
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Radical anion and dianion salts of titanyl macrocycles with acceptor substituents or an extended π-system†
Maxim A.
Faraonov
 a,
Dmitri V.
Konarev
*a,
Alexey M.
Fatalov
ab,
Salavat S.
Khasanov
c,
Sergey I.
Troyanov
b and
Rimma N.
Lyubovskaya
a
a,
Dmitri V.
Konarev
*a,
Alexey M.
Fatalov
ab,
Salavat S.
Khasanov
c,
Sergey I.
Troyanov
b and
Rimma N.
Lyubovskaya
a
aInstitute of Problems of Chemical Physics RAS, Chernogolovka, Moscow region 142432, Russia. E-mail: konarev3@yandex.ru
bMoscow State University, Leninskie Gory, 119991 Moscow, Russia
cInstitute of Solid State Physics RAS, Chernogolovka, Moscow region 142432, Russia
First published on 16th February 2017
Abstract
Crystalline anionic salts of titanyl macrocycles with acceptor substituents or an extended π-system have been obtained for the first time: (PPN+)2{O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) TiIV(PcCl84−)}2− (1), (PPN+){OTiIV(Nc˙3−)}˙−·2C6H4Cl2 (2) and (PPN+)2{OTiIV(AceTPrzPz4−)}2−·1.3C6H4Cl2·0.8C6H5CN (3) where PPN+ is the bis(triphenylphosphoranylidene)ammonium cation, PcCl8 – 2,3,9,10,16,17,23,24-octachlorophthalocyanine; Nc – 2,3-naphthalocyanine, AceTPrzPz – tetra(acenaphthenopyrazino)porphyrazine. Salts 1–3 were obtained in the reduction of the parent titanyl macrocycles by fluorenone ketyl in the presence of an excess of PPNCl in o-dichlorobenzene with following precipitation of crystals with n-hexane. Reduction of macrocycles in 1–3 is accompanied by the appearance of intense NIR bands in the solid spectra at 963–1159 nm. It has been found that the extended π-system with linear annulation in {OTiIV(Nc˙3−)}˙− provides the shift of the NIR band to smaller energies (1159 nm) in comparison with those in the spectra of {OTiIV(Pc˙3−)}˙− (995–998 nm). Reduction of macrocycles leads also to the alternation of C–Nimine bonds due to partial disruption of their aromaticity. The disruption is higher for the dianions in 1 and 3 in comparison with the radical anions in 2. One-dimensional π–π stacking chains and layers are formed in 1 and 3 with diamagnetic {OTiIV(PcCl84−)}2− and {OTiIV(AceTPrzPz4−)}2− dianions, respectively. Salt 2 contains nearly isolated [{OTiIV(Nc˙3−)}˙−]2 dimers with a strong π–π interaction between paramagnetic radical anion macrocycles. As a result, a transition from the triplet to singlet state with antiparallel ordering of spins within the dimers is observed in 2 below 200 K.
TiIV(PcCl84−)}2− (1), (PPN+){OTiIV(Nc˙3−)}˙−·2C6H4Cl2 (2) and (PPN+)2{OTiIV(AceTPrzPz4−)}2−·1.3C6H4Cl2·0.8C6H5CN (3) where PPN+ is the bis(triphenylphosphoranylidene)ammonium cation, PcCl8 – 2,3,9,10,16,17,23,24-octachlorophthalocyanine; Nc – 2,3-naphthalocyanine, AceTPrzPz – tetra(acenaphthenopyrazino)porphyrazine. Salts 1–3 were obtained in the reduction of the parent titanyl macrocycles by fluorenone ketyl in the presence of an excess of PPNCl in o-dichlorobenzene with following precipitation of crystals with n-hexane. Reduction of macrocycles in 1–3 is accompanied by the appearance of intense NIR bands in the solid spectra at 963–1159 nm. It has been found that the extended π-system with linear annulation in {OTiIV(Nc˙3−)}˙− provides the shift of the NIR band to smaller energies (1159 nm) in comparison with those in the spectra of {OTiIV(Pc˙3−)}˙− (995–998 nm). Reduction of macrocycles leads also to the alternation of C–Nimine bonds due to partial disruption of their aromaticity. The disruption is higher for the dianions in 1 and 3 in comparison with the radical anions in 2. One-dimensional π–π stacking chains and layers are formed in 1 and 3 with diamagnetic {OTiIV(PcCl84−)}2− and {OTiIV(AceTPrzPz4−)}2− dianions, respectively. Salt 2 contains nearly isolated [{OTiIV(Nc˙3−)}˙−]2 dimers with a strong π–π interaction between paramagnetic radical anion macrocycles. As a result, a transition from the triplet to singlet state with antiparallel ordering of spins within the dimers is observed in 2 below 200 K.
Introduction
Metal phthalocyanine compounds possess promising optical, conducting and magnetic properties. Some phthalocyanine derivatives can be used as sensors and materials for optical, electronic and photoelectronic devices.1,2 Conducting compounds can be obtained by oxidation of phthalocyanines or axially substituted phthalocyanine anions {MIIIL2(Pc2−)}− (M = Co, Fe; L = CN, Cl, Br) to produce crystalline salts with quasi-one-dimensional metallic behavior down to liquid helium temperatures. These salts have π–π stacking columnar arrangement of the macrocycles.3,4 Since some metal phthalocyanines contain paramagnetic metals they are also used as active components in the design of magnetic compounds. For example, oxidation of substituted manganese(II) naphthalocyanine with tetracyanoethylene or reduction of iron(II) phthalocyanine with decamethylchromocene yields compounds with the alternation of ions and ferrimagnetic ordering of spins.5,6Several radical anion and dianion salts of metal phthalocyanines were also synthesized as single crystals.7–21 Crystalline anionic salts were obtained in the reduction of MgII, ONbIV, AlIII, GeIV and ZrIV phthalocyanines with Na/Hg, potassium graphite (KC8) or LiCp*.7–11 Salts with reduced cobalt(II) and iron(II) phthalocyanines,12–16 iron(II) hexadecachlorophthalocyanine,17,18 and a large series of radical anion {M(Pc˙3−)}˙− salts, M = CuII, NiII, H2, SnII, PbII, OTiIV, OVIV, SnIVCl2 and InIIIBr, were also obtained and structurally characterized.19–22 Nevertheless, until now no anionic salts of such a type were obtained with other macrocycles except for phthalocyanines. These macrocycles can contain acceptor substituents to stabilize their radical anions in air or an extended π-system allowing the synthesis of structures with stronger π–π interactions. Reduction of metal macrocycles can also be used to obtain them in a crystalline form since in spite of the large size of the macrocycles they are well soluble in organic solvents in radical anion and dianion states.
Titanyl phthalocyanine is a well-known organic photoconductor which shows very good photoconductivity, excellent photostability, and wide absorption in the visible region with high absorbance.2,23–25 Until now several substituted titanyl and vanadyl phthalocyanines were obtained.26–28 However, the molecular structure was determined for unsubstituted titanyl and vanadyl phthalocyanines only using the Rietveld method on powdered samples29,30 or crystals,31 respectively.
In this work we chose titanyl macrocycles to introduce acceptor substituents or an extended π-system. We dissolve them in o-dichlorobenzene by their reduction and crystallize radical anion and dianion salts of titanyl octachlorophthalocyanine in (PPN+)2{OTiIV(PcCl84−)}2− (1), titanyl naphthalocyanine in (PPN+){OTiIV(Nc˙3−)}˙−·2C6H4Cl2 (2) and tetra(acenaphthenopyrazino)porphyrazine in (PPN+)2{OTiIV(AceTPrzPz4−)}2−·1.3C6H4Cl2·0.8C6H5CN (3) (Scheme 1). Preparation of single crystals of these salts allows us to determine the molecular structures of titanyl macrocycles including that of AceTPrzPz (Scheme 1) which is one of the largest macrocycles among tetrapyrazinoporphyrazines.32 We provide information on the reduction effect on the molecular structures, optical and magnetic properties of titanyl macrocycles.
 | ||
| Scheme 1 | ||
Results and discussion
Synthesis
Titanium dichloride macrocycles were synthesized by tetramerization of the corresponding dicarbonitriles in the presence of TiCl4 or TiCl4·2THF in 1-chloronaphthalene in 68–75% yield. Addition of 2-methylnaphthalene prevents the formation of products with the chlorinated periphery of the macrocycle. Substitution of chlorine atoms at titanium by oxygen with the formation of titanyl macrocycles was made by boiling of titanium dichloride macrocycles in wet pyridine (5% of H2O).33Scheme 2 of the reactions is shown below. | ||
| Scheme 2 | ||
The obtained titanyl macrocycles are reduced with sodium fluorenone ketyl in the presence of an excess of PPNCl. This leads to the dissolution of all titanyl macrocycles in the form of anions though, for example, neutral OTiIV(AceTPrzPz2−) is completely insoluble in o-dichlorobenzene. The reduction of TiIVCl2(Nc2−) with sodium fluorenone ketyl is accompanied by the substitution of chloride anions by oxygen at titanium(IV) atoms. Oxygen originates from fluorenone ketyl, and salt 2 with the {OTiIV(Nc˙3−)}˙− radical anions is formed.
According to redox potentials,34–36 the naphthalocyanine macrocycle shows weaker acceptor properties than tetrapyrazinoporphyrazine and phthalocyanine macrocycles with electron-withdrawing chloro- and pyrazino-substituents. As a result, the reduction of TiIVCl2Nc produces radical anion salt 2, whereas the reduction of OTiIV(PcCl8) and OTiIV(AceTPrzPz) under the same reaction conditions leads to the formation of dianion salts 1 and 3.
Spectra of salts in the IR and UV-visible-NIR ranges
According to the composition titanyl naphthalocyanine should have −1 charge owing to the titanyl macrocycle![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cation ratio of 1:1 in salt 2, whereas the titanyl macrocycle:cation ratio of 1:2 in salts 1 and 3 shows the formation of titanyl macrocycle dianions (2−).
:cation ratio of 1:1 in salt 2, whereas the titanyl macrocycle:cation ratio of 1:2 in salts 1 and 3 shows the formation of titanyl macrocycle dianions (2−).
The IR spectra of the parent compounds and their salts are shown in Fig. S1–S3† and the observed absorption bands are listed in Table S1.† Titanyl macrocycles show absorption bands attributed to the TiO stretching mode. The absorption band of this mode is manifested at 946 cm−1 in the spectra of OTiIV(PcCl82−) and salt 1. The TiO stretching mode is split into three bands manifested at 927, 947 and 974 cm−1 in the spectrum of OTiIV(AceTPrzPz2−). These bands are shifted to lower wavenumbers at 917, 927 and 941 cm−1 in the spectrum of 3 indicating the elongation of the TiO bonds in this salt. Salt 2 was obtained in the reduction of TiIVCl2(Nc2−) with sodium fluorenone ketyl with the formation of {OTiIV(Nc˙3−)}˙− and the appearance of a new TiO bond. That is accompanied by the manifestation of a new band at 974 cm−1. Earlier it was shown that the reduction of unsubstituted OTiIV(Pc2−) in (Bu4N+){OTiIV(Pc˙3−)}˙− and (Et4N+){OTiIV(Pc˙3−)}˙−·C6H4Cl2 is accompanied by the elongation of TiO bonds since the corresponding absorption bands are shifted from 962 to 923–928 cm−1.13
The spectra of the starting titanyl macrocycles and salts 1–3 in KBr pellets in the UV-visible-NIR range are shown in Fig. 1. Unsubstituted OTiIV(Pc2−) shows the Soret band at 347 nm and the split Q-band at 654 and 708 nm.20 The Soret and Q-bands have a close position in the spectra of OTiIV(PcCl82−) and OTiIV(AceTPrzPz2−) manifesting at 353, 660, 706 nm and 331, 652, 710 nm, respectively. Unsubstituted TiIVCl2(Pc2−) shows absorption bands at 352, 665 and 721 nm. Linear annulation in the naphthalocyanine macrocycle of TiIVCl2(Nc2−) results in a blue shift of the Soret band up to 340 nm and a strong red shift of the Q-band up to 753 and 834 nm (Fig. 1).
 | ||
| Fig. 1 UV-visible-NIR spectra of the starting titanyl macrocycles and salts 1–3 in KBr pellets prepared under anaerobic conditions. Charge transfer bands (CTB) are shown by arrows. | ||
The spectra of 1–3 (Table 1) allow one to study how the reduction affects the optical properties of titanyl macrocycles. Analysis of these spectra shows that Soret bands are blue shifted in the reduction of the macrocycle in 1 and 3 whereas maxima of the Q-bands are strongly blue shifted in the spectra of 1 and 2, but remain nearly unshifted in the spectrum of 3 (Table 1 and Fig. 1). The most pronounced changes in the formation of radical anions and dianions of titanyl macrocycles are the appearance of new intense NIR bands. The presence of these bands unambiguously allows one to determine the formation of reduced macrocycle anions. The blue shift of the Soret and Q bands and the appearance of NIR bands are characteristic of the radical anion salts of {OTiIV(Pc˙3−)}˙− and other metal phthalocyanines.20,21 It should be noted that the NIR band in the spectrum of 2 (1159 nm) is strongly red shifted in comparison with the spectra of the [OTiIV(Pc˙3−)]˙− salts (995–998 nm).20 At the same time these bands are observed almost at the same position in the spectra of 1 and 3 (999 and 963 nm, respectively). Thus, linear annulation in naphthalocyanine essentially strongly decreases the energy of the Q band and the NIR band of the radical anion even in comparison with those for tetra(acenaphthenopyrazino)porphyrazine which has essentially a larger π-system but no linear annulation. The spectra of 1 and 2 also show broad low-energy bands at 1550 and 1672 nm (Fig. 1), respectively, which can be attributed to the charge transfer between macrocycles as will be discussed in the next section. This band is not observed in the spectrum of 3 (Fig. 1), probably due to the ineffective overlapping between the {OTiIV(AceTPrzPz4−)}2− dianions.
| Compound | Position of the absorption band of phthalocyanines, nm | ||
|---|---|---|---|
| Soret band, nm | Q-band, nm | Bands in NIR, nm | |
| sh – shoulder; max – maximum. | |||
| OTiIV(Pc2−)20 |
347 | 654, 708 | — |
| (TBA+){OTiIV(Pc˙3−)}˙− (ref. 20) |
343 | 597, 630, 701 | 844 (weak), 995 |
| (TEA+){OTiIV(Pc˙3−)}˙−·C6H4Cl220 |
340 | 600, 630, 698 | 911 (weak), 998 |
| 1475 (CT band) | |||
| OTiIV(PcCl82−) |
353 | 660 (max), 706 | — |
| (PPN+)2{OTiIV(PcCl84−)}2− (1) |
340 | 637 (max), 703 | 818, 916, 999 |
| 1550 (CT band) | |||
| TiIVCl2(Pc2−) | 352 | 665(sh), 721 (max) | — |
| TiIVCl2(Nc2−) | 340 | 753, 834 (max) | — |
| (PPN+){OTiIV(Nc˙3−)}˙−·(solvent) (2) |
340 | 697 (max), 814 | 1040 (weak), 1159, 1672 (CT band) |
| OTiIV(AceTPrzPz2−) |
331 | 652 (weak), 710 (max) | — |
| (PPN+)2{OTiIV(AceTPrzPz4−)}2− (solvent) (3) |
325 | 607, 706 (max) | 963 |
Molecular and crystal structures of salts 1–3
Geometric parameters of macrocycles in 1–3 are presented in Table 2. The average lengths of the Ti–N(Pc) bonds in 1–3 are 2.068(2)–2.097(6) Å. That is slightly longer than those in {OTiIV(Pc˙3−)}˙− (2.055(1)–2.062(1) Å).20 The elongation of the Ti–N(Pc) bonds results in a stronger displacement of the Ti atoms from the 24-atom Pc plane which is maximal for the {OTiIV(AceTPrzPz4−)}2− dianions in 3. The length of the OTi bond also varies from 1.615(6) to 1.645(5) Å, and the shortest bonds are observed for the macrocycles with an extended π-system in 2 and 3.
| Compound | Average length of bonds and contacts, Å | Displacement of atoms from the 24-atom Pc plane, Å | |||
|---|---|---|---|---|---|
| M–Npyrrole | C–Npyrrole | C–Nimine short/long, difference | Metal | Npyrrole | |
| OTiIV(Pc2−) (P21/c) |
2.067(6) | 1.374(6) | 1.330(6) | 0.778 | 0.091–0.184 |
| OTi (1.628(3) Å)24 |
|||||
| OTiIV(Pc2−) (Cc) |
2.071(5) | 1.375(5) | 1.329(5) | 0.742 | 0.076–0.151 |
| OTi (1.643(3) Å)25 (both made by Rietveld analysis) |
|||||
| (Bu4N+){OTiIV(Pc˙3−)}˙− |
2.062(1) | 1.386(2) | 1.325(2)/1.350(2), 0.025 | 0.711 | 0.084–0.135 |
| OTi (1.640(1) Å)20 |
|||||
| (Et4N+){OTiIV(Pc˙3−)}˙−·C6H4Cl2 |
2.055(1) | 1.388(2) | 1.312(2)/1.351(2), 0.039 | 0.595 | 0.004–0.031 |
| (OTi (1.657(1) Å))20 |
|||||
| (PPN+)2{OTiIV(PcCl84−)}2− (1) |
2.072(3) | 1.390(4) | 1.304(4)/1.375(4), 0.071 | 0.602 | 0.015–0.019 |
| OTi (1.645(5) Å) |
|||||
| (PPN+){OTiIV(Nc˙3−)}˙−(solvent) (2) |
2.068(2) | 1.397(3) | 1.315(3)/1.353(3), 0.038 | 0.556 | 0.007–0.088 |
| OTi (1.638(2) Å) |
|||||
| (PPN+)2{OTiIV(AceTPrzPz4−)}2− (solvent) (3) (OTi (1.615(6) Å)) |
2.097(6) | 1.402(9) | 1.314(9)/1.374(9), 0.060 | 0.741 | 0.067–0.166 |
The reduction of the Pc macrocycle is accompanied by a transition from an aromatic 18 electron π-system to antiaromatic 19 and 20 electron π-systems for radical trianion and tetraanion macrocycles, respectively. As a result, partial disruption of the aromaticity of the macrocycles is observed in the reduced state leading to the alternation of the C–Nimine bonds.7–11,20,21 Such alternation is also observed for the reduced macrocycles in 1–3 (Table 2), but the difference between the shorter and longer C–Nimine bonds in 1 and 3 with tetraanion macroheterocycles is nearly two times larger than those for the radical trianion Pc˙3− (ref. 20) and Nc˙3− macrocycles (Table 2). Thus, disruption of aromaticity is more pronounced for the dianion titanyl macrocycles. Alternation of bonds is realized in such a way that shorter and longer C–Nimine bonds belong to two oppositely located isoindole units in 1–3 as is also observed in the radical anions of other metal phthalocyanines.20,21
A half of {OTiIV(PcCl84−)}2− dianion and one PPN+ cation are independent in 1. These dianions form layers in the ab plane (Fig. 2a), which are separated by the PPN+ cations (Fig. 2b). More closely packed chains can be outlined along the a axis within the phthalocyanine layers (Fig. 2a). Each phthalocyanine macrocycle has four short van der Waals (vdW) C⋯C and Cl⋯Cl contacts of about 3.5 Å length with the neighboring dianions in these chains. The interplanar distance in the chains is uniform and is rather short (3.38 Å). Most probably namely close packing of the PcCl8 macrocycles in 1 provides the appearance of an intense charge transfer band in the NIR spectrum (see above). Any vdW contacts between phthalocyanines from the neighboring chains within the layer are absent. The macrocycle is almost flat in 1 with only slight deviations of atoms from the 24-atom Pc plane (less than 0.065 Å).
 | ||
| Fig. 2 Crystal structure of (PPN+)2{OTiIV(PcCl84−)}2− (1). Views perpendicular (a) and parallel (b) to the phthalocyanine layers. Shortened vdW C,Cl⋯C,Cl contacts are shown by green dashed lines. | ||
One {OTiIV(Nc˙3−)}˙− radical anion, one PPN+ cation and two solvent C6H4Cl2 molecules are independent in 2. There are naphthalocyanine chains along the a axis (Fig. 3) which are isolated by the PPN+ cations. The {OTiIV(Nc˙3−)}˙− radical anions form dimers in these chains with multiple C,N⋯C,N vdW contacts, but macrocycles in these dimers are only slightly shifted relative to each other. As a result, there are no short vdW contacts between the neighboring dimers within the chains. The naphthalocyanine macrocycle has two adjacent naphthalene groups located strictly above the 24-atom plane, whereas two other groups are located almost in the plane. The interplanar distance of 3.169 Å is very short in the dimer indicating effective π–π interaction between the macrocycles in 2 explaining the appearance of a charge transfer (CT) band in the NIR spectrum. The interplanar distance in 2 is close to that in previously studied (Et4N+){OTiIV(Pc˙3−)}˙−·C6H4Cl2 salt (3.129 Å) which also shows an intense CT band in the NIR range.20
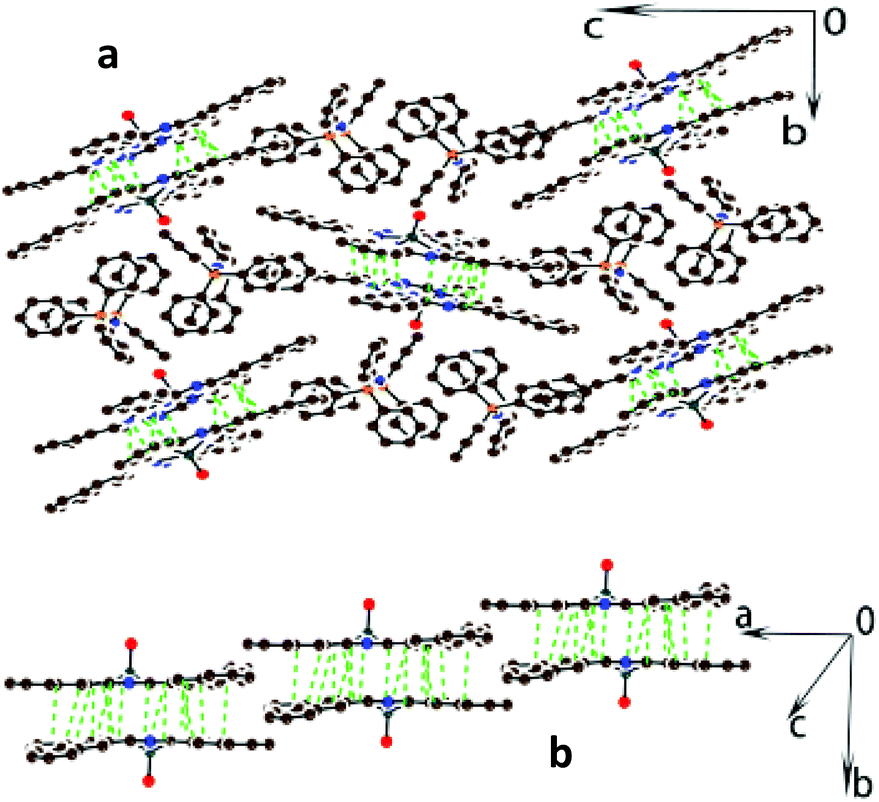 | ||
| Fig. 3 Crystal structure of (PPN+){OTiIV(Nc˙3−)}˙−·2C6H4Cl2 (2). View along the a axis and the naphthalocyanine chains (a) and view on these chains (b). Shortened vdW C,N⋯C,N contacts are shown by green dashed lines. Solvent molecules are not shown for clarity. | ||
As a result of strong disorder of solvent molecules, the structure of 3 was refined with a relatively high R-factor value (0.1158).
Nevertheless, the structures of the {OTiIV(AceTPrzPz4−)}2− dianion and the PPN+ cations were determined quite reliably. Dianions form the [{OTiIV(AceTPrzPz4−)}2−]2 dimers with a strong shift of the macrocycles but effective π–π stacking between one of four acenaphthenopyrazino groups of the {OTiIV(AceTPrzPz4−)}2− dianions (Fig. 4a and b).
 | ||
| Fig. 4 Crystal structure of (PPN+)2{OTiIV(AceTPrzPz4−)}2−·1.2C6H4Cl2·0.8C6H4Cl2 (3): view on (a) and along (b) the [{OTiIV(AceTPrzPz4−)}2−]2 dimer, view on (c) and along (d) the layers from the dimers. Shortened vdW C⋯C contacts are shown by green dashed lines. Solvent molecules are not shown for clarity. | ||
The interplanar distance between them is 3.38 Å and the shortest C,N⋯C,N vdW contacts are in the 3.19–3.36 Å range. The dihedral angle between the planes of the acenaphthenopyrazino groups of neighboring dianions in the dimer is only 0.22° showing their parallel arrangement. The strong shift of the macroheterocycles relative to each other in the dimers leads to the formation of chains arranged along the [1−1−0] direction (Fig. 4c and d). There are 4 C⋯C vdW contacts between the neighboring dimers in these chains of 3.47–3.56 Å length. These contacts are of π-type since the acenaphthene planes of two dianions involved in this interaction are nearly parallel (the dihedral angle between them is 8.12°) and the interplanar distance is only 3.42 Å. There are also several side-by-side vdW C⋯C contacts of 3.31 Å length between the [{OTiIV(AceTPrzPz4−)}2−]2 dimers from the neighboring chains (Fig. 4c, green dashed lines). The macrocycle in 3 has concave conformation and deviates strongly from planarity in contrast to the geometry of the macrocycles in 1 and 2.
EPR studies of the salts
The EPR spectra of salts 1 and 3 at 300 K contain weak asymmetric signals which can be approximated by three lines with g = 1.9975–2.0042 and the linewidth (ΔH) = 0.45–0.92 mT, which are characteristic of the radical anions of phthalocyanine species. The intensity of these signals is very low (less than 1% of spins from the total amount of titanyl macrocycle). Therefore, the signals can be most probably attributed to paramagnetic impurities. Thus, the {OTiIV(PcCl84−)}2− and {OTiIV(AceTPrzPz4−)}2− dianions are diamagnetic.
Salt 2 shows an asymmetric EPR signal which can be simulated well by two narrow lines (g1 = 2.0034 and ΔH = 0.50 mT, g2 = 2.0014 and ΔH = 0.88 mT) and one broad line with g3 = 2.0018 and ΔH = 4.67 mT at 300 K (Fig. 5).
 | ||
| Fig. 5 EPR spectrum of 2 at 300 K (a). Temperature dependence of the main component of EPR signal of salt 2 (b). | ||
Narrow lines can be attributed to paramagnetic impurities due to that their intensities are less than 3% from that of the broad signal. The broad line decreases seven times in intensity upon cooling the sample from 300 down to 200 K (Fig. 5b). Below 200 K all three lines are of low intensity and show paramagnetic temperature dependence down to 4 K. Thus, salt 2 is diamagnetic below 200 K. The crystal structure of 2 shows that the {OTiIV(Nc˙3−)}˙− radical anions form closely packed π-stacking [{OTiIV(Nc˙3−)}˙−]2 dimers that can provide strong coupling between them. As a result, a diamagnetic singlet state with an antiparallel arrangement of spins in the dimers is formed below 200 K. A triplet state of the dimers with a parallel arrangement of spins is populated above 200 K. The observed singlet–triplet transition in 2 is similar to that observed in (Et4N+){OTiIV(Pc˙3−)}˙−·C6H4Cl2 which also contains π-stacking [{OTiIV(Pc˙3−)}˙−]2 dimers. However, the temperature of the transition to the singlet state in 2 (200 K) is higher than that in (Et4N+){OTiIV(Pc˙3−)}˙−·C6H4Cl2 (150 K).20 This can be explained by stronger π–π interactions in 2, probably due to the larger π-system of the naphthalocyanine macrocycle.
Conclusions
A series of radical anion and dianion salts based on titanyl macrocycles with acceptor substituents or an extended π-system were obtained as single crystals for the first time. The reduction of titanyl macrocycles in the presence of bulky PPN+ cations allows their dissolution due to the formation of the radical anions or dianions, whereas they are very weakly soluble in organic solvents in the neutral state. Thus, titanyl macrocycles can be crystallized and their molecular structures are determined from X-ray diffraction on single crystals. Extension of a π-system by linear annulation in naphthalocyanine leads to a strong red shift of the Q-band and NIR bands of the radical anions, whereas tetra(acenaphthenopyrazino)porphyrazine with an extended π-system shows Q-bands and a NIR band of the anions nearly at the same position as in the spectra of the salts with unsubstituted titanyl phthalocyanine radical anions, {OTiIV(Pc˙3−)}˙−. Partial disruption of aromaticity is observed in the reduction of the macrocycles. This effect is more pronounced for the dianions than for the radical anions. Increased size of the macrocycle leads to the formation of π-stacked structures. In the case of 2 this provides effective magnetic coupling between the spins within the [{OTiIV(Nc˙3−)}˙−]2 dimers and the transition of the dimers to the singlet ground state even below 200 K (at a temperature higher than that in the [{OTiIV(Pc˙3−)}˙−]2 dimers). Macrocycles in 1 and 3 are packed in the π-stacked chains. However, the diamagnetic nature of the dianions does not allow one to expect the realization of conductivity or magnetic interactions in these chains. We suppose that the radical anions of these titanyl macrocycles can provide more promising magnetic properties. Preparation of these salts is now in progress.
Experimental
Materials
4,5-Dichlorophthalonitrile (Aldrich, 99%), 2,3-dicyanonaphthalene (TCI, 99%), acenaphthenequinone (Acros, 95%), and diaminomaleonitrile (Acros, 98%) were used as received. TiCl4 (Aldrich, 99%) and TiCl4·2THF (Aldrich, 97%) were used as received. Bis(triphenylphosphoranylidene)ammonium chloride (PPNCl) was purchased from Aldrich (98%). 1-Chloronaphthalene (Aldrich, technical grade) was used as received. Sodium fluorenone ketyl was obtained as described.37 Solvents were purified under an argon atmosphere. o-Dichlorobenzene (C6H4Cl2) was distilled over CaH2 under reduced pressure; hexane was distilled over Na/benzophenone, and benzonitrile (C6H5CN) was distilled over Na under reduced pressure. The solvents were degassed and stored in a glove box. The crystals of 1–3 were stored in the glove box. KBr pellets for IR- and UV-visible-NIR measurements were also prepared in the glove box. EPR measurements were performed on polycrystalline samples of 1–3 sealed in 2 mm quartz tubes under 10−5 Torr.Synthesis of titanyl macrocycles
Titanyl macrocycles (Scheme 2) were synthesized as described in ref. 33.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) TiIV(PcCl82−).
4,5-Dichlorophthalonitrile (1 g, 5.076 mmol), 2-methylnaphthalene (180 mg, 1.269 mmol) and TiCl4 (0.14 ml, 1.269 mmol) in 10 ml of 1-chloronaphthalene were heated up to 250 °C upon stirring for 5 hours under an argon atmosphere. Then the reaction mixture was cooled down to 50 °C, the precipitate was filtered under reduced pressure and washed with toluene and methanol. TiIVCl2(PcCl82−) was converted to OTiIV(PcCl82−) by boiling in wet pyridine (5% of H2O) for two hours. OTiIV(PcCl82−) was filtered, washed with water and dried at 100 °C in a vacuum for 2 hours to yield 800 mg of blue powder (68%). Anal. found: C 44.86, N 13.01, H 1.12, Cl 33.21%. Calcd for C32H8N8Cl8OTi: C 45.08, N 13.15, H 0.94, Cl 33.34%. IR (KBr), ν, cm−1: 3087m, 2922m, 2849w, 2353w, 1603m, 1471m, 1413s, 1375m, 1320m, 1069s, 980w, 945m, 890m, 829m, 775m, 705m, 659m, 463w, 431w. UV-VIS, KBr pellet λmax, nm: 353, 660, 706.
TiIV (AceTPrzPz2−).
Acenaphtho[1,2-b]pyrazine-8,9-dicarbonitrile (1 g, 3.933 mmol), 2-methylnaphthalene (140 mg, 0.983 mmol) and TiCl4 (0.11 mL, 0.983 mmol) in 10 ml of 1-chloronaphthalene were heated up to 250 °C upon stirring for 5 hours under an argon atmosphere. Then the reaction mixture was cooled down to 50 °C, the precipitate was filtered under reduced pressure and washed with toluene and methanol. TiIVCl2(AceTPrzPz2−) was converted to OTiIV(AceTPrzPz2−) by boiling in wet pyridine (5% of H2O) for two hours. OTiIV(AceTPrzPz2−) was filtered, washed with water and dried at 100 °C in a vacuum for 2 hours to yield 800 mg of black powder (75%). Anal. found: C 70.88, N 20.59, H 2.49%. Calcd for C64H24N16OTi: C 71.04, N 20.72, H 2.22%. IR (KBr), ν, cm−1: 2921m, 2851m, 1615s, 1514m, 1458m, 1431s, 1369w, 1333s, 1262m, 1229s, 1179m, 1134s, 1099m, 1033s, 974w, 947m, 927m, 828m, 772s, 716m, 675m, 660m, 630m, 559w, 464s, 449m. UV-VIS, KBr pellet λmax, nm: 331, 652, 710.
TiIV(PcCl82−).
4,5-Dichlorophthalonitrile (1 g, 5.076 mmol), 2-methylnaphthalene (180 mg, 1.269 mmol) and TiCl4 (0.14 ml, 1.269 mmol) in 10 ml of 1-chloronaphthalene were heated up to 250 °C upon stirring for 5 hours under an argon atmosphere. Then the reaction mixture was cooled down to 50 °C, the precipitate was filtered under reduced pressure and washed with toluene and methanol. TiIVCl2(PcCl82−) was converted to OTiIV(PcCl82−) by boiling in wet pyridine (5% of H2O) for two hours. OTiIV(PcCl82−) was filtered, washed with water and dried at 100 °C in a vacuum for 2 hours to yield 800 mg of blue powder (68%). Anal. found: C 44.86, N 13.01, H 1.12, Cl 33.21%. Calcd for C32H8N8Cl8OTi: C 45.08, N 13.15, H 0.94, Cl 33.34%. IR (KBr), ν, cm−1: 3087m, 2922m, 2849w, 2353w, 1603m, 1471m, 1413s, 1375m, 1320m, 1069s, 980w, 945m, 890m, 829m, 775m, 705m, 659m, 463w, 431w. UV-VIS, KBr pellet λmax, nm: 353, 660, 706.
TiIV (AceTPrzPz2−).
Acenaphtho[1,2-b]pyrazine-8,9-dicarbonitrile (1 g, 3.933 mmol), 2-methylnaphthalene (140 mg, 0.983 mmol) and TiCl4 (0.11 mL, 0.983 mmol) in 10 ml of 1-chloronaphthalene were heated up to 250 °C upon stirring for 5 hours under an argon atmosphere. Then the reaction mixture was cooled down to 50 °C, the precipitate was filtered under reduced pressure and washed with toluene and methanol. TiIVCl2(AceTPrzPz2−) was converted to OTiIV(AceTPrzPz2−) by boiling in wet pyridine (5% of H2O) for two hours. OTiIV(AceTPrzPz2−) was filtered, washed with water and dried at 100 °C in a vacuum for 2 hours to yield 800 mg of black powder (75%). Anal. found: C 70.88, N 20.59, H 2.49%. Calcd for C64H24N16OTi: C 71.04, N 20.72, H 2.22%. IR (KBr), ν, cm−1: 2921m, 2851m, 1615s, 1514m, 1458m, 1431s, 1369w, 1333s, 1262m, 1229s, 1179m, 1134s, 1099m, 1033s, 974w, 947m, 927m, 828m, 772s, 716m, 675m, 660m, 630m, 559w, 464s, 449m. UV-VIS, KBr pellet λmax, nm: 331, 652, 710.
Syntheses of crystalline salts 1–3
Crystals of 1–3 were obtained by the diffusion technique. The reaction solutions of salts 1–3 were filtered in a 50 mL glass tube of 1.8 cm diameter with a ground glass plug, and 30 mL of hexane was layered over the solution. Slow mixing of two solvents for 1.5 months provided precipitation of crystals on the walls of the tube. The solvent was decanted from the crystals and they were washed with hexane.Salts (PPN)2{OTiIVPcCl8} (1) and (PPN){OTiIVNc}·2C6H4Cl2 (2) were obtained by reduction of 34.6 mg of TiIVOPcCl8 (0.042 mmol) or 35.5 mg of TiIVCl2Nc (0.042 mmol) with sodium fluorenone ketyl (55 mg, 0.27 mmol) in the presence of PPNCl (75 mg, 0.131 mmol) in 16 ml of o-dichlorobenzene for two hours at 100 °C until complete dissolution of the starting compounds and the formation of dark green and deep-blue-green solutions, respectively. The final solutions were filtered into a tube for diffusion. Black blocks of 1 and dark violet elongated prisms of 2 were obtained in 25 and 45% yield, respectively.
Salt (PPN)2{OTiIVAceTPrzPz}·1.3C6H4Cl2·0.8C6H5CN (3) was obtained by the reduction of 36 mg of OTiIVAceTPrzPz (0.033 mmol) with sodium fluorenone ketyl (55 mg, 0.27 mmol) in the presence of PPNCl (75 mg, 0.131 mmol) in 18 ml of o-dichlorobenzene/benzonitrile mixture (15:3) for two hours at 100 °C until complete dissolution of phthalocyanine and the formation of a dark green solution. The final solution was filtered into a tube for diffusion. Black prisms were obtained in 15% yield.
The composition of 1–3 (Table 3) was determined from X-ray diffraction on single crystals. The analysis of the obtained crystals under a microscope in a glove box as well as testing of several single crystals from each synthesis by X-ray diffraction showed that only one crystalline phase is formed. Elemental analysis cannot be used to determine the composition of the obtained crystals due to their high air sensitivity and an addition of oxygen during the procedure of elemental analysis.
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| Emp. formula | C104H68Cl8N10OP4Ti | C96H62Cl4N9OP2Ti | C149.4H93.2Cl2.6N18.8OP4Ti |
| M r [g mol−1] | 1929.06 | 1609.18 | 2431.57 |
| Color and shape | Black, block | Dark-violet, prism | Black, prism |
| Crystal system | Triclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c | C2/c |
| a, Å | 11.3971(7) | 15.1213(3) | 36.695(2) |
| b, Å | 14.8799(8) | 18.0169(3) | 25.1999(8) |
| c, Å | 15.0231(9) | 29.1976(6) | 27.4198(11) |
| α, ° | 63.130(6) | 90 | 90 |
| β, ° | 77.450(8) | 98.402(2) | 101.073(5) |
| γ, ° | 78.459(8) | 90 | 90 |
| V, Å3 | 2203.1(3) | 7869.2(3) | 24883.5(19) |
| Z | 1 | 4 | 8 |
| ρ calc [g cm−3] | 1.454 | 1.358 | 1.298 |
| μ [mm−1] | 0.888 | 0.343 | 0.239 |
| F(000) | 988 | 3316 | 10043 |
| T [K] | 100(2) | 150(2) | 100(2) |
| Max. 2Θ, ° | 73.500 | 58.748 | 50.000 |
| Reflns measured | 32780 |
100772 |
68100 |
| Unique reflns | 8464 | 20327 |
20895 |
| Parameters | 586 | 1035 | 1529 |
| Restraints | 0 | 1238 | 797 |
| Reflns [Fo > 2(Fo)] | 7252 | 15449 |
11354 |
| R 1 [Fo > 2σ(Fo)] | 0.0613 | 0.0818 | 0.1158 |
| wR2 (all data) | 0.1468 | 0.1824 | 0.3277 |
| G.O.F | 0.998 | 1.105 | 1.063 |
| CCDC number | 1518488 | 1518412 | 1518411 |
General
UV-visible-NIR spectra were obtained using KBr pellets on a PerkinElmer Lambda 1050 spectrometer in the 250–2500 nm range. FT-IR spectra were obtained using KBr pellets with a PerkinElmer Spectrum 400 spectrometer (400–7800 cm−1). EPR spectra were recorded on polycrystalline samples of 1–3 from 4 up to 295 K and back from 295 to 150 K for 2 with a JEOL JES-TE 200 X-band ESR spectrometer equipped with a JEOL ES-CT470 cryostat.Crystal structure determination
Crystallographic data for 1–3 are listed in Table 3. Synchrotron X-ray data for 1 were collected at 100 K on the BL14.3 at the BESSY storage ring (Berlin, Germany) using a MAR225 detector, λ = 0.8950 Å.The data for 2 and 3 were collected on an Oxford diffraction “Gemini-R” CCD diffractometer with graphite monochromated MoKα radiation at low temperature using an Oxford Instrument Cryojet cooler. Experimental data were processed using CrysAlisPro software, Oxford Diffraction Ltd, raw data reduction to F2 was carried out using Bruker SAINT.38 The structures were solved by direct methods and refined by the full-matrix least-squares method against F2 using SHELX-2013 and 2014.39 Non-hydrogen atoms were refined in the anisotropic approximation. Positions of hydrogen atoms were calculated geometrically.
The Ti1 and O1 atoms in structure 1 are disordered around an inversion center. The structure of 2 contains one strongly disordered C6H4Cl2 molecule with the 0.355(3)/0.275(3)/0.231(3)/0.139(3) occupancies. In the structure of 3 there are completely ordered {OTiIV(AceTPrzPz4−)}2− dianion and one and two halves of independent PPN+ cations. One and half independent PPN+ cations contain one statistically disordered phenyl group. Two positions of solvent molecules contain strongly disordered C6H4Cl2 and C6H5CN molecules. Due to the strong disorder of some solvent molecules in 2 and 3 and phenyl substituents of PPN+ in 3, multiple restraints were used for the crystal structure refinement of these salts. To keep the solvent and phenyl geometry close to the ideal one in the disordered groups, the bond length restraints were applied along with the next-neighbor distances using the SADI SHELXL instruction. To keep the anisotropic thermal parameters of the disordered groups within reasonable limits the displacement components were restrained using the SIMU and DELU SHELXL instructions.
Acknowledgements
The reported study was funded by RFBR according to the research project no. 16-33-00588 mol_a.Notes and references
- C. G. Claessens, W. J. Blau, M. Cook, M. Hanack, R. J. M. Nolte, T. Torres and D. Wöhrle, Monatsh. Chem., 2001, 132, 3 CrossRef CAS.
- D. Wöhrle, G. Schnurpfeil, S. G. Makarov, A. Kazarin and O. N. Suvorova, Macroheterocycles, 2012, 5, 191 CrossRef.
- D. C. Y. Ethelbhert, M. Matsuda, H. Tajima, A. Kikuchi, T. Taketsugu, N. Hanasaki, T. Naito and T. Inabe, J. Mater. Chem., 2009, 19, 718 RSC.
- T. Inabe and H. Tajima, Chem. Rev., 2004, 104, 5503 CrossRef CAS PubMed.
- D. K. Rittenberg, L. Baars-Hibbe, A. B. Böhm and J. S. Miller, J. Mater. Chem., 2000, 10, 241 RSC.
- D. V. Konarev, L. V. Zorina, S. S. Khasanov, E. U. Hakimova and R. N. Lyubovskaya, New J. Chem., 2012, 36, 48 RSC.
- E. W. Y. Wong and D. B. Leznoff, J. Porphyrins Phthalocyanines, 2012, 16, 154 CrossRef CAS.
- J. A. Cissell, T. P. Vaid, A. G. DiPasquale and A. L. Rheingold, Inorg. Chem., 2007, 46, 7713 CrossRef CAS PubMed.
- J. A. Cissell, T. P. Vaid and A. L. Rheingold, Inorg. Chem., 2006, 45, 2367 CrossRef CAS PubMed.
- E. W. Y. Wong, C. J. Walsby, T. Storr and D. B. Leznoff, Inorg. Chem., 2010, 49, 3343 CrossRef CAS PubMed.
- W. Zhou, R. H. Platel, T. T. Tasso, T. Furuyama, N. Kobayashi and D. B. Leznoff, Dalton Trans., 2015, 44, 13955 RSC.
- M. Tahiri, P. Doppelt, J. Fischer and R. Weiss, Inorg. Chim. Acta, 1987, 127, L1 CrossRef CAS.
- D. V. Konarev, A. V. Kuzmin, S. V. Simonov, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2012, 41, 13841 RSC.
- H. Hückstädt and H. Homborg, Z. Anorg. Allg. Chem., 1998, 624, 715 CrossRef.
- D. V. Konarev, S. S. Khasanov, M. Ishikawa, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Inorg. Chem., 2013, 52, 3851 CrossRef CAS PubMed.
- D. V. Konarev, A. V. Kuzmin, S. S. Khasanov and R. N. Lyubovskaya, Dalton Trans., 2013, 42, 9870 RSC.
- D. V. Konarev, L. V. Zorina, M. Ishikawa, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Cryst. Growth Des., 2013, 13, 4930 CAS.
- D. V. Konarev, A. V. Kuzmin, M. Ishikawa, Y. Nakano, M. A. Faraonov, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Eur. J. Inorg. Chem., 2014, 3863 CrossRef CAS.
- D. V. Konarev, L. V. Zorina, S. S. Khasanov, A. L. Litvinov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2013, 42, 6810 RSC.
- D. V. Konarev, A. V. Kuzmin, M. A. Faraonov, M. Ishikawa, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Chem. – Eur. J., 2015, 21, 1014 CrossRef CAS PubMed.
- D. V. Konarev, S. I. Troyanov, M. Ishikawa, M. A. Faraonov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, J. Porphyrins Phthalocyanines, 2014, 18, 1157 CrossRef CAS.
- D. V. Konarev, A. V. Kuzmin, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2014, 43, 13061 RSC.
- M. Tanaka, in High Performance Pigments, ed. E. B. Faulkner and R. J. Schwartz, Wiley-VCH, Weinheim, 2009, p. 275 Search PubMed.
- K.-Y. Law, Chem. Rev., 1993, 93, 449 CrossRef CAS.
- Y. Wang and D. Liang, Adv. Mater., 2010, 22, 1521 CrossRef CAS PubMed.
- H.-J. Kang, E.-H. Kang, S.-W. Park, J.-W. Lee and J.-K. Lee, Macromol. Symp., 2006, 235, 195 CrossRef CAS.
- M. Mayukh, C. M. Sema, J. M. Roberts and D. V. McGrath, J. Org. Chem., 2010, 21, 7893 CrossRef PubMed.
- (a) E. A. Lukyanets and V. N. Nemykin, J. Porphyrins Phthalocyanines, 2010, 14, 1 CrossRef CAS; (b) V. N. Nemykin and E. A. Lukyanets, ARKIVOC, 2010,(Part 1), 136 CAS; (c) E. A. Lukyanets and V. N. Nemykin, in Handbook of porphyrin science with applications to chemistry, physics, materials science, engineering, biology and medicine: synthetic methodology, Series Handbook of Porphyrin Science, 2010, vol. 3, pp. 1–323 Search PubMed.
- K. Oka, O. Okada and K. Nukada, Jpn. J. Appl. Phys., 1992, 31, 2181 CrossRef CAS.
- O. Okada, K. Oka and M. Iijima, Jpn. J. Appl. Phys., 1993, 32, 3556 CrossRef CAS.
- A. J. Ramadan, L. A. Rochford, D. S. Keeble, P. Sullivan, M. P. Ryan, T. S. Jones and S. Heutz, J. Mater. Chem. C, 2015, 3, 461 RSC.
- M. P. Donzello, C. Ercolani, V. Novakova, P. Zimcik and P. A. Stuzhin, Coord. Chem. Rev., 2016, 309, 107 CrossRef CAS.
- L. A. Tomachynsk, V. Ya. Chernii and S. V. Volkov, Russ. J. Inorg. Chem., 2002, 47, 208 Search PubMed.
- A. B. P. Lever, E. R. Milaeva and G. Speier, in Phthalocyanines: Properties and Applications, ed. C. C. Leznoff and A. B. P. Lever, VCH Publishers, Inc., New York, 1993, vol. 3, p. 1 Search PubMed.
- K. Takahashi, Y. Aoki, T. Sugitani, F. Moriyama, Y. Tomita, M. Handa, K. Kasuga and K. Sogabe, Inorg. Chim. Acta, 1992, 201, 247 CrossRef CAS.
- B. L. Wheeler, G. Nagasubramanian, A. J. Bard, L. A. Schechtman, D. R. Dininny and M. E. Kenney, J. Am. Chem. Soc., 1984, 106, 7405 CrossRef.
- D. V. Konarev, S. S. Khasanov, E. I. Yudanova and R. N. Lyubovskaya, Eur. J. Inorg. Chem., 2011, 816 CrossRef CAS.
- Bruker AXS Inc., Madison, Wisconsin, USA.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The IR spectra of the starting compounds and salts 1–3. CCDC 1518488, 1518412 and 1518411. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt04896j |
| This journal is © The Royal Society of Chemistry 2017 |