
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cu2+ recognition by N,N′-benzylated bis(amino amides)†
Lingaraju
Gorla
,
Vicente
Martí-Centelles
,
Belén
Altava
*,
M. Isabel
Burguete
and
Santiago V.
Luis
 *
*
Departamento de Química Inorgánica y Orgánica, Universitat Jaume I, Avda. Sos Baynat s/n., 12071 Castellón, Spain. E-mail: altava@uji.es; luiss@uji.es; Fax: +34 964728214; Tel: +34 964728239
First published on 31st January 2017
Abstract
Two new C2-symmetric N,N′-benzylated bis(amino amides) have been synthesised and their interaction with different transition metals studied using a variety of techniques including UV-Vis and CD spectroscopy or ESI-MS. The determination of the corresponding stability constants with Cu2+ has been possible, in H2O/CH3CN 7/3 v/v, for one of these ligands (4) using potentiometric titrations. The results obtained reveal that N-benzylation affords significant changes to their properties and is accompanied by an appreciable decrease in the corresponding complexation stability constants. However, this, along with the low kinetics associated to Ni2+, facilitates the recognition of Cu2+ by 4 that can be followed by the naked-eye up to the submillimolar range. Very interestingly, the chiral nature of this ligand provides an intense and well defined CD curve for the corresponding Cu2+ complex, very sensitive to the coordination geometry, facilitating the analysis of this interaction even at the μM range. The formation by both ligands (3 and 4) of square planar complexes with Cu2+ and Ni2+ displaying a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry was confirmed by their X-ray crystal structures.
:1 stoichiometry was confirmed by their X-ray crystal structures.
Introduction
The design, preparation and study of organic ligands able to interact with specific transition metals are of great current interest for the relevant role of these metals in different areas of chemistry and biology. These ligands can be useful for the selective extraction, removal or sensing of biologically and environmentally important species.1 Transition metals are not only required for biological functions but also have great potential for industrial applications. Among the various transition metal ions, Cu2+ plays important roles in living systems as a nutrient trace element present in the active sites of some key enzymes, but, at the same time, it is also associated with some serious human physiological dysfunctions such as Alzheimer's and Prion type neurodegenerative diseases, amyotrophic lateral sclerosis, Wilson's disease etc.2 The selective interaction with Cu2+ has allowed the development of sensing systems through the quantification of photophysical signals,3 including molecular probes providing logistics of naked-eye detection through a colour change for visual perception.3e,4In this regard, nitrogen binding sites are commonly used as donor atoms for ligand design,5 and different amino acid derivatives have been shown to bind metals forming stable complexes and the results are of interest to understand the interaction of proteic structures with metals.6 Recent contributions from our group have revealed that C2-symmetrical bis(amino amides) derived from amino acids form stable complexes with Cu2+, Zn2+ and Ni2+ ions.7 These results have highlighted the importance of different structural factors for this interaction and suggested that the presence of aromatic rings at different positions can be a key structural element. In particular, the inclusion of benzyl and related groups can be very relevant for the properties of the resulting ligands as has been reported for catalytic applications,6a–c and more recently in cascade-systems for the recognition of dicarboxylates.8 Herein, we present the synthesis of two new open-chain N,N′-benzylated bis(amino amide) ligands derived from L-phenylalanine (3) and L-valine (4) and the study of their acid–base properties as well as their metal complexation capacity, in particular for Ni2+ and Cu2+ using a variety of techniques.
Results and discussion
Synthesis, characterization and acid–base properties of the ligands
Scheme 1 displays the general synthetic pathway for the preparation of ligands 3 and 4. Bis(amino amides) 1 and 2, derived from L-phenylalanine and L-valine, were prepared from the corresponding N-Cbz protected amino acid N-hydroxysuccinimide ester, via coupling with 1,3-diaminopropane and final N-deprotection using HBr/AcOH.9 Then, compounds 1 and 2 were subjected to a reductive amination process, using benzaldehyde in methanol and then sodium borohydride, to afford ligands 3 and 4 in good yields.10 A central spacer containing three methylene groups was selected as previous results had shown that this spacer is very well suited to favour the formation of strong 1:1 metal complexes with the bisdeprotonated ligand.7a,b
 | ||
| Scheme 1 Synthesis of ligands 3 and 4. (i) DME, 20 h, r.t., 70–80%; (ii) HBr/AcOH 8 h, NaOH, r.t., 60–70%; (iii) benzaldehyde, methanol, 2 h, r.t; (iv) sodium borohydride, 18 h, r.t., 70–80%. | ||
Ligands 3 and 4 were fully characterised using standard techniques (1H-NMR, 13C-NMR, ESI-MS, and FT-IR), and crystals suitable for single-crystal X-ray diffraction analysis were obtained in the case of 3.
The crystal lattice for 3 (ESI, Fig. S1†) highlights the potential of this ligand to self-associate through well-defined directional supramolecular interactions and the importance of the aromatic rings in this regard. Interestingly, one of the benzyl side chains is folded over the central spacer in such a way that all the nitrogen bound hydrogen atoms are located at relatively short distances of this aromatic ring (<4.2 Å for the shortest N–H⋯CAr distance) in particular for one of the amino groups (2.906 Å). The importance of the interactions N–H⋯Ar in peptide-like molecules has been highlighted in several examples of pseudopetidic minimalistic molecular rotors.11 The most prominent feature for the crystal organization is that a defined set of H-bonds induces a global parallel disposition of the molecules (a β-sheet-like arrangement) with carbonyl groups from the two amides of each molecule located in anti-position (Fig. 1), and H-bonding to the amide groups of two additional molecules located at the two opposite sites. A different pattern was observed for the analogous compound with a shorter aliphatic spacer, displaying a syn disposition of the carbonyl amide groups and both aromatic side chains oriented in opposite directions.8
 | ||
| Fig. 1 X-ray crystal structure of ligand 3. | ||
The proper determination of the acid–base properties of polyamines is essential for understanding their coordination properties.12,13 The protonation constants of bis(amino amides) 3 and 4 were determined by potentiometric titrations. The presence of CH3CN as co-solvent was required to guarantee the solubility of the ligands and all the titrations were carried out at 298.1 K, using 0.1 M NaCl in H2O/CH3CN 7/3 v/v to maintain a constant ionic strength. The obtained stepwise stability constants for the protonation of pseudopeptidic ligands 3 and 4, using the program HYPERQUAD,14 are presented in Table 1. For the potentiometric studies in this solvent mixture, Kw was determined to be 14.6 in good agreement with literature data.15 The starting bis(amino amides) 1 and 2 had been studied previously in 0.15 or 0.10 M NaCl.7a,b To facilitate a proper comparison, the acid–base properties of 2 in 0.1 M NaCl in H2O/CH3CN 7/3 v/v were also determined.
| Reactiona | 1 , | 2 , | 2 | 3 | 4 |
|---|---|---|---|---|---|
| a Charges omitted for clarity. b Values in parentheses are the standard deviations in the last significant figure. c Determined in 0.1 M NaCl. d Determined in 0.15 M NaCl. e 0.1 M NaCl in H2O/CH3CN 7/3 v/v. f Data taken from ref. 7b. g Data taken from ref. 7a. | |||||
| H + L ⇌ HL | 7.57(1) | 8.01(5) | 7.77(8) | 6.77(7) | 6.74(9) |
| H + HL ⇌ H2L | 6.70(1) | 6.96(7) | 7.11(8) | 5.15(1) | 6.73(9) |
As expected, the double N-benzyl substitution decreased the basicity of the resulting bis(amino amides), (i.e. logKH1 7.77 for 2 against logKH1 6.74 for 4). Regarding the effect of the amino acid residue, the values of the first protonation constants for 3 (logKH1 6.77) and 4 (logKH1 6.74) were very close, while for the second protonation constant the value obtained for 3 (logKH2 5.15) was more than one order of magnitude lower than the one for 4 (logKH2 6.73) (Table 1). The similar values for the two protonation constants of 4 are remarkable. In α,ω-alkyldiamines, the difference between the two protonation constants decreases with the length of the spacer as a consequence of the reduction in the electrostatic repulsion between the two protonated sites, but remains essentially constant after pentamethylenediamine (ΔlogK ≈ 0.9), which has been associated to statistical factors.12b The situation, however, is more complex when additional heteroatoms or functional groups are present as has been observed for large polyamines and polyazacycloalkanes.12b,16 Thus, for 1,5,9,13,17,21-hexaazaheneicosane containing propylenic spacers, identical logK values have been reported for the first two protonation steps.16a The same situation was observed for the macrocycle [32]aneN6 displaying two 1,5,9-triazanonane subunits separated by two heptamethylene chains.16b,c Very close constants have also been found for the two first protonation constants (ΔlogK ≤ 0.2) in [24]aneN6,16d [27]aneN9,16e and [36]aneN12.16f These results cannot be only explained by the changes in the statistical factors associated to large polyamines and hydrogen bonding and entropic contributions have to be considered. A similar situation had been previously observed, always in mixed solvents, in a few C2-symmetric pseudopeptides,7d,17 Solvent effects are important on protonation constants and in the case of some polyamines it has been shown that these effects can differ for the first and the second protonation steps.18 For one of the pseudopeptides previously studied, changing the solvent provided a strong reduction of the first protonation constant (ΔlogK = −1.04) while the second one slightly increased (ΔlogK = 0.19).7d The comparison of the protonation constants for the reference compound 2 in the two solvent systems in Table 1 reveals a related trend, suggesting that the same can apply here. This could reflect the involvement of the amino groups in strong hydrogen bonding in the non-protonated compound that has to be broken for the first protonation to occur. The importance of intramolecular hydrogen bonds in diamides,19 the role of aromatic fragments on the solvation and hydrogen bonding in pseudopeptidic structures,11 and the resulting effects on the acid–base properties of the groups involved,19b have been studied in detail. Overall the situation is reminiscent of the one found in polytopic systems displaying positive cooperativity,20 in particular ditopic allosteric systems for which binding of the first guest requires a reorganization of the whole system that facilitates the binding of the second guest.21 A relevant example is a tetramide-crown ether system in which hydrogen bonding inhibits anion binding that is then “switched on” through an initial cation complexation.22 The distribution diagrams (ESI, Fig. S2†) revealed the presence of H2L2+ as the predominant species at pH values lower than 5 and 4 for ligands 3 and 4 respectively while the neutral species were predominant for both ligands at pH > 8. The most relevant difference is related to the monoprotonated species HL. The lower value of the second protonation constant for 3 is reflected in the presence of HL as a more important species at pH values close to neutrality, being the major species at pH 6.
Interaction of ligands 3 and 4 with M2+species
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band to lower frequencies when the complexes were formed (i.e. 1551 cm−1 for 6a against 1629 cm−1 for 4, ESI, Fig. S3†) indicative of the deprotonation of the N–H amide groups.6h
O band to lower frequencies when the complexes were formed (i.e. 1551 cm−1 for 6a against 1629 cm−1 for 4, ESI, Fig. S3†) indicative of the deprotonation of the N–H amide groups.6h
 | ||
| Scheme 2 Synthesis of the metal complexes derived from bis(amino amides) 3 and 4. | ||
For the Cu2+·4 system a large increase was observed on the molar absorption coefficients for the bands of the ligand at low wavelengths, characteristic of charge-transfer processes when large interactions between the molecular orbitals of the ligand and those of the metal occur.23 Besides, the two absorption peaks at 490 nm 750 nm suggest the coexistence of two coordination geometries for the metal, probably square planar and octahedral respectively.24 For the Ni2+·4 system, the initial absorption peaks observed around 400 and 650 nm suggested an octahedral structure,25 but when the UV-Vis spectrum was again measured after 24 hours, a new peak at 460 nm, usually associated to square planar Ni2+ complexes,26 was detected in agreement with the slow kinetics associated to this metal cation (ESI Fig. S4†).27 For Co2+, the small absorption at 500 nm suggests the occurrence of an octahedral structure.25a
Upon addition of ca. 2 equiv. of a 0.1 M NaOH solution in water to these M2+·4 systems (ca. 2.3 mM in the metal), pH values of 9.1–9.8 were obtained (ESI, Table S2†) and for Cu2+ the color became more intense. For this system, the UV-Vis (ESI, Fig. S4†) showed the disappearance of the band at 750 nm and the increase in the intensity of the absorption at 490 nm (ε = 123 M−1 cm−1) suggesting the quantitative formation of a square planar complex (ESI Fig. S4 and S5†). The stoichiometry of the complex formed by 4 and Cu2+ was determined to be 1:1 (ligand:metal) from the Job plot of its absorbance at 490 nm as a function of the molar fraction of added Cu2+ in MeOH/H2O (ESI, Fig. S6†).28,29 The titration of Cu2+ solutions with increasing amounts of 4 provided a gradual increase in the intensity of the absorption at 490 nm and a decrease of the one at 750 nm. The isoemissive point around 640 nm was better defined under basic conditions. The non-linear fitting of the titration curves (1:1 stoichiometry) provided apparent constants (Kapp) of 562 M−1 and 3350 M−1 in the absence and presence of base (ESI, Fig. S7†).28 Similar trends were obtained when CuCl2 and CuNO3 were used instead of Cu(OAc)2 as the source of Cu2+ (ESI, Fig. S8†).
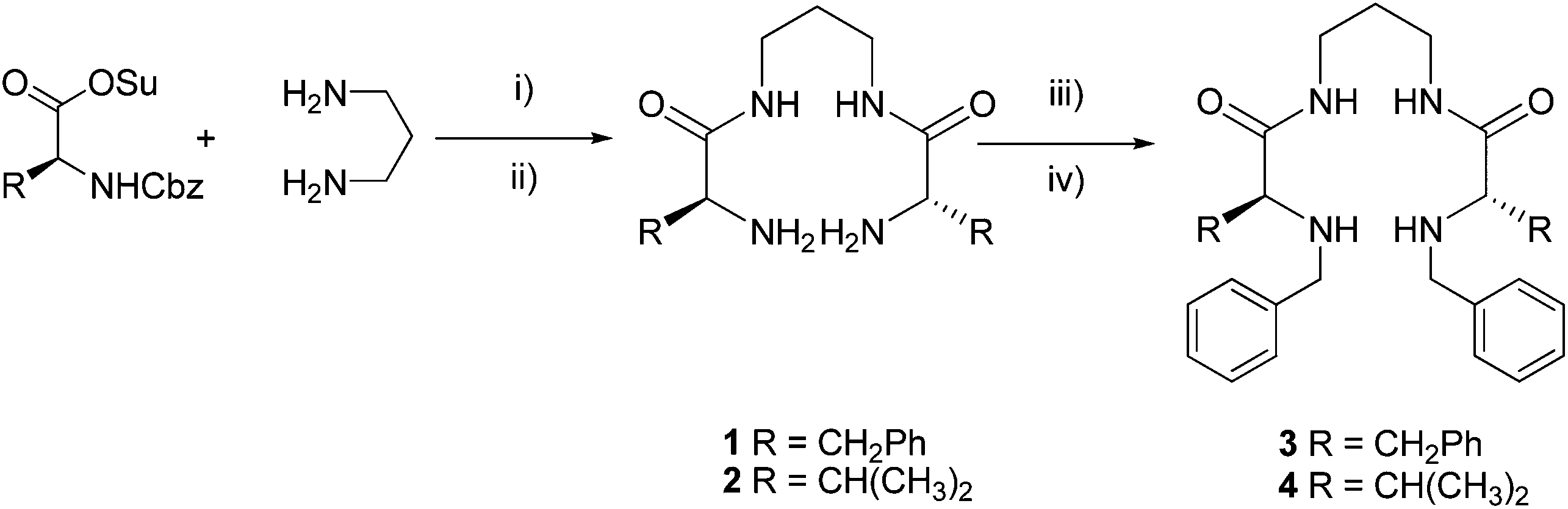
Considering the chiral nature of the ligand, the complexation of 4 with the different metal ions was also studied by CD. For this purpose, a 2.5 mM solution (MeOH/H2O 80/20 v/v) containing an equimolecular mixture of 4 and the corresponding M2+ and two equivalents of base was kept stirring for 24 hours. The CD of the free ligand 4 displayed a negative band centered at 240 nm (Fig. 2a). Very interestingly, in the presence Cu2+ (Fig. 2b, black line) a strong negative split-Cotton effect (−, +) was observed with a minimum at 529 nm (Δε = −0.44 cm2 mol−1) and a maximum at 448 nm (Δε = 0.23 cm2 mol−1), and passing through zero at 481 nm, which is the λmax for the UV band for the square planar complex. In the absence of added base, an additional weaker positive CD band was observed for the Cu2+·4 system in the 600–800 nm region (ESI, Fig. S13†) in agreement with the coexistence of two complex species and highlighting the very different 3D chiral environments present in them. This behaviour was not detected for other metals. Only for Ni2+, a weak Cotton effect (minimum at 463 nm, maximum at 284 nm) was observed (Fig. 2b). Additional CD signals were always observed below 350 nm most likely related to metal–ligand charge transfer or to intraligand transitions.23,30
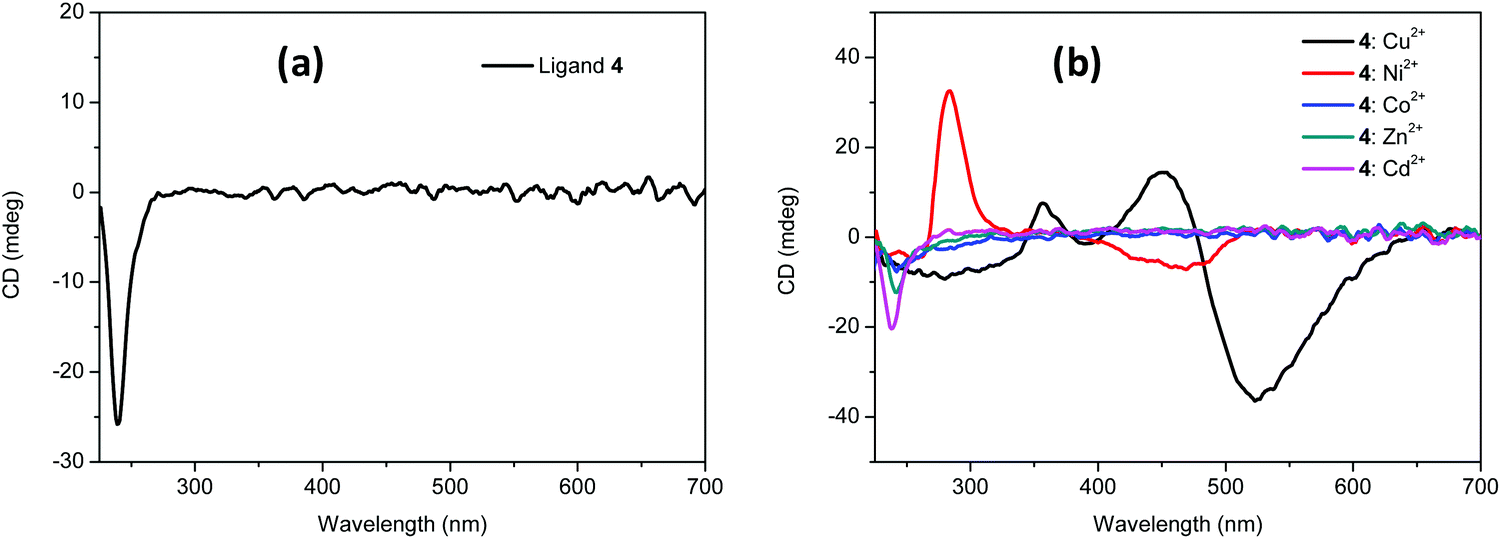 | ||
| Fig. 2 (a) CD spectra for ligand 4 in basic media (2 equiv. of NaOH). (b) CD spectra for ligand 4 in basic media (2 equiv. of NaOH) in the presence of 1 equivalent of different M2+ metals. In all cases the solvent was MeOH/H2O 80/20 (v/v) and the concentration of the metal and the ligand 2.5 mM. | ||
Taking into account that most molecular designs for the detection of Cu2+ involve the inclusion of sensitive chromogenic or fluorescent units like Rhodamine, naphthalimide, squarylium, fluorescein, etc.,3,4,31 the optical response obtained for Cu2+·4 complexes, in particular in CD, is remarkable. Nevertheless, the approximated LOD value calculated for the naked-eye detection was ca. 0.25 mM (ESI, Fig. S9†),32 while the use of UV-Vis spectra provided LOD values in the 79–91 μM range, depending on the experimental conditions (ESI, Fig. S10 to S12†). Although the naked-eye detection can compare well with some reported sensors (μM–mM range in many cases), the UV-Vis LOD values are significantly higher than those in sensors containing chromogenic fragments.31 It is worth noting that the strong CD Cotton effect observed allowed to obtain lower LOD values of 43 μM and 28 μM under neutral and basic conditions respectively by using the variation of the amplitude of the Cotton effect (ΔCD = CD440nm − CD520nm) (ESI, Fig. S13 and S14†),33 no interference by the other metals assayed was observed in any of the cases (ESI, Fig. S15†).
:1 complexation (Fig. 3). For Ni2+, Cd2+, Co2+, Zn2+ ions, the corresponding peaks associated to [MH−1L]+ species were not observed, although the peak for the [MH−1L + AcOH]+ species was present for Zn2+ and Ni2+. However, for Ni2+, the intensity of this peak was 30% of the base peak while for Zn2+ its intensity was <2% (ESI, Fig. S16†). For Co2+ and Cd2+ very minor peaks (<2%) corresponding to [2L + M]2+ and [L + M + Cl]+ species were observed.
 | ||
| Fig. 3 ESI+-MS of compound 4 (5 mM) in the presence of 1 equiv. of Cu(OAc)2 in MeOH/H2O 80/20, v/v. | ||
Potentiometric studies for Cu2+ complexes
Finally, the interaction of ligand 4 with Cu2+ was studied by potentiometric titrations over the 2–12 pH range. The stability constants for the formation of complexes were determined for a 1:1 metal–ligand ratio, using NaCl 0.1 M in H2O/CH3CN 7/3 v/v to maintain a constant ionic strength and at 298.1 K. Although the same solvent mixture used for spectroscopic measurements was also assayed here (MeOH/H2O 80/20 v/v), its use was not satisfactory and led to non-stable measurements in some instances. The results obtained are presented in Table 2 and the corresponding distribution diagrams are displayed in Fig. S17 (ESI†). The interaction of the starting bis(amino amide) 2 with Cu2+ was also studied by potentiometric titrations using the same medium to provide a proper comparison. The solubility problems associated to ligand 3 complexes again precluded carrying out accurate potentiometric titrations. When comparing the values obtained for 2 in both solvents it can be seen that the solvent plays, in this case, an important role. The formation of complex species seems to be favoured in the mixed solvent,7a although the effects coming from the stronger competition of Na+ in the medium of greater salinity need to be also considered.
β) for the Cu2+ complexes with ligands 2 and 4 at 298.1 ± 0.1 Kb
| Reactiona | 2 , | 2 | 4 |
|---|---|---|---|
| a Charges omitted for clarity. b Values in parentheses are the standard deviations in the last significant figure. c Determined in 0.15 M NaCl. d Data taken from ref. 7a. e 0.1 M NaCl in H2O/CH3CN 7/3 v/v. | |||
| Cu + L ⇌ CuL | 5.99(3) | 6.99(7) | 4.39(9) |
| Cu + L ⇌ CuH−1L + H | 0.53(1) | 1.53(7) | −0.66(3) |
| Cu + L ⇌ CuH−2L + 2H | −6.66(1) | −2.97(1) | −6.29(1) |
As can be seen in Table 2, significant differences can be observed associated to the N-benzylation. The most general and remarkable observation was the lower stability of the non-deprotonated, mono and di-deprotonated complex species for compound 4. This is easily analysed using the representation of the logKeff values vs. pH (Fig. 4). For the neutral [CuH−2L] species from 4 the formation constant (logβ = −6.29) was more than three orders of magnitude lower than the related one for ligand 2 (logβ = −2.97). In spite of this, these neutral complex species were predominant around pH 7 for 2 and 4, though [CuH−2L] starts to be formed at slightly higher pH values for 4. This confirms the formation of [CuH−2L] species at physiologic pH for 4 (ESI, Fig. S17†). On the other hand, the relative importance of [CuH−1L] and [CuL] species is reversed for both ligands. For 4, the [CuH−1L] species is more important at pH values around 6, while it is a minor species in the case of 2, and [CuL] is relatively important for 2 at pH values close to 5, whereas it is very minor for 4. The distribution diagram obtained for 4 reveals that at neutral pH values at least two species are present, [CuH−2L] and [CuH−1L], which is in good agreement with the observations obtained from spectroscopic experiments suggesting the coexistence of square planar and octahedral Cu2+ species. The decrease observed in the stability of the Cu2+ complexes upon benzylation of the terminal amino groups of 2 is higher than that observed for the benzylation of some linear polyamines, where the magnitudes of the stability constants were 1–2 orders of magnitude lower,23c although for a triethylenetetramine bearing anthracene and benzophenone instead of phenyl units an additional decrease in ca. five logarithmic units has been reported.13g
 | ||
| Fig. 4 Plot of the effective conditional constants vs. pH for the systems Cu2+·2 and Cu2+·4 in 0.1 M NaCl in H2O/CH3CN 7/3 v/v, at 298.1 ± 0.1 K. | ||
Single-crystal X-ray diffraction studies
The formation of the corresponding bisdeprotonated complexes of 3 and 4 with Cu2+ and Ni2+ and their stoichiometry and geometry were confirmed by X-ray crystallography. Crystals suitable for single-crystal X-ray diffraction analysis were obtained for all the metal complexes by the layering of a methanolic solution of the pseudopeptidic ligand over a basic aqueous solution of Cu(OAc)2 and Ni(OAc)2. The X-ray crystal structures of the copper complexes 5a and 6a are presented in Fig. 5. X-ray diffraction data confirm the formation of the metal complexes with a 1:1 stoichiometry.
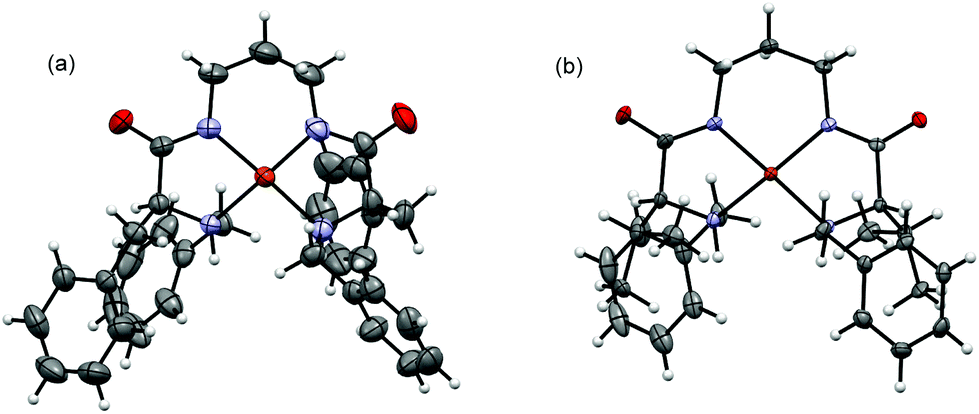 | ||
| Fig. 5 X-ray crystal structures of Cu2+ complexes 5a (a) and 6a (b). Ellipsoids represented at the 50% probability level. | ||
The Cu2+ cation in 5a is coordinated by two amine groups and two deprotonated amide groups, in a distorted square-planar geometry, and displays a torsion angle of 13.8° between the Namine–Cu–Namide planes. The Cu–Namide distances are slightly shorter than the Cu–Namine ones, being 1.921 and 2.052 Å on average, respectively.7b The N–Cu–N angles contained in the two five-membered rings Namine–Cu–Namide are ca. 13° smaller than those contained in the Namide–Cu–Namide six-membered ring, as expected for a smaller ring size with a tighter environment. The Cu2+ cation in 6a is also coordinated by two amine groups and two deprotonated amide groups, in a distorted square-planar geometry, but displays a torsion angle of 29.9°. Again the Cu–Namide distances are slightly shorter than the ones for Cu–Namine, being 1.912 and 2.034 Å on average, respectively, and both slightly shorter than in 5a. The N–Cu–N angles contained in the two five-membered rings (Namine–Cu–Namide) are ca. 14° smaller than those in the six member ring (Namide–Cu–Namide).
The X-ray crystal structures for the Ni2+ complexes 5b and 6b are presented in Fig. 6. Also for 5b and 6b the Ni2+ cation is coordinated by two amine groups and two deprotonated amide groups, in a distorted square-planar geometry displaying torsion angles of 12.9° and 14.8° respectively. In both complexes the Ni–Namide distances are slightly shorter than the corresponding Ni–Namine and the N–Ni–N angles contained in the five-membered rings are smaller than the angles contained in the six-membered ring (ca. 15° for 6b).
 | ||
| Fig. 6 X-ray crystal structure of complexes 5b (a) and 6b (b). Ellipsoids represented at the 50% probability level. | ||
Conclusions
N-Benzylation of the terminal amino nitrogen atoms in C2-symmetric pseudopeptides containing an aliphatic central spacer provides significant changes in their properties as ligands. Thus, the solubility in water of 3 and 4 and that for their respective metal complexes decreases, in particular for the phenylalanine derivative (3), which precludes a more detailed analysis of its coordination chemistry. The corresponding spectroscopic, spectrometric and potentiometric studies could be carried out with the valine derivative 4, in particular in aqueous mixed solvents, revealing that these bis(amino amides) are able to provide efficient tetradentate coordination for metal cations, in particular Cu2+. The resulting Cu2+ and Ni2+ bisdeprotonated complexes seem to represent the most important complex species for both cations, even at slightly acidic pH regions at least for the system Cu2+·4. The complexes display 1:1 stoichiometries and square planar arrangements around the metal centre, as suggested by spectroscopic data and confirmed by the corresponding X-ray structures. The presence of the additional benzyl groups in the terminal amino groups can significantly reduce the stability of the corresponding metal complexes, as has been observed when comparing the stability constants obtained potentiometrically for the system Cu2+·4 and those obtained for the non-benzylated analogue 2. The observed decrease in the stability constants for Cu2+ is significantly higher than the one observed in the case of benzylated linear polyamines. This, however, facilitates the observation of a defined spectroscopic response to the interaction of 4 with Cu2+, in particular at short times. It must be noted that the corresponding Cu2+ complexes display very strong bisigned CD signals that can be used advantageously to observe and characterize the formation of such complexes, achieving LOD values of 28 μM while using UV-Vis measurements the value was 80–90 μM.
Experimental section
All reagents and solvents were obtained from commercial sources and used as received unless otherwise stated. Deionized water was used from a “Milli-Q® Integral Water Purification System” by Millipore. Microanalyses were performed on an elemental analyzer equipped with an oxygen module. Rotatory power was determined with a digital polarimeter (Na: 589 nm). Melting points were measured using a standard apparatus and are uncorrected.General procedure for the synthesis of N,N′-benzylated bis(amino amides)
General procedure for the synthesis of M2+ complexes
Compound 3 (100 mg, 0.182 mmol) was dissolved in dry MeOH (5 mL) in a 25 mL round bottom flask and maintained under a nitrogen atmosphere. Cu(OAc)2 (36.38 mg, 0.182 mmol) dissolved in dry MeOH (5 mL) was then added and the mixture stirred for 30 min at room temperature. 1 M KOH in methanol (3.3 mL, 0.364 mmol) was added and the solution was maintained at room temperature overnight. The precipitate formed was filtered and washed with cold dichloromethane to afford the corresponding metal complex.Electromotive force measurements
The potentiometric titrations were carried out at 298.1 K using 0.1 M NaCl as the supporting electrolyte. The experimental procedure (burette, potentiometer, cell, stirrer, micro-computer, etc.) has been fully described elsewhere.34 The acquisition of the emf data was performed with the computer program CrisonCapture. The reference electrode was an Ag/AgCl electrode in saturated KCl solution. The glass electrode was calibrated as a hydrogen-ion concentration probe by titration of previously standardized amounts of HCl with CO2-free NaOH solutions and the equivalence point determined by the Gran's method,35 which gives the standard potential, E°′, and the ionic product of water 13.78, whereas in the case of the H2O/CH3CN mixture used this value was 14.6.15 The computer program HYPERQUAD was used to calculate the protonation and stability constants,14 and the HySS29 program was used to obtain the distribution diagrams.36 The pH range investigated was 2.0–12.0 and the concentration of the metal ions and the ligands 0.1 mM, with 1:1 M2+:L molar ratios. The different titration curves for each system (at least two) were treated either as a single set or as separated curves without significant variations in the values of the stability constants. Finally, the sets of data were merged together and treated simultaneously to give the final stability constants.
1H NMR experiments
The 1H spectra were recorded on a Varian INOVA 500 spectrometer (500 and 125 MHz for 1H and 13C NMR, respectively). The solvent signal was used as a reference standard.UV-Vis and CD measurements
UV-Vis absorption spectra were recorded using a Hewlett-Packard 8453 device. All measurements were performed at room temperature and spectra were recorded from 200 to 900 nm. For metal titration experiments, a M2+ solution in MeOH/H2O 80/20 (2.5 mM) and a solution of compound 4 (30 mM) in the same solvent mixture were used and aliquots of the ligand solution were gradually added to the metal solution using a micropipette. After equilibration, the corresponding absorptions were measured. For job-plot measurements stock solutions of compound 4 (5 mM in MeOH/H2O 80/20) and Cu(OAc)2 (5 mM in MeOH/H2O 80/20) were prepared and mixed in variable proportions so that the final volume of the mixture was maintained constant to 1 mL. After equilibration, the absorbance at 490 nm of each solution was independently measured. A plot of absorbance against the mole fraction of Cu2+ was used to determine the metal–ligand ratio.CD experiments were performed using a JASCO J-810 spectrometer. All measurements were performed at room temperature and spectra were recorded from 200 nm to 900 nm with 1.0 nm step and 1.0 nm bandwidth and the scanning speed was 200 nm per minute.
LOD values were calculated using the relation
| Detection limit = 3σ/k |
Mass spectrometry
Mass spectra were recorded on a Q-TOF Premier mass spectrometer with an orthogonal Z-spray electrospray interface (Micromass, Manchester, UK) by electrospray positive mode (ES+). The desolvation gas, as well as nebulizing gas, was nitrogen at a flow of 700 L h−1 and 20 L h−1, respectively. The of the source block and desolvation temperatures were set to 120 °C and 150 °C. A capillary voltage of 3.5 kV was used in the positive scan mode, respectively. The cone voltage was typically set to 20 V to control the extent of fragmentation of the identified ions. Sample solutions were infused via syringe pump directly connected to the ESI source at a flow rate of 10 mL min−1. The observed isotopic pattern of each intermediate perfectly matched the theoretical isotope pattern calculated from their elemental composition using the MassLynx 4.0 program.37IR spectroscopy
FT-IR spectra were acquired on a JASCO 6200 equipment with a MIRacle single reflection ATR diamond/ZnSe accessory. The raw IR spectral data were processed with the JASCO spectral manager software.Crystallography
Single crystals of ligand 4 and complexes 5a–6b were obtained. A suitable crystal was selected and measured on a single crystal X-ray diffractometer. Using Olex2,38 all the structures were solved with the ShelXS 2014 structure solution program using Direct Methods and refined with the ShelXL 2014 refinement package using Least Squares minimization.39 The program MERCURY was used to prepare artwork representations.40 Crystallographic data and refinement parameters are summarized in Table S1 (ESI†). Cambridge Crystallographic Data Centre files 1481178 (3), 1481179 (5a), 1481180 (5b), 1481181 (6a) and 1481182 (6b) contain the supplementary crystallographic data for this paper.Acknowledgements
Financial support from Spanish MINECO (CTQ2015-68429-R), Generalitat Valenciana (PROMETEO/2016/071) and PPI-UJI (P1-1B-2013-38) is gratefully acknowledged. L. G. thanks Generalitat Valenciana for a Grisolia fellowship (GRISOLIA 2012/015). The support of the SCIC at the Universitat Jaume I for instrumental techniques is acknowledged.Notes and references
- (a) W. Wang, Q. Wen, Y. Zhang, X. Fei, Y. Li, Q. Yang and X. Xu, Dalton Trans., 2013, 42, 1827–1833 RSC; (b) P. C. Dhar, A. Pal, P. Mohanty and B. Bag, Sens. Actuators, B, 2015, 219, 308–314 CrossRef CAS; (c) D. Singhal, N. Gupta and A. K. Singh, RSC Adv., 2015, 5, 65731–65738 RSC; (d) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed; (e) L. Pu, Chem. Rev., 2004, 104, 1687–1716 CrossRef CAS PubMed; (f) G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723–2750 CrossRef CAS PubMed; (g) P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef CAS; (h) L. Prodi, F. Bolletta, M. Montalti and N. Zaccheroni, Coord. Chem. Rev., 2000, 205, 59–83 CrossRef CAS; (i) V. Amendola, L. Fabbrizzi, F. Foti, M. Licchelli, C. Mangano, P. Pallavicini, A. Poggi, D. Sacchi and A. Taglietti, Coord. Chem. Rev., 2006, 250, 273–299 CrossRef CAS.
- (a) E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995–2044 CrossRef CAS PubMed; (b) A. Mathie, G. L. Sutton, C. E. Clarke and E. L. Veale, Pharmacol. Ther., 2006, 111, 567–583 CrossRef CAS PubMed; (c) K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205–214 CrossRef CAS PubMed; (d) D. J. Waggoner, T. B. Bartnikas and J. D. Gitlin, Neurobiol. Dis., 1999, 6, 221–230 CrossRef CAS PubMed; (e) L. I. Bruijn, T. M. Miller and D. W. Cleveland, Annu. Rev. Neurosci., 2004, 27, 723–749 CrossRef CAS PubMed; (f) C. Zhao, B. Liu, X. Bi, D. Liu, C. Pan, L. Wang and Y. Pang, Sens. Actuators, B, 2016, 229, 131–137 CrossRef CAS.
- (a) Y. Xiang, Z. Li, X. Chen and A. Tong, Talanta, 2008, 74, 1148–1153 CrossRef CAS PubMed; (b) B. High, D. Bruce and M. M. Richter, Anal. Chim. Acta, 2001, 449, 17–22 CrossRef CAS; (c) L. Tapia, M. Suazo, C. Hödar, V. Cambiazo and M. González, BioMetals, 2003, 16, 169–174 CrossRef CAS PubMed; (d) F. Chen, G. Liu, Y. Shi, P. Xi, J. Cheng, J. Hong, R. Shen, X. Yao, D. Bai and Z. Zeng, Talanta, 2014, 124, 139–145 CrossRef CAS PubMed; (e) D. Zha and L. You, ACS Appl. Mater. Interfaces, 2016, 8, 2399–2405 CrossRef CAS PubMed.
- (a) B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 205, 3–40 CrossRef CAS; (b) N. Kaur and S. Kumar, Tetrahedron, 2011, 67, 9233–9264 CrossRef CAS.
- (a) L. Fabbrizzi and A. Poggi, Chem. Soc. Rev., 2013, 42, 1681–1699 RSC; (b) A. E. Martell, R. M. Smith and R. M. Moteikaitis, NIST Critical Stability Constants Database, Texas A&M University, College Station, TX, 1993 Search PubMed.
- (a) B. Dangel, M. Clarke, J. Haley, D. Sames and R. Polt, J. Am. Chem. Soc., 1997, 119, 10865–10866 CrossRef CAS; (b) B. D. Dangel and R. Polt, Org. Lett., 2000, 2, 3003–3006 CrossRef CAS PubMed; (c) R. Polt, B. D. Kelly, B. D. Dangel, U. B. Tadikonda, R. E. Ross, A. M. Raitsimring and A. V. Astashkin, Inorg. Chem., 2003, 42, 566–574 CrossRef CAS PubMed; (d) M. Merschky and C. Schmuck, Org. Biomol. Chem., 2009, 7, 4895–4903 RSC; (e) Ł. Weseliński, E. Słyk and J. Jurczak, Tetrahedron Lett., 2011, 52, 381–384 CrossRef; (f) J. Paradowska, M. Pasternak, B. Gut, B. Gryzło and J. Mlynarski, J. Org. Chem., 2012, 77, 173–187 CrossRef CAS PubMed; (g) F. Adrián, M. I. Burguete, J. M. Fraile, J. I. García, J. García, E. García-España, S. V. Luis, J. A. Mayoral, A. J. Royo and M. C. Sánchez, Eur. J. Inorg. Chem., 1999, 2347–2354 CrossRef; (h) R. R. Wagler, Y. Fang and C. J. Burrows, J. Org. Chem., 1989, 54, 1584–1589 CrossRef.
- (a) S. Blasco, M. I. Burguete, M. P. Clares, E. García-España, J. Escorihuela and S. V. Luis, Inorg. Chem., 2010, 49, 7841–7852 CrossRef CAS PubMed; (b) I. Martí, A. Ferrer, J. Escorihuela, M. I. Burguete and S. V. Luis, Dalton Trans., 2012, 41, 6764–6776 RSC; (c) V. Martí-Centelles, D. K. Kumar, A. J. P. White, S. V. Luis and R. Vilar, CrystEngComm, 2011, 13, 6997–7008 RSC; (d) L. Gorla, V. Martí-Centelles, B. Altava, M. I. Burguete and S. V. Luis, Inorg. Chem., 2016, 55, 7617–7629 CrossRef CAS PubMed.
- E. Faggi, R. Gavara, M. Bolte, L. Fajari, L. Julia, L. Rodriguez and I. Alfonso, Dalton Trans., 2015, 44, 12700–12710 RSC.
- J. Becerril, M. Bolte, M. I. Burguete, F. Galindo, E. García-España, S. V. Luis and J. F. Miravet, J. Am. Chem. Soc., 2003, 125, 6677–6686 CrossRef CAS PubMed.
- (a) I. Alfonso, M. Bolte, M. Bru, M. I. Burguete, S. V. Luis and J. Rubio, J. Am. Chem. Soc., 2008, 130, 6137–6144 CrossRef CAS PubMed; (b) M. Bru, I. Alfonso, M. Bolte, M. I. Burguete and S. V. Luis, Chem. Commun., 2011, 47, 283–285 RSC.
- (a) I. Alfonso, M. I. Burguete and S. V. Luis, J. Org. Chem., 2006, 71, 2242–2250 CrossRef CAS PubMed; (b) I. Alfonso, M. I. Burguete, F. Galindo, S. V. Luis and L. Vigara, J. Org. Chem., 2007, 72, 7947–7956 CrossRef CAS PubMed; (c) E. Faggi, S. V. Luis and I. Alfonso, RSC Adv., 2013, 3, 11556–11565 RSC.
- (a) A. E. Martell and R. J. Motekaitis, Coord. Chem. Rev., 1990, 100, 323–361 CrossRef CAS; (b) A. Bencini, A. Blanchi, E. Garcia-España, M. Micheloni and J. A. Ramirez, Coord. Chem. Rev., 1999, 188, 97–156 CrossRef CAS.
- (a) E. García-España, P. Díaz, J. M. Llinares and A. Bianchi, Coord. Chem. Rev., 2006, 250, 2952–2986 CrossRef; (b) A. Bianchi, B. Escuder, E. García-España, S. V. Luis, V. Marcelino, J. F. Miravet and J. A. Ramírez, J. Chem. Soc., Perkin Trans. 2, 1994, 1253–1259 RSC; (c) V. J. Arán, M. Kumar, J. Molina, L. Lamarque, P. Navarro, E. García-España, J. A. Ramírez, S. V. Luis and B. Escuder, J. Org. Chem., 1999, 64, 6135–6146 CrossRef; (d) J. A. Aguilar, E. García-España, J. Guerrero, S. V. Luis, J. Llinares, J. Ramírez and C. Soriano, Inorg. Chim. Acta, 1996, 246, 287–294 CrossRef CAS; (e) A. Bencini, A. Bianchi, M. I. Burguete, P. Dappporto, A. Doménech, E. García-España, S. V. Luis, P. Paoli and J. A. Ramírez, J. Chem. Soc., Perkin Trans. 2, 1994, 569–577 RSC; (f) M. T. Albelda, M. A. Bernardo, E. García-España, M. L. Godino-Salido, S. V. Luis, M. J. Melo, F. Pina and C. Soriano, J. Chem. Soc., Perkin Trans. 2, 1999, 2545–2549 RSC; (g) G. Nishimura, K. Ishizumi, Y. Shiraishi and T. Hirai, J. Phys. Chem. B, 2006, 110, 21596–21602 CrossRef CAS PubMed.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS PubMed.
- M. A. Herrador and A. G. González, Talanta, 2002, 56, 769–775 CrossRef CAS PubMed.
- (a) B. Dietrich, M. W. Hosseini, J.-M. Lehn and R. B. Sessions, Helv. Chim. Acta, 1983, 66, 1262–1278 CrossRef CAS; (b) M. W. Hosseini and J. M. Lehn, J. Am. Chem. Soc., 1982, 104, 3525–3527 CrossRef CAS; (c) M. W. Hosseini and J. M. Lehn, Helv. Chim. Acta, 1986, 69, 587–603 CrossRef CAS; (d) B. Dietrich, M. W. Hosseini, J. M. Lehn and R. B. Sessions, J. Am. Chem. Soc., 1981, 103, 1282–1283 CrossRef CAS; (e) A. Bencini, A. Bianchi, E. Garcia-España, M. Giusti, M. Micheloni and P. Paoletti, Inorg. Chem., 1987, 26, 681–684 CrossRef CAS; (f) A. Bencini, A. Bianchi, E. Garcia-España, M. Micheloni and P. Paoletti, Inorg. Chem., 1988, 27, 176–180 CrossRef CAS.
- P. Seliger, D. Tomczyk, G. Andrijewski and E. Tomal, J. Anal. Methods Chem., 2016, 2016, 1721069 Search PubMed.
- D. B. Rorabacher, W. J. MacKellar, F. R. Shu and S. M. Bonavita, Anal. Chem., 1971, 43, 561–573 CrossRef CAS.
- (a) S. H. Gellman, G. P. Dado, C.-B. Liang and B. R. Adam, J. Am. Chem. Soc., 1991, 113, 1164–1173 CrossRef CAS; (b) T. Schneider, K. T. Halloran, J. A. Hillner, R. R. Conry and B. R. Linton, Chem. – Eur. J., 2013, 19, 15101–15104 CrossRef CAS PubMed; (c) A. Mazzanti, M. Chiarucci, L. Prati, K. W. Bentley and C. Wolf, J. Org. Chem., 2016, 81, 89–99 CrossRef CAS PubMed.
- (a) C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488–7499 CrossRef CAS PubMed; (b) A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef CAS PubMed.
- (a) J. Rebek, T. Costello, L. Marshall, R. Wattley, R. C. Gadwood and K. Onan, J. Am. Chem. Soc., 1985, 107, 7481–7487 CrossRef CAS; (b) J. Rebek, Acc. Chem. Res., 1984, 17, 258–264 CrossRef CAS; (c) F. Gaviña, S. V. Luis, A. M. Costero, M. I. Burguete and J. Rebek, J. Am. Chem. Soc., 1988, 110, 7140–7143 CrossRef.
- E. N. W. Howe, M. Bhadbhade and P. Thordarson, J. Am. Chem. Soc., 2014, 136, 7505–7516 CrossRef CAS PubMed.
- (a) The colourless H2O/MeOH solution of 4 did not absorb in the visible-UV region from 350 to 800 nm; ; (b) O. Horvath and K. L. Stevenson, Charge-transfer photochemistry of coordination compounds, VCH, Weinheim, Germany, 1993 Search PubMed; (c) M. A. Bernardo, F. Pina, E. García-España, J. Latorre, S. V. Luis, J. M. Llinares, J. A. Ramírez and C. Soriano, Inorg. Chem., 1998, 37, 3935–3942 CrossRef CAS PubMed.
- (a) M. A. Pauly, E. M. Erwin, D. R. Powell, G. T. Rowe and L. Yang, Polyhedron, 2015, 102, 722–734 CrossRef CAS; (b) W. Wang, Y. A. Lee, G. Kim, S. K. Kim, G. Y. Lee, J. Kim, Y. Kim, G. J. Park and C. Kim, J. Inorg. Biochem., 2015, 153, 143–149 CrossRef CAS PubMed; (c) P. D. Wadhavane, L. Gorla, A. Ferrer, B. Altava, M. I. Burguete, M. A. Izquierdo and S. V. Luis, RSC Adv., 2015, 5, 72579–72589 RSC.
- (a) F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, Wiley, New York, 6th edn, 1999 Search PubMed; (b) G. Bussière and C. Reber, J. Am. Chem. Soc., 1998, 120, 6306–6315 CrossRef; (c) W. Liu, A. Migdisov and A. Williams-Jones, Geochim. Cosmochim. Acta, 2012, 94, 276–290 CrossRef CAS.
- (a) M. Rajasekar, S. Sreedaran, R. Prabu, V. Narayanan, R. Jegadeesh, N. Raaman and A. K. Rahiman, J. Coord. Chem., 2010, 63, 136–146 CrossRef CAS; (b) J. Manonmani, R. Thirumuruhan, M. Kandaswamy, V. Narayanan, S. S. S. Raj, M. N. Ponnuswamy, G. Shanmugam and H. K. Fun, Polyhedron, 2001, 20, 3039–3048 CrossRef CAS.
- (a) M. G. Basallote, M. J. Fernandez-Trujillo and M. A. Manez, J. Chem. Soc., Dalton Trans., 2002, 3691–3695 RSC; (b) F. T. R. D. Almeida, B. C. S. Ferreira, A. L. D. S. L. Moreira, R. P. D. Freitas, L. F. Gil and L. V. A. Gurgel, J. Colloid Interface Sci., 2016, 466, 297–309 CrossRef PubMed.
- (a) P. Job, Anal. Chim. Appl., 1928, 9, 113–203 CAS; (b) H.-J. Schneider and A. Yatsimirsky, Principles and Methods in Supramolecular Chemistry, John Wiley & Sons, Chichester, 2000 Search PubMed; (c) P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC; (d) J. S. Renny, L. L. Tomasevich, E. H. Tallmadge and D. B. Collum, Angew. Chem., Int. Ed., 2013, 52, 11998–12013 CrossRef CAS PubMed.
- For limitations of the Job plot, in particular for weak supramolecular complexes, see, for instance: (a) F. Ulatowski, K. Dąbrowa, T. Bałakier and J. Jurczak, J. Org. Chem., 2016, 81, 1746–1756 CrossRef CAS PubMed; (b) D. B. Hibberta and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- (a) S. K. Sharma, G. Hundal and R. Gupta, Eur. J. Inorg. Chem., 2010, 2010, 621–636 CrossRef; (b) S. Kumar and R. Gupta, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2011, 50, 1369–1379 Search PubMed; (c) N. Tounsi, L. Dupont, A. Mohamadou, E. Guillon, M. Aplincourt and G. Rogez, Polyhedron, 2008, 27, 3674–3682 CrossRef CAS; (d) L.-M. López-Martínez, H. Santacruz-Ortega, R. E. Navarro, L. Machi-Lara, R. Sugich-Miranda and K. Ochoa-Lara, Polyhedron, 2014, 79, 338–346 CrossRef.
- (a) S.-P. Wu, K.-J. Du and Y.-M. Sung, Dalton Trans., 2010, 39, 4363–4368 RSC; (b) C. Liu, J. Xu, F. Yang, W. Zhou, Z. Li, L. Wei and M. Yu, Sens. Actuators, B, 2015, 212, 364–370 CrossRef CAS; (c) Y. Wang, C. Wang, S. Xue, Q. Liang, Z. Li and S. Xu, RSC Adv., 2016, 6, 6540–6550 RSC; (d) R. V. Rathod, S. Bera, M. Singh and D. Mondal, RSC Adv., 2016, 6, 34608–34615 RSC; (e) J.-J. Xiong, P.-C. Huang, C.-Y. Zhang and F.-Y. Wu, Sens. Actuators, B, 2016, 226, 30–36 CrossRef CAS; (f) Y. Ma, T. Leng, Y. Qu, C. Wang, Y. Shen and W. Zhu, Tetrahedron, 2017, 73, 14–20 CrossRef CAS.
- (a) F. L. Capitán-Vallvey, E. Arroyo-Guerrero, D. M. Fernández-Ramos and F. Santoyo-González, Microchim. Acta, 2005, 151, 93–100 CrossRef; (b) IUPAC. Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”). Compiled by A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford (1997). XML on-line corrected version: http://goldbook.iupac.org (2006) created by M. Nic, J. Jirat, B. Kosata; updates compiled by A. Jenkins. ISBN 0-9678550-9-8.
- X. Wu, L. Xu, L. Liu, W. Ma, H. Yin, H. Kuang, L. Wang, C. Xu and N. A. Kotov, J. Am. Chem. Soc., 2013, 135, 18629–18636 CrossRef CAS PubMed.
- E. Garcia-España, M.-J. Ballester, F. Lloret, J. M. Moratal, J. Faus and A. Bianchi, J. Chem. Soc., Dalton Trans., 1988, 101–104 RSC.
- (a) G. Gran, Analyst, 1952, 77, 661–671 RSC; (b) F. J. C. Rossotti and H. Rossotti, J. Chem. Educ., 1965, 42, 375 CrossRef CAS.
- L. Alderighi, P. Gans, A. Ienco, D. Peters, A. Sabatini and A. Vacca, Coord. Chem. Rev., 1999, 184, 311–318 CrossRef CAS.
- (a) MassLynx Software, version 4.0 Search PubMed; (b) S. Moco, R. J. Bino, O. Vorst, H. A. Verhoeven, J. de Groot, T. A. van Beek, J. Vervoort and C. H. R. de Vos, Plant Physiol., 2006, 141, 1205–1218 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1481178–1481182. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt04756d |
| This journal is © The Royal Society of Chemistry 2017 |