
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hard-and-soft phosphinoxide receptors for f-element binding: structure and photophysical properties of europium(III) complexes†
Nataliya E.
Borisova
 *ab,
Anastasia V.
Kharcheva
ac,
Svetlana V.
Patsaeva
c,
Leonid A.
Korotkov
a,
Sergey
Bakaev
a,
Marina D.
Reshetova
a,
Konstantin A.
Lyssenko
b,
Elena V.
Belova
d and
Boris F.
Myasoedov
d
*ab,
Anastasia V.
Kharcheva
ac,
Svetlana V.
Patsaeva
c,
Leonid A.
Korotkov
a,
Sergey
Bakaev
a,
Marina D.
Reshetova
a,
Konstantin A.
Lyssenko
b,
Elena V.
Belova
d and
Boris F.
Myasoedov
d
aDepartment of Chemistry, M.V. Lomonosov Moscow State University 1/3 Leninskie Gory, 119991 Moscow, Russian Federation. E-mail: borisova.nataliya@gmail.com; Fax: +7 495 939 0290
bN.A. Nesmeyanov Institute of Organoelement Compounds, 28 Vavilov Str., Moscow, Russian Federation. E-mail: kostya@xray.ineos.ac.ru
cFaculty of Physics, Lomonosov Moscow State University, 1/2 Leninskie Gory, 119991 Moscow, Russian Federation. E-mail: harcheva.anastasiya@physics.msu.ru
dA.N. Frumkin Institute of Physical Chemistry and Electrochemistry, RAS 31 Leninsky prospect, 119071 Moscow, Russian Federation. E-mail: mar@ipc.rssi.ru
First published on 13th January 2017
Abstract
New phosphinoyl-containing tetradentate heterocycles preorganised for metal ion binding were designed and prepared in high yields. The X-ray structures of two allied phosphinoyl-bearing 2,2′-bipyridyl and phenanthroline ligands, as well as closely related structures of 2,6-bis(diphenylphosphinoyl)pyridine and 9-(diphenylphosphinoyl)-1,10-phenanthroline-2-one, are reported. Complexes of nitrates of several lanthanides and trifluoroacetate of Eu(III) with two phosphinoyl-bearing 2,2′-bipyridyl and phenanthroline ligands were isolated and characterised. The first structures of lanthanide complexes with phosphinoyl-bearing 2,2′-bipyridyl and phenanthroline ligands are reported. The nature of the counter-ion is crucial for the coordination environment of the metal ion. The photophysical properties of the complexes differring in both the nature of the ligand and counter-ion were investigated. The photophysical properties of the complexes are strongly ligand- and counter-ion-dependent. Absorbance and luminescence excitation spectra of complexes showed main peaks in the UV range which correspond to the absorption of light by the ligand and these are ligand-dependent. Luminescence spectra of complexes show typical europium emission in the red region with a high quantum yield, which corresponds to the transitions 5D0 → 7FJ (J = 0–6). The value of deviation of the components of 5D0 → 7F2 and 5D0 → 7F1 transitions from the inversion centre shows a larger dependence on the counter-ion than on the nature of the ligand. The value of the luminescence quantum yield is larger for europium complexes with 2,2′-bipyridyl-based ligands and NO3 counter-ions than for complexes with phenanthroline-based ligands and NO3 counter-ions. A low dependence of the luminescence lifetime of Eu complexes on the nature of the ligand has been demonstrated: values in the solid state were in the range 1.1–2.0 ms.
Introduction
Growing research efforts in the field of rare earth element (REE) complexes with bipyridine-type ligands are associated with their outstanding potential from the viewpoint of their fundamental and practical applications.1–13 Extended variation capabilities of supramolecular structures, as well as modification of peripheral positions of heterocycles, allow fine tuning of the molecular properties required for high technology applications. The high coordination numbers of lanthanide ions provide alternative methods for the modification of the properties of complexes by varying the counter-ion coordinated to the metal centre. This opens several new applications for already known complexes.14,15 So the use of trifluoroacetate complexes provides a path for the preparation of several highly intense luminescent systems.15–17N,O-Polydentate hard-and-soft heterocyclic compounds attract attention as ligands for REE complexes due to their distinguished photophysical5,18–20 and magnetic21 properties and also due to their ability to discriminate ions of f-elements by their size.22–33 Over several years we have designed novel types of N,N′,O,O′-tetradentate bypyridyl-based reagents for the selective separation of actinides from lanthanides9,34–37 – a significant radiochemical problem in the field of closed nuclear fuel cycle development.38 REE complexes with this new type of ligand possess promising photophysical properties which have been studied in detail along with the structure of the complexes.39 In this work we combine hard-donor phosphinoxide groups with a soft heterocyclic framework. We investigate N,N′,O,O′-tetradentate bypyridyl-based phosphinoxide compounds as ligands for lanthanide ions. The ligands bearing phosphinoxide groups together with a pyridine structural motif show intense solvent and counter-ion dependent luminescence.40–42 In such complexes the energy transfer occurs from the ligand to the metal presumably via the pyridine (or other heterocycle) N–Ln coordination bond, but the emitted complexes have never been structurally characterised. There is one example of a structurally characterized coordination polymer of the REE complex, the pyridine-based phosphinoxide ligands, which have no metal ion coordination with the pyridine ring.43,44 The coordination of lanthanide ions with a heterocyclic five-ring moiety simultaneously with the phosphinoxide-group exists in 4,5-bis(diphenyl)phosphoranyl-1,2,3-triazolate-based complexes with REEs.45 Here we report studies on the complexation of the europium ion with two N,N′,O,O′-tetradentate planar 2,2′-bypiridine-type systems both tuned by preorganisation of the ligand. Moreover, investigation of the correlation between the nature of the counter-ion and photophysical properties was performed by the introduction of a nitrate or trifluoroacetate counter-ion.Results and discussion
The preparation of phosphinoxide-decorated 2,2′-bipyridyl starts from 6,6′-dibromo-2,2′-bipyridine which is available mostly by 2,6-dibromopyridine coupling.46 We introduced dichloro-substituted heterocycles as alternative building blocks for the construction of phosphinoxide-containing ligands. Dichloro derivatives are usually readily available for a wide range of heterocycles such as 2,2′-bipyridine47 and o-phenanthroline48,49 and can be easily transformed into corresponding bis(diphenylphosphinoxides) by the reaction with Ph2PNa (Schemes 1 and 2).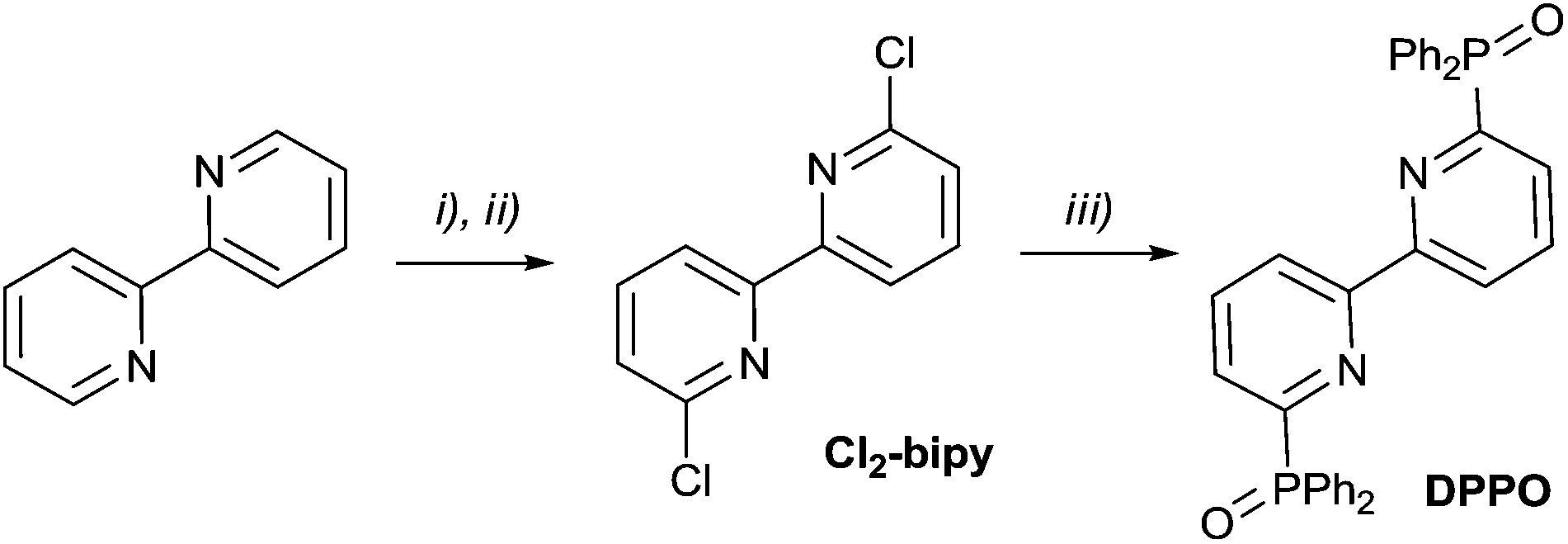 | ||
| Scheme 1 (i) H2O2, AcOH, reflux; (ii) POCl3, PCl5, reflux; then H2O, 65%; (iii) Ph2PNa, dioxane, 0 °C, RT overnight; then H2O2 (10%), 80%. | ||
 | ||
| Scheme 2 (i) PhBr, 115 °C, 4 h, 100%; (ii) air, tBuOK/tBuOH, 75%; (iii) air, EtONa/EtOH 91%; (iv) POCl3, PCl5, reflux; then H2O, 82%; (iii) Ph2PNa, dioxane, −18 °C, RT overnight, then H2O2 (5%), 70%. | ||
Oxidation of the commercially available 2,2′-bipyridyl with in situ generated peracetic acid leads to 2,2′-bipyridine-N,N′-dioxide. The latter was converted into Cl2-bipy using PCl5 in POCl3.47 The reaction of Cl2-bipy with sodium diphenylphosphide under an inert atmosphere and subsequent oxidation of the mixture with hydrogen peroxide provided the target phosphinoxide DPPO in a preparative yield of 52%.

Analytical and spectroscopic characteristics are consistent with the suggested structure. The P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O fragment in both compounds is evidenced by IR spectroscopy (1202 and 1200 cm−1) and 31P NMR. The phosphorus chemical shift (21.3 ppm in DPPO and 31.5 ppm in PnPPO) confirms the presence of the phosphine oxide group. Two doublets for the geminal carbons in the 13C NMR spectrum (156.37–155.06 ppm with 1JP–C(Py) = 132.0 Hz and 132.71–131.67 ppm with 1JP–C(Ph) = 132.0 Hz) are in keeping with the structure of DPPO. The DPPO1H NMR spectrum agrees with the spectrum described earlier.50 The 1H NMR spectrum of DPPO is simplified by protonation. Addition of a drop of strong acid (H2SO4) leads to the separation of most of the signals: the d.d. signal at 8.32 ppm corresponds to 3′-protons of bipyridyl, d.t. at 7.96 ppm is related to the 4′-proton of bipyridyl and the last 5′-proton of heteroaromatics is a part of the multiplet at 7.46 ppm. At the same time, protonation leads to a shift of the phosphorus 31P NMR signal to 21.86 ppm. The 1H NMR of PnPPO also supports the bis(phosphinoxide) structure, but the signals are more separated than those for DPPO. The protons of the common benzene ring are shifted to 7.70 ppm, and the protons at positions 3 and 8 and positions 4 and 7 form two doublets at 8.02 and 7.63 ppm, respectively. Structures are also supported by mass spectroscopy and elemental analysis.
O fragment in both compounds is evidenced by IR spectroscopy (1202 and 1200 cm−1) and 31P NMR. The phosphorus chemical shift (21.3 ppm in DPPO and 31.5 ppm in PnPPO) confirms the presence of the phosphine oxide group. Two doublets for the geminal carbons in the 13C NMR spectrum (156.37–155.06 ppm with 1JP–C(Py) = 132.0 Hz and 132.71–131.67 ppm with 1JP–C(Ph) = 132.0 Hz) are in keeping with the structure of DPPO. The DPPO1H NMR spectrum agrees with the spectrum described earlier.50 The 1H NMR spectrum of DPPO is simplified by protonation. Addition of a drop of strong acid (H2SO4) leads to the separation of most of the signals: the d.d. signal at 8.32 ppm corresponds to 3′-protons of bipyridyl, d.t. at 7.96 ppm is related to the 4′-proton of bipyridyl and the last 5′-proton of heteroaromatics is a part of the multiplet at 7.46 ppm. At the same time, protonation leads to a shift of the phosphorus 31P NMR signal to 21.86 ppm. The 1H NMR of PnPPO also supports the bis(phosphinoxide) structure, but the signals are more separated than those for DPPO. The protons of the common benzene ring are shifted to 7.70 ppm, and the protons at positions 3 and 8 and positions 4 and 7 form two doublets at 8.02 and 7.63 ppm, respectively. Structures are also supported by mass spectroscopy and elemental analysis.
The structures of the phosphinoxide-bearing ligands were studied by X-ray analysis (Fig. 1, Table 1). Structures of phosphinoxide-bearing ligands, DPPO and PnPPO, are shown in Fig. 1. A striking feature of these ligands is the unfavourable conformation for metal ion binding: both diphenylphosphine-groups are unfolded relative to the pyridine ring in anti-conformation. The dihedral angles between the PO group and the pyridine ring are close to 180° for both of the ligands (Table 1).
 | ||
| Fig. 1 ORTEPs of phosphinoxides DPPO and PnPPO with thermal ellipsoids at 50% level for all non-hydrogen atoms. Solvent molecules are omitted for clarity. Other figures are available in the ESI.† | ||
| PyPPO | DPPO | PnPPO | 1 | DPPOGd | PnPPOEu | |
|---|---|---|---|---|---|---|
| OP–CPy–NPy angle |
−173.69(12) | 169.28(14) | 176.5(3) | −176.32(18) | −4.6(6) | −9.6(7) |
| −171.02(12) | 173.8(3) | 28.1(6) | 12.3(7) | |||
| N–CPy–CPy–N angle | — | 180.0(2) | −4.4(6) | −3.3(4) | −22.4(9) | 1(1) |
| PO bond length |
1.4877(13) | 1.4877(15) | 1.491(3) | 1.4848(19) | 1.503(5) | 1.500(7) |
| 1.4923(12) | 1.490(3) | 1.507(5) | 1.495(7) | |||
| P–CPy bond length | 1.8190(17) | 1.820(2) | 1.827(4) | 1.830(3) | 1.827(7) | 1.820(8) |
| 1.8243(17) | 1.820(5) | 1.809(7) | 1.822(8) | |||
| O–P–CPy angle | 111.42(8) | 111.74(9) | 110.9(2) | 111.66(12) | 106.9(9) | 107.1(4) |
| 110.80(7) | 110.5(2) | 108.4(3) | 107.3(4) | |||
| CPy–CPy bond length | — | 1.490(3) | 1.450(6) | 1.433(3) | 1.479(10) | 1.45(1) |
This feature can be considered as a general peculiarity of the 2-pyridinephosphineoxide-type ligands. We also tested the structure of 2,6-bis(diphenylphosphine)pyridine oxide (PyPPO) prepared according to a known procedure from commercially available 2,6-dibrimopyridine,50 and the structure of 9-diphenylphosphinephenanthrolin-2-one oxide 1 (Fig. 2). Both show anti-conformation of the PO group relative to the pyridine ring (Table 1). At this moment it is still unclear if this feature is typical for all pyridine-containing phenylphosphinoxides, or whether this conformation is limited only to diphenylphosphinoxide-bearing pyridines.
 | ||
| Fig. 2 ORTEPs of 2,6-bis(diphenylphosphinoxide)pyridine and 9-diphenylphosphinephenanthrolin-2-one oxide 1 with thermal ellipsoids at 50% level for all non-hydrogen atoms. Solvent molecules are omitted for clarity. Other figures are available in the ESI.† | ||
The length of P–O bonds is approximately 1.49 Å for all investigated ligands. This distance is considerably greater than that found in Me3PO (1.44 Å),51 but is in good agreement with the P–O bond length for uncoordinated diphenylphosphinoxide groups in 4,5-bis(diphenylphosphoranyl)-1,2,3-triazolate (1.465(5) to 1.485(5) Å (ref. 45) and 1.497(2) (ref. 52)). P–CPy bond lengths are about 1.82 Å, which is close to the corresponding length of the heterocycle-phosphinoxide C–P bond found earlier.45,52 Both phosphorus atoms are near tetrahedral, and the corresponding angles are presented in Table 1 and ESI.†
The DPPO and PnPPO molecules do not form hydrogen bonded solvates in crystals. In contrast, PyPPO and phosphinoxide 1 show hydrogen bonding, either with water molecules, or during dimerization. Water molecules form strong hydrogen bonds with one of the phosphinoxide groups (2.14 Å H⋯O, O–H⋯O angle 172°, see the ESI†). Partly hydrolysed phosphinoxide 1 in the crystal form dimerises by strong hydrogen bonding (distance 2.937(3) Å, N–H⋯O angle 155(3), see in the ESI†) between the CO oxygen atom and N–H hydrogen. Phosphinoxide 1 crystal packs by C–H⋯π stacking interactions (2.488 Å) between the phenyl rings of phosphinoxide moieties of neighbouring molecules.
In spite of the unfavourable arrangement of the donor atoms, both tetradentate phosphinoxide-bearing ligands readily form complexes with lanthanides (Scheme 3).
 | ||
| Scheme 3 | ||
Diphenylphosphinoxides DPPO and PnPPO react rapidly with 4f-element salts in acetonitrile under reflux with the formation of corresponding mononuclear complexes of a general composition LLnX3, where Ln is La, Eu, Gd or Er. MALDI-TOF mass spectrometry confirms the formation of mononuclear complexes; the mass spectra possess characteristic peaks of composition LLnX2+ for both ligands and all the metal ion salts in accordance with earlier observations.39 A striking feature of europium complexes with DPPO and PnPPO is the presence of peaks corresponding to an unusual composition LLnX+, which is attributed to hypothetical reduction of the europium species. All observed signals exhibit the expected characteristic isotopic distribution patterns. The fragmentation of trifluoroacetate complexes is more complicated. The decomposition of the fluorinated counter-ion leads to a fluorine anion, which is able to replace TFA− in the complex and form mixed ions with compositions LEuTFAF+ and LEuF2+.
The IR spectra provide more information about the structures of the complexes. The intense band near 1200 cm−1, corresponding to the PO stretching, is very sensitive to metal ion coordination.18 We observed a significant lowering of the PO band frequency from near 1200 to 1144 cm−1 which confirmed the coordination of the metal ions to the phosphinoyl group. For the complexes with metal ion nitrates the NO bands for NO3 anions, which were observed near 1476 cm−1 for all complexes, are extremely informative. Less intense bands corresponding to symmetrical and asymmetrical deformation O–N–O vibrations in the NO3 anions were also observed.
The structure of the complexes of DPPO (DPPOGd(NO3)3, Fig. 3) and PnPPO (PnPPOEu(TFA)3·H2O, Fig. 4) with both nitrate and TFA counter-ions were studied by X-ray single-crystal diffraction (Tables 1 and 2). Both complexes are mononuclear and consist of one ligand coordinated to the metal ion in a tetradentate manner. Both complexes show coordination of the metal ion to the oxygen atoms of the phosphinoyl groups and to the nitrogen atoms of the pyridine heterocycle, supported by the formation of a five-membered metallocycle. The latter leads to elongation of the PO bond (from 1.4877 in DPPO and 1.490 in PnPPO to 1.503 or 1.507 in DPPOGd and 1.500 or 1.495 in PnPPOEu, see Table 1). The P–O bond lengths are within the typical range.45 A corresponding shift of IR bands was observed (vide supra). In contrast, the C–N bond in heterocycles does not undergo such changes (see the ESI†). The phosphorus atoms undergo tetrahedral distortion, which leads to a reduction in the O–P–CPy angles from about 111° to 107° (Table 1). The phosphinoyl groups are all are rotated around the CPy–P bond to adopt the conformation needed for metal ion binding. The OP–CPy–NPy torsion angles significantly diminish. The complexes are reorganised differently depending on the nature of the ligand. DPPO undergoes twisting in both the phosphinoyl group and the bipyridyl moiety. The angle between the two pyridine rings is 22.4(9)°, but the angles for the two Ph2PO-groups are significantly different. One of the phosphinoyl groups lies near the plane of the heterocycle (−4.6(6)°) but the second is twisted from the plane to form a 28.1(6)° angle. In contrast, PnPPO undergoes relatively symmetrical distortion and the two phosphinoyl groups form a dihedral angle close to each other: −9.6(7) and 12.3(7)°.
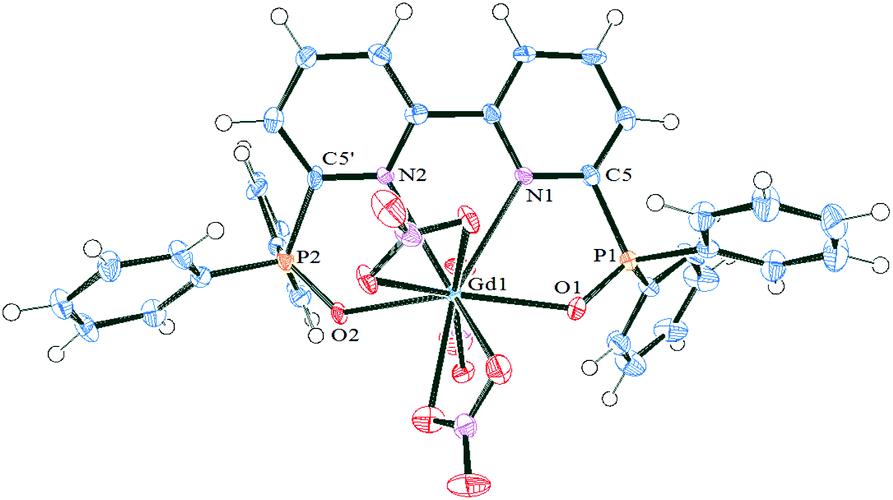 | ||
| Fig. 3 ORTEPs of complex DPPOGd(NO3)3 with thermal ellipsoids at 50% level for all non-hydrogen atoms. Hydrogen atoms and solvent molecules are omitted for clarity. Other figures are available in the ESI.† | ||
 | ||
| Fig. 4 ORTEPs of complex PnPPOEu(TFA)3·H2O with thermal ellipsoids at 50% level for all non-hydrogen atoms. Solvent molecules are omitted for clarity. Other figures are available in the ESI.† | ||
| DPPOGd(NO3)3 | PnPPOEu(TFA)3 | |
|---|---|---|
| Ln–N | 2.665(5); 2.708(6) | 2.662(7); 2.638(6) |
| Ln–Op | 2.359(5); 2.388(5) | 2.331(6); 2.345(7) |
| Ln–Oanion | 2.457(5); 2.477(5); 2.482(5); 2.498(5); 2.501(5); 2.533(5) | 2.389(4); 2.306(7); 2.319(5) |
| N–Ln–N | 60.06(17) | 61.4(2) |
| Op–Ln–Op | 154.03(16) | 162.6(2) |
| Eu–OH2 | 2.431(6) |
The nature of the counter-ion has a crucial effect on the structure of the complexes: the nitrato complex possesses the decacoordinated gadolinium ion while the trifluoroacetato-complex shows an octacoordinated europium ion in a distorted bicapped-trigonalantiprismatic environment (Fig. 4). In PnPPOEu(TFA)3 the O(2), O(1B), O(1W) and O(1A), O(1C), O(1) atoms form two bases of the prisms while the nitrogen atoms serve as two caps. The bridging nature of the nitrato groups leads to a higher coordination number of the lanthanide ion; in contrast the monodentate coordination of the trifluoroacetato-groups gives rise to additional coordination of a water molecule to the Eu ion.
The Ln–O,N distances are significantly shorter for the octacoordinated lanthanide ion than for the decacoordinated lanthanide ion, both for the ligand and the counter-ion (Table 2). So the metal ion enters deeper into the cavity of the tetradentate ligand for PnPPOEu(TFA)3 than for DPPOGd(NO3)3. The latter has an impact on the N–Ln–N and Op–Ln–Op biting angles: the angles for the Gd complex being significantly smaller (Table 2).
Such differences in coordination environments must have an impact on the metal-centred luminescence of the complexes. Due to the unique photophysical properties of REE complexes with heterocycles, the study of the influence of the nature of the ligand and the counter-ion on the photophysical properties of the complexes is of great importance.53 The difference in the coordination environment is due to the nature of the counter-ion that results in the discrepancy in photophysical properties of the complexes. Moreover, the coordinated water molecule suggests quenching of the luminescence of the corresponding complex permanently. The nature of the environment of the europium ion can be obtained through analysis of relative intensities of the transitions of its absorption and luminescence spectra. Absorbance spectra of complexes show very similar information for the complexes with the same type of ligands and are practically independent of the counter-ion (Fig. 5).
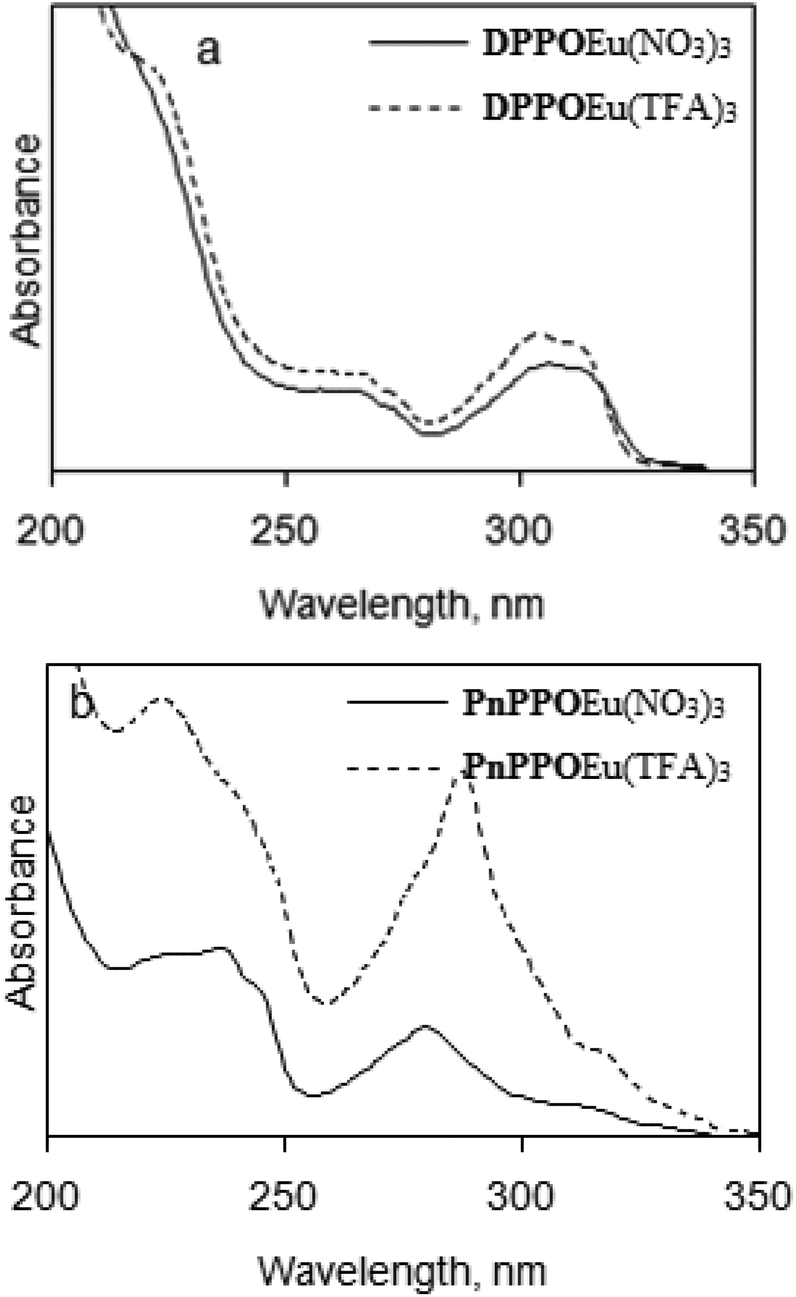 | ||
| Fig. 5 Absorbance spectra of europium complexes in acetonitrile solutions: (a) DPPOEu(NO3)3, DPPOEu(TFA)3; (b) PnPPOEu(NO3)3, PnPPOEu(TFA)3. | ||
The absorbance spectra of complexes DPPOEu(NO3)3 and DPPOEu(TFA)3 have main maxima in the spectral region of 300–310 nm that correspond to absorption by the ligand. For DPPOEu(TFA)3 this maximum shifts 2 nm towards shorter wavelengths. We can see different spectra for complexes PnPPOEu(NO3)3 and PnPPOEu(TFA)3. The absorbance maxima are located in the region of 275–290 nm and the PnPPOEu(TFA)3 maximum is shifted 10 nm towards longer wavelengths.
The luminescence spectra for the europium complexes give more information than that obtained from absorption spectra (Fig. 6 and 7). A typical europium emission spectrum shows intense luminescence signals and includes several maxima in the red region that corresponds to transitions 5D0 → 7FJ (J = 0–6).53 In this study we observed only the first five maxima: at wavelength 582 nm (5D0 → 7F0), 595 nm (5D0 → 7F1), 619 nm (5D0 → 7F2), 650 nm (5D0 → 7F3) and in the region 680–710 nm (5D0 → 7F4).
 | ||
| Fig. 6 Luminescence emission spectra of europium complexes in the solid state, λex = 270 nm: (a) DPPOEu(NO3)3; (b) DPPOEu(TFA)3. | ||
 | ||
| Fig. 7 Luminescence emission spectra of europium complexes in the solid state, λex = 270 nm: (c) PnPPOEu(NO3)3; (d) PnPPOEu(TFA)3. | ||
The first transition (5D0 → 7F0) that can be observed at the wavelength 570–585 nm is forbidden by the Judd–Ofelt theory54 and this transition is an example of a discrepancy with this theory. To determine different Eu(III) species in solution the 5D0 → 7F0 transition can also be used. In this work this line was symmetric, and so only one type of europium was present in all the crystals of the complexes studied. The correlation of the wavelength that corresponds to this transition with the ligand coordination number can be suggested.55
The second transition (5D0 → 7F1) can be observed at the wavelength 585–600 nm; this is a magnetic dipole transition. Its intensity is perfect for calibration of the luminescence intensity of the europium complex because it is almost constant. Crystal-field splitting of the 7F1 level is directly reflected by the transition and its intensity is the highest if we consider the spectra of solids with a crystal structure and with central symmetry.56
The most intensive maxima corresponds to the 5D0 → 7F2 transition for all complexes and are called “hypersensitive” due to the fact that the strongest intensity of the maximum depends on the symmetry of the europium ion and the nature of the ligand.39 This maximum can be found at the wavelength 610–630 nm. The intensity ratio of the transitions 5D0 → 7F2 and 5D0 → 7F1 is often compared with the intensities of the hypersensitive transition in different europium compounds as an alternative to measure the absolute intensity.57
In general the 5D0 → 7F3 transition can be observed at the 640–660 nm spectral range and is very weak because it is forbidden. The strong intensity of this transition is a sign of strong crystal-field perturbation.58
The 5D0 → 7F4 transition corresponds to the luminescence maxima in the spectral range of 680–710 nm.59 The intensity of the 5D0 → 7F4 transition is too low and is very little compared to other transitions, whereas the intensity of this transition is exaggerated in an overcorrected spectrum.
In this study, the 5D0 → 7F5 transition (740–770 nm) and the 5D0 → 7F6 transition (810–840 nm) were not observed because the intensities of these transitions are very low.60 The ratios of intensities corresponding to 5D0 → 7F2 and 5D0 → 7F1 transitions are different for the complexes studied: for DPPOEu(NO3)3 and PnPPOEu(NO3)3 they are 6 and 5, respectively, showing deviation from an inversion centre in the complexes.39 In complexes DPPOEu(TFA)3 and PnPPOEu(TFA)3 this ratio is ∼4, indicating that the deviation is not so strong.
The phosphorescence lifetimes of the 5D0 level in europium complexes in the solid state were 1.20 ± 0.03 ms for DPPOEu(NO3)3, 2.16 ± 0.06 ms for DPPOEu(TFA)3, 1.01 ± 0.07 ms for PnPPOEu(NO3)3 and 1.84 ± 0.05 ms for PnPPOEu(TFA)3.
Quantum yields of europium complexes were measured using the reference dye method. Rhodamine B was selected as a reference dye because of its high quantum yield value and usability.
The values of luminescence quantum yields for different concentrations were calculated (Fig. 8). For PnPPOEu(NO3)3 the luminescence quantum yield equals to 11% at the concentration 0.1–3.0 × 10−5 mol L−1 (but concentration dependence was not detected). For other complexes luminescence quenching at concentrations of more than 8.0 × 10−6 mol L−1 was observed. The maximum luminescence quantum yield was observed for DPPOEu(NO3)3 and DPPOEu(TFA)3: it equalled 85% for both complexes. So the retention of a water molecule in the coordination environment of the europium ion in solution for complex DPPOEu(TFA)3 is doubtful, while PnPPOEu(TFA)3 seems to keep the water coordinated with the metal ion even in acetonitrile solutions.
 | ||
| Fig. 8 Dependence of the value of the luminescence quantum yield on the concentration of europium complexes in acetonitrile solutions. | ||
The excitation spectra of complexes with the DPPO-type ligand have some maxima that correspond to electronic transitions: 5L6 ← 7F0 (390–405 nm), 5D2 ← 7F0 (460–470 nm), 5D1 ← 7F1 and 5D1 ← 7F0 (520–540 nm) (Fig. 9).
 | ||
| Fig. 9 Luminescence excitation spectra of europium complexes in the solid state at 300 K (λem = 618 nm). | ||
The 5L6 ← 7F0 transition is the most intense in the excitation spectra of europium complexes. Excitation of europium to induce the photoluminescence transitions mentioned above is commonly used if the ligand excitation is forbidden due to the absence of efficient energy transfer. 5L6 level excitation allows direct population of the 4f levels.61–63 The 5D1 ← 7F1 and 5D2 ← 7F0 transitions are hypersensitive (J = 2).
Conclusions
We successively designed and synthesized two related tetradentate N,N′,O,O′-ligands based on 2,2′-bipyridyl- and phenanthroline-bearing phosphinoyl-type substituents. The ligands were X-ray characterised and their complexes were studied in respect of the possible difference in metal ion coordination driven by the nature of the counter-ion or geometric rigidity of the ligand. Two examples of complexes with each ligand and every counter-ion (nitrate or trifluoroacetate) were examined by X-ray single-crystal diffraction. The coordination environment of the metal ion is strongly counter-ion dependent. But the conformations of the phosphinoyl groups relative to the pyridine rings depend on the flexibility of the ligand. As a result, the photophysical properties of the complexes are strongly ligand and counter-ion dependent:• Absorbance and luminescence excitation spectra of complexes showed main peaks in the UV range which correspond to absorption by the ligand and they are ligand-dependent. Luminescence spectra of complexes showed typical europium emissions in the red region, with high quantum yield, that correspond to the transitions 5D0 → 7FJ (J = 0–6).
• The value of deviation of the components of 5D0 → 7F2 and 5D0 → 7F1 transitions from the inversion centre showed a larger dependence on the counter-ion than on the nature of the ligand. Thus the degree of symmetry is more dependent on the counter-ion than on the ligand structure.
• The value of the luminescence quantum yield was the highest for europium complexes with the DPPO ligand and the NO3 counter-ion; the smallest for complexes with the PhenPPO ligand and the NO3 counter-ion.
• The value of phosphorescence lifetimes of the 5D0 level in the solid state was higher for complexes with the TFA counter-ion (about 2.0 ms) than for those with the NO3 counter-ion (about 1.1 ms). A small dependence of the lifetime on the nature of the ligand was demonstrated.
Experimental
General
The NMR spectra were measured with a BRUKER AVANCE-600 (600.12 MHz) NMR spectrometer at 23 °C. Chemical shifts are given in ppm relative to SiMe4 (for 1H and 13C NMR) and 85% H3PO4 (for 31P). IR spectra were recorded with an Agilent 640 FTIR spectrometer with samples in KBr pellets. Mass spectra were obtained with a MALDI-TOF Reflex 3 instrument (BRUKER) in the positive ion mode (UV laser, 337 nm). 6,6′-Dichloro-2,2′-bipyridine,47 2,9-dichlorophenanthroline23 and 2,6-bis(diphenylphosphinoyl)pyridine50 were prepared according to known procedures. All reagents and solvents were obtained from commercial sources.For X-ray structure determination the data collection was performed with an APEX II CCD diffractometer. The structure was solved and refined against F2 by full-matrix least squares techniques with the SHELXTL software package.64,65 Crystallographic data, crystal packing, hydrogen bonding information and details of data collection are listed in the ESI.† CCDC 1522002 [for PyPPO], 1522000 [for DPPO], 1521999 [for PnPPO], 1522001 [for 1], 1521997 [for DPPOGd(NO3)3] and 1521998 [for PnPPOEu(TFA)3] contain the supplementary crystallographic data for this paper.
Luminescence properties of complexes were obtained for powdered samples in the solid state and in acetonitrile solution at 300 K using a Hitachi F-7000 luminescence spectrometer. Reflection geometry was used for detecting luminescence spectra of europium complexes in the solid state. Registration of absorbance spectra of europium complex solutions was performed with a Hitachi U-1900 spectrophotometer.
N), 1550, 1436, 1420, 1200 ν(PO), 1150, 1118, 1099, 1073, 988, 807, 753, 739, 692, 566, 543, and 517. δH (CDCl3) 7.45 (8H, td, J = 7.57, 2.52 Hz, HPh-3,Ph-5), 7.51 (4H, t, J = 6.92 Hz, HPh-4), 7.91 (8H, dd, J = 11.78, 7.38 Hz, HPh-2,Ph-6), 7.96 (2H, dt, J = 7.75, 3.92 Hz, HPy-4), 8.32 (4H, dd, J = 7.34, 5.70 Hz, HPy-3,Py-5). δC (CDCl3) 156.37 (d, 1JCP = 132.0 Hz, CPy-6); 155.36 (d, 3JCP = 18.3 Hz, CPy-2); 137.39 (d, 2JCP = 9.8 Hz, CPy-5); 131.68 (d, 1JCP = 104.5 Hz, CPh-1); 132.09 (d, 2JCP = 9.2 Hz, CPh-2); 131.93 (d, 4JCP = 3.0 Hz, CPh-4); 128.63 (d, 3JCP = 20.0 Hz, CPy-4); 128.32 (d, 3JCP = 12.8 Hz, CPh-3); 122.58 (d, 4JCP = 3.0 Hz, CPy-3). δP (CDCl3) 21.3. MS (MALDI-TOF) m/z 557 ([M + H+], 100%), 579 ([M + Na+], 40%). MS (ESI) m/z 557 ([M + H+], 100%), 579 ([M + Na+], 33%), 595 ([M + K+], 40%).
Preparation of the complexes
DPPOEu(NO3)3 (71 mg, 79%). Found: C, 44.7; H, 3.3; N, 7.2. Calc. for C34H26EuN5O11P2·H2O: C, 44.75; H, 3.1; N, 7.7. νmax/cm−1 3062 ν(C–H), 2931, 2854, 3388 ν(OH), 1143 ν(PO), 1589 ν(conj. CN), 746, 728, 692 νδ(C–H), 1476 ν(NO), 1292 νa(NO2), 1027 νs(NO2). δH (acetonitrile-d3) 5.65 (1H, br.s., HPy), 6.26 (6H, br.s., HPh), 6.33 (2H, br.s., HPh), 6.80 (2H, t, J = 7.34 Hz, HPh), 9.04 (1H, br.s., HPy), 11.13 (1H, br.s., HPy). δC (acetonitrile-d3) 167.01 (br.m., CPy−2), 155.92 (br.s., CPy-4), 146.12 (d, 1JCP = 119.69 Hz, CPh-1), 133.53 (s. CPh-4), 129.12 (d, 2JCP = 9.67 Hz, CPh-2), 128.26 (d, 3JCP = 12.16 Hz, CPh-3), 112.01 (d, 1JCP = 108.08 Hz, CPy-6), 110.78 (s, CPy-3), 100.91 (d, 3JCP = 16.59 Hz, CPy-5). δP (acetonitrile-d3) 60.22. MS (MALDI-TOF) m/z 832 ([DPPOEu(NO3)2+], 100%).
DPPOGd(NO3)3 (71 mg, 79%). Found: C, 45.7; H, 3.2; N, 7.4. Calc. for C34H26GdN5O11P2: C, 45.4; H, 2.9; N, 7.8. MS (MALDI-TOF) m/z 838 ([DPPOGd(NO3)2+], 100%).
DPPOEu(TFA3) (80 mg, 76%). Found: C, 45.8; H, 2.5; N, 2.6. Calc. for C40H26EuF9N2O8P2: C, 45.9; H, 2.5; N, 2.7. νmax/cm−1 3061, 2927, 2855 ν(C–H), 1200 ν(CF3), 1144vs ν(PO), 1589vs ν(CN), 748, 729, 721, 692 νδ(C–H), 1440s νs(COO), 1707s νa(COO). δH (acetonitrile-d3) 9.36 (1H, br.s., HPy-3), 8.56 (1H, br.s., HPy-4), 7.24 (3H, br.m., HPy-5, HPh-4), 6.78 (4H, br.s., HPh-3), 5.62 (4H, br.s., HPh-2). δC (acetonitrile-d3) 169.98 (br.m., CPy-2), 153.97 (d, 3JCP = 9.18 Hz, CPy-4), 149.64 (d, 1JCP = 126.2 Hz, CPh-1), 134.32 (s, CPh-4), 131.84 (d, 2JCP = 10.32 Hz, CPh-2), 129.34 (d, 3JCP = 12.62 Hz, CPh-3), 116.46 (d, 1JCP = 108.99 Hz, CPy-2), 108.32 (s, CPy-3), 101.90 (d, 2JCP = 21.80 Hz, CPy-5). MS (MALDI-TOF) m/z 935 ([DPPOEu(TFA)2+], 15%), 822 ([DPPOEu(TFA)+], 20%), 747 ([DPPOEuF2+], 20%), 728 ([DPPOEuF+], 100%).
PnPPOEu(NO3)3 (72 mg, 78%). Found C, 47.4; H, 3.1; N, 7.6. Calc. for C36H26EuN5O11P2: C, 47.1; H, 2.85; N, 7.6. νmax/cm−1 3058, 2927, 2852 ν(C–H), 1564, 1554, 1540 ν(conj. CN, C-CAr), 1172s ν(PO), 1439vs ν(NO), 1194s νa(NO2), 1097s νs(NO2). δH (acetonitrile-d3) 8.63 (1H, br.s.), 8.08 (4H, br.s.), 7.54 (3H, br.s.), 7.35 (5H, br.s.). δP (acetonitrile-d3) 48. MS (MALDI-TOF) m/z 857 ([PnPPOEu(NO3)2+], 30%), 795 ([PnPPOEu(NO3)+], 100%).
PnPPOLa(NO3)3 (77 mg, 85%). Found C, 47.5; H, 3.0; N, 7.9. Calc. for C36H26LaN5O11P2: C, 47.75; H, 2.9; N, 7.7. δP (acetonitrile-d3) 41. MS (MALDI-TOF) m/z 843 ([PnPPOLa(NO3)2+], 100%).
PnPPOEr(NO3)3 (75 mg, 80%). Found C, 46.5; H, 2.6; N, 7.3. Calc. for C36H26ErN5O11P2: C, 46.3; H, 2.8; N, 7.5. MS (MALDI-TOF) m/z 872 ([PnPPOEr(NO3)2+], 100%).
PnPPOEu(TFA3)·H2O (74 mg, 69%) Found C, 46.8; H, 2.9; N, 2.9. Calc. for C42H26EuF9N2O8P2: C, 47.1; H, 2.45; N, 2.6. νmax/cm−1 3066, 2929, 2856 ν(C–H), 1677vs νa(COO), 1438s νs(COO), 1206s ν(CF3), 1141vs ν(PO). δH (acetonitrile-d3) 8.98 (1H, br.s., HPn-3), 8.76 (1H, br.s., HPn-5), 7.70 (1H, br.s., HPn-4), 7.37 (2H, t, J = 7.34 Hz, HPh-4), 6.94 (4H, br.s., HPh-3), 6.29 (4H, br.s., HPh-2). δC (acetonitrile-d3) 170.09 (br.s., CPn-10b), 152.56 (d, 3JCP = 4.59 Hz, CPn-2), 146.10 (d, 1JCP = 113.58 Hz, CPh-1), 133.53 (s, CPh-4), 131.48 (d, 2JCP = 11.47 Hz, CPh-2), 128.52 (d, 3JCP = 12.62 Hz, CPh-3), 116.27 (d, 1JCP = 108.99 Hz, CPn-2), 114.19 (s, CPn-4a), 93.86 (d, 2JCP = 17.21 Hz, CPn-3).
Acknowledgements
The results have been obtained under support of the RSF grant no. 16-13-10451. The NMR spectroscopic measurements were carried out in the Laboratory of Magnetic Tomography and Spectroscopy, Faculty of Fundamental Medicine, of Moscow State University.Notes and references
- J.-C. G. Bunzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- J.-C. G. Bunzli, Spectroscopic Properties of Rare Earths in Optical Materials, ed. G. K. Liu and B. Jacquier, Springer Verlag, Berlin, 2005, ch. 11, vol. 83 Search PubMed.
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- J. C. G. Bünzli, in Spectroscopic Properties of Rare Earths in Optical Materials, ed. R. Hull, J. Parisi, R. M. Osgood Jr., H. Warlimont, G. Liu and B. Jacquier, Springer, Berlin Heidelberg, 2005, ch. 9, vol. 83, pp. 462–499 Search PubMed.
- J.-C. G. Bunzli, L. J. Charbonniere and R. F. Ziessel, J. Chem. Soc., Dalton Trans., 2000, 1917–1923 RSC.
- J.-C. G. Bünzli, S. Comby, A.-S. Chauvin and C. D. B. Vandevyver, J. Rare Earths, 2007, 25, 257–274 CrossRef.
- A. Bhattacharyya, P. K. Mohapatra and V. K. Manchanda, Radiochim. Acta, 2010, 98, 141–147 CrossRef CAS.
- M. G. B. Drew, M. R. S. J. Foreman, C. Hill, M. J. Hudson and C. Madic, Inorg. Chem. Commun., 2005, 8, 239–241 CrossRef CAS.
- M. Alyapyshev, V. Babain, N. Borisova, I. Eliseev, D. Kirsanov, A. Kostin, A. Legin, M. Reshetova and Z. Smirnova, Polyhedron, 2010, 29, 1998–2005 CrossRef CAS.
- P. D. Beer, M. R. Sambrook and D. Curiel, Chem. Commun., 2006, 2105–2117 RSC.
- S. Huh, S. Jung, Y. Kim, S.-J. Kim and S. Park, Dalton Trans., 2010, 39, 1261–1265 RSC.
- Z. Kolarik, Chem. Rev., 2008, 108, 4208–4252 CrossRef CAS PubMed.
- L. N. Puntus, K. A. Lyssenko, M. Y. Antipin and J.-C. G. Bünzli, Inorg. Chem., 2008, 47, 11095–11107 CrossRef CAS PubMed.
- S. Mishra, G. Ledoux, E. Jeanneau, S. Daniele and M.-F. Joubert, Dalton Trans., 2012, 41, 1490–1502 RSC.
- Y. P. Du, Y. W. Zhang, L. D. Sun and C. H. Yan, J. Phys. Chem. C, 2008, 112, 405–415 CAS.
- J. C. Boyer, F. Vetrone, L. A. Cuccia and J. A. Capobianco, J. Am. Chem. Soc., 2006, 128, 7444–7445 CrossRef CAS PubMed.
- J. C. Boyer, L. A. Cuccia and J. A. Capobianco, Nano Lett., 2007, 7, 847–852 CrossRef CAS PubMed.
- F. de Maria Ramírez, L. Charbonnière, G. Muller, R. Scopelliti and J.-C. G. Bünzli, J. Chem. Soc., Dalton Trans., 2001, 3205–3213 RSC.
- F. Renaud, C. Piguet, G. Bernardinelli, G. Hopfgartner and J.-C. G. Bunzli, Chem. Commun., 1999, 457–458 RSC.
- S. Petoud, J.-C. G. Bünzli, T. Glanzman, C. Piguet, Q. Xiang and R. P. Thummel, J. Lumin., 1999, 82, 69–79 CrossRef CAS.
- F. Avecilla, C. Platas-Iglesias, R. Rodríguez-Cortiñas, G. Guillemot, J.-C. G. Bünzli, C. D. Brondino, C. F. G. C. Geraldes, A. de Blas and T. Rodríguez-Blas, J. Chem. Soc., Dalton Trans., 2002, 4658–4665 RSC.
- A.-S. Chauvin, J.-C. G. Bünzli, F. Bochud, R. Scopelliti and P. Froidevaux, Eur. J. Chem., 2006, 12, 6852–6864 CrossRef CAS PubMed.
- M. Galletta, S. Scaravaggi, E. Macerata, A. Famulari, A. Mele, W. Panzeri, F. Sansone, A. Casnati and M. Mariani, Dalton Trans., 2013, 42, 16930–16938 RSC.
- T. Kobayashi, T. Yaita, S. Suzuki, H. Shiwaku, Y. Okamoto, K. Akutsu, Y. Nakano and Y. Fujii, Sep. Sci. Technol., 2010, 45, 2431–2436 CrossRef CAS.
- D. Manna, S. Mula, A. Bhattacharyya, S. Chattopadhyay and T. K. Ghanty, Dalton Trans., 2015, 44, 1332–1340 RSC.
- D. Merrill and R. D. Hancock, Radiochim. Acta, 2011, 99, 161–166 CrossRef CAS.
- C.-L. Xiao, C.-Z. Wang, L.-Y. Yuan, B. Li, H. He, S. Wang, Y.-L. Zhao, Z.-F. Chai and W.-Q. Shi, Inorg. Chem., 2014, 53, 1712–1720 CrossRef CAS PubMed.
- X. Zhang, L. Yuan, Z. Chai and W. Shi, Sep. Purif. Technol., 2016, 168, 232–237 CrossRef CAS.
- C.-L. Xiao, C.-Z. Wang, L. Mei, X.-R. Zhang, N. Wall, Y.-L. Zhao, Z.-F. Chai and W.-Q. Shi, Dalton Trans., 2015, 44, 14376–14387 RSC.
- J. L. Lapka, A. Paulenova, R. S. Herbst and J. D. Law, Sep. Sci. Technol., 2010, 45, 1706–1710 CrossRef CAS.
- C. Marie, M. Miguirditchian, D. Guillaneux, J. Bisson, M. Pipelier and D. Dubreuil, Solvent Extr. Ion Exch., 2011, 29, 292–315 CrossRef CAS.
- C. Marie, M. Miguirditchian, D. Guillaumont, A. Tosseng, C. Berthon, P. Guilbaud, M. Duvail, J. Bisson, D. Guillaneux, M. Pipelier and D. Dubreuil, Inorg. Chem., 2011, 50, 6557–6566 CrossRef CAS PubMed.
- N. Boubals, M. G. B. Drew, C. Hill, M. J. Hudson, P. B. Iveson, C. Madic, M. L. Russell and T. G. A. Youngs, J. Chem. Soc., Dalton Trans., 2002, 55–62 RSC.
- N. E. Borisova and M. D. Reshetova, Russ. Chem. Bull., 2015, 64, 1882–1890 CrossRef CAS.
- M. Y. Alyapyshev, V. A. Babain, L. I. Tkachenko, A. Paulenova, A. A. Popova and N. E. Borisova, Solvent Extr. Ion Exch., 2014, 32, 138–152 CrossRef CAS.
- D. O. Kirsanov, N. E. Borisova, M. D. Reshetova, A. V. Ivanov, L. A. Korotkov, I. I. Eliseev, M. Y. Alyapyshev, I. G. Spiridonov, A. V. Legin, Y. G. Vlasov and V. A. Babain, Russ. Chem. Bull., 2012, 61, 881–890 CrossRef CAS.
- M. Y. Alyapyshev, V. A. Babain, N. E. Borisova, R. N. Kiseleva, D. V. Safronov and M. D. Reshetova, Mendeleev Commun., 2008, 18, 336–337 CrossRef CAS.
- Reprocessing and recycling of spent nuclear fuel, ed. R. Taylor, Woodhead Publishing, 80 High Street, Sawston, Cambridge, CB22 3HJ, UK, 2015 Search PubMed.
- N. E. Borisova, A. A. Kostin, E. A. Eroshkina, M. D. Reshetova, K. A. Lyssenko, E. N. Spodine and L. N. Puntus, Eur. J. Inorg. Chem., 2014, 2219–2229 CrossRef CAS.
- L. Prodi, M. Montalti, N. Zaccheroni, G. Pickaert, L. Charbonniere and R. Ziessel, New J. Chem., 2003, 27, 134–139 RSC.
- M. Pietraszkiewicz, A. Kłonkowski, K. Staniszewski, J. Karpiuk and S. Bianketti, J. Alloys Compd., 2004, 380, 241–247 CrossRef CAS.
- O. Pietraszkiewicz, M. Pietraszkiewicz, J. Karpiuk and M. Jesie, J. Rare Earths, 2009, 27, 584–587 CrossRef.
- M. A. Goni, L. M. Daniels and T. A. Siddiquee, Inorg. Chim. Acta, 2013, 394, 645–648 CrossRef CAS.
- Z. Spichal, M. Necas, J. Pinkas and J. Novosad, Inorg. Chem., 2004, 43, 2776–2778 CrossRef CAS PubMed.
- M. Correa-Ascencio, E. K. Galván-Miranda, F. Rascón-Cruz, O. Jiménez-Sandoval, S. J. Jiménez-Sandoval, R. Cea-Olivares, V. Jancik, R. A. Toscano and V. García-Montalvo, Inorg. Chem., 2010, 49, 4109–4116 CrossRef CAS PubMed.
- L. J. Charbonniere, S. Mameri, D. Flot, F. Waltz, C. Zandanel and R. F. Ziessel, Dalton Trans., 2007, 2245–2253 RSC.
- W.-J. Wang, Y.-C. Wang and H.-C. Kao, J. Chin. Chem. Soc., 2010, 57, 876–882 CrossRef CAS.
- H. C. Guo, R. H. Zheng and H. J. Jiang, Org. Prep. Proced. Int., 2012, 44, 392–396 CrossRef CAS.
- M. Yamada, Y. Nakamura, S. Kuroda and I. Shimao, Bull. Chem. Soc. Jpn., 1990, 63, 2710–2712 CrossRef CAS.
- G. R. Newkome and D. C. Hager, J. Org. Chem., 1978, 43, 947–949 CrossRef CAS.
- H. K. Wang, Acta Chem. Scand., 1965, 19, 879 CrossRef CAS.
- A. L. Rheingold, L. M. Liable-Sands and S. Trofimenko, Angew. Chem., Int. Ed., 2000, 39, 3321–3324 CrossRef CAS PubMed.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- B. M. Walsh, Judd-Ofelt theory: principles and practices, Springer, 2006 Search PubMed.
- G. R. Choppin and Z. M. Wang, Inorg. Chem., 1997, 36, 249–252 CrossRef CAS.
- M. M. Murali, M. L. Rama, D. Ramachari and C. K. Jayasankar, Spectrochim. Acta, Part A, 2014, 118, 966–971 CrossRef PubMed.
- G. P. Pires, I. F. Costa, H. F. Brito, W. N. Faustino and E. E. S. Teotonio, Dalton Trans., 2016, 45, 10960–10968 RSC.
- X. Y. Chen and G. K. Liu, J. Solid State Chem., 2005, 178, 419–428 CrossRef CAS.
- M. Xie, Y. Li and R. Li, J. Lumin., 2013, 136, 303–306 CrossRef CAS.
- V. I. Verlan, M. S. Iovu, I. Culeac, O. Bordian, V. E. Zubareva and I. Nistor, IFMBE Proc., 2016, 55, 17–20 Search PubMed.
- V. Tsaryuk, V. Zolin and J. Legendziewicz, J. Lumin., 2003, 102–103, 744–750 CrossRef CAS.
- O. L. Malta and F. R. Gonçalves e Silva, Spectrochim. Acta, Part A, 1998, 54, 1593–1599 CrossRef.
- C. A. Kodaira, H. F. Brito, O. L. Malta and O. A. Serra, J. Lumin., 2003, 101, 11–21 CrossRef CAS.
- G. M. Sheldrick, SHELXS97. Program for the Solution of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL97. Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1521997–1522002. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt04681a |
| This journal is © The Royal Society of Chemistry 2017 |