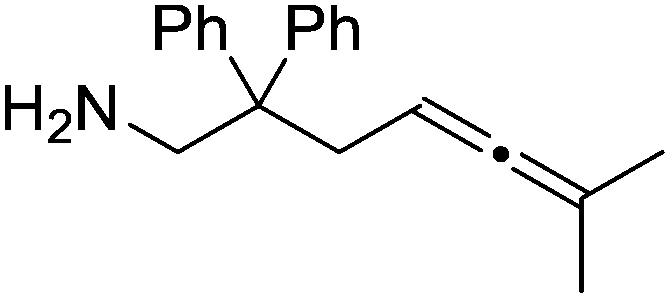
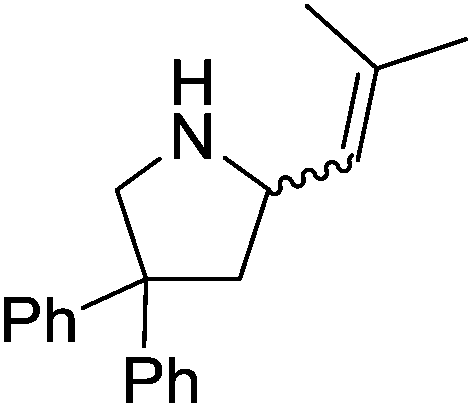
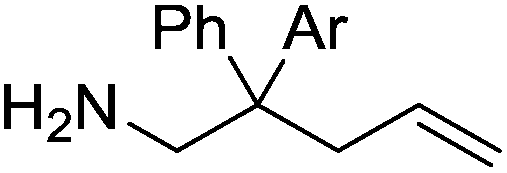
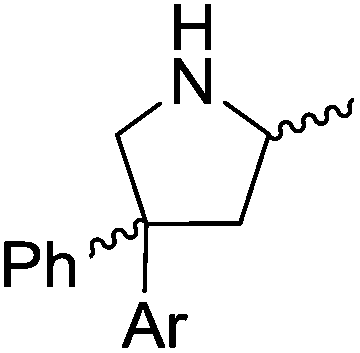
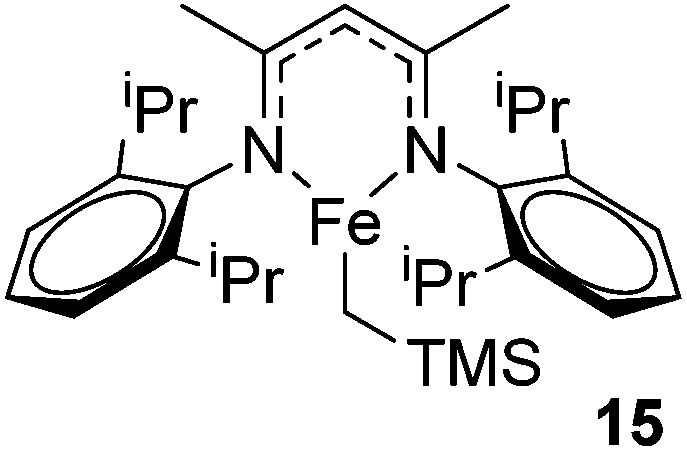
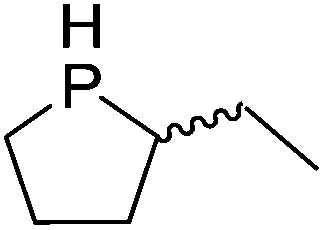
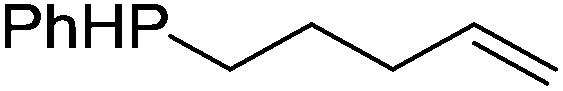
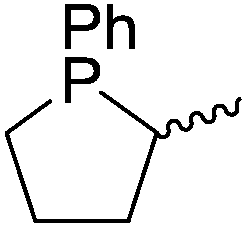
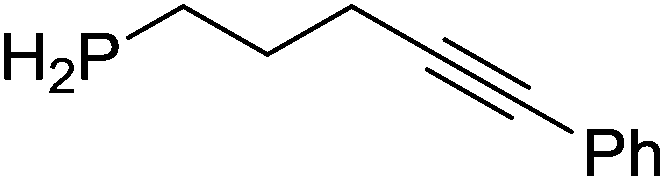
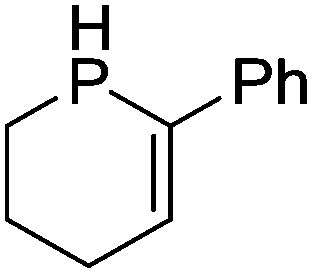
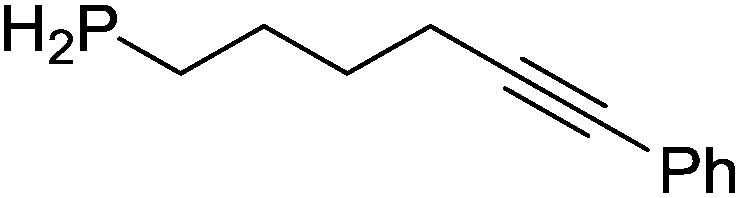
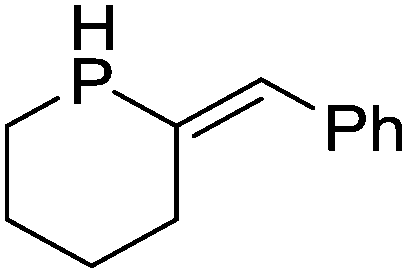
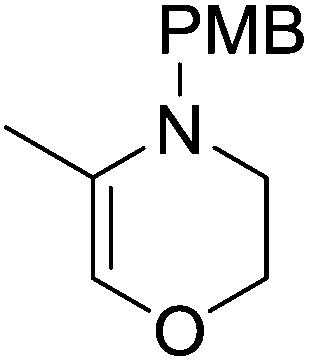
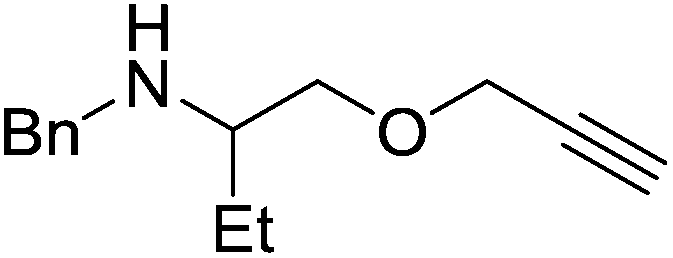
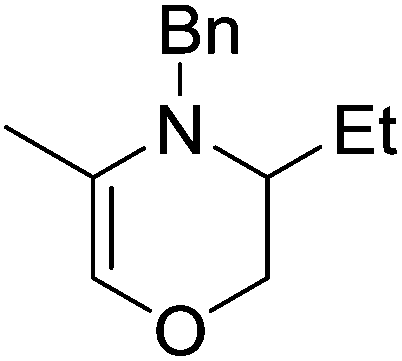
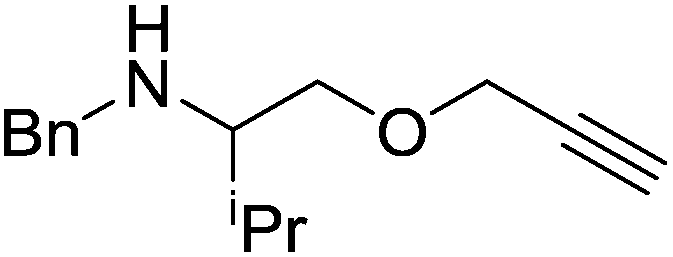
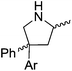
Our own research has focused on transformations with phosphorus, furnishing examples of intermolecular34 and intramolecular35 hydrophosphination. For intramolecular hydrophosphination, the challenge primarily lies in synthesis of the starting materials, but once prepared, pre-catalyst 15 (Fig. 2) can be used to afford the phospholane and phosphorane products in good yield. This is an efficient way to prepare these otherwise difficult to access cyclic phosphine structures (Table 4).
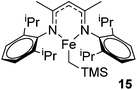 |
| | Fig. 2 Pre-catalyst 15 used in catalytic transformations involving phosphorus. | |
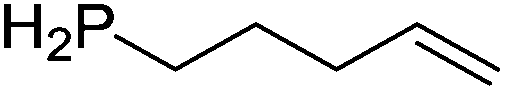
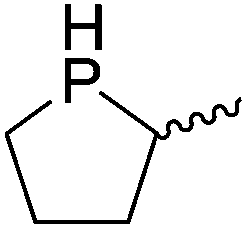
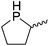
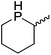
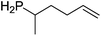
Table 4 Intramolecular hydrophosphination catalyzed by 15
| Entry |
Substrate |
Product |
Time (isomer ratio) |
Conversion (%) |
| Conditions: 0.25 mmol phosphine, 10 mol% 15, C6D6, 90 °C (entry 7, 50 °C). Conversion determined by 1H NMR. |
| 1 |

|
|
17 63:37 |
100 |
| 2 |

|
|
36 83:17 |
91 |
| 3 |
|
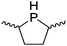
|
36 58:42 |
90 |
| 4 |
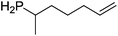
|
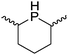
|
36 68:20:12 |
74 |
| 5 |

|
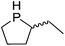
|
14 — |
20 |
| 6 |

|
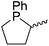
|
14 62:38 |
100 |
| 7 |
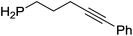
|
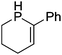
|
14 — |
98 |
| 8 |
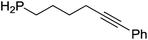
|
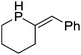
|
17 — |
100 |
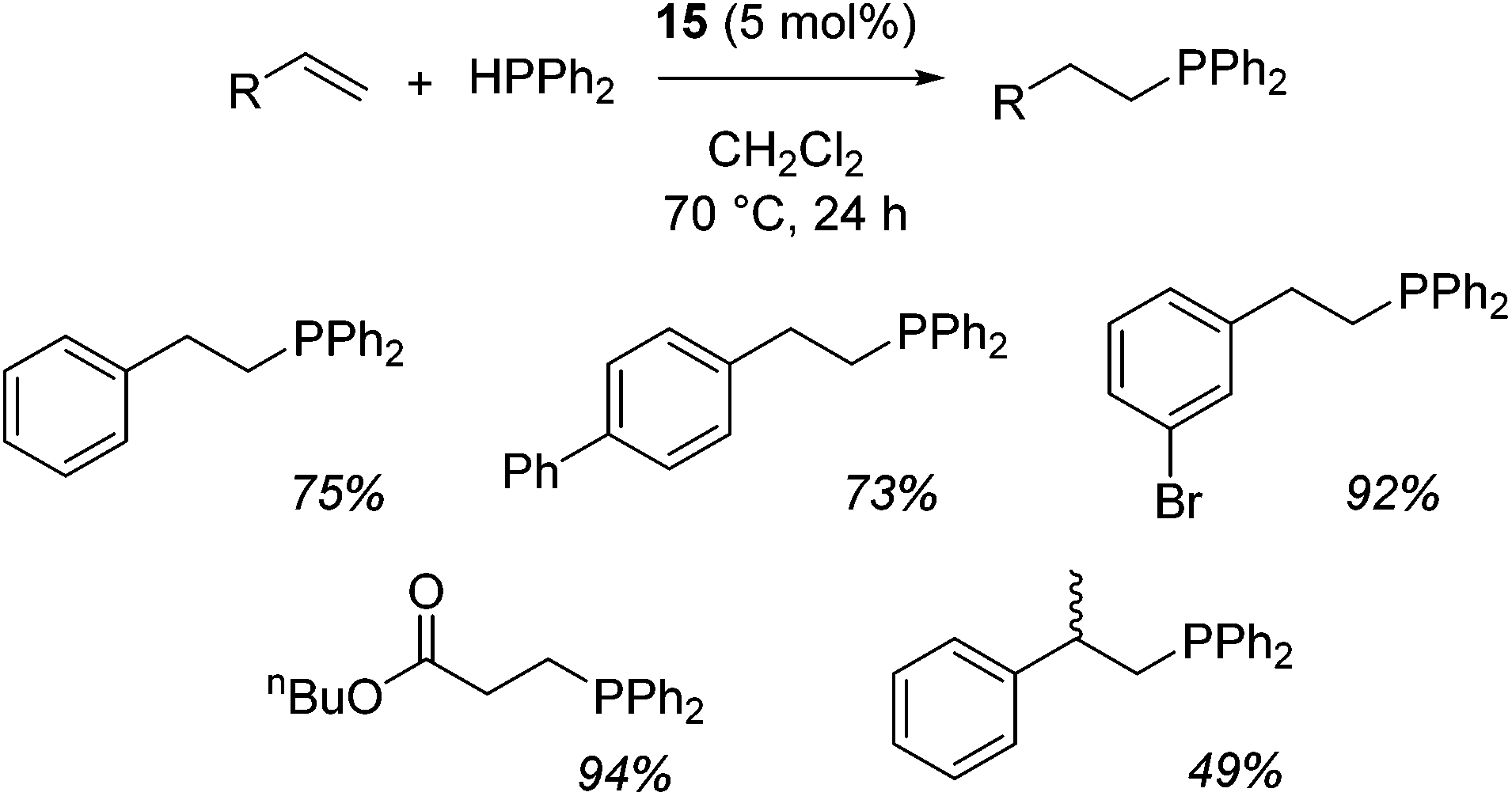
In the case of intermolecular hydrophosphination, forcing conditions are needed and the reaction requires CH2Cl2 as the solvent (Scheme 12). When the reaction solvent was changed to benzene dehydrocoupling, allowing the formation of P–P bonds, takes place (Scheme 13).34
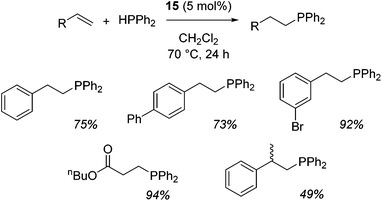 |
| | Scheme 12 Hydrophosphination of activated alkenes requires fairly forcing reaction conditions and uses CH2Cl2 as the reaction solvent. | |
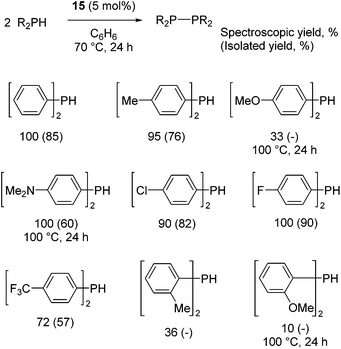 |
| | Scheme 13 Selected examples of secondary phosphines used in iron catalyzed dehydrocoupling. | |
With mechanistic study of the P–P bond forming reaction hampered by the lack of a diagnostic NMR handle (especially with the paramagnetic pre-catalyst) and with the products prone to oxidation/hydrolysis not allowing reaction sampling for kinetic analysis, it was only possible to undertake preliminary radical clock studies. This suggested, along with DFT investigations, that the dehydrocoupling reaction is radical mediated. We have subsequently extended this to the dehydrocoupling of phosphine- and amine–boranes.
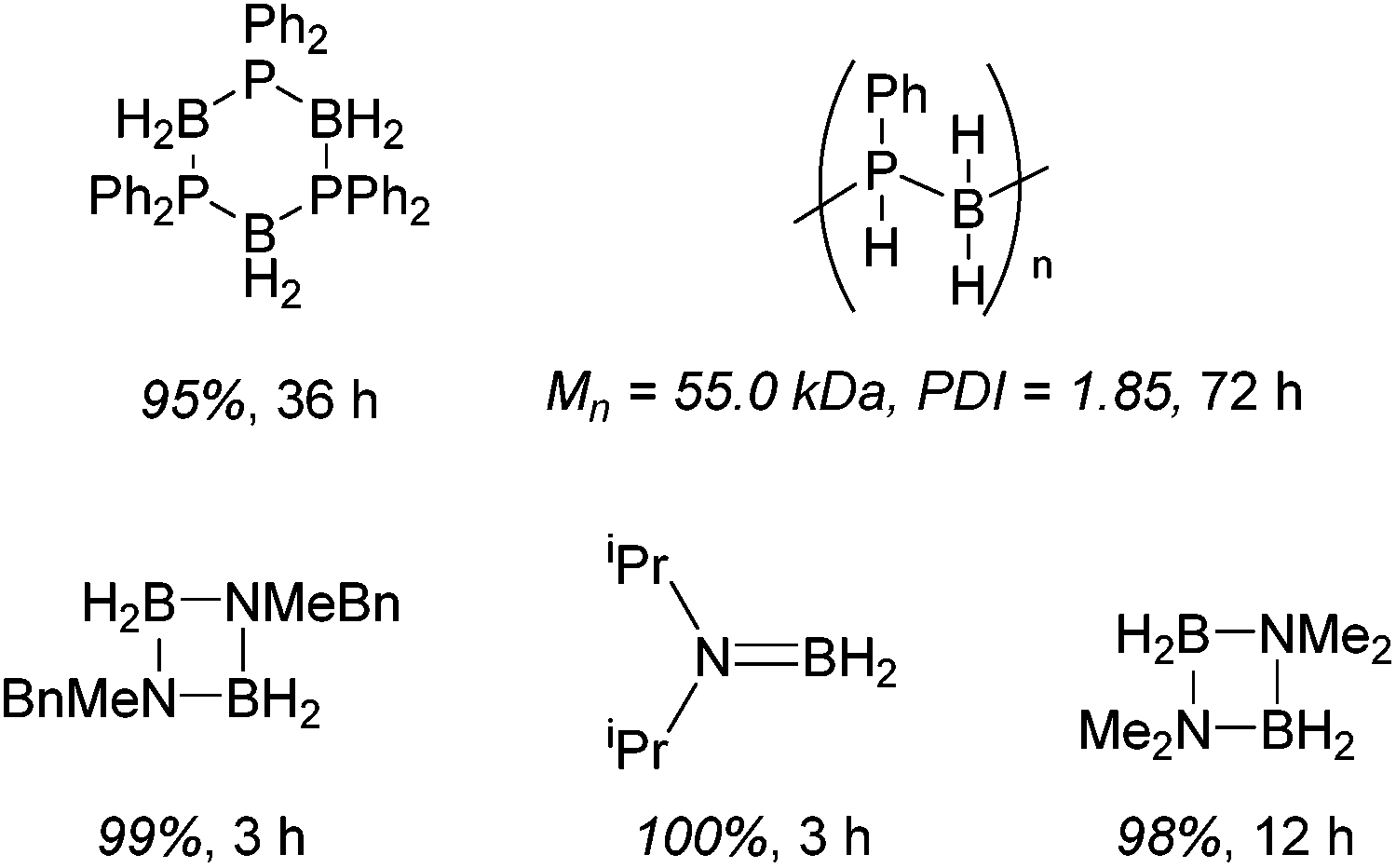
Diphenylphosphine borane can be dehydrocoupled using 15 to form the cyclic tetramer with high spectroscopic yield.36 However, a change to dicyclohexylphosphine borane does not show activity. Dehydrocoupling to form poly(phenylphosphine borane) is also achieved, with the polymer precipitating out of the reaction solvent and, upon analysis, an Mn of 55.0 kDa and PDI of 1.9 was obtained. Unfortunately ammonia borane could not be dehydropolymerized due to solubility/solvent compatibility issues, but organic functionalized amine–boranes did undergo dehydrocoupling under far milder conditions than those used for the phosphine boranes (1 mol% 15, room temperature, 3 to 12 h for amine–boranes and 10 mol% 15, 110 °C, 36 to 72 h for phosphine–boranes, Scheme 14).
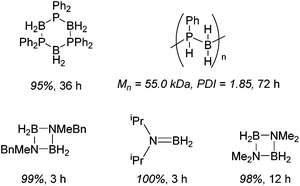 |
| | Scheme 14 Substrates that successfully dehydrocouple using 15. | |
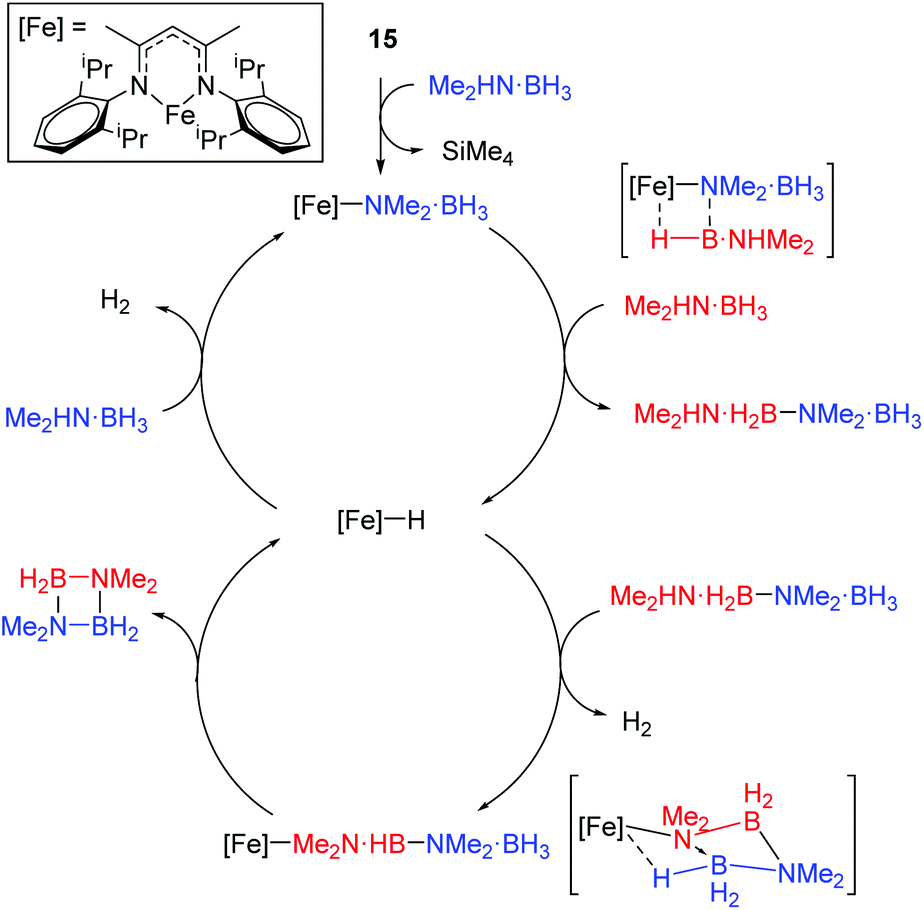
The reaction conditions for dimethylamine–borane dehydrocoupling are such that detailed mechanistic study has been possible and a catalytic cycle proposed (Scheme 15).
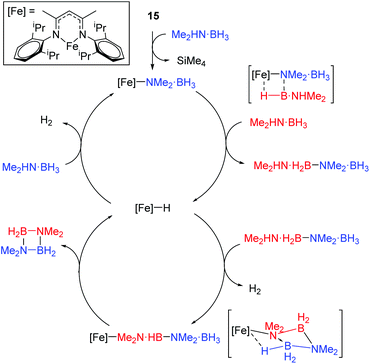 |
| | Scheme 15 Proposed catalytic cycle for amine–borane dehydrocoupling. | |
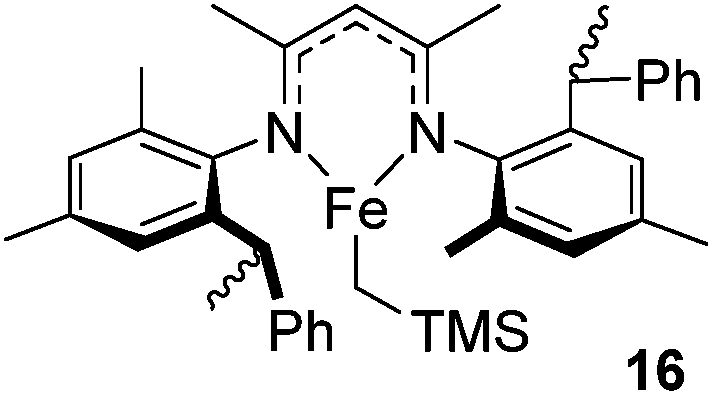
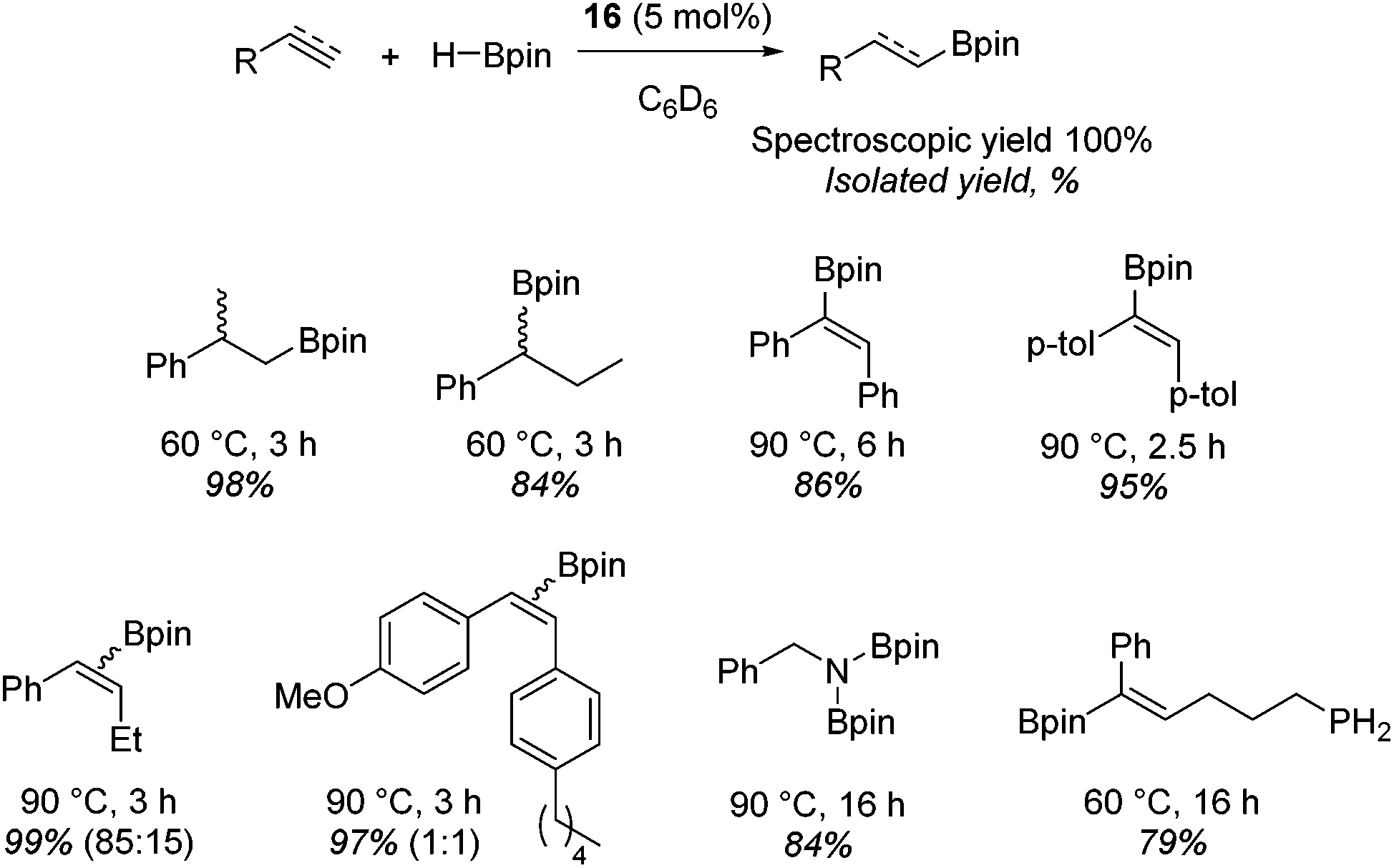
Beyond the chemistry of phosphorus, we also demonstrated that the pre-catalyst 15 can undertake hydroboration37 and that changing the ligand structure away from the classic 2,6-diisopropyl motif, to the unsymmetrical substitution pattern 16 (Fig. 3), can be beneficial for reactivity. In the presence of 16 the reaction time for the hydroboration of isoprene, (+)-valencene and β-pinene can be reduced from 7, 16 and 16 h to 2.5, 2 and 2.5 h respectively. 16 also allows for the hydroboration of more challenging substrates for example alkynes (Scheme 16). At present it is not clear why this ligand structure leads to such fast reactions. Mechanistically, we propose that the reaction proceeds via a series of Fe(II)-hydride and -alkyl intermediates, followed by boration which leads to the release of product and the regeneration of Fe(II)-hydride.
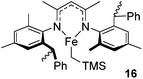 |
| | Fig. 3 Modified β-diketiminate complex 16. | |
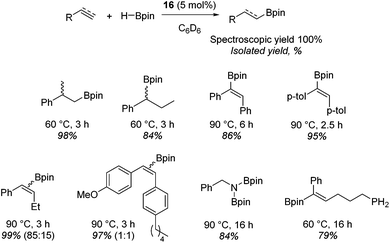 |
| | Scheme 16 Alkene and alkyne hydroboration can be carried out with 15, however, a vast improvement in reactivity is observed with 16 and this allows the hydroboration of challenging substrates. | |
Cobalt
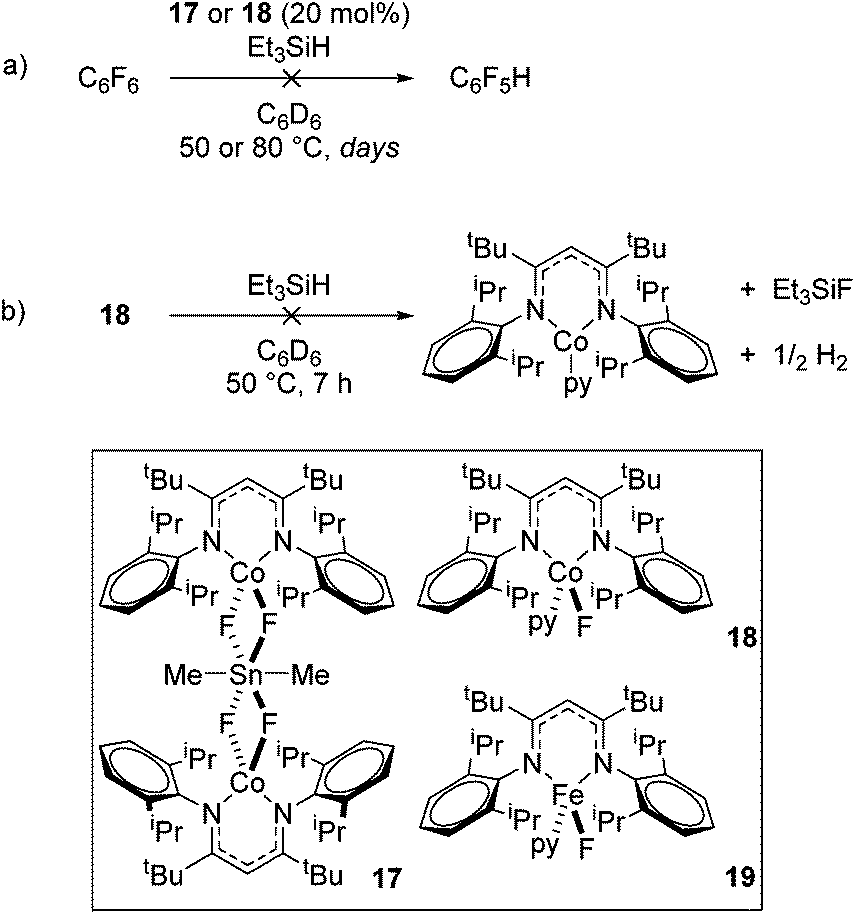
In contrast to the high catalytic reactivity obtained using iron pre-catalyst 10 in hydrodefluorination, the cobalt analogues, 17 and 18 (Scheme 17a), do not catalyze the reaction.38 This was postulated to be due to several reasons, one being the higher coordination number necessary for the cobalt complex compared to the three-coordinate iron complex. Indeed when the analogous iron-pyridine adduct (19) was trialled in catalysis using C6F6 as the substrate, no hydrodefluorination was observed. The iron complexes 10 and 11 are known to form the iron hydride dimer (of the form 9), a key intermediate in catalysis, in the presence of Et3SiH. However, when 19 is exposed to a stoichiometric amount of Et3SiH, no iron hydride forms. similarly, the cobalt hydride intermediate, necessary for hydrodefluorination to take place, is shown to be unlikely to form due to the rapid elimination of H2 when 18 is reacted with Et3SiH (Scheme 17b).
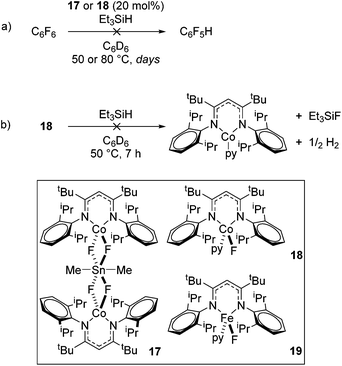 |
| | Scheme 17 (a) Hydrodefluorination is not catalytic with respect to cobalt. (b) Stoichiometric reaction with silanes does not produce Co-hydride, which is expected to be a reactive intermediate in a successful hydrodefluorination catalytic cycle. | |
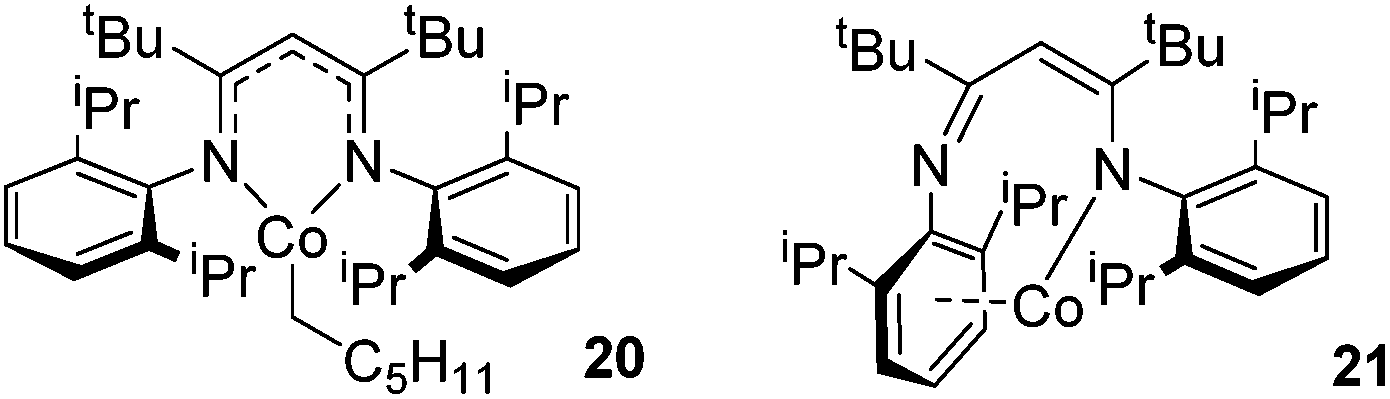
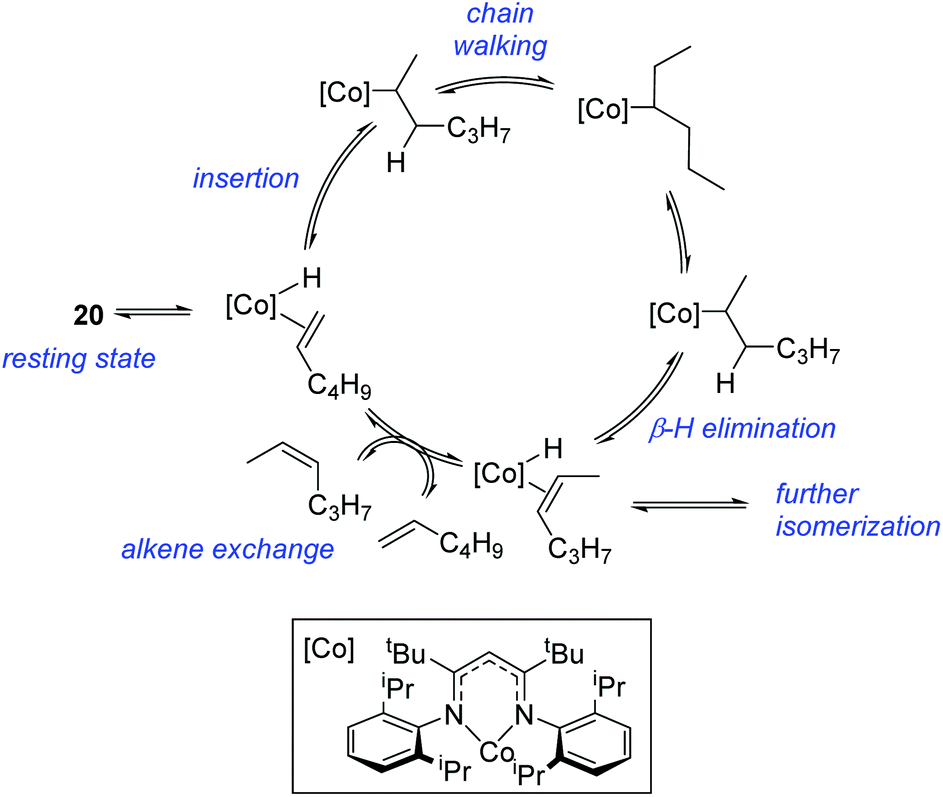
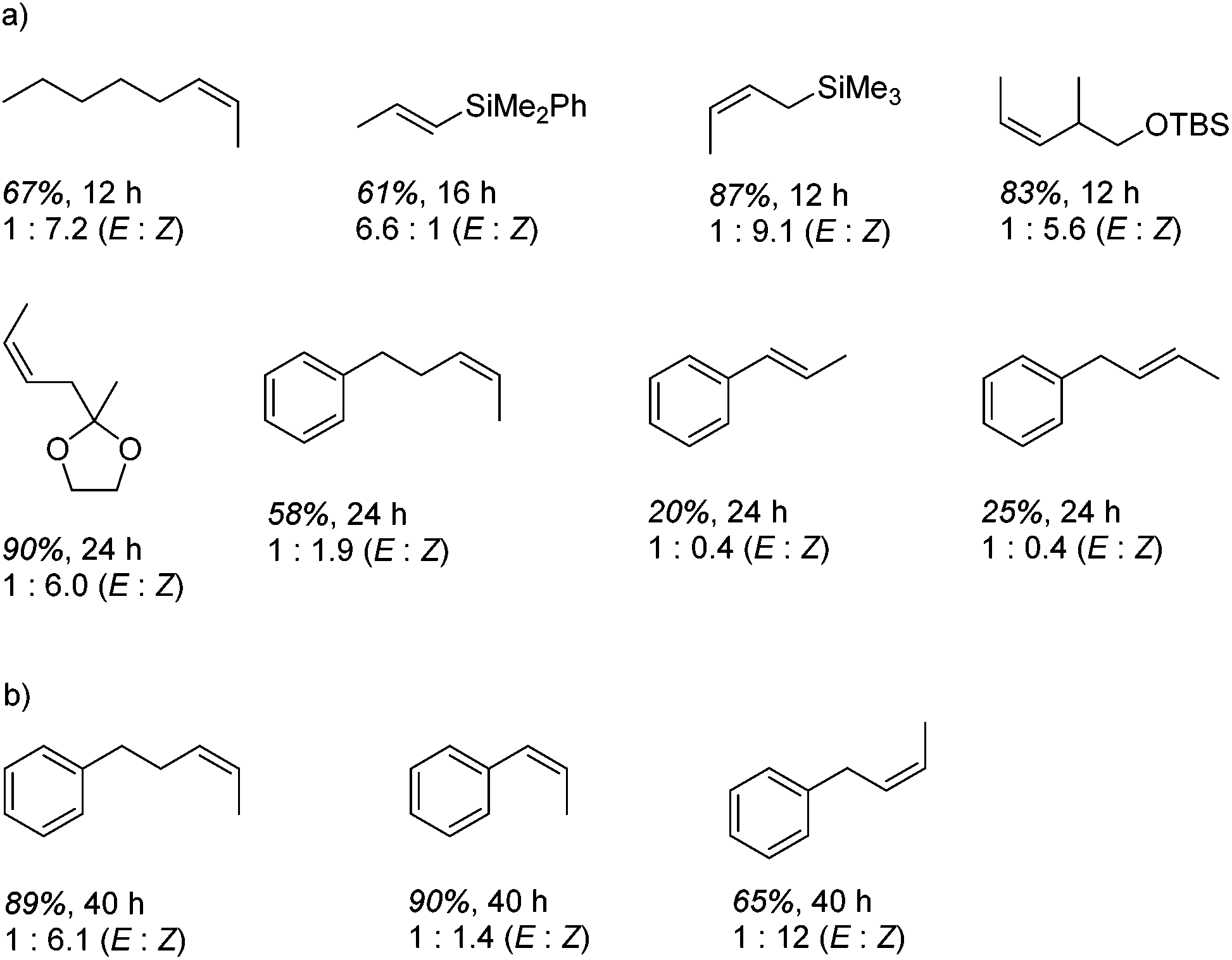
However, the cobalt β-diketiminate coordination environment can be tuned and the Co(II) alkyl complex, 20 (Fig. 4), is an excellent catalyst for double bond isomerization. It catalyzes the process under mild conditions and proceeds with a high level of selectivity for the Z-isomer; the more challenging to access, kinetic product.39 Using 1-hexene as the standard substrate, in the presence of 21, the selectivity switches to the E-alkene product, a process believed to be heterogeneous in nature due to reaction quenching on the addition of Hg (whereas Hg has no effect on the reaction catalyzed by 20). Longer reaction times when 20 is employed result in erosion of selectivity, but the addition of an alkene, such as 3,3-dimethyl-1-butene can help to improve Z-selectivity for longer reaction time periods. This is postulated to be due to the relative binding strengths of the alkenes: 3,3-dimethyl-1-butene binds more strongly to the cobalt centre than the product (Z-2-hexene) thus preventing the product binding and undergoing further isomerization. Whereas the substrate, 1-hexene, binds the most strongly, so this is allowed to react preferentially. The n-hexyl complex 20 is believed to be the catalyst resting state and detailed labeling, chain walking and reaction profiling studies allowed a catalytic cycle to be postulated which involves the interconversion of Co(II) intermediates (Scheme 18). Overall the isomerization reaction operates for a range of substrates (Fig. 5a), the majority of which favor the formation of the Z-alkene product. Detailed reaction understanding also allowed the development of isomerization to form Z-styrene products at low substrate and catalyst concentrations, which under standard reaction concentrations favour the E-styrene (Fig. 5b).
 |
| | Fig. 4 Complexes 20 and 21. | |
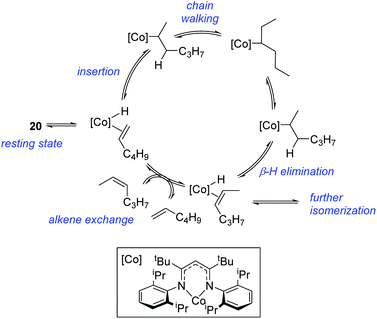 |
| | Scheme 18 Postulated mechanism for Weix and Holland's double bond isomerization. | |
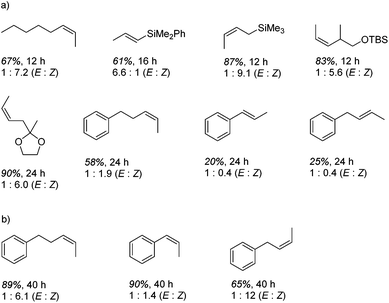 |
| | Fig. 5 (a) Selected examples of the products formed in Z-selective double bond isomerization. (b) Dilution by a factor of 20 increases the ratio of Z-product formed. Yields and ratios determined by 1H NMR. | |
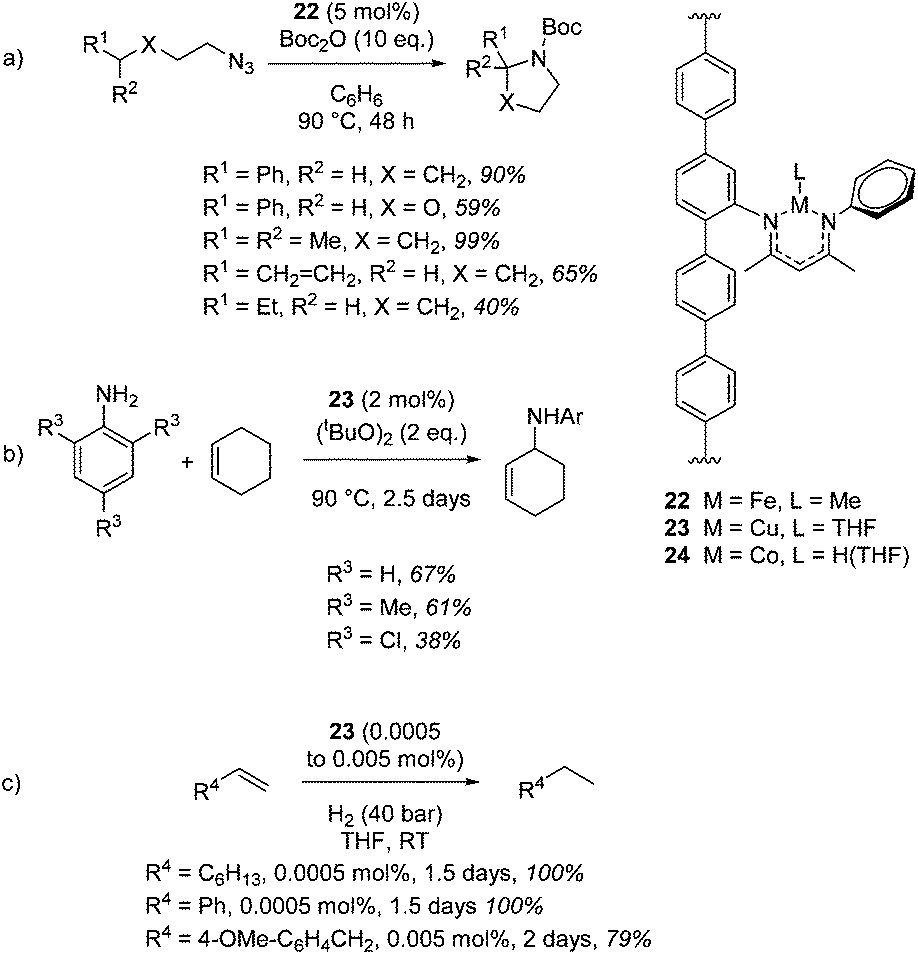
Lin and co-workers provide an elegant example of a MOF containing the β-diketiminate motif, which was installed using a post-synthetic modification of the classic UiO-type MOF topology. The β-diketiminate fragment was installed by a condensation reaction between the amine-functionalized UiO-type MOF (Zr6O4(OH)4(TPDC-NH2)6) and 4-N-phenyl-amino-3-pentene-2-one followed by complexation to an appropriate metal salt (e.g. a metal chloride) and reduction to replace unreactive chloride co-ligands with hydride or methyl groups (or in the case of copper, replace MeCN with THF). These unusual forms of β-diketiminate pre-catalysts undertake C–H amination with an iron (22, Scheme 19a) or copper (23, Scheme 19b) centred MOF and hydrogenation with a cobalt-containing MOF (24, Scheme 19c).40 Amination using 22 tolerates the presence of ethers and, notably, double bonds, with no side-reactions taking place with the latter (Scheme 19a). The copper mediated reaction (23) proceeds via a radical mechanism and 22 does not show any reactivity for this transformation. Steric bulk around the reagent nitrogen does not lead to a noticeable drop-off in yield whilst challenging substrate 2,4,6-trichloroaniline gives a moderate yield of product. similarly, only a poor yield of octane is obtained when 22 is used for hydrogenation (10% product with 0.1 mol% catalyst loading) compared to 24 (100% yield with only 0.0005 mol% catalyst loading, Scheme 19c).
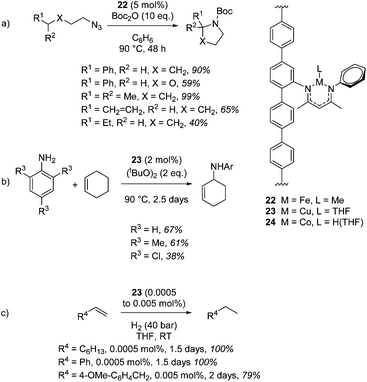 |
| | Scheme 19 A series of catalytic transformations carried out by a homologous series of MOFs. (a) C–H amination with release of N2 using an Fe–MOF. (b) C–H amination using a Cu–MOF. (c) Hydrogenation using a Co–MOF. Yields determined by 1H NMR. | |
It could be argued that the conditions used to carry out these transformations are more forcing than the conditions needed when using a discrete mononuclear homogeneous catalyst. Taking the research of Chirik and co-workers as an example, an asymmetric cobalt bis(imino)pyridine catalyst can be used to enantioselectively hydrogenate disubstituted alkenes using 4 atm H2 at RT in 24 h,41 in comparison to the MOF catalyzed (racemic) process which requires 1.5 days and 40 bar H2. This of course highlights a current limitation in β-diketiminate chemistry as well, that being the lack of transformations undertaken with enantiocontrol. However, this piece of MOF research diversifies the type of β-diketiminate complex (or reaction environment) with which to carry out catalysis, carries the benefits of heterogeneous catalysis (ease of separation, recycling, etc.) and provides an early insight in the type of progress that could be made in this area in the future.
Nickel
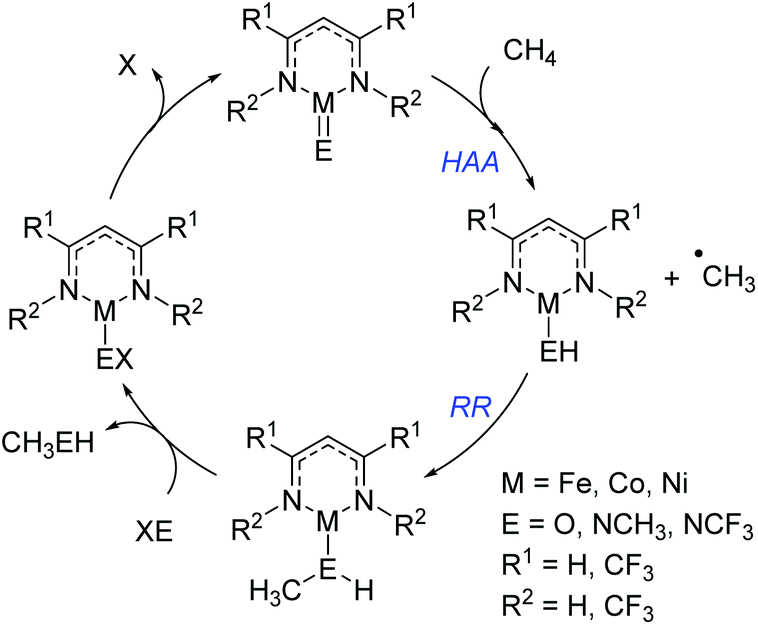
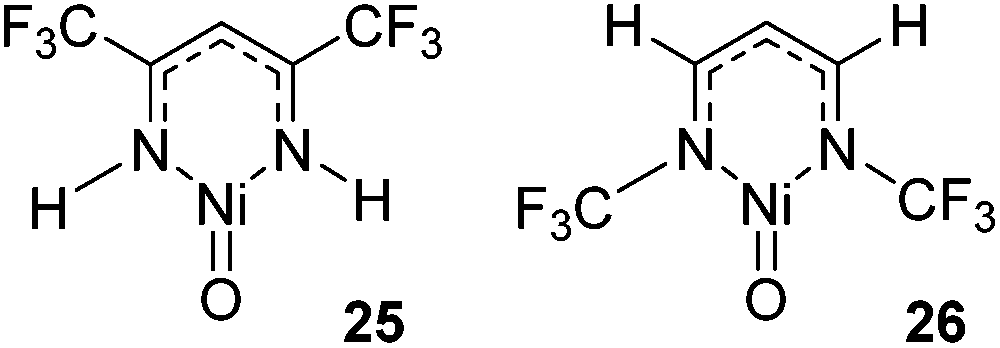
Cundari has employed computational methods to develop theoretical catalytic cycles for methane activation, comparing β-diketiminate and dihydrophosphinoethane complexes of iron, cobalt and nickel.42 The overall transformation (Scheme 20) was split into two separate reactions: hydrogen atom abstraction (HAA) and radical relay (RR), and the thermodynamics and kinetics were calculated. The study showed that the most viable metal center in terms of reaction enthalpy is nickel (in fact the reaction is less endothermic moving across the period) and so this was used as the basis for kinetic analysis. The model complexes employed used a co-ligand which is ultimately responsible for the C–H activation event. Comparison of oxide, methyl-imido and trifluoromethyl-substituted imido co-ligands (i.e. M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, MNCH3 and MNCF3) found that HAA is thermodynamically more favourable for the CF3-substituted co-ligand. In contrast RR is thermodynamically more favourable for the CH3-imido co-ligand and the energetics are such that it can off-set the benefits of the CF3-imido during HAA. Kinetically, the oxide ligand performed best and overall nickel complexes of the form 25 and 26 (Fig. 6) were demonstrated to give the lowest methane C–H activation barrier. In the case of 25 and 26, the presence of CF3 functionalized β-diketiminate ligand lowers the kinetic HAA barrier. The beneficial effects of an electron withdrawing backbone (R1) or N-functionality (R2) was also highlighted by Cramer during computational studies on Cu–oxygen complxes and the mechanism by which such β-diketiminates can undertake C–H bond hydroxylation.43
O, MNCH3 and MNCF3) found that HAA is thermodynamically more favourable for the CF3-substituted co-ligand. In contrast RR is thermodynamically more favourable for the CH3-imido co-ligand and the energetics are such that it can off-set the benefits of the CF3-imido during HAA. Kinetically, the oxide ligand performed best and overall nickel complexes of the form 25 and 26 (Fig. 6) were demonstrated to give the lowest methane C–H activation barrier. In the case of 25 and 26, the presence of CF3 functionalized β-diketiminate ligand lowers the kinetic HAA barrier. The beneficial effects of an electron withdrawing backbone (R1) or N-functionality (R2) was also highlighted by Cramer during computational studies on Cu–oxygen complxes and the mechanism by which such β-diketiminates can undertake C–H bond hydroxylation.43
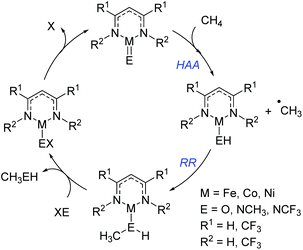 |
| | Scheme 20 Theoretical catalytic cycle for methane C–H activation and subsequent functionalization. | |
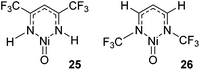 |
| | Fig. 6 Complexes that were determined by Cundari to be the most effective methane functionalization catalysts. | |
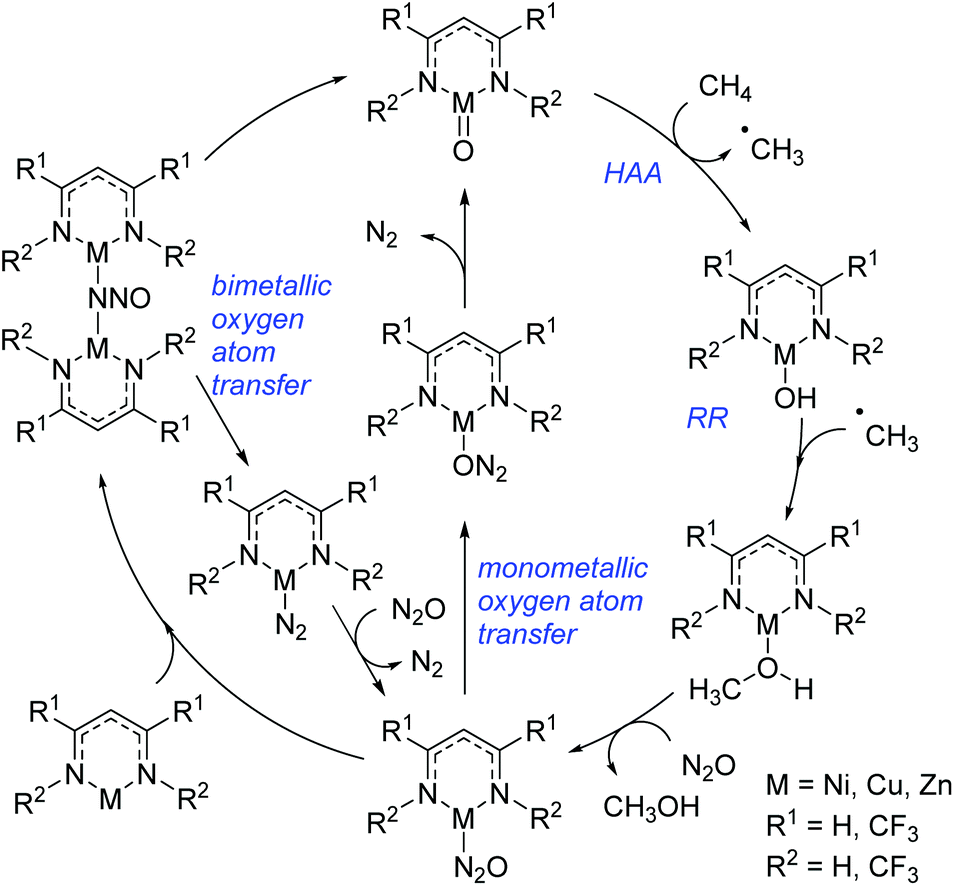
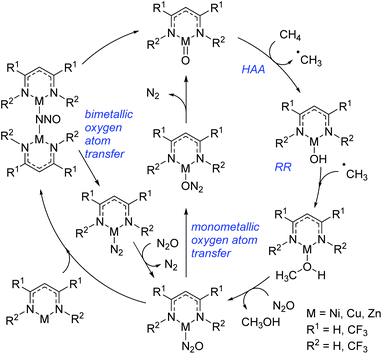
More recently Cundari extended the aforementioned study to specifically investigate the catalytic conversion of methane to form methanol (Scheme 21).44 This study looked at the late 3d metals, nickel, copper and zinc and, coupled with the trend outlined above, showed that methane HAA becomes more viable moving across the period from iron to zinc. Reactions with nickel have HAA as the rate-limiting step, whereas this shifts to RR for copper and zinc. Again, overall, the nickel complexes studied showed the greatest potential as catalysts for methane to methanol conversion, whereas the copper complex, although promising, appears to have the potential to undergo several side-reactions. The limitations of the zinc catalyst lie in the ability to return the complex to the active catalyst (with ZnO bonding motif): the formation of a stable bimetallic complex is limiting. Note that in both catalytic cycles (Schemes 20 and 21) the metal undergoes sequential reduction steps to generate the desired product, followed by re-oxidation to regenerate the first catalytic intermediate; redox reactivity of this sort, thus far, has only been shown for chromium β-diketiminate complexes.
 |
| | Scheme 21 Methane C–H activation to form methanol using N2O as the oxidant. | |
These computational studies clearly outline the outstanding potential for nickel β-diketiminate complexes to undertake fundamental and highly desirable catalytic bond transformations, but with no examples of catalytic transformations being undertaken by nickel β-diketiminate complexes, it is clear that there is much to be done in this area but the wealth of potential reactivity is huge.
Copper
Although not limited to biomimetic catalysis studies, there is a wealth of such chemistry involving copper β-diketiminate complexes.
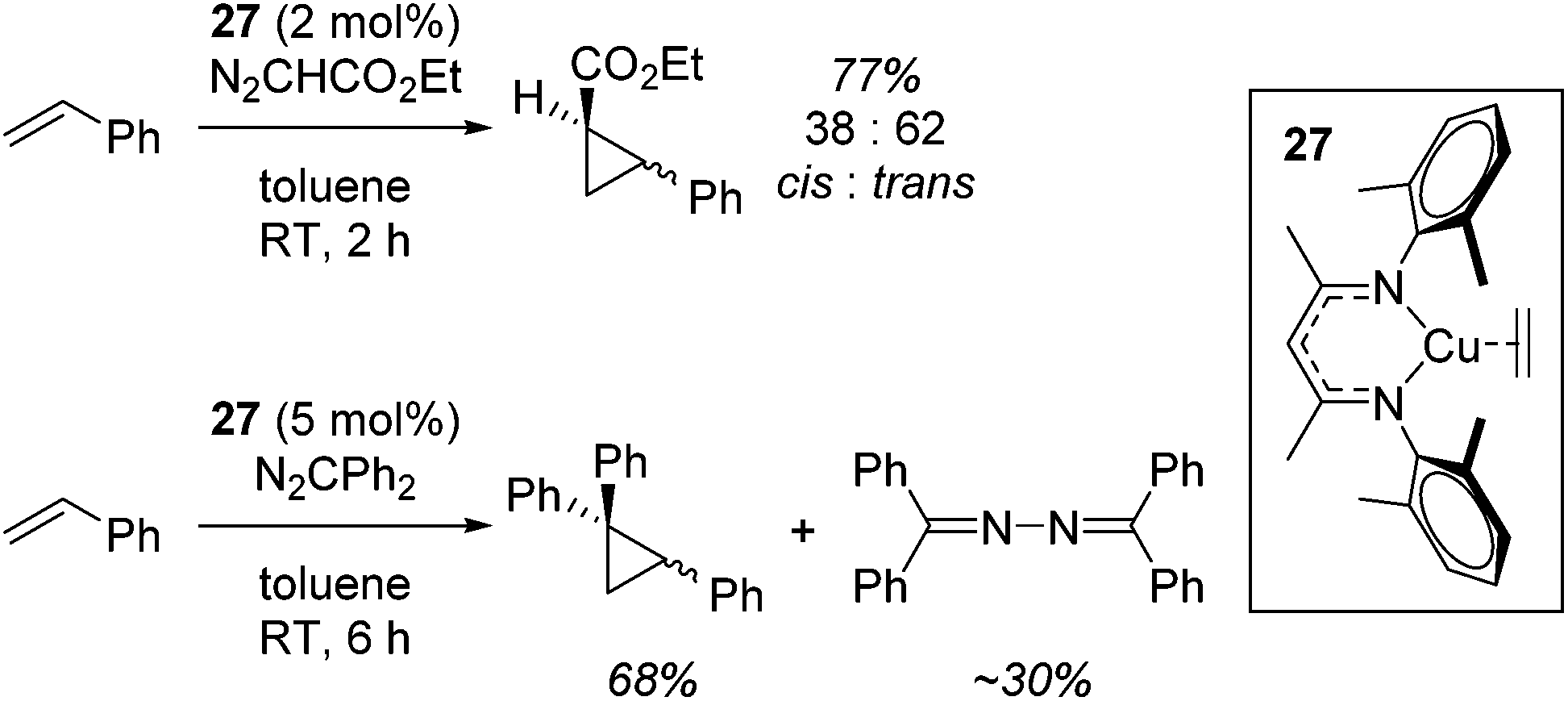
First of all, Warren used complex 27 for the cyclopropanation of styrenes using diazo compounds (N2CHCO2Et or N2CPh2, Scheme 22).45
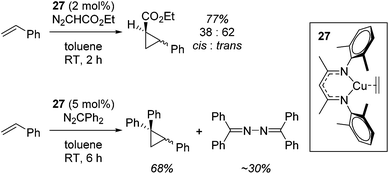 |
| | Scheme 22 Cyclopropanation using a copper β-diketiminate complex. | |
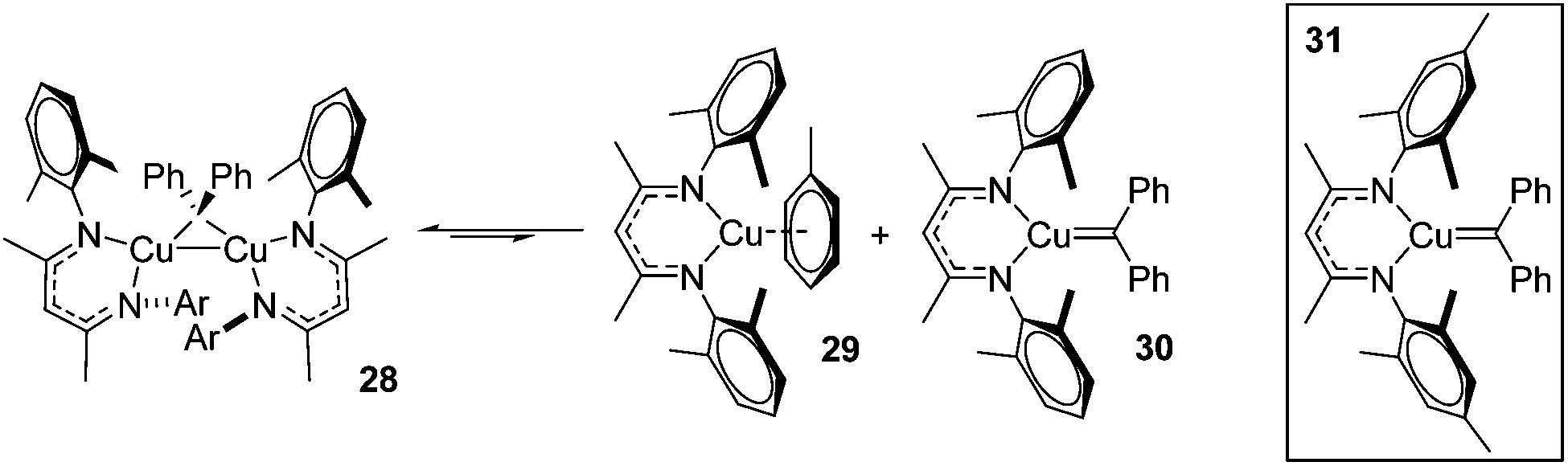
A carbene intermediate was isolated (28) which is catalytically active but, when using 28 as a catalyst, α-methyl styrene reacts cleanly at room temperature but β-methyl styrene requires heating at 45 °C and cyclopropanation occurs with competing dimerization of the carbene to form Ph2CCPh2, hinting at the important role sterics play in this transformation. 28 was shown to be in equilibrium with its monomers (29 and 30), the latter postulated to be the active catalyst (Scheme 23). The mesityl analogue of 30, 31, was fully characterized and used in a kinetic study. This showed that the reaction had a pseudo first order dependence on 31, was first order in styrene while Eyring analysis demonstrated that the reaction was likely to proceed via an associative mechanism and Hammett data confirms that 31 is electrophilic, with an increase in rate observed when electron rich styrenes are employed.
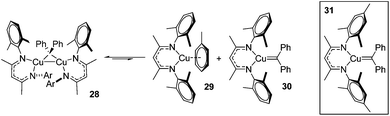 |
| | Scheme 23 Dimer 28 splits into 29 and 30, while 31 is characterized and used in kinetic studies. | |
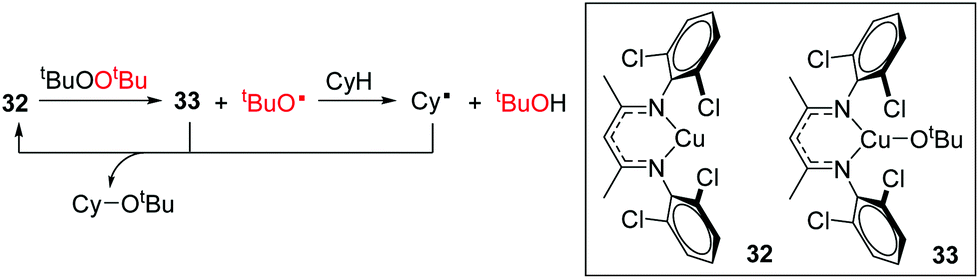
Warren and Cundari have also investigated the reaction of the Cu(I) complex 32 with alkyl peroxides, which results in the formation of alkoxide radicals for the catalytic etherification of cyclohexane.46 The combined synthetic and theoretical approach showed that tBuO˙ carries out hydrogen atom abstraction from cyclohexane, generating a cyclohexyl radical (Cy˙) which then reacts with Cu(II) intermediate 33 (Scheme 24). It would be interesting to extend the synthetic scope of this reaction to include other unactivated hydrocarbons and determine whether the product distribution/regioselectivity follows that expected for a radical mediated bond forming process.
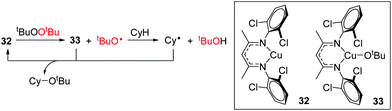 |
| | Scheme 24 Radical reaction of peroxides with cyclohexane (50% yield after 24 h, 5 mol% 32). | |
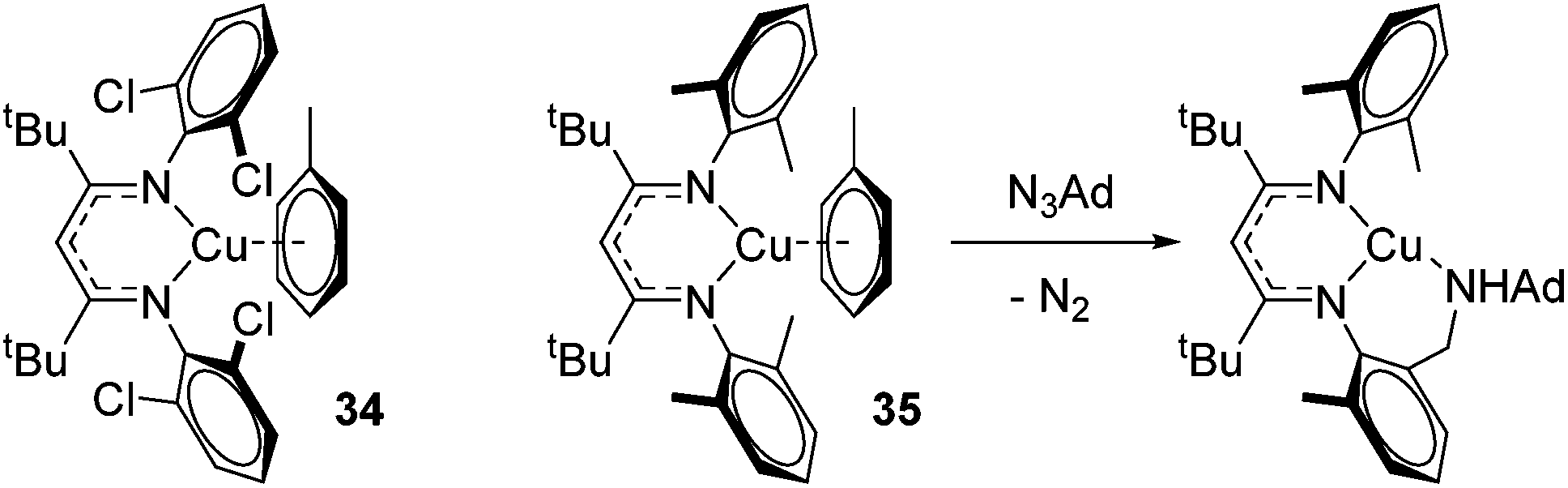
Finally, in a joint experimental and computational study, Warren and Cundari investigated catalytic C–H amination using Cu–nitrene complexes using ortho-chloro substituted β-diketiminate complex 34 (Fig. 7).47 The authors found that alkyl substituents on the aryl ligand functionality (35) led to C–H activation in the presence of an azide aminating reagent, thus preventing formation of the key nitrene intermediate need for amination of substrate (e.g. LCuNAd), whereas this was circumvented with the 2,6-dichloro ligand.
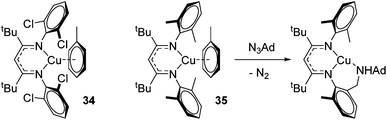 |
| | Fig. 7 Copper complexes tested in C–H amination. 35 undergoes C–H amination of the ligand preventing catalysis, whereas chloride substituents on 34 avert this. | |
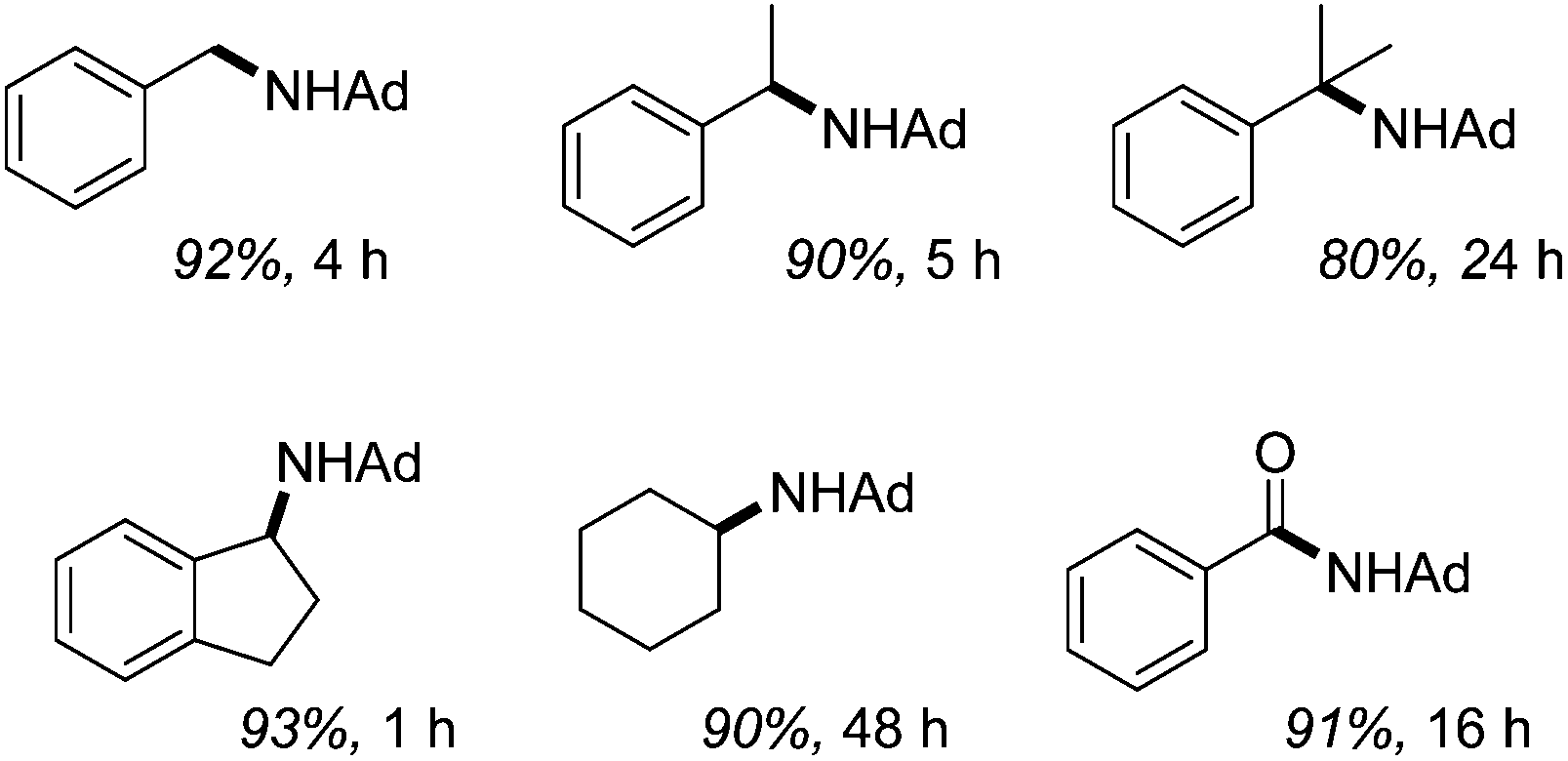
Catalytic sp3 C–H amination using 2.5 mol% 34 takes place at 110 °C within hours, Fig. 8. They also show that decreasing the alkane loading is detrimental to reactivity, favouring the formation of the diazene AdNNAd, while there is a striking relationship between increasing C–H bond dissociation energy, BDE, (for the alkane substrate) and the rate of reactivity. For example, cyclohexane has the highest BDE and the lowest rate of reactions, whereas hydrindene has the lowest BDE and can be aminated in only 1 hour.
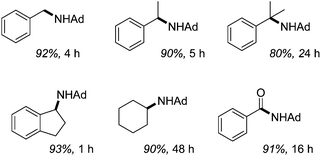 |
| | Fig. 8 C–H amination products, catalyzed by 34 using N3Ad (Ad = adamantyl). Isolated yield of HCl salt shown. | |
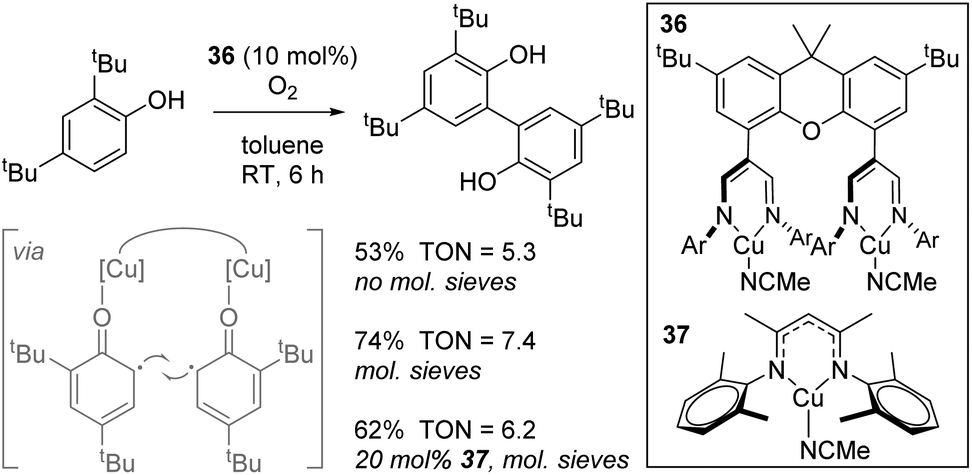
A study with biological relevance came from Limberg using complex 36 for the catalytic oxidative homocoupling of 2,4-di-tert-butylphenol to give the bisphenol product (Scheme 25).48 Although only one substrate was tested in catalysis, transformations of this general type are important in terms of the synthesis of biaryl structures for use in drug-like molecules and the biomimetic nature of this process involving O2 activation is an important transformation facilitated by copper-containing enzymes. In this manuscript, the authors use a dinuclear Cu(I) complex, 36, which can be oxidized by O2 to form a dinuclear Cu(II) species which are postulated to coordinate one molecule of phenol to each copper centre, generating phenyl radical and thus bring these reactive species in to close proximity (Scheme 25, insert). Molecular sieves are used to absorb water generated and this leads to an increase in yield. Unfortunately, 36 only leads to a marginally higher yield than mononuclear analogue 37.
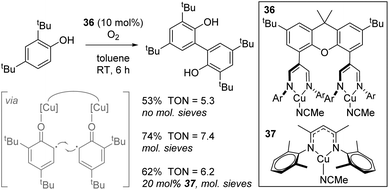 |
| | Scheme 25 Oxidative coupling reaction to form bisphenols. | |
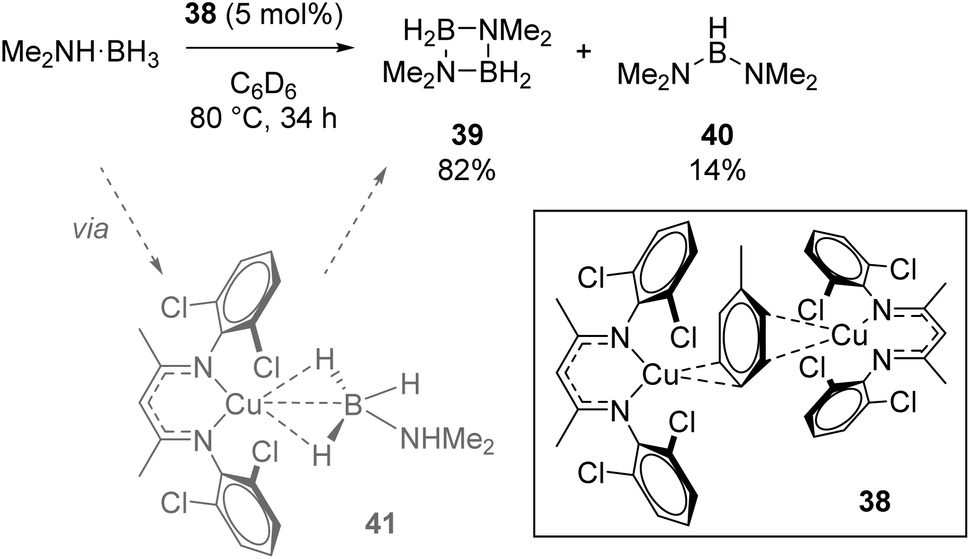
Crimmin used the Cu(I) dimer 38 for dehydrocoupling of dimethylamine borane (Scheme 26). The four-membered cycle, 39, formed as the major product, with small amounts of linear product, 40, also forming. In situ NMR monitoring provided evidence for the presence of σ-borane intermediate 41 forming during catalysis, while the authors observed reaction quenching when an Hg drop test was carried out (the conversion to 39 drops to 4% and 40 to 2.5%), which could imply a heterogeneous catalytic process.49
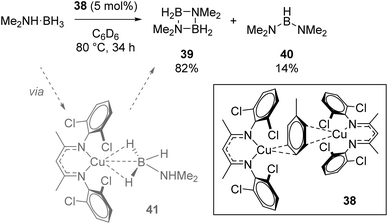 |
| | Scheme 26 Crimmin's dehydrocoupling, which is believed to be heterogeneous in nature. Conversions determined as a ratio by 11B NMR. | |
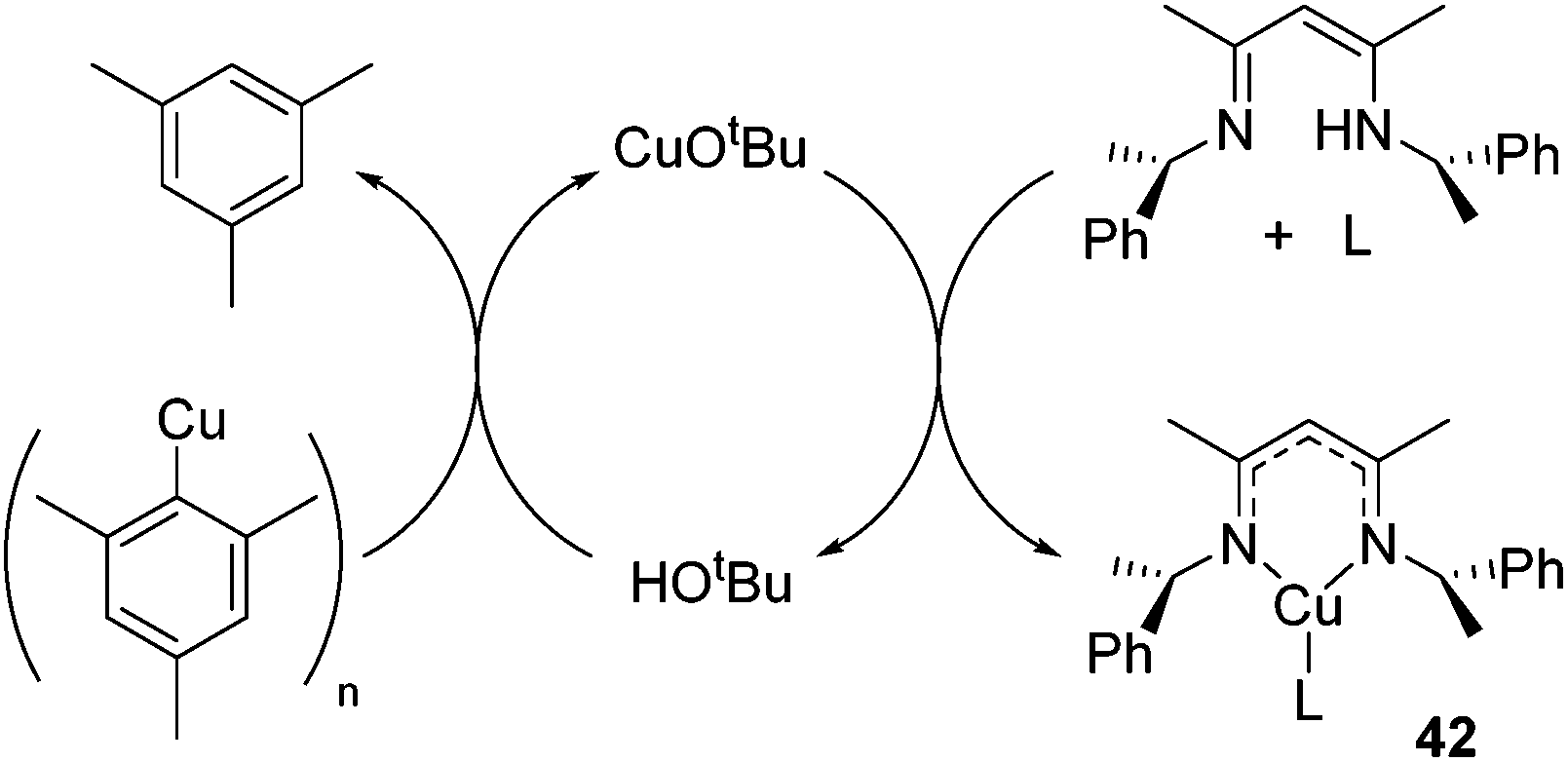
In an unusual example of catalysis whereby a catalytic method is used to form a copper β-diketiminate complex itself, Schaper showed that a catalytic amount of tBuOH or CuOtBu could be used to generate the otherwise challenging to synthesize Cu(I) alkyl β-diketiminate complexes of the general form 42 (Scheme 27).
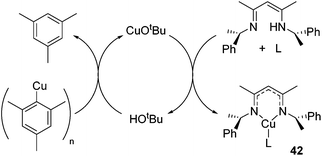 |
| | Scheme 27 Use of catalytic tert-butoxide source generates complexes of the form 42, where L includes PPh3, DMAP, MeCN, py. | |
Zinc
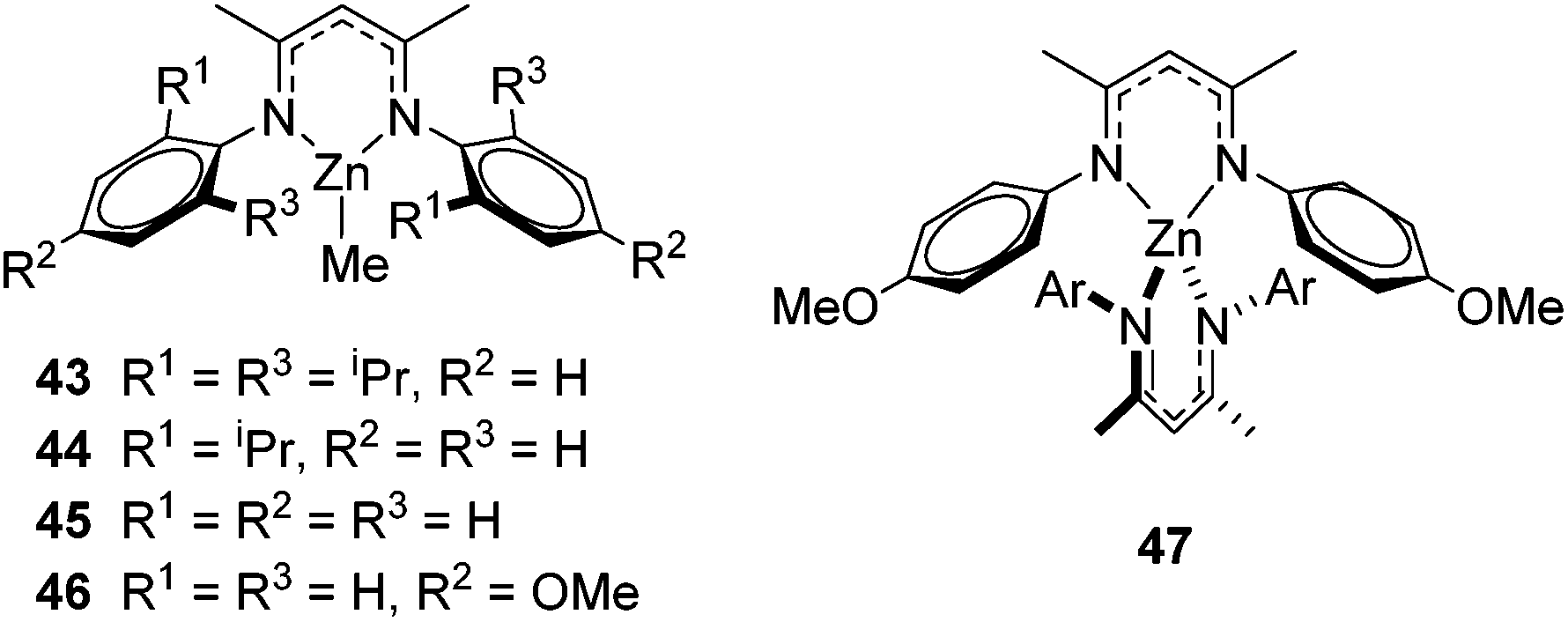
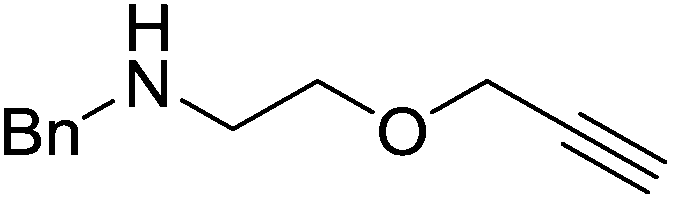
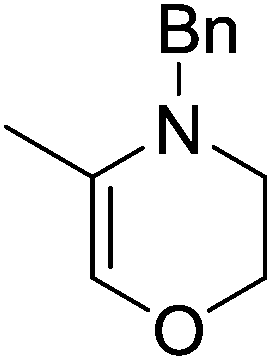
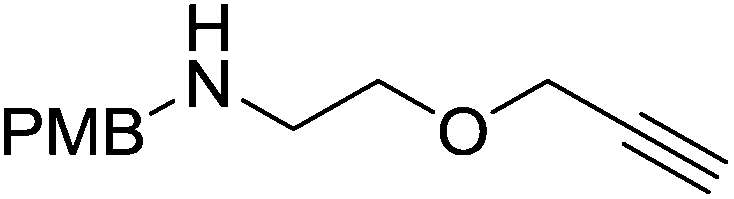
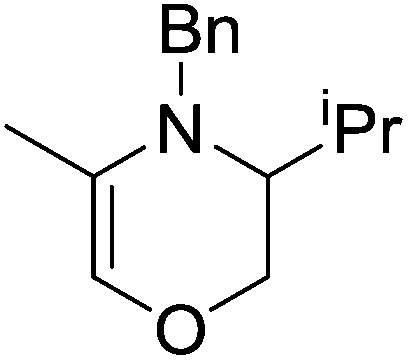
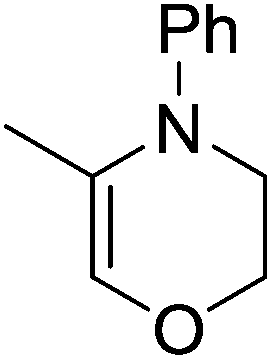
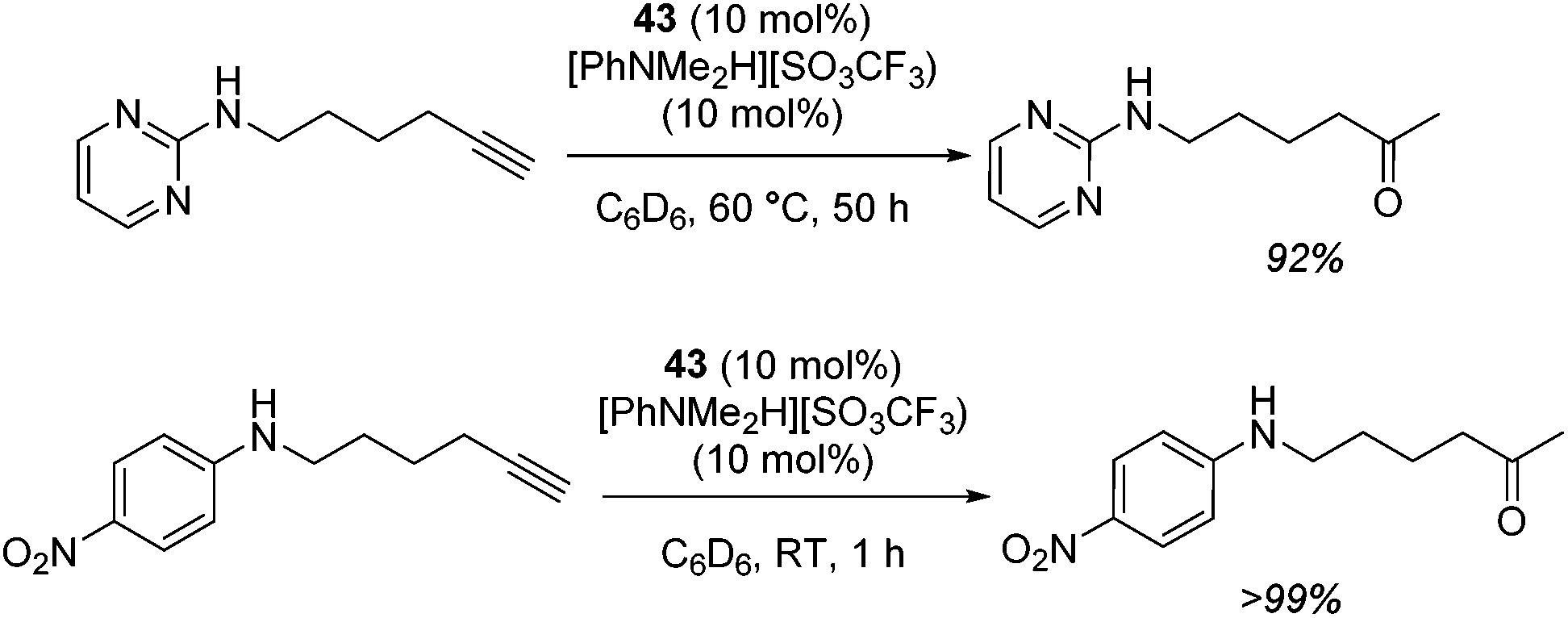
The zinc literature is dominated by polymerization chemistry, indeed, at the present time there appears to only be one example of a non-polymerization reaction catalyzed by a zinc β-diketiminate complex. Roesky showed that these complexes, which carry a methyl co-ligand with varying aryl substitution on the diketiminate fragment (43 to 46, Fig. 9), or even a bis-diketiminate complex (47), are all capable of undertaking the intramolecular hydroamination of aminoalkynes to form substituted morpholines.50
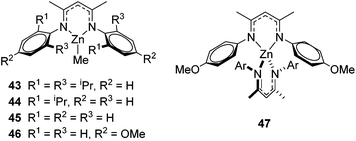 |
| | Fig. 9 Roesky's Zn complexes. | |
As seen with other examples of catalysis with β-diketiminate complexes, the 2,6-diisopropyl and, in this case, the 2-isopropyl, ligands outperform the other ligands tested (Table 5, compare reaction time and yields for 43 and 46 in entries 1–4).
Table 5 Roesky's intramolecular hydroamination of aminoalkynes catalyzed by Zn β-diketiminate complexes
| Entry |
Substrate |
Product |
Time (Catalyst) |
Conversion (%) |
| Conditions: 0.74 mmol aminoalkyne, 1 mol% pre-catalyst, 1 mol% [PhNMe2H][SO3CF3], C6D6, 60 °C. Conversion determined by 1H NMR. |
| 1 |
|
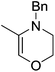
|
20 (43) |
93 |
| 20 (44) |
83 |
| 40 (45) |
83 |
| 90 (46) |
87 |
| 90 (47) |
84 |
| 2 |
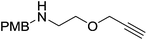
|
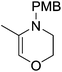
|
30 (43) |
96 |
| 23 (44) |
>99 |
| 40 (45) |
95 |
| 70 (46) |
95 |
| 40 (47) |
96 |
| 3 |
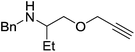
|
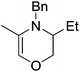
|
40 (43) |
93 |
| 90 (44) |
98 |
| 90 (45) |
97 |
| 120 (46) |
74 |
| 120 (47) |
73 |
| 4 |
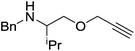
|
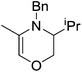
|
90 (43) |
93 |
| 90 (44) |
98 |
| 100 (45) |
84 |
| 100 (46) |
93 |
| 100 (47) |
91 |
| 5 |
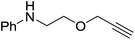
|
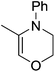
|
1 (43) |
>99 |
| 2 (44) |
>99 |
| 2 (47) |
>99 |
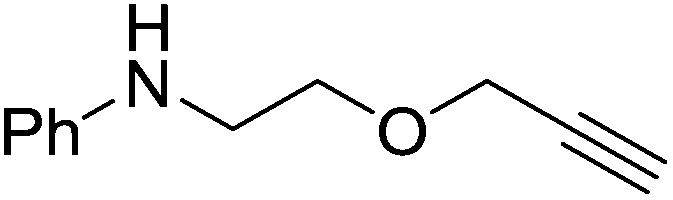
The reaction is not limited to intramolecular functionalization – it also operates well for intermolecular reactions, forming the imine which hydrolyzes on work-up (Scheme 28). It is also worth noting that the rate of the reactions increased when a co-catalytic amount of [PhNMe2H][SO3CF3] was added. It was postulated that the co-catalyst activated the Zn complex by protonating the methyl group, forming a β-diketiminate zinc triflate intermediate. This species could be prepared and characterized and showed comparable reactivity to the methyl pre-catalysts.
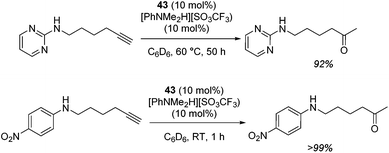 |
| | Scheme 28 Intramolecular hydroamination with isolated yield after hydrolysis. | |
Conclusions and outlook
It is clear that the β-diketiminate motif makes an excellent ligand for the 3d transition metals. Reactions range from hydrofunctionalization-type (where X–H is added across an alkene or an alkyne), hydrogenation, manipulation of alkenes and the formation of metal–element multiple bonds for the formation of nitrene and carbene intermediates prior to the preparation of C–element bonds. The diversity of the transformations achieved thus far is varied and exciting. However, there are severe limitations in the literature, with no examples beyond polymerization reported for scandium,51 no catalysis with manganese β-diketiminate complexes and, to the best of this author's knowledge, no examples of non-polymerization synthetic chemistry reported using nickel and only one or two examples of non-polymerization reactions for some of the other early 3d elements (Ti, V, Cr): it is clear that a huge range of novel and innovative catalytic reactions are yet be developed. With many of the reactions reported thus far proceeding via catalytic cycles that involve σ-bond metathesis, an entry point to developing reactivity with these as yet unexplored metal centers would simply be to employ a β-diketiminate and suitable co-ligand (e.g. an alkyl ligand) and test reactions of this sort. Beyond this, Schaper has demonstrated that inexpensive chiral β-diketiminate motifs can be readily accessed52 and are effective in lactide polymerization with tacticity control,53 but enantioselective small molecule transformations with chiral β-diketiminate complexes are yet to be achieved. It is also worth considering the range of transformations possible: although a good start has been made with many of the 3d complexes, with four of five different classes of transformation reported in the literature. If we take iron β-diketiminate catalysis as a case in point and compare to the number of catalytic transformations available with iron catalysis in general, a literature search will show transformations including cyclization, cycloaddition, carbometalation C–C and C–element cross-coupling nucleophilic substitution, carboxylation, carbonylation and ring-opening,54 to name but a few. This is an exciting thought: the untapped potential of the β-diketiminate motif as a ligand to support a huge range of catalytic transformations, not only with iron, is formidable. β-Diketiminate ligands offer many benefits when compared to other anionic ligands (e.g. Cp ligands) not least in terms of the vast range of steric and electronic variations that are possible. However, the air-sensitive nature of these low-coordinate and low oxidation state pre-catalysts, along with the need to develop two-electron chemistry, could be a barrier to their use in a wider range of synthetic organic transformations. However, with detailed mechanistic study and being able to pin down the subtle effects of the role of the ligand in catalysis (note that in the examples presented here, the non-innocent nature of the ligand is rarely discussed in the context of the catalytic cycle) it should be possible to reveal new avenues in catalysis. It is clear that when coupled with the first row transition metals we have only just started to scrape the surface.
References
-
(a) R. Bonnett, D. C. Bradley and K. J. Fisher, Chem. Commun., 1968, 886–887 RSC;
(b) R. Bonnett, D. C. Bradley, K. J. Fisher and I. F. Rendall, J. Chem. Soc. A, 1971, 1622–1627 RSC.
- J. E. Parks and R. H. Holm, Inorg. Chem., 1968, 7, 1408–1416 CrossRef CAS.
- L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031–3066 CrossRef CAS PubMed.
- Y.-C. Tsai, Coord. Chem. Rev., 2012, 256, 722–758 CrossRef CAS.
- S. P. Sarish, S. Nembenna, S. Nagendran and H. W. Roesky, Acc. Chem. Res., 2011, 44, 157–170 CrossRef CAS PubMed.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed.
-
(a)
P. W. Roesky, Molecular Catalysis of Rare-Earth Elements, Springer Berlin, Heidelberg, 2010 Search PubMed;
(b) F. T. Edelmann, Coord. Chem. Rev., 2015, 284, 124–205 CrossRef CAS;
(c) F. T. Edelmann, Coord. Chem. Rev., 2016, 318, 29–130 CrossRef CAS.
-
(a) W. E. Piers and D. J. H. Emslie, Coord. Chem. Rev., 2002, 233–234, 131–155 CrossRef CAS;
(b) A. A. Mohamed, Coord. Chem. Rev., 2010, 254, 1918–1947 CrossRef CAS;
(c) V. T. Annibale and D. Song, RSC Adv., 2013, 3, 11432–11449 RSC;
(d) D. Zhu and P. H. M. Budzelaar, Dalton Trans., 2013, 42, 11343–11354 RSC;
(e) S. J. Malthus, S. A. Cameron and S. Brooker, Coord. Chem. Rev., 2016, 316, 125–161 CrossRef CAS.
- C. Chen, S. M. Bellows and P. L. Holland, Dalton Trans., 2015, 44, 16654–16670 RSC.
- Comprehensive reviews include:
(a) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed;
(b) J. Wu, T.-L. Yu, C.-T. Chen and C.-C. Lin, Coord. Chem. Rev., 2006, 250, 602–626 CrossRef CAS;
(c) R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS.
- Comprehensive reviews include:
(a) G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed;
(b) M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC;
(c) S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS.
- Comprehensive reviews include:
(a) V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–316 CrossRef CAS PubMed;
(b) S. Arndt and J. Okuda, Adv. Synth. Catal., 2005, 347, 339–354 CrossRef CAS;
(c) P. M. Zeimentz, S. Arndt, B. R. Elvidge and J. Okuda, Chem. Rev., 2006, 106, 2404–2433 CrossRef CAS PubMed.
- Comprehensive reviews include:
(a) B. Y. Lee, H. Y. Kwon, S. Y. Lee, S. J. Na, S. I. Han, H. S. Yun, H. Lee and Y. W. Park, J. Am. Chem. Soc., 2005, 127, 3031–3037 CrossRef CAS PubMed;
(b) T. Bok, H. Yun and B. Y. Lee, Inorg. Chem., 2006, 45, 4228–4237 CrossRef CAS PubMed;
(c) D. J. Doyle, V. C. Gibson and A. J. P. White, Dalton Trans., 2007, 358–363 RSC;
(d) W. Yao, Y. Mu, A. Gao, W. Gao and L. Ye, Dalton Trans., 2008, 3199–3206 RSC;
(e) Y. H. Tsai, C. H. Lin, C. C. Lin and B. T. Ko, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4927–4936 CrossRef CAS;
(f) K. Hayashi, Y. Nakajima, F. Ozawa and T. Kawabata, Chem. Lett., 2010, 39, 643–645 CrossRef CAS;
(g) Y. C. Liu, C. H. Lin, B. T. Ko and R. M. Ho, J. Polym. Sci., Part A Polym. Chem., 2010, 48, 5339–5347 CrossRef CAS;
(h) C. H. Wang, C. Y. Li, C. H. Lin, Y. C. Liu and B. T. Ko, Inorg. Chem. Commun., 2011, 14, 1456–1460 CrossRef CAS;
(i) L. E. N. Allan, J. A. Belanger, L. M. Callaghan, D. J. A. Cameron, A. Decken and M. P. Shaver, J. Organomet. Chem., 2012, 706, 106–112 CrossRef;
(j) C. H. Wang, C. Y. Li, B. H. Huang, C. C. Lin and B. T. Ko, Dalton Trans., 2013, 42, 10875–10884 RSC;
(k) H. J. Chuang, B. H. Wu, C. Y. Li and B. T. Ko, Eur. J. Inorg. Chem., 2014, 2014, 1239–1248 CrossRef CAS;
(l) Z. Q. Hao, Y. X. Han, W. Gao, L. Xin and Y. Mu, Polyhedron, 2014, 83, 236–241 CrossRef CAS;
(m) H. V. Babu and K. Muralidharan, RSC Adv., 2014, 4, 6094–6102 RSC;
(n) C. T. Chen, M. C. Wang and T. L. Huang, Molecules, 2015, 20, 5313–5328 CrossRef CAS PubMed;
(o) X. J. Shang, W. H. Zhang and J. P. Lang, RSC Adv., 2016, 6, 11400–11406 RSC.
- Comprehensive reviews include:
(a) D. Rechavi and M. Lemaire, Chem. Rev., 2002, 102, 3467–3493 CrossRef CAS PubMed;
(b) G. Desimoni, G. Faita and K. A. Jorgensen, Chem. Rev., 2006, 106, 3561–3651 CrossRef CAS PubMed;
(c) G. C. Hargaden and P. J. Guiry, Chem. Rev., 2009, 109, 2505–2550 CrossRef CAS PubMed.
- J. R. Carney, B. R. Dillon and S. P. Thomas, Eur. J. Org. Chem., 2016, 3912–3929 CrossRef CAS.
-
(a) W. Hess, J. Treutwein and G. Hilt, Synthesis, 2008, 3537–3562 CAS;
(b) C. Gosmini and A. Moncomble, Isr. J. Chem., 2010, 50, 568–576 CrossRef CAS;
(c) P. Rose and G. Hilt, Synthesis, 2016, 463–492 Search PubMed.
-
(a) V. K. K. Praneeth, M. R. Ringenberg and T. R. Ward, Angew. Chem., Int. Ed., 2012, 51, 10228–10234 CrossRef CAS PubMed;
(b) S. Gondzik, D. Blaser, C. Wolper and S. Schulz, J. Organomet. Chem., 2015, 783, 92–95 CrossRef CAS;
(c) C. Camp and J. Arnold, Dalton Trans., 2016, 45, 14462–14498 RSC.
-
(a) K. M. Smith, Organometallics, 2005, 24, 778–784 CrossRef CAS;
(b) S. K. Ibrahim, A. V. Khvostov, M. F. Lappert, L. Maron, L. Perrin, C. J. Pickett and A. V. Protchenko, Dalton Trans., 2006, 2591–2596 RSC.
- F. Basuli, H. Aneetha, J. C. Huffman and D. J. Mindiola, J. Am. Chem. Soc., 2005, 127, 17992–17993 CrossRef CAS PubMed.
- G. Zhao, F. Basuli, U. J. Kilgore, H. Fan, H. Aneetha, J. C. Huffman, G. Wu and D. J. Mindiola, J. Am. Chem. Soc., 2006, 128, 13575–13585 CrossRef CAS PubMed.
- For a specialized review on titanium–imido complexes and their reactivity see: N. Hazari and P. Mountford, Acc. Chem. Res., 2005, 38, 839–849 CrossRef CAS PubMed.
- D. J. Mindiola, Acc. Chem. Res., 2006, 39, 813–821 CrossRef CAS PubMed.
- S. Hohloch, B. M. Kriegel, R. G. Bergman and J. Arnold, Dalton Trans., 2016, 45, 15725–15745 RSC.
- K.-C. Chang, C.-F. Lu, P.-Y. Wang, D.-Y. Lu, H.-Z. Chen, T.-S. Kuo and Y.-C. Tsai, Dalton Trans., 2011, 40, 2324–2331 RSC.
- K. C. MacLeod, B. O. Patrick and K. M. Smith, Organometallics, 2010, 29, 6639–6641 CrossRef CAS.
- K. C. MacLeod, J. L. Conway, B. O. Patrick and K. M. Smith, J. Am. Chem. Soc., 2010, 132, 17325–17334 CrossRef CAS PubMed.
- W. Zhou, K. C. MacLeod, B. O. Patrick and K. M. Smith, Organometallics, 2012, 31, 7324–7327 CrossRef CAS.
- V. C. Gibson, E. L. Marshall, D. Navarro-Llobet, A. J. P. White and D. J. Williams, J. Chem. Soc., Dalton Trans., 2002, 4321–4322 RSC.
- M. S. Zhou, S. P. Huang, L. H. Weng, W. H. Sun and D. S. Liu, J. Organomet. Chem., 2003, 665, 237–245 CrossRef CAS.
- J. Vela, J. M. Smith, Y. Yu, N. A. Ketterer, C. J. Flaschenriem, R. J. Lachicotte and P. L. Holland, J. Am. Chem. Soc., 2005, 127, 7857–7870 CrossRef CAS PubMed.
- K. Ding, F. Zannat, J. C. Morris, W. W. Brennessel and P. L. Holland, J. Organomet. Chem., 2009, 694, 4204–4208 CrossRef CAS.
- R. E. Cowley, M. R. Golder, N. A. Eckert, M. H. Al-Afyouni and P. L. Holland, Organometallics, 2013, 32, 5289–5298 CrossRef CAS.
- E. Bernoud, P. Oulié, R. Guillot, M. Mellah and J. Hannedouche, Angew. Chem., Int. Ed., 2014, 53, 4930–4934 CrossRef CAS PubMed.
- A. K. King, A. Buchard, M. F. Mahon and R. L. Webster, Chem. – Eur. J., 2015, 21, 15960–15963 CrossRef CAS PubMed.
- M. Espinal-Viguri, A. K. King, J. P. Lowe, M. F. Mahon and R. L. Webster, ACS Catal., 2016, 6, 7892–7897 CrossRef CAS.
-
N. T. Coles, M. F. Mahon and R. L. Webster, Manuscript submitted, 2017.
- M. Espinal-Viguri, C. R. Woof and R. L. Webster, Chem. – Eur. J., 2016, 22, 11605–11608 CrossRef CAS PubMed.
- K. Ding, T. R. Dugan, W. W. Brennessel, E. Bill and P. L. Holland, Organometallics, 2009, 28, 6650–6656 CrossRef CAS.
- C. Chen, T. R. Dugan, W. W. Brennessel, D. J. Weix and P. L. Holland, J. Am. Chem. Soc., 2014, 136, 945–955 CrossRef CAS PubMed.
- N. C. Thacker, Z. Lin, T. Zhang, J. C. Gilhula, C. W. Abney and W. Lin, J. Am. Chem. Soc., 2016, 138, 3501–3509 CrossRef CAS PubMed.
- S. Monfette, Z. R. Turner, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2012, 134, 4561–4564 CrossRef CAS PubMed.
- A. W. Pierpont and T. R. Cundari, Inorg. Chem., 2010, 49, 2038–2046 CrossRef CAS PubMed.
- B. F. Gherman, W. B. Tolman and C. J. Cramer, J. Comput. Chem., 2006, 27, 1950–1961 CrossRef CAS PubMed.
- C. L. McMullin, A. W. Pierpont and T. R. Cundari, Polyhedron, 2013, 52, 945–956 CrossRef CAS.
- X. L. Dai and T. H. Warren, J. Am. Chem. Soc., 2004, 126, 10085–10094 CrossRef CAS PubMed.
- R. T. Gephart, III, C. L. McMullin, N. G. Sapiezynski, E. S. Jang, M. J. B. Aguila, T. R. Cundari and T. H. Warren, J. Am. Chem. Soc., 2012, 134, 17350–17353 CrossRef PubMed.
- Y. M. Badiei, A. Dinescu, X. Dai, R. M. Palomino, F. W. Heinemann, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2008, 47, 9961–9964 CrossRef CAS PubMed.
- P. Haack, C. Limberg, K. Ray, B. Braun, U. Kuhlmann, P. Hildebrandt and C. Herwig, Inorg. Chem., 2011, 50, 2133–2142 CrossRef CAS PubMed.
- A. E. Nako, A. J. P. White and M. R. Crimmin, Dalton Trans., 2015, 44, 12530–12534 RSC.
- M. Biyikal, K. Loehnwitz, N. Meyer, M. Dochnahl, P. W. Roesky and S. Blechert, Eur. J. Inorg. Chem., 2010, 1070–1081 CrossRef CAS.
- L. Li, C. Wu, D. Liu, S. Li and D. Cui, Organometallics, 2013, 32, 3203–3209 CrossRef CAS.
- P. O. Oguadinma and F. Schaper, Organometallics, 2009, 28, 4089–4097 CrossRef CAS.
- F. Drouin, P. O. Oguadinma, T. J. J. Whitehorne, R. E. Prud'homme and F. Schaper, Organometallics, 2010, 29, 2139–2147 CrossRef CAS.
- For a recent comprehensive review see: I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a