
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface chemistry of pure tetragonal ZrO2 and gas-phase dependence of the tetragonal-to-monoclinic ZrO2 transformation†
Eva-Maria
Köck
a,
Michaela
Kogler
a,
Thomas
Götsch
 a,
Lukas
Schlicker
b,
Maged F.
Bekheet
b,
Andrew
Doran
c,
Aleksander
Gurlo
b,
Bernhard
Klötzer
a,
Benedikt
Petermüller
d,
Daniel
Schildhammer
d,
Nevzat
Yigit
e and
Simon
Penner
*a
a,
Lukas
Schlicker
b,
Maged F.
Bekheet
b,
Andrew
Doran
c,
Aleksander
Gurlo
b,
Bernhard
Klötzer
a,
Benedikt
Petermüller
d,
Daniel
Schildhammer
d,
Nevzat
Yigit
e and
Simon
Penner
*a
aInstitute of Physical Chemistry, University of Innsbruck, Innrain 80-82, A-6020 Innsbruck, Austria. E-mail: simon.penner@uibk.ac.at; Fax: +4351250758199; Tel: +4351250758003
bFachgebiet Keramische Werkstoffe/Chair of Advanced Ceramic Materials, Institut für Werkstoffwissenschaften- und technologien, Technische Universität Berlin, Hardenbergstr. 40, D-10623 Berlin, Germany
cAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
dInstitute of Inorganic and Theoretical Chemistry, University of Innsbruck, Innrain 80-82, A-6020 Innsbruck, Austria
eInstitute of Materials Chemistry, Vienna University of Technology, Getreidemarkt 9/BC, A-1060 Wien, Austria
First published on 15th March 2017
Abstract
The surface chemical properties of undoped tetragonal ZrO2 and the gas-phase dependence of the tetragonal-to-monoclinic transformation are studied using a tetragonal ZrO2 polymorph synthesized via a sol–gel method from an alkoxide precursor. The obtained phase-pure tetragonal ZrO2 is defective and strongly hydroxylated with pronounced Lewis acidic and Brønsted basic surface sites. Combined in situ FT-infrared and electrochemical impedance measurements reveal effective blocking of coordinatively unsaturated sites by both CO and CO2, as well as low conductivity. The transformation into monoclinic ZrO2 is suppressed up to temperatures of ∼723 K independent of the gas phase composition, in contrast to at higher temperatures. In inert atmospheres, the persisting structural defectivity leads to a high stability of tetragonal ZrO2, even after a heating–cooling cycle up to 1273 K. Treatments in CO2 and H2 increase the amount of monoclinic ZrO2 upon cooling (>85 wt%) and the associated formation of either Zr-surface-(oxy-)carbide or dissolved hydrogen. The transformation is strongly affected by the sintering/pressing history of the sample, due to significant agglomeration of small crystals on the surface of sintered pellets. Two factors dominate the properties of tetragonal ZrO2: defect chemistry and hydroxylation degree. In particular, moist conditions promote the phase transformation, although at significantly higher temperatures as previously reported for doped tetragonal ZrO2.
1. Introduction
The physical chemistry of different ZrO2 polymorphs has been an intensively studied research field over decades, as recent reviews prove.1–9 However, upon literature screening, open questions especially regarding phase stability, surface termination, surface activity, or gas-phase-dependent stability limits still remain. Despite this deficiency, the diversity of the application of ZrO2-based materials is considerable and directly reflects its manifold specific properties. Applications in ceramic engineering,10 as biomedical implants,11 in sensor technology,12 as microelectronic devices,13 as solid electrolytes14 and in catalysis2 have been reported. Concerning the latter, ZrO2 is used as both catalyst material and catalyst support. It is used for conversion of hydrocarbons in biomass-related reactions6 or doped with Y2O3 as an anode support and electrolyte material in fuel cells. As such, it encompasses use in catalytic processes over a wide temperature range between few hundred K up to 1273 K.15 Obviously, the respective requirements of the intrinsic properties of the used ZrO2 material are strongly depending on the specific field of application. Many empirical and fundamental research attempts have therefore been made over the last decades to resolve the nature of the active sites on these ZrO2-based materials alongside their selective generation and exact control with suitable reducibility and stability. Parameters that strongly influence the final activity are preparation routine, pre-treatment, phase (purity), termination, surface area, particle size, doping level or stoichiometry. Furthermore, when used as catalytic support material, in combination with metals like Au, Pt, Cu (methanol synthesis, water gas shift reactions) or Pt, Ni, Co, Cu (reforming reactions), also metal–oxide interfacial zones with additional multifaceted properties have to be taken into account.16 To add to the complexity, the Zr–O phase diagram exhibits five stable ZrO2 polymorphs. However, the most commonly used and best characterized are the phases with the monoclinic, the tetragonal and the cubic structure. Due to the fact that the monoclinic phase (baddeleyite) is the by far most stable polymorph under realistic experimental conditions, this phase is most widely studied among pure undoped ZrO2 materials. To access the other undoped polymorphs, specific annealing treatments are necessary. The temperature-induced phase transitions from monoclinic ZrO2 to tetragonal ZrO2 occurs at ∼1400 K, and above ∼2600 K the cubic phase is present.17 At lower temperatures the cubic and tetragonal polymorphs are usually stabilized by doping with different metal cations (e.g. Y3+, Si4+ and Zn2+). However, not only the structure, but directly also other intrinsic properties such as the amount and acidity/basicity of hydroxylation, stability of surface area or electrochemical conductivity are drastically changed. Apart from cationic doping, the tetragonal phase can also be stabilized by adding organic additives, but also by simple grain size control and additionally by interfacial effects (below a critical diameter d of d ≤ 33 nm).8 Furthermore, also anions such as OH−, SO42− are potential stabilizers for low-temperature stable tetragonal ZrO2.17 Sulfated tetragonal ZrO2 is a special case in terms of catalytic surface modification, since it is a solid superacid, e.g. it exhibits a unique activity in the catalytic isomerization of hydrocarbons. Aside from the sulfated surface termination, the presence of especially a crystalline tetragonal phase is usually reported to be crucial for the performance of the ZrO2-containing catalysts.18 Having said that, the stability of undoped and unsulfated pure tetragonal ZrO2 (in terms of thermally or gas-phase-induced transformation into monoclinic ZrO2) is still an interesting research field, since there are many preparation and pre-treatment attempts leading to different stability, but also activity and surface reactivity.In the present study, we aim to characterize the surface chemistry of a pure undoped tetragonal ZrO2 sample, prepared via a very simple sol–gel route starting from a solution of zirconium(IV)isopropoxide in isopropanol without any further additives. Calcination of the resulting Zr hydroxide aerogel leads to essentially phase pure nanocrystalline tetragonal ZrO2 (which is most likely stabilized via OH−/OH-groups, lattice anionic defects and crystallite size effect).19 Furthermore, the focus is on elucidating the gas-phase-dependent physical chemistry of this tetragonal ZrO2 phase under oxidative, reductive or inert gas atmospheres of dry and moist gases He, O2, CO, CO2 and H2. The deliberate addition of H2O is also worthwhile since phase transformation of tetragonal to monoclinic ZrO2 in H2O at room temperature has been reported.20 This includes “low temperature” studies up to 723 K to characterize the surface termination (i.e. hydroxylation, acid–base properties) and the corresponding electrochemical behavior of phase pure metastable t-ZrO2. Additionally, “high temperature” studies, to gain detailed information about the gas-phase dependent stability, up to a temperature of 1273 K were performed. For this purpose in situ/operando Fourier-Transform infrared spectroscopy, in situ/operando electrochemical impedance spectroscopy, ex situ X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS) and atomic force microscopy (AFM) studies were conducted. From a previous study,19 detailed information about the general kinetic stability of the synthesized tetragonal ZrO2 material in air has already been compiled. In fact, the aerogel precursor crystallizes at 673 K to essentially chemically and structurally phase-pure tetragonal ZrO2. Transformation to monoclinic ZrO2 in air is pronounced only upon annealing to 873 K and prolonged isothermal treatments. Less conversion takes place at isothermal treatment at higher temperatures up to 1273 K, but the main part of the sample is transformed to monoclinic (m-) ZrO2 upon re-cooling to room temperature, once the critical temperature of 873 K is overcome.
2. Experimental
2.1. Materials
Pure tetragonal ZrO2 was prepared via a sol–gel route starting with zirconium(IV)isopropoxide (isopropanol adduct, 99.9% Zr, Strem Chemicals) dissolved in isopropanol (Finne Gatt-Koller, 99.98%) and finally quenched with water. This preparation routine has been described previously in ref. 19 and is essentially a variant of the one reported by Mazdiyasni et al.21 (decomposition of zirconium alkoxides and the subsequent hydrolysis of zirconium hydroxy aerogels). The resulting aerogel was subsequently calcined at 723 K in pure O2 in the respective experimental setups. All samples were without exception checked by ex situ XRD. The surface of the calcined (at a maximum temperature of T = 723 K) tetragonal ZrO2 was 50 m2 g−1 (according to the Brunauer–Emmett–Teller (BET) method, performed using a Quantachrome Nova 2000 Surface Area and Pore Size Analyzer). To ensure chemical purity, chemical analysis of all samples was performed using a Spectro XEPOS energy dispersive X-ray fluorescence analyzer (EDXRFA) which proves purities of 99.99%. The gases were supplied by Messer (CO 4.7, CH4 4.5, O2 5.0, He 5.0, CO2 4.5, H2 5.0). For a typical experiment in the in situ/operando FT-IR/EIS setups the sample was annealed in dry O2 prior to each experiment, to ensure reproducible starting conditions (heating and cooling rates 10 K min−1, gas flow ∼1.0 mL s−1). For absolutely dry conditions, the gases were dried with a liquid N2-ethanol cooling trap at a temperature of approximately 163 K for CO, CH4, and O2, 223 K for CO2 and for He and H2 a N2 cooling trap at 77 K was used.As a structural reference commercial pure monoclinic ZrO2 (zirconium(IV) oxide, 99.99%, Alfa Aesar) subjected to the identical chemical structural and spectroscopic analysis was used (see XRD pattern in Fig. S1†).
2.2. FT-IR spectroscopy
FT-IR spectra were recorded in transmission mode on an Agilent Cary 660 spectrometer with a mid-infrared source and a DTGS detector. Transmission mode was chosen to monitor both, the surface and bulk-related infrared-detectable phenomena. The powder samples were pressed into thin pellets using a weight of ≤2 t (10 mm diameter, thickness ∼0.1 mm, sample mass about 20 mg) and then subsequently placed inside a homemade in situ/operando reactor cell,22 providing chemically inert surroundings of the sample in the heated area. In situ/operando measurements up to 1273 K under flowing and static conditions can be performed, in addition to vacuum experiments at minimum pressures of 3 × 10−7 mbar. BaF2 as window material allows to access wavelengths above 800 cm−1. Experiments in flowing mode (ambient pressure) can then be directly correlated with electrochemical impedance measurements. In static mode, the gases are pre-adsorbed on a 5 Å zeolite trap binding water sufficiently strongly, before the dry gases are desorbed into the evacuated and degassed cell. All reported spectra are corrected by the spectrum of the dry pre-oxidized oxide pellet at room temperature and under vacuum prior to exposure to the gases.2.3. Electrochemical impedance spectroscopy (EIS)
The in situ/operando impedance cell consists of an arrangement of an outer quartz tube and two smaller inner quartz tubes, to which the sample and the electrodes are attached to. For heating, a tubular Linn furnace was used. Temperature control was ensured by a thermocouple (K-element), located in the reactor about 5 mm downstream of the sample, and a Micromega PID control unit. The impedance was measured using an IM6e impedance spectrometer (Zahner Messsysteme), providing data on the impedance and the phase angle of the current as a function of voltage simultaneously. The powder samples were pressed into pellets with a weight of ≤2 t (5 mm diameter, ∼0.1 mm thick, sample mass about 20 mg) and placed between two circular Pt electrodes acting as a plate capacitor in mechanically enforced contact with the pelletized sample.For all temperature-programmed impedance measurements described in this work, an amplitude of 20 mV of the superimposed sinusoidal modulation voltage signal at an overall DC potential of 0 V and a frequency of 1 Hz were applied to the Pt electrodes, i.e. the impedance of the pellet was effectively measured in an electrochemically unpolarized state. The impedance modulus value |Z| represents the “impedance” in all temperature-programmed experiments.
For moist conditions, water vapor of 24 mbar was produced by passing He (∼1 mL s−1) through a water saturator at room temperature.
Note that the used spectrometer has a detection limit near ∼1010 Ω. In order to ensure an appropriate discussion, this phenomenon (plateau-like impedance stagnation near the detection limit) will be referred to as an “insulating” behavior in this work.
2.4. X-ray photoelectron spectroscopy (XPS)
In order to gain information about the surface-near chemistry, all samples were analyzed by X-ray photoelectron spectroscopy (XPS) using a Thermo Scientific MultiLab 2000 spectrometer with a base pressure in the low 10−10 mbar range. The instrument is equipped with a monochromated Al-Kα X-ray source (1486.6 eV), an Alpha 110 hemispherical sector analyzer. A flood gun is used for charge compensation, providing electrons with a kinetic energy of 6 eV. The energy axis shift was calibrated relative to the C–C component of the C 1s peak (set to 284.8 eV). For the analysis of the chemical states, the peak shapes used to fit the respective Zr 3d and C 1s spectra were products of Gaussian and Lorentzian peak shapes (30% Lorentzian). Several constraints were applied for the fitting procedure of the zirconium region: the splitting between the 3d5/2 and the 3d3/2 components was fixed at 2.4 eV, according to literature,23 and the peak widths were set to be the same for the spin-split components as well as for the peaks of the different oxidation states. Furthermore, to enable convergence of the fit towards a sensible minimum in the presence of multiple oxidation states, the energy shifts of the 3d5/2 peaks between the various chemical states have been constrained to literature values.242.5. X-ray powder diffraction (XRD)
The X-ray powder diffraction patterns were obtained by measuring the powder patterns in transmission geometry using a STOE STADI P powder diffractometer with Ge(111)-monochromatized MoKα1 (λ = 70.93 pm) radiation. The data were recorded in 2θ with steps of 0.1° from 2–45.875°. As a detector, a silicon microstrip solid state detector Mythen 1K was used. The structural refinement was performed with the program TOPAS 4.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25 by taking the crystal structure parameters of monoclinic26 and the tetragonal ZrO227 as starting values. The obtained crystallographic data from the associated Rietveld refinement28 are listed in Table S1.†
25 by taking the crystal structure parameters of monoclinic26 and the tetragonal ZrO227 as starting values. The obtained crystallographic data from the associated Rietveld refinement28 are listed in Table S1.†
The in situ high-temperatures synchrotron XRD experiments in H2 and CO2 flow are performed at beamline 12.2.2, Advanced Light Source, Lawrence Berkeley National Labs, California. The in situ diffraction patterns were collected in the angle-dispersive transmission mode with a focused 25 keV monochromatic beam (λ = 0.4959 Å/50 μm spot size). The sample powder was heated in 700 μm quartz capillaries under a continuous gas flow (CO2 or H2) injected through another 300 μm capillary with a cut-open end. The capillary is heated in an infrared heated SiC tube furnace we described in ref. 29. Diffraction patterns are recorded by a PerkinElmer flat panel detector (XRD 1621, dark image and strain correction) every 25 seconds during the heating and cooling cycle. As the synthesized starting material is a powdery amorphous aerogel, previous to the experiment it is in situ sintered at 723 K inside the capillary under air flow until crystallization to tetragonal ZrO2 is completed.
2.6. Atomic force microscopy (AFM)
Atomic force micrographs were obtained using a Veeco Dimension 3100 microscope in tapping mode, equipped with HI'RES cantilevers by MikroMasch (with a tip radius below 1 nm) used for high-resolution images, as well as Veeco RTESPW silicon cantilevers (about 10 nm tip radius) to obtain information on larger scales.3. Results and discussion
3.1. Surface chemistry of tetragonal ZrO2 between room temperature (RT) and 723 K
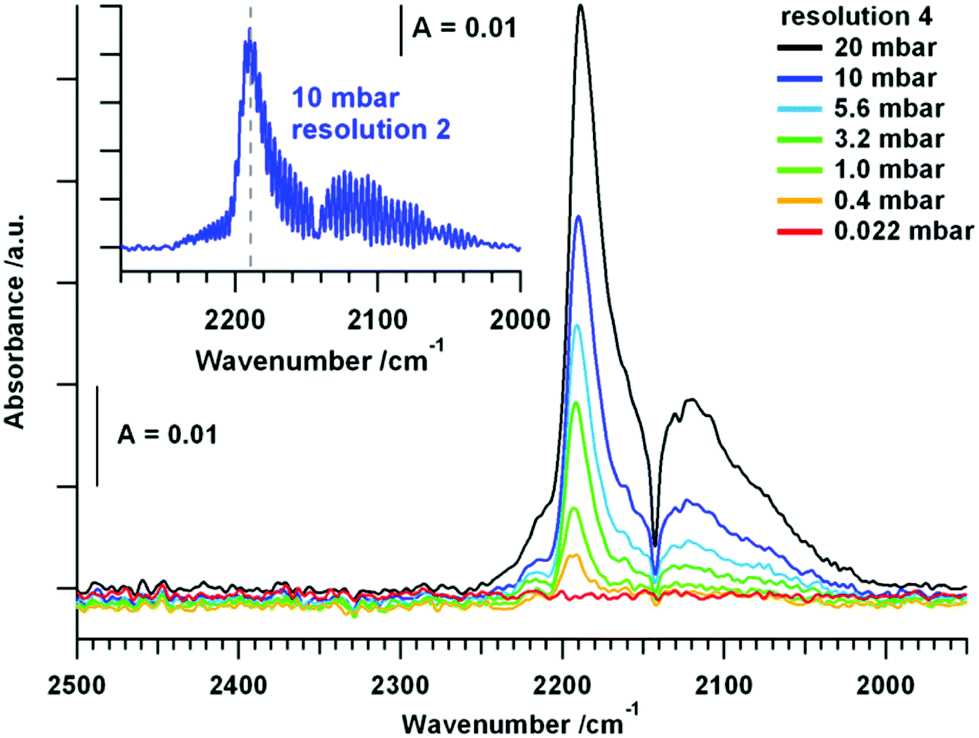 | ||
| Fig. 1 FT-IR measurements of CO adsorbed on tetragonal ZrO2 at CO pressures between 0.022 mbar and 20 mbar at room temperature. Spectra in the main panel were collected using a resolution of 4 and the inset shows a spectrum of 10 mbar CO with a resolution of 2. | ||
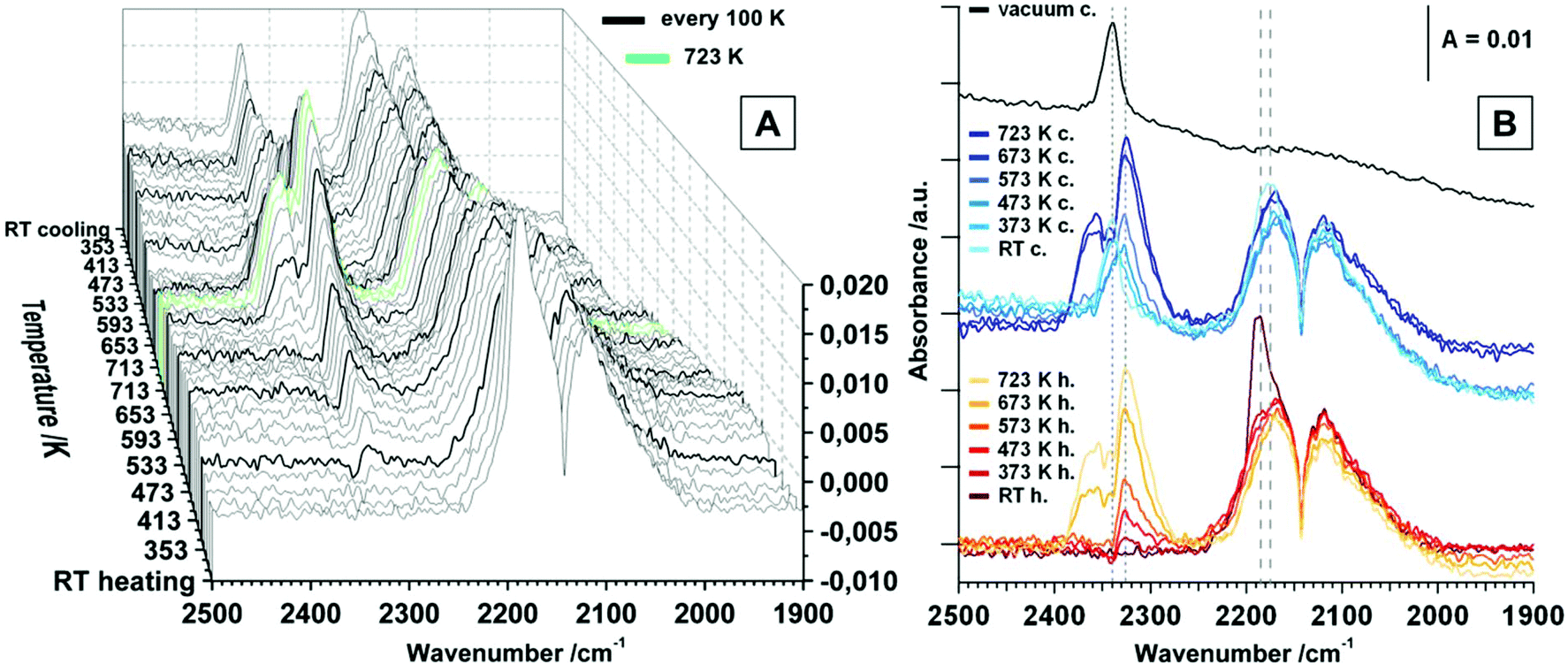 | ||
| Fig. 2 FT-IR spectra of heating and cooling tetragonal ZrO2 in a static atmosphere of 20 mbar CO; maximum temperature = 723 K, heating/cooling rates = 10 K min−1. (A) Waterfall plot – spectra are collected every 20 K step. (B) Selected spectra upon heating and cooling, including vacuum spectrum after the heating/cooling cycle. | ||
 | ||
| Fig. 3 (A) FT-IR spectra of heating and cooling tetragonal ZrO2 in flowing CO; maximum temperature = 723 K, heating/cooling rates = 10 K min−1, isothermal period at 723 K = 10 min, flow = ∼1.0 mL s−1; (B) FT-IR spectrum of tetragonal ZrO2 in flowing CO at 673 K. | ||
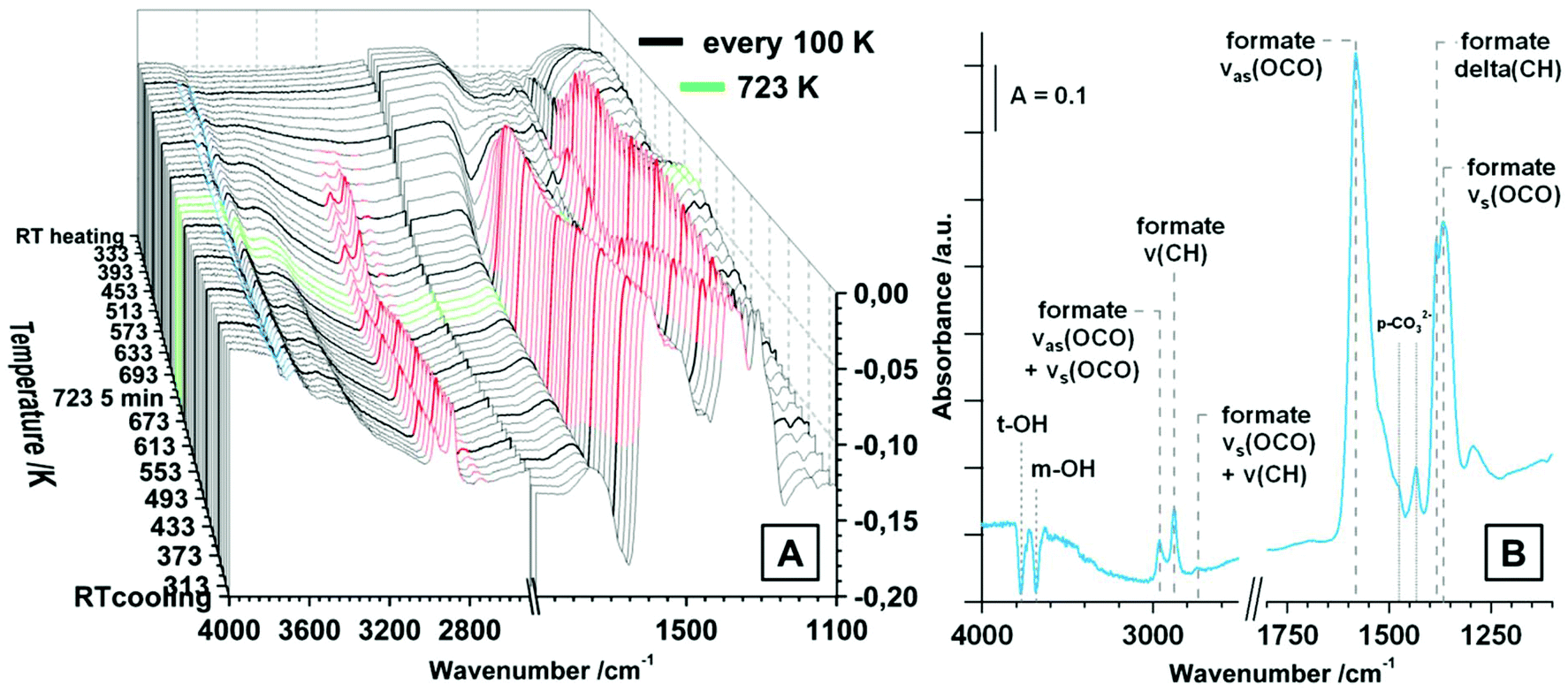
 | ||
| Fig. 4 (A) FT-IR spectra of heating and cooling tetragonal ZrO2 in 20 mbar CO2 static atmosphere; maximum temperature = 723 K, heating/cooling rates = 10 K min−1, isothermal period at 723 K = 5 min; (B) FT-IR spectra of tetragonal ZrO2 in vacuum after the heating and cooling routine in 20 mbar CO2. | ||
CO adsorption. The used pure tetragonal ZrO2 was pretreated in dry O2 up to 723 K and the spectrum of the fully oxidized sample at room temperature in vacuum was taken as background for all further spectra. The exposure of the oxide to an increasing pressure of CO leads to linearly adsorbed CO molecules, as displayed in Fig. 1.
In addition to the gas phase signal of CO between ∼2240 and 2020 cm−1 a peak at 2191 cm−1 is present, which can be assigned to CO molecules bonded to Lewis acid Zr4+ centers, most likely on a step, kink or corner due to the high shift in wavenumber.1 The evolution of the CO signal upon heating the sample in static 20 mbar CO up to 723 K is subsequently shown in Fig. 2.
The signal for physisorbed CO species decreases with increasing temperature and is no longer present above 373 K. This is correlated with the observation of a peak at 2326 cm−1 above 373 K, which is increasing up to 723 K. At first sight, this band could be assigned to a physisorbed CO2 molecule, but the molecular adsorption of CO2 on ZrO2 is usually observed at temperatures below room temperature and the presence at higher temperatures would indicate a high amount of Lewis acidic sites on the surface.18 Since this signal is actually getting more intense with increasing temperature, this interpretation can be discarded. In studies on CeO2 this phenomenon is assigned to result from electronic origin, such as oxygen vacancies or defective lattice sites.38–40 It is a well-known fact that CO can be oxidized to CO2via two reaction pathways: (i) reaction with a lattice oxygen to CO2 and an formation of oxygen vacancy and (ii) reaction with a surface hydroxyl group to form formates, which probably can be also decomposed to CO2 (see also discussion to Fig. 3 below). However, signals of gaseous CO2 (2400 cm−1–2270 cm−1) are present above 613 K. Upon re-cooling no gas phase signal for CO2 is present below 573 K and the signal referred to the oxygen vacancy shifts to 2340 cm−1 below 373 K. Molecular adsorption of CO, when re-cooling, is not as pronounced as before the heating and cooling routine and results in a less-shifted peak at 2175 cm−1. Applying vacuum leads to effective removal of all gaseous and molecularly adsorbed species. The fact that the increase of oxygen vacancies due to surface reduction with CO does not lead to an increased signal for molecularly adsorbed CO species, is consistent with the statement that such an oxygen vacancy rather is a basic adsorption spot. Interestingly, also the amount of acidic Lewis sites such as Zr4+ centers is decreased upon the heating and cooling routine. Aside from the molecular adsorption and surface reaction of CO, also a high amount of chemisorbed species is detected. This is illustrated in Fig. 3, which shows the temperature dependency of tetragonal ZrO2 in flowing CO.
The general absorbance change in Fig. 3A is originating from the pronounced reduction of the sample in pure flowing CO. For a better comprehension of this phenomenon, we refer to the discussion of Fig. 5B, where the absorbance at 2000 cm−1 was plotted versus the temperature. Derived from similar investigations on TiO2 and CeO2, an increase of the absorbance is associated with the increase of electrons in the conduction band, which, upon reduction e.g. in H2, is the case for TiO2 and CeO2 due to the possibility of donor levels near the conduction band, such as Ti3+ and Ce3+.39,41 In case of the characterized isolating tetragonal ZrO2 sample, the absorbance is decreasing with increasing reduction. Thus, CO reduction surprisingly leads to fewer donor states near the conduction band (compare also the discussion about reduction in H2, displayed in Fig. 5C). Remarkable is the onset temperature for the absorbance-affecting reduction of 373 K, which is a hundred K before the observation of carbonates, but in good correlation with the detection of oxygen vacancies at 2326 cm−1 above 373 K. In Fig. 5B, signals for bidentate formates (νas(OCO) + νs(OCO) = 2962 cm−1, ν(CH) = 2878 cm−1, νs(OCO) + ν(CH) = 2743 cm−1, νas(OCO) = 1583 cm−1, δ(CH) = 1385 cm−1 and νs(OCO) = 1367 cm−1 (ref. 32)) are observed above 473 K, increasing up to 573 K. The signals then stay more or less constant upon further heating and re-cooling. Furthermore, negative peaks (species that where present when saving the background or on the initially oxidized surface) of terminal (3774 cm−1, t-) and multi-coordinated (3686 cm−1, m-) OH groups are present above 373 K. In addition, above 373 K bands for polydentate carbonates (νas(CO3) = 1470 cm−1, νs(CO3) = 1433 cm−1) are observed in flowing CO. The formation of polydentate carbonates can arise also from direct oxidation of CO at cus Zr4+ centers34 or by chemisorption of initially formed CO2 on a neighboring O2−.16 Such reactions are believed to be catalyzed at steps of the crystals.30
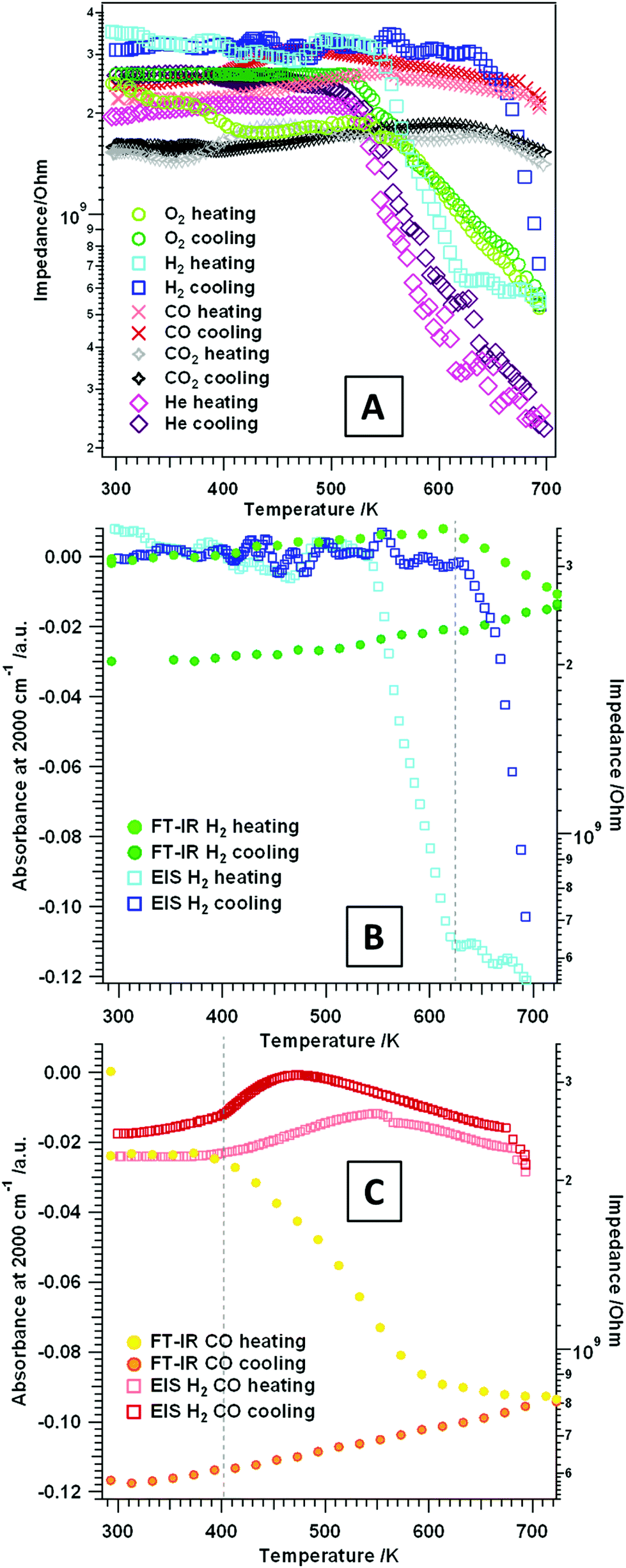 | ||
| Fig. 5 (A) Temperature-programmed electrochemical impedance results on t-ZrO2 in dry H2, CO, He, O2 and CO2. Panel (B) and (C): plot of the FT-IR absorbance at 2000 cm−1 (left y-axis) of tetragonal ZrO2 in flowing (B) H2 and (C) CO versus the temperature, and the impedance (right axis) versus the temperature. Heating/cooling rates = 10 K min−1, flow ∼1.0 mL s−1, experiments were performed between RT–723 K. | ||
Summarizing the spectroscopic results of CO on tetragonal ZrO2, two questions arise: (i) which role do the chemisorbed formate species play in terms of surface reduction and (ii) which surface sites are changed, created or blocked during the heating and cooling routine? Upon heating, the signals indicating additional oxygen vacancies, together with signals for polydentate carbonates, already rise a hundred degrees (373 K) before signals for formates (473 K) are observed. Signals for gaseous CO2 are present at significantly higher temperatures (above ∼600 K). Upon re-cooling a considerable amount of formates and polydentate carbonates is present, accompanied by a strong reduction of the amount of Lewis acidic centers capable of molecular CO adsorption. This leads to the conclusion that the chemisorbed species (especially the carbonates) block the Lewis surface activity. To verify this effect, an additional experiment with 5% CO2 seeded in flowing CO was performed (see Fig. S2†). Due to chemisorption of CO2 already at room temperature, a high amount of (bi-)carbonates is present (for detailed assignment of the species see section CO2 adsorption). Similar to the experiment in pure CO above 473 K, also formates are observed. The signals for bicarbonates and bidentate carbonates are no more present above 473 K, but the signals for polydentate carbonates persist over the whole evaluated temperature range. Striking is the fact that the decrease of the overall absorbance is strongly suppressed with respect to treatment in pure CO, indicating a much less pronounced (surface) reduction. The reasons could be the quenching/re-oxidation of CO-induced defects by CO2 or simply the “priming” of the vacancy mediated charge carriers already at low temperatures due to the presence of (bi-)carbonates. Due to the suppressed decrease of the absorbance a reversible decomposition of formates between 633 K and 723 K is visible, which can be correlated with the formation of gaseous CO2. Upon re-cooling in the 5% CO2 contaminated CO flow only very weak signals for carbonates are visible, indicating blockage of the essential active sites already during heating. For CO2 adsorption these are mainly OH groups and O2− anions. Adsorption of CO2 without the presence of CO is perfectly reversible (see section CO2 adsorption). So, clear answers to the questions raised above can be stated: (i) formates do decompose to CO2 and, thus, play a certain role in surface reduction – nevertheless, reduction is pronounced if direct CO oxidation is possible. The vacancy mediated mechanism of CO oxidation is present already at lower temperatures. (ii) A high amount of formates, together with carbonates, effectively blocks surface sites that are related to a direct oxidation mechanism of CO to CO2. These surface sites are most likely assigned to cus Zr4+ and O2− pairs on steps, edges or corners.1,30 The results in CO clearly show that the used tetragonal ZrO2 sample provides both, reactive Lewis acidic sites, alongside a high amount of hydroxylation.
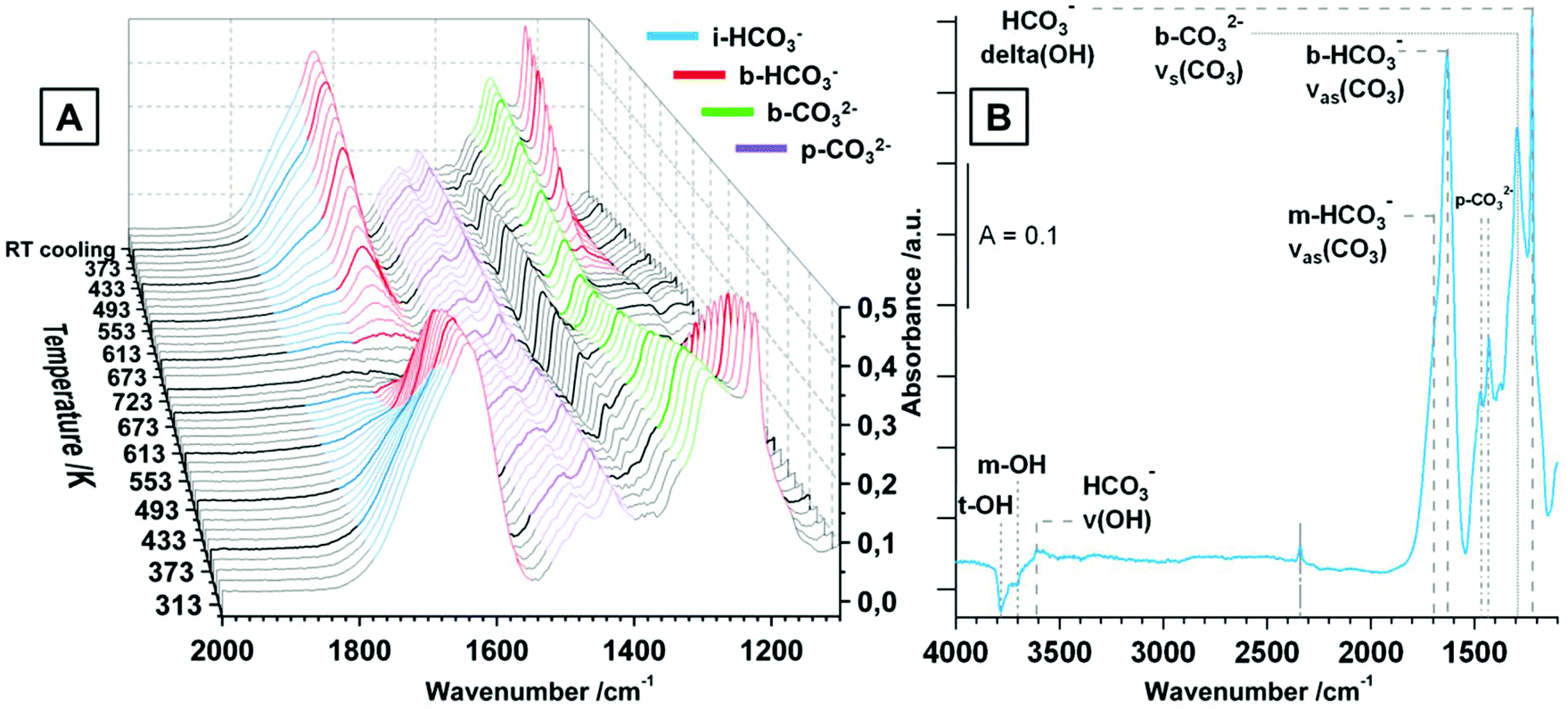
CO2 adsorption. Similar to the spectra in CO, also for the CO2 adsorption studies pure tetragonal ZrO2 was pre-treated in dry O2 up to 723 K and the spectrum of the fully oxidized sample at room temperature was taken as background for all further spectra. The increasing pressure of CO2 between 0.003 mbar and 20 mbar at room temperature is displayed in Fig. S3.† The used tetragonal ZrO2 is very sensitive to CO2 chemisorption leading to distinct signals for (bi-)carbonates already at very low pressures around 0.003 mbar. The observed species are monodentate and bidentate bicarbonates m/b-HCO3− (νas(CO3) ∼ 1700 cm−1/νas(CO3) = 1640 cm−1, νs(CO3) = superimposed, δ(OH) = 1223 cm−1), bidentate carbonates b-CO32− (νas(CO3) = superimposed, νs(CO3) = 1295 cm−1) and polydentate carbonates p-CO32− (νas(CO3) = 1470 cm−1, νs(CO3) = 1432 cm−1). The observed chemisorbed species are a product of the reaction of CO2 with basic sites of the oxides such as OH groups (leading to m- and b-bicarbonates) and other basic sites such as O2− (leading to b- and p-carbonates).16,18,30,32,35 In contrast, molecular adsorption of CO2 molecules is not observed up to a pressure of ∼1 mbar, but at higher pressures a distinct signal at 2352 cm−1 is visible, which gets superimposed by gas phase signals of CO2 above ∼8 mbar. The relatively small shift of the molecularly adsorbed CO2 peak relative to the gas phase peak vibration indicates a low geometrical distortion and a weak bond.30 Nevertheless, the principal observation of this peak corroborates the presence of a substantial amount of Lewis acid (i.e. oxygen-under-coordinated) sites even on the oxidized surface of the used tetragonal ZrO2.
The distribution of the different kinds of (bi-)carbonates is appalling, especially if compared to literature of CO2 adsorption on tetragonal ZrO2. In ref. 32, 34 and 35 already at room temperature the dominant species are polydentate carbonates. In the present case the bicarbonates are predominant, but also the amount of b-CO32− seems to be higher than reported in most literature data. This implies not only a very high hydroxylation degree, but also a generally high Brønsted basicity of the used ZrO2 compared to the reference data. The reason for these differences could be the different preparation routines, which is either precipitation from an aqueous solution of zirconyl chloride32,34 or doping with silica35 (leading to doped tetragonal ZrO2 samples). In contrast, in the present work a sol–gel method by water-quenching a solution of zirconium(IV) isopropoxide in isopropanol (without any other additives) is used.19
The evolution of the surface species upon temperature treatment of tetragonal ZrO2 in 20 mbar CO2 is presented in Fig. 4A. The same experiment was done in flowing CO2 and the general evolution of the (bi-)carbonate species is the same, but we chose to display the measurement in 20 mbar due to the better separation of the peaks.
With increasing temperature the bicarbonates are stable up to ∼473 K, and the signals for these species strongly decrease up to 673 K. In contrast, the bidentate and polydentate carbonate species are not decomposed or removed up to the maximum applied temperature of 723 K. In principal, the removal and formation of all observed (bi-)carbonate species is entirely reversible upon re-cooling and the distribution at room temperature before and after heating is exactly the same (not only in the static measurement but also under flowing conditions). Fig. 4B shows the spectrum of tetragonal ZrO2 in vacuum after the heating/cooling cycle in CO2 and the observed bands are highlighted. Additionally to the already assigned wavenumbers of the (bi-)carbonate bands, in vacuum also negative peaks for consumed OH-groups (t-OH at 3784 cm−1 and m-OH at 3700 cm−1) are visible (insertion of a CO2 molecule into an OH group leads to formation of bicarbonates). The peak at 3610 cm−1 represents the stretching mode of the OH-groups of a bicarbonate adsorbate. The peak at 2340 cm−1 is the same peak that was observed after the heating/cooling cycle in CO but is less pronounced. Due to the fact that this peak is not removed in vacuum (also in active vacuum over several hours), it is again interpreted as a feature of oxygen vacancies or defective lattice sites.
Temperature-dependent measurements between RT and 693 K. Alternating current (AC) impedance analysis was carried out on the t-ZrO2 sample in different gas atmospheres: H2 and CO, O2 and He as an inert reference gas. The aim was to detect changes in the conductivity, as well as eventual stoichiometry changes/phase transformations (especially at higher temperatures, see section 3.2) during the respective gas treatment. In general, when analyzing pressed and sintered powder samples at lower temperatures, the contribution of electronic conduction in the bulk does play a negligible role on samples with such a high band gap. In this case, at lower temperatures conduction is strongly influenced by surface-related mechanisms and grain boundary effects, such as hydroxylation, surface defects or adsorbates.42
To get a better understanding of simple temperature effects and to correlate the influence of the respective gas (oxidative or reductive properties) on the surface chemistry of the studied oxide, EIS measurements in an inert gas (He) were conducted (Fig. 5A pink traces). During treatment in dry He no heating/cooling hysteresis is observed. Furthermore, just as in O2 (heating/cooling) and H2 (heating), an insulating period between RT–520 K and semiconductive behavior (i.e. decreasing impedance values with increasing temperature) at higher temperatures is detectable. The impedance value at 693 K is interestingly lower (∼2.5 × 108 Ω) than the ones obtained at the same temperature in either dry O2 or dry H2 (∼5 × 108 Ω), but also in any other gas atmosphere. This indicates absence of any reactive surface influences in dry He and, thus, only the pure temperature effect of most likely defect-mediated conductivity is observed.
Fig. 5A also shows the impedance course during treatment of t-ZrO2 in dry O2 (green traces). Basically two distinct temperature regions are visible upon heating up to 693 K: the first one is between RT–540 K, where a plateau is visible in the temperature-dependent EIS spectra, followed by the second one starting at 540 K, where the impedance shows semiconductive behavior. The impedance value at 693 K is 5.2 × 108 Ω. Upon re-cooling to room temperature a very similar trend is shown: semiconductive behavior between 693–540 K and insulating properties at T < 540 K.
Exposure of the sample to dry H2 and heating up to ∼540 K (Fig. 5A and B light blue trace) only shows impedance values in the GΩ area, exactly similar as under O2. Upon further heating, again an impedance decrease (steeper than for dry O2) of about one order of magnitude is visible. Starting at 623 K an almost plateau-like behavior is apparent with a corresponding impedance value of ∼6.5 × 108 Ω (673 K: 5.5 × 108 Ω). A different behavior is apparent during re-cooling: between 693–630 K the impedance starts to increase (about one magnitude of order) and at temperatures <630 K up to RT the sample shows insulating properties with impedance values in the GΩ area. The correlated FT-IR spectra show a decrease in the total transmittance above 623 K which is irreversible upon re-cooling (see Fig. 5B: plot of the absorbance at 2000 cm−1versus the temperature). A decrease of the absorbance can be referred to the depletion of excitable donor states in the conduction band, which perfectly corresponds to the stagnation of the impedance decrease above 623 K. The analysis in H2 up to higher temperatures (3.2) prove a dissolution of hydrogen into the used t-ZrO2 sample and a significant change in the (at least) surface near oxidation state of Zr atoms. Thus, this distinct hysteresis between heating and cooling, in both the impedance as well as the absorbance versus temperature plots, may be an effect of the onset of this process.
A very interesting impedance trend during heating and cooling in dry CO and CO2 is visible in Fig. 5A. In summary, over the whole temperature region a plateau of the impedance in the GΩ area is apparent. This indicates that the sample is essentially an insulator in these gases and this particular temperature region. It is known from the FT-IR experiments that the sample provides a pronounced surface chemistry, but it is also proven that in both gases the acid/base properties change due to blockage of special surface sites with adsorbates. Due to the fact that especially the Lewis acid sites/cus acid–base pairs are affected, this indicates that these sites do contribute to the conductivity of the sample in an “un-blocked” state. Also the evaluation of the total absorbance at 2000 cm−1 in dry CO (Fig. 5C) confirms, that especially in CO the excitable electronic states toward the conduction band are strongly reduced with increasing temperature (T > ∼400 K) in the evaluated temperature range.
In essence, the conductivity of the characterized t-ZrO2 sample is considerably influenced by different partially counteracting effects: hydroxylation, the presence of “free” Lewis acid sites, oxygen vacancies and chemical and electronic surface blockage due to chemisorbed adsorbates capable of pinning otherwise thermally excitable states. Up to a temperature of 673 K the sample in dry He shows the highest conductivity with an essentially reversible heating/cooling cycle. In all other gases the surface is reactively influenced and/or changed, which is directly visible in the course of the impedance.
3.2. Gas-phase dependency of the phase transition tetragonal-to-monoclinic ZrO2
To further shed light on the kinetics of the transformation from tetragonal to monoclinic ZrO2, the electrochemical impedance experiments in Fig. 6 were deliberately conducted up to temperatures where the phase transformation should occur. Previous high-temperature X-ray diffraction studies revealed that the crystallization of the initially amorphous gel-precursor to the tetragonal phase is finished at 673 K if the aerogel is heated very slowly (effective heating rate: 0.01 K s−1).19 Upon further heating in air, starting at 873 K, the phase transformation to m-ZrO2 sets in ref. 19. Therefore, the experiments in Fig. 5 were limited to only 693 K to guarantee phase purity and a high surface area. Note that in the referenced study19 the in situ XRD measurements were conducted in air. To put the work into literature context, some works8,20,43–45 claim that water vapor does strongly promote the phase transformation to m-ZrO2. Up to a temperature of 693 K the absolute phase stability of the herein used ZrO2 in moist atmosphere was already proven in ref. 19. Thus, we conducted an EIS experiment where we heated the system up to the exact temperature, where phase transformation is strongly promoted in air (873 K), in dry and moist He (to also check the influence of moist/dry conditions) and routinely checked with ex situ XRD to see if the phase transformation to m-ZrO2 sets in with this comparably fast heating and cooling rates.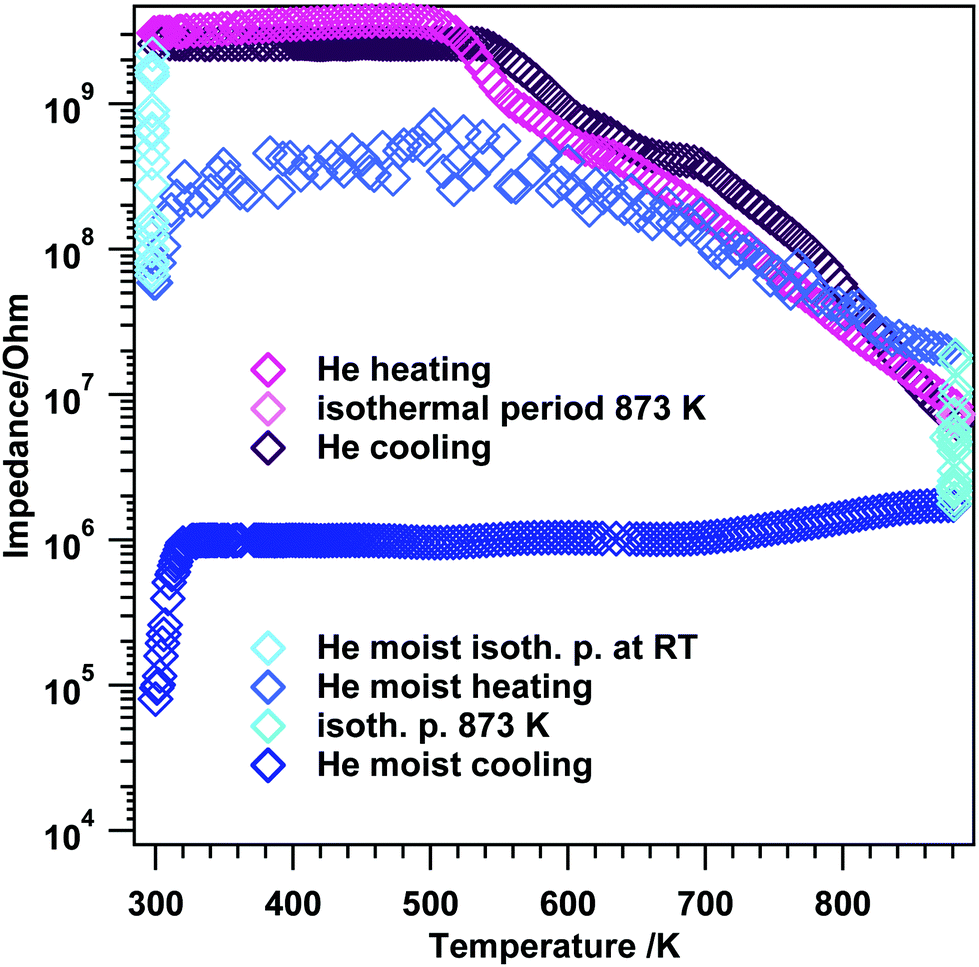 | ||
| Fig. 6 Temperature-programmed electrochemical impedance spectra on t-ZrO2 in dry and moist He. Heating/cooling rates = 10 K min−1, flow = ∼1.0 mL s−1. The experiments were performed between room temperature and 873 K. | ||
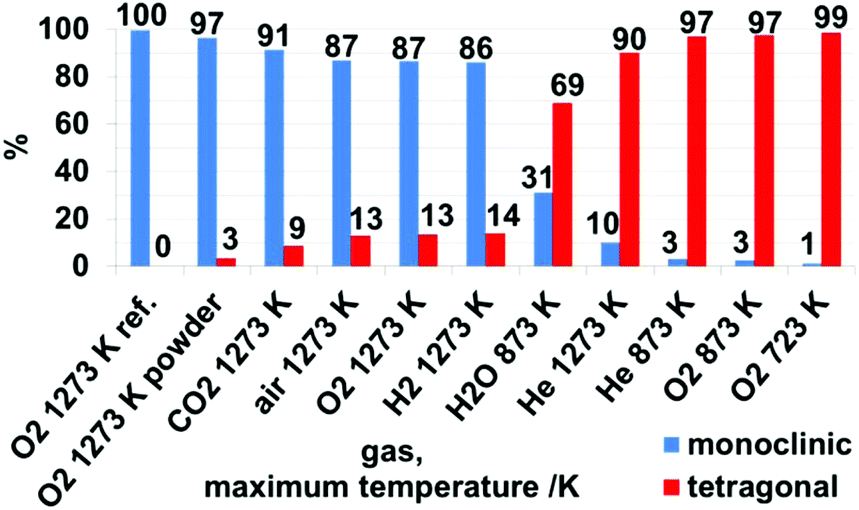 | ||
| Fig. 7 Weight-percentage of monoclinic and tetragonal phases after treatment in different gases and maximum temperatures, as derived from Rietveld analysis of X-ray patterns in Fig. S4 and S5† and summarized in Table S1.† | ||
At the beginning of the EIS experiment in moist He (Fig. 6 blue traces), the sample was kept at RT for about 10 minutes to account for eventual hydroxylation and/or hydration. During this isothermal period, the impedance decreases almost two orders of magnitude (start: 2.2 × 109 Ω, after 10 minutes: 6.6 × 107 Ω). Upon heating between room temperature and 323 K the impedance slightly increases again to a value of 3.1 × 108 Ω (de-hydration), after this an induction period up to 573 K is visible and upon further heating, semiconductive behavior leading to a value of 2.0 × 107 Ω at 873 K is apparent. The maximum temperature was again held for 10 minutes, with an impedance decrease to 1.8 × 106 Ω. Upon re-cooling to room temperature, basically an almost constant impedance value up to ∼323 K is obtained and at temperatures T < 323 K, the impedance starts to decrease again leading to a final value of 8.0 × 104 Ω at room temperature.
Rietveld analysis (Fig. 7 and S4†) show that upon heating and cooling in the impedance setup with a comparable fast rate of 10 K min−1, pronounced crystallization of the monoclinic phase is observed for the moist treated sample (31 wt% monoclinic ZrO2). In contrast, heating to lower maximum temperatures (compared to ref. 19) in dry He resulted in a comparably lower extent of transformation to monoclinic ZrO2 (3 wt% monoclinic ZrO2). Thus, these data confirm the water promoting role of the phase transformation to a certain extent above 693 K. According to ref. 8 a moist atmosphere promotes grain growth (and related phase transformation) and leads to enhanced surface diffusion. Nevertheless, note that for the tetragonal ZrO2 sample characterized in this work the impact of water vapor is not as pronounced as one could expect from literature data, since in the present case the promoted phase transformation is only observed at temperatures, where kinetically controlled phase transformation would take place anyway. The very unusual course of the impedance in this experiment also indicates a strong interaction of water vapor with the sample. In fact, a 30% increased bulk conductivity (most likely proton conduction) due to incorporation of water into doped tetragonal ZrO2 is also observed by Guo et al.45 As a matter of fact, water can replenish the oxygen vacancies, which reduces the amount of defects, subsequently leading to phase transition. The main difference to ref. 45 is that the conductivity measurement in water lasts 8 h. After this treatment, 70 wt% monoclinic ZrO2 is found in the structural mixture. Our sample, however, is chemically and structurally pure (i.e. not Y-doped) and the admission time of water is much shorter.
To focus on the phase transformation at even higher temperatures, the tetragonal ZrO2 sample used in this work was heated up to 1273 K in pure He, O2, H2 and CO2 to examine a potential gas phase dependency of the phase transformation to monoclinic ZrO2. The standard temperature program was heating and cooling with 10 K min−1 and a gas flow of ∼1 mL s−1 with a maximum temperature of 1273 K, i.e. a rather fast heating routine. This routine was performed with the in situ/operando setups (EIS and FT-IR), using pelletized samples rather than powders, but sintered and oxidatively pretreated as described in the Experimental section. Subsequently, the samples were checked by XRD and XPS.
Qualitative and quantitative phase analysis by XRD. As expected (compare kinetic phase transformation investigations in air in ref. 19) for O2, H2 and CO2 the main part of the samples was transformed to monoclinic ZrO2. As already discussed in the section above the presence of H2O vapor drastically promotes the phase transformation. The corresponding XRD data of the measurements up to 1273 K in the different gas atmospheres are shown in Fig. S5.†
Derived from Fig. 7, the samples treated in dry O2 and dry He up to 873 K show a low conversion to m-ZrO2 of 3%, in contrast to moist He up to 873 K, as discussed above. Heating the sample in dry He up to 1273 K also leads to a low amount of 10% m-ZrO2 – which is a significantly low amount. Based on the assumption that a sufficient defect concentration is crucial for the stabilization of the tetragonal phase,45 this is originating from the missing defect quenching (such as in oxidative reactants or other possible reactive interaction with gases like H2 and CO2, which is confirmed in the discussion to Fig. 8) and the absent active surface chemistry influencing the transformation kinetics upon heating in dry He. Treatment of the sample in dry O2, H2 and air up to 1273 K leads to similar amounts of 86–87% m-ZrO2. Heating/cooling in CO2 results in 91% m-ZrO2. The striking differences to He can be explained by a substantial reduction of the defect concentration of the sample because of reaction with the other gases. In the case of O2 this is simply quenching of a sub-stoichiometric oxygen-deficient lattice. Derived from subsequent XP spectra of the sample pellets (see detailed discussion below, Fig. 8) treatment in H2 and CO2 leads to formation of dissolved hydrogen and Zr-(oxy-)carbide – at least at the surface. Such reactive modifications of the sample are not present at identical treatments of the monoclinic reference sample, and this indicates an initially highly defective oxide.
 | ||
| Fig. 8 Temperature vs. 2 theta contour plots of in situ high temperature synchrotron XRD characterization of tetragonal ZrO2 heated to 1273 K and subsequently cooled down to room temperature with 20 K min−1 in carbon dioxide (a-heating and b-cooling) and hydrogen (c-heating and d-cooling). Dark colors denote high intensities, grey-white colors background intensities. ICDD reference diffraction data for t-ZrO2, m-ZrO2 and h-SiO2 shown at the bottom of each plot. | ||
For characterization in the in situ/operando setups (but also for the preparation for the ex situ XRD, XPS and AFM measurements) the starting material was pressed into pellets and in situ sintered in the respective impedance and FT-IR setups. To check for effects derived from these procedures, an identical heating/cooling routine in O2 up to 1273 K was performed for a powder of the tetragonal starting material, resulting in 97% m-ZrO2, which is 10% more than for the pressed sample. As will be discussed in detail below this can be explained by surface, crystallite sizes and agglomeration effects that enhance the stability of the tetragonal phase.8
In situ heating synchrotron XRD analysis. To study the effect of crystallite size growth on the transformation temperature of t-ZrO2 to m-ZrO2 under different gas atmosphere, the pathway of phase transformation was investigated by in situ synchrotron XRD in H2 and CO2 atmospheres. Before the in situ XRD experiments, the t-ZrO2 sample were obtained by calcination of the powdery amorphous aerogel at 723 K inside the capillary under air flow until crystallization to tetragonal ZrO2 is completed. The contour plots in the Fig. 8a–d show the recorded diffraction data for one heating/cooling cycle in H2 and CO2 atmospheres. The in situ data reveals that the initially phase-pure t-ZrO2 remains stable upon heating to 1273 K in both gases. The tetragonal to monoclinic transformation takes place during cooling down from 1273 K to room temperature at 625 and 715 K in H2 and CO2 atmosphere, respectively. The additional h-SiO2 reflections result from a partial crystallization of the fused silica quartz capillary and are not related to the zirconia sample. The final patterns of the cooled samples show m-ZrO2 as the major phase and a minor t-ZrO2 side phase. The Rietveld refinement reveals that the cooled samples to room temperature still have 14.9 wt% and 9.8 wt% of tetragonal ZrO2 in H2 and CO2 atmosphere, respectively, which in good agreement with ex situ XRD analysis (Fig. 7). Selected patterns are presented in Fig. 9.
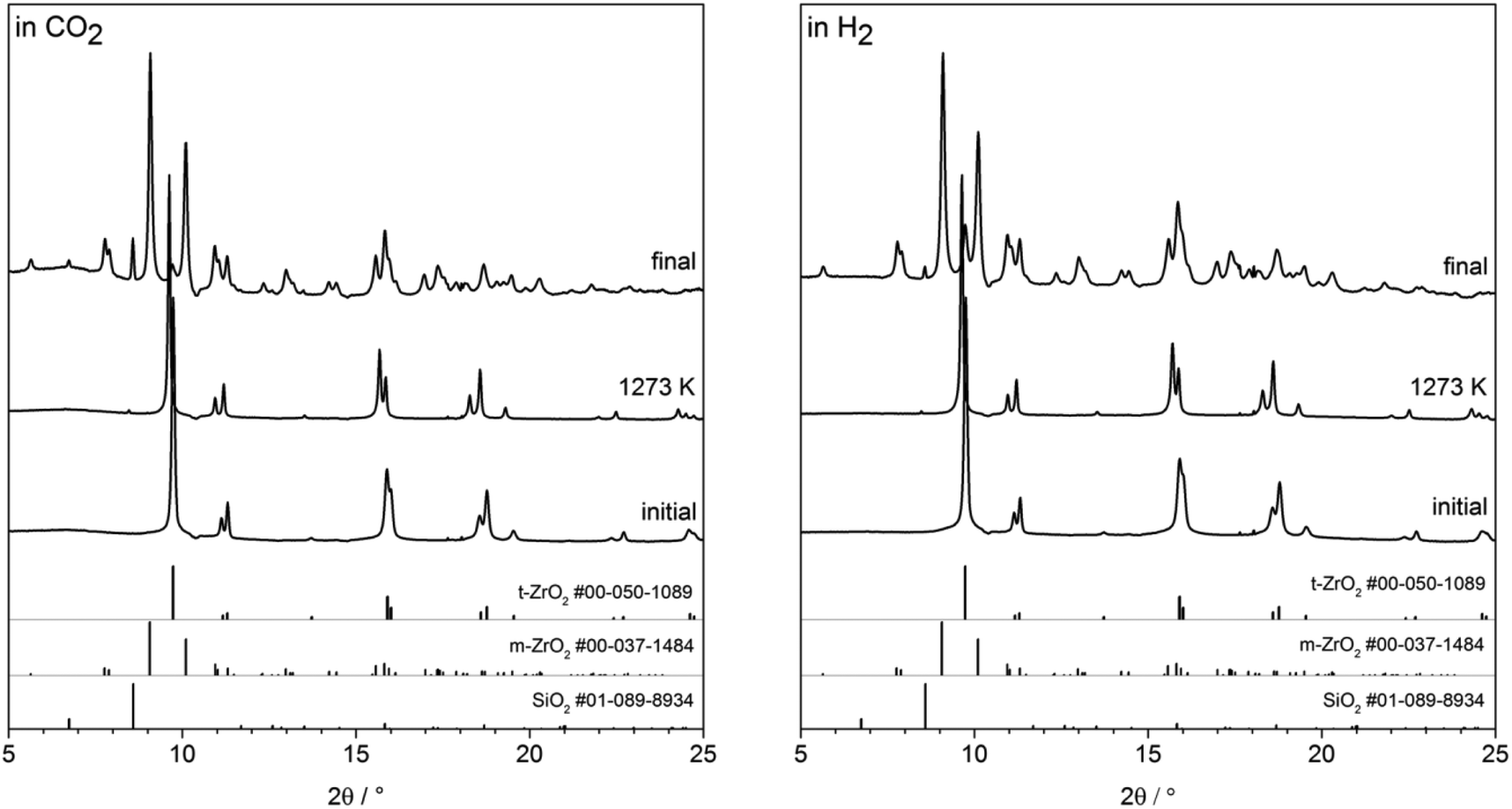 | ||
| Fig. 9 Selected XRPD patterns from the in situ series. Patterns of initial sample, at 1273 K and final sample are shown for CO2 (left) and H2 (right) atmosphere. ICDD reference diffraction data for t-ZrO2, m-ZrO2 and h-SiO2 shown at the bottom of each plot. | ||
The stabilization of the metastable t-ZrO2 phase can be explained by a combined influence of (a) a high defect density and (b) a very small crystallite size, where a decreased defect density and/or an increased crystallite size favors the stable monoclinic ZrO2. Rietveld refinement (strain was considered with Stokes-Wilson model, see strain plot in ESI†) results revealed that the initial t-ZrO2 samples have a crystallite size of 26.5–32 nm (around the critical diameter of ≈30 nm) ref. 8. The results of Rietveld refinement were further confirmed by calculating the crystallite size D (20.4 nm) from SBET surface area (50 m2 g−1) using the well-known relationship (S = 6/ρD) ref. 8 where ρ the density of t-ZrO2 (6.121 g cm−3). The crystallite size of the heated samples was almost constant up to 1073 K followed by a sharp increase above this temperature in both gases, see Fig. 10 (left panel). The maximum crystallite size of t-ZrO2 at 1273 K was found to be 41 and 43.5 nm in H2 and CO2 atmosphere, respectively. The size of crystallites remain constant during cooling in both atmospheres up to the transformation temperatures followed by a sharp decrease below transformation temperatures. Strain is released in the tetragonal phase above 1000 K when the grain size increases (see Fig. S6†). The newly formed monoclinic ZrO2 shows higher microstrains – especially in H2 – which can be attributed to a defect rich material. The higher transformation temperature in CO2 in comparison to H2 atmosphere may be due to the higher crystallite size of t-ZrO2 in CO2 than in H2 atmosphere. This finding is consistent with previous studies showed that as the crystallite size of ZrO2 increases, the monoclinic-to-tetragonal phase transformation temperature increases ref. 8, 48 and 49. However, the interaction of different gas atmosphere with site defects on ZrO2 could be also another reason to lower the transformation temperature in H2 atmosphere as will be discussed below.
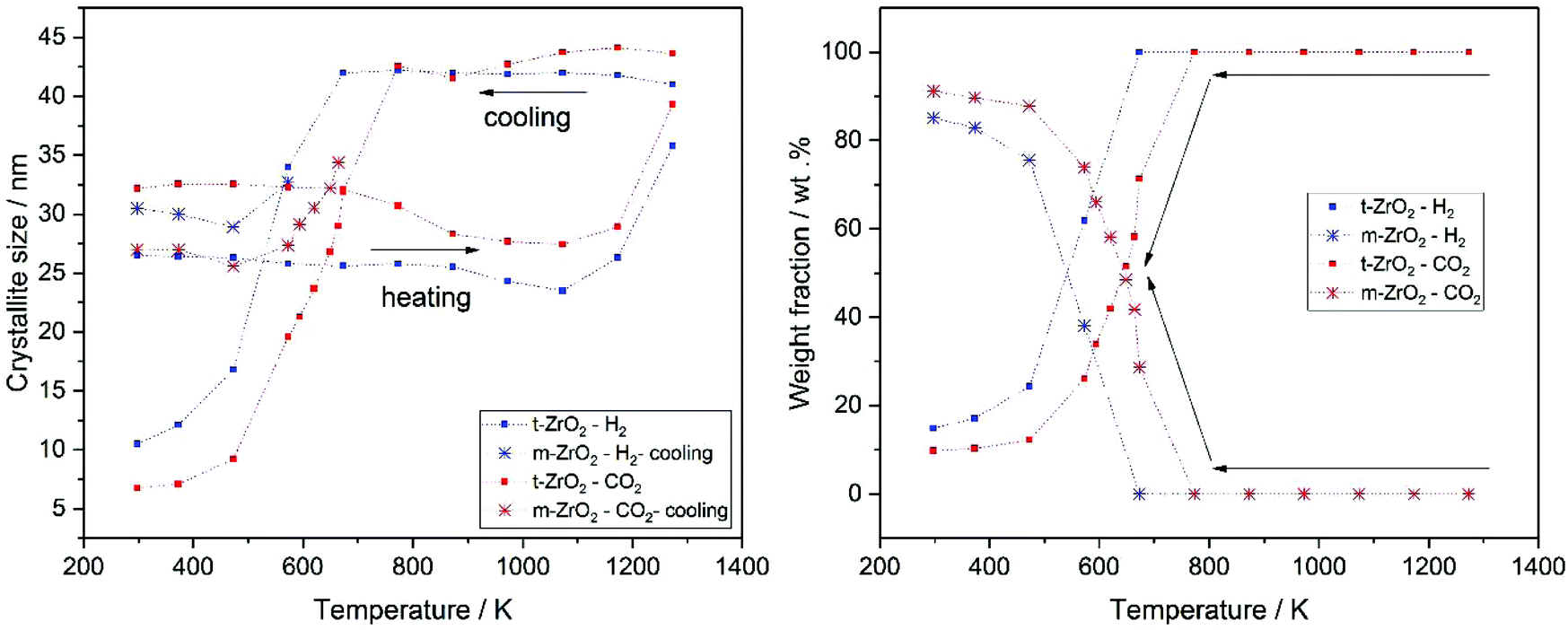 | ||
| Fig. 10 Crystallite size evolution of t-ZrO2 crystals during heating and cooling in H2 (blue) and CO2 (red) (left) and tetragonal and monoclinic phase fractions during cooling (right) as derived from Rietveld refinement of selected in situ XRPD data. | ||
Electronic structure analysis by XPS. The ex situ XP spectra in Fig. 11 reveal changes in the chemical states of the surface-near region. Mixed Gaussian/Lorentzian peak models were fitted for the 3d5/2 and 3d3/2 peaks (as outlined in the Experimental section 2.4). The assignment of the oxidation states was done according to literature values found in ref. 24 and 50. For the sample heated in dry He to 873 K (Fig. 11A), a relatively large fraction (almost 50 at%) of the total accessible Zr species (inelastic mean free path = 2.3 nm; 1304 eV kinetic energy, calculated via the G-1 predictive formula51) is found to be in a lower oxidation state than Zr(IV). This is also derived from the general peak shape, as a distinct shoulder at a binding energy of about 181 eV, as well as the apparent weaker peak splitting between the 3d5/2 and 3d3/2 components is observed. 34 at% of the Zr ions can be assigned to an oxidation state of +III, which is shifted relative to the +IV state (182.8 eV) by 1.1 eV, and the remaining 15.5 at% appear reduced to Zr(II) species (with a shift of 2.1 eV to the Zr(IV) species). After treatment at 1273 K (Fig. 11B), the amount of Zr(IV) at 63 at% is significantly larger. The fraction of Zr(III) is reduced to 25 at%, whereas the concentration of Zr(II) is only decreased slightly (to 12 at%). As no ‘external’ oxidation of the sample is possible during treatment in helium, the now larger fraction of Zr(IV) has to originate from the sample itself. As the portion of Zr(II) did not increase, a disproportionation reaction of Zr(III) to form Zr(IV) and Zr(II) must take place, possibly overlaid with a higher oxygen mobility at elevated temperatures, i.e. bulk oxygen segregates to surface-near regions. That the majority of thus oxidized species seem to be Zr(III), indicates that the reaction from Zr(III) to Zr(IV) is favored compared to the oxidation of Zr(II) to form Zr(III).
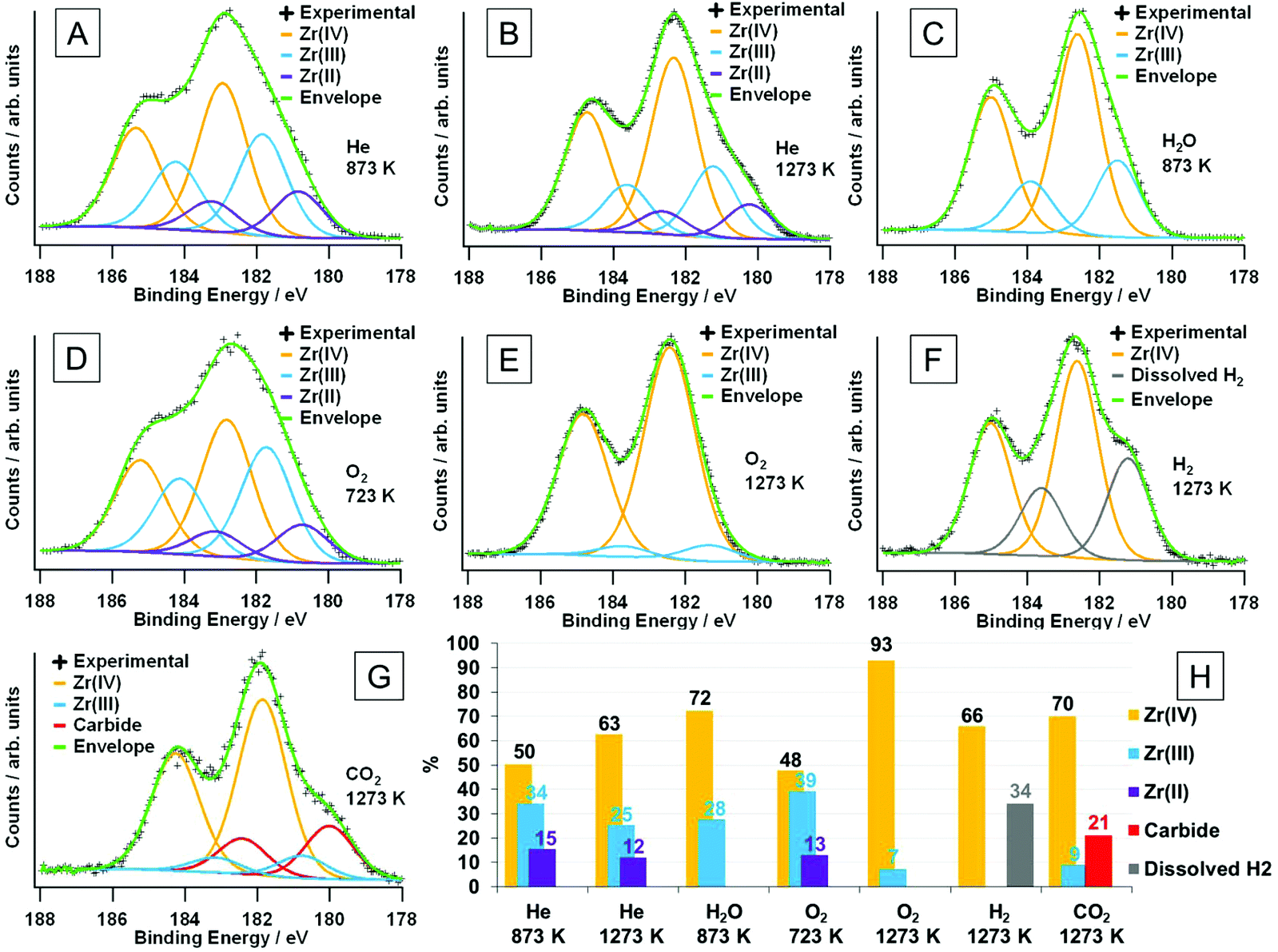 | ||
| Fig. 11 XPS Zr 3d regions for the samples after various treatments, including fits of the oxidation states. Samples were treated (heating/cooling rate of 10 K min−1) in flowing (flow ∼1 mL s−1) dry He up to (A) 873 K and (B) 1273 K, moist He up to 873 K (C), dry O2 up to 723 K (D) and 1273 K (E), and dry H2 up to 1273 K (F) and dry CO2 up to 1273 K (G). The relative at% of the observed surface species (as determined by the chemical state fits) are shown in panel (H). The maximum depth of emission for the Zr 3d peak, obtained via the inelastic mean free path for photoelectrons with this energy (1304 eV kinetic energy) calculated via the G-1 predictive formula51 is 2.3 nm. Corresponding C 1s peaks are shown in Fig. S7.† | ||
Fig. 11C shows the spectrum obtained after heating the sample to 873 K in moist He. The peak is not as broad as compared to the above-discussed spectra, without the presence of a strong shoulder. Both can be directly related to the complete absence of Zr(II). The experimental spectrum is composed of Zr(IV) (72%) and Zr(III) (28%). This suggests that water is able to oxidize the Zr(II) that was found for the dry He treatment at the same temperature, as well as to oxidize some of the Zr(III), since the amount of that state is also lower. In pure dry O2 at 723 K (Fig. 11D), the spectrum looks very similar to that for He (873 K), which indicates that the temperature is too low to effectively oxidize the sample. Only 48% are present in the form of Zr(IV), whereas nearly the same amount (39%) is present as Zr(III), and 13% are Zr(II). The Zr(IV)/Zr(III) ratio is even lower than for the helium-treated sample. Upon increasing the oxidation temperature to 1273 K the suboxides vanish almost completely, and there is only a minor fraction of 7% Zr(III) left (Fig. 11E).
After reduction in dry H2 at 1273 K (Fig. 11F), the general peak separation is comparable to treatments either in O2 at 1273 K or in H2O at 873 K with a well-defined shoulder at about 181 eV. The fits reveal that – in addition to Zr(IV) – there is a second component, which is neither Zr(III) nor Zr(II), as its shift relative to Zr(IV) is 1.4 eV, and is, thus, found in-between those two oxidation states (which would be at 1.1 eV and 2.1 eV, respectively). The shift, nevertheless, is also not large enough for it to be zirconium hydride, ZrH2, which should yield peaks at a binding energy of 178.8 eV.52 This could indicate hydrogen dissolution inside the crystal structure, which either shifts the neighboring Zr(IV) or Zr(III) energetically to lower binding energies.47 What would be in favor of the Zr(III) ions being affected is that even the spectra taken after O2 treatment contain a small fraction of suboxides. A sample reduced in pure H2 should feature an even larger portion, and the quantification of the respective spectra would yield a sum formula of ZrO1.88, which agrees with the presence of Zr(III). This hydrogen-influenced species gives rise to 34% of the total Zr 3d signal.
In Fig. 11G, the sample was annealed to 1273 K in CO2, which gives rise to another distinct shoulder, albeit at lower binding energies as compared to treatment in hydrogen. It is located at about 180 eV and is caused by a component relatively shifted to the Zr(IV) peak by 1.9 eV. The C 1s region (Fig. S7†) reveals a severely asymmetric peak being due to a Zr carbide component at 282.9 eV,53 which appears as a shoulder in the Zr 3d region. The latter peak (as the C 1s component) is shifted to higher binding energies when compared to the reference values for ZrC.53 The reason for this could be that it is not a pure ZrC layer, but rather a ZrOxCy oxy-carbide, where the remaining oxygen could influence the surrounding Zr and C atoms and cause the respective chemical shifts. This could be either because the ZrC layer itself contains oxygen or that it is indeed ZrC, but is so thin that the oxide/carbide interface is sampled as well. Taking into account the different escape depths between the Zr 3d and C 1s regions, the carbide components in both spectra yield the same absolute amounts of carbide: 21% of the surveyed zirconium and 18% of the present carbon species (the rest being adventitious carbon), with a total of Zr/C ratio of 1.2.
Surface modifications including dissolved hydrogen species and Zr-carbide in H2 and CO2 are not observed in comparable experiments of monoclinic ZrO2 (preceding study of H2 on m-ZrO2 in ref. 54 no carbide in the XPS spectra in Fig. S8†). Starting with a tetragonal sample, which is highly defective and hydroxylated and, thus, more active, seems to strongly promote such surface modifications.
The essentially different phase transformation behavior in He compared to the other gases can be explained as follows: starting with a defect concentration sufficient enough to stabilize the tetragonal phase at room temperature, simple heating only leads to an unaffected increase of the defect concentration and further stabilization of the tetragonal phase. Also upon cooling, no quenching or reaction of these defects is possible in dry He and, thus, a less pronounced phase transformation to monoclinic ZrO2 is observed. In oxidative gases such as O2 and H2O direct defect quenching is possible and phase transformation upon cooling is unavoidable. Also the formation of (oxy-)carbides or dissolved hydrogen species can be seen as a unique form of defect alteration and blockage.
A final note on the optical change of the sample color should be added at this point. The tetragonal sample annealed at 673 K exhibits a grayish/brownish color, likely due to a certain amount of reduced centers. The sample treated in He up to 1273 K is dark gray, indicating an increased amount of color centers. Treatment in dry H2 leads to an almost black pellet. The samples after treatment in water up to 873 K and O2 and CO2 up to 1273 K are white, which matches the assumption of different, but nevertheless oxidative quenching mechanisms.
Surface morphology as determined by AFM: influence of powder pressing. To sum up the XRD and XPS data, derived from the distinct surface modifications in H2 and CO2, compared to pure monoclinic ZrO2 the surface chemistry is strikingly different. Another remarkable effect is the influence of pressing and in situ sintering the sample compared to treatment of a loose powder as starting material. The XRD pattern in Fig. S5† and the corresponding Rietveld analysis in Fig. 7 and Table S1† clearly show that it makes a difference whether the original aerogel is heated in a pressed or a powderous form. Whereas the powder sample (starting with a fine milled aerogel) is 97% monoclinic after heating in dry O2 the pressed pellet still provides 13% of tetragonal phase. To further shed light on this phenomenon, AFM measurements of the pellet after treatment in O2 up to 1273 K were performed (Fig. 12). The AFM images obtained at a larger scale (5 × 5 μm, Fig. 12C) clearly show that the pellet is composed of large grains (with a grain size of about 200 nm, as obtained from a histogram). Fig. 12D makes it easier to visualize the surface, as in this 3D image, the x-, y- and z-axes all have the same scaling, revealing that the surface is rather flat, with the grains having a low profile (the z-extent of the image is 283.9 nm). Upon zooming into one such grain (Fig. 12A, z: 65.2 nm), it can be seen that a lot of small crystallites sit at its surface, which exhibit an improved contrast in the respective phase image (Fig. 12B). By measuring the size of more than 200 such grains in the phase image and fitting a Gaussian function to the resulting histogram, the most frequent crystallite size could be determined to be 16 nm (Fig. 12E). According to ref. 8 this particle size implicates stabilization of the tetragonal phase. We conclude that the main bulk of the pressed and sintered sample is transferred into monoclinic phase upon heating to 1273 K, but the surface still exhibits small tetragonal particles. In this context, Shukla and Seal8 also claim that agglomeration of many small crystals does stabilize possible μm-sized sintered tetragonal structures. Note that for the present experiments the aerogel was pressed into pellets and subsequently sintered in this “shape” inside the in situ setups. Treating such a pellet in O2 up to 1273 K leads to phase transformation to monoclinic ZrO2 but still a distinct amount of tetragonal ZrO2 is present (13 at% t-ZrO2), as derived from the AFM measurements: very likely on the surface of the pellet derived from the 16 nm size of the particles on the surface of the pellet.
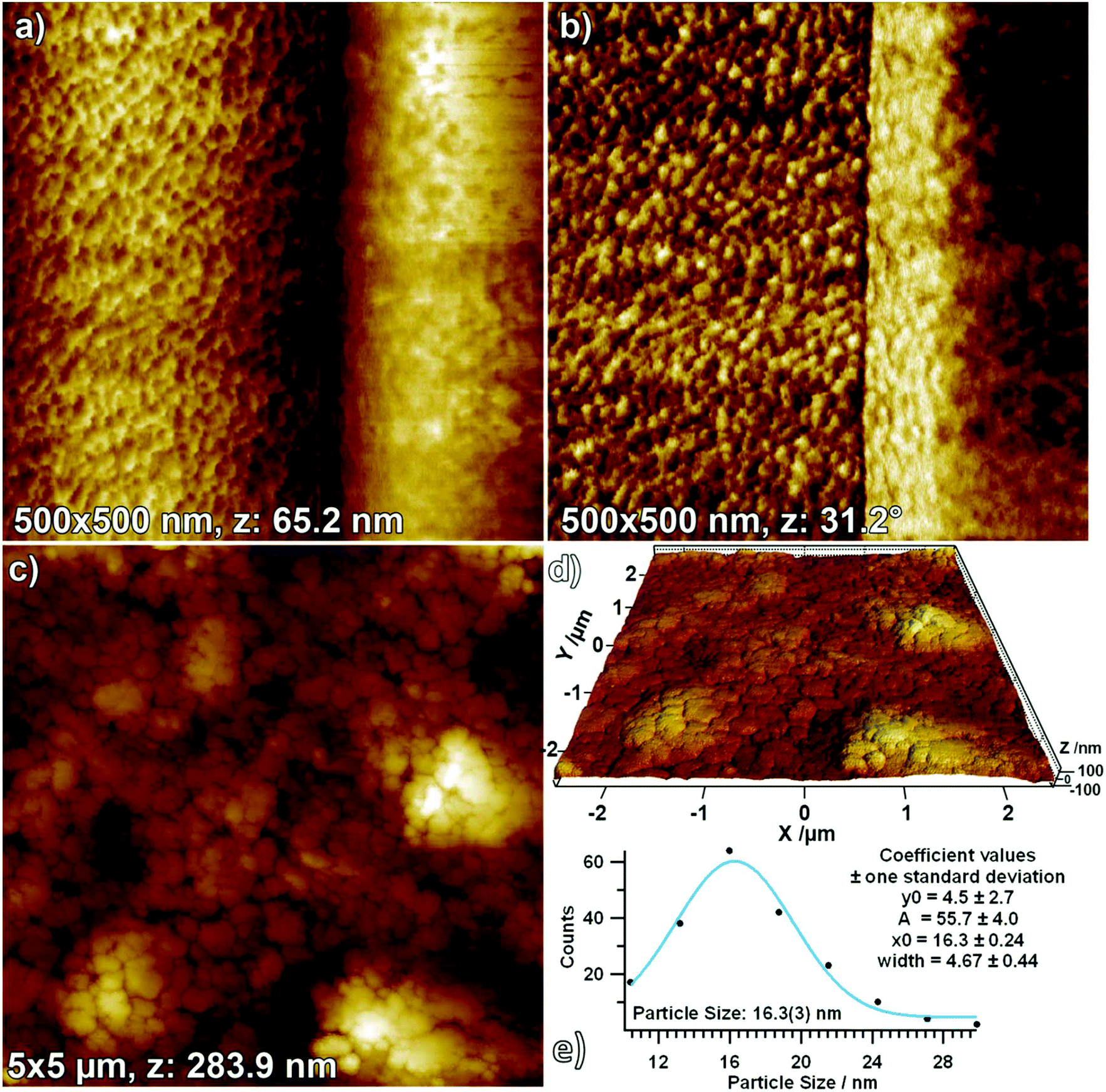 | ||
| Fig. 12 500 × 500 nm AFM topology (a) and phase (b) images, as well as a 5 × 5 μm topology image (c) with 3D rendering (d). (e) Histogram of crystallite sizes as obtained from (b) with a fitted Gaussian function. | ||
Conductivity measurements at elevated temperatures between RT–1273 K. Based on the XRD, XPS and AFM data, the corresponding EIS measurements in different gas atmospheres up to 1273 K can now be critically discussed. For detailed discussion of data obtained with a maximum temperature of 693 K, we refer to section 3.1.2. Additional comparative reference experiments were also done for monoclinic ZrO2, which are displayed in Fig. S9.†
Exposure of t-ZrO2 to dry O2 and heating up to 1273 K (Fig. 13, green trace) shows semiconductive behavior starting at T > 560 K with a step between 560–700 K, after which the impedance starts to decrease faster up to ∼1013 K. At higher temperatures, the impedance decrease flattens, leading to a value of 5.8 × 105 Ω at 1273 K. The same trend is observed during cooling with minor differences in the insulating period between 660–RT and a missing step between 560 and 700 K. Compared to m-ZrO2 during treatment in O2 (Fig. S9A†), a somewhat similar trend for both oxides is observed up to 973 K. At temperatures above 973 K the impedance decreases less strongly for t-ZrO2 than for m-ZrO2 leading to slightly different final values at 1273 K (m-ZrO2: 1.2 × 104 Ω; t-ZrO2: 5.8 × 105 Ω). This could be tentatively assigned to kinetic phase transformation/oxygen quenching processes in the tetragonal sample.
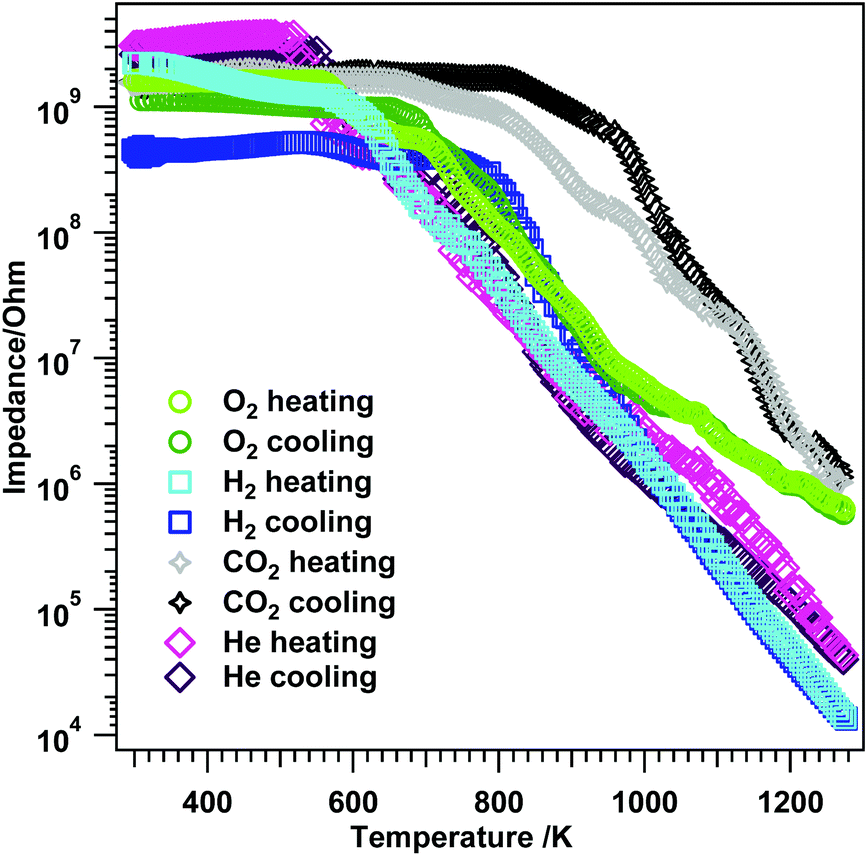 | ||
| Fig. 13 Electrochemical impedance versus temperature results on t-ZrO2 in dry O2, H2, CO2 and He. Heating/cooling rates = 10 K min−1, flow = ∼1.0 mL s−1, experiments were performed between RT–1273 K. | ||
Treatment in H2 up to 1273 K (Fig. 13 blue traces) reveals slight differences especially in the semiconductive region, leading to a final impedance value of 1.4 × 104 Ω at 1273 K. Again, re-cooling yields similar results between 1273–973 K, but at lower temperatures, differences to the heating process are evident. Two plateaus are apparent between 760–652 K (3.8 × 108 Ω) and 652–RT (4.8 × 108 Ω). In contrast, exposure of m-ZrO2 to dry H2 (Fig. S9B†) leads to semiconductive behavior at temperatures T > 415 K and a value of 1.2 × 105 Ω at 1273 K. This indicates that in dry H2 on t-ZrO2 the impedance at the highest temperature is about one order of magnitude lower than on m-ZrO2. Together with the strongly deviating behavior of re-cooling the t-ZrO2 sample in H2, this effect may be due to the above-mentioned dissolution of hydrogen in the sample, which is not the case for pure m-ZrO2.54
Striking differences during the temperature-dependent EIS analysis in CO2 are illustrated in Fig. 13 (grey and black traces). Between RT–673 K a pronounced insulating period in combination with semiconductive behavior, exhibiting several steps in the decrease of the impedance, is apparent (1.0 × 106 Ω at 1273 K). As in hydrogen, during re-cooling a similar trend as during heating in the high-temperature region 1273–1126 K is apparent, but at lower temperatures differences arise. Higher impedance values as during heating are obtained down to ∼820 K, followed by the insulating period. The monoclinic sample (Fig. S9C†) exhibits an insulating period during heating between RT–673 K and 860 K–RT during cooling as well as an impedance value of 2.5 × 105 Ω at 1273 K. From the FT-IR experiments one can deduce that the surface chemistry of the used t-ZrO2 is strongly enhanced at lower temperatures compared to the used monoclinic ZrO2 sample.42,55 The XPS investigations show that on the t-ZrO2 sample (oxy-)carbides are formed (which is not the case for the monoclinic sample – compare Fig. S9†). Since a fully percolated (oxy-)carbide layer would lead to a better conductivity, a full (oxy-)carbide surface percolation can be excluded due to the increased impedance values of t-ZrO2 at 1273 K compared to m-ZrO2.
The reference measurement in dry He again provide interesting results (pink traces Fig. 13). Upon heating to 1273 K a very short insulating period (RT–520 K) together with semiconductive behavior at temperatures >520 K is apparent. However, during He treatment the impedance starts to decrease faster as compared to O2, but comparable to H2. The value at 1273 K is lower (4.3 × 104 Ω) than in O2 and CO2 and a little bit higher than in H2 (1.4 × 104 Ω). The comparison between m- and t-ZrO2 in dry He (Fig. S9D†) again helps to provide information on pure temperature-induced conductivity effects on the samples. Basically, almost an identical impedance course is observed for both oxides with some slight differences in the value at the highest temperature (m-ZrO2: 1.1 × 105 Ω and t-ZrO2: 4.3 × 104 Ω). This again emphasizes the huge influence of the gas atmosphere to the overall electrochemical response of the sample.
To sum up the EIS investigations up to 1273 K, we note a very strong dependence of the conductivity on the nature of the gas atmosphere, which corroborates the XPS data. This might especially affect the surface and grain boundary contribution conductivity mechanisms. The phase transformation itself cannot be observed “directly”, but confirms that the latter is most likely a rather continuous process.
4. Conclusions
The conclusions derived from the presented work can be summarized as follows:• the characterization of the pure tetragonal ZrO2 compound below 723 K reveal an active surface with a high degree of hydroxylation. Lewis-acidic and Brønsted-basic centers are both found on the surface. Both CO and CO2 are effectively blocking coordinately unsaturated sites and, thus, no conductivity has been observed in these C1 gases. However, already in this temperature region, significant differences in the surface conductivity as a function of the gas atmosphere arise.
• Water promotes the transformation from tetragonal to monoclinic ZrO2 especially around 873 K. This has been worked out in a combined spectroscopic and structural study by impedance, FT-IR and X-ray diffraction measurements. Moreover, the tetragonal ZrO2 sample exhibits pronounced interactions with water, leading to additional protonic conduction. Most importantly, this transformation is observed at much higher temperatures than reported in literature.
• The high-temperature investigations up to 1273 K as a function of the gas atmosphere reveal distinct differences, ranging from being basically unaffected (He), over dissolved hydrogen (in H2) to reaction ((oxy-)carbide formation in CO2). The phase transformation is also reported to be strongly affected by the sintering and pressing history of the sample.
• Both defects and hydroxylation are the decisive parameters for steering the structural stability and reactivity of the phase-pure tetragonal ZrO2 modification. Due to critical control of these parameters during synthesis, the pure ZrO2 material shares some common behavior with the doped samples reported in literature, but also major differences. The latter is basically reflected in the much higher structural stability, especially at low temperatures and a totally different surface reactivity.
• This then enables a proper physico-chemical characterization of this prominent modification in the ZrO2 polymorphic system. These investigations demonstrate unique aspects of the used t-ZrO2 sample. Despite the fact that ZrO2, and especially in this case metastable tetragonal ZrO2, seems to be a well-investigated polymorph in literature, the unique properties point out the significant impact of sample preparation routine and sample history.
Acknowledgements
We thank Daniela Schmidmair from the Institute of Mineralogy and Petrography (University of Innsbruck, Innrain 52d, A-6020 Innsbruck, Austria) for supporting XRD measurements. We thank the FWF (Austrian Science Foundation) for financial support under the projects F4503-N16 and F4502-N16 of the SFB “Functional Oxide Surfaces and Interfaces” (FOXSI). We thank the Advanced Light Source (which is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the US Department of Energy under Contract no. DE-AC02-05CH11231) where in situ PXRD measurements are conducted at beamline 12.2.2 in the framework of AP program ALS-08865. LS appreciates the ALS for supporting his work with a doctoral fellowship. This work is part of the Cluster of Excellence “Unifying Concepts in Catalysis” coordinated by the Technische Universität Berlin. Financial support by the Deutsche Forschungsgemeinschaft (DFG) within the framework of the German Initiative for Excellence is gratefully acknowledged.References
- S. Kouva, K. Honkala, L. Lefferts and J. Kanervo, Catal. Sci. Technol., 2015, 5, 3473–3490 CAS
.
- Y. Han and J. F. Zhu, Top. Catal., 2013, 56, 1525–1541 CrossRef CAS
- O. Y. Kurapova and V. G. Konakov, Rev. Adv. Mater. Sci., 2014, 36, 177–190 Search PubMed
- N. N. Novik, V. G. Konakov and I. Y. Archakov, Rev. Adv. Mater. Sci., 2015, 40, 188–207 Search PubMed
- I. Birkby and R. Stevens, Key Eng. Mater., 1996, 122-1, 527–551 CrossRef
- X. M. Song and A. Sayari, Catal. Rev., 1996, 38, 329–412 CAS
- R. E. Juarez, D. G. Lamas, G. E. Lascalea and N. E. W. de Reca, Defect Diffus. Forum, 2000, 177-1, 1–26 CrossRef
- S. Shukla and S. Seal, Int. Mater. Rev., 2005, 50, 45–64 CrossRef CAS
- J. R. Kelly and I. Denry, Dent. Mater., 2008, 24, 289–298 CrossRef CAS PubMed
- R. H. J. Hannink, R. K. Stringe and M. V. Swain, J. Australas Ceram. Soc., 2014, 50, 1–14 CAS
- A. Afzal, Mater. Express, 2014, 4, 1–12 CrossRef CAS
- T. Liu, X. F. Zhang, L. Yuan and J. K. Yu, Solid State Ionics, 2015, 283, 91–102 CrossRef CAS
- D. Panda and T. Y. Tseng, Thin Solid Films, 2013, 531, 1–20 CrossRef CAS
- A. Atkinson, S. Barnett, R. J. Gorte, J. T. S. Irvine, A. J. McEvoy, M. Mogensen, S. C. Singhal and J. Vohs, Nat. Mater., 2004, 3, 17–27 CrossRef CAS PubMed
- M. N. Tsampas, F. M. Sapountzi and P. Vernoux, Catal. Sci. Technol., 2015, 5, 4884–4900 CAS
- S. T. Korhonen, M. Calatayud and A. O. I. Krause, J. Phys. Chem. C, 2008, 112, 16096–16102 CAS
-
W. Pies and A. Weiss, Crystal Stucture Data of Inorganic Compounds, Springer-Verlag, Berlin, Heidelberg, New York, 1975, vol. 7 Search PubMed
- V. Bolis, G. Magnacca, G. Cerrato and C. Morterra, Top. Catal., 2002, 19, 259–269 CrossRef CAS
- M. Kogler, E. M. Köck, S. Vanicek, D. Schmidmair, T. Götsch, M. Stöger-Pollach, C. Hejny, B. Klötzer and S. Penner, Inorg. Chem., 2014, 53, 13247–13257 CrossRef CAS PubMed
- S. B. Xie, E. Iglesia and A. T. Bell, Chem. Mater., 2000, 12, 2442–2447 CrossRef CAS
- K. S. Mazdiyasni, C. T. Lynch and J. S. Smith, J. Am. Ceram. Soc., 1965, 48, 372–375 CrossRef CAS
- E. M. Köck, M. Kogler, R. Pramsoler, B. Klötzer and S. Penner, Rev. Sci. Instrum., 2014, 85, 084102 CrossRef PubMed
- Thermo Fischer Scientific, XPS Interpretation of Zirconium, http://xpssimplified.com/elements/zirconium.php, status 2016.
- Y. Nishino, A. R. Krauss, Y. P. Lin and D. M. Gruen, J. Nucl. Mater., 1996, 228, 346–353 CrossRef CAS
-
Bruker AXS, Topas, Version 4.2, 2009 Search PubMed
- B. P. Mandal, N. Garg, S. M. Sharma and A. K. Tyagi, J. Nucl. Mater., 2009, 392, 95–99 CrossRef CAS
- R. D. Purohit, S. Saha and A. K. Tyagi, Mater. Sci. Eng., B, 2006, 130, 57–60 CrossRef CAS
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–72 CrossRef CAS
- A. Doran, L. Schlicker, S. Beavers, S. Bhat, M. F. Bekheet and A. Gurlo, Rev. Sci. Instrum., 2016, 88, 1–7 Search PubMed
- H.-Y. T. Chen, S. Tosoni and G. Pacchioni, Surf. Sci., 2016, 646, 230–238 CrossRef
- S. Kouva, J. Andersin, K. Honkala, J. Lehtonen, L. Lefferts and J. Kanervo, Phys. Chem. Chem. Phys., 2014, 16, 20650–20664 RSC
- K. Pokrovski, K. T. Jung and A. T. Bell, Langmuir, 2001, 17, 4297–4303 CrossRef CAS
- V. Bolis, C. Morterra, B. Fubini, P. Ugliengo and E. Garrone, Langmuir, 1993, 9, 1521–1528 CrossRef CAS
- K. T. Jung and A. T. Bell, J. Mol. Catal. A: Chem., 2000, 163, 27–42 CrossRef CAS
- B. Bachiller-Baeza, I. Rodriguez-Ramos and A. Guerrero-Ruiz, Langmuir, 1998, 14, 3556–3564 CrossRef CAS
- D. Bianchi, J. L. Gass, M. Khalfallah and S. J. Teichner, Appl. Catal., A, 1993, 101, 297–315 CrossRef CAS
- H. Wang, G. S. Li, Y. F. Xue and L. P. Li, J. Solid State Chem., 2007, 180, 2790–2797 CrossRef CAS
- C. Binet, A. Badri and J. C. Lavalley, J. Phys. Chem., 1994, 98, 6392–6398 CrossRef CAS
- C. Binet, M. Daturi and J. C. Lavalley, Catal. Today, 1999, 50, 207–225 CrossRef CAS
- F. Bozonverduraz and A. Bensalem, J. Chem. Soc., Faraday Trans., 1994, 90, 653–657 RSC
- T. L. Thompson and J. T. Yates, Chem. Rev., 2006, 106, 4428–4453 CrossRef CAS PubMed
- M. Kogler, E. M. Köck, B. Klötzer, L. Perfler and S. Penner, J. Phys. Chem. C, 2016, 120, 3882–3898 CAS
- Y. Murase and E. Kato, J. Am. Ceram. Soc., 1983, 66, 196–200 CrossRef CAS
- Y. Murase and E. Kato, J. Am. Ceram. Soc., 1979, 62, 527–527 CrossRef CAS
- X. Guo, Chem. Mater., 2004, 16, 3988–3994 CrossRef CAS
- M. Mamivand, M. A. Zaeem, H. El Kadiri and L. Q. Chen, Acta Mater., 2013, 61, 5223–5235 CrossRef CAS
- M. Youssef and B. Yildiz, Phys. Chem. Chem. Phys., 2014, 16, 1354–1365 RSC
- D. A. Ward and E. I. Ko, Chem. Mater., 1993, 5, 956–969 CrossRef CAS
- E. W. Leib, U. Vainio, R. M. Pasquarelli, J. Kus, C. Czaschke, N. Walter, R. Janssen, M. Muller, A. Schreyer, H. Weller and T. Vossmeyer, J. Colloid Interface Sci., 2015, 448, 582–592 CrossRef CAS PubMed
- I. Bespalov, M. Datler, S. Buhr, W. Drachsel, G. Rupprechter and Y. Suchorski, Ultramicroscopy, 2015, 159, 147–151 CrossRef CAS PubMed
- W. H. Gries, Surf. Interface Anal., 1996, 24, 38–50 CrossRef CAS
- J. D. Corbett and H. S. Marek, Inorg. Chem., 1983, 22, 3194–3202 CrossRef CAS
- R. Kaufmann, H. Klewenebenius, H. Moers, G. Pfennig, H. Jenett and H. J. Ache, Surf. Interface Anal., 1988, 11, 502–509 CrossRef CAS
- M. Kogler, E. M. Köck, T. Bielz, K. Pfaller, B. Klötzer, D. Schmidmair, L. Perfler and S. Penner, J. Phys. Chem. C, 2014, 118, 8435–8444 CAS
- E. M. Köck, M. Kogler, T. Bielz, B. Klötzer and S. Penner, J. Phys. Chem. C, 2013, 117, 17666–17673 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Additional EIS analysis and FT-IR experiments, XP spectra of the monoclinic reference material, additional Rietveld analysis. See DOI: 10.1039/c6dt04847a |
| This journal is © The Royal Society of Chemistry 2017 |