
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crystal structure and spin-trimer magnetism of Rb2.3(H2O)0.8Mn3[B4P6O24(O,OH)2]†
Olga V.
Yakubovich
a,
Larisa V.
Shvanskaya
ab,
Galina V.
Kiriukhina
a,
Anatoly S.
Volkov
a,
Olga V.
Dimitrova
a,
Evgeny A.
Ovchenkov
a,
Alexander A.
Tsirlin
cd,
Alexander A.
Shakin
b,
Olga S.
Volkova
abe and
Alexander N.
Vasiliev
 *abe
*abe
aM.V. Lomonosov Moscow State University, Moscow 119991, Russia. E-mail: vasil@mig.phys.msu.ru
bNational University of Science and Technology “MISiS”, 119049 Moscow, Russia
cExperimental Physics VI, Center for Electronic Correlations and Magnetism, Institute of Physics, University of Augsburg, D-86135 Augsburg, Germany
dNational Institute of Chemical Physics and Biophysics, 12618 Tallinn, Estonia
eInstitute of Physics and Technology, Ural Federal University, 620002 Ekaterinburg, Russia
First published on 31st January 2017
Abstract
The novel borophosphate Rb2.3(H2O)0.8Mn3[B4P6O24(O,OH)2] was prepared under hydrothermal conditions at 553 K. Its crystal structure was determined using single-crystal X-ray diffraction data obtained from a non-merohedral twin and refined against F2 to R = 0.057. The compound crystallizes in the orthorhombic space group Pbcn, with unit-cell parameters a = 20.076(2) Å, b = 9.151(1) Å, c = 12.257(1) Å, V = 2251.8(2) Å3, and Z = 4. The title compound is the first example of a borophosphate with manganese ions adopting both octahedral and tetrahedral coordinations. Its unique crystal structure is formed by borophosphate slabs and chains of Mn2+-centered polyhedra sharing edges and vertices. These 2D and 1D fragments interconnect into a framework with open channels that accommodate Rb+ cations and water molecules. Topological relationships between borophosphates built from three-membered rings of two borate and one phosphate tetrahedra sharing oxygen vertices, amended by additional PO4 and HPO4 tetrahedra, are discussed. The temperature dependence of the magnetic susceptibility of Rb2.3(H2O)0.8Mn3[B4P6O24(O,OH)2] reveals predominant antiferromagnetic exchange interactions and the high-temperature effective magnetic moment corresponding to the high-spin S = 5/2 state of Mn2+ ions. At 12.5 K, a magnetic transition is evidenced by ac-susceptibility and specific heat measurements. A spin-trimer model with the leading exchange interaction J ∼ 3.2 K is derived from density-functional band-structure calculations and accounts for all experimental observations.
Introduction
The chemical class of borophosphates comprises compounds with crystal structures built by PO4 and BO4/BO3 oxocomplexes sharing oxygen vertices. Similar to silicates, complex borophosphate anions may be zero-, one-, two- and three-dimensional oligomer units forming chains, layers or frameworks.1 Intensive exploration of borophosphates in the last two decades is due to their fascinating structural chemistry and numerous technological applications.2,3 Lithium iron borophosphate, Li0.8Fe(H2O)2[BP2O8]·H2O, reveals electrochemical activity as a cathode material for Li- and Na-ion batteries,4 while Snx(Ca0.05B0.975P0.975O3.95)1−x/C composites can be used as anode materials for Li-ion batteries.5Borophosphates may also have interesting connections to abiogenesis. According to ref. 6, some prebiological episodes could occur in environments with borophosphates, which arguably took part in special reactions in the form of hydrogels. Their derived chiral minerals served as drivers for prebiotic processes, thus providing conditions for the appearance of protocells.
Borophosphates with open microporous structures often comprise metal oxocomplexes and, therefore, not only exhibit adsorption, catalytic, and ion-conductive properties typical of zeolite-like compounds, but also demonstrate promising physical characteristics which depend on the nature of constituent metal atoms. The incorporation of transition metals into the borophosphate framework often leads to interesting features of forming phases.7,8
Particularly, borophosphates with Mn2+-cations are of special interest not only with respect to catalysis applications, but also as potential materials with luminescence and magnetic properties. For example, (NH4)7Mn4(H2O)[B2P4O15(OH)2]2(H2PO4)(HPO4) crystals with 16 ring pore openings show antiferromagnetic interactions between Mn2+ ions.9 KMnBP2O7(OH)2 demonstrates long-range antiferromagnetic ordering and exhibits bright orange luminescence at room temperature.10 Another Mn2+ borophosphate, (NH4)6[Mn3B6P9O36(OH)3]·4H2O, reveals canted antiferromagnetic order at low temperatures.11
In our search for functional crystals with transition metals and mixed anionic arrangement, a novel water-bearing rubidium manganese borophosphate was obtained under hydrothermal conditions. The new compound is characterized by a unique combination of six-fold and four-fold-coordinated Mn2+ cations, not mentioned before to our knowledge. We present here its crystal structure and magnetic properties in comparison with related compounds featuring borophosphate slabs of similar topology.
Experimental section
Synthesis and structure determination
Colourless transparent needle crystals of the new phase with a maximum length of 200 μm (Fig. 1) were prepared hydrothermally in the Rb2CO3–MnCl2–H3PO4–B2O3 system. A mixture of these components at the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:4 weight ratio was placed into a 4 ml stainless steel bomb with distilled water filling 80% of the volume. The experiment was conducted at a temperature of 553 K and a pressure of 70 bar over a period of 18 days followed by cooling of the furnace to room temperature. The reaction products in the form of druses of needle crystals were washed with water and dried. Their phase purity was confirmed by the agreement between the experimental powder X-ray diffraction pattern and the simulated diagram based on the single-crystal structural data.
:2:2:4 weight ratio was placed into a 4 ml stainless steel bomb with distilled water filling 80% of the volume. The experiment was conducted at a temperature of 553 K and a pressure of 70 bar over a period of 18 days followed by cooling of the furnace to room temperature. The reaction products in the form of druses of needle crystals were washed with water and dried. Their phase purity was confirmed by the agreement between the experimental powder X-ray diffraction pattern and the simulated diagram based on the single-crystal structural data.
 | ||
| Fig. 1 SEM images of the title phase, showing the druse of growing crystals (a) and the crystal morphology (b). | ||
A suitable single crystal of the title compound was analyzed with a scanning electron microscope (SEM)‡ JEOL SEM (JSM-6480LV) equipped with an INCA Energy-350 energy dispersive (EDS) detector and an INCAWave-500 four-crystal wavelength dispersive spectrometer (WDS). The measurements were made at 20 kV and 7 nA, and the sample was stable under these conditions. X-ray spectral analysis provided a semi quantitative result with the Rb:Mn:P:B:O ratio close to 1:1.5:3:2:15, which is consistent with the results of our X-ray diffraction structural study.
The single crystal X-ray diffraction data were collected at ambient temperature by using graphite-monochromated Mo-Kα radiation with an Xcalibur-S area detector diffractometer. The intensities were corrected for Lorentz and polarization effects, and a numerical absorption correction based on Gaussian integration over a multifaceted crystal model was applied. An analysis of the experimental set of X-ray reflections with CrysAlisPro12 has shown that the studied crystal was a non-merohedral twin with a twinning angle of about 8.5°. The reflections from different components of the sample were separated in the new set. 12% of reflections with partial overlap, for which individual contributions could not be revealed, were removed. All calculations were performed in the WinGX32 software package.13 Atomic scattering factors and anomalous dispersion corrections were taken from the International Tables for Crystallography.14
The crystal structure was solved via direct methods in the space group Pbcn and refined against the F2 data with SHELX programs.15 The final refinement was performed on the basis of all experimental intensities marked for each component in the HKLF5 reflection file, to the R factor of 0.057 (for 1891 unique reflections with I > 2σ(I)) with anisotropic displacement parameters for all non-hydrogen atoms. The positions of one independent H atom forming the hydroxyl group were obtained by difference-Fourier techniques and refined in an isotropic approximation. The O–H bond length was fixed by hard restraints to an empirical value of 0.85 Å in order to obtain comparable H-bond geometry not affected by arbitrary scatter of the refined O–H distance. The crystallographic characteristics of the new phase, the experimental conditions of the data collection, and the final results of the structure refinement are shown in Table 1. Table S1† presents the atomic positions and equivalent isotropic displacement parameters.§ Characteristic interatomic distances are given in Table 2. A bond-valence calculation (Table S2†) has been performed using the algorithm and parameters listed in ref. 16 and 17; its results are consistent with the assumed oxidation state of +2 for Mn and clearly confirm the assignment of the OH and H2O groups. According to our structural model, O10 corresponds to a mixed (O, OH) populated position in the 0.13:0.87 ratio. The synthesized phase is described by the following chemical formula: Rb2.26(H2O)0.76Mn3{B4P6O24[(OH)1.74O0.26]2}. Idealized formula can be written as Rb2.3(H2O)0.8Mn3[B4P6O24(O,OH)2].
| Crystal data | |
| Chemical formula, M (g mol−1) | Rb2.26(H2O)0.76Mn3{B4P6O24[(OH)1.74O0.26]2}, 1017.1 |
| Crystal system, space group | Orthorhombic, Pbcn (no. 60) |
| a, b, c (Å) | 20.0755(12), 9.1512(8), 12.2568(7) |
| V (Å3), Z | 2251.8(3), 4 |
| D c (g cm−3) | 3.000 |
| Crystal size (mm) | 0.110 × 0.040 × 0.020 |
| Crystal colour | Colourless |
| Absorption coeff. μ (mm−1) | 7.07 |
| Data collection | |
| Diffractometer | Xcalibur-S, CCD |
| Radiation | Mo-Kα (λ = 0.71073 Å), graphite monochromator |
| Temperature (K) | 298(2) |
| Scanning mode | ω |
| Measuring range | θ max = 27.493° |
| Reflections (total) | 6312 |
| h, k, l range | −25 ≤ h ≤ 26, −11 ≤ k ≤ 7, −15 ≤ l ≤ 15 |
| Refinement | |
| Reflections observed (I > 2σ(I)) | 1891 |
| Number of parameters used in the refinement | 178 |
| Absorption correction, Tmax, Tmin | Numerical, 0.574, 0.847 |
| Parameter of twinning (BASF) | 0.343(1) |
| Residuals | |
| R (observed reflections) | 0.057 |
| wR2 (all reflections) | 0.086 |
| δρ (max/min) (e Å−3) | 1.60, −1.24 |
| P1 tetrahedron | P2 tetrahedron | P3 tetrahedron |
| P1–O8 1.494(5) | P2–O6 1.510(6) | P3–O7 1.505(5) |
| P1–O2 1.548(5) | P2–O3 1.522(7) | P3–O12 1.531(6) |
| P1–O9 1.548(6) | P2–O4 1.547(6) | P3–O11 1.537(6) |
| P1–O5 1.554(6) | P2–O1 1.551(5) | P3–O10 1.556(7) |
| B1 tetrahedron | B2 tetrahedron | Mn1 octahedron |
| B1–O13 1.444(11) | B2–O13 1.445(11) | Mn1–O6 2.117(4) |
| B1–O9 1.476(11) | B2–O2 1.463(10) | Mn1–O12 2.156(5) |
| B1–O5 1.480(12) | B2–O13 1.485(11) | Mn1–O8 2.157(6) |
| B1–O1 1.491(11) | B2–O11 1.486(11) | Mn1–O3 2.189(4) |
| Mn1–O13 2.214(5) | ||
| Mn1–O12 2.259(6) | ||
| Mn2 tetrahedron | Rb1 polyhedron | Rb2 polyhedron |
| Mn2–O7 2.079(7) × 2 | Rb1–O7 2.900(6) | Rb2–O14 2.45(2) |
| Mn2–O3 2.107(5) × 2 | Rb1–O9 2.906(5) | Rb2–O9 2.964(9) |
| Rb3 polyhedron | Rb1–O2 2.931(5) | Rb2–O8 3.015(9) |
| Rb3–O10 2.604(6) × 2 | Rb1–O5 2.985(5) | Rb2–O7 3.120(10) |
| Rb3–O6 2.929(9) × 2 | Rb1–O8 2.995(5) | Rb2–O10 3.163(10) |
| Rb3–O7 3.315(9) × 2 | Rb1–O14 3.09(2) | Rb2–O2 3.256(9) |
| Rb1–O1 3.248(6) | ||
| Rb1–O3 3.278(7) | ||
| D–H⋯A | D–H | H⋯A | D⋯A | D–H⋯A |
| O10–H1⋯O10 | 0.845(16) | 1.87(7) | 2.566(15) | 139(10) |
Measurements of thermodynamic properties
The magnetic properties of Rb2.3(H2O)0.8Mn3[B4P6O24(O,OH)2] were investigated in the temperature range 2–300 K using both ac- and dc-magnetic susceptibility options of the “Quantum Design” Physical Properties Measurements System PPMS-9T. The magnetization of the title compound was measured up to 9 T in a static magnetic field and up to 30 T in a pulsed magnetic field at low temperatures. The specific heat of the pressed pellet of the sample was measured in the range 2–200 K by the relaxation method.Results and discussion
Description of the crystal structure and discussion
The asymmetric unit of the structure (Fig. 2) includes three P sites and two B sites in a strongly distorted tetrahedral coordination. Three P1–O distances of about 1.55 Å and a short one of 1.494(5) Å characterize the P1O4 polyhedron, while the P–O bond lengths in the P2O4 and P3O4 tetrahedra are in the range of 1.51–1.57 Å. In the BO4 tetrahedron, the B–O distances vary from 1.44(1) to 1.49(1) Å (Table 2). | ||
| Fig. 2 Basic structural units with the atom labeling scheme. Displacement ellipsoids are presented at the 50% probability level. Symmetry codes: (a) −x, ½ + y, z; (b) ½ − x, ½ − y, −½ + z; (c) 1 − x, y, ½ − z; (d) x, 1 − y, ½ + z; (e) x, 1 − y, −½ + z; (f) ½ + x, ½ − y, 1 − z; (h) ½ + x, ½ + y, ½ − z. | ||
Manganese atoms show a strongly distorted octahedral coordination. The Mn–O distances are in the range of 2.117(4)–2.260(6) Å for Mn1; the Mn2 atoms are on the two-fold axis and surrounded by four O atoms forming tetrahedra with the Mn2–O bond lengths of 2.079(6) and 2.107(5) Å. Two additional O atoms are situated at longer distances of 2.940(6) Å (Fig. 3). The tetrahedral coordination is relatively uncommon for Mn2+, although it has been reported for the KMnPO4 and RbMnPO4 crystal structures.18,19 The patterns of the P-, B- and Mn-polyhedra distortion are consistent with the bond-valence calculation (Table S2†). Large low-charge Rb+ cations located in the voids of the framework and statistically distributed in three positions are surrounded by eight or six oxygen atoms. The Rb1+ sites with 82% population feature six Rb1–O distances varying from 2.900(6) to 3.09(2) Å and two longer ones of 3.249(6) and 3.278(7) Å. According to our data, the hydroxyl groups substitute for one of the O atoms at the O10 vertices of the P3 tetrahedra. As a result, the [HPO4] tetrahedra, which are adjacent in the [010] direction, become linked by means of an O10–H1⋯O10 hydrogen bond interaction (Table 2). With O⋯O distances of 2.566 Å, these bonds can be classified as strong.
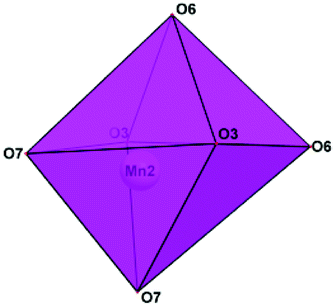 | ||
| Fig. 3 Coordination polyhedron of Mn2 (CN 4 + 2). | ||
Pairs of edge-sharing Mn1O6 octahedra and Mn2O4 tetrahedra alternate along the [001] direction and form corrugated chains parallel to the c axis of the orthorhombic unit cell (Fig. 4). A symmetrically independent fragment of the borophosphate anion presents a three-membered ring of two borate and one phosphate tetrahedra sharing oxygen vertices, amended by additional PO4 and HPO4 tetrahedra (Fig. 5a). These basic building units are reproduced by symmetry elements of the Pbcn space group and form anionic borophosphate slabs (Fig. 5b), which interchange along the a axis with the chains of Mn polyhedra (Fig. 6). The interconnection of the two-dimensional [B4P6O24(O,OH)2] sections and one-dimensional fragments of the Mn polyhedra results in thick packages with the Mn3[B4P6O24(O,OH)2] composition. The OH groups at the vertices of phosphate tetrahedra provide hydrogen bonding that has a strong influence on the structural arrangement. Thus, the bridging of the Mn3[B4P6O24(O,OH)2] packages by hydroxyl groups in the b axis direction (Fig. 4) leads to a 3D framework with open channels running along [001]. These channels accommodate Rb+ cations and H2O molecules (Fig. 7).
 | ||
| Fig. 4 Corrugated chains of edge-sharing MnO6 octahedra and MnO4 tetrahedra, associated with PO4 and BO4 tetrahedra. | ||
 | ||
| Fig. 5 The borophosphate anion built from PO4 and BO4 tetrahedra: a symmetrically independent fragment (a) and a two-dimensional slab (b). | ||
 | ||
| Fig. 6 The title crystal structure viewed along the b axis. | ||
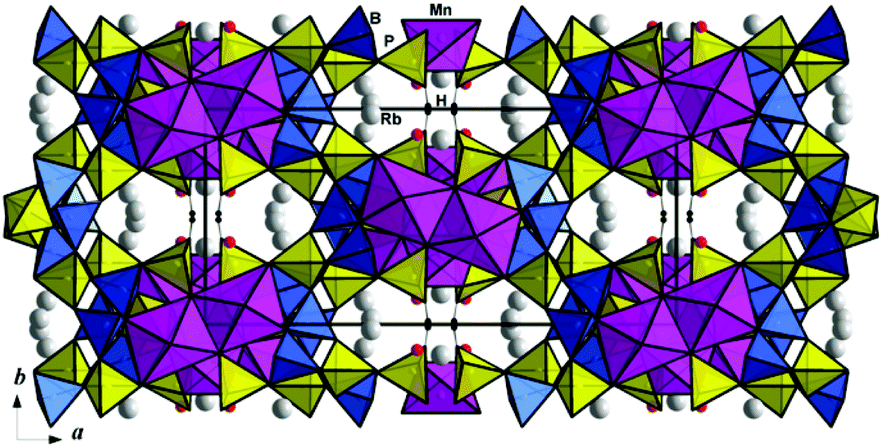 | ||
| Fig. 7 The title crystal structure viewed along the c axis. | ||
Topologically identical slabs built by phosphate and borate tetrahedra have been previously described in several other crystal structures, which are listed in Table 3. The first such cobalt borophosphate with organic molecules filling structural channels was reported by Sevov.20 In the (C2H10N2)Co[B2P3O12(OH)] crystal structure, the borophosphate slabs with three- and nine-membered rings share oxygen vertices with Co-centered octahedra to form a 3D open framework. Well-ordered protonated ethylenediamine molecules are fixed within the channels by hydrogen bonds. Later on, a series of isotypic orthorhombic compounds (C2H10N2)M2+[B2P3O12(OH)] with M = Mg, Mn, Fe, Ni, Cu, and Zn was hydrothermally synthesized and studied using single-crystal X-ray diffraction (for the Mg phase) or by Rietveld-methods.21 The Cd borophosphate, (C2H10N2)Cd[B2P3O12(OH)] (Fig. 8a), crystallizes in the same structure type.22 All these isotypic compounds reveal different unit cell volumes that obviously depend on the size of the M2+ cation (Table 3). Interestingly, using DETA as a structure-directing amine during the synthesis in the CdCl2–B2O3–H3PO4–H2O system leads to the formation of the (C4H16N3)[CdClB2P3O12(OH)] phase built by two-dimensional inorganic fragments of the same borophosphate slabs with adjacent CdO5Cl octahedra, separated by organic templates of diethylenetriamine (Fig. 8b).22 Accordingly, the c parameter of the (C4H16N3)[CdClB2P3O12(OH)] unit cell is changed significantly, and the inorganic structural fragments and organic DETA molecules alternate in the structure (Table 3).
 | ||
| Fig. 8 Crystal structures of Cd borophosphates shown in the yz projections: (C2H10N2)Cd[B2P3O12(OH)] (a) and (C4H16N3)[CdClB2P3O12(OH)] (b). | ||
| Formula, crystal structure details | Unit-cell parameters a, b, c, Å and V, Å3 | S.G., Z, ρ, g cm−3 | <M–O>, Å | <A–O>, Å | Ref. |
|---|---|---|---|---|---|
| a Crystal characteristics are given in a non-standard setting of the space group D142h = Pbcn for consistency of data. b One Cd–Cl distance is included in calculation. | |||||
| (C2H10N2)Co[B2P3O12(OH)] | 9.3501(5) | Pbca | 2.101 (M = Co2+) | (A = C2H10N2) | 20 |
| Borophosphate slabs and CoO6 octahedra form a 3D mixed anionic framework with organic molecules in channels | 12.2426(9) | 8 | |||
| 20.880(2) | 2.471 | ||||
| 2390.1(3) | |||||
| (C2H10N2)Mg[B2P3O12(OH)] | 9.3681(2) | Pbca | 2.087 (M = Mg2+) | (A = C2H10N2) | 21 |
| Borophosphate slabs and MgO6 octahedra form a 3D mixed anionic framework with organic molecules in channels | 12.2186(4) | 8 | |||
| 20.8928(5) | 2.277 | ||||
| 2391.5(1) | |||||
| (C2H10N2)Cd[B2P3O12(OH)] | 9.286(3) | Pbca | 2.271 (M = Cd2+) | (A = C2H10N2) | 22 |
| Borophosphate slabs and CdO6 octahedra form a 3D mixed anionic framework with organic molecules in channels | 12.459(3) | 8 | |||
| 21.626(6) | 2.639 | ||||
| 2502(1) | |||||
| (C4H16N3)CdCl[B2P3O12(OH)] | 9.470(2) | Pbca | 2.318b (M = Cd2+) | (A = C4H16N3) | 22 |
| Borophosphate slabs and CdO5Cl octahedra form 2D mixed anionic packages with organic molecules between | 12.307(3) | 8 | |||
| 27.311(6) | 2.411 | ||||
| 3183(1) | |||||
| K2(H2O)Co[B2P3O12(OH)] | 9.7255(7) | Pbca | 2.104 (M = Co2+) | 2.839 | 23 |
| Borophosphate slabs and CoO6 octahedra form a 3D mixed anionic framework with K atoms and water molecules in channels | 12.2075(8) | 8 | 2.866 | ||
| 19.928(2) | 2.688 | (A = K, H2O) | |||
| 2365.9(3) | |||||
| K2(H2O)Ni[B2P3O12(OH)] | 9.5543(2) | Pbca | 2.100 (M = Ni2+) | 2.988 | 23 |
| Borophosphate slabs and NiO6 octahedra form a 3D mixed anionic framework with K atoms and water molecules in channels | 12.056(2) | 8 | 3.007 | ||
| 19.814(4) | 2.681 | 3.007 | |||
| 2282.3(8) | (A = K, H2O) | ||||
| RbCo1.5(H2O)[B2P3O12(OH)] | 9.501(1) | Pbca | 2.101 (M = Co2+) | 3.219 | 24 |
| Trimeric units of CoO6 and CoO4(H2O)2 octahedra sharing edges interconnect with the borophosphate slabs in a framework that includes Rb+ ions in open channels. | 12.272(2) | 8 | 3.236 | ||
| 20.074(2) | 2.925 | 3.300 | |||
| 2340.6(5) | (A = Rb) | ||||
| CsCo1.5(H2O)[B2P3O12(OH)] | 9.5526(4) | Pbca | 2.104 | 3.331 | 25 |
| Trimeric units of CoO6 and CoO4(H2O)2 octahedra sharing edges interconnect with the borophosphate slabs in a framework that includes Cs+ ions in open channels. | 12.3190(4) | 8 | 2.145 | 3.351 | |
| 20.1123(8) | 3.16 | (M = Co2+) | 3.400 | ||
| 2366.8(1) | (A = Cs) | ||||
| Rb1.15(H2O)0.4Mn1.5[B2P3O12(O,OH)]a | 9.151(1) | Pbna | 2.182 (M = Mn2+ in octahedron) | 3.018 | Our data |
| Borophosphate slabs and chains of MnO6 and MnO4 polyhedra sharing edges and vertices, form a framework with Rb+ ions and water molecules in open channels | 12.257(1) | 8 | 2.092 (in tetrahedron) | 3.107 | |
| 20.076(2) | 3.000 | 2.954 | |||
| 2251.8(2) | (A = Rb, H2O) | ||||
| (NH4)(C4H12N2)0.5(H2O)0.5Co[B2P3O12(OH)] | 14.207(3) | I41/a | 2.097 (M = Co2+) | (A = NH4, C4H12N2, H2O) | 26 |
| Mixed anionic framework built by a 3D borophosphate network and CoO6 octahedra with NH4+ ions, organic and water molecules in intersecting channels | 24.956(6) | 8 | |||
| 5037.1(2) | 2.384 | ||||
Isostructural borophosphates, all having the same D152h = Pbca space group, may be obtained without organic templates. Two isotypic compounds, |K2(H2O)|[MB2P3O12(OH)] [M = Co or Ni], have been synthesized with K+ ions instead of ethylenediamine as the organic template. They are constructed by the connection of tetrahedral layers and MO6 octahedra, giving rise to a 3D framework with 8-ring channels aligned in the [010] direction. The negative charge of the framework is compensated by the K+ ions located in the 8-ring channels together with water molecules.23
Two more compounds, A+2Co3(H2O)2[B4P6O24(OH)2] with alkaline ions, which play a directing role in the structure formation, were formed under hydrothermal conditions.24,25 Their isotypic crystal structures include trimeric units of CoO6 and CoO4(H2O)2 octahedra sharing edges that further interconnect with the borophosphate slabs into a framework (Fig. 9). Large low-charged Rb+ or Cs+ ions occupy positions in open channels. Both orthorhombic phases are described by the same D152h = Pbca space group and similar unit cell parameters, which are naturally larger in the case of Cs2Co3(H2O)2[B4P6O24(OH)2] (Table 3).
 | ||
| Fig. 9 The Rb2Co3(H2O)2[B4P6O24(OH)2] crystal structure with three-dimensional borophosphate polyanions shown along the a axis. | ||
In contrast to the (C2H10N2)2M22+[B4P6O24(OH)2] and |K2(H2O)|[MB2P3O12(OH)] structures with isolated MO6 octahedra, or to the A+2Co3(H2O)2[B4P6O24(OH)2] structures with triplets of transition metal octahedra, the new compound Rb2.3(H2O)0.8Mn3[B4P6O24(O,OH)2] reveals Mn2+ cations with two distinct coordinations, the MnO6 octahedra and MnO4 tetrahedra that together form 1D chains. The observed structural transformation correlates with the change of the space group to D142h = Pbna. It is worth noting that, despite having different crystal structures, three groups of compounds under discussion reveal nearly identical borophosphate slabs.
An exceptional crystal structure of (NH4)2(C4H12N2)(H2O)[Co2B4P6O24(OH)2] with the three-dimensional borophosphate polyanion formed by topologically equivalent [B2P3O12(OH)] building blocks of three-membered rings of two borate and one phosphate tetrahedra features tetragonal symmetry (space group I41/a).26 The CoO6 octahedra are introduced into the borophosphate network to form a complex open framework with a three-dimensional intersecting channel system. Its voids are populated by ammonium and diprotonated piperazine ions and water molecules.
Magnetic properties
The dc-magnetic susceptibility χ in a wide temperature range follows the modified Curie–Weiss law with the temperature independent termwhere χ0 = −4 × 10−4 emu mol−1, the Curie constant C = 13.2 emu K mol−1, and the Weiss temperature Θ = −16 K (Fig. S1†). The negative value of χ0 originates from diamagnetic contributions of individual ions. The absolute value of this contribution is in quite good agreement with the sum of Pascal's constants yielding −0.427 × 10−3 emu mol−1.27 The Curie constant corresponds to the value of 8C = ng2S(S + 1) = 13.125 emu K mol−1 expected for n = 3 Mn2+ ions in the chemical formula with the g-factor g = 2 and spin S = 5/2. The negative value of the Weiss temperature indicates the predominance of antiferromagnetic exchange interactions. At low temperatures, experimental data deviate from the Curie–Weiss behavior toward larger values of the magnetic susceptibility.

The ac-magnetic susceptibility is in good agreement with the dc-data and reveals weak anomaly at TN = 12.5 K, as shown in Fig. 10. This anomaly may signal long-range magnetic ordering in the system. In contrast to conventional antiferromagnets, the magnetic susceptibility increases below the transition and displays a paramagnetic-like behavior at low temperatures. Further evidence of the magnetic transition is provided by the specific heat measurements shown in Fig. 11. A rather smeared anomaly is seen at TN = 12.5 K.
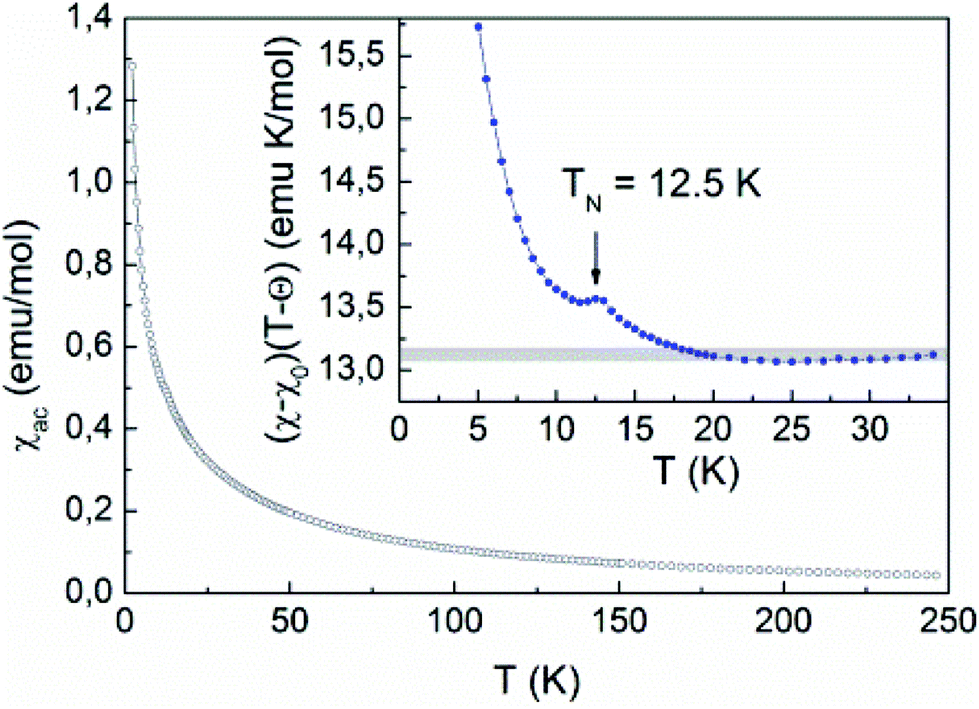 | ||
| Fig. 10 The temperature dependence of the ac-magnetic susceptibility in Rb2.3(H2O)0.8Mn3[B4P6O24(O,OH)2]. Inset: The temperature dependence of the product (χ − χ0)(T − Θ), the straight line represents the Curie constant at high temperatures. | ||
 | ||
| Fig. 11 The temperature dependence of specific heat Cp in Rb2.3(H2O)0.8Mn3[B4P6O24(O,OH)2]. | ||
The field dependence of the magnetization is shown in Fig. 12. At 2.5 K, magnetization approaches the saturation value of 15μB/f.u. around 27 T suggesting that a rather high magnetic field is required to overcome antiferromagnetic interactions between the Mn2+ spins. Additionally, deviations from linear behavior are seen around 4 T.
 | ||
| Fig. 12 The field dependences of magnetization M in Rb2.3(H2O)0.8Mn3[B4P6O24(O,OH)2] taken in a pulsed magnetic field. Inset: The field dependence of magnetization taken in a static magnetic field at 2 K. | ||
Magnetic model
Individual magnetic couplings in Rb2.3(H2O)0.8Mn3[B4P6O24(O,OH)2] were obtained from total energies of collinear spin configurations calculated using the projected augmented wave formalism implemented in the VASP code.28 For density-functional theory (DFT) band-structure calculations, the Perdew–Burke–Ernzerhof flavor of the exchange–correlation potential29 was chosen. Strong correlations in the Mn 3d shell were taken into account of the mean-field DFT+U level30 with the on-site Coulomb repulsion U = 7 eV and Hund's exchange J = 1 eV.31 Each magnetic coupling was derived from the total energies of four collinear spin configurations, as further explained in ref. 32. We used the experimental crystal structure that was modified as follows: (i) the Rb1 position was 100% occupied, while the sparsely occupied Rb2 and Rb3 positions were excluded; (ii) the O14 and O15 positions (disordered water molecules) were excluded too. These modifications change the overall composition to Rb2Mn3[B4P6(OH)2O24] without affecting the charge distribution and valence state of Mn. Since neither Rb nor water molecules are directly bonded to the Mn atoms, their effect on the magnetic couplings should be minor.Magnetic couplings Ji are calculated for pairs of spin-5/2 Mn2+ ions. Positive couplings are antiferromagnetic. In Rb2.3(H2O)0.8Mn3[B4P6O24(O,OH)2], three nearest-neighbor couplings are J1 = 0.14 K (3.478 Å, Mn1–Mn1), J2 = 3.4 K (3.632 Å, Mn1–Mn2), and J3 = −0.01 K (4.065 Å, Mn1–Mn2), where numbers in brackets stand for Mn–Mn distances (Fig. 13). The couplings beyond the nearest neighbors are below 0.02 K.
 | ||
| Fig. 13 The exchange paths for various magnetic interactions. | ||
The difference between the nearest-neighbor couplings J1 and J2 is rooted in the different Mn–O–Mn angles, 103.74° and 115.59°, respectively. According to Goodenough–Kanamori–Anderson rules, larger Mn–O–Mn angles favor antiferromagnetic couplings, hence J2 > J1. Despite its relatively short Mn–Mn distance, the coupling J3 is between the Mn1O6 octahedron and Mn2O4 tetrahedron that are not directly connected to each other. Therefore, no Mn–O–Mn superexchange pathway exists, and the coupling J3 remains weak.
The coupling J2 builds Mn1–Mn2–Mn1 trimers. The magnetic susceptibility of spin-5/2 trimers obtained from quantum Monte-Carlo simulations with J = 3.2 K, g = 1.98, and χ0 = −4 × 10−4 emu mol−1 perfectly reproduces experimental data down to 20 K (Fig. S1†). This exchange coupling is in excellent agreement with the calculated J2 = 3.4 K. The trimer model explains the increase in the susceptibility toward lower temperatures. Indeed, at low temperatures an individual trimer adopts the spin-5/2 state and behaves as a paramagnetic entity with the diverging susceptibility. Below 20 K, the magnetic transition at TN and ensuing deviations from the spin-trimer behavior are likely due to antiferromagnetic couplings between the trimers. However, these couplings are too weak for a reliable quantitative analysis.
For a spin-5/2 trimer with J = 3.2 K one expects saturation at Hs ∼ 20 T. Experimentally, the saturation feature is very broad, and the saturation field cannot be determined with sufficient accuracy. Nevertheless, at 2.5 K and 20 T the magnetization reaches around 90% of the saturated value, which means that our estimate of J is in reasonable agreement with the magnetization data.
Conclusions
Rb2.3(H2O)0.8Mn3[B4P6O24(O,OH)2] is a new borophosphate compound that reveals an original crystal structure. In contrast to other borophosphates with similar structural units, it shows an interesting combination of the magnetic high-spin Mn2+ ions in both octahedral and tetrahedral coordinations. The ensuing magnetic interactions give rise to antiferromagnetic spin trimers that manifest themselves in the paramagnetic-like behavior, with the magnetic susceptibility increasing upon cooling. Weak interactions between these trimers induce a magnetic transition (presumably, antiferromagnetic ordering) around 12.5 K.Acknowledgements
This work was supported in part by the Ministry of Education and Science of the Russian Federation in the framework of the Increase Competitiveness Program of NUST “MISiS” Grants No. K2-2016-066 and K4-2015-020, by Act 211 of the Government of Russian Federation, agreement No. 02.A03.21.0006, and by the Russian Foundation for Basic Research Grants No. 15-05-06742, 16-02-00021 and 17-02-00211. AT acknowledges financial support by the Federal Ministry for Education and Research through the Sofja Kovalevskaya Award of the Alexander von Humboldt Foundation.Notes and references
- B. Ewald, Y.-X. Huang and R. Kniep, Z. Anorg. Allg. Chem., 2007, 633, 1517 CrossRef CAS.
- O. Yakubovich, I. Steele, W. Massa and O. Dimitrova, Z. Kristallogr., 2013, 228, 509 CAS.
- H. Li and A.-V. Mudring, Cryst. Growth Des., 2016, 16, 2441 Search PubMed.
- H. Yaghoobnejad, P. Stanly, K. Ghosh and A. Choudhury, Chem. Mater., 2015, 27, 7058 CrossRef.
- M. Mouyane, J. C. Jumas, J. Olivier-Fourcade, S. Cassaignon, Ch. Jordy and P. E. Lippens, J. Solid State Chem., 2016, 233, 52 CrossRef CAS.
- J. A. L. Da Silva and N. G. J. Holm, Colloid Interface Sci., 2014, 431, 250 CrossRef CAS PubMed.
- T. Su, H. Xing, J. Xu, J. Yu and R. Xu, Inorg. Chem., 2011, 50, 1073 CrossRef CAS PubMed.
- W. Zhang, W. Cheng, H. Zhang, L. Geng, Y. Li, Ch. Lin and Zh. He, Inorg. Chem., 2010, 49, 2550 CrossRef CAS PubMed.
- M. Yang, P. Yan, F. Xu, J. Ma and U. Welz-Biermann, Microporous Mesoporous Mater., 2012, 147, 73 CrossRef.
- G. Wang, M. Valldor, Ch. Lorbeer and A.-V. Mudring, Eur. J. Inorg. Chem., 2012, 18, 3032 CrossRef.
- W. Yang, J. Li, T. Na, J. Xu, L. Wang, J. Yu and R. Xu, Dalton Trans., 2011, 40, 2549 RSC.
- CrysAlisPro, Oxford diffraction Ltd, 2014, Version 1.171.37.31 (release 14-01-2014 CrysAlis171.NET) Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- International Tables for Crystallography, ed. E. Prince, Kluwer, Dordrecht, 3rd edn, 2004, Tables 4.2.6.8 and 6.1.14 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3 CrossRef PubMed.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244 CrossRef.
- I. D. Brown, Chem. Rev., 2009, 109, 6858 CrossRef CAS PubMed.
- H. B. Yahia, E. Gaudin and J. Darriet, J. Alloys Compd., 2007, 442, 74 CrossRef.
- M. Luján, F. Kubel and H. Schmid, Z. Naturforsch., B: J. Chem. Sci., 1995, 50, 1210 Search PubMed.
- S. C. Sevov, Angew. Chem., Int. Ed. Engl., 1996, 35, 2630 CrossRef CAS.
- R. Kniep and G. Schäfer, Z. Anorg. Allg. Chem., 2000, 626, 141 CrossRef CAS.
- W. Liu, M. Ge, X. Yang, H. Chen, M. Li and J. Zhao, Inorg. Chem., 2004, 43, 3910 CrossRef CAS PubMed.
- D. Zhang, Y. Feng, Y. Lin, Y. Zhang, G. Li and H. Yuan, Dalton Trans., 2015, 44, 17100 RSC.
- H. Engelhardt, W. Schnelle and R. Kniep, Z. Anorg. Allg. Chem., 2000, 626, 1380 CrossRef CAS.
- P. W. Menezes, S. Hoffmann, Yu. Prots and R. Kniep, Z. Kristallogr. – New Cryst. Struct., 2009, 224, 1 CAS.
- W. Liu, X.-Q. Guo, G. Su, L.-X. Cao, Y.-G. Wang and J.-R. Duan, J. Solid State Chem., 2011, 184, 2538 CrossRef CAS.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- G. Kresse and J. Furthmüller, Mater. Sci., 1996, 6, 15 Search PubMed; G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- A. I. Liechtenstein, V. I. Anisimov and J. Zaane, Phys. Rev. B: Condens. Matter, 1995, 52, R5467 CrossRef CAS.
- R. Nath, K. M. Ranjith, B. Roy, D. C. Johnston, Y. Furukawa and A. A. Tsirlin, Phys. Rev. B: Condens. Matter, 2014, 90, 024431 CrossRef.
- H. Xiang, C. Lee, H.-J. Koo, X. Gong and M.-H. Whangbo, Dalton Trans., 2013, 42, 823 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic information for Rb2.3(H2O)0.8Mn3[B4P6O24(O,OH)2], the results of bond valence calculation and magnetic measurement data (PDF) and X-ray crystallographic data in CIF format (CIF). See DOI: 10.1039/c6dt04241d |
| ‡ The analysis was performed at the Laboratory of Local Methods for Studying Materials, Department of Petrology, Faculty of Geology, M.V. Lomonosov Moscow State University. |
| § Additional materials to the crystal structure investigation are obtained from Fachinformationszentrum Karlsruhe, Germany, under specification of deposit No. CSD-431432 and authors’ reference. |
| This journal is © The Royal Society of Chemistry 2017 |