
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ ligand exchange-mediated 0D/1D transformation of a polyoxovanadate†
M.
Wendt
a,
P.
Polzin
a,
J.
van Leusen
b,
C.
Näther
 a,
P.
Kögerler
*b and
W.
Bensch
*a
a,
P.
Kögerler
*b and
W.
Bensch
*a
aInstitute of Inorganic Chemistry, Christian-Albrechts University of Kiel, Max-Eyth-Str. 2, 24118 Kiel, Germany. E-mail: wbensch@ac.uni-kiel.de
bInstitute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany. E-mail: paul.koegerler@ac.rwth-aachen.de
First published on 10th January 2017
Abstract
The antimonato-polyoxovanadate {NiII(en)3}3[VIV15SbIII6O42(H2O)]·ca.15H2O was utilized as a synthon for the solvothermal in situ generation of the new compound {NiII(phen)3}2[{NiII(en)2}VIV15SbIII6O42(H2O)]·19H2O, a rearrangement induced by ligand metathesis. While in the precursor structure cations and anions are isolated, the solid-state structure of the product is characterized by 1D chains consisting of alternating [V15Sb6O42(H2O)]6− cluster shells and [Ni(en)2]2+ units covalently linked to neighboring clusters via terminal oxygen atoms. Water clusters composed of sixteen hydrogen-bonded H2O molecules are located in void spaces of the structure. The magnetic properties indicate weak antiferromagnetic interactions of the bridging Ni2+ center and adjacent polyoxovanadate anions, as well as small magnetic anisotropy of the individual Ni2+ centers.
Introduction
The structures and physico-chemical properties of high-nuclearity polyoxovanadate (POV) clusters can be significantly altered by introducing hetero-atoms like e.g. Si, Ge, As, or Sb.1 Charge compensation of the negatively charged POV clusters is realized by ammonium,2–4 alkali metals cations,5–8 protonated amine molecules9–12 or transition metal amine complexes.13–15 There are also reports that bond formation between the POV cores and cations16–18 leads to charge-neutral complexes. The main synthetic route for preparation of such hetero-POVs is the solvothermal approach applying vanadium and antimony compounds such as NH4VO3, V2O5 or Sb2O3 as educts. Hetero-POVs are synthesized at pH > 8 using VV-containing sources, and a reducing agent is required for generation of the magnetically highly interesting partially reduced or fully reduced compounds, i.e. those comprising VIV centers. Hence, the POV chemistry significantly differs from polyoxomolybdate and polyoxotungstate chemistry, where preformed cluster anions or in situ generated building blocks are used as synthons for the preparation of chemically modified high-nuclearity clusters.Recently, we discovered a Sb-POV exhibiting good solubility in water.19 In a first synthesis we reacted this compound, {Ni(en)3}3[V15Sb6O42(H2O)]·ca.15H2O (I), at T = 150 °C leading to in situ reorganization of the cluster shell and formation of the β-isomer of the [V14Sb8O42]4− anion.20 In the presence of [Ni(phen)3](ClO4)2·0.5H2O (phen = 1,10-phenanthroline) and Sb2O3 the α-isomer of [V14Sb8O42]4− could be obtained.21
To further develop this Sb-POV chemistry, we reacted I with [Ni(phen)3](ClO4)2·0.5H2O, which leads to the formation of the new compound {Ni(phen)3}2[{Ni(en)2}V15Sb6O42(H2O)]·19H2O (II). The partial replacement of the [Ni(en)3]2+ complexes in I here leads to interconnection of the {V15Sb6O42} anions into a 1D chain-based solid-state structure, in which large (H2O)16 water clusters are located. Here, we report the synthesis, crystal structure and the magnetic properties of the new compound {Ni(phen)3}2[{Ni(en)2}V15Sb6O42(H2O)]·19H2O (II) representing the first successful in situ transformation of an isolated {V15Sb6O42} moiety into a higher hierarchical structure.
Results and discussion
The title compound {Ni(phen)3}2[{Ni(en)2}V15Sb6O42(H2O)]·19H2O (II) can be prepared according to several synthesis routes. Heating I for 1 d at 150 °C in the presence of pure phenanthroline, [Ni(phen)3]2+, [Cr(phen)2Cl2]1+ or [Eu(phen)2(NO3)3] afforded formation of II (see Experimental section for details). The results of the different synthesis routes strongly indicate that an in situ ligand exchange reaction takes place.Compound II crystallizes in the monoclinic space group C2/c with four formula units per unit cell (Table S3†) with all atoms except V2, Ni1, O7, O13 and the oxygen water atoms O38–O42 located on general positions. The structure features the [V15Sb6O42(H2O)]6− cluster shell as central structural motif (Fig. 1). Two condensed V6 hexagons formed by edge-sharing VO5 polyhedra are joined by three VO5 pyramids and three Sb2O5 units forming the sequence V6–V3–V6 in the cluster anions. The terminal V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds are shorter (average: 1.619 Å) than the bridging V–μ3-O bonds (average: 1.950 Å) (Table S4†). The Sb–O bonds scatter in a narrow range (1.930–1.944 Å; average: 1.938 Å, Table S4†). All geometric parameters of the anion agree well with literature data of Sb-POVs.22–27 The oxidation states were determined by bond valance sum calculation (BVS)28 leading to the formulation [VIV15SbIII6O42]6− (Table S4†). The presence of VIV centers is supported by the strong characteristic vanadyl stretching vibration at 973 cm−1 in the IR spectrum (Fig. S3†).
O bonds are shorter (average: 1.619 Å) than the bridging V–μ3-O bonds (average: 1.950 Å) (Table S4†). The Sb–O bonds scatter in a narrow range (1.930–1.944 Å; average: 1.938 Å, Table S4†). All geometric parameters of the anion agree well with literature data of Sb-POVs.22–27 The oxidation states were determined by bond valance sum calculation (BVS)28 leading to the formulation [VIV15SbIII6O42]6− (Table S4†). The presence of VIV centers is supported by the strong characteristic vanadyl stretching vibration at 973 cm−1 in the IR spectrum (Fig. S3†).
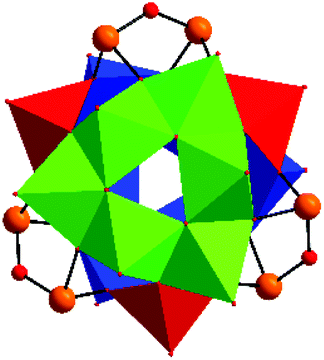 | ||
| Fig. 1 Crystal structure of the cluster anion of [V15Sb6O42]6− in II. The two V6 hexagons and the three VO5 pyramids joining the hexagons are drawn in different colors. Red: oxygen; orange: Sb (the central H2O molecule is omitted for clarity). | ||
The cluster anions are joined by [Ni(en)2]2+ complex cations into chains that elongate along [101] with the [Ni(phen)3]2+ complexes ‘decorating’ the chains (Fig. 2 and S6†). The Ni2+ cations are octahedrally coordinated by two en ligands and two O atoms from two adjacent trans-positioned polyanions. The Ni–N (2.085(3)–2.095(3) Å) and Ni–O bonds (2.158(2) Å) are in line with the literature.29 In the crystallographically independent [Ni(phen)3]2+ complex, Ni2+ is in a distorted octahedral environment with Ni–N bond lengths ranging from 2.079(3) to 2.114(3) Å, equatorial N–Ni–N angles between 79.01(12) and 98.58(13)°, and trans N–Ni–N angles from 166.96(13) to 171.66(13)° (see Table S5†).
 | ||
| Fig. 2 Crystal structure of II with view of the chains and the decorating [Ni(phen)3]2+ complexes (H atoms are omitted for clarity). | ||
Within the (101) plane a layered arrangement of alternating [Ni(phen)3]2+ complexes and chains is observed (Fig. 3).
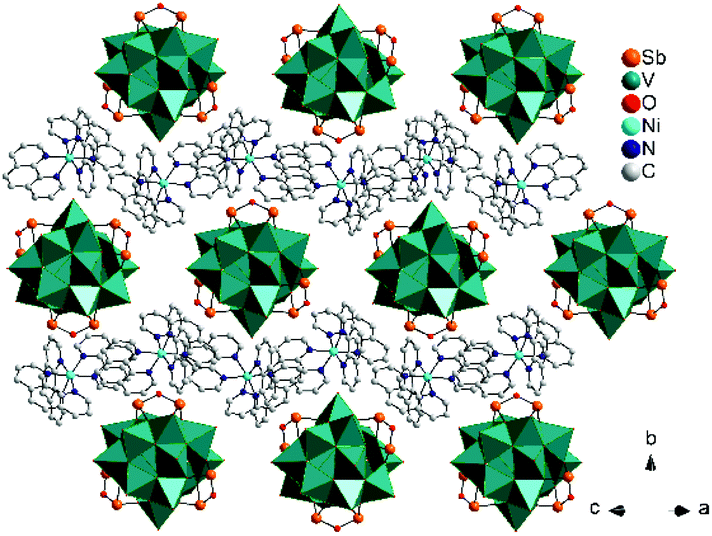 | ||
| Fig. 3 View of the arrangement of the chains in the (101) plane (H atoms are omitted for clarity). | ||
The voids between the POV clusters (Fig. 3) are occupied by (H2O)16 clusters, consisting of two unique and seven symmetry-related H2O molecules (Fig. S7†). The geometric parameters of the hydrogen bonding interactions of the water cluster (Fig. 4) are listed in Table S7.† All values are typical for strong hydrogen bonds reported for water clusters.30–34 Following the nomenclature suggested by Infantes et al.35–37 the cluster can be defined as U0 type for cross-linked discrete rings, namely U02(2)2(2)5(2)5(2)5(3)5(3)8(6). The water cluster has a stabilizing effect caused by further H bonds to oxygen atoms of the Sb-POV cluster core, to O16 connecting the {V15Sb6O42} clusters with bridging [Ni(en)2]2+, and to the N–H atoms of the [Ni(en)2]2+ complex and to the C–H atoms of the [Ni(phen)3]2+ cations (Table S6†).
 | ||
| Fig. 4 Water cluster in II constructed by hydrogen bonding interactions between the water molecules located in the free spaces in Fig. 3 (red: O, gray: H). | ||
In many structures of POVs water molecules are observed, which are frequently disordered preventing the identification of water clusters. Hence, the occurrence of the (H2O)16 cluster is unusual in Sb-POV chemistry.
The remaining water molecules (O40 and O41) are not involved in H bonding interactions. Besides the hydrogen bonding network involving the water cluster, the [Ni(en)2]2+ and the [Ni(phen)3]2+ complexes form H bonds with oxygen atoms of the POV cluster shell (Table S7†). Furthermore, pronounced π–π stacking interactions between the aromatic phenanthroline ligands are present (Fig. 5). There are three types of intermolecular distances: sandwich type arrangements (average: 3.906 Å, shortest: 3.687 Å), parallel oriented phen molecules (average: 3.841 Å; shortest: 3.666 Å) and T-shaped arrangements (3.970 Å). For such π–π interactions, energies in the order of 10 kcal mol−1 were calculated and significantly contribute to the structural arrangements.38–42
 | ||
| Fig. 5 Possible π–π stacking interactions between the aromatic phenanthroline ligands in the structure of II (T-shaped: pink, parallel: green, sandwich: orange). | ||
During the transformation of {Ni(en)3}3[V15Sb6O42(H2O)]·ca.15H2O (I) into {Ni(phen)3}2[{Ni(en)2}V15Sb6O42(H2O)]·19H2O (II), two of the [Ni(en)3]2+ complexes are formally substituted by [Ni(phen)3]2+ complexes. The third [Ni(en)3]2+ complex loses one ethylenediamine ligand and connects adjacent anions to form a chain. The observation that not all en ligands in the [Ni(en)3]2+ complexes are exchanged is somewhat surprising because [Ni(phen)3]2+ was supplied in a threefold excess. The formation of an intermediate with composition [{Ni(en)2}V15Sb6O42(H2O)]4− cannot be excluded. But based on our observations made in ref. 21 where I was transformed into the [V14Sb8O42]4− cluster, the formation of such an intermediate is not likely. It seems that an en ligand of one of the [Ni(en)3]2+ complexes is detached followed by bond formation to POV oxygen sites. Applying [Ni(phen)3]2+ in the reaction the other [Ni(en)3]2+ complexes are either exchanged or en is replaced by phen. In addition, syntheses with phen, [Cr(phen)2Cl2]1+ or [Eu(phen)2(NO3)3] also afforded crystallization of II requiring an in situ ligand exchange of en by phen.
Many Sb-POVs contain isolated cations and anions,43–45 but 1D,31,46 2D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 and 3D networks17,27 are less common. Fig. 6 summarizes the structures obtained until now using I as synthon and Ni2+-centered en and phen complexes. The compound [{Ni(en)2}2V14Sb8O42]·5.5H2O20 (III) crystallized with a Sb-richer cluster shell and the complexes join adjacent anions thus generating a layered structure. A different isomer of the [V14Sb8O42]4− anion was observed in {Ni(phen)3}2[V14Sb8O42]·phen·12H2O21 (IV), and cations and the anion are separated. In the title compound the cluster anion of the synthon is retained and the ligand exchange pattern yields a 1D structure.
16 and 3D networks17,27 are less common. Fig. 6 summarizes the structures obtained until now using I as synthon and Ni2+-centered en and phen complexes. The compound [{Ni(en)2}2V14Sb8O42]·5.5H2O20 (III) crystallized with a Sb-richer cluster shell and the complexes join adjacent anions thus generating a layered structure. A different isomer of the [V14Sb8O42]4− anion was observed in {Ni(phen)3}2[V14Sb8O42]·phen·12H2O21 (IV), and cations and the anion are separated. In the title compound the cluster anion of the synthon is retained and the ligand exchange pattern yields a 1D structure.
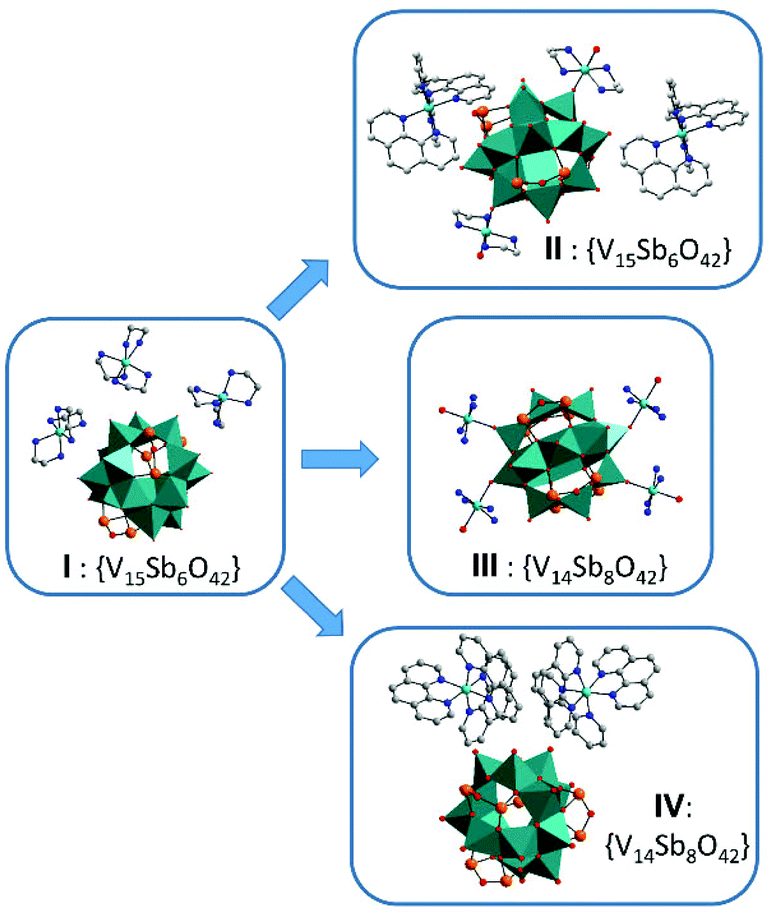 | ||
| Fig. 6 Compounds derived from the precursor {Ni(en)3}3[V15Sb6O42(H2O)]·ca.15H2O19 (I): {Ni(phen)3}2[{Ni(en)2}V15Sb6O42(H2O)]·19H2O (II), [{Ni(en)2}2V14Sb8O42]·5.5H2O20 (III) and {Ni(phen)3}2[V14Sb8O42]·phen·12H2O21 (IV). | ||
To get a better understanding of the reaction conditions the ratio of precursor compound I:[Ni(phen)3](ClO4)2·0.5H2O and the reaction temperature were altered. The lower the temperature, the lower was the yield, but II was always obtained between 80 and 140 °C. At the lowest temperatures compound II crystallized in a low yield in a mixture with the precursor I. Change of the ratio of the starting materials also leads to a lower yield, yet crystallization of II was always observed.
Magnetic measurements
The temperature-dependent magnetic susceptibility data for compound II at 0.1 Tesla and the molar magnetization Mmvs. magnetic field B at 2.0 K are shown in Fig. 7. At 290 K, the χmT value of 6.11 cm3 K mol−1 is higher than expected47 for three non-interacting high-spin Ni2+ centers (the expectation range for NiII is 2.94–4.60 cm3 K mol−1). Taking into account the contribution of 15 (hypothetically) non-interacting V4+ centers, this value is, however, significantly below the corresponding expected range 8.36–10.22 cm3 K mol−1. The deviation is readily explained by the magnetism of the {V15Sb6} polyoxovanadate anion, as it is characterized as a system of 15 spin-1/2 vanadyl groups that exhibit strong antiferromagnetic coupling,23 leading to low χmT values at room temperature. Upon decreasing the temperature, the χmT vs. T curve for II is approximatively linear down to 25 K, and subsequently sharply decreases to 4.17 cm3 K mol−1 at 2.0 K. The low-temperature characteristics of χmT are potentially due to weak antiferromagnetic interactions of the bridging Ni2+ center and the two neighboring polyoxovanadate anions as well as a small anisotropy of the single Ni2+ centers due to ligand field effects and spin–orbit coupling. For T > 25 K, non-interacting octahedrally coordinated Ni2+ centers are expected to yield an almost constant χmT vs. T curve, thus the data seem to indicate a temperature-independent paramagnetic (TIP) contribution. This assumption is, however, disproved by the fact that the remaining χmT ≈ 5.1 cm3 K mol−1 is far too large for three Ni2+ ions. In addition, we consider the magnetic data of the isolated {V15Sb6} polyoxovanadate anion23 depicted in Fig. 7 as green circles: on the one hand, the χmT vs. T curve is not a straight line through the origin (which would correspond to a TIP). On the other hand, the distinct bend of the curve at approx. 160 K does not appear in the data for II. We thus assume that the bridging Ni2+ center either affects the exchange interactions within the {V15Sb6} anion, or this effect is directly due to the {V15Sb6}–Ni–{V15Sb6} exchange pathway. The molar magnetization at 2.0 K (Fig. 7, inset) is a linear function of the applied field up to ca. 2 T, and reaches Mm = 8.4NAμB at 5.0 T without saturation. This value is larger than for the range 6.0–7.4 NAμB expected for three non-interacting (or ferromagnetically coupled) Ni2+ cations, therefore indicating a significant contribution caused by the {V15Sb6} anions at 2.0 K. Using CONDON,48,49 we explore several model scenarios to reproduce the magnetic data (see ESI†), but the simple model of additive contributions (of NiII and {V15Sb6}), which usually works well for transition metal-augmented Sb-POVs, does not satisfactorily reproduce the magnetic characteristics of II.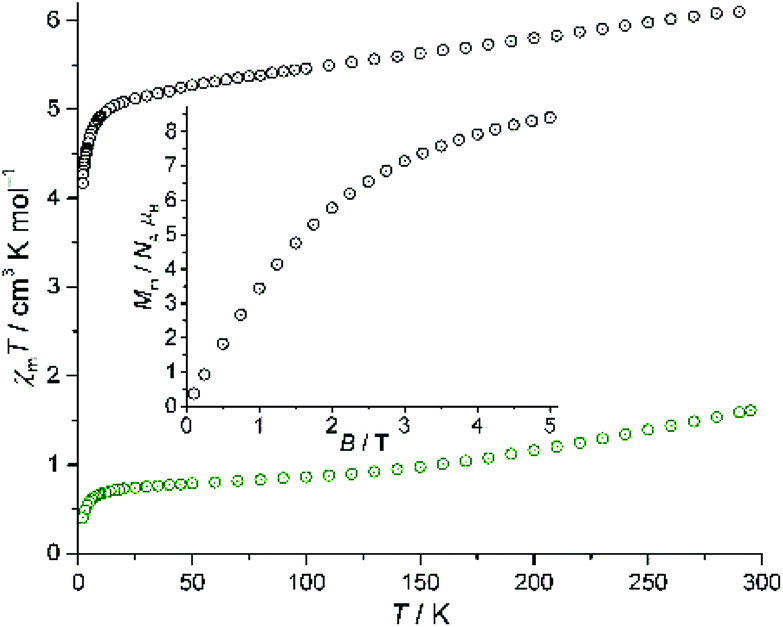 | ||
| Fig. 7 Magnetic data of II: χmT vs. temperature T at 0.1 T; inset: molar magnetization Mmvs. magnetic field B at 2.0 K. Black circles: experimental data; green circles: χmT([V15Sb6O42]6−) (scaled).23 | ||
Conclusions
We demonstrated that the soluble precursor {Ni(en)3}3[V15Sb6O42(H2O)]·ca.15H2O can be transformed in situ into {Ni(phen)3}2[{Ni(en)2}V15Sb6O42(H2O)]·19H2O in the presence of phen or phen containing complexes within 1 d, a transformation that apparently is driven by step-wise ligand exchange. This reorganization of the constituents induces formation of a chain structure consisting of Ni2+-centered complexes and anionic cluster shells, in which large and highly ordered water clusters are embedded. The usage of soluble precursors for the synthesis of new POV compounds leads to a dramatic shortening of the reaction time compared to usual POV synthesis.Experimental
Both precursor compounds {Ni(en)3}3[V15Sb6O42(H2O)]·ca.15H2O (I) and [Ni(phen)3](ClO4)2·0.5H2O were synthesized as described in literature.19,50 The compound {Ni(phen)3}2[{Ni(en)2}V15Sb6O42(H2O)]·19H2O (II) was prepared under solvothermal conditions in DURAN glass tubes (inner volume 11 mL) at 150 °C. [Eu(phen)2(NO3)3] was synthesized by dissolving 5.009 g (14.80 mmol) Eu(NO3)3·6H2O in 50 mL ethanol and adding a solution of 2.027 g phen in 20 mL ethanol. The colorless precipitate was obtained after 1 d (32% yield). The [Cr(phen)2Cl2]Cl complex was synthesized by refluxing a mixture of 0.040 g (2.5 mmol) CrCl3·6H2O, 0.010 g (1.5 mmol) Zn powder in 25 mL methanol for 20 min. Subsequently 0.901 g (5.0 mmol) phen were added and refluxed for 1 h.51Synthesis of II: 0.167 g (0.055 mmol) of I were mixed with 0.1140 g (0.165 mmol) of [Ni(phen)3](ClO4)2·0.5H2O in 4 mL dist. H2O and heated at 150 °C for 1 d. Yield: 83%, based on V. Elemental analyses: C 23.14, H 2.34, N 5.62%; calcd for C76H104Ni3N16O62Sb6V15: C 23.38, H 2.34, N 5.74%.
Further experiments were performed to investigate the ligand exchange reaction (reaction time: 1 d; T = 150 °C). Varying the amount of [Ni(phen)3](ClO4)2·0.5H2O from 50 to 150% always afforded crystallization of II with no significant changes of the yield. However, using larger concentrations of the complex leads to crystallization of the excess together with compound II. Adding en (0.1–0.5 mL) blocks the formation of II.
When using [Eu(phen)2(NO3)3] or [Cr(phen)2Cl2]Cl as precursor complexes, compound II is formed. Most importantly, adding only phen instead of any preformed complexes also leads to crystallization of II. Applying a large excess of phen (0.33–0.66 mmol) formation of II is also observed.
Compound II was characterized by elemental (CHN) analysis, energy dispersive X-ray analysis (EDX), differential thermoanalysis and thermogravimetry (DTA-TG) and IR spectroscopy as well as single crystal structure analysis (see ESI Fig. S1–S3 and Tables S1, S2†). X-ray powder diffraction patterns (XRPD) of the products obtained after the different syntheses are displayed in Fig. S4 and S5.† All chemicals were purchased (NH4VO3, NiCl2·6H2O, Sb2O3, CrCl3·6H2O, Eu(NO3)3·6H2O (Merck), ethylenediamine (Fluka), phenanthroline, Ni(ClO4)2·6H2O (ABCR)) and used without further purification.
Spectroscopic investigations
The IR spectra were measured in the 400–4000 cm−1 range using a Genesis FTIRTM spectrometer (ATI Mattson).Elemental analysis
CHN analyses were performed using an EURO EA Elemental Analyzer from EURO VECTOR Instruments.Differential thermoanalysis and thermogravimetry
Thermogravimetry analysis were performed in Al2O3 crucibles using a Netzsch STA-409CD instrument heating the sample under a nitrogen atmosphere with a constant flow rate of 75 mL min−1 and a heating rate of 1 K min−1.EDX analyses
EDX measurements were performed using a Philips Environmental Scanning Electron Microscope ESEM XL30 equipped with an EDAX detector.X-ray powder diffraction
The X-ray powder patterns were recorded using a STOE STADI-P diffractometer (transmission geometry, Cu-Kα radiation (λ = 1.540598 Å)) to determine the phase purity of the reaction product comparing the experimental pattern with that calculated from single crystal X-ray data.Magnetic measurements
A Quantum Design MPMS-5XL SQUID magnetometer was used to collect magnetic susceptibility data of II. The polycrystalline sample was compacted and immobilized into cylindrical PTFE capsules. The data were acquired as a function of the field (0.1–5.0 T at 2.0 K) and temperature (2.0–290 K at 0.1 T). The data were corrected for the diamagnetic contributions of the sample holder and the compound (χdia = −1.93 × 10−3 cm3 mol−1).Single crystal structure analysis
Data collection was performed with a STOE Imaging Plate Diffraction System (IPDS-1) with Mo-Kα radiation (λ = 0.71073 Å). A numerical absorption correction was performed (Tmin/max: 0.5232/0.7241). The crystal structure was solved with the SHELXS-9751 and refined against F2 using SHELXL-2014.52 All non H atoms were refined anisotropic. The C–H and N–H H atoms were positioned with idealized geometry and refined isotropic using a riding model. Most of the O–H H atoms were located in difference maps, their bond lengths were set to ideal values and finally, they were refined isotropic using a riding model. One water molecule is disordered over two positions and was refined using a split model. Two additional water positions are not fully occupied. For the last three O atoms the H atoms could not be located but were considered in the calculation of the formula.
Crystallographic data (excluding structure factors) for the structure in this paper have been deposited with the CCDC (No. 1504150).
Acknowledgements
We thank the state of Schleswig-Holstein for financial support. The work has been in part funded by ERC grant 308051 – MOLSPINTRON.References
- K. Y. Monakhov, W. Bensch and P. Kögerler, Chem. Soc. Rev., 2015, 44, 8443 RSC.
- G. Zhou, Y. Xu, C. Guo, Y. Liu and X. Zheng, J. Cluster Sci., 2007, 18, 388 CrossRef CAS.
- A. Müller and J. Döring, Z. Anorg. Allg. Chem., 1991, 595, 251 CrossRef.
- A. Wutkowski, N. Evers and W. Bensch, Z. Anorg. Allg. Chem., 2011, 637, 2205 CrossRef CAS.
- A. Müller, J. Döring, M. I. Khan and V. Wittneben, Angew. Chem., Int. Ed. Engl., 1991, 30, 210 CrossRef.
- A. Müller and J. Döring, Angew. Chem., Int. Ed. Engl., 1988, 27, 1721 CrossRef.
- M. I. Khan, Q. Chen and J. Zubieta, Inorg. Chim. Acta, 1993, 212, 199 CrossRef CAS.
- B. Chen, B. Wang, W. Lin, L. Fan, Y. Gao, Y. Chi and C. Hu, Dalton Trans., 2012, 41, 6910 RSC.
- G. Zhou, C. Guo, W. Liu, Y. Xu and X. Zheng, J. Coord. Chem., 2008, 61, 202 CrossRef CAS.
- X.-B. Cui, J.-Q. Xu, Y. Li, Y.-H. Sun, L. Ye and G.-Y. Yang, J. Mol. Struct., 2003, 657, 397 CrossRef CAS.
- S.-Y. Shi, Y. Chen, J.-N. Xu, Y.-C. Zou, X.-B. Cui, Y. Wang, T.-G. Wang, J.-Q. Xu and Z.-M. Gao, CrystEngComm, 2010, 12, 1949 RSC.
- S.-T. Zheng, J. Zhang and G.-Y. Yang, Z. Anorg. Allg. Chem., 2005, 631, 170 CrossRef CAS.
- S.-T. Zheng, J.-Q. Xu and G.-Y. Yang, J. Cluster Sci., 2005, 16, 23 CrossRef CAS.
- S.-T. Zheng, Y.-M. Chen, J. Zhang, J.-Q. Xu and G.-Y. Yang, Eur. J. Inorg. Chem., 2006, 397 CrossRef CAS.
- S.-T. Zheng, J. Zhang, B. Li and G.-Y. Yang, Dalton Trans., 2008, 5584 RSC.
- L. Zhang, X. Zhao, J. Xu and T. Wang, J. Chem. Soc., Dalton Trans., 2002, 3275 RSC.
- E. Antonova, C. Näther, P. Kögerler and W. Bensch, Angew. Chem., Int. Ed., 2011, 50, 764 CrossRef CAS PubMed.
- A. Wutkowski, C. Näther, P. Kögerler and W. Bensch, Inorg. Chem., 2008, 47, 1916 CrossRef CAS PubMed.
- M. Wendt, U. Warzok, C. Näther, J. van Leusen, P. Kögerler, C. A. Schalley and W. Bensch, Chem. Sci., 2016, 7, 2684 RSC.
- L. Yu, J.-P. Liu, J.-P. Wang and J.-Y. Niu, Chem. Res. Chin. Univ., 2009, 25, 426 CAS.
- M. Wendt, C. Näther and W. Bensch, Chem. – Eur. J., 2016, 22, 7747 CrossRef CAS PubMed.
- E. Antonova, C. Näther, P. Kögerler and W. Bensch, Angew. Chem., Int. Ed., 2011, 123, 790 CrossRef.
- R. Kiebach, C. Näther, P. Kögerler and W. Bensch, Dalton Trans., 2007, 3221 RSC.
- E. Antonova, C. Näther and W. Bensch, Dalton Trans., 2012, 41, 1338 RSC.
- E. Antonova, C. Näther and W. Bensch, CrystEngComm, 2012, 14, 6853 RSC.
- E. Antonova, C. Näther, P. Kögerler and W. Bensch, Inorg. Chem., 2012, 51, 2311 CrossRef CAS PubMed.
- A. Wutkowski, C. Näther, P. Kögerler and W. Bensch, Inorg. Chem., 2013, 52, 3280 CrossRef CAS PubMed.
- M. O'Keeffe and N. E. Brese, J. Am. Chem. Soc., 1991, 113, 3226 CrossRef.
- X.-X. Hu, J.-Q. Xu, X.-B. Cui, J.-F. Song and T.-G. Wang, Inorg. Chem. Commun., 2004, 7, 264 CrossRef CAS.
- A. Wutkowski, C. Näther and W. Bensch, Inorg. Chim. Acta, 2011, 379, 16 CrossRef CAS.
- M. S. Deshpande, A. S. Kumbhar and C. Näther, Dalton Trans., 2010, 39, 9146 RSC.
- G. Laus, V. Kahlenberg, K. Wurst, T. Lörting and H. Schottenberger, CrystEngComm, 2008, 10, 1638 RSC.
- E. Freire, S. Baggio, J. C. Munoz and R. Baggio, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, C58, m221 CAS.
- Z.-H. Xu, X.-F. Li and X.-W. Zhang, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2012, 42, 140 CrossRef CAS.
- L. Infantes and S. Motherwell, CrystEngComm, 2002, 4, 454 RSC.
- L. Infantes, J. Chisholm and S. Motherwell, CrystEngComm, 2003, 5, 480 RSC.
- L. Infantes, L. Fabian and W. D. S. Motherwell, CrystEngComm, 2007, 9, 65 RSC.
- J. Hilbert, C. Näther and W. Bensch, Inorg. Chem., 2015, 53, 5619 CrossRef PubMed.
- S. Grimme, Angew. Chem., Int. Ed., 2008, 47, 3430 CrossRef CAS PubMed.
- J. W. G. Bloom and S. W. Wheeler, Angew. Chem., 2011, 123, 7993 ( Angew. Chem., Int. Ed. , 2011 , 50 , 7847 ) CrossRef.
- Y.-H. Chi, J.-M. Shi, H.-N. Li, W. Wei, E. Cottrill, N. Pan, H. Chen, Y. Liang, L. Yu, Y.-Q. Zhang and C. Hou, Dalton Trans., 2013, 42, 15559 RSC.
- C. A. Hunter, K. R. Lawson, J. Perkins and C. J. Urch, J. Chem. Soc., Perkin Trans. 2, 2001, 651 RSC.
- R. Kiebach, C. Näther and W. Bensch, Solid State Sci., 2006, 8, 964 CrossRef CAS.
- E. Antonova, A. Wutkowski, C. Näther and W. Bensch, Solid State Sci., 2011, 13, 2154 CrossRef CAS.
- E. Antonova, C. Näther, P. Kögerler and W. Bensch, Dalton Trans., 2012, 41, 6957 RSC.
- Y. Gao, Z. Han, Y. Xu and C. Hu, J. Cluster Sci., 2010, 21, 163 CrossRef CAS.
- H. Lueken, Magnetochemie, Teubner, Stuttgart, 1999 Search PubMed.
- M. Speldrich, H. Schilder, H. Lueken and P. Kögerler, Isr. J. Chem., 2011, 51, 215 CrossRef CAS.
- J. van Leusen, M. Speldrich, H. Schilder and P. Kögerler, Coord. Chem. Rev., 2015, 289, 137 CrossRef.
- L. Abdel-Rahman, L. P. Battaglia, C. Rizzoli and P. Sgarabotto, J. Chem. Crystallogr., 1995, 25, 629 CrossRef CAS.
- X. Gao, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m139 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1504150. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt04412c |
| This journal is © The Royal Society of Chemistry 2017 |