
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAryl-NHC-group 13 trimethyl complexes: structural, stability and bonding insights†
Melissa M.
Wu
a,
Arran M.
Gill
b,
Lu
Yunpeng
a,
Li
Yongxin
a,
Rakesh
Ganguly
a,
Laura
Falivene
*c and
Felipe
García
 *a
*a
aSchool of Physical and Mathematical Sciences, Division of Chemistry and Biological Chemistry, Nanyang Technological University, 21 Nanyang Link, Singapore, 637371, Singapore. E-mail: Fgarcia@ntu.edu.sg; Fax: (+65)67911961
bChemistry, Faculty of Natural and Environmental Sciences, University of Southampton, Highfield Southampton, SO17 1BJ, UK
cKing Abdullah University of Science and Technology, Building 9, Level 4, Room #4358, Thuwal, 23955-6900, Saudi Arabia. E-mail: laura.falivene@kaust.edu.sa
First published on 14th December 2016
Abstract
Treatment of aromatic N-substituted N-heterocyclic carbenes (NHCs) with trimethyl-gallium and -indium yielded the new Lewis acid–base adducts, IMes·GaMe3 (1), SIMes·GaMe3 (2), IPr·GaMe3 (3), SIPr·GaMe3 (4), IMes·InMe3 (5), SIMes·InMe3 (6), IPr·InMe3 (7), and SIPr·InMe3 (8), with all complexes being identified by X-ray diffraction, IR, and multinuclear NMR analyses. Complex stability was found to be largely dependent on the nature of the constituent NHC ligands. Percent buried volume (%VBur) and topographic steric map analyses were employed to quantify and elucidate the observed trends. Additionally, a detailed bond snapping energy (BSE) decomposition analysis focusing on both steric and orbital interactions of the M–NHC bond (M = Al, Ga and In) has been performed.
Introduction
Over the past thirty years, the use of Arduengo carbenes to stabilize transition metal compounds for organic syntheses has been intensively studied.1,2 Conversely, in the case of N-heterocyclic carbene group 13 metal complexes,3 only a limited range of compounds have been applied to organic transformations,3b,4 despite having shown excellent catalytic activity in ring opening polymerization reactions (ROPs).4a,c Previous studies have shown that slight modifications within these complexes can result in drastic changes in their reactivity towards organic transformations – a good example of this being the greater product yields and selectivity displayed by IMes·AlH2Cl over IMes·AlHCl2 in hydroalumination reactions on carbonyl or epoxide containing substrates.3b Cole et al. have attributed this observation to a stronger Al–H bond and increased steric bulk in IMes·AlHCl2 resulting in poorer catalytic activity. Reports by Hevia et al.5a on the structure, stability and isomerization reactions between normal (n) and abnormal (a) NHC–gallium alkyl complexes further highlight the importance of steric and electronic factors in the stability and accessibility of both normal and abnormal NHC main group complexes. Whilst Dagorne et al.5b described the normal-to-abnormal NHC rearrangement and small molecule activation on the aluminium, gallium and indium triad. With much yet to be explored, the synthesis, characterization, and reactivity of new NHC group 13 complexes remain an exciting area for the main group and organic chemists alike. Our group has previously reported that minimal adjustment to the steric properties of the constituent NHC moiety in trimethylaluminium complexes can have a profound effect on their stability (Fig. 1, A–D). This prompted us to seek to quantify and rationalize the structure–stability–reactivity relationships of heavier group 13 NHC counterparts, using the commonly employed IMes, SIMes, IPr and SIPr carbenes as case studies.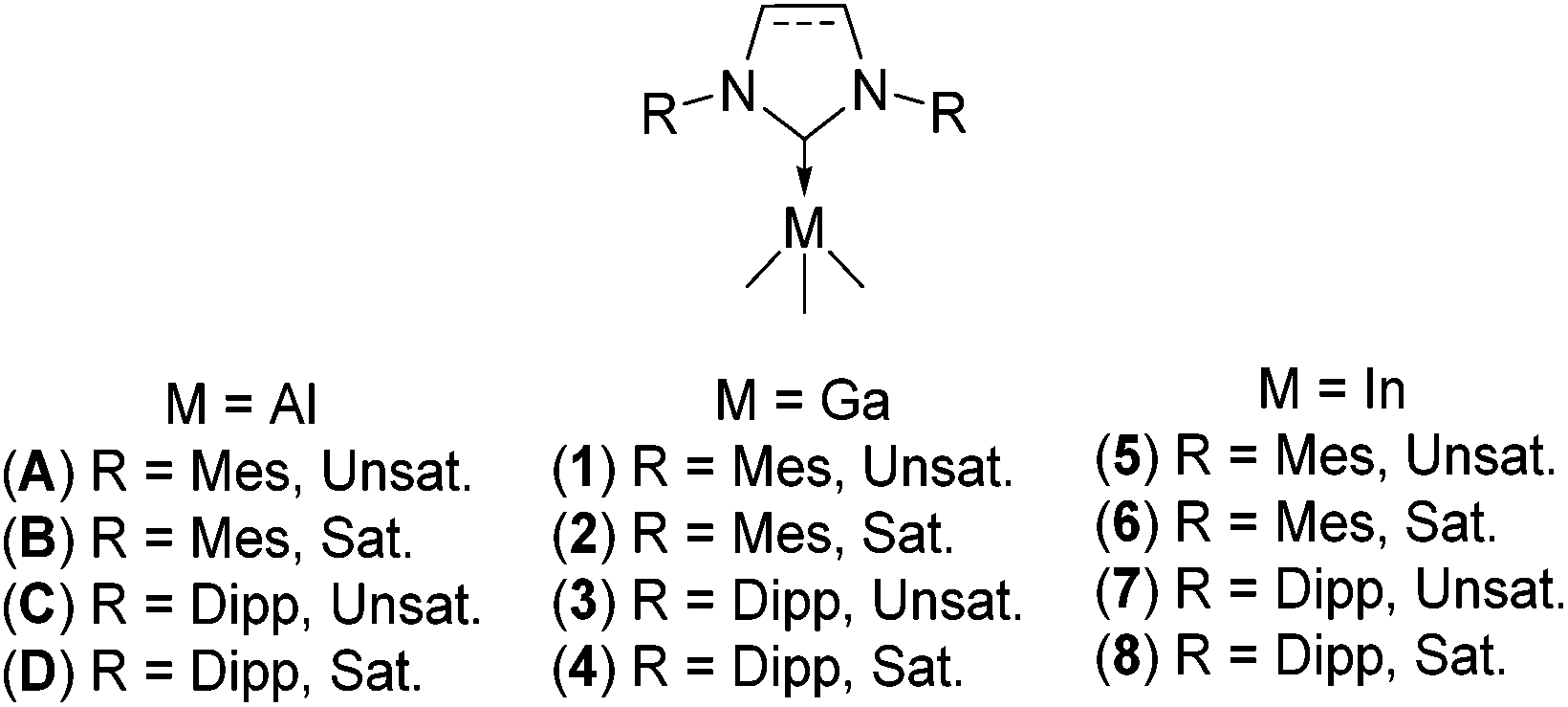 | ||
| Fig. 1 New NHC trimethyl-gallium, -indium, and -aluminium complexes. IMes·GaMe3 (1), SIMes·GaMe3 (2), IPr·GaMe3 (3), SIPr·GaMe3 (4), IMes·InMe3 (5), SIMes·InMe3 (6), IPr·InMe3 (7), SIPr·InMe3 (8), IMes·AlMe3 (A), SIMes·AlMe3 (B), IPr·AlMe3 (C), and SIPr·AlMe3 (D).6b | ||
Herein, we firstly report the synthesis and characterization of a series of aromatic N-substituted NHC gallium and indium alkyl complexes. By combining X-ray crystallographic and spectroscopic studies with theoretical calculations, we then assess the stability and bonding characteristics of these complexes. An insightful comparison between group 13-NHC complexes with transition metal–NHC and –PHC (using Al–IMes, Pd–IMes and Pd(P)–IMes as case studies) is also provided.
Results and discussion
Synthesis of complexes 1–8
The general synthetic route for the synthesis of these complexes is via the formation of Lewis acid–base adducts (Scheme 1).4–7 Hence, treatment of 1 equivalent of carbene (IMes, SIMes, IPr and SIPr) with trimethylgallium8 or indium9 resulted in the formation of their respective complexes IMes·GaMe3 (1), SIMes·GaMe3 (2), IPr·GaMe3 (3), SIPr·GaMe3 (4), IMes·InMe3 (5), SIMes·InMe3 (6), IPr·InMe3 (7), and SIPr·InMe3 (8) as shown in Fig. 1. Isolation of compounds was performed by crystallization in ether or toluene at room temperature or at 0 °C.9 | ||
| Scheme 1 Synthetic strategy for the NHC adducts. Mes (2,4,6-trimethylphenyl); Dipp (2,6-diisopropylphenyl). | ||
All compounds are highly air- and moisture-sensitive and traces of decomposition were consistently observed during their characterization. Hence, all attempts of elemental analyses were unsuccessful. Moreover, this was also observed for 4 and 8 in the solid state, where argon-gas-stored samples slowly decomposed at room temperature.
Crystallographic studies of complexes 1–8
Complexes 2–6 recrystallized from solution as two crystallographically independent, but chemically equivalent, molecules and only one molecule will be described herein. Complexes 6 and 8 are the first structurally characterized trimethylindium complexes containing saturated NHC moieties. Previously reported gallium and indium NHC species are, for the most part, heteroleptic complexes (see Fig. 2 and 3). Furthermore, only four trimethylgallium complexes have been previously structurally characterized (i.e., E,4fG,4aI![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4a and J,4c see Fig. 2).3,4,10 Generally, heavy group 13 NHC complexes adopt a four coordinate, distorted tetrahedral geometry at the metal centre, with the exception of indium complexes R, S and T (IMes·InMe2Cl, IMes·InMe2OTf and IMes·InMe(OTf)2, respectively).4d Despite being four-coordinate, the indium centre of complex R does not conform to a distorted tetrahedral geometry due to the weak carbene chloride interaction that causes the chloride to lie orthogonal to the carbene plane.4d In the case of complexes S and T, the indium centres interact with an additional neighbouring triflate substituent belonging to an adjacent molecule, hence directing the complex geometry towards a pentacoordinate trigonal bipyramidal geometry in the solid state.4d
4a and J,4c see Fig. 2).3,4,10 Generally, heavy group 13 NHC complexes adopt a four coordinate, distorted tetrahedral geometry at the metal centre, with the exception of indium complexes R, S and T (IMes·InMe2Cl, IMes·InMe2OTf and IMes·InMe(OTf)2, respectively).4d Despite being four-coordinate, the indium centre of complex R does not conform to a distorted tetrahedral geometry due to the weak carbene chloride interaction that causes the chloride to lie orthogonal to the carbene plane.4d In the case of complexes S and T, the indium centres interact with an additional neighbouring triflate substituent belonging to an adjacent molecule, hence directing the complex geometry towards a pentacoordinate trigonal bipyramidal geometry in the solid state.4d


The molecular structures of compounds 1–8 revealed the formation of four-carbon-coordinated gallium and indium atoms attached to three alkyl groups, and the presence of a neutral carbene moiety (see Fig. 4 and 5). The distorted tetrahedral geometry at the gallium and indium centres is evidenced by the C–M–C bond angles that range from 100.0° to 115.3° and 99.6° to 119.1° for gallium and indium, respectively, with metal to carbene carbon (M–Ccarbene) bond lengths ranging from 2.111 Å to 2.137 Å, and 2.301 Å to 2.342 Å for gallium and indium, respectively. In the case of 1–4, the Ga–Ccarbene bond lengths are consistent with the previously reported trimethylgallium complexes (cf. 2.130(2) Å, 2.105(2) Å, 2.132(3) Å and 2.121(3) Å and for E, G, I, and J, respectively) (see Table 1).4a,e,f In agreement with our previous observations for the lighter trimethylaluminium counterparts, similar M–Ccarbene bond distances between SIPr·AlMe3 and the less sterically encumbered IiPrMe·AlMe3 (2.127(2) Å and 2.124(6) Å, respectively)4f,6b,11,12 were observed. The Ga–Ccarbene bond distance in 4 is also comparable to that of IiPrMe·GaMe3 (E) (2.137(2) Å and 2.130(2) Å, respectively).4f
 | ||
| Fig. 4 Molecular structures of IMes·GaMe3 (1), SIMes·GaMe3 (2), IPr·GaMe3 (3) and SIPr·GaMe3 (4). Selected bond lengths (Å) and angles (°) for 1: Ga(1)–C(1) 2.111(2), C(1)–Ga(1)–C(2) 108.8(1), C(1)–Ga(1)–C(3) 105.8(1), C(2)–Ga(1)–C(3) 111.1(1), C(3)–Ga(1)–C(3A) 114.0(2). Selected bond lengths (Å) and angles (°) for 2: Ga(1)–C(1) 2.124(5), C(1)–Ga(1)–C(2) 105.0(2), C(1)–Ga(1)–C(3) 110.8(2), C(1)–Ga(1)–C(4) 106.8(2), C(2)–Ga(1)–C(3) 112.5(2), C(2)–Ga(1)–C(4) 113.1(2), C(3)–Ga(1)–C(4) 108.5(3). Selected bond lengths (Å) and angles (°) for 3: Ga(1)–C(1) 2.105(4), C(1)–Ga(1)–C(2) 101.3(1), C(1)–Ga(1)–C(3) 110.7(1), C(1)–Ga(1)–C(4) 108.4(2), C(2)–Ga(1)–C(3) 115.3(2), C(2)–Ga(1)–C(4) 112.4(2), C(3)–Ga(1)–C(4) 108.3(2). Selected bond lengths (Å) and angles (°) for 4: Ga(1)–C(1) 2.137(2), C(1)–Ga(1)–C(2) 100.0(1), C(1)–Ga(1)–C(3) 111.2(1), C(1)–Ga(1)–C(4) 107.5(1), C(2)–Ga(1)–C(3) 110.4(1), C(2)–Ga(1)–C(4) 115.1(1), C(3)–Ga(1)–C(4) 112.1(1). Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms have been omitted for clarity. | ||
 | ||
| Fig. 5 Molecular structures of IMes·InMe3 (5), SIMes·InMe3 (6), IPr·InMe3 (3) and SIPr·InMe3 (8). Selected bond lengths (Å) and angles (°) for 5: In(1)–C(1) 2.304(8), C(1)–In(1)–C(2) 105.5(3), C(1)–In(1)–C(3) 105.2(3), C(1)–In(1)–C(4) 104.3(3), C(2)–In(1)–C(3) 114.7(3), C(2)–In(1)–C(4) 111.0(4), C(3)–In(1)–C(4) 114.9(4). Selected bond lengths (Å) and angles (°) 6: In(1)–C(1) 2.316(8), C(1)–In(1)–C(2) 105.9(3), C(1)–In(1)–C(3) 109.2(3), C(1)–In(1)–C(4) 101.8(3), C(2)–In(1)–C(3) 109.7(4), C(2)–In(1)–C(4) 114.1(3), C(3)–In(1)–C(4) 115.3(3). Selected bond lengths (Å) and angles (°) for 7: In(1)–C(1) 2.309(2), C(1)–In(1)–C(2) 106.8(1), C(1)–In(1)–C(3) 108.6(1), C(1)–In(1)–C(4) 101.3(1), C(2)–In(1)–C(3) 111.4(1), C(2)–In(1)–C(4) 114.5(1), C(3)–In(1)–C(4) 113.3(1). Selected bond lengths (Å) and angles (°) for 8: In(1)–C(1) 2.342(2), C(1)–In(1)–C(2) 99.6(1), C(1)–In(1)–C(3) 104.8(1), C(1)–In(1)–C(4) 108.5(1), C(2)–In(1)–C(3) 119.1(1), C(2)–In(1)–C(4) 109.8(1), C(3)–In(1)–C(4) 113.5(1). Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms have been omitted for clarity. | ||
| Formulae | Complex | M–Ccarbene [Å] | |
|---|---|---|---|
| a See abbreviations. | |||
| 1 | IMes·GaMe3 | 1 | 2.111(2) |
| 2 | SIMes·GaMe3 | 2 | 2.124(5) |
| 3 | IPr·GaMe3 | 3 | 2.105(4) |
| 4 | SIPr·GaMe3 | 4 | 2.137(2) |
| 5 | IMes·InMe3 | 5 | 2.304(7) |
| 6 | SIMes·InMe3 | 6 | 2.316(8) |
| 7 | IPr·InMe3 | 7 | 2.309(2) |
| 8 | SIPr·InMe3 | 8 | 2.342(2) |
| 9 | IMes·AlMe3 |
A6b |
2.098(2) |
| 10 | SIMes·AlMe3 |
B6b |
2.112(6) |
| 11 | IPr·AlMe3 |
C6b |
2.103(3) |
| 12 | SIPr·AlMe3 |
D6b |
2.127(2) |
| 13 | IiPrMe·GaMe3a |
E4f |
2.130(2) |
| 14 | IMes·GaMe2OMe |
H4a |
2.089(2) |
| 15 | SIPr·GaMe3 |
I4a |
2.132(3) |
| 16 | SIMes[(CH2)2]L·GaMe2 |
L4b |
2.079(1) |
| 17 | SIMes[(CH2)3]L·GaMe2·GaMe3 |
M4b |
2.087(1) |
| 18 | SIMes[Ar’]L·GaMe2 |
N4b |
2.080(1) |
| 19 | SIMes[Ar’]L·GaMe2·GaMe3 |
N′4b |
2.070(2) |
| 20 | SIPr[Ar′]L·GaMe2 |
O4b |
2.066(1) |
| 21 | SIPr[Ar′′]L·GaMe2 |
P4b |
2.056(1) |
| 22 | IMes·InMe2Cl |
R4d |
2.267(2) |
| 23 | IMes·InMe2OTf |
S4d |
2.264(2) |
| 24 | IMes·InMe(OTf)2 |
T4d |
2.183(2) |
Spectroscopic studies of complexes 1–8
The 1H and 13C{1H} NMR spectra obtained for complexes 1–8 are consistent with the low-temperature X-ray crystallographic analyses. The 1H NMR spectra for the gallium and indium complexes display singlets ranging from δH −0.56 to −0.60, and δH −0.52 to −0.62 ppm, respectively, which is indicative of the presence of the methyl substituents on the metal centre. This is further corroborated by 13C{1H} NMR spectra which display singlets at δC −5.2 to −6.1 and δC −9.6 to −11 ppm for gallium and indium complexes, respectively. Furthermore, the IR spectra of 1–4 show a relatively strong stretching signal at around 524 cm−1, consistent with the presence of the methyl groups on the metal centre.14 Unfortunately, in the case of indium analogues, no suitable IR stretching signals were clearly observed, since the In–Me range falls within a high noise background region (i.e., ∼400 cm−1).14 An upfield shift of the Ccarbene signals provides further confirmation of the complexes, as observed with other reported trimethylgallium and indium complexes (Table 2).6b,15 Despite several attempts, no Ccarbene signal was obtained for complex 5, presumably due to the large quadrupole moment of the indium centre.4,7| Complexes | 1H [InCH3] (ppm) | 13C [MCcarbene] (ppm) | 13C [Ccarbene]a (ppm) |
|---|---|---|---|
|
a 13C NMR chemical shifts were obtained from ref. 15.
|
|||
| 1 | −0.56 | 181.7 | 219.4 |
| 2 | −0.60 | 206.1 | 243.8 |
| 3 | −0.59 | 184.5 | 220.4 |
| 4 | −0.58 | 209.0 | 244.0 |
| 5 | −0.52 | — | 219.4 |
| 6 | −0.58 | 209.3 | 243.8 |
| 7 | −0.60 | 186.8 | 220.4 |
| 8 | −0.62 | 211.7 | 244.0 |
|
E4f |
−0.10 | 176.8 | 207.5 |
|
F5b |
0.27 | 183.7 | 212.9 |
|
Q5b |
0.21 | 183.4 | 212.9 |
Lewis acid–Lewis base properties
The majority of previously reported NHC–gallium and –indium complexes comprise halide and hydride derivatives (NHC·MH3−nCln; M = Ga and In; n = 1, 2).7 For example, the chlorogallane complexes IMes·GaH2Cl and IMes·GaHCl2 displayed increased Lewis acidity of the metal centre in the presence of increasing electron withdrawing groups (i.e., chloride atoms), consequently shortening the Ga–Ccarbene bond length, and strengthening the gallium hydride bond (Table S3,† entries 10 and 11).7i The same effect has also been observed for the lighter counterparts IMes·AlH2Cl and IMes·AlHCl2, which result in significantly different catalytic activities in hydroalumination reactions (vide supra).7g With the inclusion of the herein reported trimethylgallium and indium complexes, a more complete perspective can be gained regarding substituent effects on the structural properties of group 13 NHC complexes by comparison with previously reported halide and hydride counterparts. As expected, in complexes 1 to 4, the trimethylgallium moiety proves to be a poorer Lewis acid compared to its hydride and halide analogues. This is evident from the Ga–Ccarbene bond distances reported for the IMes, SIMes and IPr compounds (see the ESI,† entries 1–3 and 9–14). A similar trend can be established in the case of indium complexes – Lewis acid strength in the increasing order: MMe3 < MH3 < MX3. The reported In–Ccarbene bond distances are shown in Table S3† (entries 5, 7 and 15–19).7The Ccarbene13C NMR chemical shift is sensitive to the extent of the metal centre Lewis acidity, which in turn is directed by the donor ability of the ligands surrounding the metal centre. The majority of previously reported NHC gallium and indium complexes failed to exhibit an M–Ccarbene signal in their 13C{1H} NMR spectra, due to the quadrupolar moment of the metal centre.4,7 Indeed, Ccarbene signals of only one gallium and one indium NHC complex (IMes·GaClH2 and IMes·InMe2Cl) have been reported (δC 172.5 and 177.5 ppm, respectively).4d,7i The Ccarbene13C{1H} NMR signal of the complexes 1–4 and 6–8 (see Table 2) are relatively downfield compared to the gallium and indium complexes IMes·GaClH2 and IMes·InMe2Cl. This is anticipated, given that the presence of the chlorido ligand(s) on the metal centre exerts a strong electron-withdrawing effect and further corroborates that the MMe3 moiety (M = Ga and In) is a poorer electron acceptor compared to MH3 and MX3. Jones et al. showed that reactions of potentially chelating bidentate bis-NHC or monodentate NHC ligands in 1:1 and 1:2 ratios, respectively, with indium halides produce pentacoordinate trigonal bipyramidal chelate or 2:1 NHC adducts, whereas hydrides only form monomeric tetracoordinate tetrahedral compounds due to the higher Lewis acidity of the former.7o,t Since trimethylindium derivatives are expected to be poorer Lewis acids than indium halides, only monomeric tetracoordinate tetrahedral species would be anticipated. Therefore, two equivalents of the IMes free carbene were reacted with trimethylindium under various experimental conditions; however, despite several attempts pentacoordinate trigonal bipyramidal adducts were unable to be isolated supporting our initial prediction.
Stability studies
We have reported that for the lighter trimethylaluminium compounds A–D, the NHC steric bulk plays a significant role in determining the resulting complex stability. Complexes containing less sterically hindered NHC moieties, i.e.A and B (IMes and SIMes, respectively), are relatively stable in their solid state and can be stored for prolonged periods under nitrogen without any signs of decomposition, whereas complexes containing bulkier NHC ligands, i.e.C and D (IPr and SIPr, respectively), slowly decompose to their respective imidazolylidene and imidazolinylidene over time.6b These stability differences were attributed to the larger percent buried volume (%VBur) occupied by the NHC ligands of C and D (IPr and SIPr), compared to those of A and B (IMes and SIMes),6b,16 indicating that subtle variations in the steric bulk of the NHC substituent (Δ%VBurca. 2–4%) profoundly impact the overall complex stability. Additional insights gleaned by Hevia et al.5a and Dagorne et al.5b showed that bulky nNHC group 13 complexes, such as IPrGa·(CH2SiMe3)3, ItBu·GaMe3 (F) and ItBu·InMe3 (Q) all isomerize to their respective aNHC counterparts, with the latter two isomerizing too rapidly to allow characterization in their normal form. Theoretical DFT calculations performed for normal and abnormal model complexes of F and Q revealed that the latter are more stable with a Gibbs free energy of −24.6 kJ mol−1 and −5.8 kJ mol−1 for nFvs. aF and nQvs. aQ, respectively.The mechanism proposed for the model complex IPrGa·(CH2SiMe3)3 involves an initial dissociation to generate the free carbene and subsequent formation of the abnormal carbene species. In the case of our complexes C and D, solid crystalline samples stored under nitrogen showed decomposition into the free carbene alongside other unidentifiable species in their NMR spectra.6b In the case of the heavier Ga and In counterparts, compounds 1–8 showed relatively greater stability compared to their lighter analogues, with signs of decomposition observed only in the case of compounds 4 and 8. However, signals indicative of the formation of abnormal species for the reported metal complexes were not observed throughout our 1H NMR studies.
In order to elucidate the stability trends within the triad, %VBur calculations were undertaken with the M–Ccarbene bond distance fixed as the value obtained from our X-ray studies, and the bond length set at 2.0 Å – to enable a comparison between the various NHC ligands unbiased by variable M–NHC bond distances (see Table 3).16 In agreement with the calculated dissociation energies, the %VBur increases gradually from 1 to 4 and 5 to 8 for the gallium and indium complexes, respectively. Consequently, the relatively low stability observed for 4 and 8 may be qualitatively rationalized by the larger volume occupied by isopropylphenyl groups compared to the mesityl groups present in the NHC moieties.6b In a quantitative comparison between stable and unstable complexes – i.e., IPr vs. SIPr, 3vs.4 and 7vs.8 – ca. 8.5% and 10.6% Δ%VBur variances are observed for Ga and In, respectively – slightly greater than those found for their lighter Al counterparts (ca. 4%).6b
| Entries | Complexes | M–Ccarbene [Å] | M–Ccarbene [Å] | %VBur | %VBur | %VBur | %VBur | E diss (kJ mol−1) |
|---|---|---|---|---|---|---|---|---|
| X-Ray | DFT | R = X-ray | R = 2.0 Å (X-ray) | R = DFT | R = 2.0 Å (DFT) | |||
| %VBur Me groups on IPr–GaR3 = 48.7. %VBur CH2SiMe3 groups on IPr–GaR3 = 64.5.a %VBur was performed on DFT optimized structure (No single crystal X-ray structures were obtained). | ||||||||
| 1 | 1 | 2.111(2) | 2.201 | 27.9 | 29.6 | 29.5 | 32.7 | 76.7 |
| 2 | 2 | 2.125(5) | 2.231 | 31.8 | 34.1 | 30.2 | 33.9 | 69.0 |
| 3 | 3 | 2.105(4) | 2.213 | 34.0 | 36.2 | 30.9 | 34.3 | 65.2 |
| 4 | 4 | 2.137(2) | 2.233 | 35.1 | 39.3 | 31.9 | 35.6 | 52.8 |
| 5 | 5 | 2.301(8) | 2.428 | 28.5 | 34.0 | 25.7 | 33.3 | 73.9 |
| 6 | 6 | 2.316(8) | 2.453 | 29.2 | 34.9 | 26.3 | 34.4 | 66.8 |
| 7 | 7 | 2.309(2) | 2.446 | 30.2 | 35.7 | 27.1 | 35.1 | 61.5 |
| 8 | 8 | 2.342(2) | 2.478 | 31.4 | 39.5 | 27.7 | 36.2 | 51.3 |
| 9 |
A6b |
2.098(2) | 2.162 | 31.7 | 33.7 | 28.9 | 32.8 | 105.2 |
| 10 |
B6b |
2.112(6) | 2.188 | 32.0 | 34.1 | 30.4 | 33.8 | 95.6 |
| 11 |
C6b |
2.103(3) | 2.164 | 33.1 | 35.0 | 31.2 | 34.3 | 93.5 |
| 12 |
D6b |
2.127(2) | 2.190 | 36.1 | 38.5 | 32.0 | 35.5 | 77.8 |
| 13 |
E4f |
2.130(2) | 2.165 | — | — | 25.5 | 27.8 | 90.3 |
| 14a |
F5b |
— | 2.316 | — | — | 31.6 | 36.7 | 34.2 |
| 15a |
Q5b |
— | 2.558 | — | — | 28.4 | 37.5 | 20.5 |
| 16 | IPr·Ga(CH2SiMe3)35a |
2.196(2) | 2.301 | 31.9 | 36.1 | 26.2 | 31.6 | 23.3 |
To gain a more thorough insight into the NHC structure–stability relationships, we calculated the DFT optimized structures for complexes 1–8 and our previously reported complexes A–D. For completeness, we extended this study to include IPr·Ga(CH2SiMe3)3 and the hypothetical ItBu trimethylgallium and indium nNHC complexes (F and Q, respectively)5b (see Table 3). Theoretical parameters were consistent with the observed experimental trends, however, the calculated Δ%VBur reduced to only ca. 3.7% and 3.1%, for gallium and indium, respectively.
Inclusion of the hypothetical complex nF produced a Δ%VBur of ca. 7% in comparison with the stable complex 3. This relatively minor discrepancy in %VBur has a pronounced effect in terms of dissociation energy, which is less than half for F than 3 (cf. 65.2 kJ mol−1 and 34.2 kJ mol−1, respectively). Furthermore, the unstable complex 4, similar in steric bulk to F (%VBur of 35.6% and 36.7%, respectively) has a significantly lower dissociation energy compared to complex 4 (cf. 34.2 kJ mol−1 and 52.8 kJ mol−1 for F and 4, respectively). These observations are in line with those we reported for Al complexes SIPr·AlMe3 (D) and ItBu·AlMe3 (cf. %VBur and Ediss for complexes D and ItBu·AlMe3 are 35.5%, 77.8 kJ mol−1 and 36.9%, 46.2 kJ mol−1, respectively) attributed to the varying electron donating properties of the SIPr and ItBu NHC moieties to the metal centre.6b
The DFT calculated %VBur between complexes 4 and 8 and their ItBu analogues nF and nQ showed comparable values (cf. 35.6%, 36.2%, 36.7% and 37.5% 4, 8, F and Q respectively). The slightly lower values observed for 4 and 8 are in accordance with their greater stability in the normal NHC form compared to nF and nQ, which readily isomerize to their abnormal form.5b Unfortunately, all attempts to isolate metal-containing species resulting from the structural decay of complexes 4 and 8 were unsuccessful.
Our calculations indicate that the facile isomerization of the previously reported IPr·Ga(CH2SiMe3)35a to its abnormal isomeric form can be attributed to the higher steric congestion imposed by the CH2SiMe3 group when compared to Me groups (%VBur 64.5% and 48.7% for CH2SiMe3 and Me groups). This is further illustrated by the longer Ga–Ccarbene bond distance and lower dissociation energies calculated for IPr·Ga(CH2SiMe3)3 and IPr·GaMe3 (3) (cf., 2.196(2) Å vs. 2.105(4) Å and 65.2 kJ mol−1vs. 23.3 kJ mol−1, respectively).
Comparative analysis of a stable vs. an unstable system using topographic steric maps of saturated complexes 2vs. 4 and 6vs. 8 showed that the distribution of steric bulk of the SIMes ligand in 2 and 6 is symmetrical around the metal, whereas for 4 and 8, localization of steric hindrance around the bulkier ortho isopropyl group was clearly observed on the steric contour map (see Fig. 6 and the ESI†). This asymmetric spatial distribution of the NHC ligand around the metal centres in 4 and 8 correlates with their reduced stability compared to 2 and 6 respectively (see the ESI†).
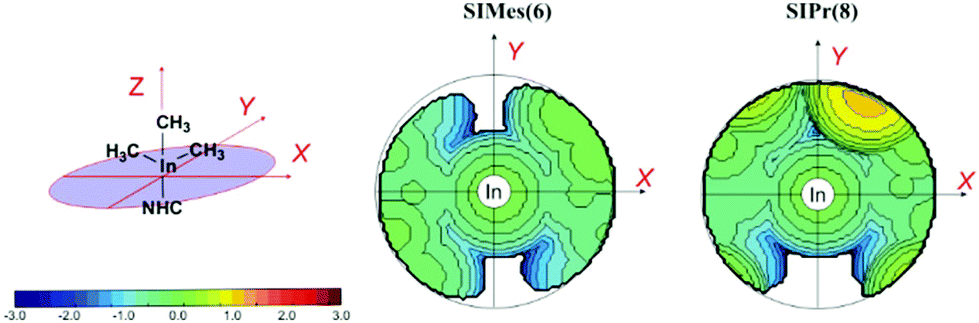 | ||
| Fig. 6 Topographic steric maps of the SIMes and SIPr ligands in 6 and 8. The iso-contour curves of the steric maps are in Å. The maps have been obtained starting from the crystallographic data of the Al–NHC complexes (CIF), with the Al–Ccarbene distance fixed at 2.0 Å. The xz plane is the mean plane of the NHC ring, whereas the yz plane is the plane orthogonal to the mean plane of the NHC ring, and passing through the Ccarbene atom of the NHC ring. | ||
Bonding studies
To gain a better understanding of the nature of M–NHC bonds with M = Al, Ga and In, bond snapping energy (BSE) analysis was performed.16 The BSE is the energy required for the dissociation of the M–L bond, analysed based on the interaction between fragments possessing both the local equilibrium geometry of the final molecule and an electronic structure suitable for bond formation. To calculate the heterolytic BSE for 1–8, the geometry of the metal fragment [M] – in this case MMe3 – was fixed, and the complex fragmented into its corresponding neutral [M] and NHC components.Although BSE does not correlate in all instances with bond dissociation enthalpies (since reorganization and relaxation of the fragments are not considered), it closely relates to bond enthalpy terms, providing a good approximation to bond strength values.
The BSE can be decomposed into two main terms, namely steric interaction (ΔE0) and orbital interaction (ΔEint) (eqn (1)):17
| BSE = −[ΔE0 + ΔEint] | (1) |
The steric interaction term ΔE0 can be further split into an electrostatic interaction term ΔEelstat and a Pauli repulsion term ΔEPauli (eqn (2)):17
| ΔE0 = ΔEelstat + ΔEPauli | (2) |
The ΔEPauli repulsion term describes the two-orbital electron interactions between the occupied orbitals of both fragments. The ΔEelstat and ΔEPauli terms constitute stabilizing and destabilizing contributions to BSE, respectively, with their relative contributions determining the overall character of ΔE0.
The ΔEint term may also be further broken down into contributions from the respective orbital interactions within the various irreducible representations τ of the overall symmetry group of the system (eqn (3)):17
| ΔEint = ΣτΔEτint | (3) |
Each complex studied in the present work, 1–8, has been optimized with a Cs imposed symmetry, where the NHC ligands are positioned in the σxy mirror plane of the molecule. Therefore, the A′ contributions to the orbital interaction energy are associated with σ-bonding, whereas the A′′ contributions represent π-interactions. The A′′ contribution of the orbital interaction energy may be further divided into NHC → M π–donation, ΔEπintC→M, and M → NHC π-backdonation, ΔEπintM→C. To estimate these two interactions, additional constrained space orbital variation (CSOV) calculations were performed.17 In particular, to assess the contribution of π-donation, bond decomposition analysis was performed by considering the interaction of a [M] fragment and an NHC ligand, excluding the set of virtual A′′ orbitals of the NHC fragment from the variational space. In this way, the A′′ contribution of the orbital interaction energy is associated only with the NHC → [M] A′′ donation, or π-donation. Similarly, the level of π back-donation was determined by explicitly excluding all virtual A′′ orbitals on the [TM] fragment.
The energy decomposition analysis (EDA) is performed in the gas phase since it refers to the intrinsic strength of the M–NHC bond, which is independent of the environment that may stabilize the two fragments. We selected one NHC ligand, namely IMes, as a case study for comparison between three metals (see Table 4). The data reported in Table 4 suggest that the greater strength of the Al–IMes bond with respect to that of the Ga–IMes bond is mainly attributable to the steric term (ΔE0). In fact, the Pauli contribution of the steric term, ΔEPauli, destabilizes the Ga system more than the electrostatic term does, with Δ(ΔEPauli(Ga) − ΔEPauli(Al)) being almost 3.5 times larger with respect to Δ(ΔEelstat(Ga) − ΔEelstat(Al)). As a result, the steric term disfavours the Ga–IMes bond by 45.7 kJ mol−1. This is in agreement with theoretical studies by Frenking et al. on a series of IMe NHC group 13 metal hydride complexes.18 Our detailed orbital analysis shows that the greater ΔEPauli term for the Ga–IMes system is related to the interactions between the occupied orbitals on the NHC and the occupied 3d orbitals on the Ga atom whereby the smaller ΔEPauli for Al is due to the lack of available d-electrons. Intuitively, the orbital interaction is primarily constituted by the σ term, which is larger in magnitude for the Ga–IMes system, as also seen for IMe·GaH318 compared to its Al counterpart (see Table 4). For the π term, the main difference is in the M → C interaction, i.e. almost 4 kJ mol−1 stronger for Al.
| A (Al) | 1 (Ga) | 5 (In) | |
|---|---|---|---|
| ΔE0 | 10.0 | 45.7 | 20.3 |
| ΔEelstat | −321.4 | −334.7 | −287.6 |
| ΔEPauli | 331.4 | 380.4 | 308.0 |
| ΔEint | −175.4 | −180.8 | −135.5 |
| σ − ΔEint | −151.3 (86.3%) | −161.4 (89.3%) | −119.8 (88.5%) |
| π − ΔEint | −24.1 (13.7%) | −19.4 (10.7%) | −15.6 (11.5%) |
| ΔEπintC→M | −8.5 | −7.3 | −5.5 |
| ΔEπintM→C | −18.1 | −14.3 | −11.4 |
| BSE | 165.3 | 135.1 | 115.2 |
In a comparison of Ga vs. In, the steric term (ΔE0) once again disfavors the Ga system. In this case, the ΔEPauli term has the greatest contribution, with Δ(ΔEPauli(Ga) − ΔEPauli(In)) being 1.5 times greater with respect to the electrostatic Δ(ΔEelstat(Ga) − ΔEelstat(In)). The orbital interaction is much greater for the Ga–IMes bond, both at the σ and π levels, compensating for the unfavored steric term. Similarly, this was reported for the computed IMe·GaH3 complex and its In analogue.18 For completeness, it is worth noting that the greater BSE observed for the Al–NHC bond, with respect to that of Ga–NHC, reflects the larger Ediss associated with Al compounds compared to Ga (see the last column in Table 3). With regard to the Ga/In trend, the dissociation energies are almost identical (varying less than 4.0 kJ mol−1), in agreement with the smaller discrepancy between the BSE for the Ga/In–NHC bonds with respect to the Al/Ga–NHC bonds (i.e. 20 kJ mol−1 and 30 kJ mol−1 for Al and Ga, respectively).
Further group 13 bonding insights can be gained from the 1H and 13C NMR spectra. Hence, DFT-NMR analyses were performed for the Ccarbene and Me group hydrogen atoms in the complexes IMes·AlMe3 (A) and SIMes·AlMe3 (B) to predict their 13C and 1H NMR spectra. The calculated chemical shielding (σC) is −3.9 and −24.3 ppm for the Ccarbene atom of the IMes and SIMes system, respectively. Decomposition of the isotropic σC into dia- and paramagnetic terms, σC = σd + σp, indicates that the change in σC is mainly due to the paramagnetic term σp, that varies by almost 21 ppm downfield from IMes to SIMes. Previous literature19 has indicated that the carbene chemical shift in NHC ligands is related to transitions between the filled σ orbitals of the M–NHC bond (HOMO) and the empty π orbital of the carbene (LUMO). Hence, we undertook analyses on the HOMO–LUMO energies of the NHC ligand showing that the energy gap decreases by almost 0.2 eV from IMes to SIMes. This can be attributed to a decreased stability of the SIMes NHC molecule HOMO, which results in a stronger magnetic coupling, and additionally accounts for the higher paramagnetic shielding (corresponding downfield shift) in Al–SIMes with respect to Al–IMes. The DFT 1H NMR analysis revealed that the upfield shift of the SIMes·AlMe3 methyl hydrogens corresponds to a reduced π back-donation [Al] → NHC (π*) that results from a smaller π orbital overlap, probably as a consequence of the slightly elongated Al–SIMes bond distance. As a result of this decreased [Al] → NHC back-donation, electron density is pushed towards neighbouring ligands on the Al centre, i.e. the methyl groups, thus leading to an upfield shift of the H atoms.
Our EDA results highlight that the interactions between the occupied orbitals on the NHC and the occupied Ga 3d orbitals destabilize this system with respect to the Al and In counterparts. However, a stronger orbital interaction for Ga compared to In sets the trend of the total M–NHC bond strength as Al > Ga > In. The overall orbital interaction is primarily constituted by the σ term, with a relatively small π term that consists mostly of a back-donation from the metal fragment to the NHC. To further extend our comparison and quantify the existing bonding discrepancies between our main group NHC complexes and well-established transition metal (TM)–NHC and –PHC systems, BSE decomposition analyses were performed, implementing IMes–Pd–IMes and IMes(P)–Pd–(P)IMes as case studies (see Table 5).
| Al–IMes | Pd–IMes | Pd–(P)IMes | |
|---|---|---|---|
| ΔEelstat | −321.4 | −589.4 | −537.3 |
| ΔEPauli | 331.4 | 578.3 | 550.6 |
| ΔE0 | 10.0 | −11.1 | 13.3 |
| ΔEint | −175.4 | −182.7 | −196.7 |
| σ − ΔEint | −151.3 (86.3%) | −129.0 (70.6%) | −143.2 (72.8%) |
| π − ΔEint | −24.1 (13.7%) | −53.8 (29.4%) | −53.5 (27.2%) |
| ΔEπintC→M | −8.5 | −5.8 | −5.6 |
| ΔEπintM→C | −18.1 | −50.6 | −49.6 |
| BSE | 165.3 | 193.8 | 183.4 |
The increased strength of the Pd–NHC bond (almost 30.0 kJ mol−1) with respect to that of Al–NHC is largely attributed to the steric term (20.0 kJ mol−1) rather than to the orbital interaction (10 kJ mol−1). For the latter contribution, despite a smaller σ term in the Pd–NHC bond, a two-fold greater π term is found due to π back-donation. In the case of the Pd–PHC system, the considered bond bears a greater resemblance to that of Al–NHC for the steric and σ terms, and as expected, a significant π back-donation term, similar to the case of Pd–NHC is found. Substitution of N with P atoms results in both strengthening of the orbital contribution (σ term) and disfavouring of the steric term (mainly Pauli term) to the M–NHC bond.
Conclusions
The present work describes the synthesis and characterization of a series of new aromatic N-substituted NHC trimethylgallium and indium species. Similarly to their aluminium counterparts, these complexes exhibit varying stabilities, which are attributed to small differences in the steric bulk of the chosen NHC. Our computational study has allowed quantification and rationalisation of discrepancies between M–NHC bond strengths for the Al, Ga, and In triad. Moreover, a quantitative comparison with well-established transition metal systems (Pd–NHC and –PHC) determines that an increase in both the electrostatic interaction and [M] → NHC π back-bonding is largely responsible for the existing differences between group 13 and transition metal NHC complexes.Experimental section
General procedures
All manipulations were carried out using standard Schlenk and glove-box techniques under a dried-argon atmosphere and using oven-dried glassware. Ether and toluene were distilled over Na/benzophenone, degassed and purged with dry argon prior to use, and stored under 4 Å molecular sieves. Deuterated benzene, C6D6, was distilled over Na and stored under a potassium mirror. Acetonitrile, for high-resolution mass spectrometry, was stirred over 4 Å molecular sieves, subsequently distilled over CaH2 prior to use, and stored under 4 Å molecular sieves. Starting materials, IMes, SIMes, IPr, and SIPr were prepared as previously described.13,22 Trimethylgallium was synthesized by first dissolving gallium trichloride (5.00 g, 28.40 mmol) in 5 mL of degassed toluene followed by dropwise addition of the mixture degassed triethylamine (4.44 g, 43.89 mmol) and trimethylaluminium (3.16 g, 43.89 mmol). Following the addition, the reaction was stirred overnight and distilled at atmospheric pressure to obtain the neat trimethylgallium.7 A solution of trimethylgallium (0.702 M) in toluene was then prepared from the distilled trimethylgallium. Trimethylindium was generated in situ by reacting MeLi (3 M in DME) with InCl3 (0.221 g, 1 mmol) dissolved in ether at −78 °C and filtered through Celite before addition to the carbene.8Instrumentation
1H (400 MHz), 13C NMR (100/125 MHz) spectra were recorded using Bruker Avance DPX400 and 500 spectrometers with the 1H, 13C NMR chemical shifts internally referenced to the residual solvent used. All NMR spectroscopic analyses were performed at room temperature (300 K). High-resolution mass spectra were obtained by using a Water Q-Tof Premier, with ESI mode. Melting points were determined on a SRS-Optimelt MPA-100 apparatus using sealed glass capillaries under argon and were uncorrected. Infrared spectra were recorded as Nujol mulls by using NaCl plates on a Shimadzu IR Prestige-21 FTIR spectrometer.Procedure for the synthesis of complexes 1–8
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 525 (ν Ga–C stretch; m). HRMS: calcd for C24H33GaN2 [M + H]+: 419.1978; found 419.1992.
= 525 (ν Ga–C stretch; s). HRMS: calcd for C24H35GaN2 [M + H]+: 421.2134; found 421.2140.
= 525 (ν Ga–C stretch; m). HRMS: calcd for C30H45GaN2 [M + H]+: 503.2917; found 503.2930.
= 521 (ν Ga–C stretch; m). HRMS: calcd for C30H47GaN2 [M + H]+: 505.3073; found 505.3090.
= 525 (ν Ga–C stretch; m). HRMS: calcd for C24H33GaN2 [M + H]+: 419.1978; found 419.1992.
= 525 (ν Ga–C stretch; s). HRMS: calcd for C24H35GaN2 [M + H]+: 421.2134; found 421.2140.
= 525 (ν Ga–C stretch; m). HRMS: calcd for C30H45GaN2 [M + H]+: 503.2917; found 503.2930.
= 521 (ν Ga–C stretch; m). HRMS: calcd for C30H47GaN2 [M + H]+: 505.3073; found 505.3090.
X-ray crystallographic studies
Diffraction-quality crystals 1–8 were obtained in toluene or ether solvent at room temperature or 0 °C. The crystals were mounted onto quartz fibres, and the X-ray diffraction intensity data were obtained at 103 K with a Bruker Kappa diffractometer equipped with a CCD detector, employing Mo Kα radiation (λ = 0.71073 Å), with the SMART suite of programs.20 All data were processed and corrected for Lorentz and polarization effects with SAINT and for absorption effects with SADABS.21 Structural solution and refinement were carried out with the SHELXTL suite of programs.22 The structures were solved by direct methods or Patterson maps to locate the heavy atoms, followed by difference maps for the light, non-hydrogen atoms. All non-hydrogen atoms were refined with anisotropic thermal parameters. The crystals of 7 had one disordered iso-propyl group modelled in two alternative sites (with ∼0.5 occupancy) and refined with appropriate restraints.Computational details
All calculations have been performed with the Amsterdam Density Functional suite of programs, ADF.23–25 Gradient-corrected density-functional calculations were based on the local density approximation with Slater exchange26 and VWN correlation.27 Gradient corrections for exchange and correlation were those proposed by Becke28 and Perdew,29 respectively. Valence electrons were described with an STO basis of triple-ζ quality, augmented by one polarization function.30 Electrons of the core shells (1s2s2p for Al, 1s2s2p3s3p for Ga, 1s2s2p3s3p3d4s4p for In, 1s2s2p for P, 1s2s2p for Si, 1s for C and N) have been treated within the frozen core approximation.23 Relativistic effects have been incorporated based on the zero-order regular approximation.Abbreviations
| IiPrMe | 1,3-Isopropyl-4,5-dimethylimidazol-2-ylide-ne |
| ItBu | 1,3-Di-tert-butylimidazol-2-ylidene |
| IMes | 1,3-Bis-(2,4,6-trimethylphenyl)imidazol-2-ylidene |
| SIMes | 1,3-Bis-(2,4,6-trimethylphenyl)imidazolin-2-ylidene |
| IPr | 1,3-Bis-(2,6-diisopropylphenyl)imidazol-2-ylidene |
| SIPr | 1,3-Bis-(2,6-diisopropylphenyl)imidazolin-2-ylidene |
| PHC | P-Heterocyclic carbene |
Acknowledgements
F. G. would like to thank NTU start-up grant (M4080552) and MOE Tier 1 grant (M4011441) for financial support. L. F. would like to thank KAUST for financial support.Notes and references
- L. Cavallo and C. S. J. Cazin, in N-heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, ed. C. S. J. Cazin, Springer, Netherlands, 2011, vol. 32, pp. 1–22 Search PubMed.
- (a) D. Zhang and G. Zi, Chem. Soc. Rev., 2015, 44, 1898–1921 RSC; (b) S. Bellemin-Laponnaz and S. Dagorne, Chem. Rev., 2014, 114, 8747–8774 CrossRef CAS PubMed; (c) S. J. Hock, L.-A. Schaper, W. A. Herrmann and F. E. Kühn, Chem. Soc. Rev., 2013, 42, 5073–5090 RSC; (d) V. P. Boyarskiy, K. V. Luzyanin and V. Y. Kukushkin, Coord. Chem. Rev., 2012, 256, 2029–2056 CrossRef CAS; (e) H. D. Velazquez and F. Verpoort, Chem. Soc. Rev., 2012, 41, 7032–7060 RSC.
- (a) C. Bour and V. Gandon, Synlett, 2015, 1427–1436 CAS; (b) C. Fliedel, G. Schnee, T. Avilés and S. Dagorne, Coord. Chem. Rev., 2014, 275, 63–86 CrossRef CAS; (c) C. Bour and V. Gandon, Coord. Chem. Rev., 2014, 279, 43–57 CrossRef CAS; (d) Y. Wang and G. H. Robinson, Inorg. Chem., 2014, 53, 11815–11832 CrossRef CAS PubMed; (e) S. Aldridge and A. J. Downs, in The Group 13 Metals Aluminium, Gallium, Indium and Thallium: Chemical Patterns and Peculiarities, ed. S. Aldridge and A. J. Downs, Wiley, UK, 2011, vol. 3 Search PubMed.
- (a) P. Horeglad, M. Cybularczyk, B. Trzaskowski, G. Z. Żukowska, M. Dranka and J. Zachara, Organometallics, 2015, 34, 3480–3496 CrossRef CAS; (b) P. Horeglad, O. Ablialimov, G. Szczepaniak, A. M. Dąbrowska, M. Dranka and J. Zachara, Organometallics, 2014, 33, 100–111 CrossRef CAS; (c) P. Horeglad, G. Szczepaniak, M. Dranka and J. Zachara, Chem. Commun., 2012, 48, 1171–1173 RSC; (d) J. H. Cotgreave, D. Colclough, G. Kociok-Köhn, G. Ruggiero, C. G. Frost and A. S. Weller, Dalton Trans., 2004, 10, 1519–1520 RSC; (e) J. D. Gorden, C. L. B. Macdonald and A. H. Cowley, J. Organomet. Chem., 2002, 643–644, 487–489 CrossRef CAS; (f) X.-W. Li, J. Su and G. H. Robinson, Chem. Commun., 1996, 23, 2683–2684 RSC.
- (a) M. Uzelac, A. Hernán-Gómez, D. R. Armstrong, A. R. Kennedy and E. Hevia, Chem. Sci., 2015, 6, 5719–5728 RSC; (b) G. Schnee, O. N. Faza, D. Specklin, B. Jacques, L. Karmazin, R. Welter, C. S. López and S. Dagorne, Chem. – Eur. J., 2015, 21, 17959–17972 CrossRef CAS PubMed.
- (a) G. Schnee, D. Specklin, J.-P. Djukic and S. Dagorne, Organometallics, 2016, 35, 1726–1734 CrossRef CAS; (b) M. M. Wu, A. M. Gill, Y. Lu, L. Falivene, Y. Li, R. Ganguly, L. Cavallo and F. García, Dalton Trans., 2015, 44, 15166–15174 RSC; (c) R. A. Musgrave, R. S. P. Turbervill, M. Irwin and J. M. Goicoechea, Angew. Chem., Int. Ed., 2012, 51, 10832–10835 CrossRef CAS PubMed; (d) Y. Zhang, G. M. Miyake and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2010, 49, 10158–10162 CrossRef CAS PubMed; (e) A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Dalton Trans., 2010, 39, 9091–9099 RSC; (f) A.-L. Schmitt, G. Schnee, R. Welter and S. Dagorne, Chem. Commun., 2010, 46, 2480–2482 RSC; (g) W.-C. Shih, C.-H. Wang, Y.-T. Chang, G. P. A. Yap and T.-G. Ong, Organometallics, 2009, 28, 1060–1067 CrossRef CAS; (h) Y. Lee, B. Li and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 11625–11633 CrossRef CAS PubMed; (i) M. Schiefer, N. D. Reddy, H.-J. Ahn, A. Stasch, H. W. Roesky, A. C. Schlicker, H.-G. Schmidt, M. Noltemeyer and D. Vidovic, Inorg. Chem., 2003, 42, 4970–4976 CrossRef CAS PubMed; (j) H. Zhou, E. J. Campbell and S. T. Nguyen, Org. Lett., 2001, 14, 2229–2231 CrossRef.
- (a) C. Bour, J. Monot, S. Tang, R. Guillot, J. Farjon and V. Gandon, Organometallics, 2014, 33, 594–599 CrossRef CAS; (b) A. El-Hellani, J. Monot, S. Tang, R. Guillot, C. Bour and V. Gandon, Inorg. Chem., 2013, 52, 11493–11502 CrossRef CAS PubMed; (c) A. Higelin, S. Keller, C. Göhringer, C. Jones and I. Krossing, Angew. Chem., Int. Ed., 2013, 52, 4941–4944 CrossRef CAS PubMed; (d) A. El-Hellani, J. Monot, R. Guillot, C. Bour and V. Gandon, Inorg. Chem., 2013, 52, 506–514 CrossRef CAS PubMed; (e) S. Tang, J. Monot, A. El-Hellani, B. Michelet, R. Guillot, C. Bour and V. Gandon, Chem. – Eur. J., 2012, 18, 10239–10243 CrossRef CAS PubMed; (f) G. E. Ball, M. L. Cole and A. I. McKay, Dalton Trans., 2012, 41, 946–952 RSC; (g) S. G. Alexander, M. L. Cole and C. M. Forsyth, Chem. – Eur. J., 2009, 15, 9201–9214 CrossRef CAS PubMed; (h) S. G. Alexander, M. L. Cole, S. K. Furfari and M. Kloth, Dalton Trans., 2009, 16, 2909–2911 RSC; (i) M. L. Cole, S. K. Furfari and M. Kloth, J. Organomet. Chem., 2009, 694, 2934–2940 CrossRef CAS; (j) N. Marion, E. C. Escudero-Adán, J. Benet-Buchholz, E. D. Stevens, L. Fensterbank, M. Malacria and S. P. Nolan, Organometallics, 2007, 26, 3256–3259 CrossRef CAS; (k) C. Jones, D. P. Mills and R. P. Rose, J. Organomet. Chem., 2006, 691, 3060–3064 CrossRef CAS; (l) R. J. Baker, H. Bettentrup and C. Jones, Eur. J. Inorg. Chem., 2003, 13, 2446–2451 CrossRef; (m) R. J. Baker and C. Jones, Appl. Organomet. Chem., 2003, 17, 807–808 CrossRef CAS; (n) R. J. Baker, R. D. Farley, C. Jones, M. Kloth and D. M. Murphy, Chem. Commun., 2002, 11, 1196–1197 RSC; (o) R. J. Baker, M. L. Cole, C. Jones and M. F. Mahon, Dalton Trans., 2002, 9, 1992–1996 RSC; (p) R. J. Baker, A. J. Davies, C. Jones and M. Kloth, J. Organomet. Chem., 2002, 656, 203–210 CrossRef CAS; (q) C. D. Abernethy, M. L. Cole and C. Jones, Organometallics, 2000, 19, 4852–4857 CrossRef CAS; (r) M. D. Francis, D. E. Hibbs, M. B. Hursthouse, C. Jones and N. A. Smithies, Dalton Trans., 1998, 19, 3249–3254 RSC; (s) D. E. Hibbs, M. B. Hursthouse, C. Jones and N. A. Smithies, Chem. Commun., 1998, 8, 869–870 RSC; (t) S. J. Black, D. E. Hibbs, M. B. Hursthouse, C. Jones, K. M. A. Malik and N. A. Smithies, Dalton Trans., 1997, 22, 4313–4319 RSC.
- Rohm and Hass Electronic Materials LLC, Synthesis of trimethylgallium, U.S. Patent2006/47132A1, 2006 Search PubMed.
- I. Pérez, J. P. Sestelo and L. A. Sarandeses, J. Am. Chem. Soc., 2001, 123, 4155–4160 CrossRef.
- Horeglad et al. previously reported the obtention of complexes 1 and 2via an indirect synthetic route,4a,b in which the reaction of 2 equivalents of a NHC alkoxy dimethylgallium complex with an equal amount of GaMe3 yielded complexes IMes·GaMe3 (G) and SIMes·GaMe3 (J) and a dimeric dialkylgallium byproduct. As for complex 4, it has been recently reported according to Scheme 1 to yield SIPrGaMe3 (I) and the structure was used to compare the coordination properties of G3c and J3e with respect to their reactivity in the polymerization of rac-LA.3c Nevertheless, these complexes have been included in this report to maintain the rigor of our studies.
- A. M. Magill, K. J. Cavell and B. F. Yates, J. Am. Chem. Soc., 2004, 126, 8717–8724 CrossRef CAS PubMed.
- H. V. Huynh, Y. Han, R. Jothibasu and J. A. Yang, Organometallics, 2009, 28, 5395–5404 CrossRef CAS.
- S. LeuthäuƁer, D. Schwarz and H. Plenio, Chem. – Eur. J., 2007, 13, 7195–7203 CrossRef PubMed.
- G. Davidson, in Characteristic Vibrations of Compounds of Main Group Elements, ed. G. Davidson, Royal Society of Chemistry, UK, 2001, vol. 24, pp. 217–267 Search PubMed.
- (a) S. J. Ryan, S. D. Schimler, D. C. Bland and M. S. Sanford, Org. Lett., 2015, 17, 1866–1869 CrossRef CAS PubMed; (b) X. Bantreil and S. P. Nolan, Nat. Protoc., 2011, 6, 69–77 CrossRef CAS PubMed; (c) M. K. Denk, J. M. Rodezno, S. Gupta and A. J. Lough, J. Organomet. Chem., 2001, 617–618, 242–253 CrossRef CAS; (d) A. J. Arduengo, R. Krafczyk and R. Schmutzler, Tetrahedron, 1999, 55, 14523–11453 CrossRef CAS.
- (a) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC; (b) A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 13, 1759–1766 CrossRef.
- H. Jacobsen, A. Correa, A. Poater, C. Costabile and L. Cavallo, Coord. Chem. Rev., 2009, 253, 687–703 CrossRef CAS.
- (a) N. Holzmann, A. Stasch, C. Jones and G. Frenking, Chem. – Eur. J., 2013, 19, 6467–6479 CrossRef CAS PubMed; (b) N. Holzmann, A. Stasch, C. Jones and G. Frenking, Chem. – Eur. J., 2011, 17, 13517–13525 CrossRef CAS PubMed.
- L. Falivene, L. Caporaso, L. Cavallo and H. Jacobsen, Dalton Trans., 2013, 42, 7281–7286 RSC.
- SMART version 5.628, Bruker AXS Inc., Madison, WI, USA, 2001 Search PubMed.
- G. M. Sheldrick, SADABS V2014/4 (Bruker AXS Inc.), University of Göttingen, Göttingen, Germany, 2014 Search PubMed.
- SHELXTL-2014/6 (Sheldrick, 2014), Bruker AXS Inc., Madison, WI, USA, 2014 Search PubMed.
- E. J. Baerends, D. E. Ellis and P. Ros, Chem. Phys., 1973, 2, 41–51 CrossRef CAS.
- L. Versluis and T. Ziegler, J. Chem. Phys., 1988, 88, 322–328 CrossRef CAS.
- (a) G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 932–942 CrossRef; (b) C. F. Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 CAS; (c) E. J. Baerends, et al., SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com; ADF2008-01 Search PubMed.
- J. C. Slater, Phys. Rev., 1951, 81, 385–390 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8827 CrossRef.
- E. van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1477674–1477679, 1477681 and 1477682. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt04448d |
| This journal is © The Royal Society of Chemistry 2017 |