
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Azido- and amido-substituted gallium hydrides supported by N-heterocyclic carbenes†
Anindya K.
Swarnakar
,
Michael J.
Ferguson
 ,
Robert
McDonald
and
Eric
Rivard
*
,
Robert
McDonald
and
Eric
Rivard
*
Department of Chemistry, University of Alberta, 11227 Saskatchewan Drive, Edmonton, Alberta, Canada T6G 2G2. E-mail: erivard@ualberta.ca
First published on 2nd January 2017
Abstract
Despite recent advances in main group N-heterocyclic carbene (NHC) coordination chemistry, gallium hydrides remain, in large part, an unexplored area of research. In this paper we outline efficient routes to azide- and amido-functionalized gallium hydrides such as NHC·GaH2N3 and NHC·GaH2N(SiMe3)2 and explore these species as potential precursors to HGaNH complexes and bulk gallium nitride (GaN).
Introduction
It is now well accepted that the coordination of main group element centers by carbon-based donors, such as N-heterocyclic carbenes (NHCs),1 cyclic(alkyl)aminocarbenes (CAACs),2 and N-heterocyclic olefins (NHOs)3 can provide access to many species that are unstable or unattainable under conventional synthetic conditions. Drawing focus to the Group 13 (triel) elements, the recent isolation of homodiatomic B2 molecular adducts4 can be viewed as a particularly salient example of the stabilization brought forth by the abovementioned carbon-based donors.Recently, our group prepared a complex containing the elusive inorganic acetylene HBNH placed between a sterically encumbered NHC donor and a large triarylfluoroborane acceptor.5 The resulting complex IPr·HB![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH·BArF3 [IPr = [(HCNDipp)2C:]; Dipp = 2,6-iPr2C6H3; ArF = 3,5-(F3C)2C6H3] was synthesized via Lewis acid-assisted N2 elimination from the non-explosive azidoborane complex IPr·BH2N3
NH·BArF3 [IPr = [(HCNDipp)2C:]; Dipp = 2,6-iPr2C6H3; ArF = 3,5-(F3C)2C6H3] was synthesized via Lewis acid-assisted N2 elimination from the non-explosive azidoborane complex IPr·BH2N3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 followed by hydride migration from B to N. This prompted us to explore parallel chemistry7 with gallium, wherein the iminogallane HGaNH could be a possible building block for the future low temperature deposition of bulk gallium nitride (GaN), a highly valued material for its luminescent and semi-conducting properties.8,9 Our preliminary investigations involving the preparation of NHC-supported azido- and amido-gallium hydrides and behavior upon heating are reported herein.
6 followed by hydride migration from B to N. This prompted us to explore parallel chemistry7 with gallium, wherein the iminogallane HGaNH could be a possible building block for the future low temperature deposition of bulk gallium nitride (GaN), a highly valued material for its luminescent and semi-conducting properties.8,9 Our preliminary investigations involving the preparation of NHC-supported azido- and amido-gallium hydrides and behavior upon heating are reported herein.
Results and discussion
Our attempted synthesis of an HGaNH complex required the discovery of a suitable route to an azidogallane adduct NHC·GaH2N3. We hoped that one analogue, IMes·GaH2N3, could be synthesized from known IMes·GaH2X (IMes = [{HCN(Mes)}2C:]; Mes = 2,4,6-Me3C6H2; X = Cl, Br or I) complexes10 by reaction with common azide sources such as Me3SiN3 or NaN3. However previous reports associated with the synthesis of the necessary starting material IMes·GaH3 involved the reaction of the N-heterocyclic carbene, IMes, with thermally unstable Li[GaH4].11 To make an eventual route to IMes·GaH2N3 more convenient, we developed a modified synthesis of IMes·GaH3 using K[HBsBu3] as a hydride source. Specifically IMes·GaCl312 was combined with three equivalents of K[HBsBu3] in THF at room temperature to give IMes·GaH3 in a 75% isolated yield via Cl/H exchange (eqn (1)). Later, IMes·GaH3 was treated with 0.5 equivalents of IMes·GaI3, according to literature procedures,10c,12 to afford IMes·GaH2I.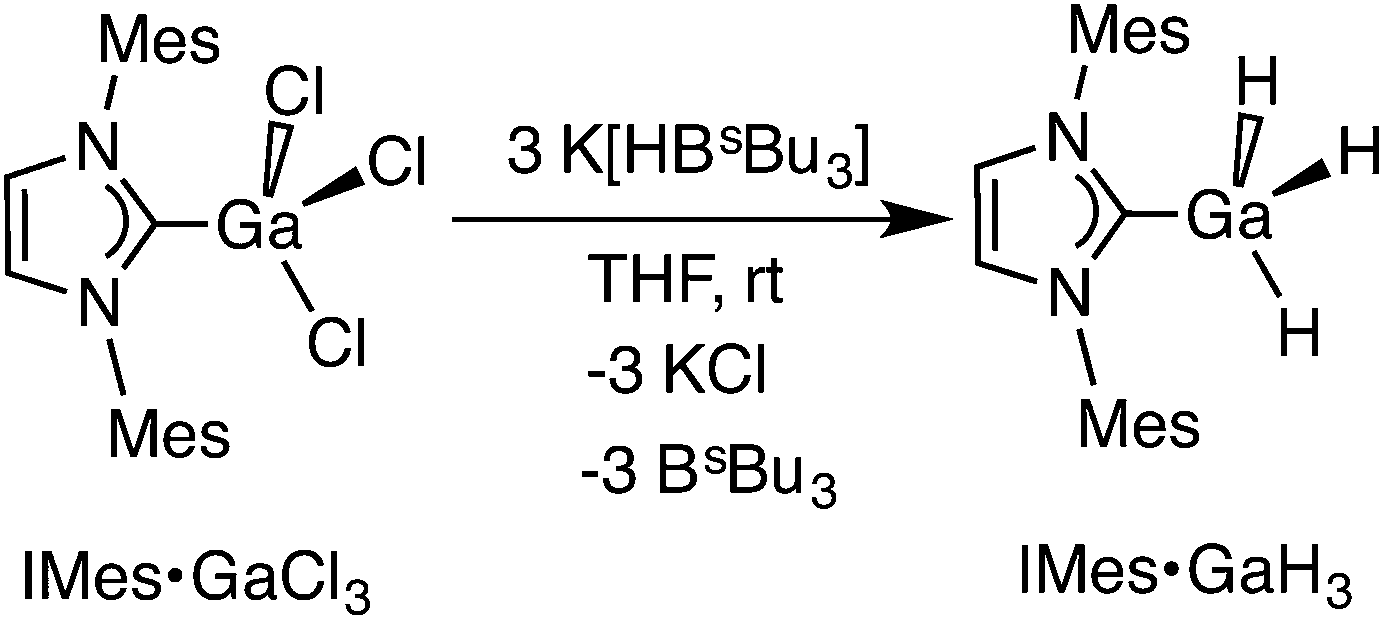 | (1) |
In order to synthesize the desired azido-gallane adduct IMes·GaH2N3, IMes·GaH2I was reacted with either Me3SiN3 or NaN3 in THF; however, no reaction transpired. A successful synthesis of IMes·GaH2N3 (1) was accomplished by combining IMes·GaH2I with the lipophilic azide salt [nBu4N]N3 in THF (Scheme 1). The 1H NMR spectrum of the resulting product (1) afforded a broad resonance at 4.52 ppm, due to the retention of two gallium-bound hydrides. Moreover, a diagnostic azide ṽ(N3) band for compound 1 was detected at 2084 cm−1, which matched well with the related asymmetric azide stretch at 2104 cm−1 found in Me3N·GaCl2N3.9b The composition of 1 was substantiated by X-ray crystallography (Fig. 1) and revealed the presence of a tetrahedral geometry about the Ga center. The C(NHC)–Ga bond distance in 1 is 2.041(4) Å and is similar to values found within known NHC-gallane complexes.10,12 The Ga–N bond length in 1 is 1.953(4) Å and comparable to the Ga–N bond distances in Christe's pentaazido gallate salt [PPh4]2[Ga(N3)5] [1.937(2)–2.049(2) Å].13
 | ||
| Scheme 1 Synthesis of IMes·GaH2N3 (1) and its conversion into IMes·GaH(OTf)2 (2). | ||
 | ||
| Fig. 1 Molecular structure of IMes·GaH2N3 (1) with thermal ellipsoids presented at a 30% probability level; all carbon bound hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Ga–C(1) 2.041(4), Ga–N(3) 1.953(4), Ga–H(1A) 1.52(5), Ga–H(1B) 1.52(5), N(3)–N(4) 1.199(6), N(4)–N(5) 1.146(6); N(3)–Ga–C(1) 101.41(18), N(3)–N(4)–N(5) 174.6(6). | ||
After the successful isolation of IMes·GaH2N3 (1), we explored the possible Lewis acid-triggered N2 elimination/hydride migration from Ga to N to form IMes·HGaNH·BArF3 (ArF = 3,5-(F3C)2C6H3); a similar transformation was previously used to yield an HBNH complex.5 Accordingly, IMes·GaH2N3 was combined with a stoichiometric amount of BArF3 followed by the heating of the reaction mixture to 80 °C in toluene. The 1H NMR spectrum of the resulting white solid indicated the formation of multiple carbene-containing products, however conclusive evidence for the formation of an HGaNH complex was not found. Instead, a salt consisting of the known [HBArF3]− anion14 was identified (Scheme 2) as one of the products in the mixture by 1H and 11B NMR spectroscopy. Attempts to isolate pure products by fractional crystallization were unsuccessful.
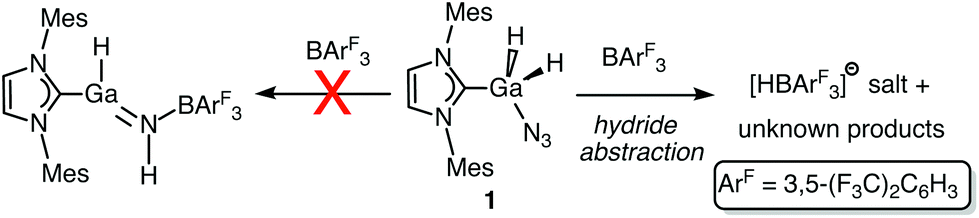 | ||
| Scheme 2 Reaction of IMes·GaH2N3 (1) with BArF3. | ||
It was previously reported that N2 loss/1,2-hydride migration in IPr·BH2N3 could also be instigated by addition of the strong electrophile MeOTf (OTf = OSO2CF3), leading to the formation of the N-methylated adduct [IPr·HBNHMe]OTf.5a However when IMes·GaH2N3 (1) was treated with one equivalent of MeOTf in CH2Cl2, multiple products were found according to 1H and 19F NMR spectroscopy. Increasing the stoichiometry of MeOTf to four molar equivalents resulted in clean formation of the new hydrido/triflate adduct IMes·GaH(OTf)2 (2) (Scheme 1) in a 80% yield as a colorless moisture-sensitive solid. Compound 2 was identified by a combination of X-ray crystallography (Fig. 2) and NMR spectroscopy. Due to the possible explosive nature of the likely by-product, MeN3 (Caution!),15 this reaction was not repeated again. However the outcome of the reaction was further confirmed by an independent synthesis of 2 by combining IMes·GaH3 with excess MeOTf (Scheme 1). The 19F NMR spectrum of IMes·GaH(OTf)2 (2) afforded a sharp resonance at −77.4 ppm that was assigned to covalently bound OTf groups. As shown in Fig. 2, the refined structure of 2 afforded the expected coordination of two OTf− substituents at gallium with corresponding Ga–O bond lengths of 1.9023(15) and 1.9186(16) Å. These bonds are significantly elongated relative to those [1.8021(1) Å] in the four-coordinate bis(hydroxy)gallium complex LGa(OH)2 (L = HC[C(Me)NDipp]2; Dipp = 2,6-iPr2C6H3),16 suggesting that the Ga-OTf interactions in 2 are weak in nature.
 | ||
| Fig. 2 Molecular structure of IMes·GaH(OTf)2 (2) with thermal ellipsoids presented at a 30% probability level; all carbon bound hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Ga–C(1) 1.9855(19), Ga–O(1) 1.9023(15), Ga–O(4) 1.9186(16), Ga–H(1) 1.45(3); O(1)–Ga–C(1) 104.12(7), C(1)–Ga–H(1) 123.2(11). | ||
The above results indicate that in the presence of a Lewis acid (BArF3) or electrophile MeOTf, IMes·GaH2N3 (1) undergoes preferential azide or hydride abstraction processes in place of N2 loss/hydride migration. The differing reactivity of the azido-gallane IMes·GaH2N3 (1) compared with IPr·BH2N3 is likely a consequence of the increased polarity and reactivity of Ga–N and Ga–H bonds due to the lower electronegativity of Ga in relation to B. Thus it appears that an alternate route to a molecular complex of HGaNH has to be devised.
In keeping with the theme of eventually generating molecular precursors to bulk gallium nitride, we prepared the gallium-silylamide complexes IMes·GaH2N(SiMe3)2 (3) and IPr·GaCl2N(SiMe3)2 (4). The amido-gallane complex IMes·GaH2N(SiMe3)2 (3) was prepared in a 93% yield as a white solid from the reaction of IMes·GaH2Cl12 with a stoichiometric amount of Li[N(SiMe3)2] (eqn (2)). The 1H NMR spectrum of 3 afforded a sharp up-field positioned resonance at 0.23 ppm due to the capping –N(SiMe3)2 group, while expected resonances for the gallium hydrides (4.51 ppm) and IMes ligand were also found. The crystallographically determined structure of 3 is found in Fig. 3; despite the presence of co-crystallized IMes·GaH(Cl)–N(SiMe3)2 as part of the crystalline lattice, bulk samples of 3 afforded both satisfactory elemental analyses and clean NMR spectra. Perhaps the most salient structural feature of 3 is the substantially longer C(NHC)–Ga length [2.0743(15) Å] in relation to that found in the bis(triflato)gallane IMes·GaH(OTf)2 (2) [1.9855(19) Å]; this is likely a consequence of the less Lewis acidic GaH2N(SiMe3)2 unit in 3 in relation to the GaH(OTf)2 moiety in 2. The nitrogen atom within the silylamido group in 3 [N(3); Fig. 3] is slightly pyramidalized as revealed by an angle sum (∑°N) value of 353.41(12)°. Due to the presence of the bulky SiMe3 groups at nitrogen, the Ga–N bond distance in 3 [1.9226(13) Å] is longer compared to the Ga–NMe2 bond length [1.816(2) Å] reported in {[(cyclopentyl)N-C6H4]2O}GaNMe2.17
 | (2) |
 | ||
| Fig. 3 Molecular structure of IMes·GaH2N(SiMe3)2 (3) with thermal ellipsoids presented at a 30% probability level; all carbon bound hydrogen atoms have been omitted for clarity; 3 co-crystallizes with ca. 15% of the mixed hydrido/chloride adduct IMes·GaH(Cl)–N(SiMe3)2. Selected bond lengths (Å) and angles (°) for 3: Ga–C(1) 2.0743(15), Ga–H(1(GA)) 1.503(18), Ga–H(2(GA)) 1.512(19), Ga–N(3) 1.9226(13); N(3)–Ga–C(1) 112.30(6), Si(1)–N(3)–Si(2) 123.25(8), Ga–N(3)–Si(1) 112.09(7), Ga–N(3)–Si(2) 118.07(7). | ||
A halogenated analogue of 3, IPr·GaCl2N(SiMe3)2 (4) was also readily prepared from the known adduct IPr·GaCl312b and one equivalent of Li[N(SiMe3)2] (eqn (2)). The molecular structure of IPr·GaCl2N(SiMe3)2 is found in Fig. 4 and displays similar overall structural features as the hydrido congener 3.
 | ||
| Fig. 4 Molecular structure of IPr·GaCl2N(SiMe2)2 (4) with thermal ellipsoids presented at a 30% probability level; all hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): Ga–C(1) 2.0570(15), Ga–N(3) 1.8932(13), Ga–Cl(1) 2.2054(4), Ga–Cl(2) 2.2047(5); N(3)–Ga–C(1) 119.05(6), Cl(1)–Ga–Cl(2) 103.54(2), Si(2)–N(3)–Si(1) 121.46(8). | ||
After the successful preparation of IMes·GaH2N(SiMe3)2 (3) and IPr·GaCl2N(SiMe3)2 (4), we decided to explore the possible formation of extended GaN structures9via thermolysis. To our surprise, both of these species are quite thermally stable and do not show any signs of HSiMe3, ClSiMe3 or HN(SiMe3)2 loss upon heating to 100 °C in toluene. Compound 3 was also stable upon microwave irradiation for 1.5 h at 130 °C in toluene, while under the same microwave conditions, compound 4 underwent partial decomposition to an [IPrH]+ salt (20%) and a new unidentified carbene-containing product (24%).
We then decided to promote possible intermolecular Ga–N bond forming processes by replacing the chloride substituents in IPr·GaCl2N(SiMe3)2 (4) with more labile OTf− groups (to form IPr·Ga(OTf)2N(SiMe3)2). Towards this goal, IPr·GaCl2N(SiMe3)2 (4) was combined with two equiv. of Ag[OTf] in CH2Cl2, however under these conditions the replacement of only one chloride transpired to form IPr·GaCl(OTf)N(SiMe3)2 (5). Compound 5 was obtained as a racemic mixture due to the presence of a chiral gallium center (eqn (3); Fig. 5). A sharp signal at −76.2 ppm in the 19F NMR spectrum of 5 indicated that the OTf group remained covalently bound to gallium in solution. Despite replacing one of the chlorine atoms with a OTf group, compound 5 was also thermally stable to 100 °C in toluene for 12 h. However, compound 5 underwent complete decomposition to several unidentified products upon microwave irradiation for 1.5 h at 130 °C in fluorobenzene.
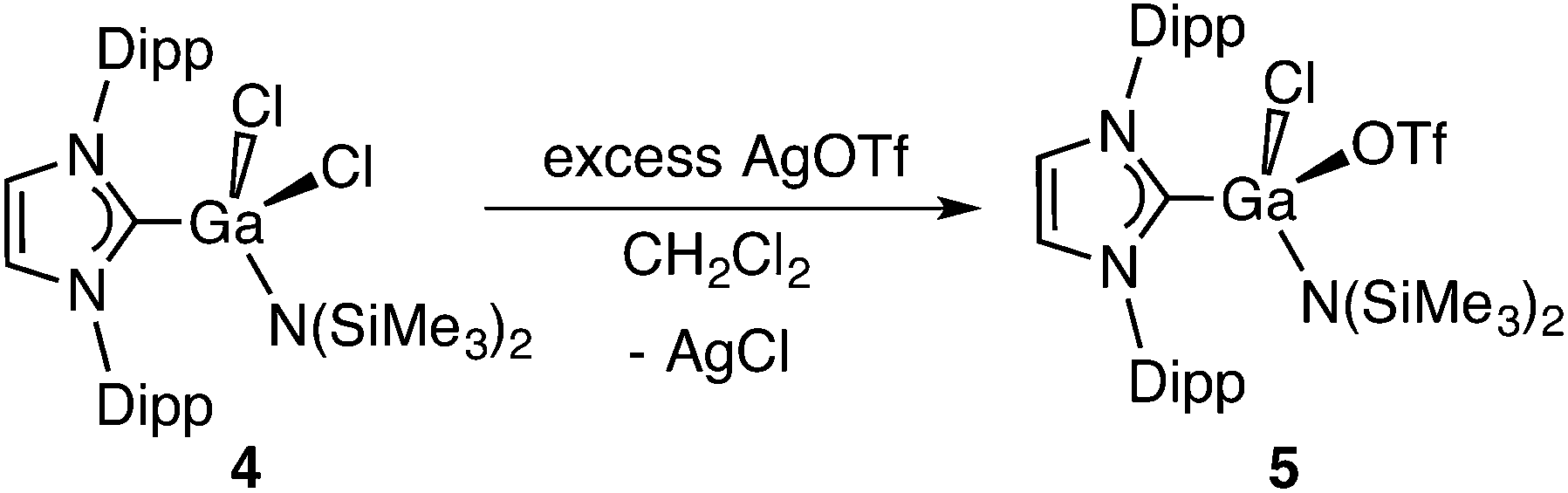 | (3) |
 | ||
| Fig. 5 Molecular structure of IPr·GaCl(OTf)N(SiMe2)2 (5) with thermal ellipsoids presented at a 30% probability level; all hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°) with parameters associated with a second molecule in the asymmetric unit listed in square brackets: Ga(A)–C(1) 2.042(3) [2.167(6)], Ga(A)–N(3A) 1.846(3) [1.840(9)], Ga(A)–Cl(A) 2.1668(11) [2.171(10)], Ga(A)–O(1) 1.969(2) [1.982(5)]; N(3A)–Ga(A)–C(1) 124.52(14) [129.1(11)], Cl(A)–Ga(A)–O(1) 102.10(9) [112.5(15)]. | ||
Conclusions
In this article, the successful isolation of N-heterocyclic carbene complexes of azido- and amido-gallanes has been described. These species represent members of a general compound class that could be eventually used to generate bulk gallium nitride under mild conditions (after suitable ligand modification). The reported gallium hydrides also have similar structural features as our recently reported active ketone hydrosilylation/borylation catalyst IPr·Zn(H)OTf·THF18a and thus we are now exploring the catalytic activity18 of these main group, NHC-supported, gallium hydrides in more detail.
Experimental
Materials and instrumentation
All reactions were performed using standard Schlenk techniques under an atmosphere of nitrogen or in an inert atmosphere glovebox (Innovative Technology, Inc.).19 Solvents were dried using a Grubbs-type solvent purification system manufactured by Innovative Technology, Inc., degassed (freeze–pump–thaw method), and stored under an atmosphere of nitrogen prior to use. K[HBsBu3] (1.0 M solution in THF), GaCl3, Li[N(SiMe3)2], and [nBu4N]N3 were purchased from Aldrich and used as received. NHC·GaX3 (NHC = IMes or IPr; X = Cl or I),12b,20 IMes·GaH2X (X = Cl or I)10a,c and BArF3 (ArF = 3,5-(F3C)2C6H3)21 were prepared according to literature procedures. 1H, 11B, 13C{1H}, and 19F NMR spectra were recorded on a Varian iNova-400 spectrometer and referenced externally to SiMe4 (1H and 13C{1H}), F3B·OEt2 (11B), and CFCl3 (19F) respectively. Elemental analyses were performed by the Analytical and Instrumentation Laboratory at the University of Alberta. Melting points were measured in sealed glass capillaries under nitrogen using a MelTemp apparatus and are uncorrected.X-ray crystallography
Crystals of suitable quality for X-ray diffraction studies were removed from a vial in a glovebox and immediately covered with a thin layer of hydrocarbon oil (Paratone-N). A suitable crystal was selected, mounted on a glass fiber, and quickly placed in a low temperature stream of nitrogen on an X-ray diffractometer.22 All data were collected at the University of Alberta using a Bruker APEX II CCD detector/D8 diffractometer using Cu Kα radiation with the crystals cooled to −100 °C. The data were corrected for absorption through Gaussian integration from the indexing of the crystal faces.23 Structures were solved using the direct methods program SHELXS-9724 (IPr·GaCl(OTf)N(SiMe3)2 (5)) or intrinsic phasing SHELXT24 (IMes·GaH2N3 (1), IMes·GaH(OTf)2 (2), IMes·GaH2N(SiMe3)2 (3), IPr·GaCl2N(SiMe3)2 (4)). Structure refinement was accomplished using either SHELXL-97 or SHELXL-2013.24 All carbon-bound hydrogen atoms were assigned positions on the basis of the sp2 or sp3 hybridization geometries of their attached carbon atoms, and were given thermal parameters 20% greater than those of their parent atoms.Synthetic procedures
Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (Discovery Grant and CREATE grants for E. R.; CREATE fellowship for A. K. S.), and the Canada Foundation for Innovation (CFI).References
- (a) C. Jones, Chem. Commun., 2001, 2293 RSC; (b) N. Kuhn and A. Al-Sheikh, Coord. Chem. Rev., 2005, 249, 829 CrossRef CAS; (c) Y. Wang and G. H. Robinson, Dalton Trans., 2012, 41, 337 RSC; (d) R. S. Ghadwal, R. Azhakar and H. W. Roesky, Acc. Chem. Res., 2013, 46, 444 CrossRef CAS PubMed; (e) L. K. Murphy, K. N. Robertson, J. D. Masuda and J. A. C. Clyburne, in N-Heterocyclic Carbenes, ed. S. P. Nolan, Wiley-VCH, Weinheim, 2014, pp. 427–497 Search PubMed; (f) E. Rivard, Dalton Trans., 2014, 43, 8577 RSC; (g) G. Prabusankar, A. Sathyanarayana, P. Suresh, C. N. Babu, K. Srinivas and B. P. R. Metla, Coord. Chem. Rev., 2014, 269, 96 CrossRef CAS; (h) C. Präsang and D. Scheschkewitz, Chem. Soc. Rev., 2016, 45, 900 RSC; (i) S. Würtemberger-Pietsch, U. Radius and T. B. Marder, Dalton Trans., 2016, 45, 5880 RSC.
- (a) D. Martin, M. Melaimi, M. Soleilhavoup and G. Bertrand, Organometallics, 2011, 30, 5304 CrossRef CAS PubMed; (b) K. C. Mondal, S. Roy and H. W. Roesky, Chem. Soc. Rev., 2016, 45, 1080 RSC; (c) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256 CrossRef CAS PubMed.
- (a) N. Kuhn, H. Bohnen, J. Kreutzberg, D. Bläser and R. Boese, J. Chem. Soc., Chem. Commun., 1993, 1136 RSC; (b) S. M. I. Al-Rafia, A. C. Malcolm, S. K. Liew, M. J. Ferguson, R. McDonald and E. Rivard, Chem. Commun., 2011, 47, 6987 RSC; (c) Y. Wang, M. Y. Abraham, R. J. Gilliard Jr., D. R. Sexton, P. Wei and G. H. Robinson, Organometallics, 2013, 32, 6639 CrossRef CAS; (d) R. S. Ghadwal, Dalton Trans., 2016, 45, 16081 RSC; (e) K. Powers, C. Hering-Junghans, R. McDonald, M. J. Ferguson and E. Rivard, Polyhedron, 2016, 108, 8 CrossRef CAS; (f) J.-S. Huang, W.-H. Lee, C.-T. Shen, Y.-F. Lin, Y.-H. Liu, S.-M. Peng and C.-W. Chiu, Inorg. Chem., 2016, 55, 12427 CrossRef CAS PubMed.
- (a) H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420 CrossRef CAS PubMed; (b) J. Böhnke, H. Braunschweig, W. C. Ewing, C. Hörl, T. Kramer, I. Krummenacher, J. Mies and A. Vargas, Angew. Chem., Int. Ed., 2014, 53, 9082 CrossRef PubMed.
- (a) A. K. Swarnakar, C. Hering-Junghans, K. Nagata, M. J. Ferguson, R. McDonald, N. Tokitoh and E. Rivard, Angew. Chem., Int. Ed., 2015, 54, 10666 CrossRef CAS PubMed; (b) A. K. Swarnakar, C. Hering-Junghans, M. J. Ferguson, R. McDonald and E. Rivard, Chem. Sci., 2016 10.1039/c6sc04893e.
- A. Solovyev, Q. Chu, S. J. Geib, L. Fensterbank, M. Malacria, E. Lacôte and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 15072 CrossRef CAS PubMed.
- For related donor–acceptor chemistry in the main group, see: (a) T. J. Marks, J. Am. Chem. Soc., 1971, 93, 7090 CrossRef CAS; (b) U. Vogel, A. Y. Timoshkin and M. Scheer, Angew. Chem., Int. Ed., 2001, 40, 4409 CrossRef CAS; (c) S. M. I. Al-Rafia, A. C. Malcolm, R. McDonald, M. J. Ferguson and E. Rivard, Angew. Chem., Int. Ed., 2011, 50, 8354 CrossRef CAS PubMed; (d) A. C. Filippou, B. Baars, O. Chernov, Y. N. Lebedev and G. Schnakenburg, Angew. Chem., Int. Ed., 2014, 53, 565 CrossRef CAS PubMed; (e) F. Dielmann, E. V. Peresypkina, B. Krämer, F. Hastreiter, B. P. Johnson, M. Zabel, C. Heindl and M. Scheer, Angew. Chem., Int. Ed., 2016, 55, 14833 CrossRef CAS PubMed; (f) Y.–P. Zhou, M. Karni, S. Yao, Y. Apeloig and M. Driess, Angew. Chem., Int. Ed., 2016, 55, 15096 CrossRef CAS PubMed.
- (a) H. Amano, N. Sawaki, I. Akasaki and Y. Toyoda, Appl. Phys. Lett., 1986, 48, 353 CrossRef CAS; (b) S. Nakamura, Y. Harada and M. Seno, Appl. Phys. Lett., 1991, 58, 2021 CrossRef CAS; (c) H. Morkoc and S. N. Mohammad, Science, 1995, 267, 51 CAS.
- (a) D. A. Neumayer, A. H. Cowley, A. Decken, R. A. Jones, V. Lakhotia and J. G. Ekerdt, J. Am. Chem. Soc., 1995, 117, 5893 CrossRef CAS; (b) J. Kouvetakis, J. McMurran, P. Matsunaga, M. O'Keeffe and J. L. Hubbard, Inorg. Chem., 1997, 36, 1792 CrossRef CAS PubMed; (c) S. D. Dingman, N. P. Rath and W. E. Buhro, Dalton Trans., 2003, 3675 RSC; (d) O. T. Beachley Jr., J. C. Pazik and M. J. Noble, Organometallics, 1998, 17, 2121 CrossRef.
- (a) M. L. Cole, S. K. Furfari and M. Kloth, J. Organomet. Chem., 2009, 694, 2934 CrossRef CAS; (b) C. D. Abernethy, M. L. Cole and C. Jones, Organometallics, 2000, 19, 4852 CrossRef CAS; (c) R. J. Baker and C. Jones, Appl. Organomet. Chem., 2003, 17, 807 CrossRef CAS; (d) M. D. Francis, D. E. Hibbs, M. B. Hursthouse, C. Jones and N. A. Smithies, J. Chem. Soc., Dalton Trans., 1998, 3249 RSC.
- A. E. Shirk, D. F. Shriver, J. A. Dilts and R. W. Nutt, Inorg. Synth., 1977, 17, 45 CrossRef CAS.
- (a) G. E. Ball, M. L. Cole and A. I. McKay, Dalton Trans., 2012, 41, 946 RSC; (b) N. Marion, E. C. Escudero-Adán, J. Benet-Buchholz, E. D. Stevens, L. Fensterbank, M. Malacria and S. P. Nolan, Organometallics, 2007, 26, 3256 CrossRef CAS.
- R. Haiges, J. A. Boatz, J. M. Williams and K. O. Christe, Angew. Chem., Int. Ed., 2011, 50, 8828 CrossRef CAS PubMed.
- (a) T. J. Herrington, A. J. W. Thom, A. J. P. White and A. E. Ashley, Dalton Trans., 2012, 41, 9019 RSC; (b) R. J. Blagg, E. J. Lawrence, K. Resner, V. S. Oganesyan, T. J. Herrington, A. E. Ashley and G. G. Wildgoose, Dalton Trans., 2016, 45, 6023 RSC.
- (a) M. E. Burns and R. H. Smith Jr., Chem. Eng. News, 1984, 62, 2 CAS; (b) A. Hassner, M. Stern, H. E. Gottlieb and F. Frolow, J. Org. Chem., 1990, 55, 2304 CrossRef CAS.
- V. Jancik, L. W. Pineda, A. C. Stückl, H. W. Roesky and R. Herbst-Irmer, Organometallics, 2005, 24, 1511 CrossRef CAS.
- F. Hild and S. Dagorne, Organometallics, 2012, 31, 1189 CrossRef CAS.
- For related work on using main group species as hydrosilylation/borylation catalysts, see: (a) P. A. Lummis, M. R. Momeni, M. W. Lui, R. McDonald, M. J. Ferguson, M. Misklozie, A. Brown and E. Rivard, Angew. Chem., Int. Ed., 2014, 53, 9347 CrossRef CAS PubMed; (b) K. Revunova and G. I. Nikonov, Dalton Trans., 2015, 44, 840 RSC; (c) C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238 CrossRef CAS; (d) A.-K. Wiegand, A. Rit and J. Okuda, Coord. Chem. Rev., 2016, 314, 71 CrossRef CAS; (e) M. M. D. Roy, M. J. Ferguson, R. McDonald and E. Rivard, Chem. – Eur. J., 2016, 22, 18236 CrossRef CAS PubMed; (f) T. J. Hadlington, M. Hermann, G. Frenking and C. Jones, J. Am. Chem. Soc., 2014, 136, 3028 CrossRef CAS PubMed; (g) A. Bismuto, S. P. Thomas and M. J. Cowley, Angew. Chem., Int. Ed., 2016, 55, 15356 CrossRef CAS PubMed Initial studies show that 2 is a competent catalyst for the hydrosilylation of benzophenone at room temperature.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518 CrossRef CAS.
- S. Tang, J. Monot, A. El-Hellani, B. Michelet, R. Guillot, C. Bour and V. Gandon, Chem. – Eur. J., 2012, 18, 10239 CrossRef CAS PubMed.
- E. L. Kolychev, T. Bannenberg, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem. – Eur. J., 2012, 18, 16938 CrossRef CAS PubMed.
- H. Hope, Prog. Inorg. Chem., 1994, 41, 1 CrossRef CAS.
- R. H. Blessing, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1995, 51, 33 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2007, 64, 112 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of crystallographic data for compounds 1–5. CCDC 1519736–1519740. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt04595b |
| This journal is © The Royal Society of Chemistry 2017 |