
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sequence of phase transitions in (NH4)3SiF7†
S. V.
Mel'nikova
a,
M. S.
Molokeev
 *ab,
N. M.
Laptash
c,
E. I.
Pogoreltsev
ad,
S. V.
Misyul‡
d and
I. N.
Flerov
ad
*ab,
N. M.
Laptash
c,
E. I.
Pogoreltsev
ad,
S. V.
Misyul‡
d and
I. N.
Flerov
ad
aLaboratory of Crystal Physics, Kirensky Institute of Physics, Federal Research Center KSC SB RAS, Krasnoyarsk, 660036, Russia. E-mail: msmolokeev@mail.ru
bDepartment of Physics, Far Eastern State Transport University, Khabarovsk, 680021, Russia
cInstitute of Chemistry, Far Eastern Branch of RAS, Vladivostok, 690022, Russia
dSiberian Federal University, Krasnoyarsk, 660074, Russia
First published on 18th January 2017
Abstract
Single crystals of silicon double salt (NH4)3SiF7 = (NH4)2SiF6·NH4F = (NH4)3[SiF6]F were grown and studied by the methods of polarization optics, X-ray diffraction and calorimetry. A sequence of symmetry transformations with the temperature change was established: P4/mbm (Z = 2) (G1) ↔ Pbam (Z = 4) (G2) ↔ P21/c (Z = 4) (G3) ↔ P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Z = 4) (G4) ↔ P21/c (Z = 8) (G5). Crystal structures of different phases were determined. The experimental data were also interpreted by a group-theoretical analysis of the complete condensate of order parameters taking into account critical and noncritical atomic displacements. Strengthening of the N–H⋯F hydrogen bonds can be a driving force of the observed phase transitions.
(Z = 4) (G4) ↔ P21/c (Z = 8) (G5). Crystal structures of different phases were determined. The experimental data were also interpreted by a group-theoretical analysis of the complete condensate of order parameters taking into account critical and noncritical atomic displacements. Strengthening of the N–H⋯F hydrogen bonds can be a driving force of the observed phase transitions.
1 Introduction
Silicon is the second most abundant element in the Earth's crust. Its double fluoride salt (NH4)3SiF7 = (NH4)3SiF7·NH4F = (NH4)3[SiF6]F is the main product of fluoride processing of natural silicon-containing raw materials (quartz and silicates of different structure and compositions) with ammonium hydrogen difluoride (NH4HF2).1–5 Fluorination reactions proceed exothermally (even at room temperature) with an entropy reserve. Elemental silicon also interacts with NH4HF2 with the formation of (NH4)3SiF7.6,7 It is accepted in the literature that this salt was first prepared by Marignac in 1859.8 He obtained it in the form of square prisms from aqueous fluoride solution. However, similar shape (‘tetrahedral prisms’) crystals were observed by Davy as early as 1812.9 The crystal structure of (NH4)3SiF7 was studied in 1942 by Hoard and Williams,10 and solved using oscillation photographs. In spite of the measurements being performed very carefully and the final model crystal structure being adequate, the authors reported large discrepancies between the calculated and observed reflection amplitudes. They associated these discrepancies with the fluorine anions, for which large temperature corrections should be performed, and pointed out that it would be of interest to obtain diffraction data for (NH4)2SiF6·NH4F at liquid air temperatures. This task has not been performed until now. Moreover, no data on phase transitions in this compound are available in the literature.Recently, we found that the isostructural compound (NH4)3GeF7 (space group P4/mbm) underwent several reversible phase transitions below room temperature with an unusual sequence P4/mbm ↔ Pbam ↔ P21/c ↔ Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) .11 Moreover, some other fluoride double salts (NH4)3M4+F7 (M4+ = Ti4+, Sn4+) undergo phase transitions but of different sequences, and a correlation with the M4+ ionic radius was found.11–16 Therefore, one might expect the temperature variation would also destabilise the room temperature structure of the silicon double fluoride salt, leading to phase transitions.
.11 Moreover, some other fluoride double salts (NH4)3M4+F7 (M4+ = Ti4+, Sn4+) undergo phase transitions but of different sequences, and a correlation with the M4+ ionic radius was found.11–16 Therefore, one might expect the temperature variation would also destabilise the room temperature structure of the silicon double fluoride salt, leading to phase transitions.
In the present paper, a complex study of (NH4)3SiF7 using optical, X-ray, and calorimetric measurements revealed that the succession of structural transformations differs from the sequences observed earlier by us in other related compounds (NH4)3M4+F7. The crystal structures of all the phases were solved and a reliable model of structural distortions associated with certain order parameters was suggested.
2 Experimental
2.1 Synthesis and crystal growth
Single crystals of a double salt (NH4)3SiF7 were grown from aqueous fluoride solution with an excess of NH4F or NH4HF2. Solid SiO2, (NH4)2SiF6, NH4F and NH4HF2 of a reagent grade were used. Silicon dioxide (amorphous or quartz) was fluorinated with NH4HF2 at 150–200 °C in a Pt or glass-carbon beaker. It should be noted that the reaction began at room temperature itself while grinding together the initial components. A double excess, at least, of NH4HF2 was taken relative to the stoichiometric reaction: SiO2 + 3.5NH4HF2 = (NH4)3SiF7 + 0.5NH3 + 2H2O. The cake obtained in this way was then water-leached, filtered and colourless prismatic single crystals of (NH4)3SiF7 were formed by slow evaporation of the final solution under ambient conditions. The crystals were recovered by vacuum filtration and air-dried. A large excess of NH4F was needed to obtain (NH4)3SiF7 in the NH4F–(NH4)2SiF6–H2O system.17 We used a triple excess relative to the stoichiometric reaction: (NH4)2SiF6 + NH4F = (NH4)3SiF7. For example, 20 g of (NH4)2SiF6 and 12–15 g of NH4F were mixed and dissolved in 100 ml of deionised water, filtered and slowly evaporated at ambient temperature with the formation of transparent tetragonal prisms. It is not surprising that Davy had observed similar crystals during the addition of excess ammonia to hexafluorosilicic acid (‘the subsilicated fluoric acid’).92.2 Calorimetric investigations
The temperature stability of the initial tetragonal phase P4/mbm of (NH4)3SiF7 was examined by measurements with a DSM-10M differential scanning microcalorimeter (DSM). Recently it has been shown that among fluorides (NH4)3M4+F7 there are at least two crystals undergoing a phase transition into the cubic phase Pmm above room temperature at ambient – (NH4)3SnF7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 – or high – (NH4)3TiF716 – pressure. Therefore the thermal investigations were performed in a wide temperature range of 100–460 K.
13 – or high – (NH4)3TiF716 – pressure. Therefore the thermal investigations were performed in a wide temperature range of 100–460 K.
A sample of the powdered compound under study was placed into an aluminium sample holder. Calorimetric measurements were carried out under a helium atmosphere on several samples with a mass of about 0.03–0.06 g. In order to gain information on the hysteretic phenomena associated with the probable structural transformations in (NH4)3SiF7, experiments were carried out using heating and cooling runs with a rate of temperature change dT/dτ of 8 to 32 K min−1. The calibration of the temperature and enthalpy scales was performed using the melting parameters of pure indium as well as tabulated data of enthalpy against temperature for Al2O3. The temperature was determined with an accuracy of ±1 K and the uncertainty on the enthalpy and entropy values was estimated to be ±5 J mol−1 and ±0.1 J mol−1 K−1, respectively.
2.3 Optical observations
Optical studies were performed using a polarizing microscope (Axioskop-40) and a Linkam LTS 350 temperature chamber. The investigations were carried out in a quasi-static regime with an accuracy of ±0.1 K in the range of 90–400 K. The birefringence was measured using a Berek compensator (Leica) with an accuracy of ≈0.00001.2.4 X-ray investigations
Optical experiments proved a complex microtwinning of the crystal under cooling. Currently it seems impossible to solve or refine the crystal structure by single crystal diffraction due to the complex picture of overlapping peaks from different domains. Moreover, a resolution of the single crystal experiment is worse than that of the powder diffraction. Subtle changes of cell parameters can be appropriately investigated by only using a powder diffraction technique, which was chosen in our case.The powder diffraction data of (NH4)3SiF7 for Rietveld analysis were collected with a Bruker D8 Advance powder diffractometer (Cu-Kα radiation) and a linear VANTEC detector. The beam was controlled by a 0.6 mm fixed divergence slit, 6 mm receiving VANTEC slit and Soller slits. An Anton Paar TTK450 attachment was used for low- and high-temperature measurements.
The diffraction data in the 2θ range 5° to 120° were collected at T = 303 K, T = 213 K, T = 183 K, and T = 133 K (Fig. 1S†). These X-ray patterns were used to solve and refine the crystal structures. Eighteen additional X-ray patterns with smaller exposition time were collected from 133 K to 303 K to identify the space group transformation at phase transition.
The crystal structures of the low-temperature phases were solved by a distortion-mode refinement of X-ray data using the TOPAS 4.218 program, with a file generated by ISODISPLACE. Amplitudes of critical symmetry-modes were used to find a structural model by the simulated anneal method. Then all atomic coordinates were refined independently by the Rietveld method. Isotropic thermal parameters of Si atoms were refined independently, but all F atoms as well as N atoms had one thermal parameter.
3 Results and discussion
3.1 Calorimetry
No anomalous behaviour of the DSM-signal was observed in experiments above room temperature until the decomposition (∼450 K) of (NH4)3SiF7. Below room temperature, calorimetric studies revealed four anomalies reproducible in the heating and cooling runs, which were suggested to be associated with the successive phase transitions G1(T1) ↔ G2(T2) ↔ G3(T3) ↔ G4(T4) ↔ G5. The background of the DSM-signal(T) dependence, considered to be the lattice contribution into the total DSM-signal, was extracted by polynomial approximation of the experimental data far from the anomalous regions. The quantity determined as the difference between the total DSM-signal and its lattice component is associated with the excess molar heat capacity ΔCp connected with the order parameters of transformations which will be discussed below.Fig. 1a depicts the temperature dependence of ΔCp for one of the samples obtained in experiments with dT/dτ = 8 K min−1. The behavior of ΔCp at the transformation G1 ↔ G2 is typical for the second order: a small asymmetric anomaly characterised by a broad temperature range of the ΔCp existence below T1 = 263 ± 1 K, which was detected as the temperature of the ΔCp maximum. A significant hysteresis in the transition temperatures T2, T3, and T4 as well as its strong dependence on the heating/cooling rate was observed during the thermal cycling. These experimental facts confirm that transformations G2 ↔ G3 ↔ G4 ↔ G5 are of the first order. Analysis of the Ti dependence on dT/dτ in the heating and cooling modes allowed the determination of the real values of phase transition temperatures and their hysteresis, which were considered as parameters under equilibrium conditions when dT/dτ → 0: T2 = 198 ± 1 K, δT2 = 1 K; T3 = 194 ± 1 K, δT3 = 2 K; T4 = 170 ± 1 K, δT4 = 5 K.
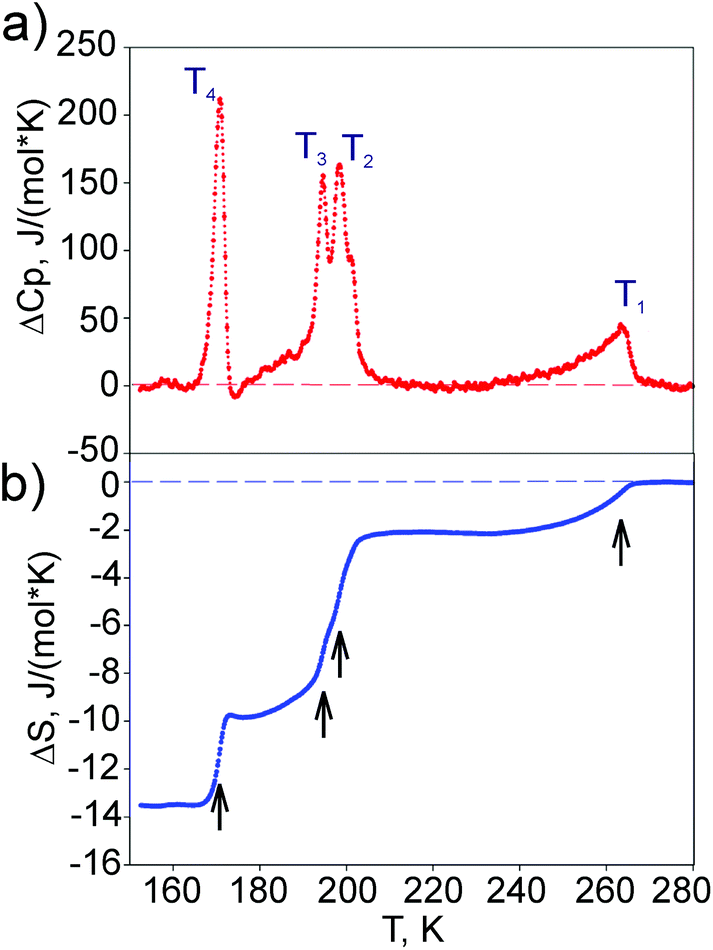 | ||
| Fig. 1 Temperature dependences of the excess heat capacity (a) and entropy (b) associated with the successive phase transitions G1 ↔ G2 ↔ G3 ↔ G4 ↔ G5. | ||
The entropy change associated with the successive phase transitions was determined by integration of the (ΔCp/T)(T) function. Fig. 1b depicts the temperature behaviour of entropy. Its total value associated with the succession of phase transitions G1 ↔ G2 ↔ G3 ↔ G4 ↔ G5 is rather large: ∑ΔSi = 13.6 ± 3.0 J mol−1 K−1, revealing some order–disorder process in the mechanism of structural distortions. Because a DSM technique is not sensitive to the small excess heat capacity which can exist far below the phase transition point, we did not discuss the value of ΔSi for individual transformations. For this we plan to perform a more detailed study of heat capacity using an adiabatic calorimeter.
Thus, the calorimetric measurements have shown that the cationic substitution Si → Ge in compounds (NH4)3M4+F7 is accompanied by a small broadening of the temperature range of the P4/mbm phase stability (T1 = 280 K in (NH4)3GeF7) and an increase in the number of the phase transformations.11
3.2 Polarization optics
The single crystals of (NH4)3SiF7, like (NH4)3GeF7, have a habitus of elongated rectangular prisms with the developed facets of (110)t. The four-fold axis is preferably located along the longest edge of the crystal, so the plate-like sample with a (001)t orientation is very rare. Two of these samples are presented in Fig. 2. The upper crystal consists of two blocks. The left side of this sample has an orientation (110)t, and extinction along the growth edges. The right-hand block side (Δnc = 0) is optically isotropic and has an orientation of (001)t. The uniaxial pressure increase (up to destruction of the sample) did not change the orientation of the fourth-order axis in (NH4)3SiF7, similar to that found earlier for (NH4)3GeF7.11 Thus, silicon and germanium compounds differ from (NH4)3TiF7, where small uniaxial pressure reoriented this axis at 90 degrees.15 | ||
| Fig. 2 Observation of crystals (NH4)3SiF7 under polarized light upon cooling: (a) phase G1, (b) G2, (c) G3, (d) G4, (e and f) G5. The upper sample consists of two blocks with different orientations of the four-fold axis in the G1 phase, the lower plate has a (110)t orientation. | ||
Fig. 2 presents photographs of the (NH4)3SiF7 plates as grown in polarised light. At room temperature the right-hand side of the upper sample is dark; and the left side has a ‘direct extinction’ in accordance with a tetragonal symmetry (G1-phase) (Fig. 2a). Upon cooling the pattern changes significantly. Below T1↓ = 263 K the dark area of the sample illuminates (Fig. 2b). The entire crystal illuminates completely or extinguishes along the growth verge (110)t when the microscope stage is rotated by 90° (G2-phase).
Further cooling leads to the appearance (disappearance) at T2↓ = 198 K (T2↑ = 199 K) of a layered structure with boundaries along [110]t in the left-hand block (Fig. 2c). Optical indicatrices in the neighbouring twins are disoriented to the direction [110]t. In the right-hand part of the specimen the direct extinction remains.
Upon cooling below T3↓ = 191 K (T3↑ = 192 K), the specimen shows a very complicated twin structure in three orthogonal directions with a ‘floating’ position of extinctions (Fig. 2d). Further cooling reveals an abrupt phase transition at T4↓ = 162 K (T4↑ = 167 K) into the tetragonal phase with, at first glance, a dark right side (Fig. 2e). But in contrast to the initial tetragonal phase (Fig. 2a) in the twins with coexistence, ‘direct’ (Fig. 2f), or ‘oblique’ (Fig. 2e) extinctions are observed in the lower (110)t plate.
The illumination of the dark area of the sample below T1↓ = 263 K (Fig. 2b) is associated with a reduction of crystal symmetry and the appearance of optical anisotropy along the [001]t direction. Fig. 3 shows the temperature behaviour of the Δnc birefringence, which appears at T1 and smoothly changes near T2 and T3, while at T4 there is a sharp disappearance of Δnc.
 | ||
| Fig. 3 The appearance of optical anisotropy in the plate (001)t at T1, and its change at T2, T3, T4: curve 1 – Δnc(T). The rotation of the optical indicatrix around [110]t and [001]t directions: curve 2 – φa(T); curve 3 – φc(T); curve 4 – φb(T). | ||
The twin structure in very thin plates (∼50 μm) consists of rather large domains in the phase G5 which allows measurement of the temperature dependence of the birefringence Δn(T) and the angle of rotation of the optical indicatrix φ(T) (Fig. 4).
 | ||
| Fig. 4 Temperature dependence of birefringence Δn(T) of the crystal (NH4)3SiF7 in the plate (110)t, measured during cooling (curve 1) and heating (curve 2). Curve 3 is Δn(T) in (NH4)3GeF7.11 | ||
At room temperature the main birefringence of tetragonal (NH4)3SiF7 (Δn = no − ne = 0.0037) is higher than that in (NH4)3GeF7 (Δn = 0.0034 (ref. 11)) and (NH4)3TiF7 (Δn = 0.0023 (ref. 12)). Fig. 4 shows that the birefringence in the phase G1 of (NH4)3SiF7 is stable, varying slightly with temperature and gradually decreasing below T1.
The DSM and polarization-optical studies as well as measuring the birefringence and the rotation angles of the indicatrix in the (NH4)3SiF7 crystal allowed the establishment of the existence of four structural phase transitions between the G1, G2, G3, G4 and G5 phases, with different symmetries. According to the temperature dependences of DSM and optical characteristics (Fig. 1, 3 and 4), the character of transitions in (NH4)3SiF7 is the following: the second order for G1 ↔ G2, and first order close to the tricritical point for G2 ↔ G3 ↔ G4. Therefore, we assume that a successive lowering of crystal symmetry taking place through three phase transitions at T1, T2, and T3 can be described in the framework of group–subgroup relationships.
The presence of the optical anisotropy Δnc and the position of extinction along the [110]t direction reveals the geometry of a new cell in the G2 phase. The group symmetry of that phase is most likely orthorhombic. Two axes of the unit cell should be perpendicular to the 4-fold axis and directed along the unit cell diagonals of the tetragonal G1 phase: [110]t = [010]or; [110]t = [100]or; [001]t = [001]or.
The geometry of twinning and rotation of the optical indicatrix only around the [110]t = [100]or direction indicates the symmetry of the G3 phase to be monoclinic P21/c11. The screw twofold axis is perpendicular to the former four-fold axis and directed along [100]or of an orthorhombic unit cell of the G2 phase. Thus, the successive lowering of symmetry during phase transitions at T1 and T2 was established, similar to that observed in (NH4)3GeF7: P4/mbm (G1) ↔ Pbam (G2) ↔ P21/c (G3).11
Further temperature lowering leads to an additional cooling decrease in the symmetry of (NH4)3SiF7 at T3. It can be seen (Fig. 3) that the angle of rotation φa is maintained at the lower phase G4, which is added by the rotation φb(T) and φc(T) around different axes of the orthorhombic unit cell. Therefore, the crystal symmetry group in this phase can be selected as triclinic P.
The phase transition G4 ↔ G5 has a first-order character: there are rapid jumps of extinction angles and birefringence values (Fig. 3). At first glance, the sample in the bottom phase G5 looks similar to that at room temperature: right-hand dark side (Δnc = 0) and orange interference colour on the left-hand side (Fig. 2e). All optical data indicate the crystal identification to be tetragonal. However, in the (110)t plane the observed twins have different positions of extinction. Simultaneous ‘direct’ and ‘oblique’ extinctions are observed (Fig. 2e and f). Thus, the crystal symmetry group in the G5 phase can be chosen as monoclinic. The above studies show the difference between the monoclinic phases G3 and G5, with respect to a two-fold axis. This axis is directed along [110]t = [100]or in G3, but along [110]t = [010]or in G5.
Thus, the polarisation-optical studies suggest the existence of a rather complicated sequence of crystal symmetry changes upon cooling: P4/mbm (G1) ↔ Pbam (G2) ↔ P21/c11 (G3) ↔ P (G4) ↔ P121/c1 (G5).
A shear deformation with component x4 (within the orthorhombic cell) appears in phase G3, causing rotation of the optical indicatrix on the angle φa (Fig. 3, curve 2). Additional components of shear deformation x5 and x6 appear in the triclinic G4 phase, causing the indicatrix rotation φb and φc (Fig. 3, curves 3, 4). A further temperature lowering results in the formation of the monoclinic phase G5, where the components x4 and x6 disappear and x5 remains, changing its sign (Fig. 3, curve 4). An unusually large angle of indicatrix rotation φb ≈ 25° is characteristic of the ‘proper’ ferroelastic transitions when a parameter of transition δ is linearly related to the shear component of the spontaneous deformation δ ≈ x5.
3.3 X-ray diffraction and group theory analysis
As the crystal structure of (NH4)3SiF7 at room temperature was solved earlier,10 we used this model (P4/mbm) to make a Rietveld refinement by using TOPAS 4.2.18 The refinement was stable and gave low R-factors (Table 1 and Fig. 1S(a)†). Atomic coordinates are given in Table 1S,† main bond lengths are given in Table 2, and the crystal structure is presented in Fig. 5a. The compound (NH4)3SiF7 was found to be very pure and all lines were indexed by the P4/mbm phase (Fig. 1S(a)†). Cell parameters after refinement were close to those from ref. 11. | ||
| Fig. 5 Crystal structures of (NH4)3SiF7: (a) G1 phase (P4/mbm) at T = 303 K; (b) G2 phase (Pbam) at T = 213 K, (c) G4 phase (P) at T = 183 K; (d) G5 phase (P21/c) at T = 133 K. | ||
| Phase | G1 | G2 | G4 | G5 |
|---|---|---|---|---|
| Temperature, K | 303 | 213 | 183 | 133 |
| Sp. Gr. | P4/mbm | Pbam |
P |
P21/c |
| a, Å | 8.0750(2) | 11.3989(5) | 5.8256(2) | 11.5710(3) |
| b, Å | 8.0750(2) | 11.3759(4) | 11.3567(4) | 11.4073(3) |
| c, Å | 5.8613(2) | 5.8343(2) | 11.4063(3) | 11.4300(3) |
| α, ° | 90 | 90 | 91.414(3) | 90 |
| β, ° | 90 | 90 | 89.808(3) | 90.745(2) |
| γ, ° | 90 | 90 | 90.052(4) | 90 |
| V, Å3 | 382.19(2) | 756.55(5) | 754.39(4) | 1508.57(6) |
| Z | 2 | 4 | 4 | 8 |
| 2θ-interval, ° | 5–120 | 5–120 | 5–120 | 5–120 |
| Number of refined coordinates and thermal parameters | 10 | 22 | 66 | 66 |
| Number of calculated independent reflections | 181 | 642 | 2286 | 2263 |
| R wp, % | 10.20 | 10.02 | 10.26 | 8.66 |
| R p, % | 7.72 | 7.64 | 7.38 | 6.21 |
| R exp, % | 3.83 | 5.94 | 5.89 | 3.42 |
| χ 2 | 2.66 | 1.69 | 1.74 | 2.54 |
| R B, % | 4.74 | 4.04 | 3.38 | 3.95 |
| P4/mbm (G1 phase) at T = 303 K | |||
| Si1–F2 × 2 | 1.623(3) | Si1–F3 × 4 | 1.656(3) |
| N1–F1 × 2 | 2.9307(1) | N1–F2 × 4 | 3.109(3) |
| N2–F3 × 2 | 2.848(4) | N2–F1 × 2 | 2.907(3) |
| N2–F2 × 2 | 3.009(1) | ||
| Pbam (G2 phase) at T = 213 K | |||
| Si1–F3 × 2 | 1.71(1) | Si1–F4 × 4 | 1.624(7) |
| Si2–F2 × 2 | 1.636(9) | Si2–5 × 4 | 1.691(7) |
| N1–F3 | 2.77(2) | N1–F2 | 2.84(2) |
| N1–F1 × 2 | 2.9207(7) | N1–F5 × 2 | 3.23(1) |
| N2–F1 | 2.88(2) | N2–F4 × 2 | 2.93(1) |
| N2–F1 | 2.94(2) | N2–F2 × 2 | 2.998(4) |
| N3–F5 × 2 | 2.81(1) | N3–F1 | 2.82(2) |
| N3–F4 × 2 | 2.88(1) | N3–F1 | 2.97(2) |
|
P (G4 phase) at T = 183 K |
|||
| Si1–F3 × 2 | 1.66(1) | Si3–F6 × 2 | 1.71(2) |
| Si1–F9 × 2 | 1.63(2) | Si3–F8 × 2 | 1.69(2) |
| Si1–F13 × 2 | 1.73(2) | Si3–F12 × 2 | 1.65(2) |
| Si2–F4 × 2 | 1.61(2) | Si4–F5 × 2 | 1.78(2) |
| Si2–F10 × 2 | 1.78(2) | Si4–F7 × 2 | 1.68(2) |
| Si2–F14 × 2 | 1.60(2) | Si4–F11 × 2 | 1.70(2) |
| N1–F6 | 2.81(3) | N1–F1 | 2.91(5) |
| N1–F1 | 2.92(5) | N1–F10 | 2.94(3) |
| N1–F7 | 3.02(3) | N1–F3 | 3.11(3) |
| N2–F5 | 2.69(3) | N2–F2 | 2.88(5) |
| N2–F4 | 2.93(3) | N2–F2 | 2.98(5) |
| N2–F8 | 3.01(3) | N2–F3 | 3.17(3) |
| N3–F2 | 2.68(3) | N3–F7 | 2.76(3) |
| N3–F13 | 2.82(3) | N3–F11 | 2.88(3) |
| N3–F3 | 2.92(4) | N3–F1 | 2.96(3) |
| N4–F8 | 2.73(4) | N4–F10 | 2.75(3) |
| N4–F1 | 2.77(3) | N4–F12 | 2.95(3) |
| N4–F4 | 2.98(4) | N4–F14 | 3.01(3) |
| N5–F12 | 2.68(3) | N5–F2 | 2.77(3) |
| N5–F6 | 2.77(4) | N5–F13 | 2.90(3) |
| N5–F9v | 2.91(4) | N5–F8 | 3.02(4) |
| N6–F2 | 2.72(3) | N6–F10 | 2.76(3) |
| N6–F11 | 2.80(3) | N6–F14 | 2.85(3) |
| N6–F5 | 2.97(4) | N6–F1 | 3.01(3) |
| P21/c (G5 phase) at T = 133 K | |||
| Si1–F5 × 2 | 1.70(1) | Si3–F3 × 2 | 1.64(2) |
| Si1–F7 × 2 | 1.66(1) | Si3–F11 × 2 | 1.66(1) |
| Si1–F9 × 2 | 1.75(1) | Si3–F13 × 2 | 1.58(1) |
| Si2–F6 × 2 | 1.68(1) | Si4–F4 × 2 | 1.57(2) |
| Si2–F8 × 2 | 1.83(1) | Si4–F12 × 2 | 1.67(1) |
| Si2–F10 × 2 | 1.71(1) | Si4–F14 × 2 | 1.79(1) |
| N1–F5 | 2.83(2) | N1–F3 | 2.84(2) |
| N1–F11 | 2.87(2) | N1–F7 | 2.89(2) |
| N1–F1 | 2.90(2) | N1–F2 | 3.06(2) |
| N2–F1 | 2.76(2) | N2–F4 | 2.85(2) |
| N2–F2 | 2.88(2) | N2–F10 | 3.02(2) |
| N2–F6 | 3.02(2) | N2–F6 | 3.16(2) |
| N3–F2 | 2.73(3) | N3–F10 | 2.78(2) |
| N3–F12 | 2.89(2) | N3–F4 | 2.95(3) |
| N3–F7 | 2.96(2) | N3–F3 | 3.00(3) |
| N4–F8 | 2.69(2) | N4–F1 | 2.72(3) |
| N4–F14 | 2.80(2) | N4–F9 | 2.88(2) |
| N4–F13 | 3.01(2) | N4–F4 | 3.01(3) |
| N5–F2 | 2.72(2) | N5–F12 | 2.79(2) |
| N5–F9 | 2.80(2) | N5–F8 | 2.84(2) |
| N5–F6 | 2.87(3) | N5–F13 | 2.94(2) |
| N6–F2 | 2.60(2) | N6–F14 | 2.73(2) |
| N6–F7 | 2.93(2) | N6–F6 | 2.97(3) |
| N6–F11 | 3.01(3) | N6–F10 | 3.06(2) |
One can see (Fig. 6 and 7a) that the first wave of superstructure peaks appears at T = 253 K. This temperature is 10 K lower than T1 = 263 K. This is because the powder sample in an X-ray experiment is cooled from the bottom, while diffraction happens only from the top surface, and the result is a temperature gradient in the powder sample. This temperature difference was further taken into account to make measurements in all other experiments. Additional peaks appearing at 253 K can be described by the emergence of instability at (1/2, 1/2, 0) k18-point (M) of the Brillouin zone of the G1 – P4/mbm unit cell (hereinafter the designation of irreducible representations (irrep) and points of the Brillouin zone are given in accordance with reference books).19,20 The observation of a similar situation in (NH4)3GeF711 allowed the conclusion that the mechanism of G1 ↔ G2 transformation in germanium and silicon compounds should be the same. The M1−⊕M4− irrep drives this phase transition, and the transformation can be written:  , where η1 and η2 are components of critical order parameters. The crystal structure for the Rietveld refinement in TOPAS 4.2 was prepared by the ISODISTORT program using the initial G1 phase (P4/mbm) and M1−⊕M4− irrep distortion. The refinement was stable and gave low R-factors (Table 1 and Fig. 1S(b)†). Atomic coordinates are given in Table 1S,† main bond lengths are given in Table 2 and the crystal structure is presented in Fig. 5b.
, where η1 and η2 are components of critical order parameters. The crystal structure for the Rietveld refinement in TOPAS 4.2 was prepared by the ISODISTORT program using the initial G1 phase (P4/mbm) and M1−⊕M4− irrep distortion. The refinement was stable and gave low R-factors (Table 1 and Fig. 1S(b)†). Atomic coordinates are given in Table 1S,† main bond lengths are given in Table 2 and the crystal structure is presented in Fig. 5b.
 | ||
| Fig. 6 X-ray powder patterns of (NH4)3SiF7 at different temperatures from 133 K to 303 K. | ||
 | ||
| Fig. 7 Fragment of X-ray powder patterns of (NH4)3SiF7 with: (a) superstructure peaks (3, 0, 3/2) and (1/2, 5/2, 2), which are prohibited in the G1 tetragonal phase (P4/mbm); (b) structure peaks (1, 1, 1) and (0, 2, 0) of the G1 tetragonal phase. | ||
Group-theoretical analysis showed that in (NH4)3SiF7, similarly to (NH4)3GeF7, the MF6 (M = Si, Ge) octahedron was split (MF6 → M1F6 + M2F6) at phase transition P4/mbm ↔ Pbam. The critical irrep M1−⊕M4− induces the greatest contribution to distortions and leads to the critical shift of fluorine ions along the a and b axes of the orthorhombic phase. These movements can be represented as rotation of the MF6 octahedron around the 4-fold axis in the tetragonal phase (Fig. 8a and b). The F–N bond lengths vary in the range of 2.907(3)–3.109(3) Å in the tetragonal phase to 2.77(2)–3.23(1) Å (Table 2) in the orthorhombic phase. The decrease of these bond lengths may be due to the formation of N–H⋯F hydrogen bonds. Since SiF6 octahedra are ordered in the G1 phase while NH4 tetrahedra are disordered due to a high local symmetry of one NH4 site (4/m), the transformation P4/mbm ↔ Pbam can be associated only with the ordering of NH4 groups. The formation of hydrogen bonds (short N⋯F contacts) after phase transition may serve as an indicator of NH4 ordering.
 | ||
| Fig. 8 The main distortions of the (NH4)3MeF7 (Me = Si, Ge) crystal structures at phase transitions, leading to rotation of MF6 octahedra during the transformations in different projections. Dashed red rectangle surrounds the formula unit of different phases. The sequence of G1 ↔ G2 ↔ G3 of the phases is the same for both (NH4)3SiF7 and (NH4)3GeF7, but their G3 ↔ G4 phase transitions are different. Further G4 ↔ G5 transition exists only in (NH4)3SiF7. | ||
It was impossible to carry out the X-ray measurements in the G3 phase due to the narrow temperature region of its existence. However, it was found from optical measurements that this phase is monoclinic.
The X-ray experiment in of the G4 phase (Fig. 1S(c),†Fig. 6 and 7b) reveals that the main peak broadening occurs, but there are no additional superstructure peaks (Fig. 7a); therefore it can be concluded that the G3 ↔ G4 phase transition is associated with the emergence of instability at the same (1/2, 1/2, 0) k18-point (M) of the Brillouin zone of P4/mbm. Optical investigations showed a triclinic symmetry of this phase. The space group P with basis of a tetragonal G1 phase (0,0,1), (1,1,0), (−1,1,0) fulfills all these conditions. The M1−⊕M4− and M5− irreps drive this phase transition and the transformation can be written as:

So we can imagine that the main rotations of SiF6 and GeF6 octahedra during G2 ↔ G3 phase transition are the same (Fig. 8c). After this transformation all similarities between (NH4)3SiF7 and (NH4)3GeF7 vanish. Under G3 ↔ G4 transition (NH4)3SiF7 shows transformation P21/c ↔ P
 (Fig. 8e), but (NH4)3GeF7 showed reconstructive phase transition P21/c ↔ Pa (Fig. 8d). Previously it was found that all NH4 groups in the Pa phase of (NH4)3GeF7 are ordered and all hydrogen atoms were localized. The crystal structure of the P phase of (NH4)3SiF7 shows several rather short F–N bonds: d(N2⋯F5) = 2.69(3) Å, d(N3⋯F2) = 2.68(3) Å, d(N4⋯F8) = 2.73(4) Å, and the range of F–N distances expands to 2.68(3)–3.17(3) Å (Table 2), which indicates the presence of N–H⋯F hydrogen bonds. However, the smallest contact over all N1⋯F contacts, d(N1⋯F6) = 2.81(3) Å, is not very short for a H-bond, therefore some NH4 disordering could remain in the crystal structure.
(Fig. 8e), but (NH4)3GeF7 showed reconstructive phase transition P21/c ↔ Pa (Fig. 8d). Previously it was found that all NH4 groups in the Pa phase of (NH4)3GeF7 are ordered and all hydrogen atoms were localized. The crystal structure of the P phase of (NH4)3SiF7 shows several rather short F–N bonds: d(N2⋯F5) = 2.69(3) Å, d(N3⋯F2) = 2.68(3) Å, d(N4⋯F8) = 2.73(4) Å, and the range of F–N distances expands to 2.68(3)–3.17(3) Å (Table 2), which indicates the presence of N–H⋯F hydrogen bonds. However, the smallest contact over all N1⋯F contacts, d(N1⋯F6) = 2.81(3) Å, is not very short for a H-bond, therefore some NH4 disordering could remain in the crystal structure.
Indeed, below T4, a new phase G5 in (NH4)3SiF7 was observed, and an additional wave of superstructure peaks appeared (Fig. 6 and 7a). All these new peaks can be described by the emergence of instability at (0, 0, 1/2) k19-point (Z) of the Brillouin zone of the G1 – P4/mbm unit cell. Indexing shows that a cell in the G5 phase becomes P21/c with twice the cell volume compared to the G4 phase (P). The M1−⊕M4−, M5− and Z1+ irreps drive this phase transition, and the transformation can be written as:  , where δ is a component of the critical order parameter. Cell parameters and the volume of the G5 phase (P21/c) (NH4)3SiF7 (Table 1 and Fig. 8f) finally became similar to those of the G4 phase (Pa) in (NH4)3GeF7 (ref. 11, Fig. 8d). The crystal structure G5 of (NH4)3SiF7 for Rietveld refinement in TOPAS 4.2 was prepared by the ISODISTORT program using the initial G1 phase (P4/mbm) and M1−⊕M4−, M5−, Z1+ irreps distortion. There are 30 amplitude modes which should be initially varied and their starting values should be found before Rietveld refinement. A simulated annealing procedure in TOPAS 4.2 was used, and a satisfactory model with low R-factors was obtained. The refinement of the obtained model was stable and gave low R-factors (Table 1 and Fig. 1S(d)†). Atomic coordinates are given in Table 1S;† the crystal structure is shown in Fig. 5d. The F–N distances are in the range of 2.60(2)–3.17(2) Å and indicate the formation of N–H⋯F hydrogen bonds. In comparison with the G4 phase their average values become smaller, and it seems that all NH4 groups are ordered in the G5 phase.
, where δ is a component of the critical order parameter. Cell parameters and the volume of the G5 phase (P21/c) (NH4)3SiF7 (Table 1 and Fig. 8f) finally became similar to those of the G4 phase (Pa) in (NH4)3GeF7 (ref. 11, Fig. 8d). The crystal structure G5 of (NH4)3SiF7 for Rietveld refinement in TOPAS 4.2 was prepared by the ISODISTORT program using the initial G1 phase (P4/mbm) and M1−⊕M4−, M5−, Z1+ irreps distortion. There are 30 amplitude modes which should be initially varied and their starting values should be found before Rietveld refinement. A simulated annealing procedure in TOPAS 4.2 was used, and a satisfactory model with low R-factors was obtained. The refinement of the obtained model was stable and gave low R-factors (Table 1 and Fig. 1S(d)†). Atomic coordinates are given in Table 1S;† the crystal structure is shown in Fig. 5d. The F–N distances are in the range of 2.60(2)–3.17(2) Å and indicate the formation of N–H⋯F hydrogen bonds. In comparison with the G4 phase their average values become smaller, and it seems that all NH4 groups are ordered in the G5 phase.
It is easy to see that the phase G1 (P4/mbm) (Fig. 8a), in fact, can be represented as a rotationally distorted antiperovskite ABX3,21 where A = [SiF6], B = [F] and X = [NH4], i.e. [SiF6][F][NH4]3. The cubic Pmm phase is a parent phase of antiperovskite21 (Fig. 9a), which is not realized in our case. However, taking Pmm as a parent phase, the analysis of sequential orientation changes of F(NH4)6 octahedra at phase transitions was performed. It should be noted that SiF6 were not taken into consideration because they were isolated and didn't form a system of linked octahedra. Therefore, using Glazer's notation22 (Fig. 9b) for the system of linked octahedral F(NH4)6, the following scheme was found:

 | ||
| Fig. 9 Relationship between perovskite-type structure and antiperovskite parent Pmm (G0) phase of [SiF6][F][NH4]3 (a). All possible octahedral tilts in neighboring layers of perovskites and corresponding Glazer's notations (b). Rotation of F(NH4)6 octahedra in a0a0c+ (c) and a0b−c+ (d), which are realized under phase transitions. | ||
One can see that the difference between (NH4)3SiF7 and (NH4)3GeF7 is not only in the number and the sequence character (G3 ↔ G4 ↔ G5) of phase transitions. Phase transition P4/mbm ↔ Pa in (NH4)3GeF7 is reconstructive and cannot be analysed by group-theoretical methods while P4/mbm ↔ P21/c transformation in (NH4)3SiF7 can be described in this way. It should be noted that K3SiF7,23 Rb3SiF7,24 and Cs3GeF725 also have the P4/mbm phase at room temperature, while Tl3SiF726 crystallizes in the P1c space group, but nothing is known about their phase transitions.
All patterns from 133 K to 303 K were treated in a similar way and the temperature dependence of cell parameters a, b, c (Fig. 2S(a)†), cell volume V (Fig. 2S(b)†) and angles α, β, γ (Fig. 2S(c)†) were plotted.
Further details of the crystal structures of G1, G2, G3 and G5 phases may be obtained from Fachinformationszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany (fax: (+49)7247-808-666; E-mail: crystdata@fiz-karlsruhe.de; http://www.fiz-karlsruhe.de/request_for_deposited_data.html on quoting the deposition numbers: CSD 432220; 432221; 432222 and 432223, respectively).
4. Conclusions
Single crystals of silicon double salt (NH4)3SiF7 = (NH4)2SiF6·NH4F = (NH4)3[SiF6]F were grown, and we should recognize that the history of the compound goes back, in fact, more than 200 years. Davy observed tetragonal prisms, which were evidently (NH4)3SiF7, in accordance with the tetragonal structure of this compound. We studied this compound by different methods. Calorimetric measurements revealed the succession of four reversible phase transitions below room temperature: T1 = 263 ± 1 K; T2 = 198 ± 1 K; T3 = 194 ± 1 K; T4 = 170 ± 1 K, and an absence of transformation into the Pmm phase up to decomposition. It was found that the application of uniaxial pressure does not change the orientation of the four-fold axis of (NH4)3SiF7, similar to (NH4)3GeF7,11 which is in agreement with the fact that they are isostructural at room temperature. Therefore, both (NH4)3SiF7 and (NH4)3GeF7 differ from (NH4)3TiF7 in which the high-pressure parent phase Pmm is attainable.
The polarization-optical studies showed lowering of symmetry during phase transitions at T1 and T2, similar to that observed in (NH4)3GeF7. Further cooling (NH4)3SiF7 revealed the existence of a complicated sequence of crystal symmetry changes with the appearance of triclinic P (G4) and new monoclinic P121/c1 (G5) phases. The large rotation angles of the indicatrix at G4 ↔ G5 phase transition is associated with the ‘proper’ ferroelastic transitions when a parameter of transition δ is linearly related to the shear component of the spontaneous deformation δ ∼ x5.
Joint XRD and optical analysis allowed us to solve and refine crystal structures of the G1, G2, G3, and G5 phases, suggest a model of the G4 phase and define the sequence of symmetry transformations: P4/mbm (Z = 2) (G1) ↔ Pbam (Z = 4) (G2) ↔ P21/c (Z = 4) (G3) ↔ P (Z = 4) (G4) ↔ P21/c (Z = 8) (G5). Indeed, the sequence of the first three phases of (NH4)3SiF7 coincides with that for (NH4)3GeF7. Then a significant difference arises between them: (NH4)3GeF7 undergoes a reconstructive phase transition to Pa, but (NH4)3SiF7 undergoes two phase transitions, which can be described by group-theoretical methods. The lowest-temperature phases in (NH4)3MF7 (M = Si, Ge) have similar cell parameters, and it seems that all NH4 groups are fully ordered in them. Therefore, the driving force of all phase transitions in (NH4)3MF7 can be associated with the emergence of N–H⋯F hydrogen bonds, because the value of N⋯F contacts decreases at phase transition under cooling. Suggested order–disorder processes are in accordance with the large values of entropy change.
The final scheme of phase transitions in (NH4)3SiF7, including irreducible representations and corresponding order parameters, can be written as:

In spite of the fact that the Pmm parent phase cannot be obtained under heating/pressure, the final scheme of phase transitions referred to the cubic phase can be expressed as:
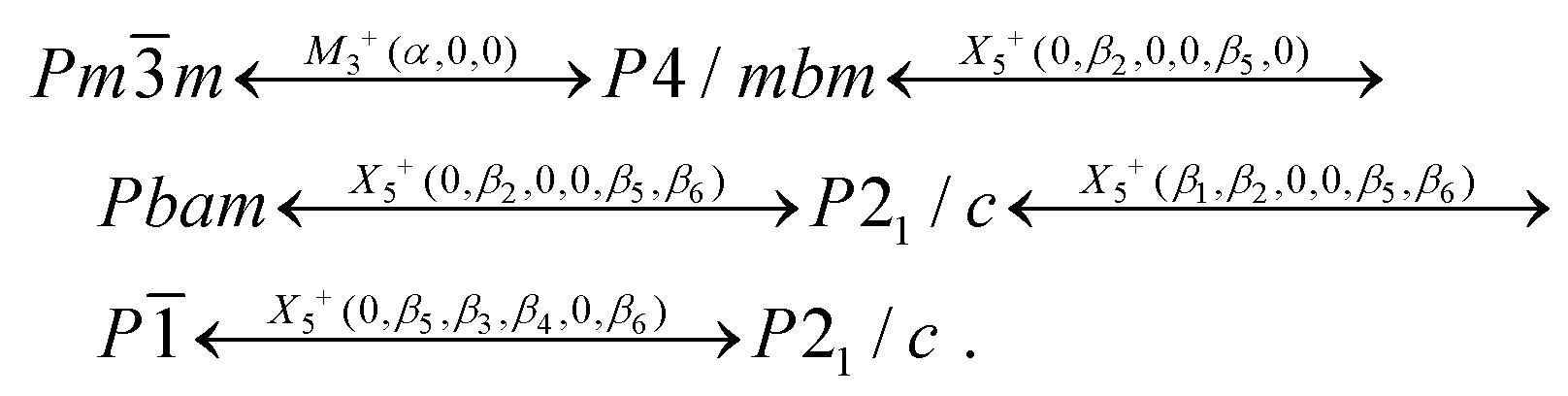
Acknowledgements
The reported study was partially supported by RFBR, research project no. 15-02-02009a.Notes and references
- N. Laptash and I. Maslennikova, IOP Conf. Ser.: Mater. Sci. Eng., 2016, 112, 012024, DOI:10.1088/1757-899X/112/1/012024.
- N. M. Laptash and L. N. Kurilenko, RU Patent, 2306546, 2007 Search PubMed.
- L. N. Kurilenko, N. M. Laptash, E. B. Merkulov and V. Yu. Glushchenko, Electron J. “Investigated in Russia”, 2002, 130/021011, 1465–1471 Search PubMed http://zhurnal.ape.relarn.ru/articles/2002/130.pdf .
- N. M. Laptash and L. N. Kurilenko, RU Patent, 2157523, 2000 Search PubMed.
- A. M. Abdel-Rehim, J. Therm. Anal., 1997, 48, 177–202 CrossRef CAS.
- G. Meyer, Z. Anorg. Allg. Chem., 2008, 634, 201–222 CrossRef CAS.
- C. Plitzko and G. Meyer, Z. Anorg. Allg. Chem., 1996, 622, 1646–1650 CrossRef CAS.
- M. C. Marignac, Ann. Mines, 1859, 15, 221–290 Search PubMed.
- J. Davy, Philos. Trans. R. Soc. London, 1812, 102, 352–369 CrossRef.
- J. L. Hoard and M. B. Williams, J. Am. Chem. Soc., 1942, 64, 633–637 CrossRef CAS.
- S. V. Mel'nikova, M. S. Molokeev, N. M. Laptash and S. V. Misyul, Dalton Trans., 2016, 45, 5321–5327 RSC.
- M. S. Molokeev, S. V. Misyul, I. N. Flerov and N. M. Laptash, Acta Crystallogr., Sect. B: Struct. Sci., 2014, 70, 924–931 CAS.
- I. N. Flerov, M. S. Molokeev, N. M. Laptash, A. A. Udovenko, E. I. Pogoreltsev, S. V. Mel'nikova and S. V. Misyul, J. Fluorine Chem., 2015, 178, 86–92 CrossRef CAS.
- E. I. Pogoreltsev, E. V. Bogdanov, A. V. Kartashev, M. S. Molokeev, I. N. Flerov and N. M. Laptash, Ferroelectrics, 2016, 501(1), 20–25 CrossRef CAS.
- S. V. Mel'nikova, E. I. Pogoreltsev, I. N. Flerov and N. M. Laptash, J. Fluorine Chem., 2014, 165, 14–19 CrossRef.
- E. I. Pogoreltsev, I. N. Flerov, A. V. Kartashev, E. V. Bogdanov and N. M. Laptash, J. Fluorine Chem., 2014, 168, 247–250 CrossRef CAS.
- J. E. Ricci and J. A. Skarulis, J. Am. Chem. Soc., 1951, 73, 3618–3627 CrossRef CAS.
- Bruker AXS TOPAS V4, General profile and structure analysis software for powder diffraction data – User's Manual, Bruker AXS, Karlsruhe, Germany, 2008 Search PubMed.
- O. V. Kovalev, Representation of Crystallographic Space Groups, CRC Press, 1993 Search PubMed.
- S. C. Miller and W. F. Love, Tables of Irreducible Representations of the Space Groups and Co-Representations of Magnetic Space Groups, Pruett Press Boulder, Colorado, 1967.
- B. V. Beznosikov and K. S. Aleksandrov, J. Struct. Chem., 2002, 43, 798–801 CrossRef CAS.
- A. M. Glazer, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 3384–3392 CrossRef CAS.
- D. L. Deadmore and W. F. Bradley, Acta Crystallogr., 1962, 15, 186–189 CrossRef CAS.
- B. Hofmann and R. Hoppe, Z. Anorg. Allg. Chem., 1979, 458, 151–162 CrossRef CAS.
- F. Averdunk and R. Hoppe, J. Less-Common Met., 1989, 146, 137–145 CrossRef CAS.
- V. A. Shreider, I. M. Vol'pin and Y. E. Gorbunova, Russ. Chem. Bull., 1988, 37, 831–835 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CSD 432220–432223. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt04874a |
| ‡ S. V. Misyul unfortunately died while this manuscript was in the process of being published. |
| This journal is © The Royal Society of Chemistry 2017 |