
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and methane cracking activity of a silicon nitride supported vanadium nitride nanoparticle composite†
Ihfaf
AlShibane
a,
Justin S. J.
Hargreaves
*a,
Andrew L.
Hector
 *b,
William
Levason
b and
Andrew
McFarlane
a
*b,
William
Levason
b and
Andrew
McFarlane
a
aSchool of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: Justin.Hargreaves@glasgow.ac.uk
bChemistry, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: A.L.Hector@soton.ac.uk
First published on 27th February 2017
Abstract
The co-ammonolysis of V(NMe2)4 and Si(NHMe)4 with ammonia in THF and in the presence of ammonium triflate ([NH4][CF3SO3]) leads to the formation of monolithic gels. Pyrolysing these gels produces mesoporous composite materials containing nanocrystalline VN and amorphous silicon imidonitride. Elemental mapping indicated a thorough distribution of VN with no evidence of large cluster segregation. Whilst not active for ammonia synthesis, the silicon nitride based materials were found to possess activity for the COx-free production of H2 from methane, which makes them candidates for applications in which the presence of low levels of CO in H2 feedstreams is detrimental.
Introduction
Gel-derived silicon imidonitrides have been examined by a number of groups as base catalysts, for example for Knoevenegal condensations1 and alkene isomerization reactions.2 Silicon nitride is also of interest as a catalyst support due to the high thermal conductivity relative to the more commonly used silicas, and in the α-Si3N4 form is oxidation resistant at typical catalytic process temperatures.3,4 Gel-based routes to silicon nitrides and imidonitrides utilise solution-phase reactions of precursor molecules, usually amides, with cross-linking groups such as ammonia5 or isocyanate.6 These make polymeric species in solution, and when the polymeric material immobilises the liquid phase, the latter can be removed to provide high surface area materials that often contain basic surface groups.7,8 These reactions provide a generic sol–gel chemistry that has been used to produce powders,9–11 films,12 membranes13 and monolithic aerogels.14,15A number of metal nitrides have been shown to have useful catalytic activities.16 Much of this work has focussed on the similarity of the activities of some metal nitrides to those of the platinum metals, with surface areas usually maximised by careful temperature control in reactions between high surface area metal oxides and ammonia.17 The highly active group 6 nitride catalysts such as Mo2N have been examined extensively, but group 5 nitrides can exhibit different selectivities, which can also vary between phases.18 VN has been shown to be a useful catalyst in ammonia decomposition reactions,19–21 amination of ethanol22 and dehydrogenation of propane.23 A limited number of studies have attempted to modify the properties of gel-derived silicon nitride materials by incorporating aluminium,24,25 boron,10 titanium26 or terbium27 into the silicon imidonitride framework, or by using it post-synthesis to support palladium nanoparticles.28 Reactions starting with mixed Si(NHMe)4/metal amide solutions have been used to obtain amorphous M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SiNx composites in which boron, titanium, zirconium or tantalum is distributed in the silicon nitride framework.29 Phase segregation occurs when these gels are fired at very high temperature (1500 °C) to yield silicon nitride supported metal nitride nanoparticles. These reactions were carried out with high metal concentrations and are not suitable to produce supported metal nitride catalysts with typical metal loadings, of a few %. We showed that similar reactions with a triflate catalyst could provide controlled incorporation of tantalum into gels, but a clear phase segregation occurred during gelation in the equivalent MoN/SiNx system.30 Here we investigate the role of the triflate catalyst, produce VN/SiNx composites and demonstrate the catalytic activity of these composite materials in methane cracking, a reaction of interest for the production of COx-free hydrogen for applications sensitive to the presence of low levels of CO, such as in PEM fuel cells.31
:SiNx composites in which boron, titanium, zirconium or tantalum is distributed in the silicon nitride framework.29 Phase segregation occurs when these gels are fired at very high temperature (1500 °C) to yield silicon nitride supported metal nitride nanoparticles. These reactions were carried out with high metal concentrations and are not suitable to produce supported metal nitride catalysts with typical metal loadings, of a few %. We showed that similar reactions with a triflate catalyst could provide controlled incorporation of tantalum into gels, but a clear phase segregation occurred during gelation in the equivalent MoN/SiNx system.30 Here we investigate the role of the triflate catalyst, produce VN/SiNx composites and demonstrate the catalytic activity of these composite materials in methane cracking, a reaction of interest for the production of COx-free hydrogen for applications sensitive to the presence of low levels of CO, such as in PEM fuel cells.31
Results and discussion
Ammonolytic gelation of Si(NHMe)4 can be facilitated by the presence of a catalytic (∼0.4 mol%) amount of ammonium triflate, and the resulting gels can be fired to produce high quality silicon nitride/imido nitride materials with mesoporosity.5 Ammonolysis of a 1:1 molar ratio of Si(NHMe)4 and V(NMe2)4 under similar conditions did not result in gelation with or without the added triflate, instead a dark precipitate would separate from a turbid solution within a few hours of allowing the mixtures to warm to ambient temperature. Removal of solvent in vacuo from these mixtures resulted in colourless, low volatility, viscous liquid in the trap, which gelled on exposure to air and was probably the unreacted silicon amide and/or small oligomers. Löffelholz et al. previously argued that similar ammonolysis rates are needed for ammonolytic gelation of such mixtures of silicon and metal amides, and in a limited number of cases achieved homogeneous gels (with no catalyst) in cases using amides with well-matched rates.29 In one of these cases (Si(NHMe)4/Ta(NMe2)5), we later showed the catalyst to be necessary for gelation when lower Ta content mixtures were employed.30 However, even with a 40:1 mixture of Si(NHMe)4:V(NMe2)4 we could not achieve gelation with the 0.4 mol% catalyst concentration used to produce gels without metal addition. Increasing the ammonium triflate concentration stepwise, the addition of sufficient ammonium triflate to match the amount of amide groups in the V(NMe2)4 precursor was found to result in the preparation of a homogeneous gel. This suggests that the triflate reacts preferentially with the metal centre, presumably by the replacement of amide groups on the vanadium with triflate.
The gels used to produce silicon–vanadium nitride composites in this study were produced from THF solutions containing a 40:1:4 ratio of Si(NHMe)4 to V(NMe2)4 to ammonium triflate. Dry liquid ammonia was added to the stirred solutions at −78 °C. On allowing the mixtures to warm to ambient temperature (unstirred) gels formed after 15–20 minutes. These were aged for around 22 hours, after which period the gels were an even green colour, suggesting a homogeneous distribution of vanadium(IV), and mechanically stable, maintaining their shape when the tube used to contain them was inverted (Fig. 1). Drying the gels in vacuo resulted in very light green xerogel powders of composition SiV0.025C0.41H1.85N1.29, with a similar thermal decomposition behaviour to that previously observed for silicon nitride precursor gels (Fig. 1).5
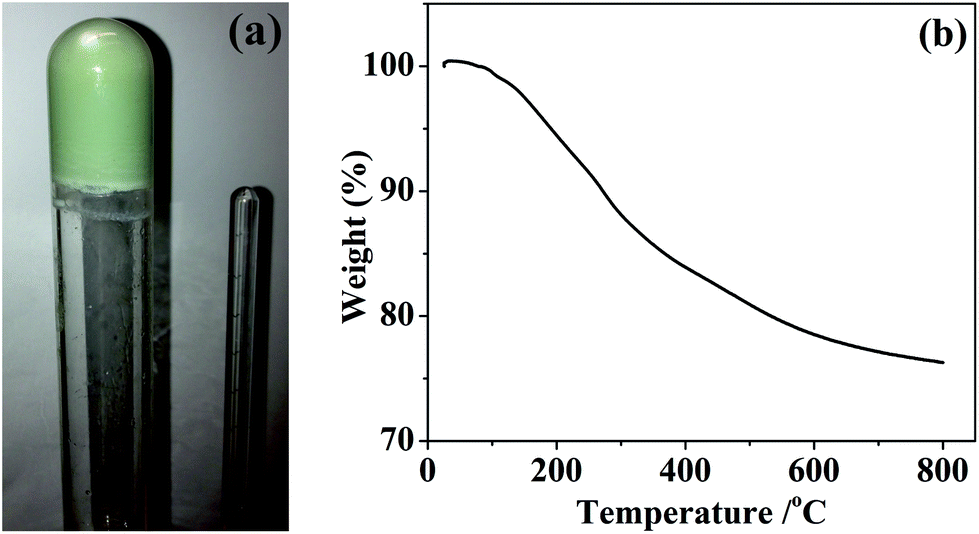 | ||
| Fig. 1 Photograph of an inverted silicon–vanadium amide gel (a) and TGA curve of vacuum dried xerogel (b). | ||
Silicon–vanadium nitride composites were produced by firing the amide-derived xerogels under dry ammonia. First they were heated for 2 hours under ammonia to maximise the removal of carbon-containing alkylamide groups by transamination, then the temperature was ramped slowly to 600 or 1000 °C and this was maintained for 2 hours before cooling to obtain the light grey powdered products. X-ray diffraction (XRD) of these materials mainly showed broad amorphous features as expected for silicon nitride at these temperatures,5 but with further broad peaks corresponding to the most intense expected reflection (the 200) for rocksalt-type VN at 600 °C and more of the VN reflections at 1000 °C (Fig. 2). Using the Scherrer equation, these features correspond to crystallite sizes of around 10 nm.
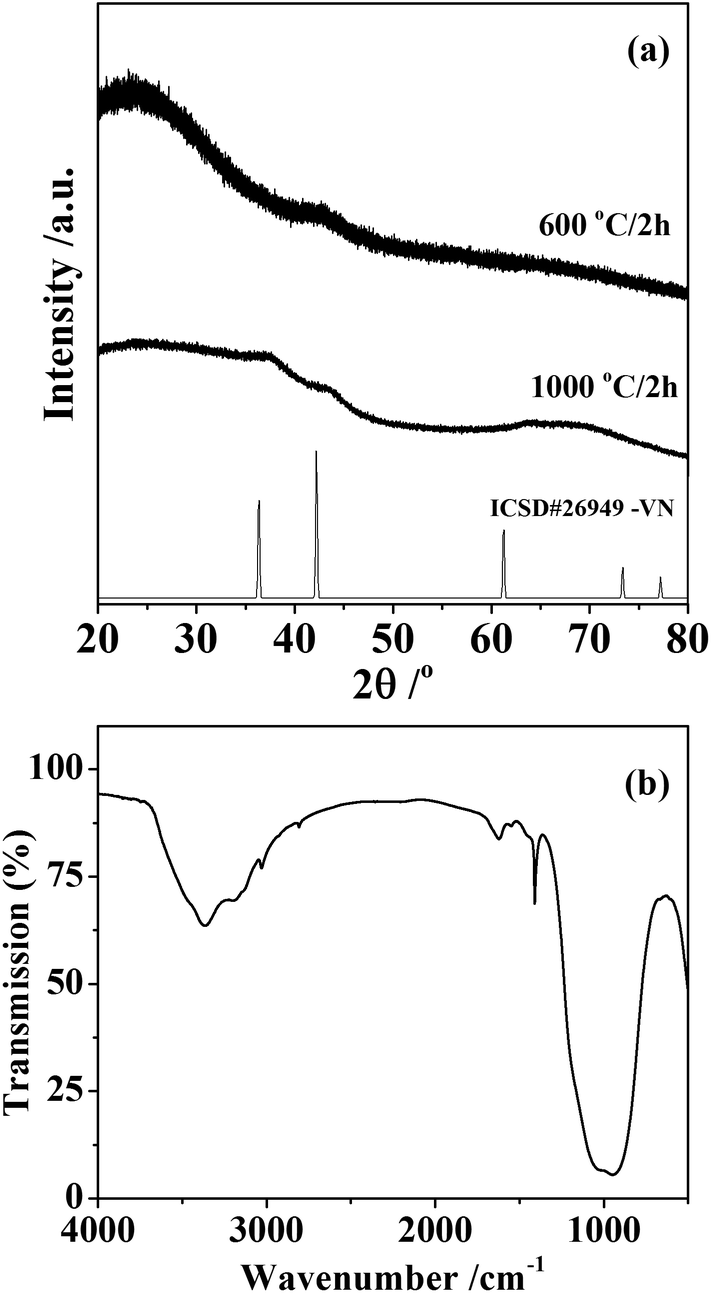 | ||
| Fig. 2 Powder XRD pattern from the silicon–vanadium nitride nano-composites (a) and infrared spectrum of the sample synthesised at 1000 °C (b). The XRD patterns are compared with crystalline VN from ICSD file 26949.32 | ||
Infrared spectra (Fig. 2) contained bands at 3365 cm−1 (νNH) and 970 cm−1 (νSiN), with additional weak features at 2808 cm−1 (νCH) and 1410 cm−1 (δNH2) consistent with materials containing mainly the expected silicon nitride but with some residual amide/imide groups in the sample. The sample produced at 1000 °C was found to have a composition of SiV0.025C0.009N0.70, although combustion analyses tend to underestimate the nitrogen contents of these materials due to the difficulty of fully combusting the oxidation resistant materials. The lack of hydrogen in the analysis suggests the amide groups seen in the IR to be a minor constituent, probably at the surface of the material.
Scanning electron microscopy (SEM, ESI: Fig. S1†) showed the presence of agglomerated particles with average size around 1 μm. Energy dispersive X-ray (EDX) analysis yielded a Si:V atomic ratio of 97.2:2.8, which is very close to the 100:2.5 ratio of the reagents, suggesting that the gel was well cross-linked with very low levels of volatile species and hence negligible loss of V or Si upon pyrolysis. The use of larger triflate concentrations compared with the non metal-containing gels increases the risk of incorporation of triflate-related impurities such as F and S in the fired product, but these were not found. Transmission electron microscopy (TEM, Fig. 3) showed the larger particles seen in the SEM to consist of loose agglomerations with at least two different microstructures: 100–200 nm spherical particles and ∼10 nm elongated particles. This is typical of silicon imidonitride/nitride samples prepared by the ammonolysis sol–gel route.5 Scanning TEM elemental mapping by EDX of the higher firing temperature sample, which is most likely to undergo phase segregation, shows a thorough distribution of vanadium and nitrogen across the sample in both particle types. We consider that on the length scales examined the distribution of transition metal catalytic active centres (in the form of nanocrystalline VN) within the catalyst is uniform.
 | ||
| Fig. 3 TEM image (top left) and EDX elemental mapping of the silicon–vanadium nitride nano-composite produced at 1000 °C. | ||
The catalytic behaviour of the silicon–vanadium nitride nanocomposite produced at 600 °C was examined. The material was found to be inactive for ammonia synthesis under our standard testing conditions (which are detailed elsewhere33). Only a very limited amount of ammonia (a total of 1.5 mmol g−1 over 12 hours at 400 °C) was produced, which corresponded to desorption of NHx species and which declined within the testing period.
In contrast, the material was found to possess activity for hydrogen production directly from methane. The reaction profile illustrating the mass normalised hydrogen formation rate over a period of 14 hours is presented in Fig. 4. Following an initial period of decay over the first 3–4 hours on stream, the activity of the material was found to stabilise to a value of around 180 μmol H2 produced gcatalyst−1 min−1. Whilst this mass normalised rate does not compare favourably with, for example, iron oxide systems in the literature (for example, a peak rate of 1 mmol H2 produced gcatalyst−1 min−1 under directly comparable reaction conditions has been reported for biogenic iron oxide34) one potential advantage is that the absence of oxide phase in the system results in the absence of CO production which is particularly deleterious for H2 streams applied to PEM fuel cells.31 Off line FTIR analysis taken at regular intervals throughout the catalyst runs showed the CO and CO2 levels to be below the limit of detection. CO2 formation, which can also occur in the presence of oxides, could also be problematic under these conditions since CO formation could occur via the reverse water gas-shift reaction, which is particularly thermodynamically favourable at such high reaction temperature. In order to determine the background activity of the silicon nitride material, directly comparable catalytic reaction runs were undertaken (ESI: Fig. S2†). This material was also reasonably active for methane cracking, again without producing COx. However, there is evidence of an induction period followed by deactivation leading to a rate of ca. 40 μmol gcatalyst−1 min−1, which is much lower than that of its vanadium containing counterpart. This is indicative of the promotional effect of vanadium upon reaction. A particularly noteworthy aspect of the vanadium containing composite is the relative stability of H2 production at extended periods upon stream.
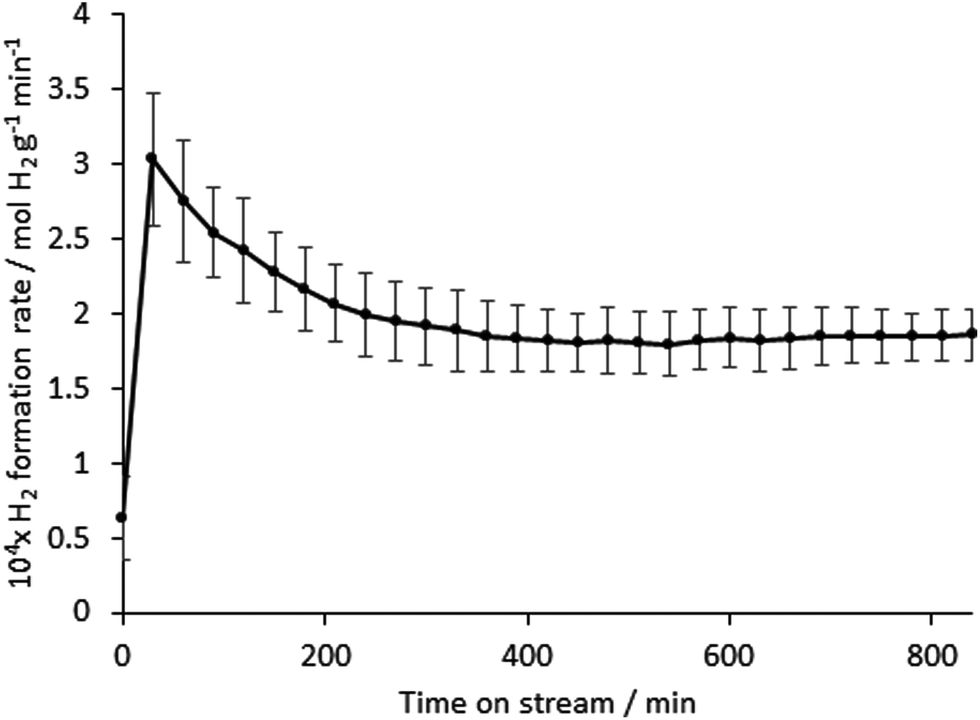 | ||
| Fig. 4 Variation in mass normalised H2 formation rate of the silicon–vanadium nitride nano-composite produced at 600 °C, measured using a 0.055 g sample with 75% CH4/25% N2 at 12 ml min−1 at 800 °C, initial ramp rate 50 °C min−1. Similar values were obtained in a repeat run with the same mass of catalyst. | ||
Nitrogen adsorption–desorption isotherms of the silicon–vanadium nitride sample used for the catalysis study were type IV in shape (ESI: Fig. S3†) indicating mesoporosity.35 Extraction of the BET surface area36 revealed a surface area of 126 m2 g−1 and an average pore radius37 of 10 nm. Comparison with the TEM images in Fig. 3 shows that this porosity is largely due to the spaces between particles in the aggregates. After 14 h on stream the surface area of the post-catalysis sample (Fig. S4†) had fallen to 18 m2 g−1 and the pore radius to 6 nm. As anticipated, the production of H2 was associated with the deposition of carbon upon the materials. The vanadium-containing sample post catalysis was found to contain 23 wt% C by combustion analysis in comparison to ca. 9 wt% deposited on the silicon nitride. The initial decay of performance evident in the first 3–4 hours on stream in Fig. 4 for the vanadium-containing material is most likely associated with deleterious carbon deposition, although beyond this period it is notable that increasing carbon content does not further adversely affect performance, since steady state activity is attained. The latter phase of activity might relate to the presence of coke resistant active sites or activity associated with the carbon itself. In both the vanadium-containing material and the silicon nitride, it is interesting to note that the amount of deposited carbon is significantly less than would be anticipated if methane cracking was the only reaction to occur. For example, in the case of a steady state production of H2 at 180 μmol gcatalyst−1 min−1 over 14 hours, as is broadly representative of the vanadium containing material, the total amount of carbon expected to be deposited would correspond to 0.90 g gcatalyst−1, equating to 47 wt%. The difference in carbon content might arise due to the occurrence of additional hydrogen producing reactions such as methane dehydroaromatisation and dehydrogenative coupling to form C2 hydrocarbons.38 The latter would not be obvious in the IR data that we used to monitor for CO.
TGA conducted under air (ESI: Fig. S5 and S6†) shows the weight loss associated with the removal of carbon to be consistent with the CHN analyses of post-reaction materials. The first derivative profiles show two mass loss features centred at around 500 and 650 °C in the case of the vanadium based system, suggesting two forms of carbon. This is in contrast with the single mass loss feature centred around 700 °C observed in the case of post-reaction silicon nitride. However caution must be exercised in this respect, as it could be possible that the presence of vanadium catalyses the oxidation of a proportion of the carbon deposited causing its loss at lower temperature. Raman spectroscopic analyses of both post-reaction samples (Fig. 5) provide evidence of both disordered and graphitic bands.
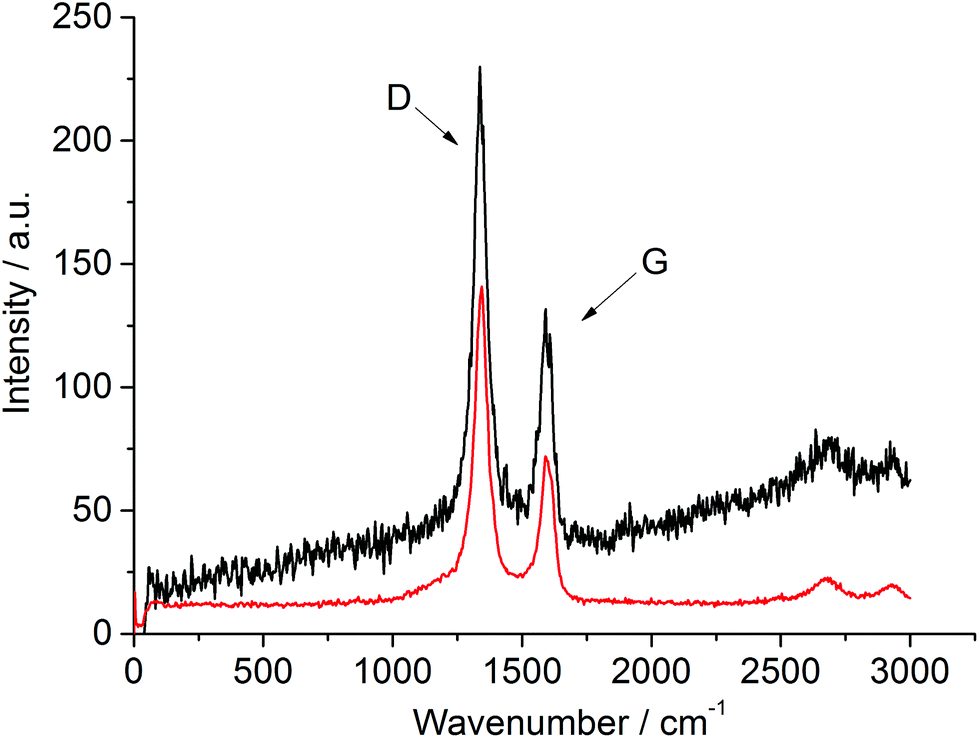 | ||
| Fig. 5 Post-catalysis Raman spectra of the silicon–vanadium nitride nano-composite (top) and the silicon nitride (bottom), both produced at 600 °C. The disordered (D) and graphitic (G) bands are labelled. | ||
Conclusions
Vanadium incorporation into silicon nitride using ammonolytic gels produced from silicon methylamide was found to require high triflate catalyst levels, suggesting that triflate reacts preferentially with the metal centre. Nonetheless homogeneous gels were produced and these could be fired to yield nanocomposites with well distributed vanadium. Silicon nitride based materials are worthy of further attention as catalysts for the production of hydrogen from methane since, although their activity is lower than that of some other systems, the absence of oxygen containing phases results in effluent streams which are free from the presence of CO, a notable downstream poison for some applications.Experimental
Most synthetic manipulations were carried out using Schlenk or glove box methods due to the high moisture sensitivity of the amides and gel intermediates. Tetrakis(methylamino)silane (Si(NHMe)4) was prepared as described elsewhere.14 For gel preparations ammonia gas was dried over Na and directly condensed into the reaction mixture, or for pyrolysis procedures it was dried by passing it through a dried molecular sieve column.V(NMe2)4 was obtained by reacting VCl4 with LiNMe2. LiNMe2 (11.2 g, 0.22 mol, obtained from 1.6 mol dm−3 nBuLi in hexanes and methylamine, Aldrich) was suspended in dry Et2O (150 cm3, distilled from sodium/benzophenone) and cooled to 0 °C. A solution of VCl4 (5 cm3, 0.05 mol, Aldrich) in dry benzene (50 cm3, distilled from sodium) was added drop wise over 25 min with stirring. The mixture was refluxed for 1 h and when cool was filtered through a ceramic frit to obtain a dark green filtrate. Solvent removal in vacuo yielded a dark oily solid, which was sublimed in vacuo at 60–80 °C to yield dark green crystals. Yield 5.6 g, 52%. Combustion analysis: 41.4% C, 9.7% H, 24.3% N (Theory for V(NMe2)4: 42.3% C, 10.7% H, 24.7% N).
To produce silicon/vanadium amide gels, V(NMe2)4 (0.020 g, 8.8 × 10−5 mol), Si(NHMe)4 (0.49 g, 3.3 × 10−3 mol) and [NH4][CF3SO3] (0.060 g, 3.6 × 10−4 mol) were transferred to a glass pressure tube with a sidearm (described previously5) for measurement of a small volume of liquid ammonia. Dry THF (6 cm3, distilled from sodium/benzophenone) was added under N2 atmosphere and the solution was stirred for 1 h at room temperature. 0.5 cm3 of dry liquid NH3 was allowed to distil into the stirred solution of the amides at −78 °C and stirring was continued for 6 hours. The stirring was stopped and the mixture was removed from the dry ice bath and allowed to warm to room temperature. Gelation occurred over 15–20 minutes and the gel was aged for a further 22 hours before solvent removal in vacuo to obtain a very light green xerogel powder, yield ∼0.28 g. Combustion analysis: 8.95% C, 3.45% H and 33.25% N. Note: a safety screen should be employed when using condensed ammonia in glass apparatus.
Pyrolysis of the xerogel used alumina combustion boats and a quartz furnace tube fitted with an arrangement of taps to allow flushing of the lines with dry ammonia for ∼20 minutes before exposing the sample to the ammonia gas. The xerogel was then heated at 200 °C for 2 h followed by ramping at 2 °C min−1 to 600 or 1000 °C, maintaining the temperature for 2 h, and then cooling to room temperature at 5 °C min−1. Ceramic yields were ∼60%. Combustion analysis: 0.3% C, <0.1% H, 24.8% N.
Catalytic testing to determine methane cracking activity followed a procedure described previously.39 0.2 g of silicon nitride or 0.55 g of the vanadium–silicon nitride composite was tested, the difference in mass reflecting the difference in density of the materials. The powders were positioned between silica wool plugs in a quartz microreactor tube held centrally in a furnace. 12 mL min−1 of pre-mixed CH4:N2 (75:25, BOC 99.98%) were flowed over the sample with product analysis being conducted by gas chromatography (Hewlett Packard 5890) using a TCD with Ar reference gas for H2 quantification following separation employing a 12′ long 1/8′′ OD Molseive 13X packed column. Additionally, the effluent was analysed periodically for COx content throughout all reaction runs by off-line FTIR analyses employing a gas-phase FTIR cell. Analysis was undertaken using a Jasco 4100 FTIR spectrometer operating in the 400–4000 cm−1 spectral range at a resolution of 4 cm−1 for 64 scans per sample. Quantification of H2 formation rates took into account molar expansion effects by reference to the N2 component of the feed as internal standard.
Powder XRD patterns were collected in Bragg–Brentano geometry with a Bruker D2 using Cu-Kα radiation. TGA of the xerogel was carried out under 50 ml min−1 of Ar and at the heating rate of 10 °C min−1 using a Mettler-Toledo TGA 851e inside a glove box, and combustion analysis was outsourced to Medac Ltd. Post-catalytic reaction TGA analysis was performed with a temperature ramp rate of 10 °C min−1 under flowing air in a TA Instruments Q500 instrument, and combustion analysis was undertaken by combustion using an Exeter Analytical CE-440 elemental analyser. Infrared spectra of solid samples were recorded as CsI pellets using a PerkinElmer Spectrum 100. SEM of gold-coated samples used a Philips XL30 ESEM with a ThermoFisher Ultradry EDX detector. TEM of samples dispersed into toluene and dropped on carbon grids was carried out with a FEI Technai 12 microscope. Raman spectroscopic analysis was conducted using a LabRAM HR system (Horiba Jobin Yvon) with a Ventus 532 laser system operating 100 mW and 532 nm. Surface areas were determined by applying the Brunauer–Emmett–Teller (BET) method to nitrogen physisorption isotherms collected on a Micromeritics Gemini III 2375 surface area analyser. Prior to analysis of the surface area, roughly 0.05 g of the material was degassed at 110 °C overnight to remove any adsorbed moisture.
Acknowledgements
The authors thank EPSRC for funding this work under EP/J019208/1 and EP/J018384/1.References
- J. S. Bradley, O. Vollmer, R. Rovai, U. Specht and F. Lefebvre, Adv. Mater., 1998, 10, 938–942 CrossRef CAS.
- D. Farrusseng, K. Schlichte, B. Spliethoff, A. Wingen, S. Kaskel, J. S. Bradley and F. Schüth, Angew. Chem., Int. Ed., 2001, 40, 4204–4207 CrossRef CAS.
- I. Kurzina, F. J. Cadete Santos Aires, G. Bergeretand and J. C. Bertolini, Chem. Eng. J., 2005, 107, 45–53 CrossRef CAS.
- F. J. Cadete Santos Aires and J. C. Bertolini, Top. Catal., 2009, 52, 1492–1505 CrossRef CAS.
- K. Sardar, R. Bounds, M. Carravetta, G. Cutts, J. S. J. Hargreaves, A. L. Hector, J. A. Hriljac, W. Levason and F. Wilson, Dalton Trans., 2016, 45, 5765–5774 RSC.
- C. Balan, K. W. Völger, E. Kroke and R. Riedel, Macromolecules, 2000, 33, 3404–3408 CrossRef CAS.
- A. L. Hector, Chem. Soc. Rev., 2007, 36, 1745–1753 RSC.
- B. Mazumder and A. L. Hector, Top. Catal., 2009, 52, 1472–1481 CrossRef CAS.
- R. Rovai, C. W. Lehmann and J. S. Bradley, Angew. Chem., Int. Ed., 1999, 38, 2036–2038 CrossRef CAS.
- F. Cheng, S. J. Archibald, S. Clark, B. Toury, S. M. Kelly and J. S. Bradley, Chem. Mater., 2003, 15, 4651–4657 CrossRef CAS.
- A. L. Hector, Coord. Chem. Rev., 2016, 323, 120–137 CrossRef CAS.
- S. Hassan, A. L. Hector and A. Kalaji, J. Mater. Chem., 2011, 21, 6370–6374 RSC.
- F. Cheng, S. M. Kelly, S. Clark, J. S. Bradley, M. Baumbach and A. Schütze, J. Membr. Sci., 2006, 280, 530–535 CrossRef CAS.
- S. Hassan, A. L. Hector, J. R. Hyde, A. Kalaji and D. C. Smith, Chem. Commun., 2008, 5304–5306 RSC.
- V. Rocher, S. M. Kelly and A. L. Hector, Microporous Mesoporous Mater., 2012, 156, 196–201 CrossRef CAS.
- J. S. J. Hargreaves, Coord. Chem. Rev., 2013, 257, 2015–2031 CrossRef CAS.
- J. S. J. Hargreaves and D. Mckay, Catalysis, 2006, 19, 84–108 CAS.
- M. K. Neylon, S. Choi, H. Kwon, K. E. Curry and L. T. Thompson, Appl. Catal., A, 1999, 183, 253–263 CrossRef CAS.
- S. T. Oyama, J. Catal., 1992, 133, 358–369 CrossRef CAS.
- J.-G. Choi, M.-K. Jung, S. Choi, T.-K. Park, I. H. Kuk, J. H. Yoo, H. S. Park, H.-S. Lee, D.-H. Ahn and H. Chung, Bull. Chem. Soc. Jpn., 1997, 70, 993–996 CrossRef CAS.
- J.-G. Choi, J. Ha and J.-W. Hong, Appl. Catal., A, 1998, 168, 47–56 CrossRef CAS.
- M. K. Neylon, S. K. Bej, C. A. Bennett and L. T. Thompson, Appl. Catal., A, 2002, 232, 13–21 CrossRef CAS.
- P. Krawiec, P. Laura De Cola, R. Gläser, J. Weitkamp, C. Weidenthaler and S. Kaskel, Adv. Mater., 2006, 18, 505–508 CrossRef CAS.
- F. Cheng, S. M. Kelly, F. Lefebvre, S. Clark, R. Supplit and J. S. Bradley, J. Mater. Chem., 2005, 15, 772–777 RSC.
- S. Kaskel, G. Chaplais and K. Schlichte, Chem. Mater., 2005, 17, 181–185 CrossRef CAS.
- F. Cheng, S. M. Kelly, S. Clark, N. A. Young, S. J. Archibald and J. S. Bradley, Chem. Mater., 2005, 17, 5594–5602 CrossRef CAS.
- S. Hassan, M. Carravetta, A. L. Hector and L. A. Stebbings, J. Am. Ceram. Soc., 2010, 93, 1069–1073 CrossRef CAS.
- F. Cheng, S. M. Kelly, N. A. Young, S. Clark, M. G. Francesconi, F. Lefebvre and J. S. Bradley, Chem. Commun., 2005, 5662–5664 RSC.
- J. Löffelholz, J. Engering and M. Jansen, Z. Anorg. Allg. Chem., 2000, 626, 963–968 CrossRef.
- S. I. U. Shah and A. L. Hector, Top. Catal., 2012, 55, 950–954 CrossRef CAS.
- T. R. Choudhary, E. Aksoylu and D. W. Goodman, Catal. Rev. Sci. Eng., 2003, 45, 151–203 CAS.
- Inorganic Crystal Structure Database (Fiz Karlsruhe) accessed via the National Chemical Database Service hosted by the Royal Society of Chemistry.
- N. Bion, F. Can, J. Cook, J. S. J. Hargreaves, A. L. Hector, W. Levason, A. R. McFarlane, M. Richard and K. Sardar, Appl. Catal., A, 2015, 504, 44–50 CrossRef CAS.
- A. Alharthi, R. A. Blackley, T. H. Flowers, J. S. J. Hargreaves, I. D. Pulford, J. Wigzell and W. Zhou, J. Chem. Technol. Biotechnol., 2014, 89, 1317–1323 CrossRef CAS PubMed.
- J. Rouquerol, D. Avnir, C. W. Fairbridge, D. H. Everett, J. M. Haynes, N. Pernicone, J. D. Ramsay, K. S. W. Sing and K. K. Unger, Pure Appl. Chem., 1994, 66, 1739–1758 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- C. Schitco, M. S. Bazarjani, R. Riedel and A. Gurlo, J. Mater. Chem. A, 2015, 3, 805–818 CAS.
- S. Burns, J. G. Gallagher, J. S. J. Hargreaves and P. J. F. Harris, Catal. Lett., 2007, 116, 122–127 CrossRef CAS.
- M. Balakrishnan, V. S. Batra, J. S. J. Hargreaves, A. Monaghan, I. D. Pulford, J. L. Rico and S. Sushil, Green Chem., 2009, 11, 42–47 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7dt00285h |
| This journal is © The Royal Society of Chemistry 2017 |