
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtremely bulky copper(I) complexes of [HB(3,5-{1-naphthyl}2pz)3]− and [HB(3,5-{2-naphthyl}2pz)3]− and their self-assembly on graphene†
Thomas F.
van Dijkman
a,
Hans M.
de Bruijn
a,
Tobias G.
Brevé
a,
Bob
van Meijeren
a,
Maxime A.
Siegler
b and
Elisabeth
Bouwman
 *a
*a
aLeiden Institute of Chemistry, Leiden University, P.O. Box 9502, 2300 RA, Leiden, The Netherlands. E-mail: bouwman@chem.leidenuniv.nl
bDepartment of Chemistry, Johns Hopkins University, MD 21218, Baltimore, USA
First published on 17th April 2017
Abstract
The synthesis and characterization, using NMR (1H and 13C), infrared spectroscopy, and X-ray crystallography, of the ethene and carbon monoxide copper(I) complexes of hydridotris(3,5-diphenylpyrazol-1-yl)borate ([TpPh2]−) and the two new ligands hydridotris(3,5-bis(1-naphthyl)pyrazol-1-yl)borate ([Tp(1Nt)2]−) and hydridotris(3,5-bis-(2-naphthyl)pyrazol-1-yl)borate ([Tp(2Nt)2]−) are described. X-ray crystal structures are presented of [Cu(TpPh2)(C2H4)] and [Cu(Tp(2Nt)2)(C2H4)]. The compound [Cu(TpPh2)(C2H4)] features interactions between the protons of the ethene ligand and the π-electron clouds of the phenyl substituents that make up the binding pocket surrounding the copper(I) center. These dipolar interactions result in strongly upfield shifted signals of the ethene protons in 1H-NMR. [Cu(Tp(1Nt)2)(CO)] and [Cu(Tp(2Nt)2)(CO)] were examined using infrared spectroscopy and were found to have CO stretching vibrations at 2076 and 2080 cm−1 respectively. The copper(I) carbonyl complexes form self-assembled monolayers when drop cast onto HOPG and thin multilayers of a few nanometers thickness when dip coated onto graphene. General macroscopic trends such as the different tendencies to crystallize observed in the complexes of the two naphthyl-substituted ligands appear to extend well to the nanoscale where a well-organized monolayer could be observed of [Cu(Tp(2Nt)2)(CO)].
Introduction
The properties of graphene can be altered and augmented through the application of self-assembled monolayers (SAMs) of functional materials and insulators.1–3 Examples include the functionalization of graphene with metal–organic frameworks and electro-catalysts through π-stacking interactions with the catalysts or with appended anchors.4–6 SAMs can alter the properties of graphene by insulating the graphene from harmful chemicals, shielding the graphene from electrical interference or inducing a field effect to produce a chemically actuated graphene field effect transistor (GFET). Because every atom in the graphene lattice is exposed to the influence of a SAM, GFETs can be used as extremely sensitive chemical sensors that respond to analytes such as ethene gas and carbon monoxide even in trace concentrations. Ethene gas is an important analyte in agricultural settings as it induces physiological responses in crops such as fruits, vegetables and flowers. In agricultural settings and particularly in nature, ethene is typically found at extremely low concentrations (10–1000 ppb). Carbon monoxide is often monitored at similarly low concentrations for safety purposes. The potential for sensitive detection of ethene and carbon monoxide using SAM-functionalized GFETs in this regard prompted us to study various aspects of such devices. In this work we use two new complexes as the models for such sensitizers and study their self-assembly on graphene surfaces to pave the way for the eventual integration of such functionalized graphene surfaces into GFET gas sensors.An interesting class of sensitizers for GFETs intended to interact with ethene and carbon monoxide consists of copper(I) complexes of hydridotrispyrazolylborate ligands. Copper(I) hydridotrispyrazolylborate complexes are a natural starting point for ethene sensing as they mimic the structure of the active sites of many biological systems. Examples of such biological systems include hemocyanin, responsible for reversible dioxygen uptake in mollusks; fungal galactose oxidase, an extracellular copper enzyme capable of catalyzing the oxidation of primary alcohols to aldehydes; and ETR-1, the proposed active site of copper(I) containing ethene-sensing membrane proteins in climacteric plants.7–9 Copper(I) is well known for its ability to reversibly bind alkenes in general and ethene in particular. Many examples of hydridotrispyrazolylborate copper(I) complexes have been reported, with ligands containing electron-withdrawing substituents like trifluoromethyl or nitro groups or bulky substituents such as phenyl or mesityl groups.10–13 By carefully selecting the substituents on the pyrazolyl rings the coordination pocket surrounding the copper(I) center can be modified electronically and structurally, allowing significant control over the electron density on the copper(I) center and the reactivity of the corresponding ethene complexes.12
So far, the interaction of the graphene plane with other molecules has typically been studied using essentially two-dimensional molecules like pentacene, pyrene and porphyrins while more structurally complex compounds are avoided.14 In order to study the interaction and the potential for self-assembly of three-dimensional complexes such as the aforementioned copper(I) compounds on the graphene surface, we set out to synthesize and study a number of copper(I) hydridotrispyrazolylborate complexes that were optimized for stacking on graphene. Current examples of non-covalent SAMs on graphene typically exploit the ability of graphene to bind to adsorbates by π-stacking interactions. For this reason the copper(I) complexes devised for the purpose were modified with naphthyl-substituents at the 3- and 5-positions of the pyrazole rings. The inclusion of 1-naphthyl and 2-naphthyl substituents offered not only the π-stacking interactions desired for self-assembly on graphene but also allowed us to study the influence of the position at which the naphthyl groups are anchored to the hydridotrispyrazolylborate core on the physical and chemical properties of the resulting complexes. In addition to their potential for self-assembly on graphene, the copper(I) complexes of new ligands [Tp(1Nt)2]− and [Tp(2Nt)2]− are some of the most sterically bulky copper(I) hydridotrispyrazolylborate complexes ever reported. Other examples of extremely bulky hydridotrispyrazolylborate ligands include: [Tp3-Ad-5-iPr]− containing the extremely bulky adamantyl group; [Tp(1Nt)]− and [Tp(2Nt)]− containing naphthyl substituents at the 3 positions of the pyrazolyl groups; [Tp3-CF3-5-(1Nt)]− and [Tp3-CF3-5-(2Nt)]− which combine naphthyl groups and trifluoromethyl groups; and [TpMs]− comprising the highly bulky mesityl group.10,15–19 The complexes described in this work have the unusual property that they include a pair of the bulky substituents at both the 3- and 5-positions of the pyrazolyl groups. The inclusion of a bulky naphthyl-substituent at the 3-position forces the naphthyl-substituents at the 3-positions towards each other, which further constricts the already crowded coordination pocket around the copper.
A less desirable aspect of the inclusion of the naphthyl-substituents is that it comes at the cost of excluding more strongly electron withdrawing substituents such as trifluoromethyl groups that are typically included to stabilize the copper(I) center against oxidation in air. To gain some initial insights we used the structurally comparable ligand [TpPh2]− as our starting point. [TpPh2]− is a ligand that has been used in biomimetic models of active sites of proteins and enzymes containing manganese, iron, nickel, cobalt and copper.7,20–28 The copper chemistry of [TpPh2]− includes relevant examples such as [Cu(TpPh2)(CO)] (which is air-stable), [Cu(TpPh2)]2, [Cu2(TpPh2)2(μ-O2)], [Cu(TpPh2)Cl] and [Cu(TpPh2)(S2CNEt)].7,22,29 Preliminary results indicated that the complex [Cu(TpPh2)(C2H4)], fortunately, is air-stable, indicating that this might also be the case for the naphthyl-substituted complexes. To the best of our knowledge the compound [Cu(TpPh2)(C2H4)] has not been reported up to now; it is included in this work as it can serve as a useful reference for the naphthyl-substituted complexes.
In this work the copper(I) chemistry of the three ligands [TpPh2]−, [Tp(1Nt)2]− and [Tp(2Nt)2]− (see Scheme 1) is explored, including the synthesis of the new ligands [Tp(1Nt)2]− and [Tp(2Nt)2]− and the characterization of the chemical properties of their ethene and CO complexes. Finally, the self-assembly, on flat carbon surfaces of graphene and HOPG, of the air-stable complexes [Cu(Tp(1Nt)2)(CO)] and [Cu(Tp(2Nt)2)(CO)] is described.
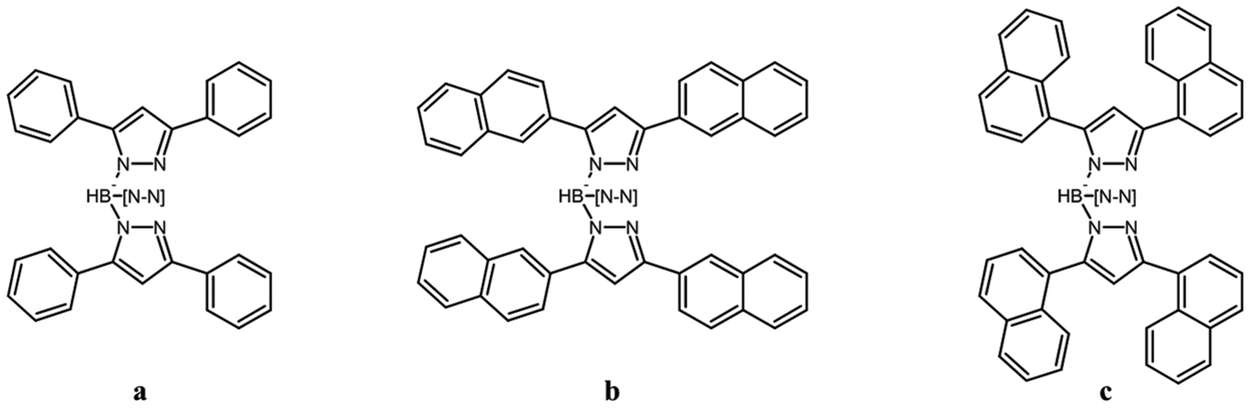 | ||
| Scheme 1 The ligands studied in this work. a. [TpPh2]−, b. [Tp(2Nt)2]−, c. [Tp(1Nt)2]−; –[N–N] represents the third substituted pyrazole ring. | ||
Results
Syntheses of the ligands
The ligands KTpPh2 and NaTp(2Nt)2 were synthesized by slow heating to reflux a small excess of the respective pyrazoles with KBH4 or NaBH4 in high-boiling solvents. The synthesis of KTpPh2 has been reported previously by Kitajima et al. who employed a solventless reaction in molten 3,5-diphenylpyrazole.7 Using the polar aprotic solvent 1,2,4-trichlorobenzene allowed us to use lower reaction temperatures and resulted in a considerably higher yield, even after an additional purification step in which a byproduct insoluble in acetone is removed. KTp(1Nt)2 was synthesized by heating KBH4 in molten 3,5-di(1-naphthyl)pyrazole, as it has a relatively low melting point (approx. 154 °C). For NaTp(2Nt)2 the boiling point of 1,2,4-trichlorobenzene was found to be insufficient for full conversion to the trisubstituted ligand. The more polar 1,3-dimethyltetrahydropyrimidin-2(1H)-one (N,N′-dimethylpropyleneurea, DMPU, b.p. 246 °C) was employed instead.In general it is our observation that polar aprotic solvents such as N,N-dimethylacetamide (b.p. 165 °C), 4-methylanisole (b.p. 174 °C), 1,2,4-trichlorobenzene (b.p. 214 °C), 1,3-dimethyl-2-imidazolidinone (b.p. 225 °C) and DMPU are excellent solvents for the synthesis of hydridopolypyrazolylborate ligands due to their polar properties and high boiling points. The exact choice of solvent depends on the solubility of the pyrazole and the alkali tetrahydridoborate in the hot solvent with reactions proceeding faster, more homogeneously and more completely in solvents capable of completely dissolving the reagents when hot. Not much difference was noted in the use of NaBH4 and KBH4 except that NaBH4 appears to dissolve more readily in the hot organic solvents, leading to a reduction in reaction times. The products were isolated by distilling off the solvents in vacuo and washing the solids with diethyl ether to remove the remaining pyrazole. In the case of KTp(1Nt)2 the impure product was obtained as a thick liquid from which the pure product could be isolated by adding a small amount of acetonitrile, causing the precipitation of an acetonitrile adduct of the potassium salt of the ligand.
An important consequence of using polar aprotic solvents like DMPU and acetonitrile is that they can become incorporated into the final product as ligands coordinating the alkali metal ion. Typical examples of coordinated solvents include small molecules such as acetone, THF, water and diethyl ether which can occur as bridging ligands in dinuclear species as found in [Na2(TpCF3,Me)2(μ-H2O)2] and [Na2(TpCF3,4-CF3Ph)2(μ-H2O)2] or as capping ligands in species such as [Cu(Tp(CF3)2)(Et2O)], [Cu(TpCF3,Ph)(H2O)] and [Na(TpCF3,Ph)(THF)].12,30 In the NMR spectra of KTp(1Nt)2·MeCN and NaTp(2Nt)2·DMPU, one equivalent of acetonitrile and DMPU respectively were observed. In addition the signals for water were found to be significantly broadened in the 1H NMR spectrum of KTp(1Nt)2 and to integrate to 2H in the 1H NMR spectrum of NaTp(2Nt)2, which indicates that DMPU and acetonitrile are not the only coordinated small molecules in the alkali salts of these ligands and that water likely completes the first coordination sphere of the alkali ions.
Synthesis of the carbonyl and ethene copper(I) complexes
The acetone adduct [Cu(TpPh2)(Me2CO)] has been reported previously and was synthesized by a salt metathesis of KTpPh2 and CuCl in acetone.7 Unfortunately, [Cu(TpPh2)(Me2CO)] was found to be too unstable in air, limiting its usefulness as a convenient intermediate for further complex synthesis. When the same synthetic procedure was applied using KTp(1Nt)2 or NaTp(2Nt)2 in acetone, deep red solutions were obtained upon even the briefest exposure of the solutions to air. Although no effort was made to identify these red products they are assumed to be copper(II) complexes resulting from oxidation upon the loss of the acetone ligand, indicating that the use of “acetone-capped intermediates” is inconvenient for such sterically demanding ligands as [Tp(1Nt)2]− and [Tp(2Nt)2]−. In contrast, the copper(I) acetonitrile complexes of [Tp(1Nt)2]− and [Tp(2Nt)2]− were found to have reasonable oxidative stability in the solid state and solution with no discoloration being seen in the solid state after several weeks of exposure to air. Solutions of the acetonitrile complexes in solvents such as benzene and dichloromethane discolored to green only after several hours of exposure to air. A downside of the use of acetonitrile as a capping ligand is that the acetonitrile is difficult to remove using ligands such as CO and ethene because the acetonitrile-adduct is always found in equilibrium with the other ligand and a small impurity of the acetonitrile complex is always found in the final product.For a more convenient synthesis of the complexes, inspiration was found in the synthesis of the carbonyl complex [Cu(TpPh2)(CO)] by a metathesis reaction between KTpPh2 and CuCl in DCM under 1 bar carbon monoxide as reported by Kitajima et al.7 We used a similar approach using CuI (which, unlike CuCl, is air-stable) and higher pressures of carbon monoxide in steel autoclaves. DCM is used as the solvent as KI and NaI are entirely insoluble in DCM which facilitates the work-up. Using this approach undesired copper complexes are not formed; the by-products are insoluble in DCM and typically the product can be purified simply through filtration and evaporation of the solvent. The use of high pressure carbon monoxide is not strictly required but was merely convenient as we had a high-pressure autoclave system available in which carbon monoxide could be safely handled. For the synthesis of the ethene complexes [Cu(Tp(1Nt)2)(C2H4)] and [Cu(Tp(2Nt)2)(C2H4)] small excesses of CuI were stirred under a 1 bar ethene atmosphere in the presence of the sodium or potassium salts of the ligands in DCM.
To prepare [Cu(TpPh2)(C2H4)], ethene was bubbled through a clear solution of [Cu(Tp(Ph)2)]2 in THF; as soon as the solution was exposed to ethene gas an equimolar mixture of KI and [Cu(TpPh2)(C2H4)] precipitated. Filtration of the precipitate afforded a white air-stable powder which was essentially insoluble in THF and diethyl ether and only slightly soluble in benzene, chloroform and DCM. The KI impurity posed no hindrance to the intended analyses and thus no attempts were made to remove it. To grow X-ray quality crystals of [Cu(TpPh2)(C2H4)] without KI we exploited the poor solubility of the complex by letting ethene gas diffuse slowly into a THF solution of KTpPh2 and CuI. As, in this way, the reaction proceeded considerably more slowly, the poorly soluble ethene complex and the KI byproduct had sufficient time to separate avoiding the inclusion of KI in the product. Compared to [Cu(TpPh2)(C2H4)] the ethene and carbonyl complexes of [Tp(2Nt)2]− have slightly higher solubilities whereas the ethene and carbonyl complexes of [Tp(1Nt)2]− are quite soluble in most solvents.
In solution in DCM or benzene the carbonyl complexes appear to be stable, allowing for short exposures to air without obvious discoloration. However, the ethene complexes are less stable in solution and the color of the solutions changes to reddish brown after exposure to air for more than a few minutes. Attempts to crystallize [Cu(Tp(1Nt)2)(C2H4)] from DCM/pentane resulted in the formation of a large amount of crystals that were found to consist entirely of pure 3,5-di(1-naphthyl)pyrazole. In the solid state all complexes are reasonably stable with respect to oxidation, allowing for short exposures to air without apparent decomposition.
Descriptions of the X-ray crystal structures
X-ray quality crystals were obtained for the compounds [Cu(TpPh2)(C2H4)] and [Cu(Tp(2Nt)2)(C2H4)]. The crystallographic data for both crystal structures are provided in Table 1; relevant bond distances and angles are given in Table 2. X-ray quality crystals of [Cu(TpPh2)(C2H4)] were grown by slow diffusion of ethene gas into a filtered THF solution prepared from an equimolar mixture of KTpPh2 and CuI. A projection of the crystal structure of [Cu(TpPh2)(C2H4)] is shown in Fig. 1. The copper(I) center is coordinated in a distorted tetrahedral geometry by three nitrogen atoms of the [TpPh2]− ligand and an ethene ligand which is coordinated in the typical η2 fashion. [Cu(TpPh2)(C2H4)] crystallized in π-stacked chains of head-to-tail rows of complexes with the phenyl-rings alternatingly rotated clockwise and anticlockwise. The compound crystallized in a trigonal space group with the copper and boron centers located on an axis of threefold-rotational symmetry. For refinement the ethene ligand was constrained to be distributed over three orientations with occupancy factors of ⅓ each. The bond lengths Cu1–C1 and Cu1–C2 are 2.05(3) and 2.02(3) Å, respectively, which fall in the range of distances observed previously in similar complexes.11 The ethene bond length C1–C2 of 1.381(18) Å is slightly longer than that in free ethene (1.3384(10) Å).31 The observed C1–C2 distance is comparable to those in [Cu(TpMs)(C2H4)] (1.345(16) Å) and [(C2H4)Cu(μ-Tp)CuCl]2 (1.347(5) Å), but somewhat longer than the distances in [Cu(TpMe2)(C2H4)] (1.329(9) Å) and [Cu(TpCF3,Ph)(C2H4)] (1.30(1) Å).11,13,32 The structure is densely packed and features π-stacking interactions between the aromatic rings of adjacent complexes. The protons of the ethene ligand and the carbon atoms in the surrounding phenyl rings feature a number of short contacts with distances in the range of 2.7–3.0 Å. The phenyl rings are rotated out-of-plane of the pyrazoles with torsion angles of 55° for the rings at the 3-positions and 51° for the rings at 5-positions.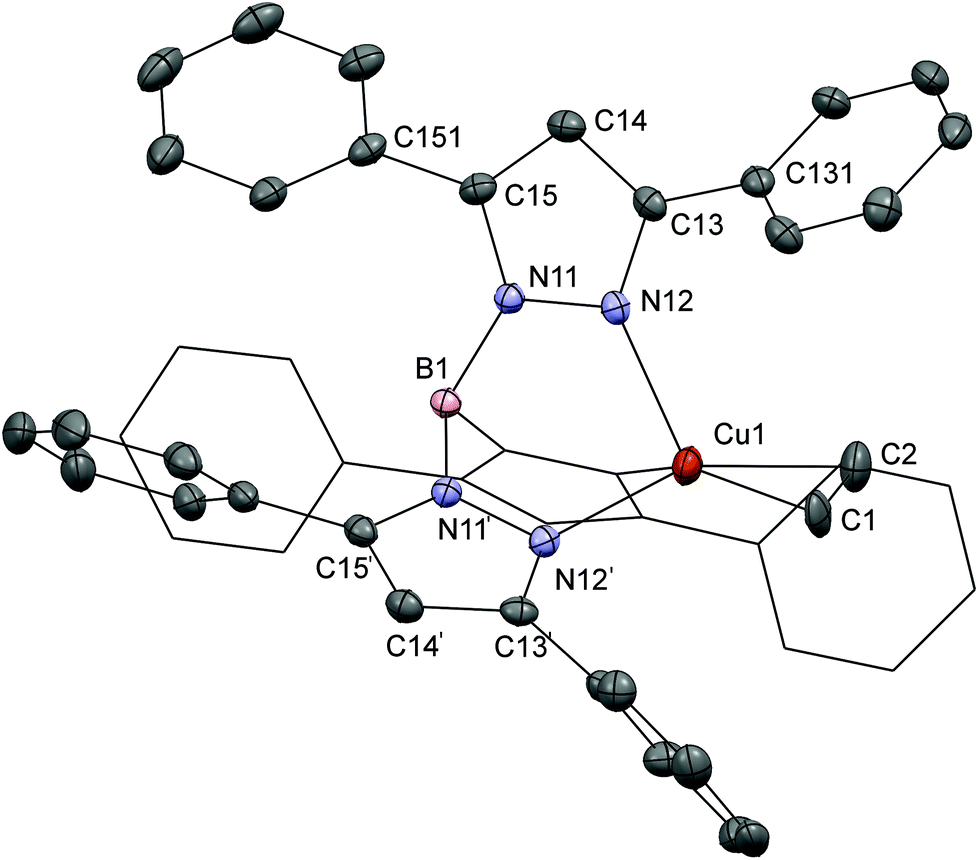 | ||
| Fig. 1 Displacement ellipsoid plot (50% probability level) of [Cu(TpPh2)(C2H4)] at 110(2) K. Hydrogen atoms have been omitted and one of the arms of the trispyrazolylborate ligand is shown as a wireframe projection for the sake of clarity; only one of the three orientations of the ethene ligand is shown. Symmetry operations: ′ = [1 − y, 2 + x − y, z], wireframe segment = [y − x − 1, 1 − x, z]. | ||
| [Cu(TpPh2)(C2H4)] | [Cu(Tp(2Nt)2)(C2H4)] | |
|---|---|---|
| w = 1/[s2(Fo2) + (0.0381P)2 + 15.2884P] where P = (Fo2 + 2Fc2)/3. | ||
| Chemical formula | C47H38BCuN6 | 2(C71H50BCuN6)·3(DCM) |
| M r | 761.18 | 2377.81 |
| Crystal system, space group | Trigonal, R3c:H | Monoclinic, C2/c |
| T (K) | 110 | 110 |
| a (Å) | 16.1559(3) | 33.0952(9) |
| b (Å) | — | 10.6204(3) |
| c (Å) | 25.6568(6) | 35.1414(10) |
| α (°) | — | — |
| β (°) | — | 111.348(3) |
| γ (°) | 120 | — |
| V (Å3) | 5799.6(3) | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 504.2(6) 504.2(6) |
| Z | 6 | 4 |
| ρ (g cm−3) | 1.308 | 1.373 |
| Radiation type | Cu Kα | Cu Kα |
| μ (mm−1) | 1.117 | 2.21 |
| Crystal size (mm) | 0.47 × 0.32 × 0.23 | 0.27 × 0.21 × 0.13 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 10907, 2459, 2452 |
34168, 11268, 10091 |
| R int | 0.017 | 0.026 |
| (sinθ/λ)max (Å−1) |
0.616 | 0.617 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.020, 0.054, 1.06 | 0.035, 0.089, 1.04 |
| No. of reflections | 2459 | 11268 |
| No. of parameters | 190 | 802 |
| No. of restraints | 27 | 87 |
| Residual e density (e Å−3) | 0.15, −0.31 | 0.62, −0.62 |
| [Cu(TpPh2)(C2H4)] | [Cu(Tp(2Nt)2)(C2H4)] | |
|---|---|---|
| a For [Cu(TpPh2)(C2H4)] the angle N12–Cu–N12′ is given. | ||
| C1–C2 | 1.381(18) | 1.356(3) |
| Cu1–C1 | 2.02(3) | 2.0379(15) |
| Cu1–C2 | 2.05(3) | 2.0626(16) |
| Cu1–N12 | 2.0959(16) | 2.0720(13) |
| Cu1–N22 | 2.0216(13) | |
| Cu1–N32 | 2.1440(13) | |
| N12–Cu1–N22a | 90.05(6) | 90.65(5) |
| N22–Cu1–N32 | 89.66(5) | |
| N32–Cu1–N12 | 102.98(6) | |
| Cu1⋯B1 | 3.066(1) | 3.052(2) |
Colorless single crystals of [Cu(Tp(2Nt)2)(C2H4)] were obtained by diffusion of diethyl ether into a DCM solution of the complex. The complex crystallizes in the space group C2/c with two independent complex molecules and three solvent molecules in the asymmetric unit. The crystal structure of [Cu(Tp(2Nt)2)(C2H4)] (Fig. 2) shows a distorted tetrahedral copper(I) center coordinated by three nitrogen atoms of the [Tp(2Nt)2]− ligand and an η2-coordinated ethene ligand. The ethene ligand in [Cu(Tp(2Nt)2)(C2H4)] is found in one orientation sandwiched between two naphthyl groups. The Cu1–C1, Cu1–C2 and C1–C2 distances are 2.0379(15), 2.0626(16) and 1.356(3) Å respectively. The Cu1–Cethene bond distances in [Cu(Tp(2Nt)2)(C2H4)] are comparable to those in [Cu(TpPh2)(C2H4)], while the C1–C2 bond is slightly shorter. The ethene ligand is oriented approximately along the plane formed by Cu1, N21 and B1 and tilted away from the approximately coplanar pyrazole ring. The angle between the axis Cu1–B1 and the centroid of the ethene ligand is 165°.
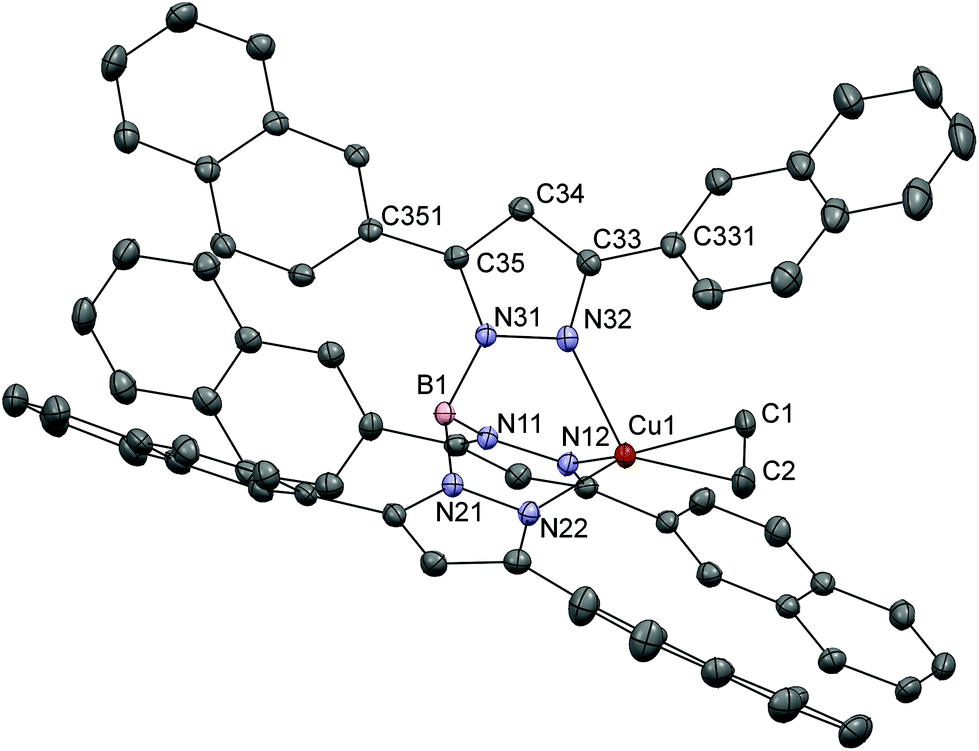 | ||
| Fig. 2 Displacement ellipsoid plot (50% probability level) of one of the independent molecules [Cu(Tp(2Nt)2)(C2H4)] at 110(2) K. Hydrogen atoms and solvent molecules have been omitted for the sake of clarity. | ||
The dihedral angle between conjoined aromatic rings is typically ∼45° as a result of the competition between steric and electronic effects. For instance, biphenyl has a dihedral angle of 44.4°.33 In [Cu(Tp(2Nt)2)(C2H4)] the dihedral angles between the pyrazoles and the naphthyl groups are in the range of 37°–52°. The deflections of the naphthyl rings away from the approximate 45° angle found in biphenyl are likely the effect of crystal packing interactions indicating relatively low rotational barriers in accordance with the low rotational barrier of 6.2(2.3) kJ mol−1 calculated for biphenyl.33 The crystal packing is dominated by π-stacking interactions, but also includes a short contact of 3.05 Å between the ethene ligand and a proton of a naphthyl group from a nearby complex.
NMR spectroscopy
The sodium or potassium salts of the ligands as well as the copper complexes were analyzed using 1H and 13C NMR; relevant chemical shifts are collected in Table 3. The sodium and potassium salts of the ligands are quite soluble in polar aprotic solvents like THF, acetone and acetonitrile and somewhat soluble in DCM and chloroform. In protic solvents such as C2D5OD and CD3OD noticeable solvolysis of the B–H bond occurs within only a few minutes as evident from the appearance of a new sharp peak in 11B NMR with a small downfield shift of 5–10 ppm compared to the 11B signal of the original ligand. The sharpness of the peak observed after solvolysis indicates the absence of JBH-coupling interactions and suggests that the initial reaction in the degradation pathway of the trispyrazolylborate ligands is protonation and subsequent solvolysis of the hydride and not of the B–N bonds.| 1H NMR (ppm, L = C2H4) | 13C NMR (ppm, L = C2H4) | ν(CO) (cm−1, L = CO) | Ref. | |
|---|---|---|---|---|
| a T = room temperature unless otherwise specified. b [Cu(Tp)(C2H4)] was observed as [Cu(Tp)(C2H4)]2·[CuCl]2. | ||||
| Free C2H4 | 5.40 (CD2Cl2) | 123.20 (CD2Cl2) | 35 | |
| 5.40 (CDCl3) | 123.13 (CDCl3) | |||
| 5.25 (C6D6) | 122.96 (C6D6) | |||
| 5.36 (THF-d8) | 123.09 (THF-d8) | |||
| Free CO | 2143 | |||
| [Cu(TpPh2)(L)] | 3.53 (CDCl3) | 81.6 (CDCl3) | 2080 | This work |
| 17 | ||||
| [Cu(Tp(1Nt)2)(L)] | 2.64 (C6D6, 297 K) | 81.2 (C6D6, 343 K) | 2076 | This work |
| 2.57 (C6D6, 343 K) | 78.8 (CD2Cl2, 203 K) | |||
| 2.15 (CD2Cl2, 297 K) | ||||
| [Cu(Tp(2Nt)2)(L)] | 3.53 (CDCl3) | 81.91 (C6D6) | 2080 | This work |
| 3.53 (THF-d8) | ||||
| 3.78 (C6D6) | ||||
| [Cu(TpMs)(L)] | 2.72 (CDCl3) | 77.4 (C6D6) | 11 | |
| 3.08 (C6D6) | ||||
| [Cu(Tp)(L)]b | 4.43 (CD2Cl2) | 2083 | 32 and 36 | |
| [Cu(TpMe2)(L)] | 4.41 (CD2Cl2) | 2066 | 32 and 36 | |
| [Cu(TpCF3,Ph)(L)] | 4.91 (CD2Cl2) | 85.8 (CD2Cl2) | 2103 | 13 and 30 |
| 5.20 (C6D6) | ||||
| [Cu(TpCF3)(L)] | 4.98 (C6D6) | 85.8 (C6D6) | 2100 | 13 and 37 |
| 4.80 (CDCl3) | 85.7 (CDCl3) | |||
| [Cu(Tp(CF3)2)(L)] | 4.96 (CDCl3) | 89.1 (CDCl3) | 2137 | 13 and 38 |
| 4.98 (C6D12) | 89.5 (C6D12) | |||
| 4.94 (C6D6) | ||||
The 1H-NMR spectra of [Cu(TpPh2)(C2H4)], [Cu(Tp(1Nt)2)(C2H4)] and [Cu(Tp(2Nt)2)(C2H4)] were recorded in CD2Cl2, CDCl3 and C6D6. In all cases the spectra did not show the presence of free ethene. The ethene protons of [Cu(TpPh2)(C2H4)] are present as a sharp singlet at 3.53 ppm (CDCl3, 298 K). The ethene protons for [Cu(Tp(2Nt)2)(C2H4)] are located at 3.53 ppm in CDCl3 and THF-d8 (298 K) but at 3.78 ppm in C6D6 (298 K). The ethene signal in [Cu(Tp(1Nt)2)(C2H4)] was observed at the unexpectedly low chemical shifts of 2.64 ppm (C6D6, 297 K see Fig. 3) and even at 2.15 ppm in CD2Cl2 (297 K). The surprisingly large shift for [Cu(Tp(1Nt)2)(C2H4)] even significantly exceeds that reported for [Cu(TpMs)(C2H4)] (2.72 ppm, CD2Cl2), a complex featuring a somewhat similar coordination environment for the ethene molecule.11 The unusually large upfield shifts of the ethene protons in [Cu(Tp(1Nt)2)(C2H4)] are attributed to additional shielding by the naphthyl groups, most likely enforced by restricted rotation of the naphthyl rings; the same explanation was offered for [Cu(TpMs)(C2H4)].11,34
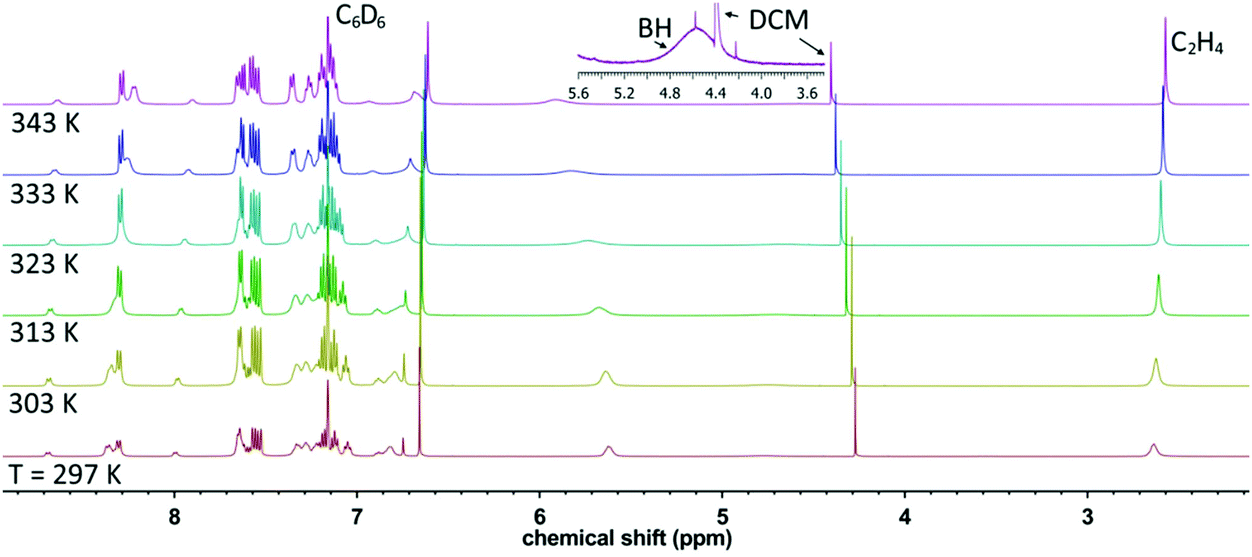 | ||
| Fig. 3 Variable temperature 1H-NMR spectra of [Cu(Tp(1Nt)2)(C2H4)] in C6D6. The inset shows the borohydride peak; the small singlet on top of the BH peak is a 13C-satellite peak of the DCM. | ||
As may be expected, the upfield shifts are more pronounced than those observed in similar complexes featuring strongly electron-withdrawing ligands such as [Cu(TpCF3,Ph)(C2H4)] (4.91 ppm, CD2Cl2) and [Cu(Tp(CF3)2)(C2H4)] (4.96 ppm, CDCl3).13 More unexpected is that the shifts exceed those observed in copper(I) complexes featuring electron-donating ligands like [Cu(Tp)(C2H4)] and [Cu(TpMe2)(C2H4)] (respectively 4.41 ppm and 4.43 ppm in CD2Cl2).32 The 13C-shifts of the ethene carbons in [Cu(Tp(1Nt)2)(C2H4)] and [Cu(Tp(2Nt)2)(C2H4)] in benzene-d6 were observed at 81.2 ppm and 81.9 ppm respectively which compare well with the signal observed for [Cu(TpPh2)(C2H4)] (81.6 ppm in CDCl3). The lack of a strong upfield shift in the carbon atoms of the ethene ligands indicates that the cause of the upfield shifts of the proton signals is very local, which corroborates the hypothesis that interaction between the ethene protons and the π-electrons of the aryl-substituents on the ligands is responsible.
In contrast to [Cu(TpPh2)(C2H4)] and [Cu(Tp(2Nt)2)(C2H4)] the ethene signal in [Cu(Tp(1Nt)2)(C2H4)] is not observed as a sharp peak but rather as a broadened singlet which sharpens as the temperature rises (see Fig. 3). The peak broadening observed in [Cu(Tp(1Nt)2)(C2H4)] also extends to a number of resonances for naphthyl protons, such as the prominent broad signal at ∼5.5 ppm, indicating restriction of movement of these protons in the complex. When the temperature was raised to 70 °C, the broad signals broadened further while the ethene signal sharpened, indicating that the restricted movement of the 1-naphthyl substituents does not become significantly less restricted at elevated temperatures. A similar observation was reported by Rheingold et al. for the structurally comparable ligand [Tp1Nt]−, which, in the complex [Co(Tp1Nt)(TpMe2)], produced NMR spectra aptly referred to as “baffling”.17 The underlying cause of the complicated 1H-NMR spectrum of [Co(Tp1Nt)(TpMe2)] was found to be a lack of symmetry in the complex which is retained in solution. The 1H-NMR spectrum of [Cu(Tp(1Nt)2)(C2H4)] is further complicated by the decomposition of the ligand which releases free pyrazole upon hydrolysis of the B–N bonds.
Initially the free pyrazole was assumed to be an impurity carried over from the ligand, but further purification of the ligand did not result in pyrazole-free NMR spectra for [Cu(Tp(1Nt)2)(C2H4)]. This observation is in agreement with the crystallization experiments with [Cu(Tp(1Nt)2)(C2H4)], which resulted in the formation of crystals of the free pyrazole and confirm that over long periods of time [Cu(Tp(1Nt)2)(C2H4)] is unstable in solution even in the absence of air. Only when the integration was corrected for the presence of free pyrazole satisfactory values were obtained.
Peak broadening is not seen in the 1H-NMR spectrum of [Cu(Tp(1Nt)2)(CO)], nor in [Cu(Tp(2Nt)2)(CO)]; clearly, the structural asymmetry present in [Cu(Tp(1Nt)2)(C2H4)] is not present in the carbonyl complexes. The 13CO signals were not observed in 13C{1H}-NMR; their absence is attributed to peak broadening of the carbonyl signal. The absence of clear signals for the carbonyl ligands is not unusual; unless 13C-labeled CO is used it is generally very difficult to observe these signals.39
In all cases the hydride was observed as a broadened singlet around 4.5 ppm. The BH signals showed no visible splitting patterns as a consequence of high quadrupole moments of the 10B (S = 3) and 11B (S = 3/2) nuclei. Typically, splitting of the BH peaks in polypyrazolylborate ligands and their complexes is only observed if the local symmetry around the B nuclei is high, as asymmetry increases the quadrupole moments of the boron nuclei. Though the BH signals in copper(I) hydridotrispyrazolylborate complexes are a typical feature of such complexes they are rarely discussed in much detail.12
Infrared spectroscopy
Previously, we reported a series of tunable copper(I) hydridotrispyrazolylborate complexes which showed a strong correlation between the CO stretching frequency and the electronic properties of the copper(I) ion, a trend that has been observed for a wide range of [Cu(TpR,R)(CO)]-type complexes.12,39–41 To gauge the electronic properties of the copper(I) centers in [Cu(Tp(1Nt)2)(CO)] and [Cu(Tp(2Nt)2)(CO)], IR spectra of solid samples were recorded; their CO-stretching frequencies were found to be 2076 and 2080 cm−1 respectively. This places the electronic properties of [Cu(Tp(1Nt)2)(CO)] and [Cu(Tp(2Nt)2)(CO)] close to the structurally similar compound [Cu(TpPh2)(CO)] for which a frequency of 2080 cm−1 has been reported.42 These CO stretching frequencies are significantly lower than those reported for electron-poor copper carbonyl complexes like [Cu(Tp(CF3)2)(CO)] and [Cu(TpCF3,Ph)(CO)] (2137 and 2103 cm−1) and higher than the CO frequency reported for an electron-rich carbonyl complex such as [Cu(TpMe2)(CO)].28,36,38 The CO stretching frequencies of [Cu(Tp(1Nt)2)(CO)] and [Cu(Tp(2Nt)2)(CO)] are therefore best considered to indicate copper(I) centers that are neither particularly electron rich nor electron poor. The CO stretching frequencies of a number of carbonyl copper(I) hydridotrispyrazolylborate complexes are listed in Table 3.Self-assembly on HOPG and graphene
To study the behavior of the carbonyl complexes on graphene surfaces two approaches were used. In the first approach we exploited the characteristic ability of carbon allotropes such as graphite and amorphous carbon (e.g. charcoal) to adsorb dissolved compounds. Our aim was to adsorb the carbonyl complexes as monolayers on the surface of graphene by immersing samples of graphene (on silicon wafer substrates) in concentrated solutions of the carbonyl complexes in dichloromethane. In the second approach a dilute (∼0.5 μM) solution of either [Cu(Tp(1Nt)2)(CO)] or [Cu(Tp(2Nt)2)(CO)] was drop cast on freshly cleaved highly-oriented pyrolytic graphite (HOPG) in the hope of forming domains of monolayers on the HOPG surface. The concentration of the solutions was chosen so as to result in a less-than-complete surface coverage of the complexes on the HOPG surface.The layer thicknesses obtained by dip coating were estimated using ellipsometry. The obtained layers were found to be of homogeneous thickness throughout each sample with only small variations (see Fig. 4b); the obtained layer thicknesses signify layers of 3–5 complex molecules (Fig. 5).
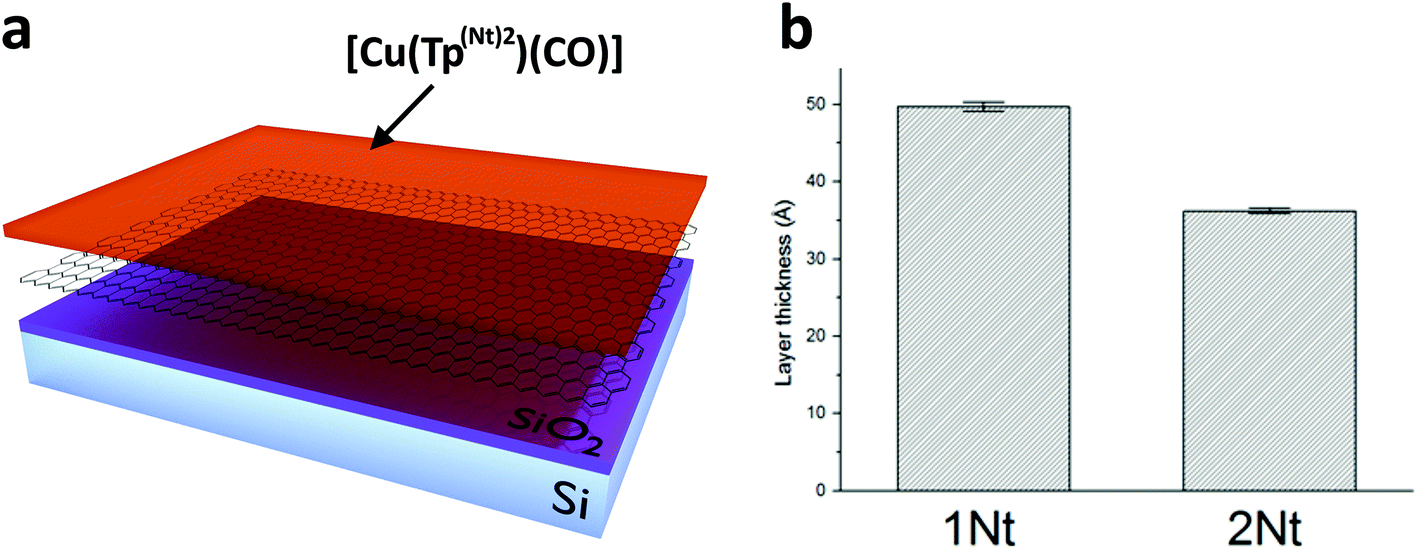 | ||
| Fig. 4 a. Schematic exploded view of the graphene samples. CVD graphene is placed atop a silicon dioxide layer on a silicon wafer and covered in a layer of [Cu(Tp(Nt)2)(CO)]. b. Estimated layer thicknesses as determined using ellipsometry of the [Cu(Tp(1Nt)2)(CO)] and [Cu(Tp(2Nt)2)(CO)] layers on graphene obtained by dip-coating in concentrated solutions of the complexes in DCM. | ||
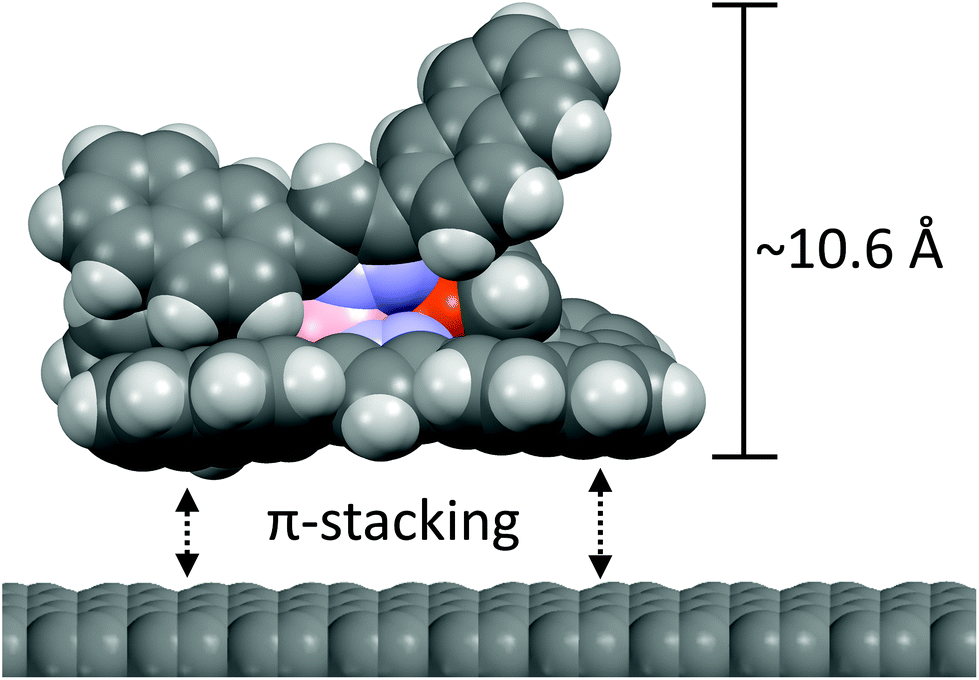 | ||
| Fig. 5 Illustration of the proposed stacking of the complex [Cu(Tp(2Nt)2)(C2H4)] onto graphene. The X-ray crystal structure of [Cu(Tp(2Nt)2)(CO)] has not been determined but the compound is assumed to have roughly the same dimensions. | ||
Ellipsometry could not be used for the samples prepared by drop casting of [Cu(Tp(1Nt)2)(CO)] and [Cu(Tp(2Nt)2)(CO)] as HOPG is not sufficiently flat. The HOPG samples and the graphene samples were studied using atomic force microscopy (AFM). A clear difference was observed between the two compounds in the HOPG samples. The sample drop cast with [Cu(Tp(2Nt)2)(CO)] showed domain-like areas delineated by steps of approximately 10 Å height while the sample drop cast with [Cu(Tp(1Nt)2)(CO)] showed no obvious surface structures (see Fig. 6). The step height of the domains corresponds with the expected height of a monolayer of [Cu(Tp(2Nt)2)(CO)]. The angles between the domains are approximately 60° or 120°, indicating that they are possibly aligned with the underlying graphene lattice (Fig. 6b).
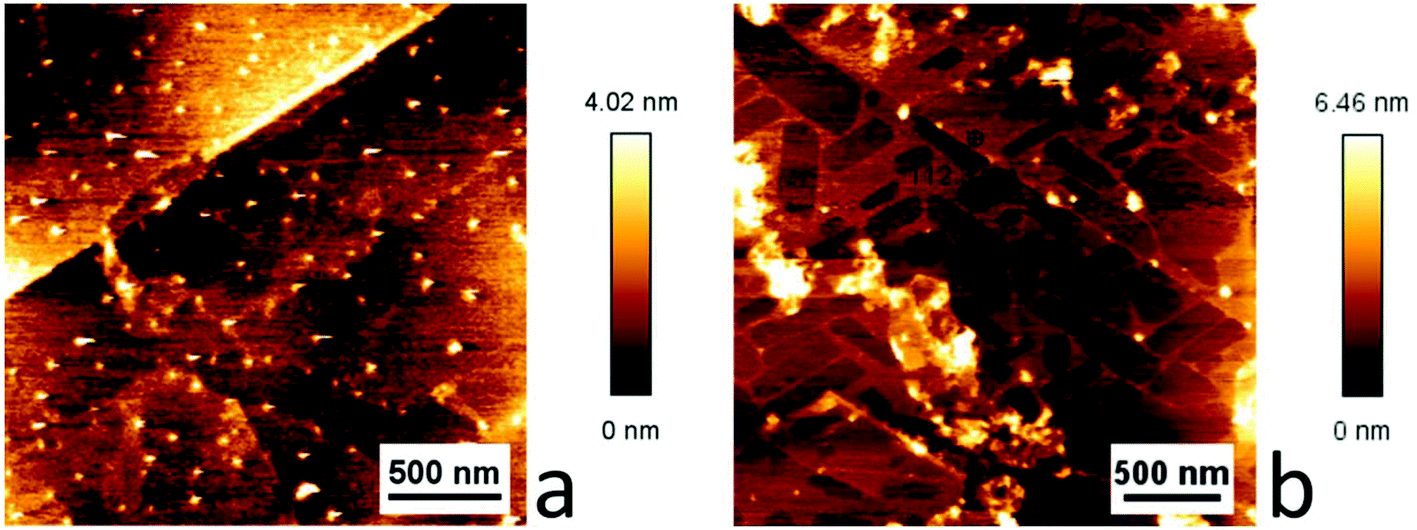 | ||
| Fig. 6 AFM scans of [Cu(Tp(1Nt)2)(CO)] (a) and [Cu(Tp(2Nt)2)(CO)] (b) drop cast on HOPG. The domains visible in (b) have lengths and widths of hundreds of nm and a step height of approximately 10 Å corresponding to monolayer coverage. | ||
The graphene samples dip coated with [Cu(Tp(1Nt)2)(CO)] and [Cu(Tp(2Nt)2)(CO)] did not show the domains observed in the HOPG samples. Instead, fibrous structures were observed with average heights that correspond well with the layer thicknesses determined using ellipsometry. The larger structures on the dip-coated sample of [Cu(Tp(1Nt)2)(CO)] appear to align along similar axes; the structures are much smaller than those observed in the dip-coated sample of [Cu(Tp(2Nt)2)(CO)] (see Fig. 7).
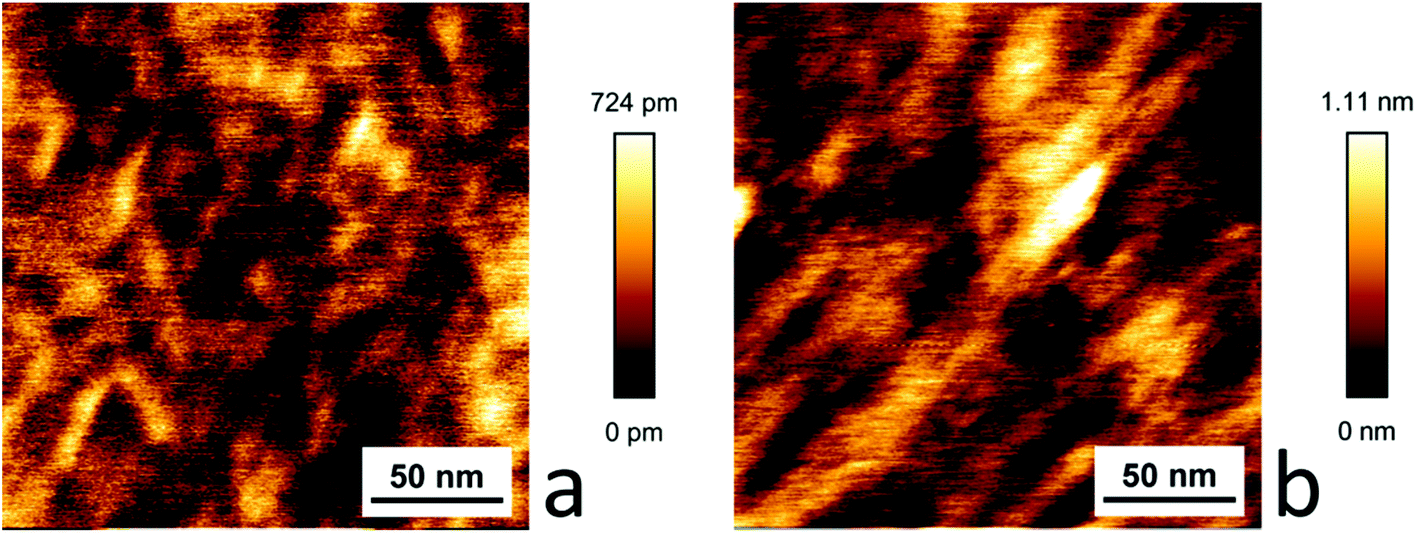 | ||
| Fig. 7 AFM scans of dip-coated samples of [Cu(Tp(1Nt)2)(CO)] (a) and [Cu(Tp(2Nt)2)(CO)] (b) on graphene. | ||
As AFM does not normally offer atomic resolution, scanning tunneling microscopy (STM) was used to study the ordering on the surface in greater detail. To maximize the odds of observing self-assembly on HOPG the use of the typically non-crystalline [Cu(Tp(1Nt)2)(CO)] was forgone in favor of [Cu(Tp(2Nt)2)(CO)]. In order to improve the ordering on the surface the solvent used to drop cast [Cu(Tp(2Nt)2)(CO)] was not DCM but cyclohexane. Cylcohexane offers the advantage of a lower rate of evaporation compared to DCM, which potentially leads to the formation of larger, more ordered domains as the self-assembly of [Cu(Tp(2Nt)2)(CO)] has more time to proceed. Cyclohexane was not used for dip coating experiments as the solubility of the complex in cyclohexane is quite low, i.e. the dissolution of [Cu(Tp(2Nt)2)(CO)] in cyclohexane to form a 0.5 mM solution proceeded only very slowly and a 1 mM solution could not be prepared due to saturation.
The domain of [Cu(Tp(2Nt)2)(CO)] on HOPG in the lower half of Fig. 8 shows square-packed regions. The maxima of the regions have a lattice constant of approximately 2.6 nm which corresponds to slightly more than two widths of [Cu(Tp(2Nt)2)(CO)] (based on the crystal structure of [Cu(Tp(2Nt)2)(C2H4)]) which would be 2.4 nm. We assume the regions that make up the domain to consist of [Cu(Tp(2Nt)2)(CO)] molecules packed anti-parallel to each other with naphthyl groups rotated to accommodate π-stacking interactions with the graphene substrate.
 | ||
| Fig. 8 STM scan of [Cu(Tp(2Nt)2)(CO)] on HOPG. In the lower half of the image a discrete domain of self-assembled [Cu(Tp(2Nt)2)(CO)] is clearly distinguishable from the bare HOPG planes in the top half of the image. | ||
Discussion
We set out in this work to study the properties of copper(I) complexes of the new, extremely bulky naphthyl-substituted hydridotrispyrazolylborate ligands [Tp(1Nt)2]− and [Tp(1Nt)2]−. The resulting complexes were expected to have properties similar to those of the structurally related complexes [Cu(TpPh2)(C2H4)], [Cu(TpPh2)(CO)] and [Cu(TpMs)(C2H4)]. Instead, an apparent mismatch was found between the observations from infrared spectroscopy and NMR. Whereas the IR spectra for [Cu(Tp(1Nt)2)(CO)] and [Cu(Tp(2Nt)2)(CO)] conformed reasonably to expectations with CO frequencies close to those reported for [Cu(TpPh2)(CO)], the large upfield shifts in the 1H NMR spectra of [Cu(Tp(1Nt)2)(C2H4)] and [Cu(Tp(2Nt)2)(C2H4)] for the ethene protons seemingly indicate the copper(I) centers to be more electron rich. This apparent mismatch between the results from the IR and NMR experiments is ascribed to shielding of the ethene protons by the π-electrons of nearby aromatic groups. Indeed the crystal structures of [Cu(TpPh2)(C2H4)] and [Cu(Tp(2Nt)2)(C2H4)] show the ethene protons to be in close proximity to the nearby π-systems. Further supporting this explanation are the observations that the protons of the ethene ligand appear as a broadened singlet in the 1H-NMR spectrum of [Cu(Tp(1Nt)2)(C2H4)], whereas peak broadening is essentially absent in the 1H-NMR spectrum of [Cu(Tp(2Nt)2)(CO)] while it dominates the spectrum of [Cu(Tp(2Nt)2)(C2H4)]. The peak broadening observed in the ethene signal in [Cu(Tp(1Nt)2)(C2H4)] is likely the result of the limited ability of 1-naphthyl groups to rotate with respect to the adjacent pyrazole plane, an effect that was observed before in other naphthyl-substituted hydridotrispyrazolylborate ligands such as [TpCF3,1Nt]− and [Tp1Nt]−.17The influence of the conformational freedom of the [Tp(1Nt)2]− and [Tp(2Nt)2]− ligands extends beyond electronic effects as observed in 1H-NMR spectra. Notably, the complexes of [Tp(2Nt)2]− show a pronounced tendency to crystallize and have low solubilities in most solvents while the complexes containing the ligand [Tp(1Nt)2]− show barely any tendency to crystallize. Although the complexes of [Tp(1Nt)2]− form solids (rather than oils) all attempts at crystallization failed. The inability of complexes containing the ligand [Tp(1Nt)2]− to form well-defined crystals likely stems from the formation of a number of different conformational isomers of the complexes due to the limited rotational freedom of the naphthyl groups in the [Tp(1Nt)2]− ligand.
This important difference between the naphthyl-substituted complexes reported in this work can be observed even on the nanoscale in the self-assembled layers on HOPG and graphene using AFM and STM. Whereas [Cu(Tp(2Nt)2)(CO)] formed clearly visible domains the surface of a similarly prepared sample bearing [Cu(Tp(1Nt)2)(CO)] showed no evidence of ordered assembly. When samples were prepared using dip coating the resulting layers were found to be quite even in thickness, forming layers of between three and five monolayers. The difference in the layer thicknesses observed in the samples of [Cu(Tp(1Nt)2)(CO)] and [Cu(Tp(2Nt)2)(CO)] may have been caused by the difference in the concentrations of the solutions of [Cu(Tp(1Nt)2)(CO)] and [Cu(Tp(2Nt)2)(CO)] used to dip the samples. Drop casting dilute solutions of the complexes [Cu(Tp(2Nt)2)(CO)] on HOPG was found to be an effective means by which to apply a monolayer. The fact that discrete, ordered monolayers in drop-cast samples of [Cu(Tp(2Nt)2)(CO)] were observed on STM indicates that for [Cu(Tp(2Nt)2)(CO)] on graphene, Volmer–Weber growth does not apply. The epitaxial growth on graphene of complex molecules such as [Cu(Tp(2Nt)2)(CO)] is probably described best as Stranski–Krastanov growth in which the formation of two-dimensional and three-dimensional islands occurs simultaneously after a certain number of monolayers have built up.43
In AFM, multilayer surfaces of [Cu(Tp(1Nt)2)(CO)] and [Cu(Tp(2Nt)2)(CO)] were observed with undulating structures where both the thinner and thicker parts appeared to be amorphous. As the multilayers have average thicknesses corresponding to approximately 3–5 monolayers the thinner zones observed on AFM were likely only two or three monolayers in thickness, which probably places the critical layer thickness of the graphene/[Cu(Tp(2Nt)2)(CO)] composite at two or three monolayers. The critical layer thickness is a result of lattice mismatch between the substrate and the adsorbate, which, in the case of [Cu(Tp(2Nt)2)(CO)] adsorbed on graphene, must be substantial as graphene has a hexagonal lattice while [Cu(Tp(2Nt)2)(CO)] adsorbed in a square packing.
Conclusions
In this work the complexes [Cu(TpPh2)(C2H4)], [Cu(Tp(1Nt)2)(C2H4)], [Cu(Tp(2Nt)2)(C2H4)], [Cu(Tp(1Nt)2)(CO)], and [Cu(Tp(2Nt)2)(CO)] were synthesized. The new naphthyl-substituted hydridotrispyrazolylborate ligands [Tp(1Nt)2]− and [Tp(2Nt)2]− constitute some of the bulkiest hydridotrispyrazolylborate ligands reported to date. The ethene complexes were found to show particularly strong shielding of the ethene protons in NMR spectra due to the limited rotational freedom of the naphthyl groups combined with the steric pressure caused by the 3,5-disubstitution of the pyrazole rings which forces the naphthyl groups at the 3-positions towards the ancillary ligands. The conformational freedom of the naphthyl groups in the carbonyl complexes is considerably larger than that in the ethene complexes, which indicates, together with the information gained from the X-ray crystal structures of [Cu(TpPh2)(C2H4)] and [Cu(Tp(2Nt)2)(C2H4)], that the interactions between the π electron clouds of the naphthyl groups around the copper(I) center and the ethene protons play an important role in the properties of the resulting ethene complexes. The carbonyl complexes were used to prepare monolayers or thin multilayers on graphene and HOPG and showed that a bulk property such as a pronounced ability or inability to crystallize on the macroscale translates well to the nanoscale. The findings presented in this work offer insight into the exciting chemistry and behavior of bulky, π-stacking copper(I) complexes that likely also apply to other complex molecules incorporating π-stacking ligands and opens the way for further application-oriented studies of such complexes in combination with graphene.Experimental
General considerations
All manipulations of air-sensitive compounds were performed under an atmosphere of purified argon gas using standard Schlenk techniques. All solvents were purchased from commercial sources and were reagent grade. Solvents used for air-sensitive manipulations were dried and deaerated using a PureSolv MD 5 Solvent Purification System and stored on 3 Å molecular sieves under argon. Where appropriate, glassware was flame dried in vacuo immediately before use. 3,5-Diphenyl-1H-pyrazole was synthesized following a literature procedure.71H and 13C NMR spectra were recorded on a Bruker DPX300 spectrometer (300 MHz for 1H, 75.44 MHz for 13C), a Bruker DMX400 spectrometer (400 MHz for 1H, 128.3 MHz for 11B and 100.6 MHz for 13C) or a Bruker Avance AV500 spectrometer (500 MHz for 1H and 126 MHz for 13C). Chemical shifts are given in ppm and referenced using the deuterated solvents as the internal references for 1H and 13C.3513C spectra were recorded using 1H-decoupling. Elemental analyses were performed by Mikrolab Kolbe in Germany. IR spectra were recorded on a PerkinElmer UATR Two FT-IR spectrometer set to a resolution of 2 cm−1. ESI-MS spectra were recorded on a Thermo Finnigan AQA ESI-MS system in MeCN. HRMS spectra were recorded on a Thermo Scientific LTQ Orbitrap XL high resolution FT-MS system in MeCN. AFM experiments were performed using a Nanoworld USC-FO.3-KO.3 tip in a JPK NanoWizard 4a NanoScience AFM. STM experiments were performed using a freshly cut Pt/Ir wire tip at 298 K. The samples of graphene on SiO2@Si were prepared by wet-transfer of CVD graphene which was purchased from Graphenea.44X-ray crystallography
All reflection intensities were measured at 110(2) K using a SuperNova diffractometer (equipped with an Atlas detector) with Cu Kα radiation (λ = 1.54178 Å) under the program CrysAlisPro (Version 1.171.36.32, Agilent Technologies, 2013). The same program was used to refine the cell dimensions and for data reduction. The structure was solved with the program SHELXS-2013 and was refined on F2 with SHELXL-2013.45 Analytical numeric absorption correction based on a multifaceted crystal model was applied using CrysAlisPro. The temperature of the data collection was controlled using the system Cryojet (manufactured by Oxford Instruments). The H atoms were placed at calculated positions (unless otherwise specified) using the instructions AFIX 13 or AFIX 43 with isotropic displacement parameters having values 1.2 or 1.5 times the Ueq of the attached B or C atoms.Sample preparation for AFM and STM
Solutions of [Cu(Tp(1Nt)2)(CO)] and [Cu(Tp(2Nt)2)(CO)] (0.5 mM in cyclohexane) were drop-cast in drops of 5 μL on freshly cleaved HOPG mounted on steel sample holding plates. The samples were left to dry in a stream of filtered argon for at least thirty minutes before being scanned. For the samples that were dip coat 11 mg (10 μmol) of [Cu(Tp(1Nt)2)(CO)] or [Cu(Tp(2Nt)2)(CO)] were added to 1 mL dry, degassed DCM. [Cu(Tp(1Nt)2)(CO)] dissolved completely while [Cu(Tp(2Nt)2)(CO)] did not; the solution was filtered (0.45 μm PTFE) before use. Samples of 288 nm thick SiO2 on Si coated with CVD graphene were then immersed completely in the solution/suspension for 10 minutes after which the samples were removed from the solutions and thoroughly rinsed using a flow of DCM from a syringe (2 × 20 mL). The samples were left to dry in air for at least 60 minutes before being used for ellipsometry and AFM. The STM sample was prepared by drop casting 10 μL of a 0.5 μM solution of [Cu(Tp(2Nt)2)(CO)] in cyclohexane onto a ∼25 mm2 freshly cleaved HOPG surface. After drying under argon, the sample was dried further in a stream of argon for 10 minutes and used immediately afterwards.Syntheses
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C69H46BN6–) 969.38715; found 969.38715.
C69H46BN6–) 969.38715; found 969.38928.
C69H46BN6–) 969.38715; found 969.38715.
C69H46BN6–) 969.38715; found 969.38928.
Synthesis of the copper compounds
Acknowledgements
We thank Gerwin Spijksma for HRMS and Fons Lefeber and Karthick Sai Sankar Gupta for their assistance with the NMR experiments. We thank Dr Wangyang Fu for providing the graphene samples.References
- S. A. DiBenedetto, A. Facchetti, M. A. Ratner and T. J. Marks, Adv. Mater., 2009, 21, 1407–1433 CrossRef CAS.
- S. Kobayashi, T. Nishikawa, T. Takenobu, S. Mori, T. Shimoda, T. Mitani, H. Shimotani, N. Yoshimoto, S. Ogawa and Y. Iwasa, Nat. Mater., 2004, 3, 317–322 CrossRef CAS PubMed.
- I. H. Campbell, J. D. Kress, R. L. Martin, D. L. Smith, N. N. Barashkov and J. P. Ferraris, Appl. Phys. Lett., 1997, 71, 3528–3530 CrossRef CAS.
- A. J. Clough, J. W. Yoo, M. H. Mecklenburg and S. C. Marinescu, J. Am. Chem. Soc., 2015, 137, 118–121 CrossRef CAS PubMed.
- J. D. Blakemore, A. Gupta, J. J. Warren, B. S. Brunschwig and H. B. Gray, J. Am. Chem. Soc., 2013, 135, 18288–18291 CrossRef CAS PubMed.
- S. C. Eady, M. M. MacInnes and N. Lehnert, ACS Appl. Mater. Interfaces, 2016, 8, 23624–23634 CAS.
- N. Kitajima, K. Fujisawa, C. Fujimoto, Y. Morooka, S. Hashimoto, T. Kitagawa, K. Toriumi, K. Tatsumi and A. Nakamura, J. Am. Chem. Soc., 1992, 114, 1277–1291 CrossRef CAS.
- J. W. Whittaker, Arch. Biochem. Biophys., 2005, 433, 227–239 CrossRef CAS PubMed.
- F. I. Rodríguez, J. J. Esch, A. E. Hall, B. M. Binder, G. E. Schaller and A. B. Bleecker, Science, 1999, 283, 996–998 CrossRef.
- C. Martin, M. Sierra, E. Alvarez, T. R. Belderrain and P. J. Perez, Dalton Trans., 2012, 41, 5319–5325 RSC.
- C. Martîn, J. M. a. Muñoz-Molina, A. Locati, E. Alvarez, F. Maseras, T. s. R. Belderrain and P. J. Pérez, Organometallics, 2010, 29, 3481–3489 CrossRef.
- T. F. van Dijkman, M. A. Siegler and E. Bouwman, Dalton Trans., 2015, 44, 21109–21123 RSC.
- H. V. R. Dias, H. L. Lu, H. J. Kim, S. A. Polach, T. Goh, R. G. Browning and C. J. Lovely, Organometallics, 2002, 21, 1466–1473 CrossRef CAS.
- J. M. MacLeod and F. Rosei, Small, 2014, 10, 1038–1049 CrossRef CAS PubMed.
- K. Fujisawa, N. Tada, Y. Ishikawa, H. Higashimura, Y. Miyashita and K.-i. Okamoto, Inorg. Chem. Commun., 2004, 7, 209–212 CrossRef CAS.
- K. Fujisawa and H. Takisawa, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 986–989 CAS.
- A. L. Rheingold, L. M. Liable-Sands and S. Trofimenko, Inorg. Chem., 2001, 40, 6509–6513 CrossRef CAS PubMed.
- T. F. van Dijkman, M. A. Siegler and E. Bouwman, Eur. J. Inorg. Chem., 2016, 2586–2594 CrossRef CAS.
- P. Chen, D. E. Root, C. Campochiaro, K. Fujisawa and E. I. Solomon, J. Am. Chem. Soc., 2003, 125, 466–474 CrossRef CAS PubMed.
- A. E. Baum, H. Park, D. Wang, S. V. Lindeman and A. T. Fiedler, Dalton Trans., 2012, 41, 12244–12253 RSC.
- A. Mukherjee, M. Martinho, E. L. Bominaar, E. Münck and L. Que, Angew. Chem., Int. Ed., 2009, 48, 1780–1783 CrossRef CAS PubMed.
- D. J. Harding, W. Phonsri, P. Harding, J. Sirirak, Y. Tangtirungrotechai, R. D. Webster and H. Adams, New J. Chem., 2015, 39, 1498–1505 RSC.
- S. T. Kleespies, W. N. Oloo, A. Mukherjee and L. Que, Inorg. Chem., 2015, 54, 5053–5064 CrossRef CAS PubMed.
- J. A. Schofield, W. W. Brennessel, E. Urnezius, D. Rokhsana, M. D. Boshart, D. H. Juers, P. L. Holland and T. E. Machonkin, Eur. J. Inorg. Chem., 2015, 4643–4647 CrossRef CAS.
- S. Paria, S. Chatterjee and T. K. Paine, Inorg. Chem., 2014, 53, 2810–2821 CrossRef CAS PubMed.
- T. E. Machonkin, M. D. Boshart, J. A. Schofield, M. M. Rodriguez, K. Grubel, D. Rokhsana, W. W. Brennessel and P. L. Holland, Inorg. Chem., 2014, 53, 9837–9848 CrossRef CAS PubMed.
- H. Park, M. M. Bittner, J. S. Baus, S. V. Lindeman and A. T. Fiedler, Inorg. Chem., 2012, 51, 10279–10289 CrossRef CAS PubMed.
- R.-M. Zhu, L. Hu, Y.-Z. Li, Y. Song and J.-L. Zuo, Inorg. Chem. Commun., 2013, 35, 79–82 CrossRef CAS.
- S. M. Carrier, C. E. Ruggiero, R. P. Houser and W. B. Tolman, Inorg. Chem., 1993, 32, 4889–4899 CrossRef CAS.
- H. V. R. Dias and T. Goh, Polyhedron, 2004, 23, 273–282 CrossRef CAS.
- J. L. Duncan, Mol. Phys., 1974, 28, 1177–1191 CrossRef CAS.
- J. S. Thompson, R. L. Harlow and J. F. Whitney, J. Am. Chem. Soc., 1983, 105, 3522–3527 CrossRef CAS.
- A. Almenningen, O. Bastiansen, L. Fernholt, B. N. Cyvin, S. J. Cyvin and S. Samdal, J. Mol. Struct., 1985, 128, 59–76 CrossRef CAS.
- R. J. Abraham, M. Canton and L. Griffiths, Magn. Reson. Chem., 2001, 39, 421–431 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- M. I. Bruce and A. P. P. Ostazewski, J. Chem. Soc., Dalton Trans., 1973, 2433–2436 RSC.
- H. V. R. Dias, H.-J. Kim, H.-L. Lu, K. Rajeshwar, N. R. de Tacconi, A. Derecskei-Kovacs and D. S. Marynick, Organometallics, 1996, 15, 2994–3003 CrossRef CAS.
- H. V. R. Dias and H.-L. Lu, Inorg. Chem., 1995, 34, 5380–5382 CrossRef CAS.
- K. Fujisawa, T. Ono, Y. Ishikawa, N. Amir, Y. Miyashita, K.-i. Okamoto and N. Lehnert, Inorg. Chem., 2006, 45, 1698–1713 CrossRef CAS PubMed.
- J. L. Schneider, S. M. Carrier, C. E. Ruggiero, V. G. Young and W. B. Tolman, J. Am. Chem. Soc., 1998, 120, 11408–11418 CrossRef CAS.
- H. V. R. Dias, W. Jin, H.-J. Kim and H.-L. Lu, Inorg. Chem., 1996, 35, 2317–2328 CrossRef CAS PubMed.
- S. Imai, K. Fujisawa, T. Kobayashi, N. Shirasawa, H. Fujii, T. Yoshimura, N. Kitajima and Y. Moro-oka, Inorg. Chem., 1998, 37, 3066–3070 CrossRef CAS.
- E. Bauer, Z. Kristallogr. - New Cryst. Struct., 1958, 110, 372–394 CrossRef.
- J. W. Suk, A. Kitt, C. W. Magnuson, Y. Hao, S. Ahmed, J. An, A. K. Swan, B. B. Goldberg and R. S. Ruoff, ACS Nano, 2011, 5, 6916–6924 CrossRef CAS PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- T. Yamada, T. Nagata, K. D. Sugi, K. Yorozu, T. Ikeno, Y. Ohtsuka, D. Miyazaki and T. Mukaiyama, Chem. – Eur. J., 2003, 9, 4485–4509 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1524294 and 1524295. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt04851j |
| This journal is © The Royal Society of Chemistry 2017 |