
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMononuclear ruthenium compounds bearing N-donor and N-heterocyclic carbene ligands: structure and oxidative catalysis†
Hai-Jie
Liu
a,
Marcos
Gil-Sepulcre
a,
Laia
Francàs
b,
Pau
Nolis
 c,
Teodor
Parella
c,
Jordi
Benet-Buchholz
b,
Xavier
Fontrodona
d,
Jordi
García-Antón
a,
Nuria
Romero
a,
Antoni
Llobet
ab,
Lluís
Escriche
*a,
Roger
Bofill
*a and
Xavier
Sala
*a
c,
Teodor
Parella
c,
Jordi
Benet-Buchholz
b,
Xavier
Fontrodona
d,
Jordi
García-Antón
a,
Nuria
Romero
a,
Antoni
Llobet
ab,
Lluís
Escriche
*a,
Roger
Bofill
*a and
Xavier
Sala
*a
aDepartament de Química, Facultat de Ciències, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Barcelona, Catalonia, Spain. E-mail: roger.bofill@uab.cat; lluis.escriche@uab.cat; xavier.sala@uab.cat; Fax: +34 93 581 24 77
bInstitute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans 16, 43007 Tarragona, Catalonia, Spain
cServei de Ressonància Magnètica Nuclear, Facultat de Ciències, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Barcelona, Catalonia, Spain
dServeis Tècnics de Recerca, Edifici P-II, Campus Montilivi, Universitat de Girona, 17071 Girona, Catalonia, Spain
First published on 25th January 2017
Abstract
A new CNNC carbene-phthalazine tetradentate ligand has been synthesised, which in the reaction with [Ru(T)Cl3] (T = trpy, tpm, bpea; trpy = 2,2′;6′,2′′-terpyridine; tpm = tris(pyrazol-1-yl)methane; bpea = N,N-bis(pyridin-2-ylmethyl)ethanamine) in MeOH or iPrOH undergoes a C–N bond scission due to the nucleophilic attack of a solvent molecule, with the subsequent formation of the mononuclear complexes cis-[Ru(PhthaPz-OR)(trpy)X]n+, [Ru(PhthaPz-OMe)(tpm)X]n+ and trans,fac-[Ru(PhthaPz-OMe)(bpea)X]n+ (X = Cl, n = 1; X = H2O, n = 2; PhthaPz-OR = 1-(4-alkoxyphthalazin-1-yl)-3-methyl-1H-imidazol-3-ium), named 1a+/2a2+ (R = Me), 1b+/2b2+ (R = iPr), 3+/42+ and 5+/62+, respectively. Interestingly, regulation of the stability regions of different Ru oxidation states is obtained by different ligand combinations, going from 62+, where Ru(III) is clearly stable and mono-electronic transfers are favoured, to 2a2+/2b2+, where Ru(III) is almost unstable with regard to its disproportionation. The catalytic performance of the Ru–OH2 complexes in chemical water oxidation at pH 1.0 points to poor stability (ligand oxidation), with subsequent evolution of CO2 together with O2, especially for 42+ and 62+. In electrochemically driven water oxidation, the highest TOF values are obtained for 2a2+ at pH 1.0. In alkene epoxidation, complexes favouring bi-electronic transfer processes show better performances and selectivities than those favouring mono-electronic transfers, while alkenes containing electron-donor groups show better performances than those bearing electron-withdrawing groups. Finally, when cis-β-methylstyrene is employed as the substrate, no cis/trans isomerization takes place, thus indicating the existence of a stereospecific process.
Introduction
N-Heterocyclic carbenes (NHCs) are neutral compounds featuring a divalent C atom that possesses six electrons in its valence shell and is inserted into an N-heterocycle. NHCs are excellent ligands for transition metal ions (M), forming rather strong M–C bonds and often stable complexes under ambient conditions.1 Transition metal complexes containing NHCs have found multiple applications in important catalytic transformations, such as hydrogenation, transfer hydrogenation, water reduction and water oxidation.2When designing catalysts for redox processes, controlling the oxidative power, the accessibility and stability of the oxidation states involved in the catalytic cycle is of paramount importance for the selectivity of the catalysed reaction. In general, in the presence of electron-donating ligands (such as carbenes) high oxidation states of the central metal ion will be stabilised, and hence its redox potentials decrease,3 thus facilitating oxidative catalytic processes. Additionally, when a water molecule is directly coordinated to the metal centre, the redox properties of the complex will be affected by proton exchange. The successive 1e− oxidations taking place are accompanied by a sequential loss of protons favoured by the enhanced acidity of the bonded aqua ligand. This phenomenon, known as proton coupled electron transfer (PCET), allows transition metals to achieve high oxidation states quite easily, since the successive loss of protons – going from the aqua to the hydroxo and finally oxo ligand – allows the maintenance of the total charge of the complex.4 In addition, the σ and π donation of the oxo ligand present at high oxidation states further stabilises high oxidation states at the metal centre. Thus, promising examples of water oxidation catalysis have been reported within the last 6 years with Ir5 and Ru6 NHC complexes, most of which are monometallic, although a few are multimetallic. Interestingly, during the past few years researchers have emphasised the distinctive and sometimes superior performance of bimetallic catalysts because of the possible cooperative interactions existing between both M–OH2 active sites thanks to their relative disposition imposed by the bridging ligand.7
Furthermore, Ru NHC complexes have also found relevant applications in alkene epoxidation catalysis.8 A remarkable example is the use of Ru–aqua complexes with an increasing number of NHC units that stabilise the Ru(IV)/Ru(III) redox potential to a much higher extent than the Ru(III)/Ru(II) one, thus favouring the disproportionation of the Ru(III) oxidation state. As a consequence, the Ru(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species becomes a powerful two-electron oxidant. This is interesting because it avoids radical reaction pathways associated with 1 electron oxidation processes,9 and becomes particularly attractive for the olefin epoxidation reactions, since it will favour a concerted pathway that will generate a stereoselective product.8
O species becomes a powerful two-electron oxidant. This is interesting because it avoids radical reaction pathways associated with 1 electron oxidation processes,9 and becomes particularly attractive for the olefin epoxidation reactions, since it will favour a concerted pathway that will generate a stereoselective product.8
Within this context, and given the feasible preparation of thermodynamically stable NHCs and the interest in using them as ligands in oxidative catalytic systems, we have synthesised and characterised a new tetradentate imidazolium precursor ligand (1,4-bis(1-methylimidazolium-1-yl)phthalazine; H2L12+) and evaluated its effect on the electrochemical properties and oxidative catalytic activity of the corresponding Ru complexes obtained in combination with an additional auxiliary tri-N-dentate ligand such as the meridional ligand trpy, the facial ligand tpm and either the meridional or facial ligand bpea (trpy = 2,2′:6′,2′′-terpyridine, tpm = tris(pyrazol-1-yl)methane, bpea = N,N-bis(pyridin-2-ylmethyl)ethanamine) (Chart 1). However, this new ligand H2L12+ loses a carbene moiety upon reacting with Ru(III) precursors under reflux in MeOH or iPrOH, generating two new imidazolium precursor ligands 1-(4-alkoxyphthalazin-1-yl)-3-methyl-1H-imidazol-3-ium (PhthaPz-OR; R = Me, iPr), named, respectively, Me-HL2+ and iPr-HL2+ (see Chart 1). As a consequence, we have obtained the mononuclear complexes cis-[Ru(Me-L2)(trpy)X]n+, cis-[Ru(iPr-L2)(trpy)X]n+, [Ru(Me-L2)(tpm)X]n+ and trans,fac-[Ru(Me-L2)(bpea)X]n+ (X = Cl, n = 1; X = H2O, n = 2), named, respectively, 1a+/2a2+, 1b+/2b2+, 3+/42+ and 5+/62+, which show interesting redox properties when employed in water oxidation and alkene epoxidation catalysis.
 | ||
| Chart 1 Drawing of the imidazolium precursor ligands (H2L12+, Me-HL2+, iPr-HL2+) and the auxiliary tri-N-dentate ligands (tpm, bpea and trpy) used in this work. | ||
Results and discussion
Synthesis of the ligand H2L12+
H2L1(Cl)2 and H2L1(PF6)2 were obtained following a one-step nucleophilic attack of 1-methylimidazole to 1,4-dichlorophthalazine (dcp) in DMF (Scheme 1). The insolubility of H2L1(Cl)2 in DMF allowed the easy isolation of the ligand by simple filtration and subsequent washing with diethyl ether (yield 70%). Subsequent treatment of H2L1(Cl)2 with a NH4PF6 saturated solution in MeOH allowed the exchange of the chloride by the PF6− counterion (H2L1(PF6)2). | ||
| Scheme 1 Synthetic procedure for the synthesis of H2L1(Cl)2 and H2L1(PF6)2. | ||
Characterization of the ligand H2L12+
NMR spectroscopy for L12+ has been carried out both in acetone-d6 (H2L1(PF6)2) and methanol-d4 (H2L1(Cl)2). Both 1D (1H, 13C{1H}) and 2D (HSQC and HMBC) experiments were necessary to characterise the structure of the ligand in solution (Fig. 1 and S1 in the ESI†). All resonances could be unambiguously assigned based on their integrals, multiplicity and the C2v symmetry of the ligand in solution. For H2L1(PF6)2, both H9 and H10 (or H9′ and H10′) display a doublet of doublets with a mirror effect, which is in agreement with the typical AA′BB′ (9 9′10 10′ in our case) pattern of this kind of systems,10 as shown in the inset of Fig. 1. The singlet appearing at very low fields in acetone-d6 (Fig. 1a) can be assigned to the imidazolic protons 6 and 6′ in accordance with the high electron-withdrawing effect of the two heteroatoms present in the α position, as previously reported for similar ligands.11 However, the integral of this resonance at 9.9 ppm sharply decreases (up to only 5% of the expected value) when the 1H NMR spectrum of H2L1(Cl)2 is recorded in methanol-d4 (Fig. 1b), showing the fast exchange rate of these acidic protons with a protic solvent. | ||
| Fig. 1 600 MHz 1H NMR spectrum of H2L1(PF6)2 in acetone-d6 (a) and of H2L1(Cl)2 in MeOD (b). Inset: Zoomed image of the aromatic region of H2L1(PF6)2. Note the sharp decrease in the intensity of the acidic protons H6 when recorded in MeOD due to fast exchange with the solvent. | ||
Suitable crystals for X-ray diffraction analysis were obtained by slow diffusion of diethyl ether into a solution of H2L1(PF6)2 in acetone (Fig. 2). It is worth mentioning that the steric congestion of both five membered rings with the central phthalazine moiety (especially protons H6′–H9′ and H4–H9, at 2.40–2.45 Å) place the three scaffolds in different planes, with the left-side imidazole ring 42.5° below the phthalazine plane and the right-side imidazole ring 44.3° above (Fig. 2). The ORTEP plot for the cationic moiety of H2L12+ and the acquisition and crystallographic data for H2L1(PF6)2 can be found in Fig. S2 and Table S1 in the ESI,† respectively.
 | ||
| Fig. 2 ORTEP plot of the crystal structure of H2L12+. The hydrogen atoms at closer distances have been labelled, and the angles between the plane of two imidazoles and the phthalazine scaffold are included. Color code: nitrogen, violet; carbon, black; hydrogen, blue. | ||
Reaction of H2L12+ with [Ru(T)Cl3] (T = trpy, tpm, bpea). Breakage of ligand H2L12+ and synthesis of complexes 1a+/2a2+, 1b+/2b2+, 3+/42+ and 5+/62+
Following synthetic strategies previously reported by our group7b,10,12 2 molar equivalents of [RuIII(T)Cl3] (T = trpy, tpm, bpea) were mixed with H2L12+, triethylamine (Et3N) as the reducing agent and LiCl to ensure the presence of a labile site in the generated complexes, and refluxed in MeOH for 16 h. After hot filtration, addition of a few drops of a saturated aqueous solution of NH4PF6 to the crude solution and partial solvent evaporation under vacuum, a brown precipitate appeared in all cases. However, despite the expected bimetallic species with the general formula [RuII2(T)2(μ-Cl)(μ-L1)]3+ or [RuII2(T)2(Cl)2(μ-L1)]2+, when the obtained compounds were subjected to 1H NMR analysis, their integrals matched those of a mononuclear Ru complex (see, for example, the 1H NMR spectrum shown in Fig. S3 in the ESI† for the mononuclear compound obtained after reflux of [RuIII(trpy)Cl3] with H2L1(Cl)2 in MeOH, where the sum of the integrals of the aromatic protons is a multiple of 17 instead of the expected number of 30 for a dinuclear complex).As a consequence, although H2L12+ shows excellent stability in air and also dissolves in acetone or methanol at room temperature, it decomposes when refluxed overnight in methanol, thus pointing to the replacement of one imidazole ring of H2L12+ by a methoxy group due to the nucleophilic attack of the solvent (Scheme S1 in the ESI† and Chart 1). Similar phenomena have already been reported by other authors when using related tetradentate CNNC or tridentate CNN ligands under similar conditions.13 Then, isopropanol, with increased steric hindrance compared to methanol, was also tested as the solvent for the coordination of H2L12+ to Ru. However, the same process occurred, with decomposition of the tetradentate ligand and formation of a mononuclear complex (Scheme 2). As a result, the new ligands PhthaPz-OMe (Me-HL2+) and PhthaPz-OiPr (iPr-HL2+) have been obtained from H2L12+ (Chart 1 and Scheme S1†), which can only act as CN bidentate ligands towards Ru. The breakage of H2L12+ can also be explained from an electronic point of view, since when H2L12+ coordinates to the first electrophilic Ru ion, there is a flow of electron-density from the ligand to the metal centre and, therefore, the nucleophilic attack of a MeOH or iPrOH solvent molecule becomes still more favourable.
 | ||
| Scheme 2 Synthetic procedures used for the synthesis of 1a+/2a2+, 1b+/2b2+, 3+/42+ and 5+/62+. Note the breakage of H2L12+ when refluxed in MeOH or iPrOH. | ||
As a consequence, due to the breakage of H2L12+ under the conditions used, we adjusted the [RuIII(T)Cl3]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2L12+ molar ratio to 1.5:1 in order to maximise the yield of formation of the Ru mononuclear species. Therefore, complexes 1a+ (cis-[RuII(Me-L2)(trpy)Cl]PF6), 1b+ (cis-[RuII(iPr-L2)(trpy)Cl]PF6), 3+ ([RuII(Me-L2)(tpm)Cl]PF6) and 5+ (trans,fac-[RuII(Me-L2)(bpea)Cl]PF6) were obtained in good yields. The subsequent synthesis of the corresponding aqua complexes involved the presence of AgBF4 in acetone/H2O, which promotes the decoordination of the chlorido ligand by formation of an AgCl precipitate and allows the coordination of a water molecule. After AgCl filtration, acetone was slowly evaporated under vacuum. The counter ion could be easily exchanged from BF4− to PF6− by adding excess NH4PF6(aq) into the aqueous solution, obtaining the whole set of Ru–aqua complexes [Ru(R-HL2)(T)(H2O)](PF6)2 (R = Me, T = trpy, 2a2+; R = iPr, T = trpy, 2b2+; R = Me, T = tpm, 42+; R = Me, T = bpea, 62+) as red (or brown) precipitates in yields ranging from 35 to 68% (Scheme 2).
:H2L12+ molar ratio to 1.5:1 in order to maximise the yield of formation of the Ru mononuclear species. Therefore, complexes 1a+ (cis-[RuII(Me-L2)(trpy)Cl]PF6), 1b+ (cis-[RuII(iPr-L2)(trpy)Cl]PF6), 3+ ([RuII(Me-L2)(tpm)Cl]PF6) and 5+ (trans,fac-[RuII(Me-L2)(bpea)Cl]PF6) were obtained in good yields. The subsequent synthesis of the corresponding aqua complexes involved the presence of AgBF4 in acetone/H2O, which promotes the decoordination of the chlorido ligand by formation of an AgCl precipitate and allows the coordination of a water molecule. After AgCl filtration, acetone was slowly evaporated under vacuum. The counter ion could be easily exchanged from BF4− to PF6− by adding excess NH4PF6(aq) into the aqueous solution, obtaining the whole set of Ru–aqua complexes [Ru(R-HL2)(T)(H2O)](PF6)2 (R = Me, T = trpy, 2a2+; R = iPr, T = trpy, 2b2+; R = Me, T = tpm, 42+; R = Me, T = bpea, 62+) as red (or brown) precipitates in yields ranging from 35 to 68% (Scheme 2).
Structural characterisation of complexes 1a+/2a2+, 1b+/2b2+, 3+/42+ and 5+/62+
All mononuclear complexes have been characterised by spectroscopic (1D and 2D NMR) and spectrometric (ESI-MS) techniques and by elemental analysis (EA).In the 1H NMR spectrum of 1a+ (Fig. 3), the loss of the “ABBA” spin–spin coupling pattern perfectly agrees with the reduced symmetry of H2L12+ after nucleophilic decomposition. Furthermore, the two singlets integrating three protons each at 4.78 and 3.47 ppm can be assigned to the methyl group of the intact imidazole ring and the methyl group of the new methoxy substituent formed, respectively. Additional 13C NMR and 2D-NMR spectra allowed full assignment of all resonances (see Fig. S4 in the ESI†).
 | ||
| Fig. 3 600 MHz 1H NMR spectrum of 1a+ in CD2Cl2 and its corresponding proton assignment. | ||
As expected, a similar 1H NMR spectrum to 1a+ was obtained for 1b+. However, now the singlet at 3.47 ppm assigned to the methoxy substituent in 1a+ is replaced by a doublet and a septuplet (at 1.09 and 4.54 ppm, integrating six and one protons, respectively) due to the presence of the isopropoxy substituent (Fig. S5a in the ESI†). Furthermore, the integrity and purity of 1a+ and 1b+ were confirmed by EA and ESI-MS (Fig. S8a and b in the ESI†).
The chlorido compounds 1a+ and 1b+ display Cs symmetry in solution, with the symmetry plane passing through the PhthaPz-OMe (1a+) and PhthaPz-OiPr (1b+) ligand, the Ru centre, the chlorido ligand and carbons C(27) (1a+) or C(28) (1b+) of the trpy ligand, interconverting the two sides of the molecule. Thus, with respect to the relative position of the chlorido ligand in relation to the Ru carbene bond, both the cis and trans isomers could be formed either for 1a+ or 1b+. However, only one isomer was obtained in the reaction crude for both 1a+ and 1b+, as determined by 1H NMR (Fig. 3 and S5a†). 2D ROESY NMR spectra were then obtained to identify the cis or trans nature of the obtained compounds. As shown in Fig. 4, in the case of 1b+ (see Fig. S5e† for the ROESY NMR spectra of the aromatic region of 1b+) strong interactions were observed between the isopropyl group and H24, H27 and H28 of the trpy ligand as well as between the methyl group of the imidazole ring and H21 of the trpy ligand, which clearly allow the identification of the cis disposition of the 1b+ complex. The same conclusion could be extracted from the ROESY NMR spectra of 1a+ (Fig. S4e†), and therefore the only obtained isomer is also cis in nature. Again, additional 13C NMR and 2D-NMR spectra allowed full assignment of all resonances of 1b+ (Fig. S5 in the ESI†).
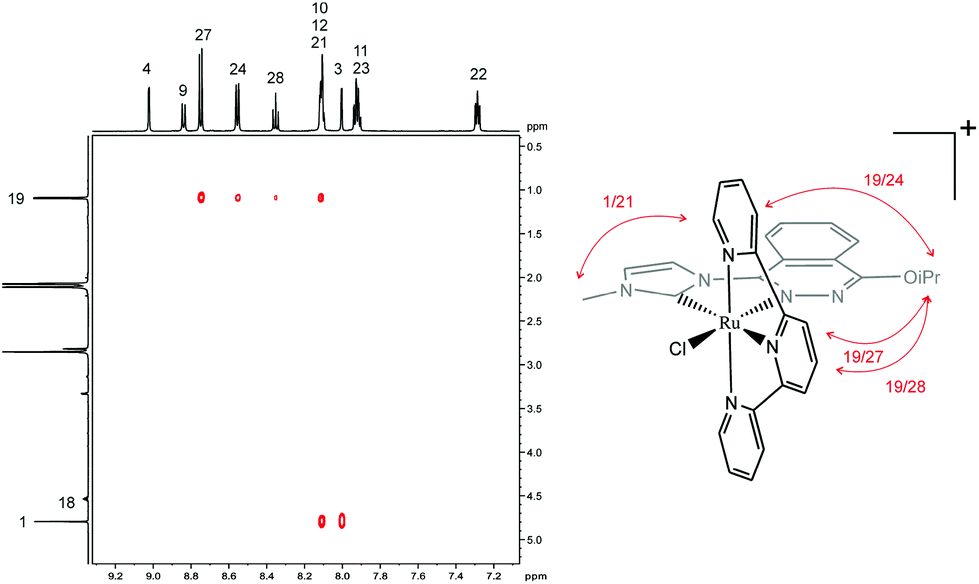 | ||
| Fig. 4 Expanded area of 2D ROESY NMR spectrum of 1b+ in acetone-d6 and schematic drawing of the observed interactions. | ||
With regard to 3+, due to the C3 symmetry of the tpm ligand and its characteristic facial coordination mode, no isomeric mixtures are expected. This has been corroborated by its 1H NMR spectrum (Fig. S6a†). The lack of symmetry of 3+ (C1 group) converts the whole set of protons in different resonances, and a complex spectrum is obtained. The assignment of each resonance to a single proton and carbon was carried out by 2D NMR experiments (HSQC, HMBC, ROESY and TOCSY), while the integrity and purity of 3+ were confirmed by EA and ESI-MS (see Fig. S6 and S8c, respectively, in the ESI†).
Concerning 5+, due to the flexibility of the tridentate bpea ligand, able to potentially coordinate the Ru metal ion either facially or meridionally,14 seven diastereomers could be potentially formed when combining bpea with the non-symmetric bidentate CN ligand Me-L2 (Fig. 5).15 The notation fac and mer refers to the facial or meridional disposition of the bpea ligand, respectively, whereas up and down indicate the relative orientation of the ethyl group of bpea with regard to the chlorido ligand upon coordination. In the fac complexes, the cis/trans notation refers to the position of the chlorido ligand with respect to the aliphatic N atom of the bpea ligand, while in the mer cases the cis/trans notation refers to the position of the chlorido ligand with respect to the carbene atom of Me-L2. Both steric and electronic interactions between the ligands coordinated to the Ru metal ion play a key role in the formation of the synthetically obtainable isomeric mixture. However, in the synthesis of 5+, only the trans,fac isomer is formed (see below). Hydrogen bonding interactions between the protons α to the pyridylic nitrogens of bpea and the chlorido ligand dramatically stabilise the trans,fac conformation, lowering the energy of the system. This strong stabilisation of the trans,fac isomer has already been reported and thoroughly studied by means of theoretical DFT calculations for similar Ru-based systems,16 and the predominance of these hydrogen-bonding interactions over other factors for stabilising and selectively obtaining the trans,fac isomer in a series of related complexes has already been established by several research groups.17 Furthermore, the preference of bpea for the facial coordination upon heating (thermodynamic conditions) has also been reported.14
 | ||
| Fig. 5 Possible diastereomers for 5+. The Me-L2 ligand is represented as a CN connector for the sake of clarity. | ||
Effectively, the trans,fac nature of the 5+ complex was confirmed by selective NOESY NMR experiments, whose key interactions unambiguously revealed its stereoisomeric nature (Fig. S7e in the ESI†). Thus, interactions between H1 and H20–H21 and between H18 and H34 are observed, confirming its trans,fac configuration. As a consequence, analogously to what happened with 3+, no symmetry is observed in its 1H NMR spectrum (Fig. 6). Finally, the assignment of each resonance to a single proton and carbon was carried out by 2D NMR experiments (HSQC, ROESY), while the integrity and purity of 5+ were confirmed by EA and ESI-MS (see Fig. S7 and S8d, respectively, in the ESI†).
 | ||
| Fig. 6 600 MHz 1H NMR spectrum of 5+ in acetone-d6 and its corresponding proton assignment. | ||
Suitable crystals for X-ray diffraction analysis of 5+ were obtained by slow diffusion of diethyl ether into a solution of the complex in methanol (Fig. 7), and a selection of the more relevant bond distances and angles is reported in Table S2.† An ORTEP plot for the cationic moiety of this complex as well as that corresponding to its unit cell can be found in Fig. S9 of the ESI.† Thus, 5+ crystallises in a small unit cell containing one PF6− anion and one independent complex molecule. Additionally, a complete description of the acquisition and crystallographic data can be found in Table S3 of the ESI.†
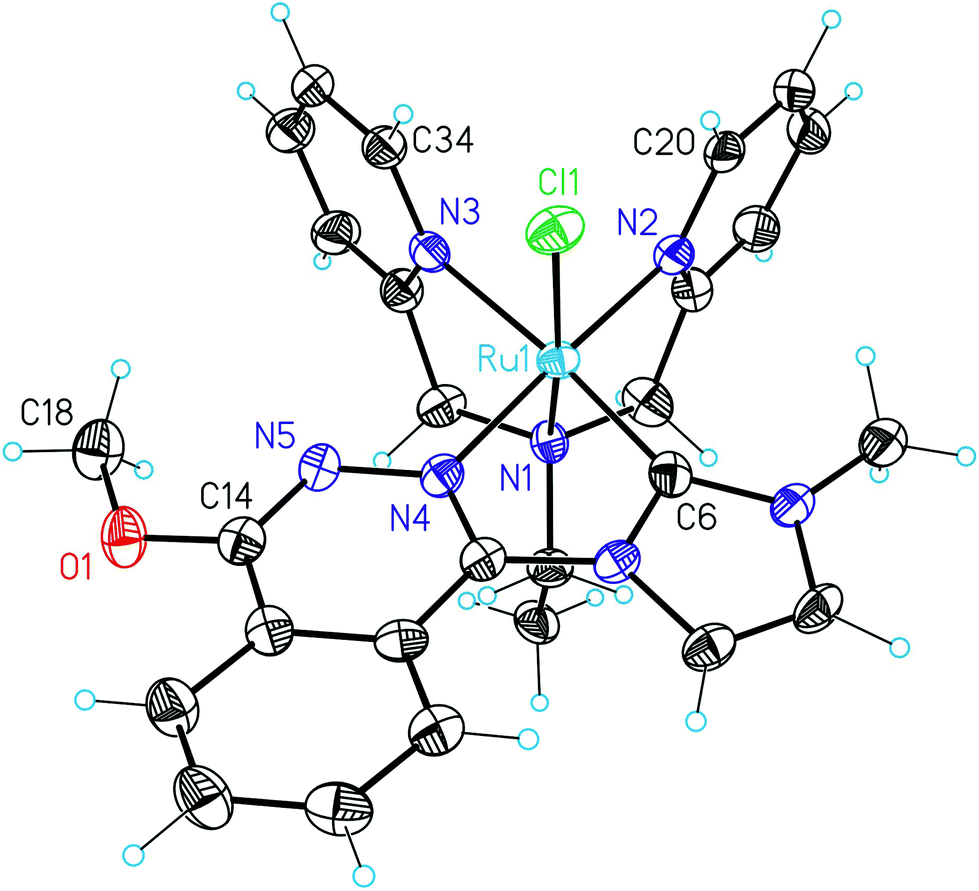 | ||
| Fig. 7 ORTEP plot of the crystal structure of the cationic part of 5+. Color code: ruthenium, blue; nitrogen, violet; oxygen, red; chlorine, green; carbon, black; hydrogen, blue. Atoms appearing in Table S2† or throughout the text have been labelled accordingly. | ||
The Ru(II) ion adopts a distorted octahedral geometry with bond distances and angles that resemble those of analogous complexes reported in the previous literature.15b,18 The Ru carbene bond distance (1.962 ± 0.004 Å) is shorter than the Ru–N bonds, which range between 2.0 and 2.1 Å. The N1–Ru–Cl (171.63° ± 0.10°), N2–Ru–Cl (94.92° ± 0.12°) and N3–Ru–Cl (90.90° ± 0.11°) bond angles clearly confirm the facial coordination of bpea to Ru. In addition, the Ru–Cl bond appears trans to the aliphatic N atom of bpea, confirming again the trans,fac nature of 5+. Furthermore, the imidazole and the phthalazine rings do not lie exactly on the same plane. Instead, there is a torsion angle of 10.6° ± 0.7°. However, this angle is obviously shorter with regard to the one observed for the free ligand, which is around 43° (Fig. 2). The methoxy group is nearly on the same plane of the phthalazine skeleton, since the observed torsion angle C18–O–C14–N5 is only 1.9° ± 0.6°. Finally, the N1–Ru–N3 and N1–Ru–N2 angles are, respectively, 81.15° ± 0.14° and 81.68° ± 0.15°, away from the 90° angle for an ideal octahedral geometry, due to the formation of two five-membered rings when bpea coordinates to the central Ru ion. In addition, clear hydrogen-bonding interactions are observed between the pyridyl protons of bpea on C20 and C34 and the chlorido ligand (2.68–2.71 Å). This electronic interaction is responsible for the strong stabilisation of the trans,fac configuration of 5+, as stated before.16,17
Replacement of the chlorido ligand by a water molecule in this family of complexes induces significant chemical shift displacements. This is exemplified by the 1a+/2a2+ 1H NMR comparison shown in Fig. S10,† where mainly protons close to these monodentate ligands such as H22, H26 and H27 are affected. Similar displacements of the chemical shifts were obviously observed for the very similar 1b+/2b2+ couple, and both complexes maintain their cis conformation after the coordination of the aqua ligand (see Fig. S11 and S12,† respectively, for a full NMR assignment of all proton and carbon resonances of 2a2+ and 2b2+).
Complexes 42+ and 62+ also maintain their original conformation in solution after chloride displacement, as can be deduced from the NMR spectra shown in Fig. S13 and S14 in the ESI,† respectively. Furthermore, the integrity and purity of all four aqua complexes were confirmed by EA and ESI-MS (Fig. S15 in the ESI†).
Electrochemical and spectrophotometric characterisation of complexes 1a+/2a2+, 1b+/2b2+, 3+/42+ and 5+/62+
CV and DPV techniques have been used to determine the electrochemical properties of all complexes. The CVs of complexes 1a+, 1b+, 3+ and 5+ in dichloromethane are depicted in Fig. S16 in the ESI.† All chlorido complexes exhibit a single reversible wave corresponding to the RuIII/RuII process. The redox potentials vs. SCE are very close for 1a+ (0.79 V) and 1b+ (0.78 V) given the high structural and chemical similarity of Ru in both meridional complexes, while a clear downshift of E1/2 is observed for the facial derivatives 3+ (0.71 V) and 5+ (0.68 V). This is in agreement with the higher σ-donating and lower π-acceptor capacity of both the pyrazolyl rings (3+) and the aliphatic N (5+) with regard to the pyridyl units of the trpy scaffold. The observed decrease in the redox potentials lies within a 70–110 mV range and is in agreement with previous results obtained for analogous Ru carbene complexes containing trpy or bpea.15bThe redox behaviour of the four Ru–OH2 complexes has been extensively investigated in aqueous media and their redox potentials and pKa values are summarised in Table 1, together with those of related aqua complexes containing the bpy ligand instead of the carbene bidentate scaffold for the sake of comparison.
| Entry | E III/II1/2 | E IV/III1/2 | E III/II1/2 | ΔE1/2c |
E
IV/II1/2
|
E V/IV1/2 | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|
| pH 1a | pH 7b | pKa1 | pKa2 | |||||||
| a 0.1 M triflic acid. b Phosphate buffer solution (μ = 0.1 M). c ΔE1/2 = (EIV/III1/2 − EIII/II1/2). d Calculated as (EIV/III1/2 + EIII/II1/2)/2. e This work. | ||||||||||
| 1 | 2a 2+ | 0.74 | 0.52 | 0.49 | 0.03 | 0.50 | 1.29 | 3.0 | 11.5 | |
| 2 | 2b 2+ | 0.73 | 0.51 | 0.48 | 0.03 | 0.49 | — | 2.8 | 11.0 | |
| 3 | 4 2+ | 0.62 | — | 0.35 | — | — | 1.33 | 1.8 | 11.2 | |
| 4 | 6 2+ | 0.61 | 0.52 | 0.32 | 0.20 | 0.42 | 1.28 | 1.2 | 11.7 | |
| 5 | [Ru(trpy)(bpy)(OH2)]2+ | 0.81 | 0.62 | 0.49 | 0.13 | 0.55 | — | 1.7 | 9.7 | 19 |
| 6 | [Ru(tpm)(bpy)(OH2)]2+ | 0.70 | 0.71 | 0.40 | 0.31 | 0.55 | — | 1.9 | 10.8 | 20 |
| 7 | [Ru(bpea)(bpy)(OH2)]2+ | 0.70 | 0.46 | 0.34 | 0.12 | 0.40 | — | 1.2 | 11.1 | 21 |
At pH 1, a single reversible wave corresponding to the RuIII–OH2/RuII–OH2 process is observed for all aqua complexes (black lines in Fig. S17, S19, S21 and S22 in the ESI†), in which again a cathodic shift of E1/2 (110–130 mV) takes place when introducing the facial ligands (entries 3 and 4 vs. entries 1 and 2, Table 1), following the same trend observed for the Ru–aqua complexes bearing bpy instead of the bidentate carbene ligand (entries 5–7, Table 1).
At neutral pH, two very close redox processes separated by only 30 mV can be observed for 2a2+ and 2b2+, corresponding to the RuIV–O/RuIII–OH and RuIII–OH/RuII–OH2 processes (red lines in Fig. S17 and S19†), thus making the stability region of the Ru(III) species very small (ΔE1/2 = 30 mV, Table 1). The decrease in the stability region of Ru(III) when introducing carbene ligands into Ru polypyridilic complexes has already been described,8,15b which can be confirmed in our case when comparing with the ΔE1/2 value for [Ru(trpy)(bpy)(OH2)]2+ (130 mV, Table 1). This tendency, however, can be reversed when replacing the trpy ligand in 2a2+ and 2b2+ by the facial aliphatic ligand bpea (Fig. S22†), since the higher σ-donating and lower π-acceptor capacity of bpea provoke a stabilisation of the Ru(III) state21 (lowering the EIII/II1/2 potential by 160–170 mV while keeping EIV/III1/2 unaltered, entries 1, 2 and 4, Table 1). Consequently, ΔE1/2 is 200 mV for 62+. Unfortunately, for the tpm derivative 42+ the RuIV–O/RuIII–OH process could not be detected (Fig. S21†). The absence of the Ru(IV/III) redox couple in CV experiments is quite common for aqua complexes and is due to slow heterogeneous electron-transfer kinetics from the solution to the electrode surface.22 Finally, the effect of the higher σ-donating character of the carbene ligand compared to bpy is evidenced when comparing the EIII/II1/2 values of 42+ and [Ru(tpm)(bpy)(OH2)]2+ (cathodic shift of 50 mV, entries 3 and 6, Table 1). The simultaneous removal of protons and electrons (PCET processes) taking place for the four aqua-complexes can be observed in their Pourbaix diagrams (Fig. 8 and S20†), which allows the measurement of their pKa1 (RuIII–OH2) and pKa2 (RuII–OH2) values. Thus, the aqua groups of 62+ (bpea) and 42+ (tpm) for the Ru(III) state are more acidic than those corresponding to their meridional (trpy) counterparts (pKa1 values of 1.2 and 1.8 vs. 3.0–2.8, Table 1), while no significant differences are observed among the pKa2 values. Finally, higher acidities are observed for their non-carbene analogues (lower pKa1 and especially pKa2 values, entries 5–7, Table 1), given the lower σ-donating character of bpy compared to the carbene bidentate ligand.
 | ||
| Fig. 8 Plot of E1/2vs. pH (Pourbaix diagram) for complexes 2a2+ (a), 42+ (b) and 62+ (c). The pH/potential regions of stability for the various oxidation states and their dominant proton compositions are indicated by using abbreviations such as RuII–OH2, for example, for [RuII(Me-L2)(OH2)(T)]2+ (T = trpy, tpm, bpea). The vertical lines in the various E/pH regions show the pKa values. | ||
Also, in order to confirm the correspondence of all observed redox waves to mono-electronic electrochemical processes, bulk electrolysis experiments were carried out at pH 4.9 for the aqua complexes (Fig. S18 in the ESI†). Thus, for 2a2+ at 0.75 V vs. SCE (just after the predicted potential of the second redox wave) a value of 2.06 electrons per complex molecule was obtained (Fig. S18a†), while for 42+ at 0.6 V (after the potential of the unique redox wave was observed) a value of 0.97 electrons per molecule was obtained (Fig. S18b†). Finally, the stability of the RuIII–OH species and the stepwise mono-electronic nature of both RuIII/II and RuIV/III processes have been confirmed for 62+, since after applying a potential of 0.57 V vs. SCE (just after the expected potential of the first redox process), a value of 0.91 electrons per molecule was obtained (Fig. S18c†), while when the potential was set at 0.75 V, 1.87 electrons were transferred per molecule (Fig. S18d†). In summary, from an electronic point of view all aqua complexes favour mono-electronic transfers between Ru(II), Ru(III) and Ru(IV). However, for 2a+ and 2b+ their tendency for a bielectronic RuIV/II process is very similar to the one electron transfer process RuIV/III, whereas in all the other cases the 1 electron transfer process is clearly favoured, as evidenced when comparing their respective EIV/II1/2 and EIV/III1/2 values (Table 1).
The UV-vis spectra of the eight complexes described in this work have been recorded in methanol and are displayed in Fig. S23 in the ESI.† Two regions can be observed in all cases: one region between 260 nm and 350 nm (or 325 nm for 5+/62+) with very intense bands due to intra ligand π–π* transitions, and a second one between 350 nm (or 325 nm for 5+/62+) and 550 nm, where typical broad unsymmetrical metal-to-ligand charge transfer (MLCT) bands appear, which could be tentatively assigned to Ru(dπ)–N ligand(π*) transitions.19,21 Also, the electronic nature of the monodentate ligand influences to some extent the energies of the transitions involving Ru d orbitals. Thus, the MLCT bands for the Ru–aqua complexes are blue-shifted with regard to those of their Ru–Cl counterparts due to the relative stabilisation of the Ru(dπ) levels provoked by the almost non-π-donor character of the aqua ligand.
Electrochemical and chemical water oxidation by complexes 2a2+, 2b2+, 42+ and 62+
The capacity of the aqua complexes to oxidise water into dioxygen was initially tested electrochemically. For this purpose, the CVs of 2a2+, 42+ and 62+ were recorded in aqueous solution at pH 1.0 until redox potentials were high enough to reach the oxidation states potentially able to oxidise water. Accordingly, a large electrocatalytic wave above 1.4 V vs. SCE corresponding to the oxidation of water to dioxygen was observed in all cases (see below).In order to obtain kinetic information about the catalytic process, a “foot of the wave analysis” (FOWA)23 was carried out to calculate the apparent rate constant kobs. For this purpose we followed the equations adopted for water oxidation recently reported by some of us.24 Thus, under catalytic conditions, eqn (1) is operative:
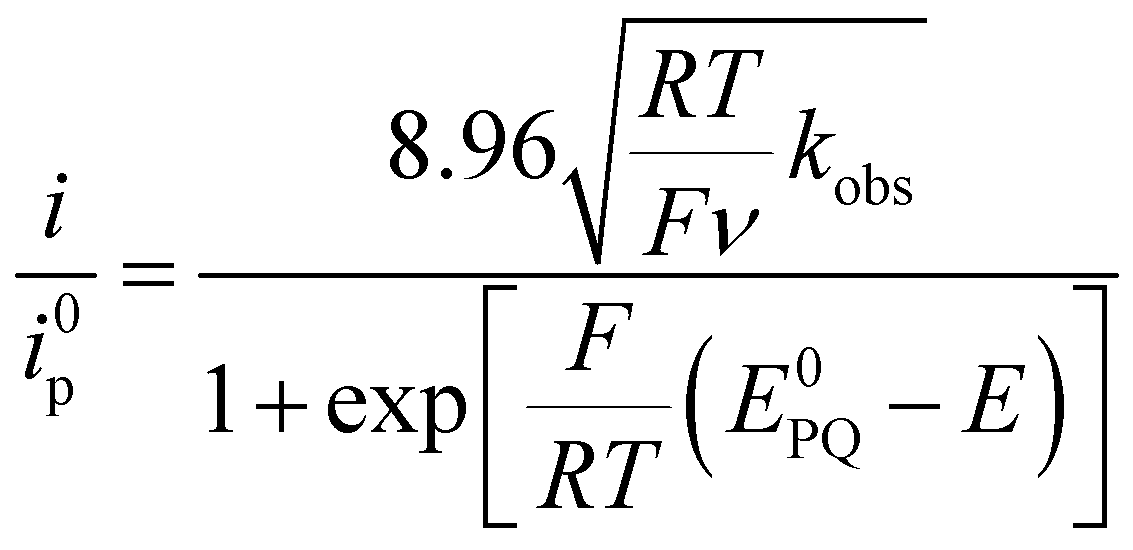 | (1) |
 | ||
| Fig. 9 Background corrected CV of a 0.68 mM solution of 42+ in 0.1 M aqueous triflic acid (pH 1.0) at 100 mV s−1 scan rate (a), and “foot of the wave analysis” of 42+ by plotting i/i0pvs. 1/{1 + exp[(F/RT)(E0PQ − E)]} (b). Inset: Plot of different kobs values extracted from the “foot of the wave analysis” at each concentration (the dotted line represents the trend of the kobs values). | ||
In all cases, the largest slope at the very beginning of the catalytic process (which translates to the foot of the wave in the original CVs) gives the value of kobs, which is independent of the catalyst concentration, indicating the existence of a water nucleophilic attack (WNA) mechanism.25 Moreover, under the used electrocatalytic scheme, kobs is equivalent to the maximum turnover frequency (TOFmax) that a catalyst molecule can operate the water oxidation reaction when the applied potential tends to infinite.23 At pH 1.0, the obtained kobs values (expressed in s−1) follow the trend 2a2+ (0.570) > 62+ (0.051) > 42+ (0.015).
Finally, the relationship between the turnover frequency TOF and the overpotential (η), defined as the difference between the applied potential E and the thermodynamic potential of the catalysed reaction E0AC, in this case water oxidation, is governed by eqn (2), whose logarithms for all three aqua compounds at pH 1.0 are plotted in Fig. 10 (catalytic Tafel plots).
 | (2) |
 | ||
| Fig. 10 Catalytic Tafel plots for 2a2+ (green), 42+ (red) and 62+ (blue) at pH 1.0. | ||
Fig. 10 shows how the higher value of E0PQ for 42+ (1.33 V, Table 1) translates in lower turnover frequencies when η is low (red line before reaching the plateau, when η makes the TOF reach its maximum and equals kobs). Also, the higher performance of 2a2+ (green line) is evident, in concordance with the higher kobs values deduced by the “foot of the wave analysis”. However, it should be noted that the kinetic parameters for catalytic reactions derived from electrochemical measurements depend on various details of the experimental procedures, and therefore values from different studies should be compared carefully.26
The four aqua complexes were also tested as chemically triggered water oxidation catalysts in the presence of Ce(IV) as the sacrificial oxidant. The total gas evolved was manometrically measured (Fig. S26 in the ESI†) and its composition in terms of the O2:CO2 ratio was analysed by means of on-line mass spectrometry (Fig. S27†). In the presence of 100 equivalents of Ce(IV) at pH 1, 42+ generated more gas (≈ 15 mBar) after 30 min of reaction than the other three complexes (Fig. S26†). In general, only considering the amount of generated gas, facial complexes are superior to their meridional counterparts. However, when the composition of the generated gases is analysed by on-line MS (Fig. S27†), 42+ has the lowest O2:CO2 ratio (1:5.5), followed by 62+, with a 1:1.4 ratio, while the O2:CO2 ratio is much higher for 2a2+ and 2b2+ (1:0.6). Therefore, despite being poor, the stability of the meridional trpy-based complexes 2a2+/2b2+ is clearly higher than that of their facial (tpm or bpea) counterparts 42+/62+, which easily get oxidised under the harsh reaction conditions of chemical water oxidation by Ce(IV) at pH 1.0. This is clearly reflected in Fig. S28,† where the profile of O2 evolution of the four aqua complexes has been compared. Therefore, taking into account the volume of the vial (16.04 mL) and the amount of catalyst used (2.0 μmol), the turnover numbers (TN) at 298 K for 2b2+ and 2a2+ (2.39 and 2.17, respectively) are higher than those of 62+ (1.63) and 42+ (0.75). Moreover, this behaviour is consistent with the results obtained during the electrochemically triggered water oxidation at pH 1.0, with the highest TOF value corresponding to the trpy derivatives (2a2+/2b2+) and the lowest one to the tpm complex (42+).
Catalyst–catalyst intermolecular oxidative degradation involving RuIVO species27 or the direct degradation of the complexes by the highly oxidant Ce(IV) species are considered as the potential origin of the evolved CO2. In our system, the only relevant differences between the four evaluated complexes are the tridentate ligands employed. Therefore, tpm and bpea (both containing aliphatic carbon atoms prone to be easily oxidised under the harsh catalytic conditions employed) quickly decompose, generating large amounts of CO2 that arise from ligand oxidation. Given that a great number of robust water oxidation catalysts containing the trpy ligand have been reported,28 the observed evolution of CO2 from 2a2+/2b2+ clearly reflects a relative weakness of the PhthaPz-OR family of ligands under oxidative conditions.
Alkene epoxidation by complexes 2a2+, 2b2+, 42+ and 62+
Complexes 2a2+, 2b2+, 42+ and 62+ have been tested with regard to their ability to chemically oxidise alkenes. The catalytic reactions have been carried out using a catalyst:substrate:oxidant:water ratio of 1:1000:2000:2000 after a 120 min mixing period of the catalysts in the absence of the substrate (see the Experimental section for further details), during which the excess of water ensures the generation of the oxidant PhIO species from PhI(OAc)2.12,29 This mixing period before substrate addition is crucial in order to improve the rate of the catalytic reaction. Scheme S2† summarises the set of reactions that take place during the catalytic epoxidation of alkenes for the proposed systems. All products of each catalytic experiment have been identified by GC-MS, and all gathered results are shown in Table 2. For instance, the system: 1.7 mM of 2a2+, 1.7 M of cis-β-methylstyrene, 3.4 M of PhI(OAc)2, and 3.4 M of H2O in 1,2-dichloroethane (DCE, entry 2) gives 1.42 M of cis-β-methylstyrene oxide in 525 minutes, which represents a TN value of 840 and a TOF value of 1.6 min−1, and since the conversion of the initial substrate is complete the selectivity in the epoxide formation is 84%.
| Cat. | Entry | Alkene | Conv.b (%) | Selec.c (%) | TN/TOFd |
|---|---|---|---|---|---|
|
a Catalyst:substrate:oxidant:water ratio of 1:1000:2000:2000. See the Experimental section for further details.
b Substrate conversion = {[substrate]initial − [substrate]final}/[substrate]initial × 100.
c Epoxide selectivity = [epoxide]final/{[substrate]initial − [substrate]final} × 100.
d TN is the turnover number with regard to the total epoxide obtained. TOF is the turnover frequency expressed in epoxide cycles per minute (TN min−1).
e
cis-Epoxide.
f DCE volume is 5 mL.
|
|||||
| 2a 2+ | 1 | Styrene | 42 | 46 | 194/0.8 |
| 2 | cis-β-Methylstyrene | >99 | 84e | 840/1.6 | |
| 3 | trans-Stilbenef | >99 | 68 | 680/1.3 | |
| 4 | Cyclooctene | >99 | 93 | 930/1.9 | |
| 2b 2+ | 5 | Styrene | 29 | 66 | 191/1.1 |
| 6 | cis-β-Methylstyrene | >99 | 82e | 816/1.3 | |
| 7 | trans-Stilbenef | >99 | 60 | 596/1.1 | |
| 8 | Cyclooctene | 99 | 96 | 946/2.2 | |
| 4 2+ | 9 | Styrene | 23 | 26 | 60/0.5 |
| 10 | cis-β-Methylstyrene | 97 | 56e | 545/0.4 | |
| 11 | trans-Stilbenef | 90 | 16 | 148/0.3 | |
| 12 | Cyclooctene | >99 | 76 | 756/0.3 | |
| 6 2+ | 13 | Styrene | 21 | 13 | 27/0.1 |
| 14 | cis-β-Methylstyrene | 99 | 69e | 687/0.7 | |
| 15 | trans-Stilbenef | 91 | 15 | 136/0.2 | |
| 16 | Cyclooctene | >99 | 94 | 940/0.4 | |
Similar figures are obtained for both trpy-based aqua-complexes (2a2+/2b2+) on the one hand and for both facial derivatives (42+/62+) on the other. Also, when comparing both sets of catalyst pairs, a clearly higher epoxidation capacity (higher conversion and selectivity) is observed for 2a2+/2b2+ compared to 42+/62+. For example, styrene 2a2+ yields a 42% conversion (entry 1), while 42+ and 62+ only reach 23 and 21% conversion, respectively (entries 9 and 13), and selectivity for 2b2+ is 66% (entry 5) while it is only 26% and 13% for 42+ and 62+, respectively. Also, for cis-β-methylstyrene selectivities above 80% are obtained for 2a2+ and 2b2+ (entries 2 and 6), while for 42+ and 62+ they are below 60% and 70%, respectively (entries 10 and 14), and for trans-stilbene complete conversion and selectivities above 60% are obtained for 2a2+ and 2b2+ (entries 3 and 7), while for 42+ and 62+ conversion is around 90% and selectivity is close to 15% (entries 11 and 15). This behaviour can be rationalised on the basis of the electronic nature of the two pairs of complexes. Thus, while for 2a2+/2b2+ bi-electronic transfers between the Ru(II) and Ru(IV) species are thermodynamically almost as favourable as the mono-electronic processes (Ru(III) stability region is minimal with regard to its disproportionation, Fig. 8a and S20†), for 42+/62+ clearly mono-electronic processes take place (Fig. 8b and c). It is well known that catalysts favouring bi-electronic processes drive epoxidation reactions to concerted pathways and mono-electronic ones drive them to radical mechanisms, the latter usually ending up reducing the selectivity of the whole process by the generation of a wide set of by-products (Scheme S3†).8,15b,30 Therefore, the existence of bi-electronic processes for 2a2+/2b2+ could explain the higher selectivity observed with regard to their mono-electronic counterparts 42+ and 62+. Also, together with these electronic arguments, other conceivable reasons for the reduced epoxidation capacity of 42+/62+ may arise due to the chemical nature of their facial ligands, since tpm and bpea are prone to be oxidised under oxidative conditions (they possess aliphatic C atoms), and their steric bulkiness may also hinder the interaction between the substrates and the catalyst active site. Interestingly, different results have been obtained with related Ru–N5C complexes containing the same auxiliary trpy or bpea ligands but the smaller NHC ligand N-methyl-N′-2-pyridylimidazolium, where the bpea-containing complex yields higher selectivity in front of styrene and higher conversion efficiency and selectivity towards trans-stilbene than its corresponding trpy-complex.15b Therefore, these results demonstrate again the dramatic influence of the electronic and steric properties of the carbene ligand on the catalytic performance of the Ru complexes.
Table 2 also shows that the studied aqua complexes perform much better with substrates containing electron-donor groups than with those bearing electron-withdrawing substituents, indicating the strong electrophilic character of the RuIVO group in all cases. Therefore, the best results are gathered for cyclooctene (entries 4, 8, 12 and 16) whereas the poorest values are obtained for styrene (entries 1, 5, 9 and 13) and trans-stilbene (entries 3, 7, 11 and 15), the latter also suffering from potential steric effects due to the bulkiness of its two phenyl rings.
Finally, another interesting feature observed is the stereospecific nature of the catalytic epoxidation process. For the whole set of aqua complexes when cis-β-methylstyrene is employed as the substrate no cis/trans isomerisation takes place. Therefore, for 42+/62+ ring closure must be faster than C–C rotation for the radical intermediates proposed to be formed (Scheme S3,† top), while for 2a2+/2b2+ the stereospecificity could be explained on the basis of the proposed concerted bi-electronic oxene insertion into the double bond (Scheme S3,† bottom).
Conclusions
A new tetradentate imidazolium precursor ligand has been synthesised and fully characterised by NMR and X-ray diffraction analysis. This ligand decomposes in nucleophilic solvents at high temperatures due to C–N bond cleavage, generating a bidentate NHC-phthalazine scaffold (R-L2) during the synthesis of the corresponding four Ru chlorido and aqua complexes [Ru(R-L2)(T)X]n+ (X = Cl, n = 1, X = H2O, n = 2; R = Me, iPr; T = trpy, tpm, bpea), which have been fully characterised electronically and spectroscopically.Modulation of the thermodynamic stability in aqueous media of the Ru(III) oxidation state has been observed for the four aqua compounds. Thus, while for 42+/62+ (T = tpm/bpea) the Ru(III) state is clearly stable at moderately high potentials and they increase their oxidation state from Ru(II) through mono-electronic processes (ΔE1/2 = 200 mV for the latter), for the trpy-based complexes 2a2+/2b2+ the Ru(III) state is almost unstable with regard to its disproportionation (ΔE1/2 = 30 mV). This divergence in the electronic behaviour has direct implications in the epoxidation capacity of alkenes with PhI(OAc)2, since the higher conversion and selectivity observed for 2a2+/2b2+ can be rationalised on the basis of the existence of bi-electronic transfers that avoid the generation of radical intermediates of high energy that could reduce the selectivity of the whole process. Additionally, the absence of cis/trans isomerisation in all cases – therefore leading to stereospecific epoxidation processes – may be explained on the basis of either a concerted bi-electronic process (2a2+/2b2+) or a radical mechanism in which the ring closure is much faster than C–C rotation (42+/62+).
We have also shown that the four aqua complexes are moderately unstable during catalytic water oxidation triggered by Ce(IV) addition due to ligand oxidation under the harsh conditions employed, especially those containing aliphatic carbon atoms (42+/62+). Also, under electrochemically triggered conditions 2a2+ is the fastest catalyst at pH 1.0.
In conclusion, in this work we have evidenced that it is possible to modulate the electronic and catalytic properties of Ru NHC complexes by using different auxiliary meridional or facial N-tridentate ligands.
Experimental section
Materials and instrumentation
All reagents used in the present work were obtained from Sigma Aldrich Chemical Co. and were used without further purification. Reagent-grade organic solvents were obtained from Scharlab. RuCl3·3H2O was supplied by Alfa Aesar. The starting ligands tri(1H-pyrazol-1-yl)methane (tpm) and N,N-bis(pyridin-2-ylmethyl)ethanamine (bpea) were prepared as described in the literature.31,32 The synthetic manipulations were routinely performed under a nitrogen atmosphere using a Schlenk flask and vacuum-line techniques.UV-vis spectroscopy was carried out with a HP8453 spectrometer using 1 cm quartz cells. NMR spectroscopy was performed on a Bruker DPX 250 MHz, DPX 360 MHz, Avance-II 400 MHz, DPX 500 MHz or a Avance-II 600 MHz spectrometer. Samples were run in MeOD, DCM-d2 or acetone-d6 with internal references. Elemental analyses were performed using a Carlo Erba CHMS EA-1108 instrument from the Chemical Analysis Service of the Universitat Autònoma de Barcelona (SAQ-UAB). Electrospray ionisation Mass Spectrometry (ESI-MS) experiments were performed on an HP298s gas chromatography (GC-MS) system from the SAQ-UAB. Cyclic voltammetry and differential pulse voltammetry experiments were performed on a Bio Logic Science Instrument SP-150 potentiostat using a three-electrode cell. A glassy carbon electrode (7 mm diameter) was employed as the working electrode while a platinum wire as the auxiliary electrode and a SCE as the reference electrode. Working electrodes were polished with 0.05 micron alumina paste and washed with distilled water and acetone before each measurement. The complexes were dissolved in acetonitrile, methanol or dichloromethane solutions of 0.1 M ionic strength containing the necessary amount of n-Bu4NPF6 (TABH) as the supporting electrolyte. For electrochemical analysis performed in water, the complexes were dissolved in pH 1 triflic acid solution or solutions of phosphate buffer for other pH values, with a 0.1 M ionic strength. The pH values were increased or reduced by adding drops of 0.1 M NaOH solution or the pH 1 triflic acid solution. E1/2 values here presented were estimated from CV experiments from the average of the oxidative and reductive peak potentials (Ep,a + Ep,c)/2. For the epoxidation catalytic studies, experiments were performed as follows. First, mixing for a period of 120 min was carried out by adding in a vial 1 mL of 1,2-dichloroethane (DCE) as the solvent, 1.60 g (5.0 mmol) of (diacetoxyiodo)benzene (PhI(OAc)2) as the oxidant, 1 mmol of 1,1′-biphenyl as the internal standard, 2.5 × 10−3 mmol of catalyst (2a2+ to 62+) and 90 μL (5.0 mmol) of water. This mixing period before substrate addition was observed to be important in order to improve the rate of the catalytic reaction. Then, the substrate (2.5 mmol) was added to the previous mixture, achieving a final volume of approx. 1.47 mL and the corresponding initial concentrations: catalyst, 1.7 mM; substrate, 1.7 M; biphenyl, 0.68 M; PhI(OAc)2, 3.4 M; water, 3.4 M. These concentrations correspond to a catalyst:substrate:oxidant:water ratio of 1:1000:2000:2000. Aliquots were taken every 5, 10, 15, 20, 25 or 30 min until completion of the reaction. Each aliquot was filtered through a Pasteur pipette filled with Celite; after that diethyl ether was added in order to elute the organic compounds and the filtrate was analysed in an HP 5890 PACKARD SERIES II Gas Chromatograph (GC) coupled to a mass selective detector with ionisation by electronic impact. The characterisation of the reaction products was carried out by comparison with commercial products or by GC-MS spectrometry. GC conditions: initial temperature 40 °C for 10 min, ramp rate variable for each substrate (typically from 10 °C min−1 to 20 °C min−1), final temperature 250 °C, injection temperature 220 °C, detector temperature 250 °C. Yield of epoxide and substrate conversion were calculated with regard to the initial concentration of the substrate.

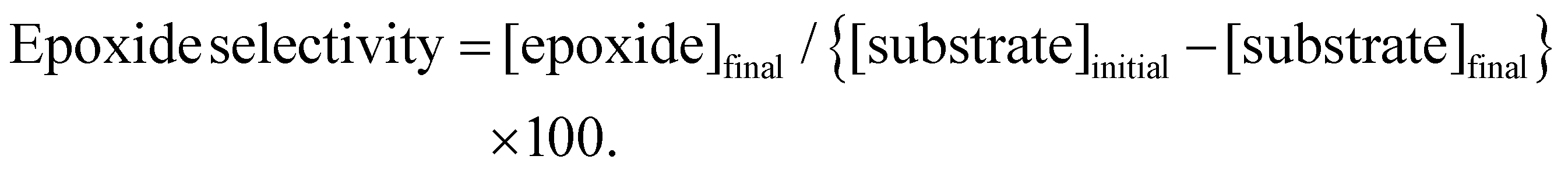
On-line manometry measurements were performed on a Testo 521 differential pressure manometer with an operating range of 1 to 100 hPa and a measurement accuracy of 0.5%, coupled to thermostatted reaction vessels for dynamic monitoring of the headspace pressure above each reaction. On-line monitoring of the gas evolution was carried out on a Pfeiffer Omnistar GSD 301C mass spectrometer. Typically, a degassed vial of 16.04 mL containing 1.5 mL of a 1.33 mM solution of the catalysts in 0.1 M triflic acid was connected to a capillary tubing apparatus. Subsequently, 0.5 mL of an Ar degassed solution of 400 mM (NH4)2CeIV(NO3)6 in 0.1 M triflic acid (100 equiv.) were injected by using a Hamilton gastight syringe, and the reaction was dynamically monitored at 25 °C. A response ratio of 1:2 was observed when equal concentrations of dioxygen and carbon dioxide were injected, which was used for the calculation of their relative concentrations.
X-ray crystal structure determination
Crystals of H2L12+ were grown by slow diffusion of diethyl ether into a solution of H2L1(PF6)2 in acetone. Crystals of 5+ were prepared by slow diffusion of diethyl ether into a solution of 5+ in methanol.Structure solution and refinement were performed using SHELXTL. The crystal data parameters of H2L12+ and 5+ are listed in Tables S1 and S3.† The structures of H2L12+ and 5+ were analysed using the programs ORTEP and Mercury. All information related to the structures can be found in the deposited CIF-files.
Synthetic preparations
988), 313 (14247), 413 (4700), 475 (4332). ESI-MS (MeOH): m/z = 610.1 ([M − PF6]). Elemental analysis (% found): C, 44.58; H, 3.10; N, 12.95. Calcd for C28H23ClF6N7OPRu: C, 44.54; H, 3.07; N, 12.99.
315), 314 (14616), 413 (5036), 479 (3889). ESI-MS (MeOH): m/z = 638.1 ([M − PF6]). Elemental analysis (% found): C, 46.07; H, 3.52; N, 12.49. Calcd for C30H27ClF6N7OPRu: C, 46.01; H, 3.48; N, 12.52.
:water = 1:3, 40 mL). AgBF4 (109 mg, 0.56 mmol) was added into the solution, which was then refluxed at 90 °C for 4 hours. After cooling to room temperature, the reaction crude was filtered through Celite® to remove the black solid formed. The red-brown solution was concentrated under vacuum until about 20 mL was left, followed by centrifugation (10000 rpm, 10 min) to remove the potential colloidal silver still remaining. To the clear red solution 20 drops of saturated aqueous NH4PF6 solution were added and the precipitate formed was filtered off, washed with diethylether and dried under vacuum. Yield: 91 mg (65%). 1H-NMR (600 MHz, acetone-d6, 298 K) δ 9.01 (d, 1H, J4–3 = 2.4 Hz, H4), 8.80 (d, 2H, J26–27 = 7.4 Hz, H26), 8.78 (d, J9–10 = 8.7 Hz, H9), 8.62 (d, 2H, J23–22 = 8.0 Hz, H23), 8.44 (t, 2H, J27–26,26′ = 8.1 Hz, H27), 8.19 (d, 2H, J20–21 = 5.0 Hz, H20), 8.12 (t, 1H, J10–9,11 = 8.7 Hz, H10), 8.04–8.00 (m, 4H, H12, H3, H22), 7.91 (t, 1H, J11–10,12 = 7.5 Hz, H11), 7.37 (m, 2H, H21), 4.56 (s, 3H, H1), 3.46 (s, 3H, H18). 13C-NMR (151 MHz, acetone-d6, 298 K) δ 200.62 (C6), 159.47 (C24), 158.01 (C14), 157.82 (C20), 156.41 (C25), 153.08 (C7), 138.22 (C22), 137.65 (C27), 134.20 (C10), 132.83 (C11), 127.53 (C21), 126.29 (C3), 124.05 (C12), 123.85 (C23), 122.38 (C26), 121.12 (C9), 120.90 (C8), 119.57 (C14), 119.21 (C3), 54.43 (C18), 36.55 (C1). UV/vis (methanol): λmax, nm (ε, M−1 cm−1) = 275 (12189), 309 (13040), 388 (4338), 467 (4474). ESI-MS (MeOH): m/z = 594.1 ([M − 2PF6 + 1]). Elemental analysis (% found): C, 38.14; H, 2.89; N, 11.06. Calcd for C28H25F12N7O2P2Ru: C, 38.11; H, 2.86; N, 11.11.
:3). AgBF4 (109 mg, 0.56 mmol) was then added to the solution, which was then refluxed at 90 °C for 4 hours. After cooling to room temperature, the reaction crude was filtered through Celite® to remove the silver chloride formed. The brown filtrate was then concentrated in a rotary evaporator until about 20 mL, followed by centrifugation (10000 rpm, 10 min) to remove the remaining solids. To the clear red solution 20 drops of a saturated aqueous NH4PF6 solution were added. The brown precipitate formed was filtered off, washed with diethyl ether and dried under vacuum. Yield: 91 mg (65%). 1H-NMR (600 MHz, acetone-d6, 298 K) δ 9.03 (d, 1H, J4–3 = 2.4 Hz, H4), 8.84 (d, 2H, J27–28 = 6.7 Hz, H27), 8.81 (d, 1H, J9–10 = 8.7 Hz, H9), 8.63 (d, 2H, J24–23 = 8.0 Hz, H24), 8.50 (t, 1H, J28–27,27′ = 8.1 Hz, H28), 8.23 (dd, 2H, J21–22,23 = 10.5, 5.6 Hz, H21), 8.13 (t, 1H, J10–9,11 = 8.5 Hz, H10), 8.10 (d, 1H, J12–11 = 8.7 Hz, H12), 8.06–8.01 (m, 3H, H3, H23), 7.93 (t, 1H, J11–10,12 = 7.6 Hz, H11), 7.38 (ddd, 1H, J22–21,23,24 = 7.0, 5.6, 1.2 Hz, H22), 4.58 (s, 3H, H1), 4.49 (dt, 1H, J18–19,19′ = 12.3, 6.2 Hz, H18), 1.07 (d, 6H, J19–18 = 6.2 Hz, H19). 13C-NMR (151 MHz, acetone-d6, 298 K) δ 200.66 (C6), 159.47 (C25), 157.96 (C21), 157.31 (C14), 156.32 (C26), 152.73 (C7), 138.23 (C23), 137.53 (C28), 134.05 (C10), 132.74 (C11), 127.63 (C22), 126.16 (C3), 124.24 (C12), 123.77 (C24), 122.47 (C27), 121.15 (C8), 121.07 (C9), 119.80 (C13), 119.62 (C4), 70.98 (C18), 36.39 (C1), 20.90 (C19). UV/vis (methanol): λmax, nm (ε, M−1 cm−1) = 280 (12006), 311 (14895), 392 (4700), 463 (4220). ESI-MS (MeOH): m/z = 622.1 ([M − 2PF6 + 1]). Elemental analysis (% found): C, 39.63; H, 3.24; N, 10.74. Calcd for C30H29F12N7O2P2Ru: C, 39.57; H, 3.21; N, 10.77.
:3). AgBF4 (109 mg, 0.56 mmol) was added into the solution that was then refluxed at 90 °C for 4 hours. After cooling to room temperature, the reaction crude was filtered through Celite® to remove the silver chloride formed. The brown filtrate was then concentrated in a rotary evaporator until about 20 mL, followed by centrifugation (10000 rpm, 10 min) in order to remove the remaining solids. To the clear red solution 20 drops of saturated aqueous NH4PF6 solution were added. The red precipitate formed was filtered off, washed with diethyl ether and dried under vacuum. Yield: 76 mg (55%). 1H-NMR (600 MHz, acetone-d6, 298 K) δ 9.90 (s, 1H, H24), 8.99 (d, 1H, J4–3 = 2.4 Hz, H4), 8.97 (d, 1H, J9–10 = 8.5 Hz, H9), 8.83 (d, 1H, J20–21 = 1.7 Hz, H20), 8.72 (d, 1H, J31–32 = 2.9 Hz, H31), 8.67 (d, 1H, J22–21 = 2.7 Hz, H22), 8.58 (d, 1H, J33–32 = 2.0 Hz, H33), 8.53 (d, 1H, J26–27 = 5.5 Hz, H26), 8.47 (d, 1H, J12–11 = 8.1 Hz, H12), 8.29 (dd, 1H, J10–9,11 = 8.2, 7.7 Hz, H10), 8.17 (t, 1H, J11–10,12 = 7.7 Hz, H11), 7.74 (d, 1H, J3–4 = 2.3 Hz, H3), 6.85 (m, J21–20,22 = 2.4 Hz, J28–27 = 2.2 Hz, H21, H28), 6.80 (t, 1H, J32–31,33 = 2.5 Hz, H32), 6.34 (t, 1H, J27–26,28 = 2.5 Hz, H27), 4.20 (s, 3H, H18), 3.74 (s, 1H, H1). 13C-NMR (151 MHz, acetone-d6, 298 K) δ 200.22 (C6), 158.58 (C14), 152.76 (C7), 148.70 (C33), 148.04 (C28), 147.06 (C20), 135.74 (C26), 134.89 (C31), 134.17 (C10), 133.64 (C22), 133.40 (C11), 125.84 (C3), 124.48 (C12), 122.02 (C9), 121.75 (C8), 120.96 (C13), 120.61 (C4), 109.06 (C32), 108.69 (C27), 108.04 (C21), 76.61 (C24), 55.38 (C18), 36.65 (C1). UV/vis (methanol): λmax, nm (ε, M−1 cm−1) = 295 (8297), 392 (5315). ESI-MS (MeOH): m/z = 575.1 ([M − 2PF6 + 1]). Elemental analysis (% found): C, 32.02; H, 2.81; N, 16.19. Calcd for C23H24F12N10O2P2Ru: C, 31.99; H, 2.80; N, 16.22.
:3). AgBF4 (109 mg, 0.56 mmol) was then added into the solution, which was refluxed at 90 °C for 4 hours. After cooling to room temperature, the reaction crude was filtered through Celite® to remove the silver chloride formed. The red-brown solution was concentrated in a rotary evaporator until about 20 mL, followed by centrifugation (10000 rpm, 10 min) to remove the remaining solids. To the clear red solution 20 drops of saturated aqueous NH4PF6 solution were added. The precipitate formed was filtered off, washed with diethyl ether and dried under vacuum. Yield: 96 mg (68%). 1H-NMR (600 MHz, acetone-d6, 298 K) δ 8.99 (d, 1H, J4–3 = 2.4 Hz, H4), 8.96 (d, 1H, J34–33 = 5.3 Hz, H34), 8.93 (m, 2H, H20, H9), 8.36 (d, 1H, J12–11 = 8.1 Hz, H12), 8.23 (t, 1H, J10–9,11 = 7.7 Hz, H10), 8.08 (t, 1H, J11–10,12 = 7.6 Hz, H11), 7.99 (td, 1H, J32–33,31 = 7.8, J32–34 = 1.4 Hz, H32), 7.88 (td, 1H, J22–23,21 = 7.4, J22–20 = 1.7 Hz, H22), 7.72 (d, 1H, J3–4 = 2.4 Hz, H3), 7.67 (d, 1H, J31–32 = 7.9 Hz, H31), 7.57 (m, 1H, H33), 7.55 (d, 1H, J23–22 = 7.9 Hz, H23), 7.50 (t, 1H, J21–22,20 = 6.6 Hz, H21), 4.57–4.40 (m, 4H, H25, H29), 3.71 (s, 3H, H1), 3.65 (s, 3H, H18), 2.40 (m, 1H, J27–27′,28 = 9.2, 5.0 Hz, H27), 2.30 (m, 1H, J27′–27,28 = 9.2, 5.0 Hz, H27′), 0.91 (t, 3H, J28–27,27′ = 7.0 Hz, H28). 13C-NMR (151 MHz, acetone-d6, 298 K) δ 202.85 (C6), 161.20 (C24), 159.56 (C30), 158.81 (C14), 151.85 (C7), 149.37 (C20), 147.67 (C34), 137.42 (C32), 136.72 (C22), 134.06 (C10), 132.67 (C11), 125.82 (C3), 124.41 (C12), 124.30 (C21), 123.78 (C33), 121.63 (C8), 121.54 (C23), 121.49 (C31), 121.35 (C9), 120.47 (C13), 119.89 (C4), 67.89 (C25), 67.29 (C29), 62.80 (C27), 54.19 (C18), 35.89 (C1), 7.97 (C28). UV/vis (methanol): λmax, nm (ε, M−1 cm−1) = 299 (5810), 423 (5753). ESI-MS (MeOH): m/z = 586.1 ([M − 2PF6]). Elemental analysis (% found): C, 37.06; H, 3.60; N, 11.15. Calcd for C27H31F12N7O2P2Ru: C, 36.99; H, 3.56; N, 11.19.
Acknowledgements
Support from MINECO (CTQ2011-26440, CTQ2015-64261-R, CTQ2016-80058-R and CTQ2015-64436-P) is gratefully acknowledged. M. G.-S. is grateful for the award of a PIF doctoral grant from UAB. J. G.-A. acknowledges the Serra Húnter Program. We also thank the Servei de Ressonància Magnètica Nuclear, Universitat Autònoma de Barcelona, for allocating instrument time.Notes and references
- W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162 CrossRef CAS.
- (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorious, Nature, 2014, 510, 485 CrossRef CAS PubMed; (b) S. Kaufhold, L. Petermann, R. Staehle and S. Rau, Coord. Chem. Rev., 2015, 304–305, 73 CrossRef CAS.
- A. Dovletoglou, S. A. Adeyemi and T. J. Meyer, Inorg. Chem., 1996, 35, 4120 CrossRef CAS PubMed.
- (a) X. Sala, S. Maji, R. Bofill, J. García-Antón, L. Escriche and A. Llobet, Acc. Chem. Res., 2014, 47, 504 CrossRef CAS PubMed; (b) M. Dakkach, M. I. López, I. Romero, M. Rodríguez, A. Atlamsan, T. Parella, X. Fontrodona and A. Llobet, Inorg. Chem., 2010, 49, 7072 CrossRef CAS PubMed; (c) T. J. Meyer and M. H. V. Huynh, Inorg. Chem., 2003, 42, 8140 CrossRef CAS PubMed.
- (a) R. Lalrempuia, N. D. McDaniel, H. Müller-Bunz, S. Bernhard and M. Albrecht, Angew. Chem., Int. Ed., 2010, 49, 9765 CrossRef CAS PubMed; (b) A. Petronilho, M. Rahman, J. A. Woods, H. Al-Sayyed, H. Müller-Bunz, J. M. Don MacElroy, S. Bernhard and M. Albrecht, Dalton Trans., 2012, 41, 13074 RSC; (c) A. Petronilho, J. A. Woods, S. Bernhard and M. Albrecht, Eur. J. Inorg. Chem., 2014, 708 CrossRef CAS; (d) Z. Codolà, J. M. S. Cardoso, B. Royo, M. Costas and J. Lloret-Fillol, Chem. – Eur. J., 2013, 19, 7203 CrossRef PubMed; (e) A. Volpe, A. Sartorel, C. Tubaro, L. Meneghini, M. Di Valentin, C. Graiff and M. Bonchio, Eur. J. Inorg. Chem., 2014, 665 CrossRef CAS; (f) I. Corbucci, A. Petronilho, H. Müller-Bunz, L. Rocchigiani, M. Albrecht and A. Macchioni, ACS Catal., 2015, 5, 2714 CrossRef CAS.
- (a) Z. Chen, J. J. Concepcion and T. J. Meyer, Dalton Trans., 2011, 40, 3789 RSC; (b) R. Staehle, L. Tong, L. Wang, L. Duan, A. Fischer, M. S. G. Ahlquist, L. Sun and S. Rau, Inorg. Chem., 2014, 53, 1307 CrossRef CAS PubMed.
- (a) L. Francàs, R. Bofill, J. García-Antón, L. Escriche, X. Sala and A. Llobet, Ru-Based Water Oxidation Catalysts, in Molecular Water Oxidation Catalysts: A Key Topic for New Sustainable Energy Conversion Schemes, ed. A. Llobet, John Wiley & Sons, Ltd, 2014, pp. 29–50 Search PubMed; (b) J. García-Antón, R. Bofill, L. Escriche, A. Llobet and X. Sala, Eur. J. Inorg. Chem., 2012, 4775 CrossRef; (c) N. Planas, G. J. Christian, E. Mas-Marza, X. Sala, X. Fontrodona, F. Maseras and A. Llobet, Chem. – Eur. J., 2010, 16, 7965–7968 CrossRef CAS PubMed; (d) L. Mognon, S. Mandal, C. E. Castillo, J. Fortage, F. Molton, G. Aromí, J. Benet-Buchhlolz, M.-N. Collomb and A. Llobet, Chem. Sci., 2016, 7, 3304 RSC.
- (a) E. Masllorens, M. Rodríguez, I. Romero, A. Roglans, T. Parella, J. Benet-Buchholz, M. Poyatos and A. Llobet, J. Am. Chem. Soc., 2006, 128, 5306 CrossRef CAS PubMed; (b) L. Vaquer, P. Miró, X. Sala, F. Bozoglian, E. Masllorens, J. Benet-Bucholz, X. Fontrodona, T. Parella, I. Romero, A. Roglans, M. Rodríguez, C. Bo and A. Llobet, ChemPlusChem, 2013, 78, 235 CrossRef CAS.
- (a) F. R. Keene, Coord. Chem. Rev., 1999, 187, 121 CrossRef CAS; (b) T. J. Meyer, J. Electrochem. Soc., 1984, 131, 221C CrossRef CAS.
- J. Aguiló, A. Naeimi, R. Bofill, H. Mueller-Bunz, A. Llobet, L. Escriche, X. Sala and M. Albrecht, New J. Chem., 2014, 38, 1980 RSC.
- (a) J. Van Veldhuizen, J. Campbell, R. Giudici and A. Hoveyda, J. Am. Chem. Soc., 2005, 127, 6877 CrossRef CAS PubMed; (b) B. J. Truscott, R. Klein and P. T. Kaye, Tetrahedron Lett., 2010, 51, 5041 CrossRef CAS.
- J. Aguiló, L. Francàs, R. Bofill, M. Gil-Sepulcre, J. García-Antón, A. Poater, A. Llobet, L. Escriche, F. Meyer and X. Sala, Inorg. Chem., 2015, 54, 6782 CrossRef PubMed.
- (a) C. Chen, H. Y. Qiu, W. Z. Chen and D. Q. Wang, J. Organomet. Chem., 2008, 693, 3273 CrossRef CAS; (b) J. S. Ye, X. M. Zhang, W. Z. Chen and S. Shimada, Organometallics, 2008, 27, 4166 CrossRef CAS; (c) X. L. Liu and W. Z. Chen, Organometallics, 2012, 31, 6614 CrossRef CAS; (d) B. Tong, J. Qiang, Q. Mei, H. Wang and Q. Zhang, Inorg. Chem. Commun., 2012, 17, 113 CrossRef CAS.
- J. Mola, I. Romero, M. Rodríguez, F. Bozoglian, A. Poater, M. Solà, T. Parella, J. Benet-Buchholz, X. Fontrodona and A. Llobet, Inorg. Chem., 2007, 46, 10707 CrossRef CAS PubMed.
- (a) M. Dakkach, T. Parella, A. Atlamsani, I. Romero and M. Rodríguez, Adv. Synth. Catal., 2011, 353, 231 CrossRef CAS; (b) M. Dakkach, A. Atlamsani, T. Parella, I. Romero and M. Rodríguez, Inorg. Chem., 2013, 52, 5077 CrossRef CAS PubMed.
- L. Vaquer, J. De Tovar, J. García-Antón, A. Llobet and X. Sala, Inorg. Chem., 2013, 52, 4985 CrossRef CAS PubMed.
- (a) X. Sala, A. Poater, A. Von Zelewsky, T. Parella, X. Fontrodona, I. Romero, M. Solà, M. Rodríguez and A. Llobet, Inorg. Chem., 2008, 47, 8016 CrossRef CAS PubMed; (b) J. S. Ye, X. M. Zhang, W. Z. Chen and S. Shimada, Organometallics, 2008, 27, 4166 CrossRef CAS.
- F. Laurent, E. Plantalech, B. Donnadieu, A. Jiménez, F. Hernández, M. Martínez-Ripoll, M. Biner and A. Llobet, Polyhedron, 1999, 18, 3321 CrossRef CAS.
- K. J. Takeuchi, M. S. Thompson, D. W. Pipes and T. J. Meyer, Inorg. Chem., 1984, 23, 1845 CrossRef CAS.
- A. Llobet, P. Doppelt and T. J. Meyer, Inorg. Chem., 1988, 27, 514 CrossRef CAS.
- M. Rodríguez, I. Romero and A. Llobet, Inorg. Chem., 2001, 40, 4150 CrossRef.
- (a) J. C. Dobson and T. J. Meyer, Inorg. Chem., 1988, 27, 3283 CrossRef CAS; (b) G. E. Cabaniss, A. A. Diamantis, W. R. Murphy, R. W. Linton and T. J. Meyer, J. Am. Chem. Soc., 1985, 107, 1845 CrossRef CAS.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235 CrossRef CAS PubMed.
- R. Matheu, S. Neudeck, F. Meyer, X. Sala and A. Llobet, ChemSusChem, 2016, 9, 3361 CrossRef CAS PubMed.
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, J. Am. Chem. Soc., 2015, 137, 10786 CrossRef CAS PubMed.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983 CrossRef CAS PubMed.
- L. Francàs, X. Sala, E. Escudero-Adan, J. Benet-Buchholz, L. Escriche and A. Llobet, Inorg. Chem., 2011, 50, 2771 CrossRef PubMed.
- J. Aguiló, L. Francàs, H. J. Liu, R. Bofill, J. García-Antón, J. Benet-Buchholz, A. Llobet, L. Escriche and X. Sala, Catal. Sci. Technol., 2014, 4, 190 Search PubMed.
- J.-H. In, S.-E. Park, R. Song and W. Nam, Inorg. Chim. Acta, 2003, 343, 373 CrossRef CAS.
- (a) L. K. Stultz, R. A. Binstead, M. S. Reynolds and T. J. Meyer, J. Am. Chem. Soc., 1995, 117, 2520 CrossRef CAS; (b) W.-H. Fung, W.-Y. Yu and C.-M. Che, J. Org. Chem., 1998, 63, 7715 CrossRef CAS.
- D. L. Reger, T. C. Grattan, K. J. Brown, C. A. Little, J. J. S. Lamba, A. L. Rheingold and R. D. Sommer, J. Organomet. Chem., 2000, 607, 120 CrossRef CAS.
- S. Pal, M. K. Chan and W. H. Armstrong, J. Am. Chem. Soc., 1992, 114, 6398 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic (NMR, UV-Vis), spectrometric (ESI-MS), electrochemical (CV, DPV, bulk electrolysis, Pourbaix diagrams), catalytic (manometries) and structural (X-ray diffraction) data. CCDC 1522633 and 1522634. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt04729g |
| This journal is © The Royal Society of Chemistry 2017 |