
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spontaneous Si–C bond cleavage in (TriphosSi)-nickel complexes†
Anette
Petuker
a,
Stefan
Mebs
b,
Nils
Schuth
b,
Philipp
Gerschel
a,
Matthew L.
Reback
a,
Bert
Mallick
a,
Maurice
van Gastel
*c,
Michael
Haumann
*b and
Ulf-Peter
Apfel
 *a
*a
aRuhr University Bochum, Inorganic Chemistry I, Universitätsstraße 150, 44801 Bochum, Germany. E-mail: ulf.apfel@rub.de
bFreie Universität Berlin, Department of Physics, Arnimallee 14, 14195 Berlin, Germany. E-mail: michael.haumann@fuberlin.de
cMax-Planck-Institut für chemische Energiekonversion, Stiftstrasse 34-36, 45470 Mülheim a. d. Ruhr, Germany. E-mail: maurice.vangastel@cec.mpg.de
First published on 8th December 2016
Abstract
Herein, we report on the versatile reactions of CH3C(CH2PPh2)3 as well as CH3Si(CH2PPh2)3 derived Ni-complexes. While Ni[CH3C(CH2PPh2)3] complexes reveal high stability, the Ni[CH3Si(CH2PPh2)3] analogs show rapid decomposition at room temperature and afford the unprecedented pseudo-tetrahedral phosphino methanide complex 5. We provide a detailed electronic structure of 5 from X-ray absorption and emission spectroscopy data analysis in combination with DFT calculations, as well as from comparison with structurally related complexes. A mechanistic study for the formation of complex 5 by reaction with BF4− is presented, based on a comparison of experimental data with quantum chemical calculations. We also show a simple route towards isolable Ni(I)-complexes on the gram scale.
Introduction
The utility of metal phosphine complexes for catalysis has been well documented for more than half a century.1,2 These complexes often participate in catalytic transformations by controlling steric properties as well as the stability of reactive intermediates. While the P–C bond of phosphine ligands is quite stable, there are reports of thermally oxidative P–C bond cleavages.3 For example, 1,1-bis(diphenylphosphino)methane (dppm) undergoes thermally oxidative P–C(sp3) bond cleavage to afford Ph2P and CH2PPh2 bond fragments.3–5 Likewise, the deprotonation of methylene-bridged bis-phosphines, the oxidative addition of R2PCH3 or its deprotonated congener R2PCH2Li to metals was reported to afford phosphino methanide (R2PCH2) complexes.6,7 Moreover, Caulton and co-workers reported a transannular Si–C(sp3) coordination in [(PNP)]NiCl (PNP = (tBu2PCH2SiMe2)2N)8 and selective bond cleavage leading to a phosphino methanide fragment coordinated to nickel. The R2PCH2 unit was suggested to possess different mesomeric structures (Fig. 1).9–12 As a result of the different possible mesomeric structures, the R2PCH2 fragment can act as a redox-non-innocent ligand allowing for different oxidation states of the metal center.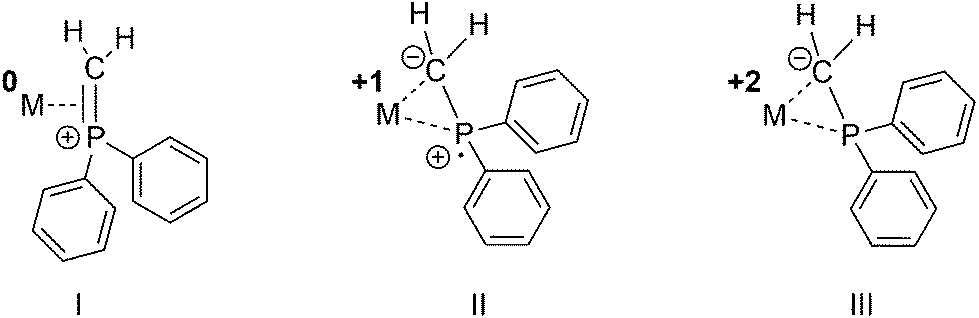 | ||
| Fig. 1 Hypothetical binding modes of a Ph2PCH2 fragment with concomitant formal cationic, neutral or anionic oxidation states of the Ph2PCH2 ligand. | ||
Thus, owing to its chemical versatility, the η2-coordinated R2PCH2 ligand was even suggested to play a role in catalytic transformations such as the hydrodesulfurization reaction of thiophenes,13 cyclopropanation reactions14 in the metathesis of C–C triple bonds or as a source of Fe(0)(bis-NHC) complexes (NHC = N-heterocyclic carbene) in the reduction of organic amides.15 Besides the possibility to act as a redox-non-innocent ligand, they are also able to alter the coordination environment of metals, in particular their ligand fields, and thus play a pivotal role in controlling catalytic conversions including selectivity.16,17
We recently investigated the different catalytic properties for Sonogashira cross-coupling and hydrosilylation reactions of CH3C(CH2PPh2)3 (TriphosC, 1a) and CH3Si(CH2PPh2)3 (TriphosSi, 1b) derived Fe(II)- and Ni(II)-complexes due to different flexible coordination behavior.18,19
To extend our investigations, we set out to synthesize bimetallic nickel-complexes. Herein, we report on the reaction of (TriphosSi)NiCl2 and (TriphosC)NiCl2 and their Fe-homologues with different nickel(II)-salts. While our attempt at forming bimetallic nickel species was unsuccessful, we unveiled a rich chemistry of TriphosC/Si complexes resulting in the serendipitous formation of an unprecedented pseudo-tetrahedral nickel phosphino methanide complex 5 (Scheme 1). EXAFS, XANES and EPR spectroscopy techniques in combination with DFT calculations were used to determine the electronic structures for direct comparison with structurally related Ni(II) and Ni(I) complexes.
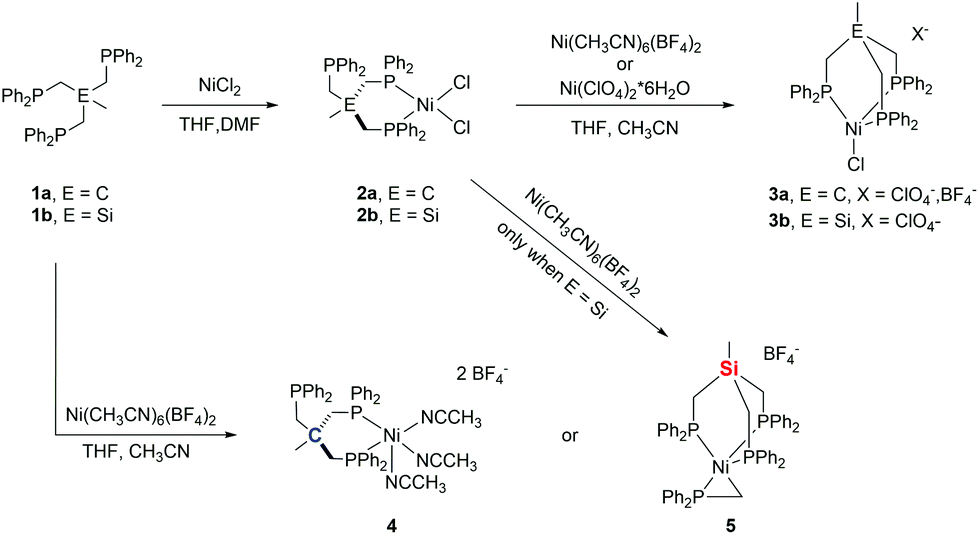 | ||
| Scheme 1 Schematic overview of the herein performed reactions. | ||
Results and discussion
Synthesis and characterization
We recently reported the synthesis of the square planar Ni-complex 2a and tetrahedral Ni-complex 2b from TriphosC (1a) and TriphosSi (1b) in the presence of NiCl2 and described the reasons for their different structures.18 In an attempt to establish a route for bimetallic nickel systems containing either a square planar or tetrahedral Ni-moiety, we set out to utilize the dangling phosphine as an additional metal binding site as was reported in mixed NiII/Sn,20 Ni0/Ni0,21 as well as Ni0/AuI complexes22 also comprising the TriphosC ligand.We thus treated complexes 2a and 2b with [Ni(CH3CN)6](BF4)2 as well as Ni(ClO4)2. In neither case were we able to observe a bimetallic complex. Instead, the reactions led to the immediate binding of the dangling arm to the nickel center thus affording complexes 3a(ClO4), 3a(BF4) and 3b(ClO4) in good yields (Scheme 1). The formation of complexes 3a(BF4) and 3b(ClO4) was unequivocally confirmed by X-ray structure analyses (Fig. 2 and S1†). The Ni–Cl bond length in 3b(ClO4) 2.193(5) Å and the P–Ni–P bond angles (95.50(1) Å) are in good agreement with the structural parameters of 2b.
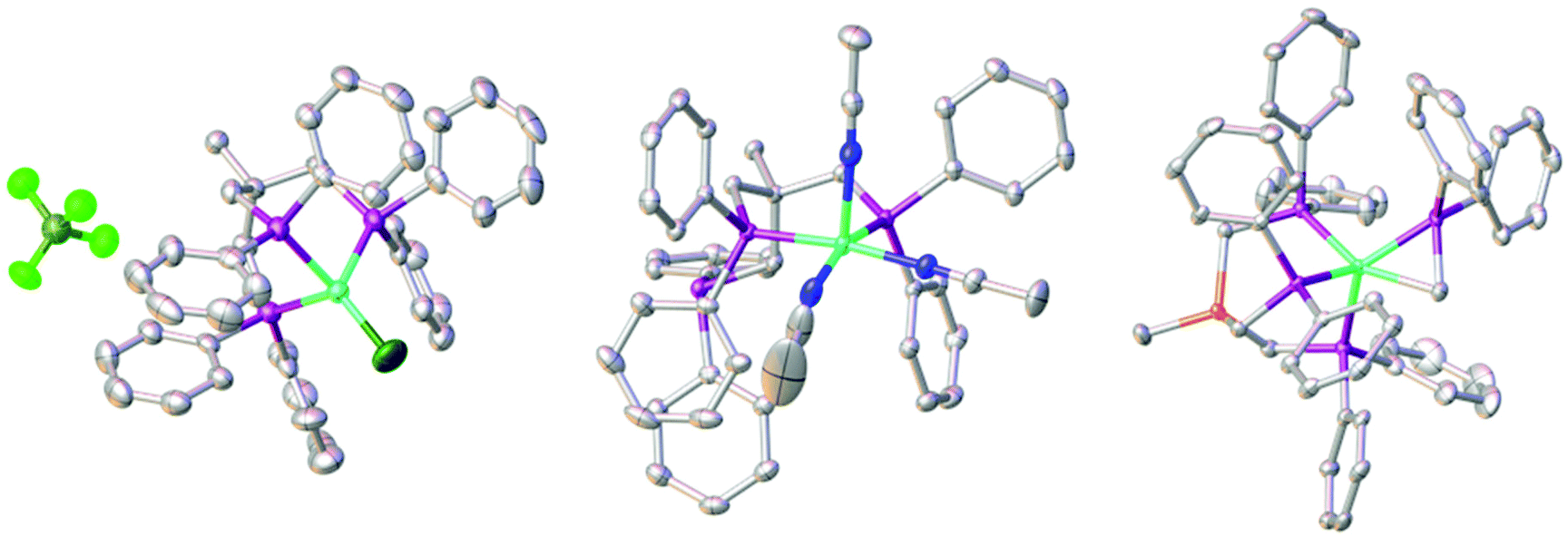 | ||
| Fig. 2 Molecular structures of compounds 3a(BF4) (left), 4 (middle) and 5 (right) with thermal ellipsoids drawn at the 50% probability level (hydrogen atoms are omitted for clarity). Color scheme: turquois: nickel; fern: boron; light green: fluorine; dark green: chlorine; purple: phosphorus; blue: nitrogen; grey: carbon; orange: silicon. | ||
As a consequence of the change in coordination, the Ni–P distances in complex 3b(ClO4) decrease by about 0.15 Å to 2.282(3) Å compared to 2b. In contrast, the changes of the bond length in 3a(BF4) compared to 2a are negligibly small.
Complexes 3a(ClO4) and 3a(BF4) both show UV-vis spectra absorption bands at 425 and 676 nm. In contrast, the analogous complex 3b(ClO4) shows a bathochromically-shifted UV-vis spectrum with bands at 437 and 708 nm (Fig. S2†). This observation is in line with our previous findings, showing different structural and electronic properties for Ni- as well as Fe-derived TriphosC and TriphosSi complexes.18,19 Surprisingly, neither the addition of thallium nor silver salts allowed for the formation of any of the complexes 3. No reaction was observed with thallium salts and the precipitation of metallic silver was observed with silver salts.
We wondered if the formation of the κ3-coordinated TriphosC/Si-ligand is an intrinsic result of the non-coordinating perchlorate or tetrafluoroborate anions. To investigate this possibility, compounds 1a and 1b were reacted with Ni(ClO4)2. In this case, we were unable to isolate any reaction product. On the other hand, the reaction of compound 1a with [Ni(CH3CN)6](BF4)2 afforded complex 4 (Scheme 1) in 80% yield and reveals two absorption bands in its UV-vis spectrum at 422 nm (ε = 1490 L mol−1 cm−1) and 728 nm (ε = 460 L mol−1 cm−1) (Fig. S3†). The composition of complex 4 was subsequently confirmed by structure analysis (Fig. 2). The coordination mode of 4 is best described as square pyramidal having a κ2-bound TriphosC ligand with Ni–P distances of 2.1717 (7) and 2.1951(7) Å. Additionally, three acetonitrile ligands are bound to the nickel center. The axial Ni–N distance is 2.268(3) Å, which is significantly longer than those observed for the equatorial Ni–N bonds (1.925(2) and 1.928(3) Å).
Contrary to the reaction of compound 1a, the reaction of 1b and [Ni(CH3CN)6](BF4)2 under the same reaction conditions afforded a dark red crystalline solid with characteristic UV-vis bands at 520 (ε = 1990 L mol−1 cm−1) and 429 nm (ε = 671 L mol−1 cm−1) (Fig. S3/S4†). Furthermore, compound 5 shows an ESI-MS signal at m/z = 897.1 and a characteristic 31P{1H} NMR spectrum (Fig. S5/S6†). A doublet at 7.25 (3JP–P = 49 MHz) and a quartet centered at −26.4 (3JP–P = 49 MHz) clearly indicate a different overall structure as reported for complex 4, which did not reveal any 31P{1H} NMR signal. The final confirmation of the composition of 5 was provided by X-ray structure analysis (Fig. 2). The Ni-center shows a pseudo square-pyramidal coordination environment comprised by the κ3-bound TriphosSi ligand 1b and a bidentate CH2PPh2 ligand. The average Ni–P(TriphosSi) bond length is 2.247 Å. In contrast, the Ni–P(CH2PPh2) bond is significantly shorter (2.1322(5) Å). The Ni–C distance is 2.060(2) Å revealing a symmetric π-binding of the CH2PPh2 ligand. The substantial π-character of the P–C bond in the CH2PPh2 unit is further supported by the short C–P distance (1.725(2) Å), which is significantly less than expected for a C–P single bond (∼1.83 Å) and in good agreement with commonly observed P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond lengths (∼1.73 Å).23 Notably, complex 5 can also be obtained in good yield (74%) when complex 2b reacts with [Ni(CH3CN)6](BF4)2. Since no additional source of phosphino methanide is present and the formation of a phosphino methanide was never observed for the C-derived counterpart 1a, the CH2PPh2 ligand must stem from a selective and rapid decomposition of 1b.
C double bond lengths (∼1.73 Å).23 Notably, complex 5 can also be obtained in good yield (74%) when complex 2b reacts with [Ni(CH3CN)6](BF4)2. Since no additional source of phosphino methanide is present and the formation of a phosphino methanide was never observed for the C-derived counterpart 1a, the CH2PPh2 ligand must stem from a selective and rapid decomposition of 1b.
Complex 5 is furthermore stable to air and moisture and soluble in a wide range of solvents, including pentane, chlorinated solvents and acetonitrile without any notable decomposition even at higher temperatures (Fig. S4†). The uniqueness of complex 5 is further supported by the fact that only 11 structurally characterized substances containing the η2-R2PCH2 ligand are listed in the Cambridge Structural Database and none of them reveals a coordination environment as found in complex 5.23 While the oxidative addition of R2PMe ligands to generate a η2-CH2PR2 has been reported7,12,15,24,25 and a TriphosSi derived ruthenium complex26 was shown to afford the phosphino methanide structural motif in the presence of LiBEt3H, to the best of our knowledge, a spontaneous Si–C(sp3) cleavage of TriphosSi under such mild conditions has not been reported in the literature.
However, it is known that the Si–C bond of TriphosSi can be cleaved to afford the CH3–PPh2 motif but only at elevated temperatures.27 We therefore assume that a Ni(BF4)2-induced cleavage of the Si–C bond is responsible for the formation of the CH2PPh2 ligand. Although this hypothesis is similar to the BF3 catalyzed selective Si–C bond cleavage in (PNP)NiF complexes reported by Caulton and coworkers,28,29 a different cleavage mechanism has to be assumed. This stems from the following observations:
(1) In contrast to the significantly elongated Si–C bond in (PNP)Ni+ (2.069(4) Å), the Si–C bonds in complex 2b, 3b(ClO4) and 5 are not activated and all reveal Si–C bond lengths of about 1.89 Å.
(2) The treatment of complexes 2b and 3b(ClO4) with BF3·Et2O or BF4− does not lead to the formation of complex 5 and highlights the critical importance of labile ligands, such as acetonitrile.
(3) The formation of 5 is induced by nickel. This is in line with the previous findings on the reactivity of 1b and [Fe(CH3CN)6](BF4)2 that did not allow for the formation of the homologous phosphine methanide iron complex.19,30
In a control experiment, the reaction of the iron complex 6b with [Ni(CH3CN)6](BF4)2 was shown to yield complex 5. This was unequivocally established by ESI-MS, the missing quadrupole doublets in the Mössbauer spectra of crystalline product samples, as well as structural analysis. In contrast, when iron-complexes 6b were reacted with Ni(ClO4)2 or 6a with [Ni(CH3CN)6](BF4)2 under the same experimental conditions, complexes 3b(ClO4) and 3a(BF4), respectively, were obtained along with FeCl2 (Scheme 2). The Ni/Fe scrambling was unequivocally confirmed by X-ray structure analysis, Mössbauer spectroscopy, as well as ESI-MS experiments and the same molecular structure and ESI-MS datasets were obtained as for 3b(ClO4) and 3a(BF4).
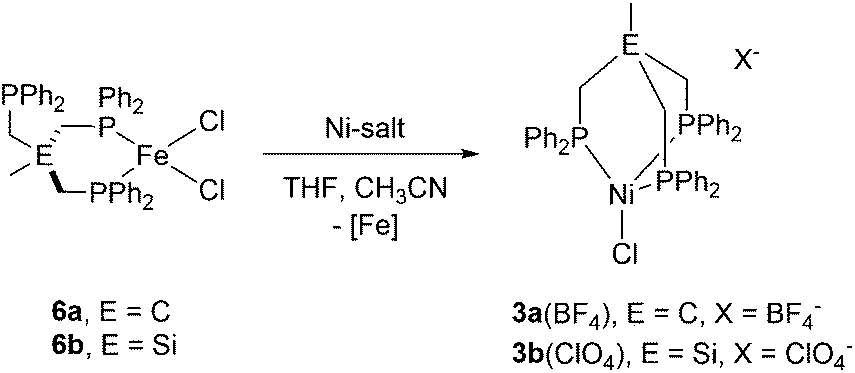 | ||
| Scheme 2 Synthesis of the nickel-complex 3a(BF4) and 3b(ClO4) via reaction of tripodal Fe-complexes 6a with [Ni(CH3CN)6](BF4)2 and 6b with Ni(ClO4)2. | ||
This suggests that both the BF4− counter ion as well as the nickel(II) center play a synergistic role in the controlled Si–C cleavage. Although time dependent 31P NMR spectroscopic investigations are in line with a rapid and highly specific reaction revealing solely the formation of complex 5 immediately after addition of [Ni(CH3CN)6](BF4)2 to 1b (Fig. S5†), we were not able to shed light on the mechanism of this cleavage experimentally or to isolate any side product.
Reactivity of complex 5
The high reactivity of L4M(CH2PMe2) complexes (M = Co, Fe) and other phosphino methanide complexes3,6,7,13,15,25,31–35 prompted us to investigate the reactivity of the unprecedented complex 5. Most notable is the W(PMe3)3(η2-CH2PMe2)H complex, which was reported as an electron-rich and highly reactive complex for the hydrodesulfurization of thiophenes.13,32 Due to the high π-character of the P–C bond, we expected that 5 would react with heterocumulenes and examined the reaction of 5 with CO2 under 10 bar/70 °C as was reported by Bennett et al. for [(dcpe)NiCl(C(CHCO2Me)PPh2-κP,C1)] (dcpe = bis(dicyclohexylphosphino)ethane).36
Contrary to our expectations, no reaction was observed. Likewise, no reaction was observed when CS2, CO, styrene or 1-octene was added to complex 5 at elevated temperatures. Subsequently, the Wittig–Horner like reaction with benzophenone and acetophenone was attempted but did not show any conversion of complex 5. Even refluxing under atmospheric conditions or treatment with HCl did not reveal any visible decomposition and complex 5 was quantitatively recovered.
Next, complex 5 was reacted with 2 equivalents of (phenylethynyl)magnesium bromide to give an immediate reaction affording a single product (7b) that was isolated in 65% yield. Although this is an unexpected result, Grignard reagents are very well known for their potential to also act as reductants.37,38 The composition of 7b was confirmed by X-ray structure analysis (Fig. 3). The magnetic moments of complexes 7a and 7b were measured using the Evans method and found to be 1.9μB and 1.7μB respectively, which is in line with literature findings for Ni(I) complexes.39 The Ni(I)-center is tetrahedrally coordinated by the κ3-bound TriphosSi ligand 1b and one bromide ligand. The phosphorus atoms are not equivalent, which is shown by the three observed Ni–P distances (2.246(2) Å, 2.258(2) Å and 2.219(2) Å). The Ni–Br distance is 2.403(1) Å. Although a structurally related (TriphosC)Ni(I)-complex was previously reported by Huttner as well as Sacconi and co-workers, their synthetic procedure failed in our hands.39,40 We subsequently attempted a different synthetic route towards such Ni(I)-complexes that allows for a simple straightforward gram scale synthesis. The reaction of 2a with cobaltocene gave the analogous C-derived Ni(I)-compound 7a and was confirmed by X-ray structure analysis (Scheme 3 and Fig. 3). Notably, the Ni(I)-compounds can be obtained on the gram scale and stored under an inert atmosphere at room temperature without any decomposition. Compound 7a is isostructural to 7b with a Ni–Cl distance of 2.2442(2) Å and three different Ni–P distances of 2.214(1), 2.221(1) and 2.237(1) Å. Although bearing different halides, the electronic properties are similar, which is clearly visible from the similar UV-vis spectra, showing bands at 402 nm and 388 nm for compounds 7a and 7b, respectively (Fig. S8†). The replacement of CH2PPh2 by a bromide ligand directed our attention towards the replacement with neutral ligands. Inspired by independent reports by Ozerov as well as Shiu et al. on ligand substitutions with PMe3, we examined the reactivity of complex 5 with PMe3.3,41
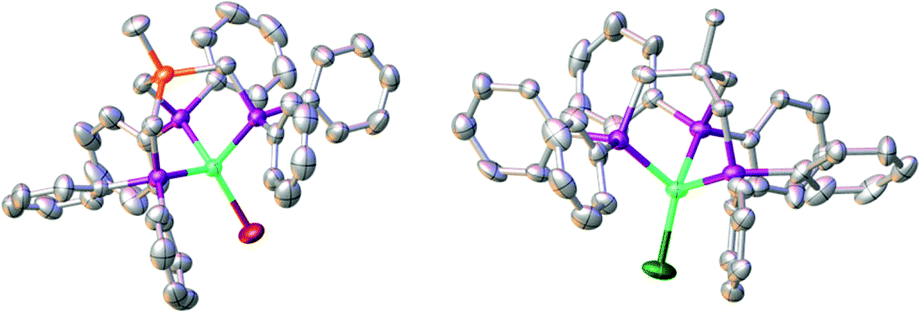 | ||
| Fig. 3 Molecular structures of compounds 7a (right) and 7b (left) with thermal ellipsoids drawn at the 50% probability level (hydrogen atoms are omitted for clarity). | ||
 | ||
| Scheme 3 Synthesis of nickel(I)-complexes 7a and 7b. | ||
An immediate color change to dark purple was observed when an excess of PMe3 was added yielding complex 8 as a dark red compound (Fig. S6†). The reaction led to the complete detachment of the TriphosSi1b and the phosphino methanide ligands, affording the trigonal pyramidal Ni(II)-complex 8 and uncoordinated TriphosSi. We were unable to determine the fate of the phosphino methanide ligand. The nickel center is coordinated by four equatorial trimethylphosphines as well as one acetonitrile ligand. When the reaction instead was carried out under atmospheric conditions, complex 9 was isolated as the main species in 35% yield (Scheme 4).
 | ||
| Scheme 4 Synthesis of nickel complexes 8 and 9 starting from complex 5. | ||
Electronic structures of compound 5
Contrary to our initial expectations, complex 5 is thermodynamically and kinetically robust revealing its unprecedented electronic structure. Besides the steric shielding effects of the PPh2 group as well as the TriphosSi ligand we assumed electronic properties to have a major contribution towards the low reactivity compared to other known phosphino methanide complexes. Although EPR spectroscopy is a powerful tool to elucidate the electronic structure of metal complexes, it is of no use to elucidate such properties since complexes 3a(ClO4), 3a(BF4) and 3b(ClO4) and 5 are EPR silent. The EPR spectrum of 7a is shown in Fig. S9.† As is clear from the figure, the spectrum displays an unusually rich albeit greatly overlapping hyperfine structure. The closest related literature data that could be found are for (PMe3)2NiCl2,42 earlier attempts to obtain the EPR spectrum of 7a in frozen solution were not successful.39 The trigonal coordination of the former complex is too far removed from the ligand field present in 7a. Thus, in order not to overinterpret the data by fitting with many species, the spectrum has been carefully analyzed by hand to retrieve data that we feel is safe. In particular, a repeating pattern of 4 equidistant signals in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 ratio is observed on the left-most and right-most edges of the spectrum (Fig. S9,† inset). The pattern of the 4 signals is characterized by a hyperfine splitting of 0.52 mT (14.5 MHz), and is assigned to the axially coordinated chloride ion. The larger hyperfine coupling constant present between two consecutive repeating patterns of 4 signals amounts to 2.5 mT (70 MHz). This can be classified as a strong hyperfine interaction, which is typical of 31P (I = 1/2) in σ interactions with the spin-carrying 3d orbital of the metal.43 Although the sheer multitude of overlapping signals in the center of the spectrum unfortunately hampers further quantitative analysis, the observed repeating 31P structure of roughly equal intensity between the repeating patterns indicates that the three phosphorus atoms of TriphosC are magnetically not equivalent, since three equivalent phosphorus atoms would have resulted in a more regular pattern of 5 lines with a 1:4:6:4:1 ratio. The frozen-solution EPR spectrum thus fully corroborates the finding by X-ray crystallography of three distinct Ni–P bond lengths.
:1:1:1 ratio is observed on the left-most and right-most edges of the spectrum (Fig. S9,† inset). The pattern of the 4 signals is characterized by a hyperfine splitting of 0.52 mT (14.5 MHz), and is assigned to the axially coordinated chloride ion. The larger hyperfine coupling constant present between two consecutive repeating patterns of 4 signals amounts to 2.5 mT (70 MHz). This can be classified as a strong hyperfine interaction, which is typical of 31P (I = 1/2) in σ interactions with the spin-carrying 3d orbital of the metal.43 Although the sheer multitude of overlapping signals in the center of the spectrum unfortunately hampers further quantitative analysis, the observed repeating 31P structure of roughly equal intensity between the repeating patterns indicates that the three phosphorus atoms of TriphosC are magnetically not equivalent, since three equivalent phosphorus atoms would have resulted in a more regular pattern of 5 lines with a 1:4:6:4:1 ratio. The frozen-solution EPR spectrum thus fully corroborates the finding by X-ray crystallography of three distinct Ni–P bond lengths.
We subsequently used X-ray absorption and emission spectroscopy at the Ni K-edge to further characterize the molecular and electronic structure of the complexes 2a, 3a(BF4), 7a, and 5 in powder samples. The nickel-ligand bond lengths obtained from extended X-ray absorption fine structure (EXAFS) analysis (Fig. S11 and Table S1†) were in good agreement with the values from XRD (Tables S3–S5†). EXAFS showed a similar Ni–P and Ni–Cl bond length in 2a, 3a(BF4), and 7a, including overall slightly longer Ni–P bonds in the more reduced 7a, and confirmed the Ni–P bond length heterogeneity (about 2.14–2.32 Å) and shorter Ni–C bond length (∼2.06 Å) in 5. This indicates that the nickel site geometry of the complexes in powder material was similar to the single crystals.
The X-ray absorption near edge structure (XANES) spectra (Fig. S12†) showed pronounced Ni K-edge shape differences between the compounds reflecting nickel site geometry changes. The ∼1.5 eV lower edge energy for the Ni(I) compound 7a (8340.6 eV) compared to the Ni(II) compounds 2a and 3a (BF4) (∼8341.7 eV) is due to a significantly increased charge density at the metal in the more reduced compound. The edge energy (∼8342.1 eV) close to the Ni(II) level suggested a divalent nickel ion also in 5 and essentially ruled out a Ni(0) or Ni(I) species. The steeper edge rise compared to the spectra of the complexes with four-coordinated (tetrahedral, 3a(BF4); square-planar, 2a) nickel sites in the other complexes likely results from the additional equatorial and more ionic Ni–C bond at the distorted square-pyramidal nickel in complex 5.
Non-resonantly excited Kβ main-line emission spectra (radiative 3p → 1s electron decay) were recorded (Fig. S13†), which are sensitive to the metal spin and oxidation state.44 Compounds 2a, 3a(BF4), and 5 showed similar Kβ1,3 line energies (∼8265.7 eV), suggesting the same Ni(II) oxidation state. The Ni(I) in 7a shifted the line to ∼0.5 eV lower energy. The Kβ′ spin-polarization feature gained intensity due to the exchange coupling of 3p with increasing numbers of unpaired Ni(d) electrons.45 Compound 3a(BF4) showed a larger Kβ′ feature than 2a and 5, suggesting an increased number of unpaired Ni(d) spins in 3a(BF4) and therefore a high-spin (hs, M = 3) Ni(II) ion in 3a(BF4), but a low-spin (ls, M = 1) Ni(II) ion in 2a and 5.
The pre-edge absorption feature in the XANES probes resonant core-level (1s) electron excitation into unoccupied valence levels with Ni(3d) and ligand characters (core-to-valence transitions, ctv) whereas the Kβ satellite emission feature (Kβ2,5) probes the reverse process, namely radiative valence electron decay to the core hole (valence-to-core transitions, vtc). Fig. 4 shows ctv and (non-resonantly excited) vtc spectra of 2a, 3a(BF4), 5, and 7a. Theoretical spectra were obtained from single-point calculations on XRD structures. Good agreement between the experimental and calculated ctv and vtc spectra (Fig. 4, solid lines) was only obtained for a Ni(I) ion in 7a, a high-spin-Ni(II) ion in 3a(BF4), and a low-spin-Ni(II) ion in 2a and 5. The spin and oxidation state assignment was further corroborated by electronic energies obtained for single-point calculations on XRD structures or with gas-phase geometry-optimized model structures (Table S2†).
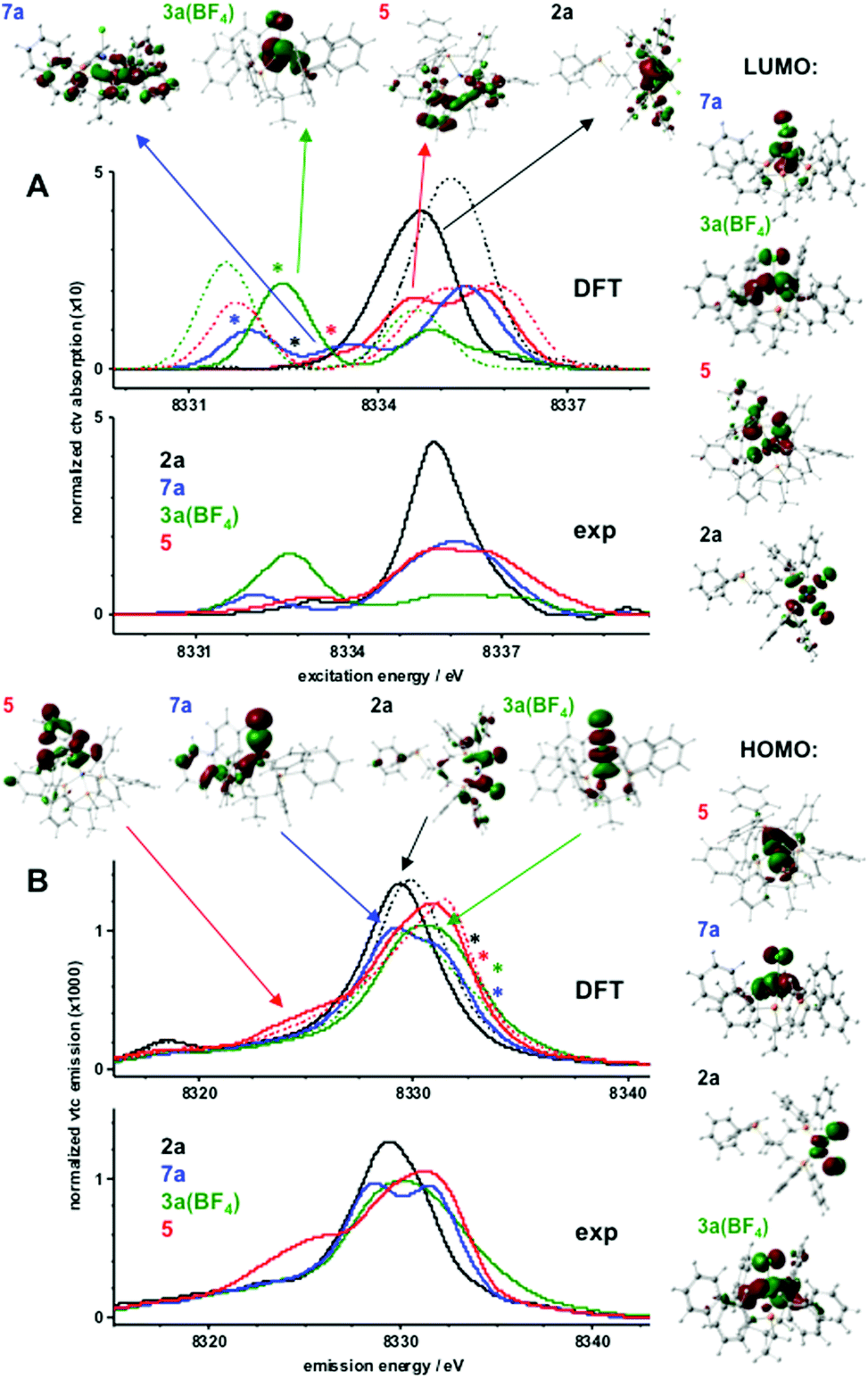 | ||
| Fig. 4 Electronic excitation (core-to-valence, ctv) and decay (valence-to-core, vtc) transitions. Experimental spectra were derived from XANES and Kβ emission data in Fig. S12 and S13;† theoretical data correspond to single-point DFT calculations (BP86/TZVP) using crystallographic coordinates. (A) Experimental (bottom) and calculated (middle) pre-edge absorption spectra due to resonant electronic excitation (ctv) into unoccupied target MOs. Spectral regions and target MOs (top) for transitions with prominent oscillator strength are marked by arrows (dominant MO characters: 7a, Ni(d); 3a(BF4), Ni(d); 5, ligand; 2a, P); approximate energy regions marked by asterisks correspond to transitions into respective LUMOs shown on the right. (B) Experimental (bottom) and calculated (middle) Kβ2,5 spectra due to decay from occupied MOs to the core hole (vtc). Spectral regions and source MOs (top) for transitions with prominent oscillator strength are marked by arrows (dominant MO characters: 5, P–C; 7a, Cl; 2a, Cl; 3a(BF4), Ni(d)); approximate energy regions marked by asterisks correspond to decay transitions from respective HOMOs shown on the right. Calculated spectra in A and B (solid lines) correspond to nickel species (hs/ls = high/low spin; Ni(d) electron count, spin state, multiplicity (M = 2S + 1) in parenthesis): 2a, ls-Ni(II) (8, 0, 1); 3a(BF4), hs-Ni(II) (8, 2/2, 3); 5, ls-Ni(II) (8, 0, 1); 7a, Ni(I) (9, 1/2, 2); dashed lines show spectra for respective alternative spin states of 2a (hs, M = 3), 3a(BF4) (ls, M = 1), and 5 (hs, M = 3). | ||
Electronic excitation and decay transitions underlying the ctv and vtc spectra were assigned from the DFT calculations, which provided information on the electronic structure of the compounds (Fig. 4 and S14–S16†).
The prominent ctv feature of 2a was dominated by excitations into phosphorus-centered MOs whereas the transition into the α- and β-spin LUMOs contributed very weakly at the lowest ctv energies and the strongest low-energy vtc peak reflected the two Cl− ligands. Decay from the HOMOs on the nickel and chlorine atoms contributed very weakly at the highest vtc energies. The Ni(d) degeneracy was close to an ideal square-planar case with the d(x2 − y2) dominating the LUMOs at the highest energies. The HOMO–LUMO energy gap was ∼2 eV. The negative charge within the complex resided largely on the Cl− ions and was compensated by positive charges mostly on nickel and the ligands. Little charge was located on the P-atoms.
For 3a(BF4), the prominent lowest-energy ctv peak was due to the excitation into the β-spin LUMO with mixed Ni-d(xy, xz, yz) character and considerable delocalization onto the Cl− ion and P-ligands. Whereas weaker ctv features at higher energies reflected transitions into ligand-based MOs, relatively strong contributions from the decay from the α- and β-spin HOMOs caused significant vtc intensities at the highest energies for 3a(BF4). The Ni(3d) degeneracy reflected the distorted tetrahedral geometry with mixed d(xy, xz, yz) dominated LUMOs. The HOMO–LUMO gap was ∼3 eV. A significant positive charge on nickel was compensated by negative charge on Cl− so that the complex charge was due to almost one positive charge on the phosphine ligands. The two unpaired spins resided in mixed d(xy, xz, yz)-dominated and delocalized HOMOs, with close to one spin on Ni, significant spin density on P and ligand species, and few spins on Cl−.
For 7a, the prominent lowest-energy ctv peak was exclusively due to the excitation into the β-spin LUMO with mixed d(xy, xz, yz) character and delocalization onto the Cl− ligand whereas transitions at higher ctv energies occurred into phosphine-based MOs; decay from MOs on the single Cl− ligand accounted for the low-energy vtc feature and decay from both HOMOs contributed significantly at the highest vtc energies. Decreased Ni(d) degeneracy compared to 3a(BF4) reflected increased distortion of tetrahedral symmetry in 7a, which is in line with the observations from our EPR measurements. The HOMO–LUMO gap was decreased to ∼1.6 eV. The surplus negative charge in 7a was shared between the Ni ion, the Cl−, and mostly the phosphine ligands. The unpaired spin resided mostly on nickel (∼70%) and the remainder was equally distributed over the ligand species.
For 5, transitions into the α- and β-spin LUMOs with dominant d(x2 − y2) character and strong delocalization onto the C–P group corresponded very weakly at the lowest ctv energies and the higher energy ctv features reflected transitions mostly into phosphine-based MOs; decay from the d(z2) dominated HOMOs contributed significantly at the highest vtc energies. The most pronounced vtc intensity at the lowest energies observed only for 5 was indicative of decay from C–P ligand-centered MOs, thereby detecting this ligand species. The Ni(d) degeneracy reflected the distorted square-pyramidal geometry with d(x2 − y2) LUMOs at the highest energies followed by d(z2)-dominated HOMOs. The HOMO–LUMO gap (∼2 eV) was similar to 2a. Positive charge was located on nickel (∼0.3), the phosphine ligands (∼0.8), and also on the Si atom (∼0.2); a low negative charge (about −0.1) resided on the C–P group.
Formation of 5 – theoretical insight
The cleavage of the Si–C bond in TriphosSi has been investigated theoretically, by allowing TriphosSi1b to interact with nickel and fluoride. The fast reaction of TriphosSi1b and [Ni(CH3CN)6](BF4)2, which we were not able to investigate on the 1H and 31P{1H} NMR time scale at room temperature, led us initially to aim at developing a general hypothesis of the reaction pathway. The Si–C cleavage is most likely initiated by a fluoride-induced cleavage from the BF4− counter ion. This hypothesis is in accordance with literature reports on the decomposition of BF4− to afford F− and not HF, which is not feasible under the herein described experimental conditions.46–48 Notably, this assumption is also supported by our experimental findings that complex 5 can be formed as the sole product when 1b reacts with Ni(ClO4)2 in the presence of additional NaF added to the reaction mixture (Fig. S17†). Likewise, complex 5 is not observed when 1b reacts with NiCl2 and NaF. Similarly, the reaction of 1b with [Ni(CH3CN)6](BF4)2 and Ag(OTf) (Fig. S18/S19†) does not afford 5. Therefore, we assume that the simultaneous nickel–phosphine coordination, the presence of non-coordinating counter ions as well as the presence of fluoride ions are of eminent importance for the formation of compound 5.Taking this into account, we calculated the reaction enthalpy for the formation of a Si–F bond concomitant with the formation of the phosphino methanide according to:
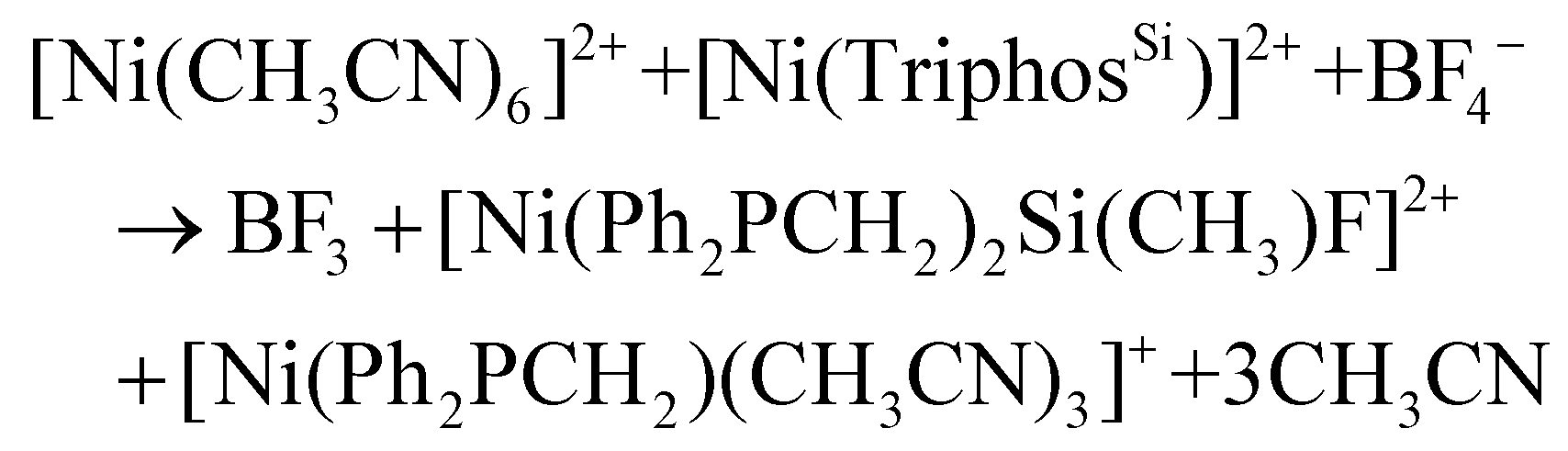
The reaction is very exothermic (−173 kcal mol−1) and supports our assumption of the formation of a Si–F bond. Subsequently, multiple attempts have been made to search for a reasonable transition state that leads to low activation energy for such a reaction and to elucidate the tentative mechanistic pathway (Scheme 5). Of critical importance is that Ni2+ as opposed to Fe2+ initially binds to only two phosphorus atoms of the TriphosSi ligand, leaving one arm free, which is in accordance with the formation of complex 4. A second Ni2+–F complex, formed from BF4− and [Ni(CH3CN)6]2+, was then found to coordinate with the dangling phosphine, leaving Si prone to nucleophilic attack by F−. This attack happens in a barrierless fashion during geometry optimization upon inclusion of F− near the Si center. The Si atom transiently becomes five-coordinate in the coordination complex, and attains a trigonal bi-pyramidal ligand field (Fig. 5), weakening the Si–C bond trans to F− from 1.96 Å to 2.14 Å. Interestingly, firstly the weakened Si–C bond does not correspond to the dangling phosphine arm, but rather to one of the two arms of TriphosSi-coordinated Ni2+.
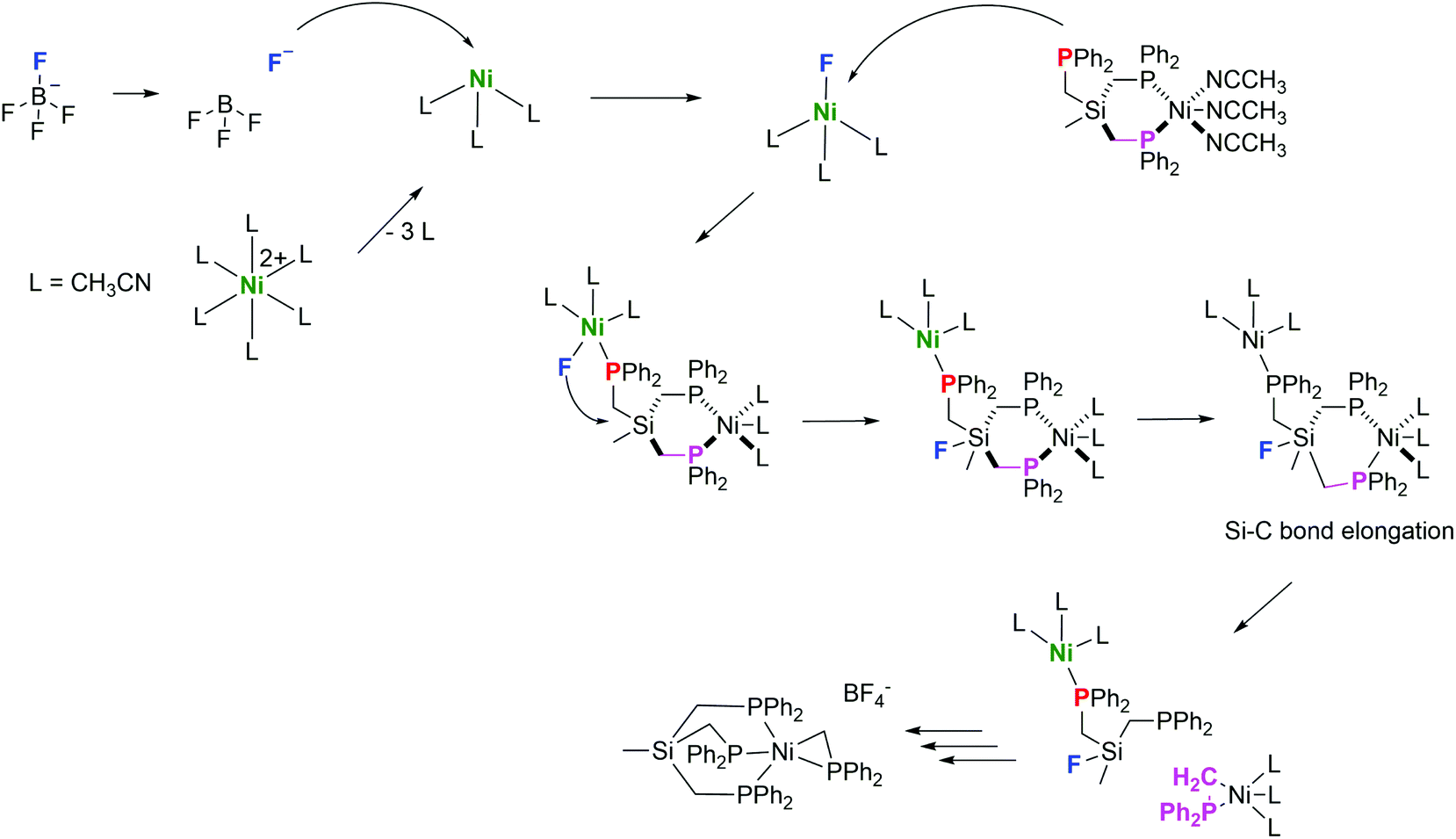 | ||
| Scheme 5 Calculated mechanism towards the formation of complex 5. | ||
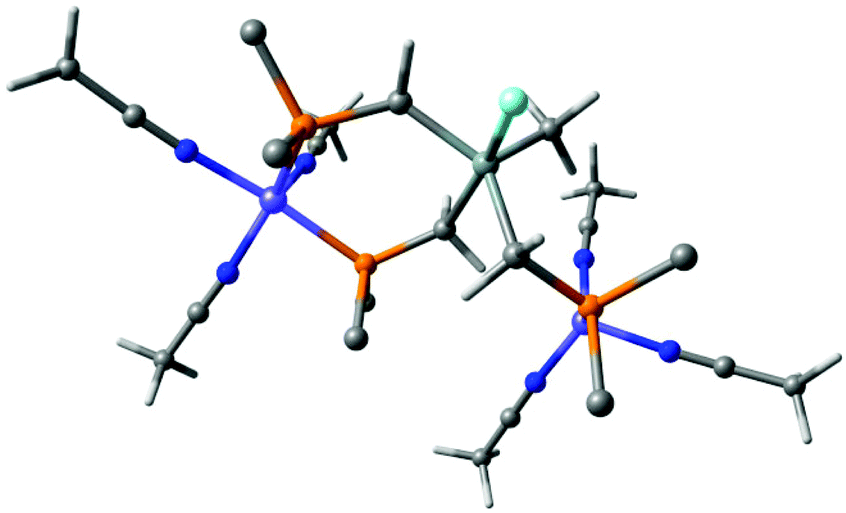 | ||
| Fig. 5 Coordination complex of TriphosSi to two Ni2+ cations and subsequent attack by F− leaving the Si atom five-coordinate and trigonal bipyramidal. The Si–C bond trans to F− elongates to 2.14 Å. Phenyl groups have been truncated by carbon atoms for clarity. Color code: nickel (purple); phorphorus (orange); nitrogen (blue), fluoride (cyan), carbon (grey), hydrogen (white). | ||
Secondly, the transient five-coordinate ligation of Si, being a third-row element, is not attainable for C, which has to strictly adhere to the octet rule, thus explaining the difference in the reactivity of TriphosC and TriphosSi ligands. Subsequently, further elongation of the Si–C bond leads to a transition state 5 kcal mol−1 above that of the coordination complex in Fig. 5 and Table S6.†
After passing the transition state, Si–C bond cleavage is complete and the ligand fragments rearrange as indicated in the above reaction. We assume that the (Ph2PCH2)Ni-fragment is immediately stabilized by a free TriphosSi ligand, while the Si–F fragment fully decomposes and affords two additional Ph2PCH2 fragments. Notably, the concomitant coordination of two independent nickel ions seems to be pivotal for the initiation of the TriphosSi decomposition and allows for the specific formation of complex 5.
Conclusion
We have shown that the substitution of a quaternary carbon atom by a silicon atom within the ligand scaffold of TriphosC leads to interesting differences in the reactivity with Ni(II) salts. While we were unable to establish a bimetallic complex system, a rich and versatile chemistry of TriphosC and TriphosSi with respect to Ni-complexes is presented. Most interestingly, the reaction of TriphosSi and [Ni(CH3CN)6](BF4)2 afforded the phosphino methanide complex 5 under ambient conditions as the sole product. Due to the unexpected reactivity of 5, the electronic structure of this complex was analyzed by X-ray absorption and emission spectroscopy in comparison with the structurally related complexes 2, 3a(BF4), and 7a. Although the phosphino methanide ligand was reported to be a non-innocent ligand and structural parameters suggest a PC double bond, complex 5 can be best described as an Ni2+ complex comprising an anionic Ph2P–CH2− unit. A detailed mechanism towards the formation of complex 5 is suggested based on DFT calculations and suggests the initial formation of an unstable bimetallic intermediate. The theoretical investigation revealed a fluoride induced decomposition of TriphosSi that is bound to a [Ni(CH3CN)3]2+ fragment. In addition, the reactivity of complex 5 was tested and allowed for the generation of the “NiI–Br” complex 7b in good yields. Subsequently, a simple synthetic route towards the Ni(I) complex 7a was established, allowing for the straightforward synthesis of this complex on the gram scale.
Experimental section
Materials and methods
All reactions were performed under a dry argon atmosphere using standard Schlenk techniques or in a glovebox. TriphosC (1a),49 TriphosSi,18 [(TriphosC)NiCl2] (2a),18 [(TriphosSi)NiCl2] (2b),18 [(TriphosC)FeCl2] (6a),19 [(TriphosSi)FeCl2] (6b)19 and [Ni(CH3CN)6](BF4)250 were synthesized according to literature procedures. All other compounds were obtained from commercial vendors and used without further purification. All solvents were dried prior to use according to standard methods. Magnetic moments were obtained by the Evans method51 applying temperature corrected densities52 and using 2%-TMS solution as the reference in CD3CN.
EPR experiments have been performed at T = 30 K on a Bruker Elexsys E500 spectrometer and a ST9402 resonator. The microwave frequency amounted to 9.65 GHz; a microwave power of 2 mW and a modulation amplitude of 0.5 mT were used. Lower modulation amplitudes have been used as well, but did not lead to the observation of an additional hyperfine structure.
Method 2: Compound 6a (50 mg, 0.067 mmol) was dissolved in 5 ml THF followed by addition of [Ni(CH3CN)6](BF4)2 (16 mg, 0.033 mmol). The obtained green solution was stirred for 24 h at room temperature and subsequently evaporated to dryness. The residue was then recrystallized from DCM to afford compound 3a(BF4) as a green solid (20 mg, 75%). FAB-MS. Calcd for [C41H39ClNiP4]+: 717.13. Found: 717.1. IR (KBr, cm−1): 3385, 3055, 2960, 1647, 1482, 1459, 1435, 1092, 1060, 832, 742, 694. Anal. Calcd for C41H39BClF4NiP4 + H2O: C, 59.79; H, 5.02. Found: C, 59.4; H, 5.26. UV-vis (λ, nm): 425, 676.
Method 2: In analogy to method 1, compound 6b (50 mg, 0.065 mmol) and [Ni(ClO4)2]·6H2O (12 mg, 0.033 mmol) were reacted to yield a green solid 3b(ClO4) (20 mg, 80%). ESI-MS. Cacld for [C40H39ClNiP3Si]+: 733.11. Found: 733.0. IR (KBr, cm−1): 3400, 3056, 2950, 1483, 1436, 1264, 1094, 990, 810, 744, 693, 623. UV-vis (λ, nm): 437, 708.
:1) followed by the addition of [Ni(CH3CN)6](BF4)2 (70 mg, 0.16 mmol) dissolved in 2 mL MeCN and stirred for 24 h at room temperature. The resulting dark red solution was evaporated to dryness. The residue was then redissolved in acetonitrile and recrystallized by slow diffusion of diethyl ether into the acetonitrile solution of 4. The dark red crystals obtained were filtered off, washed with hexane and diethyl ether and dried in a vacuum to yield 140 mg (89%) of compound 4. ESI-MS. Calcd for [C41H39NiP3 + H2O]+: 701.4. Found: 701.1. IR (KBr, cm−1): 3053, 1483, 1435, 1284, 1121, 1060, 993, 797, 744, 695.
:1) followed by addition of [Ni(CH3CN)6](BF4)2 (410 mg, 0.937 mmol) in 5 ml MeCN. The resulting dark red solution was stirred for 24 h at room temperature. Subsequently, the solvent was removed and the residue was redissolved in acetonitrile. Crystallization via diffusion of diethyl ether into a solution of 5, dissolved in acetonitrile, afforded red crystals that were filtered off, washed with hexane and diethyl ether and dried in a vacuum to yield 680 mg (74%) of compound 5.
Method 2: Compound 2b (50 mg, 0.065 mmol) and [Ni(CH3CN)6](BF4)2 (16 mg, 0.033 mmol) dissolved in 4 ml THF/MeCN (1:1) were treated according to method 1 to yield 5 as a dark red solid (18 mg, 28%). 31P{1H} NMR (100 MHz, CD3CN): δ 7.00 (d, 3 P, PPh2), 26.50 (q, 1 P, CH2PPh2). (PPh2). ESI-MS. Calcd for [C53H5NiP4Si]+: 897.21. Found: 897.0. IR (KBr, cm−1): 3051, 2926, 1578, 1479, 1432, 1092, 1056, 995, 818, 777, 745, 696, 640. Anal. Calcd for C53H51BF4NiP4Si + CH3CN: C, 64.35; H, 5.30; N, 1.36. Found: C, 64.44; H, 5.15; N, 1.35.
X-ray data collection and structure solution refinement
Single crystals suitable for X-ray analysis were coated with Paratone-N oil, mounted on a fiber loop, and placed in a cold, gaseous N2 stream on the diffractometer. For 9: an Oxford XCalibur diffractometer performing φ and ω scans at 170(2) K. Diffraction intensities were measured using graphite-monochromatic Mo Kα radiation (λ = 0.71073 Å). For 3b(ClO4), 3a(BF4) and 7b: an IPDS I diffractometer performing ω scans at 170(2) K. Diffraction intensities were measured using graphite-monochromatic Mo Kα radiation (λ = 0.71073 Å). Data collection, indexing, initial cell refinements, frame integration, final cell refinements and absorption corrections were accomplished with the programs X-Area, X-Red and X-Shape. For 4, 5, and 7a: a SuperNova diffractometer performing φ and ω scans at 100(2) K. Diffraction intensities were measured using graphite-monochromatic Cu Kα radiation (λ = 1.54184 Å). Data collection, indexing, initial cell refinements, frame integration, final cell refinements, and absorption corrections were accomplished with the program CrystalClear.53 Space groups were assigned by analysis of the metric symmetry and systematic absences (determined by XPREP) and were further checked by PLATON54,55 for additional symmetry. Structures were solved by direct methods and refined against all data in the reported 2Θ ranges by full-matrix least squares on F2 with the SHELXL program suite56 using the OLEX2 interface.57 Crystallographic data as well as refinement parameters are presented in Tables S3–S5 in the ESI.†X-ray spectroscopy experiments
X-ray absorption (XAS) and emission (XES) spectra at the Ni K-edge were collected at the triple-undulator beamline ID26 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France). Powder samples of nickel complexes (ground under anaerobic conditions with BN, 1:10 w/w ratio) were placed in Kapton-covered acrylic glass holders in a liquid-He cryostat (Cryovac) at 20 K. The incident energy was varied by using a Si[311] double-crystal monochromator (energy bandwidth ∼0.2 eV at the Ni K-edge). XAS spectra were collected using the rapid-scan mode of the beamline (XANES, 10 s; EXAFS, 30 s scan duration; energy step size 0.1 eV or 1 eV) simultaneously in transmission mode, by total-fluorescence monitoring (>95% Kα X-ray fluorescence) using a 20 cm2 area scintillation detector placed at 90° and ∼1 m distance to the sample and shielded by 10 μM Co foil against scattered incident X-rays, and by narrow-band detection of the Kβ X-ray fluorescence at the Kβ1,3 line maximum (8265.7 eV). Fluorescence-detected XAS spectra were corrected for self-absorption flattening using the transmission spectra (Fig. S10†). For XES data collection a vertical-plane Rowland-circle spectrometer was used with an avalanche photodiode (APD) for fluorescence detection. An energy bandwidth of ∼1 eV at the Ni Kβ emission lines was achieved using the [551] Bragg reflections of 5 spherically-bent Si wafers (R = 1000 mm). Kβ main and satellite line spectra were obtained using non-resonant excitation (8900 eV) with an energy step size of 0.35 eV. The X-ray beam was attenuated using stacked Al foils (20 μm) to avoid radiation induced effects on samples whenever necessary and the absence of radiation effects was verified using our previously established procedures.58 Energy axes of the monochromator and emission spectrometer were calibrated (precision ±0.1 eV) as outlined elsewhere.59,60 The signal-to-noise ratio of XAS and XES spectra was increased by the averaging of spectra collected on separate sample spots. XAS spectra were normalized to yield XANES spectra and EXAFS oscillations were extracted as described previously.61 The pre-edge region of XANES spectra and Kβ2,5 emission lines were isolated by subtraction of polynomial spline functions through the main K-edge rise or the Kβ main line high-energy tail using the program XANDA.62
Density functional theory (DFT) calculations
DFT calculations of the electronic excitation and decay transitions were carried out on the Soroban computer cluster of Freie Universität Berlin using Gaussian 0963 for geometry-optimization of XRD structures and the ORCA program64 for calculation of ctv and vtc transition spectra. The BP86/TZVP functional/basis-set combination65,66 was used for both procedures. Geometry optimization of XRD structures was carried out in a vacuum. The calculated stick spectra in the lowest (ctv) or highest (vtc) energy ranges were shifted, broadened by Gaussians, and scaled (transition-energy-range/energy-shift/Gaussian-FWHM/scaling-factor: ∼5 eV/215.0 eV/1 eV/1000, ctv; ∼25 eV/214.5 eV/3/3 eV, vtc) for comparison with experimental data. Calculations of the mechanism towards the formation of complex 5 as well as a relaxed surface scan of the Si–C bond to find the activation energy for Si–C bond cleavage have also been carried out in ORCA,64 using the BP86 functional and the Def2-SVP65,66 basis set. Cartesian coordinates of the geometry-optimized coordination complex as well as a graph of the potential energy surface of the relaxed surface scan are included in the ESI.†
Acknowledgements
We thank Manuel Seidel for experimental support. This work was supported by the Fonds of the Chemical Industry (Liebig grant to U.-P. A.), the Max Planck Gesellschaft, the Deutsche Forschungsgemeinschaft (Emmy Noether grant to U.-P. A., AP242/2-1 and grant Ha3265/6-1 to M. H.), and the Bundesministerium für Bildung und Forschung (grant 05K14KE1 to M. H.). Support from the Research Department Interfacial Systems Chemistry is gratefully acknowledged. M. H. thanks the beamline staff at ID26 for excellent technical support and the ESRF for a Long Term Project.Notes and references
- L. C. Misal Castro, H. Li, J.-B. Sortais and C. Darcel, Green Chem., 2015, 17, 2283–2303 RSC.
- D. Herault, D. H. Nguyen, D. Nuel and G. Buono, Chem. Soc. Rev., 2015, 44, 2508–2528 RSC.
- K.-B. Shiu, S.-W. Jean, H.-J. Wang, S.-L. Wang, F.-L. Liao, J.-C. Wang and L.-S. Liou, Organometallics, 1997, 16, 114–119 CrossRef CAS.
- M. I. Bruce, J. R. Hinchliffe, R. Surynt, B. W. Skelton and A. H. White, J. Organomet. Chem., 1994, 469, 89–97 CrossRef CAS.
- S. Miranda, E. Cerrada, A. Mendía and M. Laguna, Inorg. Chem. Commun., 2012, 21, 151–154 CrossRef CAS.
- R. Robinson, J. M. Clarkson, M. A. Moody and P. R. Sharp, Organometallics, 2011, 30, 1730–1733 CrossRef CAS.
- H. H. Karsch, Chem. Ber., 1984, 117, 783–796 CrossRef CAS.
- H. Fan, B. C. Fullmer, M. Pink and K. G. Caulton, Angew. Chem., Int. Ed., 2008, 47, 9112–9114 CrossRef CAS PubMed.
- E. Lindner, E. U. Küster, W. Hiller and R. Fawzi, Chem. Ber., 1984, 117, 127–141 CrossRef CAS.
- M. D. Fryzuk, K. Joshi, R. K. Chadha and S. J. Rettig, J. Am. Chem. Soc., 1991, 113, 8724–8736 CrossRef CAS.
- H. H. Karsch, G. Grauvogl, M. Kawecki, P. Bissinger, O. Kumberger, A. Schier and G. Mueller, Organometallics, 1994, 13, 610–618 CrossRef CAS.
- A. Sattler and G. Parkin, Chem. Commun., 2011, 47, 12828 RSC.
- A. Sattler and G. Parkin, J. Am. Chem. Soc., 2011, 133, 3748–3751 CrossRef CAS PubMed.
- X. Zhang, Z.-Y. Geng, Y.-C. Wang, W.-Q. Li, Z. Wang and F.-X. Liu, J. Mol. Struct. (THEOCHEM), 2009, 893, 56–66 CrossRef CAS.
- B. Blom, G. Tan, S. Enthaler, S. Inoue, J. D. Epping and M. Driess, J. Am. Chem. Soc., 2013, 135, 18108–18120 CrossRef CAS PubMed.
- A. B. Chaplin and P. J. Dyson, Eur. J. Inorg. Chem., 2007, 2007, 4973–4979 CrossRef.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC.
- A. Petuker, C. Merten and U.-P. Apfel, Eur. J. Inorg. Chem., 2015, 2139–2144 CrossRef CAS.
- A. Petuker, K. Merz, C. Merten and U.-P. Apfel, Inorg. Chem., 2016, 55, 1183–1191 CrossRef CAS PubMed.
- E. F. de Assis and C. A. Filgueiras, Transition Met. Chem., 1994, 19, 484–486 CrossRef CAS.
- J. Mautz, K. Heinze, H. Wadepohl and G. Huttner, Eur. J. Inorg. Chem., 2008, 2008, 1413–1422 CrossRef.
- K. J. Cluff, N. Bhuvanesh and J. Blümel, Chem. – Eur. J., 2015, 21, 10138–10148 CrossRef CAS PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 2002, 58, 389–397 CrossRef.
- H. H. Karsch, H.-F. Klein and H. Schmidbaur, Chem. Ber., 1977, 110, 2200–2212 CrossRef CAS.
- S. Freitag, J. Henning, H. Schubert and L. Wesemann, Angew. Chem., Int. Ed., 2013, 52, 5640–5643 CrossRef CAS PubMed.
- A. W. Holland and R. G. Bergman, Organometallics, 2002, 21, 2149–2152 CrossRef CAS.
- F. Wittkamp, C. Nagel, P. Lauterjung, B. Mallick, U. Schatzschneider and U.-P. Apfel, Dalton Trans., 2016, 45, 10271–10279 RSC.
- B. C. Fullmer, H. Fan, M. Pink, J. C. Huffman, N. P. Tsvetkov and K. G. Caulton, J. Am. Chem. Soc., 2011, 133, 2571–2582 CrossRef CAS PubMed.
- M. F. Laird, N. P. Tsvetkov, M. Pink, T. He, R. W. Buell and K. G. Caulton, Inorg. Chim. Acta, 2011, 374, 79–87 CrossRef CAS.
- A. Aloisi, J.-C. Berthet, C. Genre, P. Thuéry and T. Cantat, Dalton Trans., 2016, 45, 14774–14788 RSC.
- K.-R. Pörschke, G. Wilke and R. Mynott, Chem. Ber., 1985, 118, 298–312 CrossRef.
- K. E. Janak, J. M. Tanski, D. G. Churchill and G. Parkin, J. Am. Chem. Soc., 2002, 124, 4182–4183 CrossRef CAS PubMed.
- L. Turculet, J. D. Feldman and T. D. Tilley, Organometallics, 2004, 23, 2488–2502 CrossRef CAS.
- W. W. N. O. X. Kang, Y. Luo and Z. Hou, Organometallics, 2014, 33, 1030–1043 CrossRef.
- R. Ramírez-Contreras, N. Bhuvanesh and O. V. Ozerov, Organometallics, 2015, 34, 1143–1146 CrossRef.
- M. A. Bennett, J. Castro, A. J. Edwards, M. R. Kopp, E. Wenger and A. C. Willis, Organometallics, 2001, 20, 980–989 CrossRef CAS.
- M. S. Kharasch and S. Weinhouse, J. Org. Chem., 1936, 1, 209–230 CrossRef CAS.
- L. Wu, Q. Liu, I. Fleischer, R. Jackstell and M. Beller, Nat. Commun., 2014, 5, 3091 Search PubMed.
- B. C. Janssen, V. Sernau, G. Huttner, A. Asam, O. Walter, M. Büchner and L. Zsolnai, Chem. Ber., 1995, 128, 63–70 CrossRef CAS.
- A. Bencini, C. Benelli, D. Gatteschi and L. Sacconi, Inorg. Chim. Acta, 1979, 195–199 CrossRef CAS.
- C. M. Palit, D. J. Graham, C.-H. Chen, B. M. Foxman and O. V. Ozerov, Chem. Commun., 2014, 50, 12840–12842 RSC.
- M. J. Nilges, E. K. Barefield, R. L. Belford and P. H. Davis, J. Am. Chem. Soc., 1977, 99, 755–760 CrossRef CAS.
- A. Özbolat-Schön, M. Bode, G. Schnakenburg, A. Anoop, M. van Gastel, F. Neese and R. Streubel, Angew. Chem., Int. Ed., 2010, 49, 6894–6898 CrossRef PubMed.
- U. Bergmann and P. Glatzel, Photosynth. Res., 2009, 102, 255–266 CrossRef CAS PubMed.
- S. Mebs, B. Braun, R. Kositzki, C. Limberg and M. Haumann, Inorg. Chem., 2015, 54, 11606–11624 CrossRef CAS PubMed.
- D. L. Reger, R. P. Watson, J. R. Gardinier, M. D. Smith and P. J. Pellechia, Inorg. Chem., 2006, 45, 10088–10097 CrossRef CAS PubMed.
- E. Tomat, L. Cuesta, V. M. Lynch and J. L. Sessler, Inorg. Chem., 2007, 46, 6224–6226 CrossRef CAS PubMed.
- Q. Zhu, K. J. Nelson, W. W. Shum, A. DiPasquale, A. L. Rheingold and J. S. Miller, Inorg. Chim. Acta, 2009, 362, 595–598 CrossRef CAS.
- S. Herold, A. Mezzetti, L. M. Venanzi, A. Albinati, F. Lianza, T. Gerfin and V. Gramlich, Inorg. Chim. Acta, 1995, 235, 215–231 CrossRef CAS.
- B. J. Hathaway, D. G. Holah and A. E. Underhill, J. Chem. Soc., 1962, 2444–2448 RSC.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- L. W. Reeves and W. G. Schneider, Can. J. Chem., 1957, 35, 251–261 CrossRef CAS.
- Rigaku/MSC, CrystalClear and CrystalStructure, Rigaku/MSC Inc., The Woodlands, Texas, USA, 2006 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- K. G. V. Sigfridsson, P. Chernev, N. Leidel, A. Popović-Bijelić, A. Gräslund and M. Haumann, J. Biol. Chem., 2013, 288, 9648–9661 CrossRef CAS PubMed.
- N. Leidel, P. Chernev, K. G. V. Havelius, S. Ezzaher, S. Ott and M. Haumann, Inorg. Chem., 2012, 51, 4546–4559 CrossRef CAS PubMed.
- P. Chernev, C. Lambertz, A. Brünje, N. Leidel, K. G. V. Sigfridsson, R. Kositzki, C.-H. Hsieh, S. Yao, R. Schiwon and M. Driess, et al. , Inorg. Chem., 2014, 53, 12164–12177 CrossRef CAS PubMed.
- H. Dau, P. Liebisch and M. Haumann, Anal. Bioanal. Chem., 2003, 376, 562–583 CrossRef CAS PubMed.
- K. V. Klementiev, freeware: http://www.cells.es/Beamlines/CLAESS/software/xanda.html.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Son-nenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hase-gawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Starov-erov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Strat-mann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian09 Revision E.01, Inc, Wallingford CT, 2013 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray structural data, UV-vis, X-ray emission and absorption spectra and DFT calculations. CCDC 1486081–1486088. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt04048a |
| This journal is © The Royal Society of Chemistry 2017 |