DOI:
10.1039/C7DT00292K
(Paper)
Dalton Trans., 2017,
46, 6853-6869
Design, structural diversity and properties of novel zwitterionic metal–organic frameworks†
Received
24th January 2017
, Accepted 3rd March 2017
First published on 9th March 2017
Abstract
Seven new zwitterionic metal–organic frameworks (ZW MOFs) of compositions {[Cd(L1)(OH2)]·2H2O}n (1), {[Mn(L1)(OH2)2]·H2O}n (2), {[Cu(HL1)2(OH2)3]·9H2O}n (3), {[Mn2(L2)2(OH2)4]·3H2O}n (4), [Co(L2)(OH2)4]·H2O (5), [Ni(L2)(OH2)3]n (6), and {[Cd(L2)(OH2)3]·4H2O}n (7), where H3L1Br = 3-carboxy-1-(3,5-dicarboxybenzyl)pyridinium bromide and H3L2Br = 4-carboxy-1-(3,5-dicarboxybenzyl)pyridinium bromide, have been synthesized under hydrothermal conditions. We demonstrate that the diversity of these crystal structures suggests that the tridentate and flexible nature of ZW ligands L1 and L2 make them excellent candidates for the synthesis of new ZW MOFs. A multi-charged anionic nature is a common feature of L1 and L2, and therefore, allows the rational design of ZW MOFs without the presence of additional counterions for charge compensation. All materials were structurally characterized by single-crystal X-ray diffraction and further characterized by elemental analyses, infrared spectroscopy (IR), powder X-ray diffraction (PXRD), thermogravimetric analyses (TGA), differential scanning calorimetry (DSC) and adsorption measurements. Most interestingly, permanent porosity could be observed for 1, originated from 4 Å channel pores and confirmed by methanol adsorption experiments, which yielded an uptake of 7.43 wt% at 25 °C; and respectively, anhydrates of 1, 2, 4 and 6 can be rehydrated upon exposure to ambient air, as evidenced by TGA and PXRD measurements. In addition, we report an in-depth CSD analysis of selected structural parameters, coordination modes and topologies exhibited by MOFs based on ZW ligands L1 and L2 along with the regio-isomeric analogue L3, where H3L3Br = N-(4-carboxybenzyl)-(3,5-dicarboxyl)pyridinium bromide.
Introduction
Over the past few decades, synthesis, crystal structures and properties of metal–organic frameworks (MOFs) have been widely researched. These porous materials, composed of metal clusters and polytopic organic linkers, can be tailored to specific needs because of their structural and functional tunability, a feature that is not available with conventional porous materials (e.g., zeolites, activated carbons).1–3 This “tunability” allows for various applications, such as gas storage, chemical separation, sensing and catalysis,4–18 with a focus on CO2 capture in our laboratory.19–25 We previously reported a new design strategy for accessing MOFs built from zwitterionic (ZW) ligands with adsorption properties based on multi-point host guest interactions.26,27 In a continuation of this work, we synthesized the new pyridinium based multicharged anionic ZW ligand, H3L1Br = 3-carboxy-1-(3,5-dicarboxybenzyl) pyridinium bromide (Fig. 1). The positively charged pyridinium center is known to potentially enhance interactions with gas molecules,12 and thus, frameworks based on pyridinium carboxylate ligands have been deployed for various applications such as gas adsorption, sensing etc.28–31 In addition, ZW ligand L1 permits the rational design of ZW MOFs without the presence of additional anions for charge compensation. L1 contains three carboxylate groups at the terminal positions that can undergo deprotonation, completely or partially, and therefore, can coordinate to the metal ions in various coordination modes yielding a variety of interesting frameworks. The –CH2– motif linking the pyridine and phenyl rings can freely rotate in the assembly process to meet requirements of various coordination geometries. To study the structural influence of ligand L1 on the resulting frameworks, a similar ligand, H3L2Br = 4-carboxy-1-(3,5-dicarboxybenzyl) pyridinium bromide, was used to synthesize MOFs under matching reaction conditions (Fig. 1). To the best of our knowledge only two studies have reported MOFs designed through this pyridinium based ZW ligand L2.32,33 Mak et al. used L2 to synthesize three families of lanthanide coordination polymers luminescence behavior which could be tuned by doping the concentration of lanthanide ions.32 Li et al. studied the photoluminescent properties of seven complexes with diverse structural features synthesized using L2.33 In addition, a variety of framework structures have been reported with the regio-isomeric ligand H3L3Br (N-(4-carboxybenzyl)-(3,5-dicarboxyl)pyridinium bromide), differing only in the position of the nitrogen atom (Fig. 1).34,35
 |
| | Fig. 1 Lewis structures of ligand L1 and regio-isomeric ligands L2 and L3. | |
A fundamental study probing their structure–property relationships has not been reported, and is imperative for the optimization of these materials for various applications. In this contribution, the systematic investigations on the synthesis and structural characterization of seven new ZW MOF materials based on zwitterionic ligands L1 and L2 that have been synthesized under hydrothermal conditions, namely, {[Cd(L1)(OH2)]·2H2O}n (1), {[Mn(L1)(OH2)2]·H2O}n (2), {[Cu(HL1)2(OH2)3]·9H2O}n (3), [Mn2(L2)2(OH2)4]·3H2O (4), [Co(L2)(OH2)4]·H2O (5), [Ni(L2)(OH2)3]n (6), and {[Cd(L2)(OH2)3]·4H2O}n (7) will be reported. All compounds were characterized by single-crystal X-ray diffraction, infrared spectra (IR), elemental analyses, powder X-ray diffraction (PXRD), thermogravimetric analyses (TGA), differential scanning calorimetry (DSC) and adsorption measurements.
Experimental
General information
Commercially available reagents were used as received without further purification. Ligands L1 3-carboxy-1-(3,5-dicarboxybenzyl)pyridinium bromide (H3L1Br) and L2 4-carboxy-1-(3,5-dicarboxybenzyl)pyridinium bromide (H3L2Br) were synthesized as described below.
Synthesis of 3-carboxy-1-(3,5-dicarboxybenzyl)pyridinium bromide (H3L1Br)
Pyridine-3-carboxylic acid (2.87 g, 10 mmol) and dimethyl 5-(bromomethyl)isopthalate (2.73 g, 10 mmol) were dissolved in 20 mL of acetonitrile and refluxed for 24 hours at 100 °C. After the mixture was cooled to room temperature, the resulting white precipitate was filtered-off, washed with acetonitrile and dried in vacuum to give Me2HL1Br (3.06 g, yield 93%). 1H-NMR (400 MHz, D2O): δ = 9.28 (s, 1H); 8.92 (d, 1H); 8.85 (d, 1H); 8.39 (s, 1H); 8.18 (s, 2H); 8.06 (t, 1H); 5.95 (s, 2H); 3.80 (s, 6H) (Fig. S1,† top).
Deprotection was performed by dissolving Me2HL1Br (2.0 g) in acetic acid (50 mL) and aqueous HBr (44%, 50 mL). The solution was refluxed for 12 hours and slowly poured into 500 mL ice water. The resulting precipitate was collected by filtration and washed with distilled water (3 × 30 mL). The filter cake was dried in vacuum and recrystallized in CH3CN to give the product (1.64 g, yield 82%), main IR bands (KBr pellet, cm−1): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3435 (w), 3079 (s), 1701 (m), 1636 (s), 1560 (m), 1389 (s), 753 (m), 573 (s) (Fig. S2†). 1H-NMR (400 MHz, d6-DMSO): δ = 9.82 (s, 1H); 9.49 (d, 1H); 8.97 (d, 1H); 8.50 (s, 3H); 8.23 (t, 1H); 6.11 (s, 2H) (Fig. S1,† bottom).
= 3435 (w), 3079 (s), 1701 (m), 1636 (s), 1560 (m), 1389 (s), 753 (m), 573 (s) (Fig. S2†). 1H-NMR (400 MHz, d6-DMSO): δ = 9.82 (s, 1H); 9.49 (d, 1H); 8.97 (d, 1H); 8.50 (s, 3H); 8.23 (t, 1H); 6.11 (s, 2H) (Fig. S1,† bottom).
Synthesis of 4-carboxy-1-(3,5-dicarboxybenzyl)pyridinium bromide (H3L2Br)
This ligand was synthesized in a similar procedure as H3L1Br above, but substituting pyridine-4-carboxylic acid for pyridine-3-carboxylic acid. Me2HL2Br (2.98 g, yield 91%). 1H-NMR (400 MHz, D2O): δ = 8.95 (d, 2H); 8.54 (s, 1H); 8.27 (d, 2H); 8.12 (s, 2H); 5.86 (s, 2H); 3.83 (s, 6H) (Fig. S3,† top). H3L2Br (1.79 g, yield 75%), main IR bands (KBr pellet, cm−1): = 3353 (w), 3066 (s), 1718 (s), 1641 (s), 1573 (m), 1391 (s), 751 (s), 585 (m) (Fig. S4†).1H-NMR (400 MHz, d6-DMSO): δ = 9.18 (d, 2H); 8.48 (s, 1H); 8.31 (s, 2H); 8.22 (d, 2H); 5.91 (s, 2H) (Fig. S3,† bottom).
Synthesis of {[Cd(L1)(OH2)]·2H2O}n (1)
Polycrystalline bulk material was synthesised by dissolving a mixture of Cd(CH3COO)2 (682 mg, 2.56 mmol) and H3L1Br (78 mg, 0.27 mmol) in 5 mL of water. 400 μL of concentrated aqueous HNO3 was added to the solution followed by heating to 95 °C with continuous stirring for 12 hours. The white precipitate was filtered off and washed with water and diethyl ether and dried in air. The purity was checked by X-ray powder diffraction (Fig. S5†). Yield 62% based on L1. Calculated for C30H30Cd2N2O18 (931.38): C 38.69, H 3.25, N 3.01; found: C 38.43, H 3.16, N 2.98. Main IR bands (KBr pellet, cm−1): = 3431 (w), 3056 (m), 1644 (s), 1560 (w), 1381 (m), 731 (s), 550 (w) (Fig. S2†).
Single crystals suitable for X-ray structure determination were prepared similar as described above without stirring in a closed snap vial at 85 °C for 3 days. White block-shaped crystals were obtained after cooling as a major phase.
Synthesis of {[Mn(L1)(OH2)2]·H2O}n (2)
Polycrystalline bulk material and clear white block-shaped single crystals were prepared similar as described for 1, but using Mn(CH3COO)2·2H2O (442 mg, 2.56 mmol) instead of Cd(CH3COO)2. The purity was checked by X-ray powder diffraction (Fig. S6†). Yield 74% based on L1. Calculated for C15H15MnNO9 (408.22): C 44.13, H 3.70, N 3.43; found: C 44.13, H 3.96, N 3.32. Main IR bands (KBr pellet, cm−1): = 3450 (w), 3072 (m), 1651 (s), 1549 (w), 1390 (s), 722 (m), 520 (m) (Fig. S2†).
Synthesis of {[Cu(HL1)2(OH2)3]·9H2O}n (3)
Blue needle-shaped single crystals were prepared similar as described for 1, but using Cu(CH3COO)2·2H2O (464 mg, 2.56 mmol) instead of Cd(CH3COO)2. The synthesis of phase pure bulk material was not successful owing to the presence of different phases in the resulting heterogeneous mixture which could not be separated.
Synthesis of {[Mn2(L2)2(OH2)4]·3H2O}n (4)
Polycrystalline bulk material was synthesised by dissolving a mixture of Mn(CH3COO)2·2H2O (442 mg, 2.56 mmol) and H3L2Br (78 mg, 0.27 mmol) in 5 mL of water. 800 μL of concentrated aqueous HCl was added to the solution followed by heating to 85 °C with continuous stirring for 12 hours. The ivory colored precipitate was filtered off, washed with water and diethyl ether and dried in air. The purity was checked by X-ray powder diffraction (Fig. S7†). Yield 61% based on L2. Calculated for C15H15MnNO9 (408.22): C 44.13, H 3.70, N 3.43; found: C 41.36, H 3.72, N 3.06. Main IR bands (KBr pellet, cm−1): = 3302 (w), 3073 (m), 1639 (m), 1548 (m), 1366 (m), 725 (s), 520 (s) (Fig. S4†).
Single crystals suitable for X-ray structure determination were prepared similar to process described above without stirring, and in a closed snap vial at 85 °C for 3 days. White clear plate-shaped crystals were obtained upon cooling as a major phase.
Synthesis of [Co(L2)(OH2)4]·H2O (5)
Plate-shaped pink single crystals were prepared similar as described for 4, but using Co(CH3COO)2·4H2O (124.5 mg, 0.5 mmol) instead of Mn(CH3COO)2·2H2O. The synthesis of phase pure bulk material was not successful owing to the presence of different phases in the resulting heterogeneous mixture which could not be separated.
Synthesis of [Ni(L2)(OH2)3]n (6)
Polycrystalline bulk material and green block-shaped single crystals were prepared similar as described for 4 but using Ni(CH3COO)2 (88.4 mg, 0.5 mmol) instead of Mn(CH3COO)2·2H2O. The purity of the bulk material was checked by X-ray powder diffraction (Fig. S8†). Yield 42% based on L2. Calculated for C15H15NiNO9 (411.97): C 43.73, H 3.67, N 3.4; found: C 41.39, H 3.17, N 3.08. Main IR bands (KBr pellet, cm−1): = 3374 (w), 2997 (w), 1619 (s), 1541 (s), 1375 (s), 715 (s), 496 (s) (Fig. S4†).
Synthesis of {[Cd(L2)(OH2)3]·4H2O}n (7)
White clear block-shaped single crystals were prepared similar as described for 4, but using Cd(CH3COO)2·2H2O (133.3 mg, 0.5 mmol) instead of Mn(CH3COO)2·2H2O. The synthesis of phase pure bulk material was not successful owing to the presence of different phases in the resulting heterogeneous mixture which could not be separated.
Single-crystal structure analysis
Data collections were performed on single crystals coated with Paratone-N oil and mounted on Kapton loops. Single crystal X-ray data of all compounds were collected on a Bruker Kappa Apex II X-ray diffractometer outfitted with a Mo X-ray source (sealed tube, λ = 0.71073 Å) and an APEX II CCD detector equipped with an Oxford Cryosystems Desktop Cooler low temperature device. The APEX-II software suite was used for data collection, cell refinement, and reduction.36 Absorption corrections were applied using SADABS.37 Space group assignments were determined by examination of systematic absences, E-statistics, and successive refinement of the structures. Structure solutions were performed using intrinsic phasing methods implemented with ShelXT and structure refinements were performed by least-squares refinements against |F|2 followed by difference Fourier synthesis using ShelXL, both softwares are part of the ShelX program package.38–40 All non-hydrogen atoms were refined with anisotropic displacement parameters. If not stated differently, the O–H-hydrogen atoms were located in the difference map, where the bond lengths were set to ideal values with dO–H = 0.84 Å and were refined using a riding model. The C–H atoms were positioned with idealized geometry and were refined with fixed isotropic displacement parameters [Ueq(H) = −1.2Ueq(C)] using a riding model with dC–H = 0.95 Å (aromatic) and 0.99 Å (methylene). Details of the structure determination are given in Table 1. In 3, the coordinating water molecule is disordered over two positions (O3W/O3W′) with SOFs of 0.5. Part of the non-coordinating structural fragments in 3 could not be refined and thus, the data were corrected using the SQUEEZE option in PLATON.41 SQUEEZE estimated a total count of 57.6 electrons per 206.5 Å3 of solvent accessible volume in 3, which is in good agreement to be 5.5 water molecules (Z = 1; H2O = 10e−; 57.6e−/1 = 57.6e− ≈ 5.5H2O).
Table 1 Selected crystal data and details on the structure determinations from single crystal data for ZW MOFs 1–7
| Compound |
1
|
2
|
3
|
4
|
5
|
6
|
7
|
| Formula |
C30H30Cd2N2O18 |
C15H15MnNO9 |
C60H66Cu2N4O37 |
C15H16MnNO9.5 |
C15H19CoNO11 |
C15H15NiNO9 |
C15H21CdNO12 |
| MW [g mol−1] |
931.36 |
408.22 |
1562.24 |
417.23 |
448.24 |
411.99 |
519.73 |
| Crystal system |
Triclinic |
Triclinic |
Triclinic |
Triclinic |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P |
P21/c |
P21/n |
P |
|
a [Å] |
7.4650(3) |
7.7335(3) |
11.7780(7) |
8.6474(4) |
11.2047(8) |
7.4619(8) |
9.0302(5) |
|
b [Å] |
10.1512(4) |
10.1906(4) |
12.1842(8) |
10.0521(5) |
14.5919(9) |
22.051(2) |
10.2284(5) |
|
c [Å] |
11.7742(5) |
11.7904(4) |
13.5056(9) |
10.3075(5) |
11.9668(7) |
10.1124(12) |
11.2165(6) |
|
α [°] |
104.3926(10) |
66.3080(11) |
95.876(2) |
75.8767(14) |
90 |
90 |
74.5063(14) |
|
β [°] |
108.4324(10) |
77.4597(11) |
98.626(2) |
66.3576(13) |
116.9244(18) |
102.376(4) |
71.6148(14) |
|
γ [°] |
101.8028(10) |
77.4597(11) |
112.384(2) |
83.4926(15) |
90 |
90 |
73.4060(15) |
|
V [Å3] |
780.00(6) |
803.26(5) |
1744.6(2) |
795.84(7) |
1744.5(2) |
1625.3(3) |
924.15(9) |
|
T [K] |
170.15 |
170(2) |
170.15 |
170(2) |
170.15 |
170.15 |
170.15 |
|
Z
|
1 |
2 |
1 |
2 |
4 |
4 |
2 |
|
D
calc. [g cm−3] |
1.983 |
1.688 |
1.487 |
1.741 |
1.707 |
1.684 |
1.868 |
|
μ [mm−1] |
1.455 |
0.875 |
0.709 |
0.887 |
1.048 |
1.246 |
1.250 |
| Min/max transmission |
0.816/0.909 |
0.889/0.926 |
0.907/0.959 |
0.909/0.935 |
0.863/0.926 |
0.767/0.869 |
0.796/0.922 |
|
θ
max [°] |
28.375 |
28.339 |
28.380 |
28.354 |
28.288 |
28.357 |
28.302 |
| Measured reflections |
23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 527 527 |
23326 |
68686 |
24545 |
22831 |
20684 |
36479 |
| Unique reflections |
3873 |
4011 |
8703 |
3977 |
4328 |
4044 |
4586 |
| Reflections [F0 > 4σ(F0)] |
3838 |
3072 |
7533 |
3622 |
3857 |
3281 |
4424 |
| Parameter |
241 |
241 |
500 |
259 |
254 |
236 |
273 |
|
R
int
|
0.0356 |
0.0379 |
0.0430 |
0.0367 |
0.0370 |
0.0646 |
0.0360 |
|
R
1 [F0 > 4σ(F0)] |
0.0202 |
0.0248 |
0.0418 |
0.0279 |
0.0267 |
0.0393 |
0.0177 |
| wR2 [all data] |
0.0533 |
0.0652 |
0.1213 |
0.0734 |
0.0709 |
0.0896 |
0.0454 |
| GOF |
1.109 |
1.066 |
1.054 |
1.026 |
1.032 |
1.046 |
1.069 |
| Δρmax/Δρmin [e Å−3] |
1.077/−0.999 |
0.432/−0.289 |
0.990/−0.757 |
0.670/−0.594 |
0.594/−0.348 |
0.570/−0.462 |
0.758/−0.828 |
CCDC 1428987–1428992 (1–6) and 1428994 (7) contain the supplementary crystallographic data for this paper.
Powder X-ray diffraction (PXRD)
PXRD data was collected on a Bruker D2 Phaser diffractometer equipped with a Cu sealed tube (λ = 1.54178 Å). Powder samples were dispersed on low-background discs for analyses. PXRD data of the anhydrates 1′, 2′, 4′ and 6′ was collected using an airtight sample holder. Simulation of the PXRD data was performed by using single crystal data and the powder pattern module of the Mercury CSD software package.42
Thermogravimetric analysis (TGA)
TG data was collected with a TGA Q50 from TA instruments. All measurements were performed using platinum crucibles in a dynamic nitrogen atmosphere (50 mL min−1) and a heating rate of 3 °C min−1. The instrument was corrected for buoyancy and current effects and was calibrated using standard reference materials.
Differential scanning calorimetry (DSC)
DSC data was collected using a TGA Q20 from TA instruments. All measurements were performed using Tzero aluminum pans, a dynamic nitrogen atmosphere (50 mL min−1) and a heating rate of 3 °C min−1. The instrument was calibrated using standard reference materials.
Adsorption analysis
Methanol adsorption measurements were performed using a modified TG analyzer setup. A sample of activated 1′ was obtained by heating the as-synthesized 1 under nitrogen atmosphere to 120 °C@3 °C min−1. The weight loss was monitored to confirm the loss of solvent molecules (Δmstep1 = 7.73%, uncoordinated H2O; and Δmstep2 = 3.86%, coordinated H2O). Followed by cooling to 30 °C, 1′ was subjected to methanol vapors until the mass gain stabilized. To confirm reversibility, the sample was heated back to 120 °C.
Elemental analysis
Elemental data (C, H, and N) was collected on PerkinElmer Series II CHNS/O analyzer.
Spectroscopy
FT-IR data were recorded on a Nicolet iS10 from Thermo Scientific. 1H-NMR data were recorded on Avance DMX-400 from Bruker.
Results and discussion
Ligand design strategy
Continuing with our earlier efforts exploring the use of anionic ZW ligands for the synthesis of new MOF materials with absent additional non-ZW anions, we designed the new ZW ligand H3L1Br which upon deprotonation yields the anionic ZW ligand L1-bearing a total of two negative charges, sufficient to balance two positive charges of metal centers during MOF synthesis. A two-step reaction starting with pyridine-3-carboxylic acid leads to the formation of H3L1Br as depicted in Fig. 2. It should be noted that similar reactions with pyridine-4-carboxylic acid leads to the formation of H3L2Br.
 |
| | Fig. 2 Two-step reaction scheme for synthesis of H3L1Br (H3L2Br). Ligand in its deprotonated anionic form L1−2(L2−2) is also displayed with charges highlighted by dashed ellipsoids. | |
Three new ZW MOFs of composition {[Cd(L1)(OH2)]·2H2O}n (1), {[Mn(L1)(OH2)2]·H2O}n (2), {[Cu(L1)2(OH2)3]·9H2O}n (3) were obtained with H3L1Br and four with H3L2Br, {[Mn2(L2)2(OH2)4]·3H2O}n (4), [Co(L2)(OH2)4]·H2O (5), [Ni(L2)(OH2)3]n (6), and {[Cd(L2)(OH2)3]·4H2O}n (7). Different coordination modes exhibited by ligands L1 and L2 can be observed in 1–7 as shown in Fig. 3. Please note that generally, the carboxylate groups of the 3,5-dicarboxybenzyl units can be identified as the major metal coordination sites. Interestingly, the carboxylate group of the 4(3)-carboxy-pyridinium units is only coordinating in 1.
 |
| | Fig. 3 Different coordination modes exhibited by ligands L1 and L2 as observed in MOFs 1–7. Color scheme: Cd(II), orange; Mn(II), majenta; Cu(II), turquoise; Co(II), brown; Ni(II), pink; carbon, grey; nitrogen, blue; oxygen, red and hydrogen, white. | |
Crystal structure of {[Cd(L1)(OH2)]·2H2O}n (1)
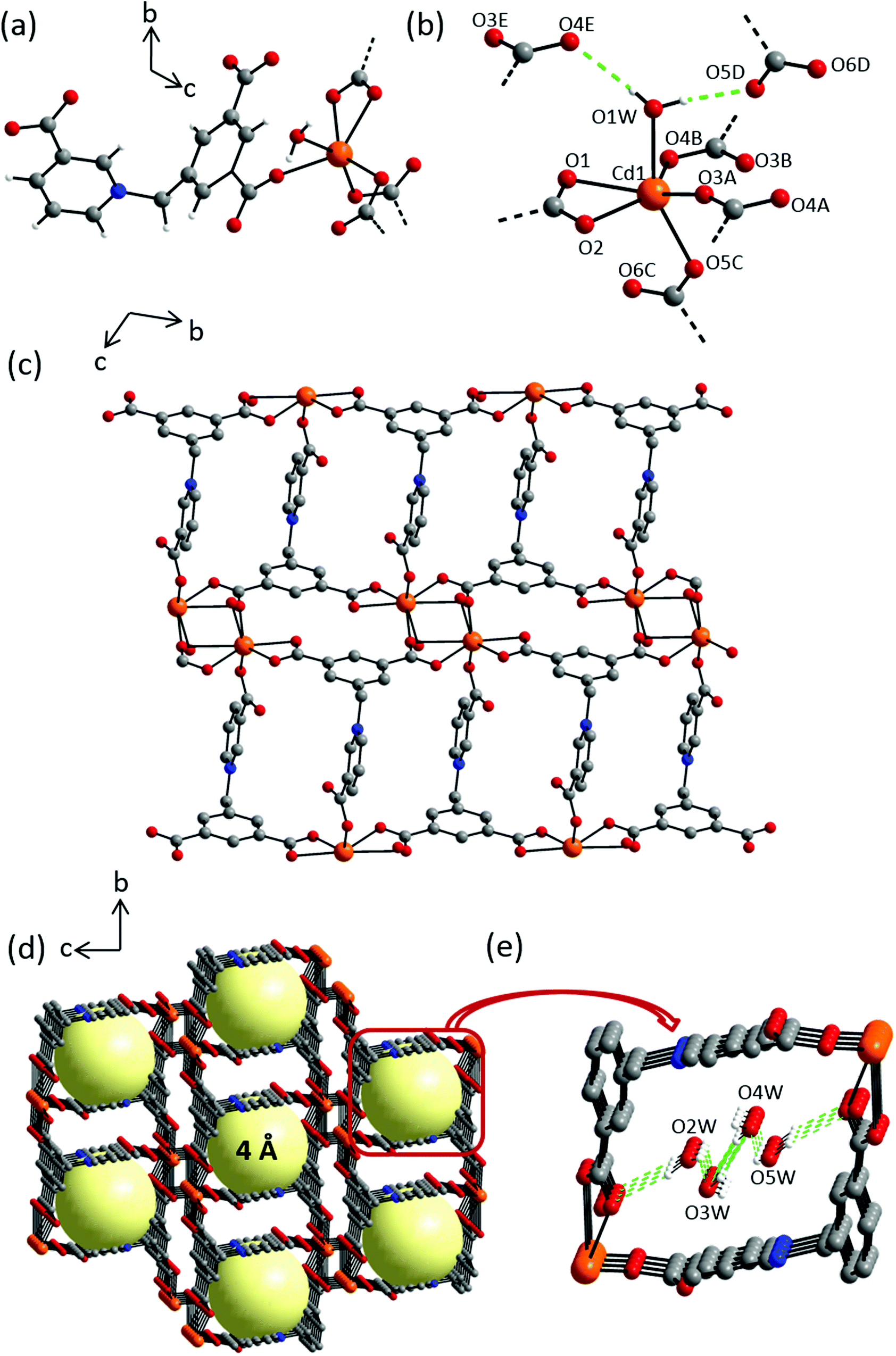
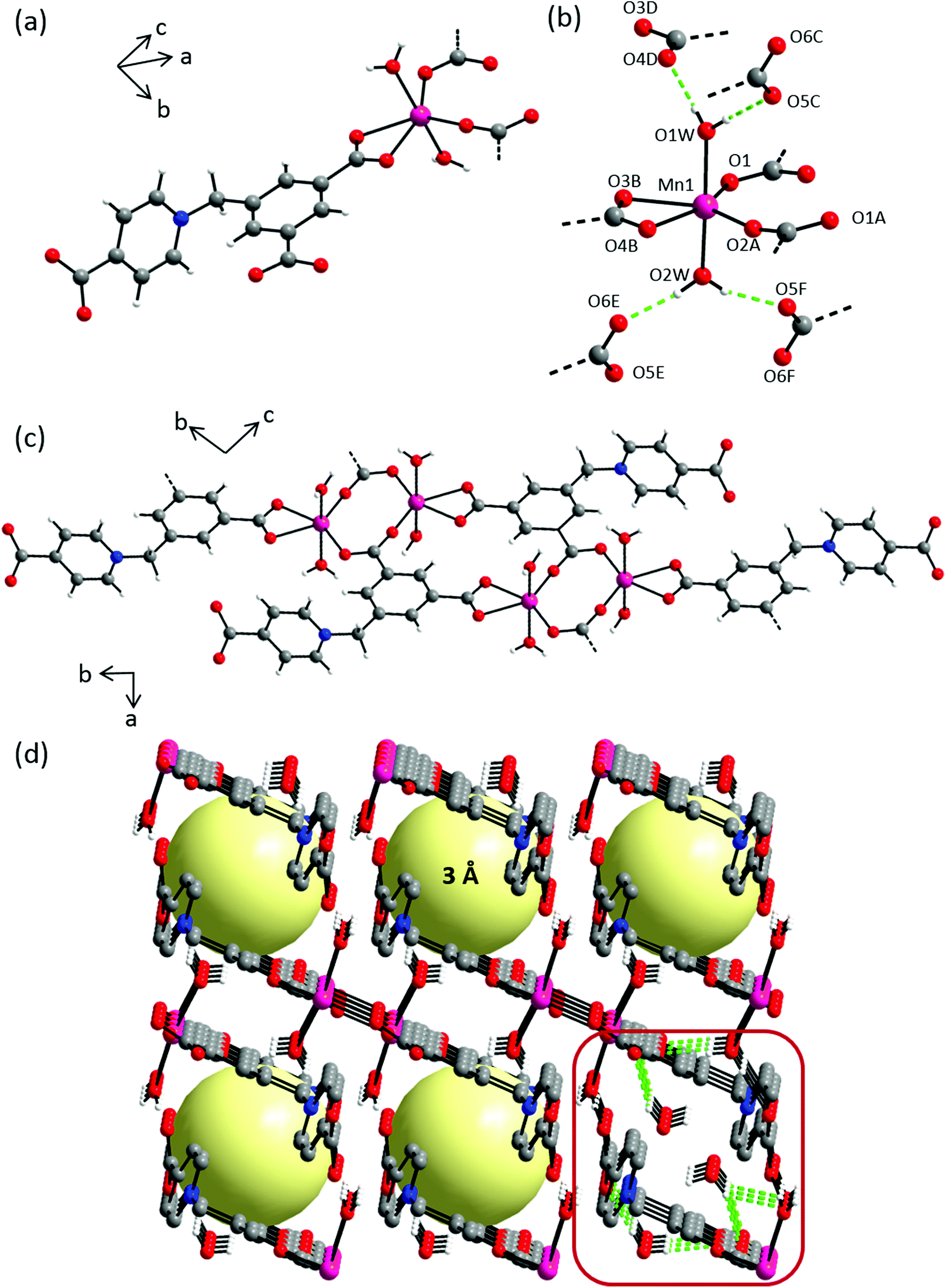
Compound 1 crystallizes in the centrosymmetric triclinic space group P. The asymmetric unit consists of one Cd(II) cation, one L1 ligand, one coordinating and two non-coordinating water molecules (Fig. S9†). All atoms are located in general positions. In the crystal structure, each Cd(II) cation is coordinated by four symmetry-related L1 ligands and one water molecule in a distorted octahedral geometry (Fig. 4a). The Cd(OL1)5Owater octahedron is markedly stretched with long Cd–Owater distance of 2.388(1) Å and five short Cd–OL1 distances ranging 2.270(1)–2.320(2) Å (Fig. 4b, Table S1†). L1 connects four metal ions through carboxylate oxygen atoms in a μ4–κ6, η1:η2:η2:η1 bridging mode to give rise to layers extending along the bc plane (Fig. 4c). Intra- and inter-molecular hydrogen bonding can be found involving hydrogens of the coordinating water molecules and the nearest non-coordinating carboxylate O atoms around the metal centers (Fig. 4e, Table S2†). Also, distinct aromatic π–π stacking interactions can be found (Table S2†). Taking all of these extensive interchain interactions into account, the overall crystal packing can be regarded as a 3D supramolecular coordination network forming 1D channel pores along the crystallographic a-axis with pore diameters of approx. 4 Å (Fig. 4d).
 |
| | Fig. 4 Crystal structure of 1 depicting: (a) coordination environment of the Cd(II) cation; (b) hydrogen bonding interactions exhibited by coordinating water molecules, dashed green lines indicate hydrogen bonding interactions, selected atoms are labelled. Symmetry codes: A = −x + 1, −y + 1, −z; B = x, y − 1, z; C = −x + 1, −y + 1, −z + 1; D = −x + 1, −y + 2, −z + 2; E = x + 1, y, z + 1; (c) an independent chain along a-axis; (d) crystal packing along a-axis; yellow spheres represent potential solvent accessible voids, hydrogen atoms and the solvent molecules are omitted for clarity; (e) hydrogen bond interactions among the water molecules occupying the pores. Color scheme: Cd(II), orange; carbon, grey; nitrogen, blue; oxygen, red and hydrogen, white. | |
Crystal structure of {[Mn(L1)(OH2)2]·H2O}n (2)
Compound 2 crystallizes in triclinic space group P with two formula units in the unit cell. The asymmetric unit consists of one Mn(II) cation, one L1 ligand, two coordinating and one non-coordinating water molecule, with all atoms located in general positions (Fig. S10a†). In the crystal structure, each Mn(II) cation is coordinated by three symmetry-related L1 ligands in a pentagonal bipyramidal geometry (Fig. 5a), exhibiting two long Mn–Owater distances (2.208(1), 2.212(1) Å) and one short, and two long Mn–OL1 distances (2.073(1), 2.147(2), 2.203(1) Å); whereas the angles around the metal cation range between 56.86(15)–92.64(16)° (Fig. 5b, Table S3†). L1 connects Mn(II) cations in a μ3–κ4, η1:η1:η2 bridging mode into chains that extend along the b-axis (Fig. 5c). These infinite chains are extended into a 3D supramolecular architecture by π–π stacking interactions between benzene rings of neighbouring chains with interplanar separation of 3.346(1) Å (Fig. 5d). These chains are further stabilized by hydrogen bonding interactions between the coordinating water molecules and carboxylic groups from adjacent chains, namely, O2W⋯O6D/O4C = 1.842(1)/2.025(2) Å and O1W⋯O6E = 1.809(1) Å (Fig. S10b, Table S4†). The smallest interchain separation of two adjacent Mn(II) cations amounts to 5.297(3) Å. The pyridinium rings are roughly perpendicular to benzene rings with an angle of 74.9° between planes. Benzene rings adopt mirror configurations in adjacent chains and are alternately arranged.
 |
| | Fig. 5 Crystal structure of 2 depicting: (a) coordination environment of Mn(II) cation; (b) hydrogen bonding interactions exhibited by coordinated water molecules, dashed green lines indicate intermolecular hydrogen bonding interactions, selected atoms are labelled. Symmetry codes: A = −x + 2, −y + 1, −z + 2; B = −x + 2, −y + 2, −z + 2; C = x − 1, y + 1, z; D = x − 1, y, z; E = −x + 3, −y + 2, −z + 2; (c) an independent chain along c-axis; (d) perspective view of crystal packing along b-axis showing 3 Å pores; hydrogen atoms and the solvent molecules occupying pores are omitted for clarity, yellow spheres represent potential solvent accessible voids. Color scheme: Mn(II), majenta; carbon, grey; nitrogen, blue; oxygen, red and hydrogen, white. | |
Crystal structure of {[Cu(HL1)2(OH2)3]·9H2O}n (3)
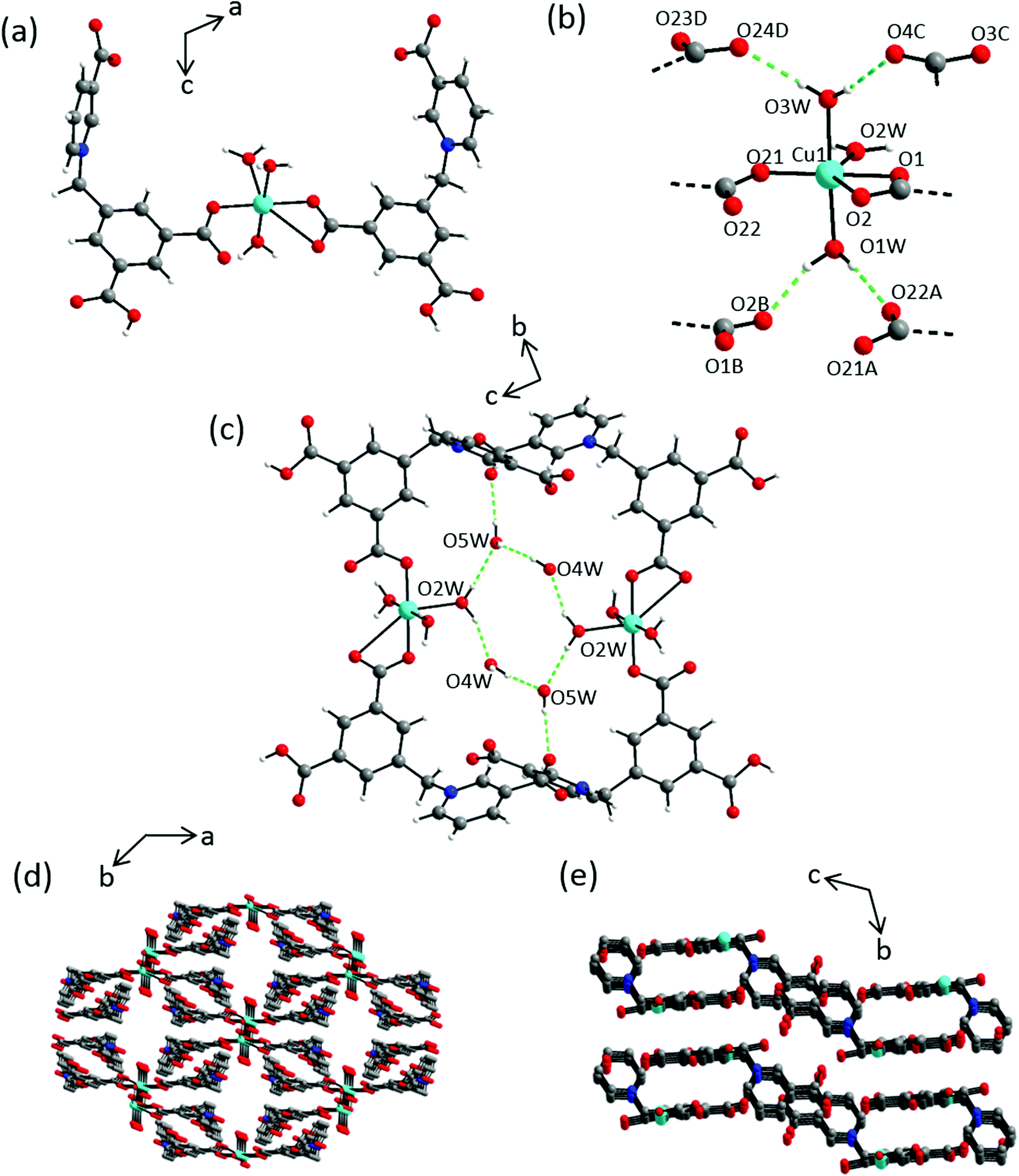
Compound 3 crystallizes in the triclinic space group P with one Cu(II) cation located on a center of inversion, two partially protonated L1 ligands, three coordinating and 3.5 non-coordinating water molecules in the asymmetric unit, with all atoms in general positions (Fig. S11a†). Both ligands are protonated at one carboxylic group each to give an overall charge of −1, thus compensating the Cu2+ charge in 1:2 metal:ligand ratio. In the crystal structure, ligands L1 coordinate to the metal center in a mono- and bidentate fashion to give an overall distorted octahedral geometry with three coordinated water molecules (Fig. 6a). The resulting CuO6 hexagon is composed of two short M–OL1 distances of ∼1.9 Å and a relatively longer M–OL1 bond of 2.804(2) Å, along with three M–Owater distances in the range 1.988(1)–2.037(2) Å (Table S5†). These coordinating water molecules give rise to intermolecular hydrogen bonding interactions with the nearby carboxylate hydrogen acceptors: O1W–HA⋯O22A (1.823 Å; 168.8°), O1W–HB⋯O2B (1.838 Å; 161.9°), O3W′–HA⋯O24D (1.909 Å, 160.0°), O2W–HB⋯O4C (2.049 Å; 171.1°) (Fig. 6b, S11b and Table S6†). Additional hydrogen bonding interactions are observed between the coordinating and non-coordinating water molecules: O2W–HA⋯O5 W (2.085 Å; 172.5°), O4W–HA⋯O5 W (1.919 Å; 170.7°), O2W–HB⋯O4 W (1.893 Å, 160.1°) (Fig. 6c). Also, stabilizing aromatic π–π interactions can be found between benzene rings of L1 in adjacent dimeric units with an interplanar separation of 3.619(1) Å. The smallest interlayer separation of two adjacent Mn(II) cations amounts to 5.133(3) Å. A perspective view of the resulting supramolecular 3D network is shown in Fig. 6d and e.
 |
| | Fig. 6 Crystal structure of 3 depicting: (a) full coordination sphere of the Cu(II) cation along the b-axis; (b) coordination environment of the Cu(II) cation with adjacent hydrogen bonded carboxylate groups; dashed green lines indicate hydrogen bonding interactions, selected atoms are labelled. Symmetry codes: A = −x + 2, −y + 2, −z + 1; B = x + 2, −y, −z + 1; C = −x + 3, −y + 2, −z + 1; D = −x + 2, −y + 3, −z + 1; (c) intermolecular water–water hydrogen-bond network; (d) 3D supramolecular network along c-axis; (e) and along a-axis, hydrogen atoms and the solvent molecules occupying pores are omitted for clarity. Color scheme: Cu(II), turquiose; carbon, grey; nitrogen, blue; oxygen, red and hydrogen, white. | |
Crystal structure of {[Mn2(L2)2(OH2)4]·3H2O}n (4)
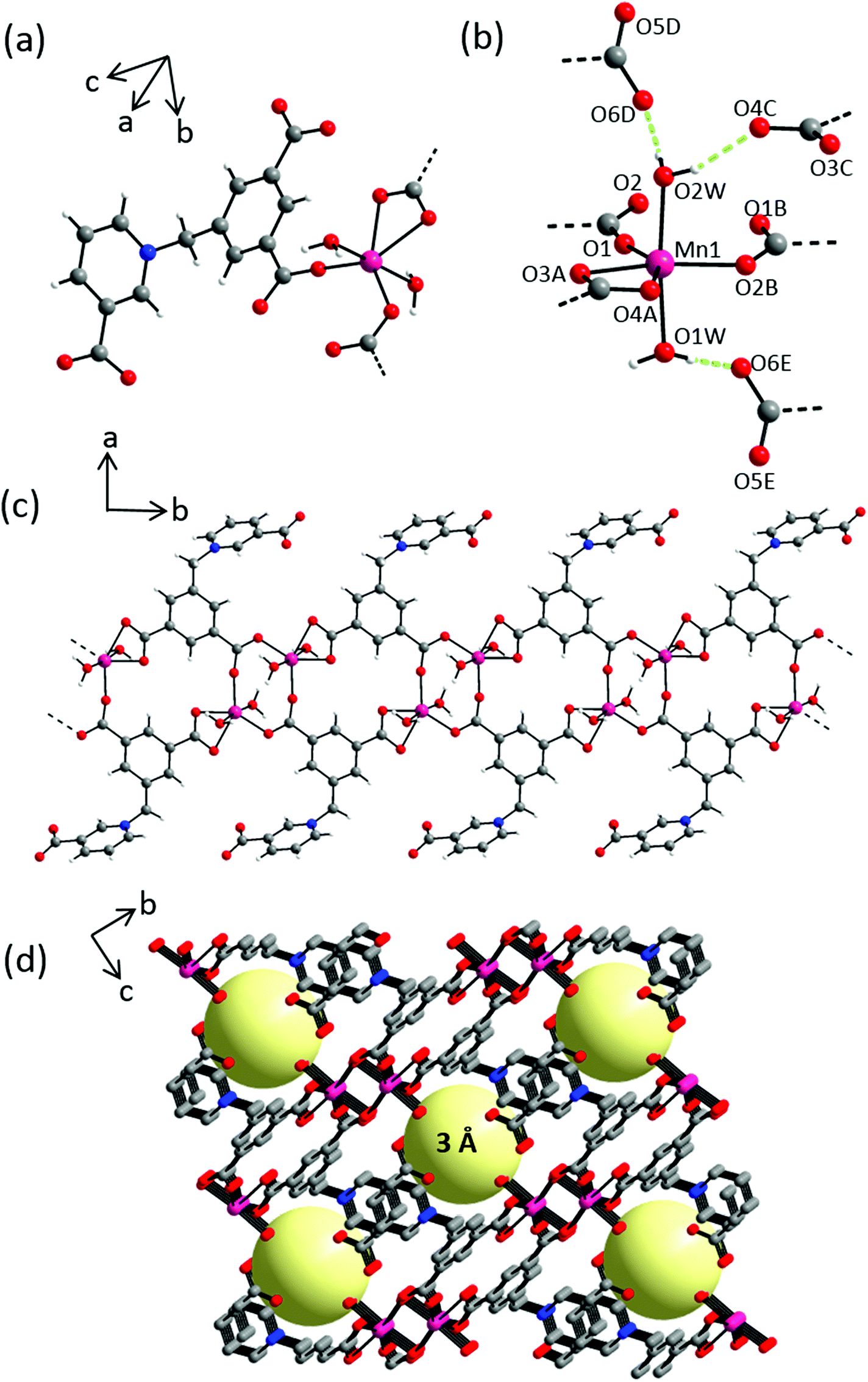
Compound 4 crystallizes in the triclinic space group P with two formula units in the unit cell. The asymmetric unit consists of one Mn(II) cation, one L2 ligand, two coordinating and one and half non-coordinating water molecules, all in general positions (Fig. 7a and S12a†). In the crystal structure, each Mn(II) cation is hexacoordinated by three symmetry related L2 and two water molecules in axial positions in a distorted octahedral geometry (Fig. 7b). The resulting MnO6 hexagon is formed by two similar Mn–Owater distances of 2.162(1) and 2.107(1) Å, two equal Mn–OL2 distances of 2.121(2) Å and two comparatively longer Mn–OL2 distances of 2.251(1) and 2.325(2) Å (Table S7†). L2 coordinates to the metal centers in a μ3–κ4, η1:η1:η2 fashion giving rise to 1D chains along the c-axis (Fig. 7c). This coordination mode is similar to that exhibited by L1 in 2. Interchain hydrogen bonding interactions between axial coordinating water molecules and carboxylate oxygen atoms of L2 stabilize the crystal packing (Fig. 7d). Taking these interactions into account, namely O1W–HA⋯O5C (1.887 Å; 177.6°), O1W–HB⋯O4D (1.941 Å; 175.4°), O2W–HA⋯O6E (1.861 Å, 161.1°), O2W–HB⋯O5F (1.917 Å; 164.1°), the overall crystal packing can be regarded as a 3D supramolecular coordination network with smallest interchain separations between two adjacent Mn(II) centers being 5.665(3) Å, resulting in 1D channel pores of ∼3 Å (Fig. 7d). Non-coordinating water molecules occupy these inter-chain voids and are stabilized by hydrogen bonding interactions with adjacent carboxylic groups (Fig. S12b, Table S8†). Additional stabilizing aromatic π–π stacking interactions can be found as evidenced by close interchain centroid–centroid separations of 3.556(3) Å between adjacent pyridinium units.
 |
| | Fig. 7 Crystal structure of 4 depicting: (a) coordination environment of Mn(II) cation; (b) hydrogen bonding interactions exhibited by coordinated water molecules; dashed green lines indicate hydrogen bonding interactions, selected atoms are labelled. Symmetry codes: A = −x, −y, −z; B = −x, −y, −z + 1; C = x + 1, y, z; D = x + 1, y, z − 1; E = x + 1, y + 1, z; F = −x − 1, −y, −z; (c) an independent chain along a-axis; (d) 3D network showing 3 Å 1-D channel pores along c-axis, solvent molecules occupying pores are omitted from all but one pore for clarity, yellow spheres represent potential solvent accessible voids. Color scheme: Mn(II), majenta; carbon, grey; nitrogen, blue; oxygen, red and hydrogen, white. | |
Crystal structure of ZW MOF [Co(L2)(OH2)4]·H2O (5)
Compound 5 crystallizes in the monoclinic space group P21/c with four formula units in the unit cell (Table 1). The asymmetric unit is composed of discrete units featuring one Co(II) cation, one L2 ligand, four coordinating and one non-coordinating water molecule, with all the atoms located in general positions (Fig. S13†). In the crystal structure, each Co(II) cation is hexa-coordinated by the ligand and two water molecules in equatorial positions and two further water molecules in axial positions in a distorted octahedral fashion (Fig. 8a). The resulting MO6 hexagon is composed of two M–L2 bonds with distances of 2.095(2) and 2.246(1) Å, two equatorial M–Owater distances of ∼2.02 Å and two relatively longer axial M–Owater bonds with distances of 2.113(2) and 2.130(2) Å with a roughly linear O3W–M–O4 W angle of 176.1(2)° (Table S9†). L2 exhibits a μ1–η2 coordination mode (Fig. 8a) with one carboxylate group coordinating in a bidentate mode whereas the remaining carboxylate groups are non-coordinating. In the crystal packing, discrete units of 5 are stabilized by intermolecular hydrogen bonding interactions between water molecules and carboxylate groups, such as O1W–HB⋯O4E (1.911 Å; 172.3°), O2W–HA⋯O6C (1.900 Å, 143.4°), O2W–HB⋯O3B (1.775 Å; 165.8°), O3W–HA⋯O5A (1.875 Å, 167.7°), O3W–HB⋯O4B (1.964 Å; 171.8°), O4W–HA⋯O4D (2.066 Å, 159.4°), O4W–HB⋯O5C (1.868 Å; 175.4°) (Table S10†). Additional stabilizing intermolecular aromatic π–π stacking interactions can be found between adjacent benzene rings with centroid–centroid separations of 3.692(2) Å. The shortest intermolecular distance between the metal cations amounts to 6.835(1) Å. Perspective view of crystal packing along b-axis is shown in Fig. 8b.
 |
| | Fig. 8 (a) Crystal structure of 5 showing coordination environment of Co(II) cation, dashed green lines indicate intermolecular hydrogen bonding interactions. Selected atoms are labelled. Symmetry codes: A = −x − 1, −y + 2, −z + 1; B = x + 1, −y + 1.5, z + 0.5; C = x + 1, −y + 2.5, z + 0.5; D = −x − 1, −y + 2, −z + 1; E = x + 1, y, z; (b) perspective view of the framework along b-axis, hydrogen atoms and solvent molecules occupying pores are omitted for the sake of clarity. Color scheme: Co(II), brown; carbon, grey; nitrogen, blue; oxygen, red and hydrogen, white. | |
Crystal structure of [Ni(L2)(OH2)3]n (6)
Compound 6 crystallizes in monoclinic space group P21/n with four formula units in unit cell (Table 1). The asymmetric unit is comprised of one Ni(II) cation, one L2 ligand, and three coordinating water molecules, all in general positions (Fig. S14a†). In the crystal structure, each Ni(II) cation is hexacoordinated by two symmetry related L2 ligands and three water molecules, two of them in axial positions, in a distorted octahedral geometry (Fig. 9b). The resulting MO6 hexagon is composed of three M–L2 bonds with bond distances in the range 2.028(2)–2.156(1) Å, an equatorial M–Owater distance of 2.035(2) Å and two approximately equal axial M–Owater distances of 2.065(2) and 2.042(2) Å (Table S11†). The L2 ligands bridge the metal centers in a μ2–η1:η2 mode yielding 1D chains along the c-axis (Fig. 9c). These chains are interpenetrated in the crystal packing and stabilized by inter- and intrachain hydrogen bonding interactions: O1W–HA⋯O1B (1.989 Å; 169.3°), O1W–HB⋯O5D (1.890 Å; 167.9°), O2W–HA⋯O4E (1.883 Å, 177.6°), O2W–HB⋯O5D (2.063 Å; 157.7°), O3W–HA⋯O6C (1.874 Å, 164.1°), O3W–HB⋯O2 (1.871 Å; 145.3°) (Fig. S14b and Table S12†). These hydrogen bond interactions give rise to a 3D supramolecular coordination network with smallest interchain separation between two adjacent Ni(II) centers being 5.216(4) Å (Fig. 9d).
 |
| | Fig. 9 Crystal structure of 6 depicting: (a) coordination environment of Ni(II) cation; (b) hydrogen bonding interactions exhibited by coordinated water molecules, dashed green lines indicate hydrogen bonding interactions, selected atoms are labelled. Symmetry codes: A = x, y, z + 1; B = −x + 1, −y + 2, z; C = −x + 1.5, y − 0.5, −z − 0.5; D = −x, −y + 2, −z − 1; E = −x + 0.5, y − 0.5, −z − 0.5; (c) an independent chain along the a-axis; (d) perspective view of framework showing the interpenetration of chains along c-axis, hydrogen atoms are omitted for clarity. Color scheme: Ni(II), pink; carbon, grey; nitrogen, blue; oxygen, red and hydrogen, white. | |
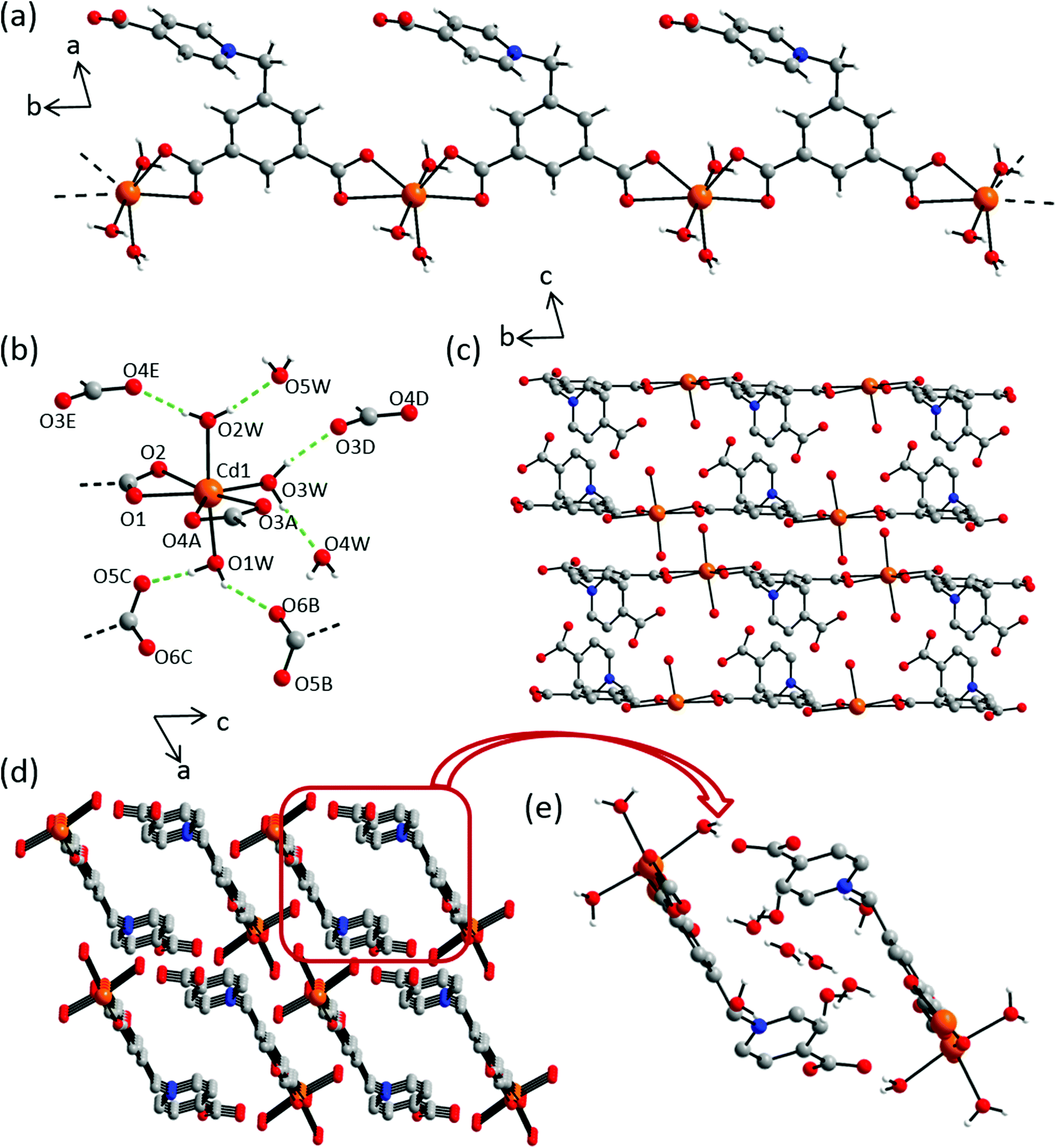
Crystal structure of {[Cd(L2)(OH2)3]·4H2O}n (7)
Compound 7 crystallizes in the centrosymmetric triclinic space group P with two formula units in the unit cell. The asymmetric unit consists of one Cd(II) metal cation, one L2 ligand, three coordinating and four non-coordinating water molecules, with all atoms in general positions (Fig. S15†). In the crystal structure, each metal center is hepta-coordinated by two L2 ligands and one water molecule in the equatorial position along with two additional water molecules in axial positions. The L2 ligands exhibit a μ2–κ4, η2:η2 bridging mode with a non-coordinating terminal carboxylic group, thereby leading to the formation of chains along the crystallographic b-axis. The MO7 cluster consist of M–OL2 bond distances ranging 2.305(2)–2.719(2) Å, whereas the axial and equatorial M–Owater bond distances measure 2.264(3), 2.281(2) Å and 2.279(1) Å respectively (Table S13†). Intermolecular hydrogen bonding interactions can be found between coordinating water molecules and carboxylate oxygen atoms, along with the non-coordinating water molecules, namely, O1W–HA⋯O5C (1.835 Å; 169.2°), O1W–HB⋯O6B (2.007 Å; 148.8°), O2W–HA⋯O6WF (1.900 Å, 169.8°), O2W–HB⋯O4E (1.914 Å; 173.3°), O3W–HA⋯O3D (1.857 Å, 168.5°) and O3W–HB⋯O6WF (1.929 Å; 164.0°) (Fig. 10b and e and Table S14†). The pyridinium and phenyl ring of L2 are almost perpendicular to each other with an angle of 80.1(5)° between the two in respective chains (Fig. 10d). In the crystal packing, these chains form a parallelogram along the b-axis, with metal centers at two opposite corners. The non-coordinating water molecules occupy the interchain voids (Fig. 10e).
 |
| | Fig. 10 Crystal structure of 7 depicting: (a) an independent chain along the c-axis; (b) coordination environment of Cd(II) cation with adjacent hydrogen bonded carboxylate groups, dashed green lines indicate hydrogen bonding interactions, selected atoms are labelled. Symmetry codes: A = x, y − 1, z; B = x − 1, y − 1, z, C = −x, −y + 1, −z + 1; D = −x, −y + 1, z; E = −x + 1, −y + 1, −z; F = −x + 1, −y, −z; G = x − 1, y, z; (c) crystal packing along a- and (c) b-axes; (d) hydrogen atoms and solvent molecules are omitted for the sake of clarity; (e) interchain pore environment with non-coordinating water molecules. Color scheme: Cd(II), orange; carbon, grey; nitrogen, blue; oxygen, red and hydrogen, white. | |
Ligand–metal coordination modes
The above detailed crystal structure analyses reveal diverse coordination modes of ZW ligands L1 and L2 with μ2–κ3, η1:η2 and μ3–κ4, η1:η1:η2 present in two structures each (6/7 and 2/4 respectively). Coordination modes μ4–κ6, η1:η2:η2:η1, μ1–η1 and μ1–η2 are present in one structure each, 1, 3 and 5 respectively. A thorough bridging mode analysis (CSD 2017, Version 5.38) of ligand L2 and the regioisomeric ligand L3, differing only in the position of a nitrogen atom, revealed μ5–κ6, η1:η1:η2:η1:η1 to be the most common mode (10 hits) followed by μ2–κ3, η1:η2 (3 hits). Additional, less frequently observed modes, along with their structural dimensionality and overall architectures are listed in Fig. 11 and Table 2.
 |
| | Fig. 11 Structural analysis of L2 as representative example for L1 and L3. α denotes the dihedral angle between the pyridinium and benzene planes and β represents the bond angle as labelled. Color scheme: carbon, grey; nitrogen, blue and oxygen, red. | |
Table 2 Overview of important structural parameters found in compounds based on ligands L1, L2 and L3. Refer to the labelling scheme in Fig. 11
| Compound |
α [°] |
β [°] |
Coordination mode |
Dimensionality & architecture |
Ref. |
| 4,4′-bpy = 4,4′-bipyridine, 1,4-bbi = 1,1′-(1,4-butanediyl)bis(imidazole), Bpe = 1,2-di(pyridine-4-yl)-ethylene. |
| {[Cd(L1)(OH2)]·2H2O}n (1) |
83.86 |
115.21 |
μ4–κ6, η1:η1:η2:η2 |
3D, ∼4 Å 1D pores |
This work |
| {[Mn(L1)(OH2)2]·H2O}n (2) |
74.92 |
110.72 |
μ3–κ4, η1:η1:η2 |
1D chains |
This work |
| [Cu(HL1)2(OH2)3]n·9H2O (3) |
70.37 |
112.76 |
Mono-, bidentate |
Dimeric units |
This work |
| [Mn2(L2)2(OH2)4]·3H2O (4) |
85.71 |
112.49 |
μ3–κ4, η1:η1:η2 |
1D chains |
This work |
| [Co(L2)(OH2)4]·H2O (5) |
73.44 |
111.16 |
Bidentate |
Discrete units |
This work |
| [Ni(L2)(OH2)3]n (6) |
89.01 |
110.45 |
μ2–κ3, η1:η2 |
Dimeric units |
This work |
| {[Cd(L2)(OH2)3]·4H2O}n (7) |
80.06 |
112.58 |
μ2–κ4, η2:η2 |
1D chains |
This work |
| {[M(L2)(OH2)2]·NO3·2H2O}n [M = La, Pr] |
87.92 |
111.15 |
μ6–κ7, η1:η1:η2:η1:η1:η1 |
3D |
32
|
| {[Nd2(L2)2(NO3)(OH2)2]·Cl·6H2O}n |
84.94 |
115.10 |
μ6–κ7, η1:η1:η2:η1:η1:η1μ5–κ7, η1:η1:η2:η1:η2 |
3D |
32
|
| {[M(L2)(OH2)2]·Cl·3H2O}n [M = Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu] |
73.37 |
114.10 |
μ5–κ6, η1:η1:η2:η1:η1 |
3D |
32
|
| {[Zn(L2)(OH2)3]·H2O}n |
71.29 |
110.74 |
μ2–κ3, η1:η2 |
3D layered meso-helix |
33
|
| {[Zn(L2)]·H2O}n |
65.56 |
112.31 |
μ4–κ5, η2:η1:η1:η1 |
Pillar layered 3D reticular |
33
|
| {[Z(L2)(4,4′-bpy)0.5]·2H2O}n |
87.45 |
115.21 |
μ3–κ4, η2:η1:η1 |
Triply interpenetrated 3D → 3D |
33
|
| {[Cd(L2)]·H2O}n |
50.04 |
114.49 |
μ6–κ6, η1:η1:η1:η1:η1:η1 |
3D pillared-layer |
33
|
| {[Cd(L2)(1,4-bbi)(OH2)]·H2O}n |
78.78 |
110.26 |
μ2–κ3, η1:η2 |
2D polythreaded network |
33
|
| {[Cu(L)(4,4′-bpy) (OH2)]·5H2O}n |
81.06 |
113.13 |
μ2–κ3, η1:η2 |
3D |
33
|
| {[Cu(L2)(bpe)]·6H2O}n |
76.54 |
110.92 |
μ2–κ4, η2:η1:η1 |
2D layer |
33
|
| {[Cu3(HL3)4(azopy)3(OH2)2]·L3}n |
72.97 |
110.02 |
μ2–κ3, η2:η1 |
2D grids |
34
|
| {[Cu3(L3)2(bpe)3(OH2)3]·(NO3)2}n |
83.28 |
109.75 |
μ2–κ2, η1:η1 |
2D → 3D interpenetration |
34
|
| {Na3[Na9(L3)6(OH2)18]}n |
83.79 |
111.87 |
μ3–κ3, η1:η1:η1 |
3D |
35
|
| [Cu6(L3)6(OH2)18] |
76.14 |
117.56 |
μ2–κ2, η1:η1 |
3D |
35
|
| [Cu3(L3)2(OH)2(H2O)2]n |
87.71 |
112.12 |
μ4–κ4, η1:η1:η1:η1 |
3D |
35
|
| {[Cu3(L3)2(OH)2]·2H2O}n |
85.83 |
111.89 |
μ5–κ5, η1:η1:η1:η1:η1 |
3D |
35
|
| [Cu(L3)2(OH2)3]·H2O |
73.66 |
111.16 |
Monodentate |
Dimeric units |
35
|
Thermal stabilities
Bulk materials of compounds 1, 2, 4 and 6 were characterized by TGA and DSC measurements (Fig. 12). Upon heating 1 to 100 °C, a two-step decomposition can be observed with weight losses of 6.4 and 3.5% respectively, with both steps being accompanied by endothermic events in the DSC curve. Both steps are in good agreement with calculations for the release of two non-coordinating [Δmcalc(2H2O) = 7.7%] and one coordinating water molecule [Δmcalc(H2O) = 3.9%]. One-step weight loss of 13.2% was observed on heating 2 to 110 °C, which corresponds to the release of one non-coordinating and two coordinating water molecules [Δmcalc(3H2O) = 13.2%]. As found from elemental analysis, compound 4 partially dehydrates instantly after synthesis and workup at room temperature. It can be assumed that during this process one of the volatile non-coordinating water molecules is removed from the framework giving the new composition {[Mn(L2)(OH2)2]·H2O}n. This trihydrate shows a one-step mass loss of 12.9% upon heating, which is in very good agreement with the calculated mass loss of 12.7% for three water molecules. Note that additional heating rate-dependent TGA measurements of compounds 2 and 4 could not distinguish non-coordinating from coordinating water molecules (Fig. S16†). Upon heating 6 to 160 °C, a mass loss of 12.6% was observed, which corresponds to the loss of three coordinating water molecules in [Ni(L2)(OH2)3]n. This solvent removal process was found to be at comparatively higher temperatures, as each of the water molecules is stabilized by intermolecular hydrogen bonding with adjacent carboxylate oxygen atoms.
 |
| | Fig. 12 TGA and DSC curves for compounds (a) 1; (b) 2; (c) 4; and (d) 6. Heating rate = 3 °C min−1; peak temperatures depicted Tp [°C]. | |
Additional PXRD studies were performed to acquire additional insight into the thermal water removal processes of compounds 1, 2, 4 and 6 (Fig. 13). Upon heating 1 and 2 at 120 °C for 12 h, no structural changes can be observed, as evidenced in unchanged reflection positions of anhydrates 1′ and 2′vs. hydrates 1 and 2. However, the significant broadening of reflections indicates a slight loss in the crystallinity of the samples (Fig. 13a and b). In addition, the rehydration process was investigated by exposing 1′ and 2′ to regular laboratory air at room temperature for 1–2 h, yielding rehydrated samples 1′′ and 2′′. It was found that the PXRD patterns (Fig. 13a and b,† top patterns) and TGA curves (Fig. S17†) of 1′′ and 2′′ match those of pristine compounds 1 and 2, indicating the reversibility of the overall dehydration–rehydration processes. A similar characteristic was found for compound 6, but no loss in crystallinity of 6′ could be observed (Fig. 13d).
 |
| | Fig. 13 Powder X-ray diffraction patterns for simulated, as-synthesized, dehydrated (X′) and rehydrated samples (X′′) of (a) 1, (b) 2, (c) 4 and (d) 6. | |
Compound 4 shows a significantly different powder pattern upon dehydration, indicating substantial structural changes during anhydrate 4′ formation. Investigations on the rehydration process show no changes in the pattern of 4′′vs. 4′, indicating a non-reversible rehydration process (Fig. 13c). Noteworthy however, further TGA studies of 4′′ resulted in a similar weight loss as compared to pristine sample 4 (Fig. S17†). Thus, taking the PXRD and TGA results into account, it can be concluded that rehydration of 4′ results in the formation of a new polymorphic hydrate modification 4′′. To verify which of the two hydrate modifications represents the thermodynamically stable form at room temperature, equivalent amounts of both forms were stirred in water for 24 h. PXRD studies of the resulting solid yielded a powder pattern with reflections corresponding entirely to phase 4 (Fig. S18†). Thus, it can be concluded that a dynamic equilibrium exists between both forms with 4 being the thermodynamically stable form at room temperature whereas 4′′ is the metastable form.
Adsorption properties
Structural analysis of 1 revealed a permanent porosity of 16.2% solvent accessible voids originating from the presence of 4 Å channel pores. To analyze this microporosity,43–47 N2 gas adsorption isotherms were collected at 77 K, but surprisingly no affinity towards nitrogen could be observed. Follow-up high-pressure CO2 adsorption experiments at 295 K also showed a negligible gas uptake, excluding any gate-opening processes. Nevertheless, the permanent porosity of 1 could be confirmed by using methanol as the probe showing an uptake of 7.4 wt% at 25 °C (Fig. 14). This process was found to be fully reversible upon heating to 110 °C. Since the kinetic diameters of methanol and nitrogen are very similar (3.60 and 3.64 Å respectively), the absence of nitrogen adsorption can be attributed to either kinetic effects or to pore shrinkage at low temperatures due to framework flexibility.
 |
| | Fig. 14 Gravimetric adsorption of methanol in 1 with experimental mass changes (red) shown as a function of temperature (black). | |
Conclusions
A series of ZW MOFs based on 3-carboxy-1-(3,5-dicarboxybenzyl)pyridinium bromide [H3L1Br] and 4-carboxy-1-(3,5-dicarboxybenzyl)pyridinium bromide [H3L2Br] were synthesized under hydrothermal conditions exhibiting versatile coordination features. The diversity of herein reported crystal structures marks ZW ligands L1 and L2 as excellent candidates for the synthesis of new ZW MOFs. This work also contains an in-depth CSD analysis of selected structural parameters, namely coordination modes and topologies that are exhibited by MOFs based on L1 and L2, along with the regio-isomeric counterpart L3. μ5–κ6, η1:η1:η2:η1:η1 was found to be the most prevalent coordination mode present in majority of structures followed by μ2–κ3, η1:η2. However, this general trend should be further probed through systematic investigations on similar ZW ligands to get additional insights into their structure–property relationships.
Acknowledgements
We gratefully acknowledge Clarkson University for generous start-up funding and the donors of the American Chemical Society Petroleum Research Fund (56295-DNI10) for support of this research.
References
- G. R. Ferey, Chem. Soc. Rev., 2008, 37, 191 RSC.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561 RSC.
- Y. He, B. Li, M. O'Keeffe and B. Chen, Chem. Soc. Rev., 2014, 43, 5618 RSC.
- P. Ramaswamy, N. E. Wong and G. K. H. Shimizu, Chem. Soc. Rev., 2014, 43, 5913 RSC.
- V. Stavila, A. A. Talin and M. D. Allendorf, Chem. Soc. Rev., 2014, 43, 5994 RSC.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC.
- Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657 RSC.
- J. B. DeCoste and G. W. Peterson, Chem. Rev., 2014, 114, 5695 CrossRef CAS PubMed.
- C. K. Brozek and M. Dinca, Chem. Soc. Rev., 2014, 43, 5456 RSC.
- E. Barea, C. Montoro and J. A. R. Navarro, Chem. Soc. Rev., 2014, 43, 5419 RSC.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS PubMed , all articles of this special issue.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed.
- T. A. Makal, J.-R. Li, W. Lu and H.-C. Zhou, Chem. Soc. Rev., 2012, 41, 7761 RSC.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2011, 112, 869 CrossRef PubMed , all articles of this special issue.
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1201 RSC , all articles of this special issue.
- D. Aulakh, J. B. Pyser, X. Zhang, A. A. Yakovenko, K. R. Dunbar and M. Wriedt, J. Am. Chem. Soc., 2015, 137, 9254 CrossRef CAS PubMed.
- D. Aulakh, J. R. Varghese and M. Wriedt, Inorg. Chem., 2015, 54, 8679 CrossRef CAS PubMed.
- M. Wriedt, A. A. Yakovenko, G. J. Halder, A. V. Prosvirin, K. R. Dunbar and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 4040 CrossRef CAS PubMed.
- J. P. Sculley, W. M. Verdegaal, W. Lu, M. Wriedt and H.-C. Zhou, Adv. Mater., 2013, 25, 3957 CrossRef CAS PubMed.
- M. Wriedt, J. P. Sculley, A. A. Yakovenko, Y. Ma, G. J. Halder, P. B. Balbuena and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 9804 CrossRef CAS PubMed.
- W. An, D. Aulakh, X. Zhang, W. Verdegaal, K. R. Dunbar and M. Wriedt, Chem. Mater., 2016, 28, 7825 CrossRef CAS.
- W. M. Verdegaal, K. Wang, J. P. Sculley, M. Wriedt and H.-C. Zhou, ChemSusChem, 2016, 9, 636 CrossRef CAS PubMed.
- M. Wriedt, J. P. Sculley, W. M. Verdegaal, A. A. Yakovenko and H.-C. Zhou, Chem. Commun., 2013, 49, 9612 RSC.
- A. A. Yakovenko, Z. Wei, M. Wriedt, J.-R. Li, G. J. Halder and H.-C. Zhou, Cryst. Growth Des., 2014, 14, 5397 CAS.
- M. Wriedt, J. P. Sculley, D. Aulakh and H.-C. Zhou, J. Chem. Educ., 2016, 93, 2068 CrossRef CAS.
- D. Aulakh, J. R. Varghese and M. Wriedt, Inorg. Chem., 2015, 54, 1756 CrossRef CAS PubMed.
- D. Aulakh, A. P. Nicoletta, J. R. Varghese and M. Wriedt, CrystEngComm, 2016, 18, 2189 RSC.
- M. Higuchi, D. Tanaka, S. Horike, H. Sakamoto, K. Nakamura, Y. Takashima, Y. Hijikata, N. Yanai, J. Kim, K. Kato, Y. Kubota, M. Takata and S. Kitagawa, J. Am. Chem. Soc., 2009, 131, 10336 CrossRef CAS PubMed.
- Q.-X. Yao, Z.-F. Ju, X.-H. Jin and J. Zhang, Inorg. Chem., 2009, 48, 1266 CrossRef CAS PubMed.
- X.-M. Zhang, Y.-Q. Wang, K. Wang, E.-Q. Gao and C.-M. Liu, Chem. Commun., 2011, 47, 1815 RSC.
- M. Higuchi, K. Nakamura, S. Horike, Y. Hijikata, N. Yanai, T. Fukushima, J. Kim, K. Kato, M. Takata, D. Watanabe, S. Oshima and S. Kitagawa, Angew. Chem., Int. Ed., 2012, 51, 8369 CrossRef CAS PubMed.
- H.-N. Li, H.-Y. Li, L.-K. Li, L. Xu, K. Hou, S.-Q. Zang and T. C. W. Mak, Cryst. Growth Des., 2015, 15, 4331 CAS.
- H.-Y. Li, L.-H. Cao, Y.-L. Wei, H. Xu and S.-Q. Zang, CrystEngComm, 2015, 17, 6297 RSC.
- J.-X. Chen, H.-Q. Zhao, H.-H. Li, S.-L. Huang, N.-N. Ding, W.-H. Chen, D. J. Young, W.-H. Zhang and T. S. Andy Hor, CrystEngComm, 2014, 16, 7722 RSC.
- J.-X. Chen, M. Chen, N.-N. Ding, W.-H. Chen, W.-H. Zhang, T. S. A. Hor and D. J. Young, Inorg. Chem., 2014, 53, 7446 CrossRef CAS PubMed.
-
SAINT and APEX 2 Software for CCD Diffractometers, Bruker AXS Inc., Madison, WI, 2014 Search PubMed.
-
G. M. Sheldrick, SADABS, version 2014/4, University of Göttingen, Germany, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- B. Chen, S. Ma, F. Zapata, F. R. Fronczek, E. B. Lobkovsky and H.-C. Zhou, Inorg. Chem., 2007, 46, 1233–1236 CrossRef CAS PubMed.
- Y.-W. Li, J.-R. Li, L.-F. Wang, B.-Y. Zhou, Q. Chen and X.-H. Bu, J. Mater. Chem. A, 2013, 1, 495–499 CAS.
- Y.-W. Li, J. Xu, D.-C. Li, J.-M. Dou, H. Yan, T.-L. Hu and X.-H. Bu, Chem. Commun., 2015, 51, 14211–14214 RSC.
- Y.-W. Li, H. Yan, T.-L. Hu, H.-Y. Ma, D.-C. Li, S.-N. Wang, Q.-X. Yao, J.-M. Dou, J. Xu and X.-H. Bu, Chem. Commun., 2017, 53, 2394–2397 RSC.
- O. Toma, N. Mercier, M. Allain, A. A. Kassiba, J.-P. Bellat, G. Weber and I. Bezverkhyy, Inorg. Chem., 2015, 54, 8923–8930 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data of Me2HL1, H3L1, Me2HL2 and H3L2; IR data of H3L1, 1 and 2; IR data of H3L2, 4, and 6; PXRD patterns for phase pure 1, 2, 4, and 6; temperature ellipsoid plots for MOFs 1–7, coordination networks exhibiting hydrogen bonding interactions, characteristic bond distances and angles responsible for intra and intermolecular hydrogen bonding interactions for 1–7; heating rate dependent TGA measurements on MOFs 2 and 4; PXRD pattern for dehydrated 4′ and rehydrated 4′′ upon exposure to regular air. CCDC 1428987–1428992 and 1428994. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt00292k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*