
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailoring the local environment around metal ions: a solution chemical and structural study of some multidentate tripodal ligands†
Ferenc
Matyuska
a,
Attila
Szorcsik
b,
Nóra V.
May
c,
Ágnes
Dancs
a,
Éva
Kováts
d,
Attila
Bényei
e and
Tamás
Gajda
 *ab
*ab
aDepartment of Inorganic and Analytical Chemistry, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary. E-mail: gajda@chem.u-szeged.hu
bMTA-SZTE Bioinorganic Chemistry Research Group, Dóm tér 7, H-6720 Szeged, Hungary
cResearch Centre for Natural Sciences HAS, Magyar tudósok körútja 2, H-1117 Budapest, Hungary
dInstitute for Solid State Physics and Optics, Wigner Research Centre for Physics HAS, Konkoly Thege Miklós u. 29-33, H-1121 Budapest, Hungary
eDepartment of Pharmaceutical Chemistry, University of Debrecen, Egyetem tér 1, Debrecen H-4032, Hungary
First published on 6th June 2017
Abstract
Manganese(II), copper(II) and zinc(II) complexes of four polydentate tripodal ligands (tachpyr (N,N′,N′′-tris(2-pyridylmethyl)-cis,cis-1,3,5-triaminocyclohexane), trenpyr (tris[2-(2-pyridylmethyl)aminoethyl]amine, tach3pyr (N,N′,N′′-tris(3-pyridylmethyl)-cis,cis-1,3,5-triaminocyclohexane) and tren3pyr (tris[2-(2-pyridylmethyl)aminoethyl]amine)) were characterized in both solution and solid states. A combined evaluation of potentiometric, UV-VIS, NMR and EPR data allowed the conclusion of both thermodynamic and structural information about the complexes formed in solution. The four tailored polydentate tripodal ligands studied here exhibit a high thermodynamic stability, and a variety of coordination environments/geometries for the studied transition metal ions. Our data indicate that tachpyr is a more efficient zinc(II) chelator and a similar copper(II) chelator compared to trenpyr. Considering the higher number of N-donors and conformational flexibility of trenpyr, as well as the energy demanding switch to the triaxial conformation required for metal ion binding of tachpyr, the above observation is surprising and is very likely due to the encapsulating effect of the more rigid tachpyr skeleton. This relative binding preference of tachpyr for zinc(II) may be related to the observation that zinc(II) is one of the principal metals targeted by tachpyr in cells. In contrast, trenpyr is a considerably more efficient manganese(II) chelator, since it acts as a heptadentate ligand in the aqueous Mn(trenpyr) complex. The crystal structures of copper(II) and zinc(II) complexes of tachpyr indicated important differences in the ligand conformation, induced by the position of counter ions, as compared to earlier reports. The closely related new ligands, tach3pyr and tren3pyr, have been designed to form oligonuclear complexes. Indeed, we obtained a three dimensional polymer with a copper(II)/tren3pyr ratio of 11/6. Within this metal–organic framework, three distinctly different copper geometries can be identified: square pyramidal, trigonal bipyramidal and tetrahedral. Two square pyramidal and four trigonal bipyramidal copper centres create a hexanuclear subunit with a large inside cavity. These moieties are linked by tetrahedral copper(II) centres, constructing the three-dimensional polymer structure. The formation of such polynuclear complexes was not detected in solution. Both tach3pyr and tren3pyr form only mononuclear complexes with square pyramidal and trigonal bipyramidal geometries, respectively.
Introduction
The favourable and preorganized spatial distribution of donor atoms in tripodal ligands has been for a long time recognised1 and applied to develop for example, structural/functional models of metalloproteins2–6 or supramolecular assemblies.7 Polydentate (n > 4) tripodal ligands are generally synthesized by the derivatization of (tri/tetra)dentate tripodal platforms, such as cis,cis-1,3,5-triaminocyclohexane (tach), tris-(2-aminoethyl)amine (tren) or nitrilotriacetic acid (nta). Such modifications may provide additional donor site(s), may influence the steric environment around the metal centre or may help introduce further metal binding site(s) in order to create polynuclear complexes. Accordingly, these polydentate tripodal ligands are efficient metal sequestering agents,8 artificial receptors,9 metal ion sensors10 or artificial enzymes.11,12Tachpyr (N,N′,N′′-tris(2-pyridylmethyl)-cis,cis-1,3,5-triaminocyclohexane) and trenpyr (tris[2-(2-pyridylmethyl)aminoethyl]amine) are versatile polydentate tripodal ligands (Scheme 1). Tachpyr and trenpyr bind strongly to most transition metal ions.13–22 The Fe(II/III) and Mn(II/III) complexes of trenpyr are highly active SOD-mimicking compounds.13,14 Tachpyr and some of its derivatives are promising chelators of 64/67Cu for radiotherapeutic uses.23,24 Moreover, both tachpyr and trenpyr, as well as some of their methyl-substituted analogues, are cytotoxic metal chelators with potential anti-tumor activity.17,25–28 Tachpyr induces inhibition of ferritin synthesis,25 and triggers activation of CHK kinases, leading to cell-cycle arrest in G2, a radiosensitive phase of the cell cycle.26 The intracellular chelation of zinc as well as iron, but not copper, may play a fundamental role in the apoptosis induced by tachpyr and its derivatives. Accordingly, the apoptotic caspases 9 and 3 were blocked in cells pre-treated with either iron or zinc.27,28
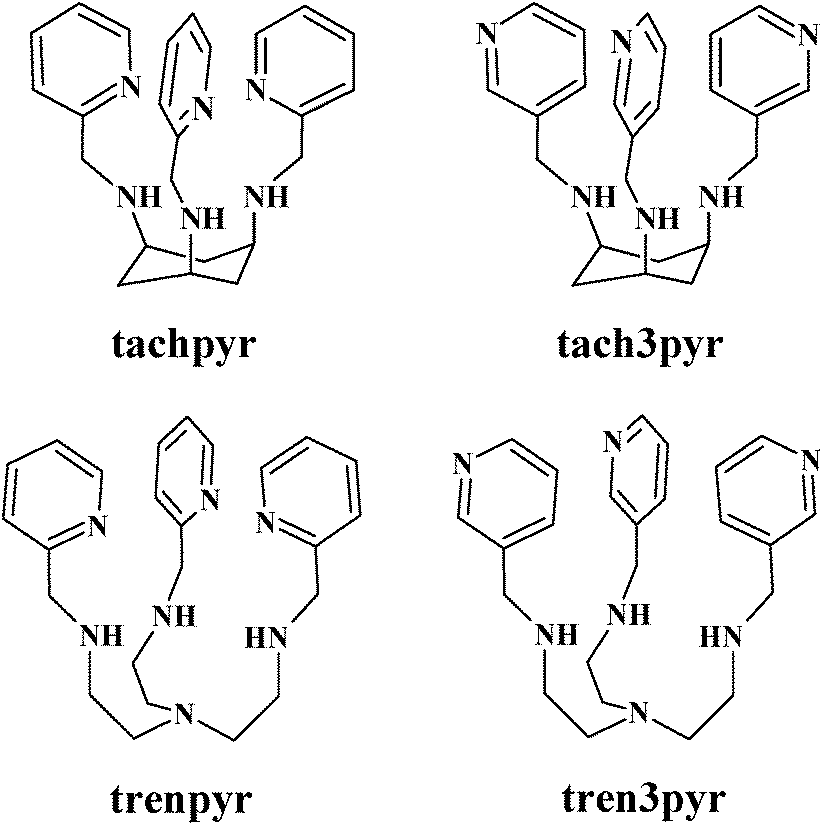 | ||
| Scheme 1 Schematic structures of the studied ligands. | ||
Although both tachpyr and trenpyr have been designed for efficient metal ion binding, and a number of crystal structures are available for their metal complexes,13–22 thermodynamic data on the stability of these complexes are very scarce. To our knowledge, no solution equilibrium or detailed solution structural study is reported for tachpyr, and in the case of trenpyr stability constants are available only for the Cu(II) and Zn(II) complexes at I = 1 M.20,21
Due to the favourable position of pyridine nitrogens of tachpyr and trenpyr, these ligands encapsulate the metal ions by fused chelate rings, which is probably one of the main reasons for their efficient metal binding ability. This encapsulating effect is not present in the case of 3-pyridylmethyl-substituted derivatives of tach and tren (tach3pyr and tren3pyr, see Scheme 1). Although the positions of pyridine nitrogens in these ligands are not appropriate for the coordination to metal ions already bound to the tripodal platform, it may be advantageous in the construction of oligonuclear complexes. Indeed, the position of the pyridine nitrogens in tach3pyr and tren3pyr may allow the formation of supramolecular structures or metal–organic frameworks. In addition, the steric and aromatic properties of pyridine rings of tach3pyr and tren3pyr can be also advantageous to develop enzyme mimics. The Cu(tach)(OH) complex is an efficient model of hydrolytic enzymes,29–32 but it readily transforms into the inactive dihydroxo-bridged [Cu2(tach)2(OH)2] dimer.29N-Alkylation of tach suppresses the formation of inactive dimer species, therefore these complexes efficiently promote the hydrolysis of both bis(4-nitrophenyl)phosphate (bnpp)33 and DNA.32 In the Cu(II)-tach3pyr system, the formation of the inactive dimer would be unfavourable, too, and the non-coordinating pyridine rings may enhance the substrate binding to the hydrolytically active species.
To this end, beside tachpyr and trenpyr, we also synthesized two new tripodal ligands (tach3pyr and tren3pyr, Scheme 1). Here, we report the characterization of their Mn(II), Cu(II) and Zn(II) complexes in both solution and solid states.
Materials and methods
Materials
All reagents were of analytical grade and used without further purification. A copper(II) perchlorate solution was prepared from analytically pure compounds obtained from Sigma-Aldrich and standardized complexometrically. A 0.1 M NaOH standard solution (Sigma) was used for pH titrations. The compounds tris-(2-aminoethyl)amine (98%), pyridine-2-carboxaldehyde (99%), pyridine-3-carboxaldehyde (98%) and cis,cis-l,3,5-cyclohexane-tricarboxylic acid (98%) were purchased from Sigma-Aldrich and TCI, respectively. The buffers 2-([N-morpholino]ethanesulfonic acid) (MES), N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES) and 2-(cyclohexylamino)-ethanesulfonic acid (CHES) were purchased from Sigma-Aldrich. Tach·3HBr34 and trenpyr14,20 were prepared as previously reported.Synthesis of N,N′,N′′-tris(2-pyridylmethyl)-1,3,5-cis,cis-triamino-cyclohexane (tachpyr, L1)
Tachpyr was prepared by a slight modification of the previously reported method.34 Tach(HBr)3 (1.032 g, 2.775 mmol) was dissolved in H2O (5 mL) with NaOH (0.333 g, 8.325 mmol) to form a clear solution. Benzene (100 mL) was added and the water was removed by azeotropic distillation with a Dean–Stark trap. Pyridine-2-carboxaldehyde (0.905 g, 8.45 mmol) was added during this process. After 24 h reflux the solvent was evaporated. The resulting white imine was dissolved in MeOH (40 mL) and NaBH4 (1.09 g, 29 mmol) was added in small portions. The reaction mixture was stirred for 24 h, then the solvent was removed by rotary evaporation. The residue was dissolved in CHCl3 (40 mL) and stirred vigorously with a 40 mL 5% aqueous NaHCO3 solution. The layers were separated, and the CHCl3 solution was further washed with saturated NaCl solutions (2 × 40 mL), then dried over Na2SO4. After the filtration, the organic phase was treated with dry HCl gas to form the hydrochloric salt of the ligand (tachpyr·4HCl). The product was washed with CHCl3 (60 mL) and dry Et2O (60 mL), (1.37 g, 92%). The purity was checked by 1H-NMR in 10%–90% D2O/H2O at pH = 3.25 (8.52, d, 3H; 7.88, t, 3H; 7.47, d, 3H; 7.43, t, 3H; 4.42, s, 6H; 3.50, t, 3H; 2.71, d, 3H; 1.77, dt, 3H, no other signals were detected, see Fig. S1 in the ESI†). (HR)ESI-MS m/z 403![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2534, calculated for C24H31N6 [M + H]+ = 403.2532.
2534, calculated for C24H31N6 [M + H]+ = 403.2532.
Synthesis of N,N′,N′′-tris(3-pyridylmethyl)-1,3,5-cis,cis-triamino-cyclohexane (tach3pyr, L2)
The synthesis was similar as described above for tachpyr. The ligand was obtained as a hydrochloride salt (tach3pyr·6HCl, yield 87%). Purity was checked by 1H-NMR in 10%–90% D2O/H2O at pH = 3.0 (8.90, s, 3H; 8.79, d, 3H; 8.60, d, 3H; 8.02, dd, 3H; 4.58, s, 6H; 3.68, t, 3H; 2.83, d, 3H; 1.84, q, 3H). (HR)ESI-MS m/z 4032608, calculated for C24H31N6 [M + H]+ = 403.2532.
Synthesis of tris[2-(3-pyridylmethyl)aminoethyl]amine (tren3pyr, L4)
Tren3pyr was prepared by a slight modification of the previously reported method for the synthesis of trenpyr.14,20 In brief, 349 mg (2.38 mmol) of tris-(2-aminoethyl)amine (tren) and 841 mg (7.85 mmol) of the corresponding aldehyde were mixed in 50 mL water-free methanol with 3 Å molecular sieves, and refluxed for 4 h. After cooling, 1.0 g (26.6 mmol) NaBH4 was added to the solution in small portions and stirred overnight. Then the methanol was evaporated, the crude product was dissolved in 50 mL chloroform, extracted with 3 × 20 mL 10% aqueous Na2CO3 solution, and dried over Na2SO4. After filtering, the filtrate was treated with dry HCl gas, which resulted in the precipitation of hydrochloride salts (tren3pyr·6HCl). Purity was checked by 1H-NMR in 10%/90% D2O/H2O at pH = 2.98 (8.74, s. 3H; 8.69, d, 3H; 8.28, d, 3H; 7.77, dd, 3H; 4.39, s, 6H; 3.24, t, 6H; 2.90, t, 6H, no other signals were detected, see Fig. S2 in the ESI†). (HR)ESI-MS m/z = 420.2715, calculated for C24H34N7 [M + H]+ = 420.2876.Potentiometric measurements
The protonation and coordination equilibria were investigated by potentiometric titrations in aqueous solution (I = 0.1 M NaCl, and T = 298.0 ± 0.1 K) under Ar using an automatic titration set including a PC controlled Dosimat 665 (Metrohm) autoburette and an Orion 710A precision digital pH-meter. The Metrohm Micro pH glass electrode (125 mm) was calibrated35via the modified Nernst equation (1):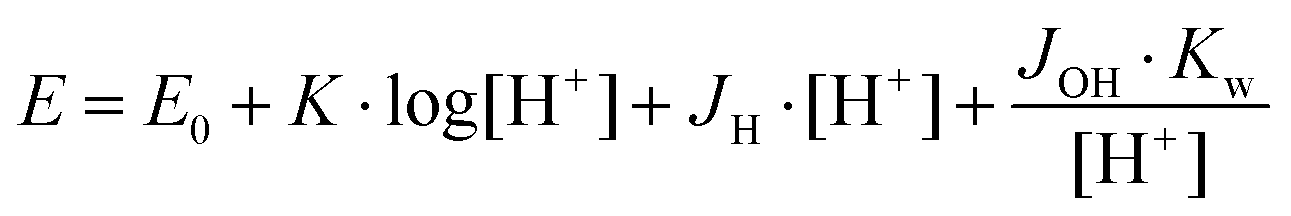 | (1) |
In a 60% (w/w) dimethylsulfoxide (dmso)–water mixture (I = 0.1 M NaCl, and T = 298.0 ± 0.1 K) the electrode system was calibrated to the pH = log[H+] scale by means of blank titrations (HCl versus NaOH), which is similar to the method suggested by Irving et al.37 in pure aqueous solutions. The obtained ionization constant of water under the conditions used pKw = 15.586 ± 0.05, which corresponds well to the literature data.38 The reproducibility of the titration points included in the calculations was within 0.005 pH units. In a 60% (w/w) DMSO–water mixture the copper(II)–tach3pyr 1/1 and zinc(II)–tach3pyr 1/1 systems were clear between pH 1.9–10 and pH 1.9–7.6, respectively.
The complex formation was described by a general equilibrium process as follows:
 | (2) |
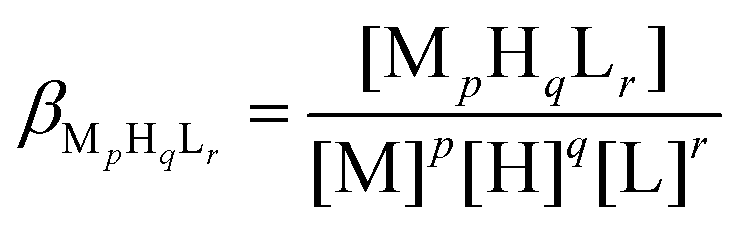 | (3) |
Using the above mentioned calibration protocol, the protonation and formation constants obtained in a 60% (w/w) dimethylsulfoxide (dmso)–water mixture are considered to be βmixed (or practical) constants40 and are valid only under the given conditions.
The protonation constants were determined from four independent titrations (90 data points per titration), with a ligand concentration of 2–3 × 10−3 M. The complex formation constants were evaluated from 5–7 independent titrations (∼90 data points per titration). The metal-to-ligand ratios were 3:2, 1:1, and 1:2. The metal ion concentrations varied between 1.0–2.9 × 10−3 M, depending on the metal-to-ligand ratio.
UV-Vis, EPR and NMR measurements
UV-Vis spectra were measured on Unicam Helios α or Thermo Scientific Evolution 200 spectrophotometers using a cell with a 1 cm optical pathlength. Similar concentrations were used as described above for the potentiometric titrations. The individual UV-Vis spectra of the complexes were calculated by PSEQUAD.39 The EPR spectra were recorded at room temperature and at 77 K using a BRUKER EleXsys E500 spectrometer (microwave frequency, 9.81 GHz; microwave power, 13 mW; modulation amplitude, 5 G; modulation frequency, 100 kHz). The EPR spectra were recorded in aqueous solution (or in 60% (w/w) dmso–water), and were simulated by a spectral decomposition algorithm.41 Since the copper(II) salt used to make the stock solution was a natural mixture of isotopes, the spectrum of each species was calculated as the sum of spectra containing 63Cu and 65Cu weighed by their natural abundance. The copper and ligand coupling constants are given in units of gauss (1 G = 10−4 T).The 1H NMR measurements were performed on a Bruker Avance DRX 500 spectrometer. The spectra were recorded at 25 °C in 10% D2O/H2O or 100% D2O solution, at ligand concentrations of ∼3.0 × 10−3 M with a tube diameter of 5 mm. The chemical shifts (δ) were measured relative to dioxane as an internal reference and converted to a SiMe4 reference using δdioxane = 3.70. Data were processed using the Topspin 2.0 software package (Bruker).
Hydrolysis of bis(p-nitrophenyl)phosphate (bnpp)
The reaction was performed in 60% (w/w) dmso–water (I = 0.1 M NaCl, T = 298 K). The increase in the absorbance maximum at 405 nm of the p-nitrophenolate anion (ε = 18900 M−1 cm−1) was monitored in a buffered solution (20 mM CHES or HEPES). The pK of p-nitrophenol was determined independently for our conditions (pK = 7.50 ± 0.05). The initial concentration of bnpp varied from 0.5 mM to 6.5 mM. The initial slope method (∼4% conversion) was used to determine the pseudo first-order rate constants. The reported data are the average of triplicate measurements (reproducibility ∼10%).
X-ray data collection, structure solution and refinement for compounds 1, 2, 3 and 4
Single crystals suitable for X-ray crystallography of compounds 1 and 2 were grown in water solution containing Zn(ClO4)2 and tachpyr in molar ratios of 3/2 and 1/1, respectively. The solutions also contained NaCl, and the pH was adjusted to pH = 8 by a NaOH solution. Colourless single crystals were mounted on loops and transferred to a goniometer. X-ray diffraction data were collected at room temperature (20 °C) on a Rigaku RAXIS-RAPID II diffractometer (using a Cu-Kα radiation for crystal 1 and Mo-Kα for crystal 2). For crystals 1 and 2 with a high volume a very careful multi-scan absorption correction was carried out using the program CrystalClear.423 was obtained from aqueous solution at pH 7 standing at room temperature for three months, and single crystal X-ray diffraction data were recorded using a Bruker-Nonius MACH3 diffractometer at room temperature (20 °C). A ψ scan absorption correction was applied,43 the number of ψ scan sets was 3. Theta correction and averaged transmission function were applied. For Fourier smoothing the window value was 5. Sir201444 and SHELXL45 under WinGX46 software were used for solution and refinement, respectively. The structures were solved by direct methods. The models were refined by full-matrix least squares on F2. A summary of data collection parameters is given in Table 1. In the structures 1 and 2 hydrogen atom positions were located in different electron density maps or placed in geometric positions. They were included in structure factor calculations but they were not refined. The isotropic displacement parameters of the hydrogen atoms were approximated from the U(eq) value of the atom they were bonded to. The refinement of non-hydrogen atoms was carried out with anisotropic temperature factors except for the disordered oxygen of water in structure 1 (for which the occupancy was set to 0.5 and hydrogen atoms were not defined) and ClO4 in structure 2.
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| Color/shape | Colourless/platelet | Colourless/chunk | Blue/block | Blue/block |
| Empirical formula | C24H30N6O9Cl2Zn | C24H30N6O4Cl2Zn | C24H32N6O9Cl2Cu | C156H246N42O12Cl22Cu11 |
| Moiety formula | [Zn(C24H30N6)](ClO4)2(O) | [Zn(C24H30N6)](Cl)(ClO4) | [Cu(C24H30N6)](ClO4)2(H2O) | [Cu11(C24H33N7)6(Cl)11](Cl)11(C2H6O)6(H2O)6 |
| Formula weight | 682.81 | 602.81 | 682.99 | 4380.77 |
| Temperature (K) | 293(2) | 293(2) | 293(2) | 298(2) |
| Wavelength (Å) | 1.54178 | 0.71073 | 0.71073 Å | 1.54178 |
| Crystal system | Monoclinic | Trigonal | Monoclinic | Cubic |
| Space group | P21/n |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c c |
P21/n | I23 |
| Unit cell dimensions | 8.5454(2), 14.1957(3), 24.2629(6) | 13.9000(5), 13.9000(5), 45.0290(14) | 8.5315(16), 14.200(6), 24.011(6) | 26.8501(1) |
| a, b, c (Å) | ||||
| α, β, γ (°) | 90, 100.298(1), 90 | 90, 90, 120 | 90, 99.230(10), 90 | 90, 90, 90 |
| Volume (Å3) | 2895.87(12) | 7534.5(6) | 2871.3(15) | 19357.0(2) |
| Z/Z′ | 4/1 | 12/1 | 4/1 | 4/0.1667 |
| Density (calc.) (Mg m−3) | 1.566 | 1.594 | 1.580 | 1.503 |
| Absorption coeff. (mm−1) | 3.415 | 1.236 | 1.009 | 4.60 |
| F(000) | 1408 | 3744 | 1412 | 9060 |
| Crystal size (mm) | 0.50 × 0.30 × 0.05 | 0.25 × 0.25 × 0.20 | 0.55 × 0.45 × 0.15 | 0.30 × 0.26 × 0.25 |
| Diffractometer | Rigaku RAXIS-RAPID II | Rigaku RAXIS-RAPID II | Enraf Nonius MACH3 | Rigaku-Oxford SuperNova |
| Absorption correction | Multi-scan | Multi-scan | ψ scan | Multi-scan |
| Min. and max. transmission | 0.5159, 1.0000 | 0.8646, 1.0000 | 0.736, 0.863 | 0.63, 1.0000 |
| θ angle for data collection (°) | 6.506 ≤ θ ≤ 58.920 | 3.230 ≤ θ ≤ 27.472 | 2.694 ≤ θ ≤ 25.975 | 3.29 ≤ θ ≤ 67.68 |
| Index ranges | −9 ≤ h ≤ 8, −15 ≤ k ≤ 15, −24 ≤ l ≤ 26 | −18 ≤ h ≤ 18, −18 ≤ k ≤ 18, −58 ≤ l ≤ 58 | 0 ≤ h ≤ 10; −7 ≤ k ≤ 17, −29 ≤ l ≤ 29 | −33 ≤ h ≤ 33, −31 ≤ k ≤ 133, −32 ≤ l ≤ 24 |
| No. of reflections collected | 20821 |
72365 |
8665 | 73532 |
| Completeness to 2θ | 0.977 | 0.997 | 0.995 | 0.995 |
| No. of independent reflections (Rint) | 4076 (0.0455) | 1924 (0.0377) | 5615 (0.0325) | 6505 (0.03) |
| Reflections I > 2σ(I) | 3202 | 1614 | 4436 | 6034 |
| Refinement method | Full-matrix least-squares on F2 | |||
| Data/restraints/parameters | 4076/7/406 | 1924/0/107 | 5615/5/394 | 6505/5/381 |
| Goodness-of-fit on F2 | 1.148 | 1.148 | 1.060 | 1.09 |
| Final R indices [I > 2σ(I)]R1, wR2 | 0.0493, 0.1262 | 0.0531, 0.1697 | 0.0875, 0.2192 | 0.0837 |
| R indices (all data) R1, wR2 | 0.0635, 0.1537 | 0.0595, 0.1812 | 0.1049, 0.2353 | 0.0866, 0.2415 |
| Max. and mean shift/esd | 0.006, 0.000 | 0.000, 0.000 | 0.001;0.000 | 2.593, 0.02 |
| Largest diff. peak and hole (e Å−3) | 0.415 and −0.369 | 0.711 and −1.145 | 1.187 and −0.852 | 1.69 and −1.81 |
Suitable single crystals of 4 were prepared by dissolving the precipitate formed in 0.1 M aqueous NaCl solution (at a [Cu(II)]/[tren3pyr] ratio 2/1 and at pH 4–5) in ethanol, and allowed for slow evaporation of the solvent for several weeks at room temperature. X-ray diffraction data were collected on a Rigaku-Oxford Diffraction SuperNova Dual source diffractometer equipped with an Atlas detector using Cu–Kα radiation. For data collection and absorption correction the CrysAlis software was used.47 The summary of data collection and refinement parameters is also collected in Table 1.
Selected bond lengths and angles of compounds were calculated by PLATON software.48 The graphical representation and the edition of CIF files were done by the Mercury49 and PublCif50 software, respectively.
Results and discussion
Crystal structures of tachpyr complexes
The crystal structure of [Zn(tachpyr)]·(ClO4)2·(CH3OH) has been reported (CSD ref. code DOSVAI) by Planalp and co-workers18 and is crystallized in the cubic space group P213 with unit cell dimensions a, b, c = 14.3088 Å. The ZnN6 coordination sphere was described as a distorted octahedron. In this study, two new crystal forms of the [Zn(tachpyr)] complex cation were crystallized and their structure determined: [Zn(tachpyr)]·(ClO4)2·O (1) crystallized in a monoclinic P21/n space group and [Zn(tachpyr)]·(ClO4)·Cl (2) in the trigonal crystal system, crystallized in the Rc space group. The copper(II) complex [Cu(tachpyr)]·(ClO4)2·H2O (3) crystallized in the P21/n space group (Table 1), which was found to be isostructural with the known crystal structure of [Cu(tachpyr)]·(ClO4)2·0.5(C2H3N) (unit cell a/b/c = 8.4921/14.3418/23.9735 Å and β = 99.874°) for which the CSD ref. code is CAFXEN.19Fig. 1 depicts the ORTEP representation of the complex cations from 1, 2 and 3. In all cases, the coordination sphere of metal ions is best described as a distorted octahedron.
 | ||
| Fig. 1 Molecular structures of the metal complex cations of 1 (with the indication of rings A–G), 2 and 3 (from left to right). Displacement parameters are drawn at the 30% probability level and hydrogens are omitted for clarity. | ||
The different counter ions affected not only the arrangement of the crystal lattice but also the conformation of the tachpyr molecule around the metal ions. In the crystals of 1, 3 and CAFXEN, the three 2-pyridylmethyl arms have different conformation. In contrast, in 2 and DOSVAI, the three-fold rotation axis goes through the Zn(II) ion and the asymmetric unit contains one pyridine arm of the ligand. Though crystal 1 contains a Zn(II) ion, its conformation is much more similar to the two copper(II) complexes (3 and CAFXEN, see Fig. S3†) than the other two zinc(II) complexes (2 and DOSVAI). Fig. 2 shows overlaid structures of the metal complex cations of 1, 2, and 3 together with DOSVAI18 and CAFXEN.19
 | ||
| Fig. 2 Conformational comparison for the metal complex cations [Zn(tachpyr)] in crystal structures 1 (colored by element), 2 (blue), and DOSVAI18 (pink), and [Cu(tachpyr)] in crystal structures 3 (yellow) and CAFXEN19 (green). The twist angle defined for the coordination sphere of [M(tachpyr)] complexes18 is also shown. | ||
For conformational comparison, the root mean square of the distances between the pair of atoms (RMSD) and maximum difference (max.D) was calculated by using the Mercury program.49 The closest geometrical similarity is between 3 and CAFXEN (RMSD = 0.042 and max.D = 0.089), but high similarities are between 1/3 (RMSD = 0.124 and max.D = 0.220) and 2/DOSVAI (RMSD = 0.172 and max.D = 0.267), too. Despite the conformational and cell similarities (cell similarity index51,52 = 0.00566), the relatively low isostructurality index51,52 (68.4%) suggests that the position of the complexes is slightly shifted in 3 as compared to CAFXEN. The cell similarity and isostructurality indices are somewhat higher for structures 1/3 (0.0072 and 82.1%, respectively). The calculation of these indices can be found in the legend of Fig. S3.†
For comparison, some selected bond lengths and angles together with angles between different ring planes are collected in Table 2. The asymmetric pyridine arms resulted in three significantly different Zn–Npyr bond distances and Npyr–Zn–Npyr angles for [Zn(tachpyr)] in crystal 1. Similarly, different angles can be measured between the ring planes (Table 2) in this complex. The twist angle (α) (Fig. 2) defined for the comparison of a [M(tachpyr)] coordination sphere15,18 also shows significant difference in the conformation of the three arms. Though in crystal 2 and DOSVAI the [Zn(tachpyr)] complex has the same C3 symmetry, the Zn–Npyr bond distance is significantly longer and the twist angle is smaller for 2 than for DOSVAI (Table 2 and Fig. 1). The positions of the counter ions around the metal complexes are accounted for the conformational differences. In crystal 2 the perchlorate anions positioned exactly on the C3 axis, above the pyridine rings of tachpyr, however in DOSVAI they localized aside of the pyridine rings (Fig. S4 and S5†). In crystals 1, 3 and CAFXEN the perchlorate anions are in different hydrogen bond connections with the three pyridine arms (Fig. S5†). Bond distances and angles of the determining hydrogen bonds are collected in Table S1.†
| [Zn(tachpyr)] | [Cu(tachpyr)] | ||||
|---|---|---|---|---|---|
| (1) | (2) | DOSVAI18 | (3) | CAFXEN19 | |
| a Angles between planes calculated for the rings A–G drawn in Fig. 1. | |||||
| M–Ntach (Å) | 2.164(3) | 2.157(3) | 2.160(3) | 2.022(5) | 2.033(5) |
| 2.174(4) | 2.223(5) | 2.245(5) | |||
| 2.158(4) | 2.105(5) | 2.099(5) | |||
| M–Npyr (Å) | 2.160(4) | 2.228(3) | 2.165(4) | 2.061(5) | 2.053(5) |
| 2.137(3) | 2.031(5) | 2.032(5) | |||
| 2.219(4) | 2.438(5) | 2.458(5) | |||
| Ntach–M–Npyr (°) | 78.7(1) | 77.3(1) | 79.01(14) | 81.8(2) | 81.9(2) |
| 79.2(1) | 76.9(2) | 77.4(2) | |||
| 78.5(1) | 80.3(2) | 79.8(2) | |||
| Ntach–M–Ntach (°) | 87.1(1) | 88.9(1) | 90.21(14) | 89.2(2) | 89.1(2) |
| 90.1(1) | 91.7(2) | 90.6(2) | |||
| 91.0(1) | 92.5(2) | 93.0(2) | |||
| Npyr–M–Npyr (°) | 97.1(1) | 92.95(9) | 93.97(14) | 97.4(2) | 97.2(2) |
| 102.9(1) | 99.9(2) | 100.5(2) | |||
| 89.9(1) | 86.6(2) | 86.5(2) | |||
| Cg(A)–Cg(B), Cg(A)–Cg(C), Cg(A)–Cg(D) (°)a | 69.6(2) | 68.78(15) | 63.44(14) | 72.7(3) | 73.1(2) |
| 58.5(2) | 55.2(3) | 55.9(2) | |||
| 53.6(2) | 51.2(3) | 50.7(2) | |||
| Cg(B)–Cg(E), Cg(C)–Cg(F), Cg(D)–Cg(G) (°)a | 13.8(2) | 10.83(18) | 9.85(14) | 13.9(3) | 13.3(2) |
| 1.0(2) | 1.1(3) | 2.5(2) | |||
| 14.0(2) | 20.1(3) | 21.3(2) | |||
| Twist angle α (°) | 49.1(2) | 36.8(2) | 43.7(2) | 49.3(3) | 49.8(2) |
| 43.6(2) | 44.9(3) | 44.1(2) | |||
| 44.9(2) | 47.3(3) | 47.5(2) | |||
All compounds were racemates. Structures 1, 2, 3 and CAFXEN have a centrosymmetric space group and the relative configurations of N1, N2 and N3 are S,S,S. Structure DOSVAI is in a chiral space group and in the deposited structure the secondary amines have the chirality R,R,R. For comparison, the structure of 4 (see later) was solved and refined well in a non-centrosymmetric space group as an inversion twin, and the relative configuration of the secondary nitrogen atoms was found to be S,S,R.
Crystal structure of [Cu11(tren3pyr)6Cl11](Cl)11·6H2O·6EtOH (4)
In the solid state tren3pyr forms a 3D metal–organic framework (MOF) with copper(II). This is the first published crystal structure with the novel tren3pyr ligand. The cubic structure is based on a distinct copper(II) containing secondary building units (SBU) in the nodes and (3-pyridylmethyl)amino groups at the edges forming a high symmetry MOF. The formation of this MOF is explained by the strong metal binding ability of all nitrogens of tren3pyr, and by the position of pyridine nitrogens, which are not able to form chelate rings with the secondary/tertiary amines. In this way, all tren3pyr molecules are bound to four different copper(II) ions (Fig. 3a and 4b). This structural feature results in the formation of an infinite (Cu11L6)n polymer, in which each copper(II) has a coordinated chloride, and the counter ions are also chlorides. Two of these polymers create an interpenetrated network in the crystal structure. Beside the rigid framework, more than 20% of the unit cell volume consists of separated voids (Fig. S6†). In these cavities the remaining electron density could be well modelled by chloride ions, solvent ethanol and water molecules hence the void volume of the unit cell is 1.7% and 326 Å3 in the refined complete structure. | ||
| Fig. 3 Structure of the three types of Cu(II) centres in 4 and their immediate environments (type 1 (a), type 2 (b) and type 3 (c)). Displacement parameters are drawn at the 30% probability level, and hydrogen atoms are omitted for clarity. | ||
 | ||
| Fig. 4 Top (a) and side view (b) of the Cu6(tren3pyr)4 subunit in 4 with a large inside cavity. For clarity, the third 3-pyridylmethyl leg is shown only for one of the ligand molecules, together with a type 3 copper, which connects the hexanuclear subunits. Displacement parameters are drawn at the 30% probability level, hydrogen atoms, solvent water and ethanol molecules as well as chloride counter ions are also omitted for clarity. | ||
Within this metal–organic framework three distinctly different copper centres or SBUs can be identified. The first one has a slightly distorted trigonal bipyramidal geometry (τ = 0.885), and this type of copper is bound to all tren-like moieties (Fig. 3a). The second has a nearly perfect square pyramidal geometry (τ = 0.053) with four pyridine nitrogens in its basal plane (Fig. 3b). The distance of the metal ion from the mean plane of the four nitrogens is 0.3 Å. All pyridine rings originated from different ligand molecules, i.e. this copper(II) centre connects four type 1 SBUs, and is therefore responsible for the formation of the hexanuclear subunits with a large inside cavity (Fig. 4 and Fig. S6†). The third type of copper centre has a distorted tetrahedral geometry (Fig. 3c) constituted by three pyridine nitrogens and a chloride ion. The distances between the metal ion and the centroid of the tetragon to the N3 plane are 0.4 and 0.7 Å, respectively. The three coordinated pyridine rings belong to three different hexanuclear subunits, consequently these copper centres create the 3D MOF. Two of these 3D polymers are interlaced and form an interpenetrated network with a special topology in the crystal structure (Fig. 6S†).
Altogether the unit cell (Fig. S6†) contains 24 tren3pyr ligands with 168 nitrogen donors, all of them are coordinated, and 44 copper(II) ions (24, 12 and 8 of type 1, 2 and 3, respectively). Selected data for the coordination are shown in Table 3, some of the values are equal because of the lattice symmetry. The shortest Cu⋯Cu distances are 6.796 Å. Several mononuclear structures related to type 1 and 2 copper(II) centres can be found in the literature,53,54 but only copper(I) complexes55 are reported to have a similar coordination sphere to type 3 SBU. The structures of individual SBUs are close to their mononuclear counterparts,53–55i.e. despite the extensive coordination connectivity within this MOF (Fig. 4) the whole structure is surprisingly unstrained.
| Type 1 (tbp) | Type 2 (sp) | Type 3 (Td) | |
|---|---|---|---|
| Cu–Ntertiary (Å) | 2.021(4) | — | — |
| Cu–Nsecondary (Å) | 2.132(6), 2.125(8) | — | — |
| 2.131(8) | |||
| Cu–Npyridine (Å) | — | 2.051(8), 2.037(8) | 2.109(13) |
| 2.051(8), 2.037(8) | |||
| Cu–Cl (Å) | 2.2159(15) | 2.479(2) | 2.443(11) |
| N–Cu–N (°) | 85.0(2), 83.7(3) 84.8(3), 109.6(5) | 88.0(2), 89.6(2) | 116.1(2) |
| 122.1(5), 125.5(3) | 161.5(4), 164.7(6) | ||
| Cl–Cu–N (°) | 178.6(2), 94.9(2), 95.0(2), 96.3(2) | 97.6(2), 99.3(2) | 101.6(3) |
The solid state structure is further stabilized by weak C–H⋯Cl and strong N–H⋯Cl or O–H⋯Cl hydrogen bonds involving solvent molecules and chloride counter ions, too. The large cavity in the structure caused uncertainty in the coordinates of the solvent water molecules and one of the chloride ions leading to errors in the CIF but the overall structure is considered to be correct. Data collection was performed both at room temperature and 100 K, but no improvement in the refinement could be reached using the low temperature dataset, because of considerable disorder of the solvent molecules in the voids.
Solution chemical studies
| logβpqra |
tachpyr | tach3pyr [in 60% w/w dmso–H2O] | tachb | trenpyr [in 1 M KNO3]c | tren3pyr | trend |
|---|---|---|---|---|---|---|
| a p, q and r are the stoichiometric numbers of metal ions, protons and ligands, respectively, in the given species. b In 0.1 M NaClO4.29 c Ref. 21. d In 0.1 M NaClO4.56 | ||||||
| logβ011 |
8.48(1) | 8.36(1) [8.05(1)] | 10.21 | 8.92(1) [9.12] | 8.63(1) | 10.12 |
| logβ021 |
15.27(1) | 15.01(1) [14.47(2)] | 18.88 | 16.70(1) [17.26] | 16.20(1) | 19.53 |
| logβ031 |
20.54(1) | 20.20(3) [19.44(4)] | 25.93 | 23.04(1) [24.17] | 22.60(2) | 27.95 |
| logβ041 |
22.40(6) | 23.84(5) [22.15(10)] | 25.21(3) [26.67] | 26.34(3) | ||
| logβ051 |
26.89(5) [23.95(20)] | — [27.8] | 29.55(4) | |||
| logβ061 |
29.40(7) [25.85(25)] | 32.24(5) | ||||
| pqr | tachpyr | tach3pyr | tach | |||||
|---|---|---|---|---|---|---|---|---|
| Cu(II) | Zn(II) | Mn(II) | Cu(II) [in 60% w/w dmso–H2O] | Zn(II) [in 60% w/w dmso–H2O] | Mn(II) | Cu(II)b | Zn(II)c | |
| 111 | 21.8(2)a | 18.02(5) | 11.97(6) | 11.75(1) [12.01(9)] | 10.30(6) [10.54(2)] | 10.26(7) | 15.95 | |
| 101 | 20.0(2)a | 15.18(3) | 6.47(5) | 5.26(1) [7.49(4)] | 3.15(5) [3.87(1)] | 2.34(5) | 10.86 | 6.95 |
| 1−11 | [−0.43(6)] | 2.36 | ||||||
| 1−20 | [−10.11(7)] | |||||||
| 2−22 | 8.48 | |||||||
| pqr | Trenpyr | tren3pyr | Tren | |||||
|---|---|---|---|---|---|---|---|---|
| Cu(II) [in 1 M KNO3]d | Zn(II) [in 1 M KNO3]f | Mn(II) | Cu(II) | Zn(II) | Mn(II) | Cu(II)g | Zn(II) | |
|
a Determined from the spectrophotometric data.
b In 0.1 M NaClO4.29
c In 0.1 M NaClO4.57
d
Ref. 20.
e Determined from both the spectrophotometric data (logβ111 = 26.7 ± 0.1, logβ101 = 21.2 ± 0.1) as well as the ligand displacement and pH-metric study (logβ111 = 26.8 ± 0.1, logβ101 = 21.2 ± 0.1).
f
Ref. 21.
g In 0.1 M NaClO4.56
h In 1 M NaClO4.58
|
||||||||
| 131 | 27.35(3) | |||||||
| 121 | 23.10(6) [23.92] | 23.56(3) | ||||||
| 111 | 26.75(15)e [27.7] | 20.81(3) [21.38] | 15.56(3) | 19.39(3) | 14.07(3) | |||
| 101 | 21.2(1)e [21.4] | 15.52(3) [15.62] | 10.48(2) | 14.63(3) | 8.96(1) | 18.50 | 14.50g | |
| 1−11 | 6.22(3) | −0.38(2) | 9.33 | 4.10h | ||||
The formation constants of the Cu(Htachpyr) and Cu(tachpyr) complexes (Table 5) were determined by pH-dependent spectrophotometric titration (Fig. 5), allowing 6 days for equilibration. The estimated errors of the formation constants were increased ±0.2 due to the uncertainties induced by I > 0.1 M at the acidic end of the titration. The speciation of the Cu(Htachpyr) complex shows a maximum around pH 1.7, while Cu(tachpyr) is the sole species in the solution above pH 4 (Fig. 6). The pH-dependent Vis/NIR and EPR spectra of the copper(II)–tachpyr system are depicted in Fig. 6 and 7. In Cu(Htachpyz) one of the secondary amino groups is still protonated, i.e. the metal ion is either 4N or 5N coordinated. The Vis/NIR spectrum detected around pH 1.7 (Fig. 5) shows a transition at 620 nm with a relatively strong low-energy shoulder at 840 nm, indicating a square pyramidal geometry with a strong apical coordination, similarly to the copper(II) complexes of several related tach derivatives.12,17,59
 | ||
| Fig. 5 Effect of pH on the Vis/NIR spectra of the copper(II)–tachpyr systems (T = 298 K, I = 0.1 M NaCl, [Cu2+] = [tachpyr] = 0.00226 M, the blue spectrum corresponds to CuL, the red and green ones mostly to CuHL and Cu2+(aq)). The insert shows the changes of absorbance at 620 nm. | ||
 | ||
| Fig. 6 Speciation diagram of the M(II)–tachpyr complexes (T = 298 K, I = 0.1 M NaCl, [M2+] = [tachpyr] = 0.002 M, the curves of the uncomplexed metal ions have been omitted for clarity). | ||
 | ||
| Fig. 7 Experimental (black) and simulated (red) EPR spectra of the copper(II)–tachpyr system at room temperature (A) and at 77 K (B) ([Cu2+] = [tachpyr] = 0.003 M). The calculated component spectra of the main species are also shown on the top. At pH = 6.05 the solution was not fully equilibrated and contained ∼20% Cu(Htachpyr) complex, too. | ||
The EPR spectrum recorded in acidic solution (Fig. 7, Table 6) is also consistent with a distorted square pyramidal structure. The basicity corrected formation constants of the Cu(Htachpyr) and Cu(Htach) complexes allow an approximate comparison of their stability (logβ* = logβ111 − (logβ01(4/3) − logβ011) = 8.08 and 0.23 for tachpyr and tach, respectively, Table 5). Considering the nearly eight orders of magnitude difference, the Cu(Htachpyz) complex is very likely 5N coordinated.
| Complex | g o | A o (G) | g ⊥ or gx, gy | g ‖ or gz | A ⊥ or Ax,Ay (G) | A ‖ or Az (G) |
|---|---|---|---|---|---|---|
| a See the text. | ||||||
| Cu(II)–tachpyr | ||||||
| CuHL | 2.1065(4) | 66.1(5) | 2.0439(3) | 2.2082(1) | 25.0(4) | 167.2(1) |
| 2.0662(2) | 52.8(3) | |||||
| CuL | 2.1141(8) | 58(1) | 2.0745(1) | 2.2455(2) | 35.2(2) | 146.4(2) |
| Cu(II)–tach3pyr | ||||||
| CuL | 2.1144(4) | 51.5(3) | 2.0442(5) | 2.2555(3) | 37.7(9) | 147.2(3) |
| 2.0824(8) | 38.7(6) | |||||
| CuL(OH) | 2.1207(2) | 58.2(2) | 2.0593(1) | 2.2546(1) | 35.7(1) | 148.8(1) |
| CuL(OH)2 | 2.1222(4) | 60.2(4) | 2.0527(1) | 2.2532(1) | 20.8(1) | 157.7(1) |
| Cu(II)–tren3pyr | ||||||
| CuH4L | — | — | 2.060(2) | 2.424(1) | 15(2) | 130(1) |
| CuH3L | 2.1272(5) | 59.1(3) | 2.182(2) | 2.029(1) | 101(2) | 80(2) |
| CuH2L | 2.144(2) | 71(2) | ||||
| CuHL | 2.1272(5) | 59.1(3) | 2.188(2) | 2.020(1) | 103(2) | 72(1) |
| CuL | 2.145(2) | 77(2) | ||||
| CuL(OH) | 2.1329(4) | 40.3(3) | 2.187(2) | 1.998(1) | 90(2) | 78(1) |
| Isotropic componenta | — | — | 2.113(2) | 20(2) | ||
The formation of Cu(tachpyr) results in a red shift of d–d bands (620 nm → 650 nm, 840 nm → 940 nm), indicating an even stronger coordination along the z-axis.60 This is also confirmed by the nearly 13 orders of magnitude difference in the basicity corrected formation constants of Cu(tachpyr) and Cu(tach) (logβ* = logβ101 − logβ01(4/3) = −2.3 and −15.07 for tachpyr and tach, respectively, Table 5). The complex Cu(tachpyr) has higher g and smaller A values as compared to its protonated form (Table 6), which also suggests stronger axial binding, i.e. 6N coordination in a distorted octahedral environment, as was observed in the crystal structure of complex 3. The broad room temperature spectra of this complex (Fig. 7A) are probably due to the dynamic Jahn–Teller effect, which is known to operate in 6N-coordinated copper(II) complexes.61
In the zinc(II)–tachpyr system two complexes can be identified too (Fig. 6). The species Zn(Htachpyr) is only a minor complex around pH 3, and Zn(tachpyr) is a dominant one above pH 5, which indicates high thermodynamic stability. Indeed, its basicity corrected stability constant is 12 orders of magnitude higher than that of the corresponding Zn(tach) complex (Tables 4 and 5). The ligand exchange process of Zn(tachpyr) is slow on the NMR time-scale (Fig. 8). The important differences in the spin coupling pattern between the free and bound ligands (considerably decreased coupling constants between the CH and CH2 protons of the cyclohexane ring, which results in a broad singlet and two doublets, respectively), clearly show the switch between the two chair conformations (triequatorial NH → triaxial NH).62 Although the triaxial conformer is energetically unfavoured in the free ligand, it is the preferred one in metal complexes of all tach derivatives.15–19,63 In accordance with earlier reports,15 the single set of signals observed for this slow exchanging species indicate a C3 symmetry, the chemical inequivalence of benzylic protons (∼4.16 ppm) shows conformational rigidity even within the 2-pyridylmethyl arms. All these facts, together with the significant chemical shift difference of protons close to the nitrogen atoms in the bound and unbound ligands (Fig. 9) suggest tight 6N-coordination around zinc(II), as seen in the crystal structures of 1 and 2.
 | ||
| Fig. 8 The pH-dependence of 1H-NMR spectra of tachpyr in the presence of zinc(II) ([Zn2+] = 0.002 M; [tachpyr] = 0.003 M; squares: aromatic protons next to Npyr; circles: benzylic protons; rhombus: CH of a cyclohexane ring; triangles: inequivalent CH2 of a cyclohexane ring of the free (open) and bound (filled) ligands). | ||
 | ||
| Fig. 9 Effect of pH on the Vis/NIR spectra of the copper(II)–trenpyr systems (T = 298 K, I = 0.1 M NaCl, [Cu2+] = [tachpyr] = 0.00184, the blue and red spectra correspond to CuL and CuHL, respectively) The insert show the changes of absorbance at 640 nm. | ||
An earlier RP-HPLC study on the aqueous Mn(II)–tachpyr system indicated nearly complete dissociation at pH 5.5.15 In agreement with this finding, our potentiometric data indicated that the formation of Mn(Htachpyr) and Mn(tachpyr) started at pH 4.6 (at metal ion and ligand concentrations of 2 mM), and the latter species is the sole complex only above pH 8 (Fig. 6). Consequently, the Mn(tachpyr) complex is considerably less stable than the corresponding copper(II) or zinc(II) species (Δlogβ101 = 13.21 and 8.66, respectively). This is in accordance with the Irwing–Williams series, although the complete transformation of Mn(II)(aq) into Mn(tachpyr) at pH 8 in equimolar solution confirms the high metal ion sequestering ability of tachpyr.
The formation constants of copper(II)– and zinc(II)–trenpyr complexes have been already published for the 1 M KNO3 background electrolyte.20,21 In order to have comparable data with other formation constants reported in this work, we repeated the solution equilibrium study of these systems in 0.1 M NaCl solutions, and complemented it by pH-dependent Vis/NIR and 1H NMR studies. The formation constants of the Cu(Htrenpyr) and Cu(trenpyr) complexes (Table 5) were determined by pH-dependent spectrophotometric titration (Fig. 9). Since slow kinetics, comparable with the Cu(II)-tachpyr system, was not observed, the stability of the Cu(trenpyr) complex was also determined by the ligand displacement method at pH 10 using diethylenetriamine pentaacetic acid (dtpa) (Fig. S7†). The formation constants determined by the two methods are in agreement with each other within ±0.1 log units (Table 5).
In our background electrolyte, too, Cu(Htrenpyr) and Cu(trenpyr) are the unique species between pH 2–4 and 7–11, respectively (Fig. 10). Taking into account the ionic strength difference, our formation constants (Table 5) are in good agreement with those reported earlier.21 The Vis/NIR spectrum of Cu(Htrenpyr) is close to that of Cu(Htachpyr), including the low energy transitions around 850 nm, indicating a square pyramidal geometry in both species. On the other hand, the spectrum of Cu(trenpyr) (λd–dmax = 650 and 720 nm with nearly equal intensities, Fig. 9) is completely different from that of the 6N coordinated Cu(tachpyr), and indicates an intermediate geometry between square pyramidal and trigonal bipyramidal.64,65
 | ||
| Fig. 10 Speciation diagram of the M(II)–trenpyr complexes (T = 298 K, I = 0.1 M NaCl, [M2+] = [trenpyr] = 0.002 M, the curves of the uncomplexed metal ions have been deleted for clarity). | ||
In the presence of zinc(II), the formation of Zn(H2trenpyr), Zn(Htrenpyr) and Zn(trenpyr) was detected, similarly to the earlier report.20 Zn(Htrenpyr) is a dominant species at pH 4, its deprotonation is complete at pH 7. The 1H NMR data revealed a slow ligand exchange on the NMR time-scale for both Zn(Htrenpyr) and Zn(trenpyr) complexes (Fig. 11). The proton signals, especially the methylene protons of Zn(Htrenpyr), are considerably broadened. Since the signals of the free ligand are sharp in the Zn(II)–trenpyr system at any pH, the observed broadening for Zn(Htrenpyr) is due to the conformational changes, likely induced by intramolecular proton-transfer processes, i.e. the proton ‘hops’ between the secondary nitrogens.
 | ||
| Fig. 11 The pH-dependence of 1H-NMR spectra of trenpyr in the absence and presence of zinc(II) ([Zn2+] = 0.002 mM, [tachpyr] = 0.0024 M, 298 K). | ||
The signals belonging to the Zn(trenpyr) complex are rather sharp (Fig. 11). Since in the crystal structure of [Zn(trenpyr)](ClO4)2 one of the pyridine rings is not coordinated, and therefore the ligand is asymmetrically bound,20 it is surprising that only one set of signals is present for the three ‘legs’ on the NMR spectrum of the slow exchanging Zn(trenpyr). This indicates C3 symmetry of the complex, and either seven-coordinated {Ntert,3NH,3Npyr} or six-coordinated {3NH,3Npyr} type binding mode of trenpyr. In the case of zinc(II) the latter is more likely, and this binding mode is present in the Zn(II) complex of the closely related Schiff-base derivative of trenpyr.66 Furthermore, the aromatic protons beside the pyridine N (C6H), despite the expectations, are considerably more upfield shifted (7.48 ppm) in Zn(Htrenpyr) than in Zn(trenpyr) (8.07 ppm), which indicates notable changes in the coordination environment of zinc(II) during the deprotonation. All these facts may support the {3NH,3Npyr} type coordination in Zn(trenpyr).
In the presence of manganese(II) only two species were detected (Fig. 9). The complex Mn(Htrenpyr) is a minor complex around pH 5, its deprotonation results in Mn(trenpyr), which is the sole species above pH 7 (at metal ion and ligand concentrations of 2 mM).
The significant differences between the deprotonation of CuHL/ZnHL complexes of tachpyr and trenpyr are noteworthy (compare Fig. 6 and 10), which, however, does not apply for the MnHL species. This is due to the different denticity of tachpyr and trenpyr, as well as the different coordination geometries of their metal complexes. In the case of tachpyr complexes, the deprotonation of the 5N coordinated MHL complexes results in the increase of coordinated nitrogen donors, i.e. the metal promoted deprotonation is favoured. In contrast, in the copper(II)– and zinc(II)–trenpyr complexes, the coordination number is unchanged during the MHL → ML deprotonation, therefore it is less favoured since the incoming secondary NH should displace an already bound nitrogen from the coordination sphere. On the other hand, our equilibrium data indicate that the process M(Htrenpyr) = M(trenpyr) + H+ is more favoured for Mn(II) than for Zn(II) or Cu(II) (pK = 5.08, 5.23 and 5.56, respectively), despite the striking differences in the stability of the corresponding M(trenpyr) complexes (Table 5). Since manganese(II) is the weakest Lewis-acid among these metal ions, the above order of pKs is unusual, and suggests that the coordination of the secondary NH-group induces some additional stabilization in the case of Mn(II). In this respect, it is worth noting that in the crystal structure of [Mn(trenpyr)](PF6)2 the metal ion is seven coordinated.14 The above mentioned additional stabilization is most likely provided by the chelate coordination of the third NH-group and pyridine nitrogen on the same ‘leg’, i.e. the complex Mn(trenpyr) is probably seven-coordinated ({Ntert,3NH,3Npyr}) in solution, too.
The basicity corrected formation constants allow an approximate comparison of M(tachpyr)/M(tach) or M(trenpyr)/M(tren) complexes, but it would provide erroneous results in the case of M(tachpyr)/M(trenpyr) pairs. It is more appropriate to calculate the conditional stability constants at a given pH (Kcond(pH) = ∑[MHxL]/([M2+]free × (∑[HxL]free)) in order to compare their metal ion binding abilities. In the case of tachpyr logKcond(pH 7.4) = 18.9, 14.0 and 5.3 for copper(II), zinc(II) and manganese(II), respectively. The analogous values for trenpyr are logKcond(pH 7.4) = 19.1, 13.4 and 8.4, respectively. These data indicate that tachpyr is a more efficient zinc(II) chelator, a nearly similar copper(II) chelator, and a less efficient manganese(II) chelator than trenpyr. Considering the energy demanding switch between the (triequatorial NH → triaxial NH) conformations of tachpyr and the conformational flexibility of trenpyr, these data are somewhat surprising, and is very likely due to the encapsulating effect of the more rigid tachpyr skeleton. On the other hand, the observed relative binding preference of tachpyr for zinc(II) is probably related to the observation that zinc(II) is one of the principal metals targeted by tachpyr in cells.27
Since one of our aims has been to use tach3pyr as a catalyst for phosphodiester hydrolysis, we applied a dmso–water mixture in order to increase the solubility of complexes. In a 60% (w/w) dmso–water mixed solvent the copper(II)–tach3pyr systems were clear up to pH 10. Although, in the presence of zinc(II) a precipitate was observed, too, further investigations were performed in this medium. The formation constants of the copper(II) complexes in 60% (w/w) dmso–water are also listed in Table 5 and their speciation diagrams are depicted in Fig. 12.
 | ||
| Fig. 12 Speciation diagram of the copper(II)–tach3pyr complexes ([Cu2+] = [tach3pyr] = 0.001 M, T = 298 K, I = 0.1 M NaCl in 60% (w/w) dmso–water) and the pH−kobs profile (■) of the bnpp hydrolysis ([bnpp]ini = 0.002 M). | ||
In this medium, the Cu(tach3pyr) complex has considerably higher stability than in pure water, especially considering the somewhat lower basicity of the ligand. The Vis/NIR and EPR parameters of the Cu(tach3pyr) complex (d–d transitions at 692 and ∼1050 nm (Fig. S8,†Table 6 and Fig. S9†)), are very similar to those of some N-alkylated tach derivatives,59 and indicate a square pyramidal geometry, which is enforced by the facial coordination of the ligand.
As expected, the presence of 3-pyridylmethyl legs prevent the formation of the dihydroxo-bridged dicopper complex formed with tach,29 and above pH 7 two mononuclear mixed hydroxo-complexes (Cu(tach3pyr)(OH) and Cu(tach3pyr)(OH)2) are formed (Fig. 12). The deprotonation of the coordinated water molecules (pK = 7.94 and 9.77) resulted in a slight blue-shift of the Vis/NIR (Cu(tach3pyr)(OH): 650 and ∼900 nm; Cu(tach3pyr)(OH)2: 640 and ∼900 nm) and EPR spectra (Table 6 and Fig. S9†), suggesting that the square pyramidal geometry is retained in these species, too.
The relatively open coordination sphere of Cu(tach3pyr)(OH) species is favourable from the point of view of hydrolytic catalysis. Indeed, we observed a notable hydrolytic activity for the mono-hydroxo species against bis(p-nitrophenyl) phosphate (bnpp), an activated phosphodiester (Fig. 12). The water molecule in the fifth position of copper(II) can be easily replaced by the substrate, and the copper-bound hydroxide ion may act as an intramolecular nucleophile (Fig. S10†). The initial rate of hydrolysis as a function of bnpp concentration (Fig. S11†) shows saturation kinetics. The treatment of these data, using the Michaelis–Menten model, yielded the following parameters: kcat = (3.0 ± 0.4) × 10−5 s−1 and KM = (4.2 ± 0.8) mM (Fig. S11†). It is rather difficult to compare these values with those of the related copper(II) complex of N,N′,N′′-trimethyl-cis,cis-1,3,5-triaminocyclohexane,33 due to the different temperature and solvent used, as well as the unlike mechanism. Although it seems that the Cu(tach3pyr)(OH) species is somewhat more active, the above KM value does not indicate a notable increase of substrate binding due to the expected π–π interaction between pyridyl and nitrophenyl rings. Therefore, further kinetic studies were not performed.
Tren3pyr forms more stable complexes with copper(II) and zinc(II) than tach3pyr (Table 5), due to the four available nitrogens for metal ion binding of the tripodal platform. In the solution containing copper(II) and tren3pyr only mononuclear complexes (Cu(Hxtren3pyr), x = 3, 2, 1, 0, −1) have been observed (Fig. 13). At twofold metal ion excess over tren3pyr, a precipitate was detected at pH 4–5, which prevented the equilibrium study. The precipitate was than recrystallized from ethanol to obtain complex 4. In equimolar solutions, precipitation and formation of polymeric species were not observed at 298 K, only mononuclear complexes were detected (see the MS spectrum in Fig. S12†).
 | ||
| Fig. 13 Speciation diagram of the copper(II)–tren3pyr complexes ([Cu2+] = [tren3pyr] = 0.002 M, T = 298 K, I = 0.1 M NaCl). | ||
According to the relatively high stability of tren3pyr complexes, their formation start at pH 2 and after three overlapped deprotonations the Cu(tren3pyr) species formed, a dominant complex around pH 7 (Fig. 13). During these processes, the Vis/NIR spectra show gradually increasing d–d bands at 870 nm with a shoulder at 700 nm (Fig. 14). The calculated individual spectra of these complexes (Fig. S13†) indicate invariable band positions.
 | ||
| Fig. 14 Effect of pH on the Vis/NIR spectra of the copper(II)–tren3pyr systems (T = 298 K, I = 0.1 M NaCl, [Cu2+] = [tren3pyr] = 0.003 M). | ||
The room temperature EPR spectra are also identical between pH 2–7 (Table 6, Fig. S14†). Although, the parameters of EPR spectra measured at 77 K (Fig. 15) show some subtle variations with pH (Table 6), they are essentially similar. Both the typical d–d bands and the ‘reversed’ order of g-values (gz < gx ∼ gy (Table 6), i.e. the complexes have a ground state with an unpaired electron in the dz2 orbital67) indicate a trigonal bipyramidal geometry in Cu(Hxtren3pyr), x = 3, 2, 1, 0. It means that the metal ion is bound to the four nitrogens of the tren-like subunit already in Cu(H3tren3pyr), and the successive deprotonations are related to non-coordinating pyridine rings. Although, the pK values of these deprotonations (pK = 3.79, 4.17 and 4.76) are somewhat higher than those of the free ligand (Table 4), this is due to the higher electron withdrawing effect of the protonated secondary amino nitrogens as compared to the neutral (and metal ion coordinated) ones.
 | ||
| Fig. 15 Experimental (black) and simulated (red) EPR spectra of the copper(II)–tren3pyr systems at 77 K ([Cu2+] = [tren3pyr] = 0.003 M). The calculated component spectra of the main species are also shown on the right. | ||
The low temperature EPR spectra indicate the presence of two additional species, which appeared during the fast-freezing process. Between pH 2.5–3.2 a weak axial spectrum (dx2−y2 ground state, g⊥ > g‖, Table 6) can be detected which probably belongs to Cu(H4tren3pyr), undetectable at room temperature. The other appeared between pH 3.5–5 as a broad isotropic signal, due to a small amount (20%) of magnetically coupled copper(II) centres. This is probably related to the polynuclear complex 4, which became thermodynamically more stable at low temperature.
Above pH 7 a single deprotonation was observed which resulted in the formation of Cu(tren3pyr)(OH). Both Vis/NIR and EPR spectra (Fig. 14 and 15) show considerable differences between Cu(tren3pyr) and Cu(tren3pyr)(OH), although the typical features of a trigonal bipyramidal geometry are retained. The low temperature EPR parameters (Table 6) indicate the absence of rhombic distortion (a more symmetric coordination environment) in the xy-plane of the mixed hydroxo complex.
In the presence of zinc(II) two major species form, Zn(tren3pyr) between pH 6–8 and Zn(tren3pyr)(OH) above pH 8 (Fig. S15†). Both complexes have slow ligand exchange processes on the NMR timescale (Fig. 16). In Zn(tren3pyr) the signals of ethylene protons are considerably broadened and those of the benzylic protons completely disappeared from the spectra. On the other hand, at higher pH the signals are narrowed, and the benzylic protons (∼4.1 ppm) are clearly visible on the spectrum of Zn(tren3pyr)(OH), indicating conformationally less labile pyridine rings in this species (see later). The signals of aromatic protons are less affected by such dynamic processes, implying that zinc(II) is coordinated only by the four nitrogens of the tren-like subunit, similarly to the corresponding copper(II) complexes.
 | ||
| Fig. 16 The pH-dependence of 1H-NMR spectra of tren3pyr in the presence of zinc(II) ([Zn2+] = 0.0024 M, [tachpyr] = 0.003 M, pH (bottom up) = 2.30, 5.44, 5.7, 7.8, 8.73, 9.63, 10.66). | ||
It is interesting to compare the pKs of coordinated water deprotonation in tren (pK = 9.17 (Cu), 10.4 (Zn)) and tren3pyr (pK = 8.41 (Cu), 9.34 (Zn)) complexes. Although, several reasons may lead to the higher water acidity in the present systems,68 the conformationally less labile pyridine rings in Zn(tren3pyr)(OH) (see above) may indicate a H-bonding network between the hydroxide ion and pyridine nitrogen(s), which stabilize the position of the ring(s) and facilitate the formation of a M − OH− moiety.
Conclusion
The comparative evaluation of our solution chemical data indicated that the four polydentate tripodal ligands studied here exhibit a high thermodynamic stability, and a variety of coordination environments/geometries for manganese(II), copper(II) and zinc(II). Tachpyr is a more efficient zinc(II) chelator and a similar copper(II) chelator compared to trenpyr. Considering the higher number of N-donors and conformational flexibility of trenpyr, as well as the energy demanding switch to the triaxial conformation required for metal ion binding of tachpyr, the above observation is surprising and is very likely due to the encapsulating effect of the more rigid tachpyr skeleton. This relative binding preference of tachpyr for zinc(II) may be related to the observation that zinc(II) is one of the principal metals targeted by tachpyr in cells. In contrast, trenpyr is a considerably more efficient manganese(II) chelator, since it acts as a heptadentate ligand in the aqueous Mn(trenpyr) complex. The crystal structures of copper(II) and zinc(II) complexes of tachpyr indicated important differences in the ligand conformation, induced by the position of counter ions, as compared to earlier reports.The closely related new ligands, tach3pyr and tren3pyr, have been designed to form oligonuclear complexes. Indeed, we obtained a three dimensional polymer with a copper(II)/tren3pyr ratio of 11/6. Within this metal–organic framework three distinctly different copper geometries can be identified. Two square pyramidal and four trigonal bipyramidal copper centres create a hexanuclear subunit with a large inside cavity. These moieties are linked by tetrahedral copper(II) centres, forming the three-dimensional polymer structure.
In solution, only mononuclear complexes are formed with tach3pyr and tren3pyr, and the formation of polynuclear complexes was not detected. According to its facial donor set tach3pyr forms square pyramidal complexes. The copper(II) core in Cu(tach3pyr)(OH) surrounded by aromatic rings does not facilitate the binding of a bnpp substrate, i.e. this complex has a comparable hydrolytic activity to copper(II) complexes of other tach-derivatives. The absence of pyridine nitrogens in chelatable positions allows the tren-subunit to enforce trigonal bipyramidal geometry in tren3pyr complexes.
Acknowledgements
The research was supported by the National Research, Development and Innovation Office (NKFIH) through projects GINOP-2.3.2-15-2016-00038, OTKA K101541 and OTKA NK105691.References
- A. G. Blackman, Polyhedron, 2005, 24, 1–39 CrossRef CAS.
- L. Q. Hatcher and K. D. Karlin, Advances in Inorganic Chemistry Including Bioinorganic Studies, 2006, vol. 58, pp. 131–184 Search PubMed.
- L. M. Berreau, Eur. J. Inorg. Chem., 2006, 273–283 CrossRef CAS.
- B. A. Jazdzewski and W. B. Tolman, Coord. Chem. Rev., 2000, 200, 633–685 CrossRef.
- G. Parkin, Chem. Rev., 2004, 104, 699–767 CrossRef CAS PubMed.
- A. Jancso, I. Torok, L. Korecz, A. Rockenbauer and T. Gajda, J. Chem. Soc., Dalton Trans., 2002, 2601–2607 RSC.
- Y. E. Alexeev, B. I. Kharisov, T. C. H. Garcia and A. D. Garnovskii, Coord. Chem. Rev., 2010, 254, 794–831 CrossRef CAS.
- E. Wong, S. Liu, S. J. Rettig and C. Orvig, Inorg. Chem., 1995, 34, 3057–3064 CrossRef CAS.
- B. Kuswandi, Nuriman, W. Verboom and D. N. Reinhoudt, Sensors, 2006, 6, 978–1017 CrossRef CAS.
- Z. Dai and J. W. Canary, New J. Chem., 2007, 31, 1708–1718 RSC.
- E. A. Lewis, H. H. Khodr, R. C. Hider, J. R. L. Smith and P. H. Walton, Dalton Trans., 2004, 187–188 RSC.
- A. Szorcsik, F. Matyuska, A. Benyei, N. V. Nagy, R. K. Szilagyie and T. Gajda, Dalton Trans., 2016, 45, 14998–15012 RSC.
- T. Nagano, T. Hirano and M. Hirobe, J. Biol. Chem., 1989, 264, 9243–9249 CAS.
- A. Deroche, I. MorgensternBadarau, M. Cesario, J. Guilhem, B. Keita, L. Nadjo and C. HoueeLevin, J. Am. Chem. Soc., 1996, 118, 4567–4573 CrossRef CAS.
- G. Park, A. M. Przyborowska, N. Ye, N. M. Tsoupas, C. B. Bauer, G. A. Broker, R. D. Rogers, M. W. Brechbiel and R. P. Planalp, Dalton Trans., 2003, 318–324 RSC.
- G. Park, F. H. Lu, N. Ye, M. W. Brechbiel, S. V. Torti, F. M. Torti and R. P. Planalp, J. Biol. Inorg. Chem., 1998, 3, 449–457 CrossRef CAS.
- M. L. Childers, F. Su, A. M. Przyborowska, B. Bishwokarma, G. Park, M. W. Brechbiel, S. V. Torti, F. M. Torti, G. Broker, J. S. Alexander, R. D. Rogers, K. Ruhlandt-Senge and R. P. Planalp, Eur. J. Inorg. Chem., 2005, 3971–3982 CrossRef CAS.
- G. Park, N. Ye, R. D. Rogers, M. W. Brechbiel and R. P. Planalp, Polyhedron, 2000, 19, 1155–1161 CrossRef CAS.
- G. Park, E. Dadachova, A. Przyborowska, S. J. Lai, D. S. Ma, G. Broker, R. D. Rogers, R. P. Planalp and M. W. Brechbiel, Polyhedron, 2001, 20, 3155–3163 CrossRef CAS.
- A. Mohamadou and C. Gerard, J. Chem. Soc., Dalton Trans., 2001, 3320–3328 RSC.
- C. Gerard, A. Mohamadou, J. Marrot, S. Brandes and A. Tabard, Helv. Chim. Acta, 2005, 88, 2397–2412 CrossRef CAS.
- A. Mohamadou, C. Gerard and J. Marrot, Polyhedron, 2008, 27, 3036–3040 CrossRef CAS.
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS PubMed.
- D. S. Ma, F. Lu, T. Overstreet, D. E. Milenic and M. W. Brechbiel, Nucl. Med. Biol., 2002, 29, 91–105 CrossRef CAS PubMed.
- S. V. Torti, F. M. Torti, S. P. Whitman, M. W. Brechbiel, G. Park and R. P. Planalp, Blood, 1998, 92, 1384–1389 CAS.
- J. Turner, C. Koumenis, T. E. Kute, R. P. Planalp, M. W. Brechbiel, D. Beardsley, B. Cody, K. D. Brown, F. M. Torti and S. V. Torti, Blood, 2005, 106, 3191–3199 CrossRef CAS PubMed.
- R. Zhao, R. P. Planalp, R. Ma, B. T. Greene, B. T. Jones, M. W. Brechbiel, F. M. Torti and S. V. Torti, Biochem. Pharmacol., 2004, 67, 1677–1688 CrossRef CAS PubMed.
- S. V. Torti, R. Ma, V. J. Venditto, F. M. Torti, R. P. Planalp and M. W. Brechbiel, Bioorg. Med. Chem., 2005, 13, 5961–5967 CrossRef CAS PubMed.
- Y. Fujii, T. Kiss, T. Gajda, X. S. Tan, T. Sato, Y. Nakano, Y. Hayashi and M. Yashiro, J. Biol. Inorg. Chem., 2002, 7, 843–851 CrossRef CAS PubMed.
- C. Sissi, F. Mancin, M. Palumbo, P. Scrimin, P. Tecilla and U. Tonellato, Nucleosides, Nucleotides, Nucleic Acids, 2000, 19, 1265–1271 CAS.
- C. Sissi, F. Mancin, M. Gatos, M. Palumbo, P. Tecilla and U. Tonellato, Inorg. Chem., 2005, 44, 2310–2317 CrossRef CAS PubMed.
- T. Kobayashi, S. Tobita, M. Kobayashi, T. Imajyo, M. Chikira, M. Yashiro and Y. Fujii, J. Inorg. Biochem., 2007, 101, 348–361 CrossRef CAS PubMed.
- K. A. Deal, G. Park, J. Shao, N. D. Chasteen, M. W. Brechbiel and R. P. Planalp, Inorg. Chem., 2001, 40, 4176–4182 CrossRef CAS PubMed.
- T. Bowen, R. P. Planalp and M. W. Brechbiel, Bioorg. Med. Chem. Lett., 1996, 6, 807–810 CrossRef CAS.
- H. Rosotti and F. J. C. Rosotti, The determination of stability constants, McGraw-Hill Book Co., New York, 1962, p. 149 Search PubMed.
- E. Högfeldt, Stability Constants of Metal-Ion Complexes. Part A. Inorganic Ligands, Pergamon, New York, 1982, p. 32 Search PubMed.
- H. M. Irving, M. G. Miles and L. D. Pettit, Anal. Chim. Acta, 1967, 38, 475–488 CrossRef CAS.
- SCQuery: The IUPAC Stability Constants Database, Royal Society of Chemistry, Sourby Old Farm, Timble, Otley, Yorks, 1993–2005.
- L. Zékány, I. Nagypál and G. Peintler, PSEQUAD for chemical equilibria, Technical Software Distributors, Baltimore, MD, 1991 Search PubMed.
- H. Sigel, A. D. Zuberbuhler and O. Yamauchi, Anal. Chim. Acta, 1991, 255, 63–72 CrossRef CAS.
- A. Rockenbauer and L. Korecz, Appl. Magn. Reson., 1996, 10, 29–43 CrossRef CAS.
- CrystalClear SM 1.4.0, Rigaku/MSC Inc., 2008 Search PubMed.
- A. C. T. North, D. C. Phillips and F. S. Mathews, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1968, 24, 351–359 CrossRef.
- M. C. Burla, R. Caliandro, B. Carrozzini, G. L. Cascarano, C. Cuocci, C. Giacovazzo, M. Mallamo, A. Mazzone and G. Polidori, J. Appl. Crystallogr., 2015, 48, 306–309 CrossRef CAS.
- SHELXL-2013 Program for Crystal Structure Solution, University of Göttingen, Germany, 2013 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- CrysAlis CCD, Oxford Diffraction Ltd, Abingdon, Oxfordshire, England, 2006 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van De Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- S. P. Westrip, J. Appl. Crystallogr., 2010, 43, 920–925 CrossRef CAS.
- A. Kálmán, L. Párkányi and Gy. Argay, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 1039–1049 CrossRef.
- A. Kálmán and L. Párkányi, Isostructurality of Organic Crystals in Advances in Molecular Structure Research, ed. M. Hargittai and I. Hargittai, JAI Press Inc., 1997, vol. 3, pp. 189–226 Search PubMed.
- M. Schatz, M. Becker, O. Walter, G. Liehr and S. Schindler, Inorg. Chim. Acta, 2001, 324, 173–179 CrossRef CAS.
- J. Moncol, M. Mudra, P. Lonnecke, M. Koman and M. Melnik, J. Chem. Crystallogr., 2004, 34, 423–431 CrossRef CAS.
- E. W. Ainscough, A. G. Bingham and A. M. Brodie, J. Chem. Soc., Dalton Trans., 1984, 989–991 RSC.
- R. J. Motekaitis, A. E. Martell, J. M. Lehn and E. I. Watanabe, Inorg. Chem., 1982, 21, 4253–4257 CrossRef CAS.
- Y. Fujii, T. Itoh, K. Onodera and T. Tada, Chem. Lett., 1995, 305–306 CrossRef CAS.
- G. Anderegg and V. Gramlich, Helv. Chim. Acta, 1994, 77, 685–690 CrossRef CAS.
- G. Park, J. Shao, F. H. Lu, R. D. Rogers, N. D. Chasteen, M. W. Brechbiel and R. P. Planalp, Inorg. Chem., 2001, 40, 4167–4175 CrossRef CAS PubMed.
- E. Prenesti, P. G. Daniele, S. Berto and S. Toso, Polyhedron, 2006, 25, 2815–2823 CrossRef CAS.
- M. Noack, G. F. Kokoszka and G. Gordon, J. Chem. Phys., 1971, 54, 1342–1350 CrossRef CAS.
- C. Yu, C. L. Dumoulin and G. C. Levy, Magn. Reson. Chem., 1985, 23, 952–958 CrossRef CAS.
- R. A. D. Wentworth, R. F. Childers and L. J. Zompa, Inorg. Chem., 1971, 10, 302–306 CrossRef CAS.
- D. H. Lee, N. N. Murthy and K. D. Karlin, Inorg. Chem., 1997, 36, 5785–5792 CrossRef CAS PubMed.
- N. Wei, N. N. Murthy and K. D. Karlin, Inorg. Chem., 1994, 33, 6093–6100 CrossRef CAS.
- R. M. Kirchner, C. Mealli, M. Bailey, N. Howe, L. P. Torre, L. J. Wilson, L. C. Andrews, N. J. Rose and E. C. Lingafelter, Coord. Chem. Rev., 1987, 77, 89–163 CrossRef CAS.
- M. Q. Ehsan, Y. Ohba, S. Yamauchi and M. Iwaizumi, Bull. Chem. Soc. Jpn., 1996, 69, 2201–2209 CrossRef CAS.
- J. W. Canary, J. Xu, J. M. Castagnetto, D. Rentzeperis and L. A. Marky, J. Am. Chem. Soc., 1995, 117, 11545–11547 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of the ligands, crystallographic data, figures depicting the arrangement of hydrogen bonds and unit cells, UV-VIS, EPR, MS spectra and speciation curves of the complexes studied, proposed mechanism as well as saturation kinetic data for the hydrolysis of bnpp. CCDC 1526243–1526246 for structures 1–4, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt00104e |
| This journal is © The Royal Society of Chemistry 2017 |