
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Determination of ZFS parameters from the EPR spectra of mono-, di- and trinuclear MnII complexes: impact of magnetic coupling†
Luis
Escriche-Tur
 *ab,
Mercè
Font-Bardia
c,
Belén
Albela
b and
Montserrat
Corbella
*ad
*ab,
Mercè
Font-Bardia
c,
Belén
Albela
b and
Montserrat
Corbella
*ad
aDepartament de Química Inorgànica i Orgànica (Secció inorgánica), Universitat de Barcelona, C/Martí i Franquès 1-11, 08028 Barcelona, Spain. E-mail: luis.escrichetur@gmail.com; montse.corbella@qi.ub.es
bLaboratoire de Chimie, ENS de Lyon, Université de Lyon, 46 Allée d'Italie, 69364 Lyon Cedex 07, France
cCristal·lografia, Mineralogia i Dipòsits Minerals, Universitat de Barcelona, Martí i Franquès s/n, 08028 Barcelona, Spain
dInstitud de Nanociència i Nanotecnologia de la Universitat de Barcelona (IN2UB), Av. Joan XXIII s/n, 08028 Barcelona, Spain
First published on 17th January 2017
Abstract
A family of new MnII compounds, consisting of seven dinuclear, three mononuclear, and four trinuclear ones, were synthesised using benzoic acid derivatives n-RC6H4COOH, where n-R = 2-MeO, 3-MeO, 4-MeO, or 4-tBu, and 2,2′-bipyridine (bpy) or 1,10-phenantroline (phen) as blocking ligands. The crystal structures of nine of these compounds and the magnetic studies of all of them are reported here. Each type of compound was formed depending on the presence or absence of ClO4− ions, the solvent used, and/or the presence of a small amount of water in the reaction medium. The use of the tert-buthylbenzoate ligand gave unexpected results, very likely due to the steric hindrance caused by the voluminous tBu groups. The EPR spectra of each type of compound give some peculiar features that allow its identification. Attempts to fit these spectra have been made in order to determine the ZFS parameters, D and E, of the MnII ion (for mononuclear and dinuclear systems) or of the ground state (for trinuclear systems). For trinuclear systems, the single-ion ZFS parameters estimated from those of the ground state provided a good simulation of the EPR spectra of these compounds. The EPR signals observed in each case have been rationalised according to the energy level distribution and the plausible population in the excited states. In some particular situations, the sign of DMn could be determined from the fit of the EPR spectra of the antiferromagnetic dinuclear compounds, the source of the difference between the spectra lying in the second excited state.
Introduction
MnII complexes are widely present in numerous enzymes, as an essential ingredient of their active site.1 MnII ions can be implicated in both redox and non-redox processes, acting as catalysts or having a structural role.2–5 Owing to its single-ion high spin (S = 5/2) and comparable size, MnII is commonly used as a probe to replace diamagnetic ions, such as Mg2+,6,7 in other biological systems. Additionally, some MnII compounds can be used as catalysts in several industrial processes, commonly for the epoxidation of olefins,8–12 making them good candidates to replace 2nd and 3rd row transition metal ions in oxidation catalysis.From the analyses of EPR spectra of MnII compounds, important information can be extracted, since the splitting of the magnetic sublevels will depend on the zero-field splitting (ZFS) parameters, D and E. Indeed, even a slight distortion from a regular octahedral environment can result in significant ZFS parameters and highly complicated EPR spectra.13 Furthermore, it is well known that, for several transition metal ions, the ZFS can probe their structural and electrostatic environments.14 For example, the ZFS for MnII ions is larger when it is bonded to halide ligands.13,15–22 It also depends on the coordination number, since pentacoordinated ions display greater axial ZFS parameters (|D| = 0.25–0.30 cm−1) than hexacoordinated ones (|D| = 0.0008–0.1750 cm−1).23,24 Moreover, the ZFS parameters could also be affected by the ratio between the N- and O-based ligands.25
The reaction between MnII carboxylate and bidentate ligands (NN), such as 2,2′-bipyridine (bpy) and 1,10-phenanthroline (phen), leads to the formation of MnII compounds with different nuclearity: mononuclear, dinuclear, trinuclear, or 1D systems.26,27 The carboxylate groups display a wide variety of coordination modes, such as monodentate terminal, chelating, bidentate bridging, and monodentate bridging modes. Particularly in these compounds, the carboxylate ligands could bridge the MnII ions in μ1,1 or μ1,3 coordination modes, and in the latter case in either a syn–syn or syn–anti conformation.
The magnetic interaction between neighboring MnII ions depends on the coordination mode of the carboxylate ligand. For instance, dinuclear compounds with μ1,1-carboxylate bridges present a ferromagnetic interaction (S = 5 ground state), while those with μ1,3-carboxylate bridges show an antiferromagnetic interaction (S = 0 ground state).26
We herein present three kinds of compounds: dinuclear [{Mn(NN)2}2(μ-n-RC6H4COO)2](ClO4)2 (1–7), mononuclear [Mn(NN)3-m(n-RC6H4COO)m(H2O)x](ClO4)2-m (with m = 1 or 2 and x = 0, 1, or 2) (8–10), and trinuclear [Mn3(NN)2(μ-n-RC6H4COO)6] (11–14) compounds, synthesised with n-MeOC6H4COO− (n = 2, 3, 4) or 4-tBuC6H4COO− ligands, and NN = bpy or phen. The magnetic properties of these compounds have been studied and their EPR spectra have been deeply analysed. Moreover a qualitative rationalisation of the displayed EPR signals has been performed. Fits of these spectra have been attempted, leading to a reasonable determination of the zero field splitting parameters, D and E. The Zeeman plots of the dinuclear compounds have been deeply analysed to provide a better understanding of the EPR spectra.
Experimental section
Synthesis
All manipulations were performed under aerobic conditions. Reagents and solvents were obtained from commercial sources and used without further purification. Caution! Perchlorate salts of compounds containing organic ligands are potentially explosive. Only small quantities of these compounds should be prepared. Mn(n-MeOC6H4COO)2·xH2O (n = 2 and 4) was obtained from the reaction between MnCO3 and n-MeOC6H4COOH in boiling water. After several hours, the solution was filtered and concentrated to give a pale pink precipitate of the desired product. For n = 2, C16H14MnO6 (M.W. = 357.16 g mol−1). Anal. Calcd (%): C, 53.80; H, 3.95. Found (%): C, 52.20; H, 3.90. For n = 4, C16H14MnO6·2H2O (M.W. = 393.25 g mol−1). Anal. Calcd (%): C, 48.87; H, 4.61. Found (%): C, 49.2; H, 4.70. Mn(3-MeOC6H4COO)2·2/3EtOH was obtained following the same procedure but assisting the crystallisation by adding EtOH to the mother liquor and leaving it undisturbed in the refrigerator. C16H14MnO6·2/3EtOH (M.W. = 387.93 g mol−1). Anal. Calcd (%): C, 53.67; H, 4.67. Found (%): C, 53.37; H, 4.80. Mn(4-tBuC6H4COO)2·3H2O was obtained following the same procedure as for n-R = 2-MeO and 4-MeO but using Mn(AcO)2 instead of MnCO3. C22H26MnO4 (M.W. = 463.42 g mol−1). Anal. Calcd (%): C, 57.02; H, 6.96. Found (%): C, 57.70; H, 6.80.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O 1:1 (v/v) mixture (30 mL) by stirring for around 24 h. Then, a solution of NaClO4 (0.33 mmol, 0.041 g) in EtOH:H2O 1:1 (v/v) (10 mL) was added to the previous one. Meanwhile, 2,2′-bipyridine (bpy) (0.72 mmol, 0.11 g) was dissolved in EtOH (10 mL) and added to the previous solution. The resulting yellow solution (solvent: 50 mL of EtOH:H2O 3:2) was stirred for 15 minutes and filtered to separate any possible impurity. Then, the solution was left undisturbed at room temperature. The crystallisation of the product begins after a month of slow evaporation and may last up to six months. During this time, neither oxidation nor decomposition of the sample was observed. Yield: 65%. X-ray suitable single-crystals were obtained from the mother liquor. C56H46Cl2Mn2N8O14 (M.W. = 1235.79 g mol−1). Anal. Calcd (%): C, 54.43; H, 3.75; N, 9.07. Found (%): C, 54.49; H, 3.90; N, 9.19. IR (cm−1): 3435 (br), 3114 (w), 3078 (w), 3007 (w), 2981 (w), 2950 (w), 2841 (w), 1592 (s), 1567 (s), 1489 (m), 1472 (m), 1438 (s), 1403 (s), 1313 (m), 1280 (m), 1245 (m), 1193 (w) 1171 (m), 1092 (br,s), 1015 (s), 847 (m), 774 (m), 750 (m), 737(m), 647 (w), 622 (m), 572 (w), 520 (w), 410 (m).
:H2O 1:1 (v/v) (20 mL), to which a solution of NaClO4 (0.50 mmol, 0.073 g) in CH3CN:H2O 1:1 (10 mL) was added. Then, another solution of bpy (1.20 mmol, 0.19 g) in CH3CN:H2O 1:1 (10 mL) was also added. The resulting yellow solution was left undisturbed at room temperature to obtain yellow needles after 3 weeks of slow evaporation. Yield: 82%. Single-crystals suitable for X-ray characterisation were obtained after two months of keeping the solution in the fridge. C56H46Cl2Mn2N8O14 (M.W. = 1235.79 g mol−1). Anal. Calcd (%): C, 54.43; H, 3.75; N, 9.07. Found (%): C, 54.31; H, 3.81; N, 9.18.
:H2O 3:2 (v/v) mixture (25 mL) by stirring for around 24 h. Then, a solution of NaClO4 (0.12 mmol, 0.014 g) in EtOH:H2O (v/v) (10 mL) was added to the previous one. Meanwhile, 2,2′-bipyridine (bpy) (0.23 mmol, 0.046 g) was dissolved in EtOH (10 mL) and added to the other solution. The resulting yellow solution (solvent: 45 mL of EtOH:H2O 4:5) was stirred for 15 minutes and filtered to separate any possible impurity. After 2 weeks of slow evaporation at room temperature, big yellow crystals were collected by filtration and washed with EtOH. Yield: 80%. C64H46Cl2Mn2N8O14 (M.W. = 1331.88 g mol−1). Anal. Calcd (%): C, 57.71; H, 3.48; N, 8.41. Found (%): C, 56.47; H, 3.37; N, 8.20. IR (cm−1): 3442 (br), 3065 (w), 2964 (w), 2832 (w), 1610 (s), 1588 (s), 1564 (s), 1513 (m), 1492 (w), 1458 (w), 1223 (m), 1390 (m), 1340 (w), 1300 (w), 1274 (w), 1241 (w), 1153 (m) 1100 (vs), 1015 (m), 847 (s), 778 (w), 759 (w), 730 (m), 660 (w), 642 (w), 615 (m), 567 (w), 419 (w).
:H2O 2:1 mixture (15 mL). Then, a solution of phen (0.60 mmol, 0.12 g) in CH3CN (5 mL) was added to the previous solution and the mixture was stirred for 15 minutes. Afterwards, MeOH (30 mL) was added. After a month of slow evaporation yellow single-crystals were filtered and washed with MeOH. Yield: 95%. C64H46Cl2Mn2N8O14 (M.W. = 1331.88 g mol−1). Anal. Calcd (%): C, 57.71; H, 3.48; N, 8.41. Found (%): C, 57.34; H, 3.59; N, 8.48. IR (cm−1): 3435 (br), 3062 (w), 2999 (w), 2941 (w), 2840 (w), 1618 (s), 1578 (s), 1521 (s), 1456 (w), 1482 (s), 1306 (s), 1231 (m), 1150 (m), 1086 (vs), 1042 (m), 852 (s), 776 (m), 731 (s), 722 (s), 423 (w).
:H2O 4:3 (35 mL). The resulting pale yellow solution was stirred for 15 minutes. After 1 month of slow evaporation, yellow crystals were filtered and washed with EtOH. Yield: 65%. X-ray suitable single-crystals were obtained from the mother liquor. C28H26MnN2O8 (M.W. = 573.45 g mol−1). Anal. Calcd (%): C, 58.64; H, 4.57; N, 4.89. Found (%): C, 58.56; H, 4.69; N, 5.03. IR (cm−1): 3448 (s), 2967 (w), 2939 (w), 2837 (w), 1603 (s), 1544 (s), 1513 (m), 1423 (m), 1376 (s), 1292 (w), 1270 (m), 1236 (m), 1186 (w), 1163 (w), 1099 (m), 1057 (w), 1019 (m), 859 (m), 832 (m), 767 (s), 733 (m), 660 (w), 637 (w), 554 (w), 443 (w).
:H2O 1:1 (v/v) mixture (30 mL) by stirring for around 24 h. Then, a solution of NaClO4 (0.33 mmol, 0.041 g) in EtOH:H2O 1:1 (v/v) (10 mL) was added to the previous one. Meanwhile, 2,2′-bipyridine (bpy) (0.72 mmol, 0.11 g) was dissolved in EtOH (10 mL) and added to the previous solution. The resulting yellow solution (solvent: 50 mL of EtOH:H2O 3:2) was stirred for 15 minutes and filtered to separate any possible impurity. Then, the solution was left undisturbed at room temperature. The crystallisation of the product begins after a month of slow evaporation and may last up to six months. During this time, neither oxidation nor decomposition of the sample was observed. Yield: 65%. X-ray suitable single-crystals were obtained from the mother liquor. C56H46Cl2Mn2N8O14 (M.W. = 1235.79 g mol−1). Anal. Calcd (%): C, 54.43; H, 3.75; N, 9.07. Found (%): C, 54.49; H, 3.90; N, 9.19. IR (cm−1): 3435 (br), 3114 (w), 3078 (w), 3007 (w), 2981 (w), 2950 (w), 2841 (w), 1592 (s), 1567 (s), 1489 (m), 1472 (m), 1438 (s), 1403 (s), 1313 (m), 1280 (m), 1245 (m), 1193 (w) 1171 (m), 1092 (br,s), 1015 (s), 847 (m), 774 (m), 750 (m), 737(m), 647 (w), 622 (m), 572 (w), 520 (w), 410 (m).
:H2O 1:1 (v/v) (20 mL), to which a solution of NaClO4 (0.50 mmol, 0.073 g) in CH3CN:H2O 1:1 (10 mL) was added. Then, another solution of bpy (1.20 mmol, 0.19 g) in CH3CN:H2O 1:1 (10 mL) was also added. The resulting yellow solution was left undisturbed at room temperature to obtain yellow needles after 3 weeks of slow evaporation. Yield: 82%. Single-crystals suitable for X-ray characterisation were obtained after two months of keeping the solution in the fridge. C56H46Cl2Mn2N8O14 (M.W. = 1235.79 g mol−1). Anal. Calcd (%): C, 54.43; H, 3.75; N, 9.07. Found (%): C, 54.31; H, 3.81; N, 9.18.
:H2O 3:2 (v/v) mixture (25 mL) by stirring for around 24 h. Then, a solution of NaClO4 (0.12 mmol, 0.014 g) in EtOH:H2O (v/v) (10 mL) was added to the previous one. Meanwhile, 2,2′-bipyridine (bpy) (0.23 mmol, 0.046 g) was dissolved in EtOH (10 mL) and added to the other solution. The resulting yellow solution (solvent: 45 mL of EtOH:H2O 4:5) was stirred for 15 minutes and filtered to separate any possible impurity. After 2 weeks of slow evaporation at room temperature, big yellow crystals were collected by filtration and washed with EtOH. Yield: 80%. C64H46Cl2Mn2N8O14 (M.W. = 1331.88 g mol−1). Anal. Calcd (%): C, 57.71; H, 3.48; N, 8.41. Found (%): C, 56.47; H, 3.37; N, 8.20. IR (cm−1): 3442 (br), 3065 (w), 2964 (w), 2832 (w), 1610 (s), 1588 (s), 1564 (s), 1513 (m), 1492 (w), 1458 (w), 1223 (m), 1390 (m), 1340 (w), 1300 (w), 1274 (w), 1241 (w), 1153 (m) 1100 (vs), 1015 (m), 847 (s), 778 (w), 759 (w), 730 (m), 660 (w), 642 (w), 615 (m), 567 (w), 419 (w).
:H2O 2:1 mixture (15 mL). Then, a solution of phen (0.60 mmol, 0.12 g) in CH3CN (5 mL) was added to the previous solution and the mixture was stirred for 15 minutes. Afterwards, MeOH (30 mL) was added. After a month of slow evaporation yellow single-crystals were filtered and washed with MeOH. Yield: 95%. C64H46Cl2Mn2N8O14 (M.W. = 1331.88 g mol−1). Anal. Calcd (%): C, 57.71; H, 3.48; N, 8.41. Found (%): C, 57.34; H, 3.59; N, 8.48. IR (cm−1): 3435 (br), 3062 (w), 2999 (w), 2941 (w), 2840 (w), 1618 (s), 1578 (s), 1521 (s), 1456 (w), 1482 (s), 1306 (s), 1231 (m), 1150 (m), 1086 (vs), 1042 (m), 852 (s), 776 (m), 731 (s), 722 (s), 423 (w).
:H2O 4:3 (35 mL). The resulting pale yellow solution was stirred for 15 minutes. After 1 month of slow evaporation, yellow crystals were filtered and washed with EtOH. Yield: 65%. X-ray suitable single-crystals were obtained from the mother liquor. C28H26MnN2O8 (M.W. = 573.45 g mol−1). Anal. Calcd (%): C, 58.64; H, 4.57; N, 4.89. Found (%): C, 58.56; H, 4.69; N, 5.03. IR (cm−1): 3448 (s), 2967 (w), 2939 (w), 2837 (w), 1603 (s), 1544 (s), 1513 (m), 1423 (m), 1376 (s), 1292 (w), 1270 (m), 1236 (m), 1186 (w), 1163 (w), 1099 (m), 1057 (w), 1019 (m), 859 (m), 832 (m), 767 (s), 733 (m), 660 (w), 637 (w), 554 (w), 443 (w).
Physical characterisation
C, H and N analyses were carried out by the “Centres Científics i Tecnològics” of the Universitat de Barcelona. Infrared spectra were recorded on KBr pellets in the 4000–400 cm−1 range with a Thermo Nicolet Avatar 330 FTIR spectrometer. Magnetic measurements were performed on microcrystalline samples in a Quantum Design MPMS XL5 SQUID Magnetometer at the “Unitat de Mesures Magnètiques” (Universitat de Barcelona). Magnetic susceptibility was measured between 2 and 300 K with a magnetic field of 0.02 T. Magnetisation measurements were performed at 2 K from 0 to 5.0 T. Pascal's constant was used to estimate the diamagnetic corrections for the compound. The fit of the experimental magnetic data was performed by minimizing the function R = ∑[(χMT)exp − (χMT)calcd]2/∑[(χMT)exp]2. Solid-state EPR spectra were recorded at X-band (9.4 GHz) frequency using a Bruker ESP-300E spectrometer from room temperature to 4 K at the “Unitat de Mesures Magnètiques” (Universitat de Barcelona).Single-crystal X-ray crystallography
The data collection for compounds 2, 6, 7, 8 (at 90–100 K), 5, and 9 (at 302 K) was performed on a Bruker Apex-II diffractometer, whereas for 1, 3, and 12, it was performed at room temperature on a MAR345 diffractometer, both equipped with graphite monochromatic Mo Kα radiation (λ = 0.71073 Å). Unit-cell parameters were determined using 230–9972 reflections and refined by the least-squares method. 6090– 127139 reflections were collected using the Φ- and ω-scan (Bruker Apex-II) or Φ-scan (MAR345) method. Data were corrected for absorption effects using multi-scan (2, 6, 8, 7, 5, and 9) or empirical (1, 3, and 12) methods (SADABS).28 Tables containing crystallographic data collection and structure refinement details are summarised in the ESI (Tables S1–S3†).
The structures were solved by direct methods and refined by full-matrix least-squares using SHELXL-97.29 Non-hydrogen atoms were refined anisotropically, whereas hydrogen atoms were computed and refined with isotropic thermal parameters riding on their respective carbon or oxygen atoms. The crystal structures of compounds 1, 3, 5, and 12 were isotropically refined without any complication, but they showed high R1 and wR2 values and, in some cases, the presence of spurious peaks very close to other atoms. These peaks not only complicate the refinement of the anisotropic displacements but also altered the results. Therefore, the reflections showing high standard deviations were removed from the HKL files of 1, 3, 5, and 12, enabling us to better refine the structures and providing much better results. Even though some reflections were removed, the relationship between parameters to refine and reflections were more than enough to refine the structure in an appropriate way.
Compounds 1, 3, 5, and 6 crystallise in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetric unit consists of half a [{Mn(NN)2}2(μ-n-RC6H4COO)2]2+ complex and one ClO4− anion. The other half of the complex is generated by an inversion centre. Around 400 parameters were refined in the final cycle of refinement on F2 using no more than 15 restraints.
. The asymmetric unit consists of half a [{Mn(NN)2}2(μ-n-RC6H4COO)2]2+ complex and one ClO4− anion. The other half of the complex is generated by an inversion centre. Around 400 parameters were refined in the final cycle of refinement on F2 using no more than 15 restraints.
Compound 2 crystallises in the monoclinic space group P21/c. The asymmetric unit consists of half a [{Mn(bpy)2}2(μ-3-MeOC6H4COO)2]2+ complex and a ClO4− anion. The other half of the complex is generated by an inversion centre. A total of 370 parameters were refined in the final cycle of refinement on F2 without using any restraint.
Compound 7 crystallises in the monoclinic space group C2/m. The asymmetric unit consists of a fourth part of a [{Mn(phen)2}2(μ-4-MeOC6H4COO)2]2+ complex and half a ClO4− anion. The rest of the complex is generated by an inversion centre and a mirror plane. A total of 239 parameters were refined in the final cycle of refinement on F2 using 36 restraints.
Compound 8 crystallises in the triclinic space group P. The asymmetric unit consists of a [Mn(H2O)(3-MeOC6H4COO)(phen)2]+ complex and a ClO4− anion. A total of 424 parameters were refined in the final cycle of refinement on F2 without using any restraint.
Compound 9 crystallises in the monoclinic space group Cc. The asymmetric unit consists of four slightly different complexes with the formula [Mn(H2O)2(2-MeOC6H4COO)2(phen)]. The H atoms of the water ligands were fixed in logical positions considering the plausible H bonds, with isotropic thermal parameters riding on their respective O atoms. A total of 448 parameters were refined in the final cycle of refinement on F2 without using any restraint.
Single crystals of the mononuclear compound 10 were isolated and mounted on a diffractometer. However, the crystals were very thin, so they did not provide good diffraction frames and the structure could not be determined.
Compound 12 crystallises in the triclinic space group P. The asymmetric unit consists of half a [Mn3(bpy)2(μ-4-MeOC6H4COO)6] complex. The rest of the complex is generated by an inversion centre. A total of 448 parameters were refined in the final cycle of refinement on F2 without using any restraint.
Results and discussion
Synthesis
The reaction between MnII salts, carboxylate ligands, and nitrogen-based ligands (NN) is well described in the literature.26,27,30–42 As a function of the MnII:carboxylate:NN ratio, the solvent, the presence or absence of certain counter-anions, and the degree of deprotonation of some ligands, different kinds of compounds with diverse nuclearity can be obtained consisting of ionic or neutral complexes. Moreover, carboxylate ligands can display a wide number of coordination modes.
In this work, the reaction between the corresponding MnII carboxylate, Mn(n-RC6H4COO)2 (n-R = 2-MeO, 3-MeO, 4-MeO, and 4-tBu), and a bidentate nitrogen-based ligand (NN), 2,2′-bipyridine (bpy) or 1,10-phenantroline (phen), led to the formation of seven ionic dinuclear (1–7), three mononuclear (ionic, 8, or neutral, 9 and 10), and four neutral trinuclear (11–14) compounds. The kinds of compounds obtained for each couple of ligands are summarised in Table 1. Fig. 1 shows a concise diagram of the reactions and the obtained compounds, depending on the presence or absence of ClO4− ions and the solvent used when these were determining factors.
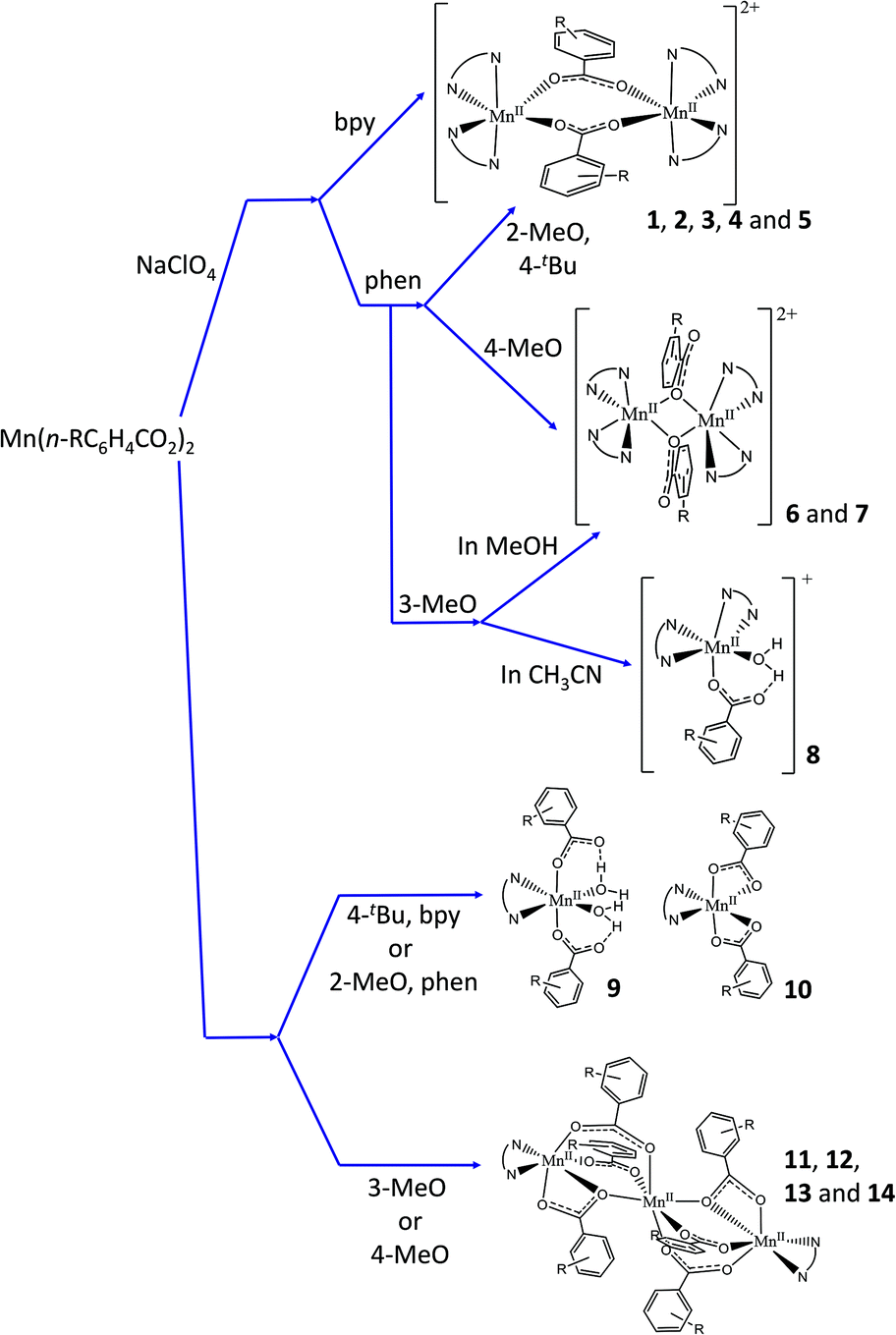 | ||
| Fig. 1 Diagram showing compounds formed depending on the reagents, stoichiometry, and the solvent. | ||
| R | NN | [Mn2] ionic | [Mn] | [Mn3] |
|---|---|---|---|---|
| a Compounds from which the crystal structures have been determined. | ||||
| 2-MeO | bpy | 1 (μ1,3)a | — | — |
| 3-MeO | bpy | 2 (μ1,3)a | — | 11 |
| 4-MeO | bpy | 3 (μ1,3)a | — | 12 |
| 4-tBu | bpy | — | 10 (neutral) | — |
| 2-MeO | phen | 4 (μ1,3) | 9 (neutral)a | — |
| 3-MeO | phen | 6 (μ1,1)a | 8 (ionic)a | 13 |
| 4-MeO | phen | 7 (μ1,1)a | — | 14 |
| 4-tBu | phen | 5 (μ1,3)a | — | — |
When NaClO4 was added besides the MnII carboxylate and the NN ligand, ionic dinuclear compounds with the formula [{Mn(NN)2}2(μ-n-RC6H4COO)2](ClO4)2 (1–3, with NN = bpy; 4–7, with NN = phen) were obtained. It was not possible to obtain this kind of compound for n-R = 4-tBu and NN = bpy. In most cases, the use of CH3CN or EtOH led to the formation of the same compound. However, for n-R = 3-MeO and NN = phen, a mononuclear compound with the formula [Mn(H2O)(3-MeOC6H4COO)(phen)2](ClO4) (8) was obtained in these solvents in the presence of a small amount of water. The formation of the dinuclear compound 6 (3-MeO/phen) was only attained in MeOH.
As reported previously, when no perchlorate salt is added, neutral compounds of different nuclearities could be obtained as a function of the Mn:NN ratio (3:2 or 1:1) and/or solvent.27,30 With the manganese carboxylates used in this work, the Mn:NN ratio and the solvent (CH3CN or EtOH) made no difference in the type of product that was formed, but they did for the conditions of crystallisation (time, crystallinity, etc.) and/or the purity of the sample. For instance, nice yellow crystals of 9 were obtained in EtOH, while the sample contaminated with some brown product precipitated in CH3CN. All attempts to obtain some neutral compounds with n-R = 2-MeO and NN = bpy were unsuccessful; nevertheless, when NN = phen, the mononuclear compound 9 was obtained. Similarly, for n-R = 4-tBu and NN = bpy, a mononuclear compound was obtained (10), while we did not succeed in establishing any reproducible synthesis using phen. The formation of trinuclear compounds was attained when n-R = 3-MeO and 4-MeO with both NN ligands.
The presence of water in the reaction medium was required in nearly all cases, since it assists the dissolution of the MnII carboxylates. Nevertheless, H2O also guaranteed the formation of the dinuclear compounds 3 and 5 because it prevented the fast precipitation of the corresponding neutral compounds, observed after several minutes of stirring when no additional H2O was added.
Regarding the dinuclear compounds, two different coordination modes of the carboxylate ligands can be found, even though the synthetic method is the same. Fortunately, these coordination modes can be easily differentiated with the 1620–1300 cm−1 window of the IR spectra of these compounds. Although the nitrogen-based ligands (bpy and phen) show some bands in this region, it is possible to identify the two characteristic bands of the carboxylate groups, assigned to the asymmetric (νa) and symmetric (νs) vibrations. Values of Δν = νa(COO) − νs(COO) < 200 cm−1 indicate that the carboxylate ligands show a μ1,3-coordination mode, whereas values of Δν > 200 cm−1 are indicative of a μ1,1-coordination mode.26,43 For compounds 1–5, the bands assigned to the carboxylate ligands arise at ∼1550 and ∼1390 cm−1, with Δν ≈ 160 cm−1, consistent with a μ1,3-coordination mode. In contrast, 6 and 7 show two bands at ∼1570 and ∼1310 cm−1 (with Δν ≈ 260 cm−1), consistent with a μ1,1-coordination mode.
The reason why one type of bridge is preferentially formed is very difficult to rationalise. As explained by Gómez et al., two factors could contribute to this different behaviour: the steric and the electronic effects. In the referred work, only compounds containing phen and the R group in meta or para position show a μ1,1-coordination mode.26 Compounds with R = MeO follow the same tendency: while compounds 6 and 7, with the respective n-R = 3-MeO and 4-MeO, have μ1,1-brigdes, compound 4, with n-R = 2-MeO, exhibits a μ1,3-coordination mode. However, compound 5 (n-R = 4-tBu), whose carboxylate shows a μ1,3-coordination mode, does not follow the expected trend. The cause of this variation may lie in the presence of the highly voluminous tBu group that, in spite of being in para position, can be the source of steric hindrance. Indeed, the formation of the μ1,1-brigde shortens the Mn⋯Mn distance from ∼4.65 (for a μ1,3-brigde) to ∼3.46 Å, also setting the facing phen ligands closer (see below).
Description of structures
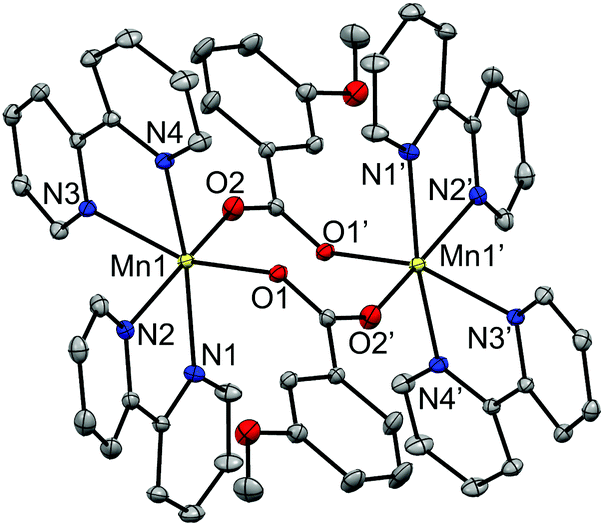 | ||
| Fig. 2 Crystal structures of the cationic complexes of compound 2, showing the anisotropic displacements as ellipsoids at 50% probability. Hydrogen atoms are omitted for clarity. | ||
The two MnII ions are bridged by two μ1,3-n-RC6H4COO− ligands in syn–anti mode, with Mn⋯Mn distances ranging from 4.54 to 4.75 Å. The hexacoordination of each Mn ion is completed by two bpy (1, 2 and 3) or phen (5) ligands. The Mn–O and Mn–N distances are in the ranges 2.09–2.14 Å and 2.26–2.36 Å, respectively, leading to distorted octahedral environments around the MnII ions. All these structural parameters are in agreement with those reported for analogous compounds.26,30–34,44,45
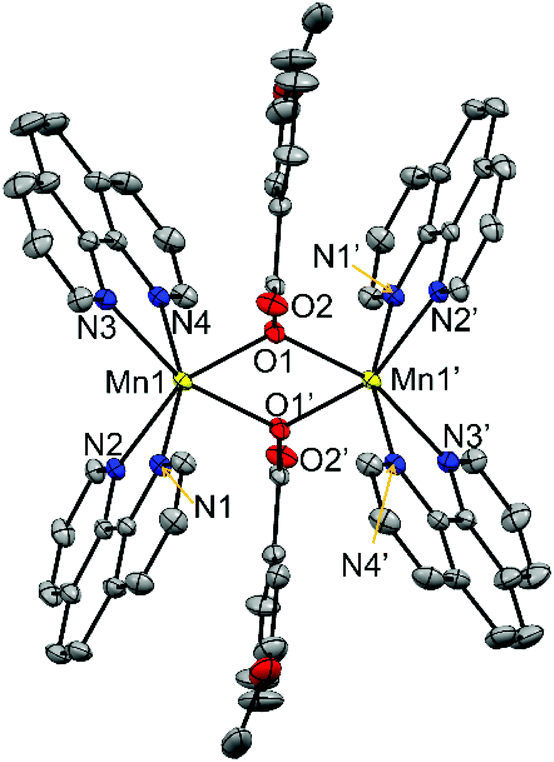 | ||
| Fig. 3 Crystal structures of the cationic complexes of compounds 6, showing the anisotropic displacements as ellipsoids at 50% probability. Hydrogen atoms are omitted for clarity. | ||
The two MnII ions are linked through two μ1,1-n-RC6H4COO− bridges, with Mn⋯Mn distances of ∼3.45 Å. Two phen ligands are linked to each MnII ion, leading to distorted octahedral geometry. The Mn–O distances are in the range 2.16–2.18 Å while the Mn–N are 2.26 Å. The Mn2O2 ring is planar, and the Mn–O–Mn ring is close to 105°. The structural parameters for compounds 6 and 7 are in agreement with those reported for analogous compounds with the [Mn2(μ1,1-R′CCO)2]2+ core.26,35,36,46
In these compounds the phenyl ring and the carboxylate groups of the benzoate derivative bridge are almost coplanar and are perpendicular to the Mn2O2 ring. As may be seen comparing the structures of compounds containing μ1,3- and μ1,1-brigdes (Fig. 2 and 3, respectively), the shorter Mn⋯Mn distance in the latter keeps the NN ligands closer to the aromatic ring of the carboxylate bridge and, consequently, this aromatic ring, which is found between two phen ligands, is forced to twist and become placed almost parallel to these ligands. For compounds 6 and 7, the R group (3-MeO and 4-MeO, respectively) remains in the same plane as the phenyl ring, so their presence does not disturb the formation of the μ1,1-bridges. On the other hand, the bulky tBu group has two methyl groups that stand out from the plane of the phenyl group, being able to cause steric hindrance. Hence, the formation of a dinuclear compound with μ1,1-4-tBuC6H4COO− bridges could be tentatively considered improvable. The obtained dinuclear compound with this ligand shows μ1,3-bridges (5).
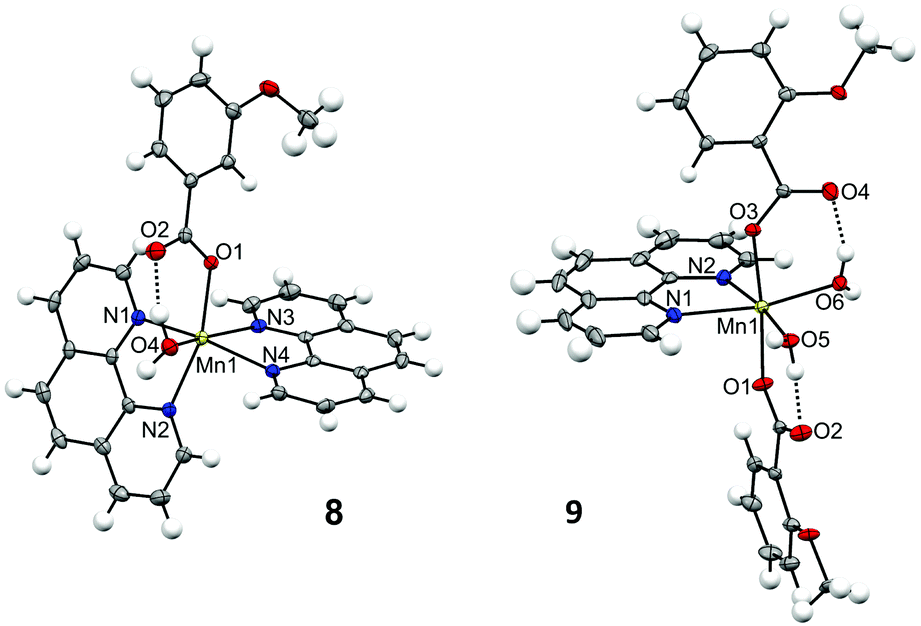 | ||
| Fig. 4 Crystal structures for the cationic complex of 8 and for one of the subunits of compound 9, showing the anisotropic displacements as ellipsoids at 50% probability. | ||
The structure of compound 8 consists of a cationic complex with the formula [Mn(H2O)(3-MeOC6H4COO)(phen)2]+ and a perchlorate anion. The carboxylate ligand is coordinated in a monodentate mode, with Mn–O distances of ∼2.15 Å. There are hydrogen bonds between the H2O molecule and the hanging oxygen atom of the carboxylate ligand (O2). The hexacoordination of the MnII ion is completed by two phen ligands, with Mn–N distances of ∼2.26 Å. The structure of compound 9 comprises four neutral complexes with the formula [Mn(H2O)2(2-MeOC6H4COO)2(phen)] with very similar structural parameters (Fig. S3 and Table S7†). Fig. 4 and Table S6† only concern the structure of one of these complexes. In this compound, the two carboxylate ligands are coordinated in monodentate mode. Each hanging oxygen atom of these ligands is interacting with one of the H2O molecules through hydrogen bonds. The hexacoordination of the MnII ion is completed by one phen ligand, with Mn–N distances of ∼2.28 Å. The MnII ions in these compounds show a distorted octahedral geometry.
Crystals of compound 10 were poorly diffracting and its crystal structure could not be refined. However, some atoms could be assigned to several Q peaks. Apparently, the structure of this compound consists of a neutral complex with the formula [Mn(bpy)(4-tBuC6H4COO)2], where the carboxylate ligands are coordinated in bidentate mode.
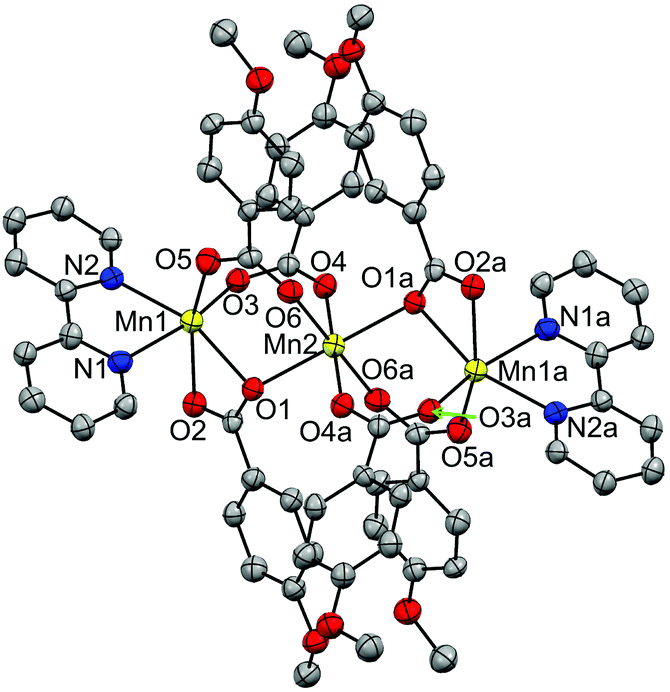 | ||
| Fig. 5 Crystal structure of compound 12, showing the anisotropic displacements as ellipsoids at 50% probability. Hydrogen atoms are omitted for clarity. | ||
The structure of this compound consists of three MnII ions in a linear array, where the central Mn ion is located at a crystallographic inversion centre. This central ion (Mn2) is coordinated by six oxygen atoms of six 4-methoxybenzoate ligands and is linked to each terminal Mn ion (Mn1) through three 4-methoxybenzoate ligands, with Mn⋯Mn distances of ∼3.60 Å. While two of these ligands link the Mn ions with both oxygen atoms (μ1,3) in a syn–syn mode, the third one links them through just one oxygen atom (μ1,1). The Mn–O distances range from 2.15 to 2.25 Å. The other oxygen atom (O2) of this latter ligand is weakly bonded to the terminal Mn ion, with Mn–O distances of 2.33 Å. The hexacoordination of the terminal ions is completed by a phen ligand, with Mn–N distances of ∼2.27 Å. These structural parameters are in agreement with those reported for analogous compounds.27,30,37–42
The terminal Mn ions show a substantial elongation in the direction of the μ1,1 bridge. The central ion, in spite of having angles of 180° between facing bonds, displays a certain elongation in the direction of the μ1,1 bridge.
Magnetic properties
Magnetic susceptibility data were recorded for all polynuclear compounds (1–7 and 11–14) from room temperature to 2 K. Magnetisation measurements were performed at 2 K from 0 to 5.0 T for the compounds having S ≠ 0 ground states (6, 7 and 11–14).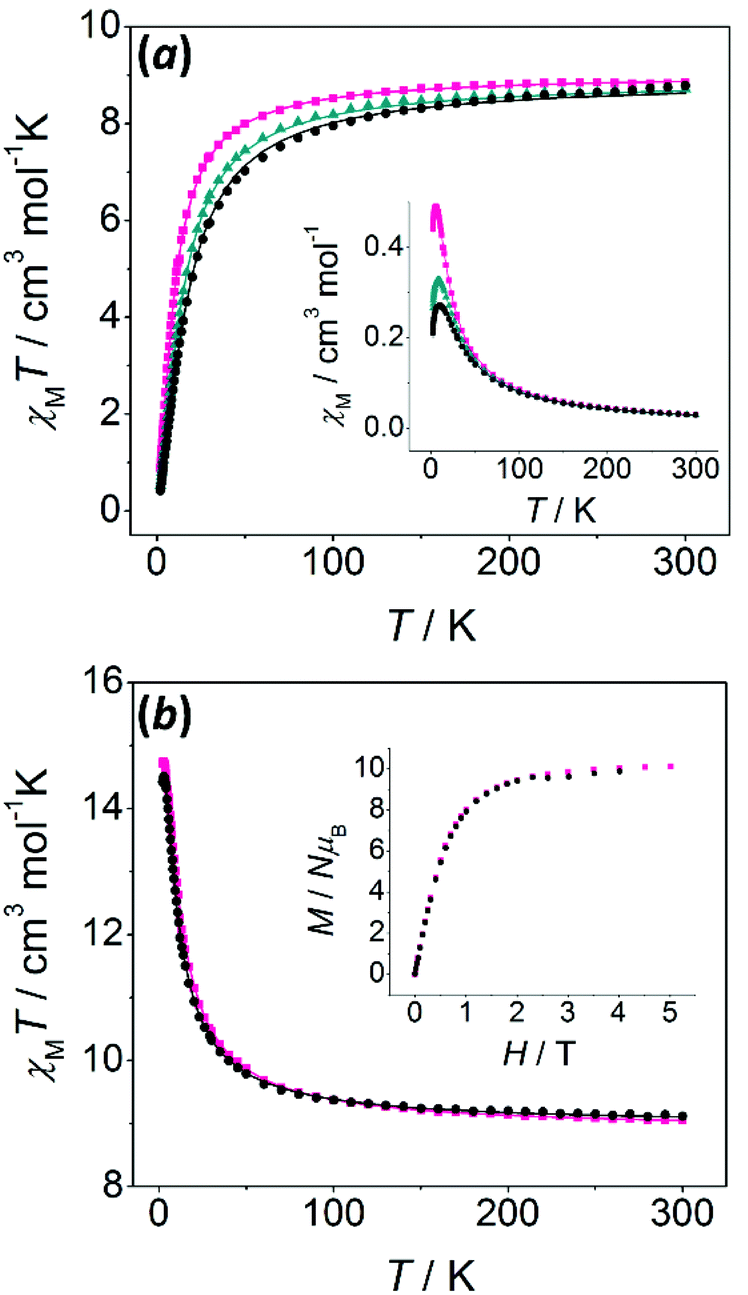 | ||
| Fig. 6 (a) χMT versus T and χMversus T (inset) plots for compounds 1 (black circles), 2 (dark cyan triangles), and 3 (pink squares); (b) χMT versus T and M/Nμβversus H plots (inset) for compounds 6 (black circles) and 7 (pink squares). The solid lines correspond to the best fit of the experimental data. | ||
χ M T values at room temperature are close to the value expected for two uncoupled MnII ions. Two different behaviours may be found: (a) for compounds with μ1,3-bridges (1–5), χMT values decrease as the temperature falls, indicative of an antiferromagnetic behaviour; (b) for compounds with μ1,1-bridges (6 and 7), χMT values increase as the temperature falls, indicative of a ferromagnetic behaviour. For these compounds, the S = 5 ground state is confirmed by the field dependence of the magnetisation at 2 K (inset of Fig. 6b), which shows M/Nμβ values consistent with 10 unpaired electrons.
χ M T versus T plots were fitted using the PHI program, which uses the Hamiltonian H = −2JS1S2.47 The results of the fit are collected in Table 2. The two ferromagnetic compounds (6 and 7) were fitted taking into account intermolecular interactions (zJ′), which improved the fit and reproduced the slight decrease of the χMT values below 2.5 K.
| g | 2Ja/cm−1 |
zJ′b/cm−1 |
R | |
|---|---|---|---|---|
| a Refers to H = −2JS1S2, uncertainties of which were found to be smaller than 0.01 cm−1. b Intermolecular interactions. c Fitted considering TIP = 2 × 10−3 cm3 mol−1; R = ∑[(χMT)exp − (χMT)calcd]2/∑[(χMT)exp]2. | ||||
| 1 | 2.02 | −2.4 | — | 1.0 × 10−4 |
| 2 | 2.02 | −2.0 | — | 2.5 × 10−5 |
| 3 | 2.03 | −1.4 | — | 1.9 × 10−5 |
| 4c | 2.02 | −1.9 | — | 2.0 × 10−5 |
| 5 | 2.00 | −2.0 | — | 3.8 × 10−5 |
| 6 | 2.02 | +1.1 | −0.002 | 3.3 × 10−6 |
| 7 | 2.01 | +1.3 | −0.001 | 9.2 × 10−5 |
These results are in accord with the study reported by Gómez et al., where analogous compounds containing μ1,3-n-RC6H4COO− bridges show antiferromagnetic interactions whereas those with μ1,1-n-RC6H4COO− bridges display ferromagnetic interactions.26 The ferromagnetic interaction mediated by the μ1,1-bridge is principally observed in MnII compounds having Mn–(μ1,1-O)–Mn angles of around 104°. However, other compounds with Mn–O–Mn angles ranging from 104° to 108° displaying antiferromagnetic behaviour have been reported.35,36,46
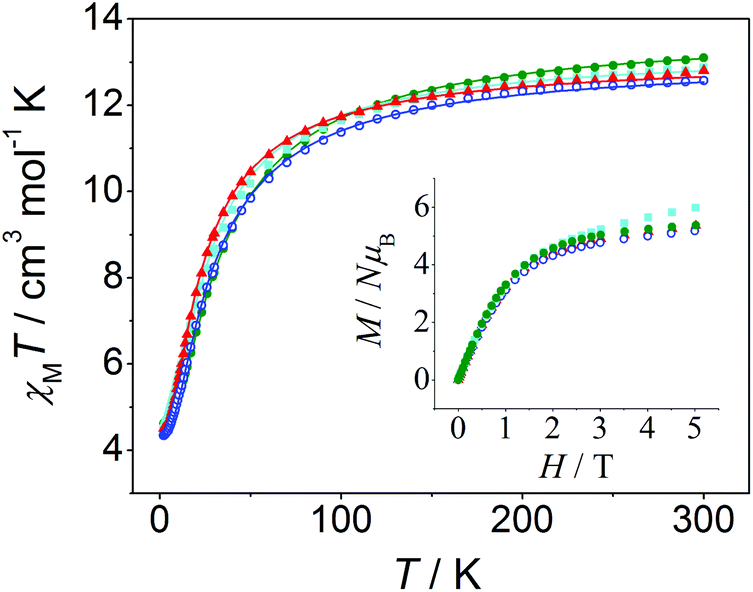 | ||
| Fig. 7 χ M T versus T and M/Nμβversus H plots (inset) for compounds 11 (red triangles), 12 (blue open circles), 13 (cyan squares), and 14 (green filled circles). The solid lines correspond to the best fit of the experimental data. | ||
χ M T versus T plots were fitted with the PHI program47 considering the Hamiltonian H = −2J(S1S2 + S2S3) − 2J13(S1S3) (see Fig. 8), where it is assumed that J13 = 0 due to the large Mn⋯Mn distance between the terminal ions (∼7.2 Å). The results of these fits are collected in Table 3, these being consistent with those found in the literature for analogous trinuclear compounds.27,30,37–42 Moreover, the results obtained for 12, with a marked μ1,1-mode of the carboxylate bridge and a weak antiferromagnetic interaction (2J = −2.5 cm−1), are in agreement with the magneto-structural correlations previously reported.27 However, the structural data of the compounds reported here are too scarce to provide new insights into these correlations, since only the crystal structure of compound 12 could have been determined.
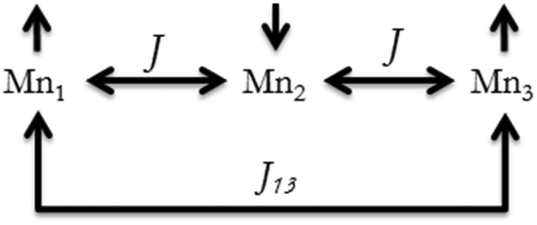 | ||
| Fig. 8 Possible magnetic interactions in the linear trinuclear compounds. | ||
| Compound | g | 2Ja/cm−1 |
R SUS | |
|---|---|---|---|---|
| a Refers to H = −2J(S1S2 + S2S3) − 2J13(S1S3), with J3 = 0, uncertainties of which were found to be smaller than 0.01 cm−1; RSUS = ∑[(χMT)exp − (χMT)calcd]2/∑[(χMT)exp]2. | ||||
| 11 | 3-MeO/bpy | 2.04 | −2.1 | 5.9 × 10−5 |
| 12 | 4-MeO/bpy | 2.00 | −2.5 | 3.4 × 10−5 |
| 13 | 3-MeO/phen | 2.02 | −2.2 | 3.2 × 10−4 |
| 14 | 4-MeO/phen | 2.05 | −2.9 | 3.9 × 10−5 |
EPR spectroscopy
The X-band EPR spectra of all MnII compounds herein reported (1–14) were recorded on powdered samples at different temperatures. At room temperature, all of them show a band centred at g ≈ 2; however, the spectra become more complicated as the temperature falls. To exemplify, Fig. 9 shows the spectra at different temperatures for a dinuclear (5, 4-tBu/phen) and a trinuclear (12, 4-MeO/bpy) compound. At 4 K, the spectra of each type of compound show peculiar features that allow its identification.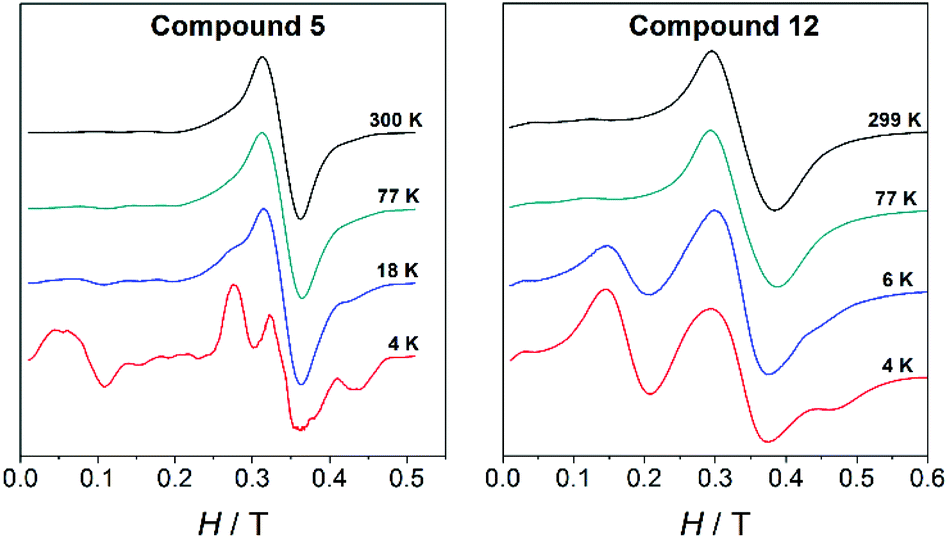 | ||
| Fig. 9 Variable-temperature X-band EPR spectra for the dinuclear compound 5 and the trinuclear compound 12. | ||
The fit of the EPR spectra was performed with the PHI program.47 It is worth noting that, even though this program is able to fit EPR spectra, the initial values from which the fit will begin are of importance for the final result. Hence, we performed several simulations screening different D and E values, within the logical range that one may expect for hexacoordinated MnII ions, in order to choose the best initial values. All spectra were fitted considering a linewidth between 1.0 and 1.8 cm−1.
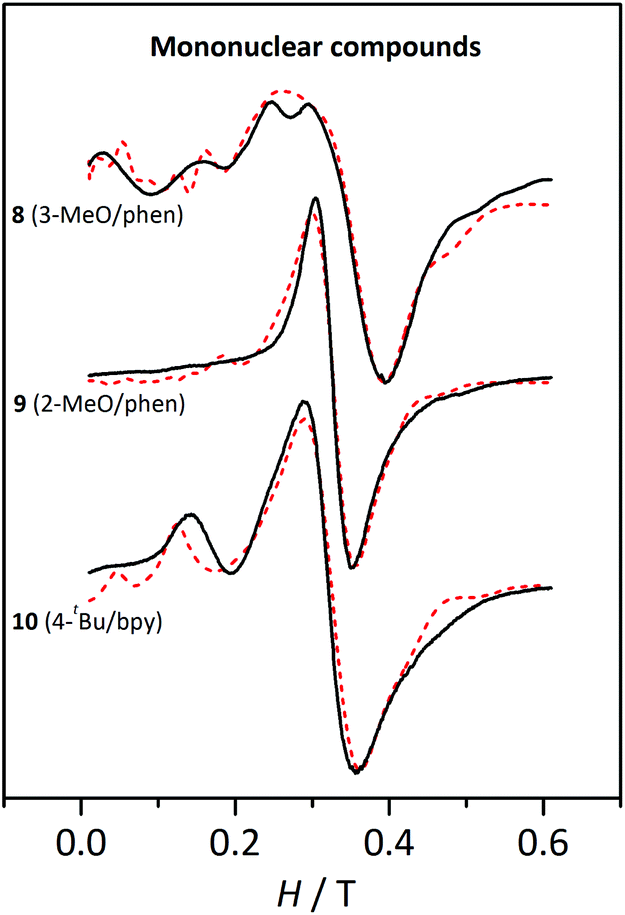 | ||
| Fig. 10 X-band EPR spectra for the mononuclear compounds 8, 9, and 10 at 4 K. The dashed lines correspond to the best fit of the experimental data. | ||
The three compounds display a broad and intense band at g ≈ 2 and more features more or less pronounced at lower fields. These spectra were fitted with the PHI program47 considering a high-spin MnII ion and the single-ion ZFS parameters (DMn and EMn). The results from these fits are listed in Table 4.
| Compound | g | |DMn|/cm−1 | |EMn|/cm−1 | |EMn|/|DMn| |
|---|---|---|---|---|
| 8, [Mn(H2O)(3-MeOC6H4COO)(phen)2](ClO4); 9, [Mn(H2O)2(2-MeOC6H4COO)2(phen)]; 10, [Mn(bpy)(4-tBuC6H4COO)2(H2O)]. | ||||
| 8 | 2.0 | 0.064 | 0.010 | 0.16 |
| 9 | 2.08 | 0.033 | 0.010 | 0.30 |
| 10 | 2.10 | 0.045 | 0.015 | 0.33 |
The DMn values obtained from these fits are consistent with MnII ions with distorted octahedral geometry.24 Several simulations were performed by changing the sign of DMn and EMn. While the shape of the spectra only depends on the |EMn|, they vary with the sign and magnitude of DMn. However, such differences were too small to unambiguously establish the sign of this parameter.
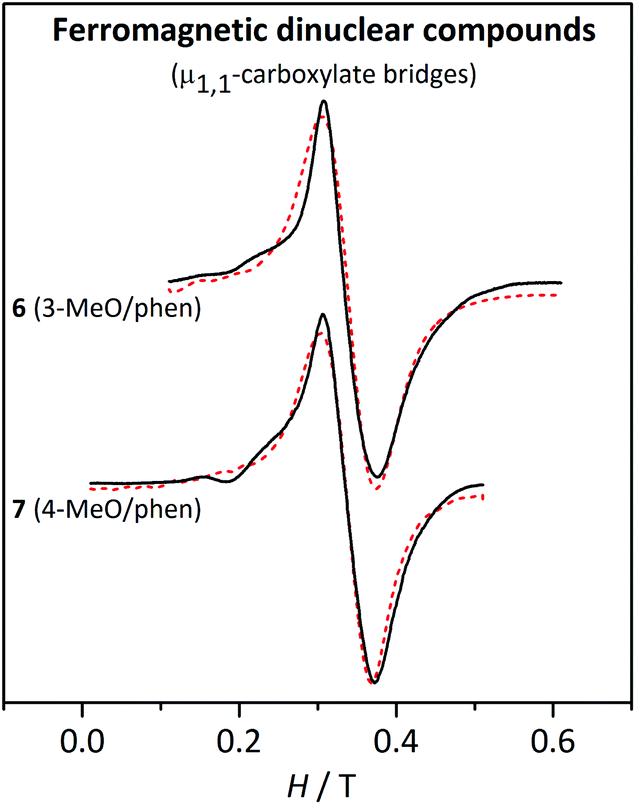 | ||
| Fig. 11 X-band EPR spectra for the ferromagnetic dinuclear compounds 6 and 7 at 4 K. The dashed lines correspond to the best fit of the experimental data. | ||
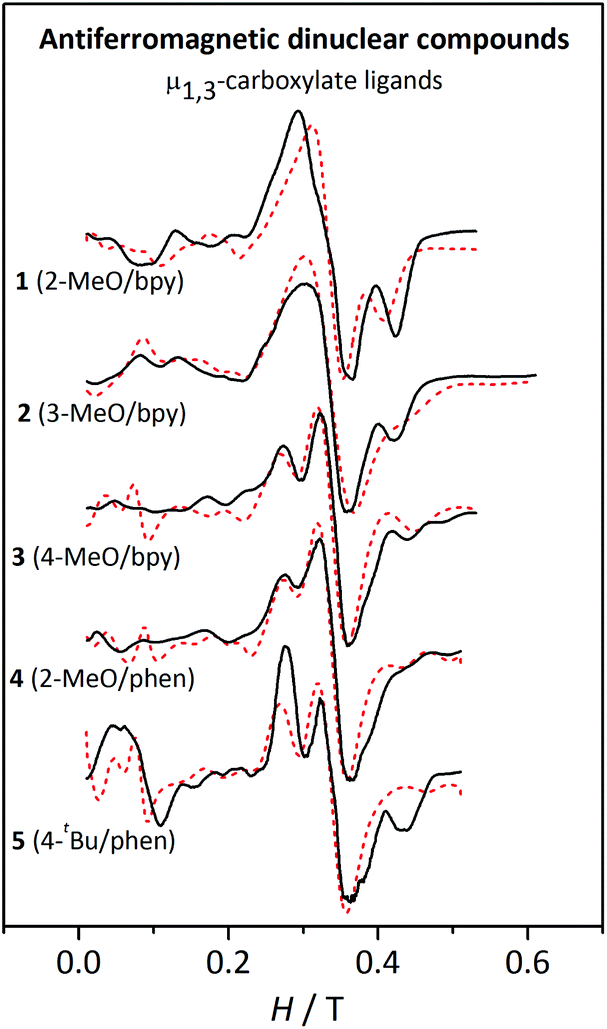 | ||
| Fig. 12 X-band EPR spectra for the antiferromagnetic dinuclear compounds 1–5 at 4 K. The dashed lines correspond to the best fit of the experimental data. | ||
The cause of the difference between the EPR spectra of anti- or ferromagnetic compounds relies on the values of the zero-field splitting parameters, |DS| and |ES|, of the states populated at low temperature. In fact, |DS| and |ES| become smaller as the spin increases.48 Accordingly, the zero-field splitting parameters of the S = 5 ground state of the ferromagnetic compounds are much smaller than those of the first excited states, S = 1 and S = 2, of the antiferromagnetic compounds.
The EPR spectra at 4 K of the dinuclear compounds were fitted with the PHI program47 with the aim of evaluating the single-ion ZFS parameters (DMn and EMn), considering two high-spin MnII ions, with either parallel or antiparallel disposition of their spin moments, using the magnetic coupling constant (2J) obtained from the fit of the magnetic data, and keeping g = 2.00. The results of these fits are collected in Table 5. Finding a good simulation of the EPR spectra of the dinuclear compounds was far more complicated than that for the mononuclear ones. Indeed, before starting the fits, it was necessary to perform several simulations screening different DMn and EMn/DMn values in order to find the best initial values for the fits.
| Compound | 2J/cm−1 | D Mn/cm−1 | |EMn|/cm−1 | |EMn|/|DMn| |
|---|---|---|---|---|
| 1 | −2.4 | ±0.070 | 0.020 | 0.29 |
| 2 | −2.0 | ±0.083 | 0.017 | 0.20 |
| 3 | −1.4 | +0.125 | 0.006 | 0.05 |
| 4 | −1.9 | +0.104 | 0.013 | 0.13 |
| 5 | −2.0 | +0.121 | 0.017 | 0.14 |
| 6 | +1.1 | ±0.030–0.048 | 0.009–0.012 | 0.19–0.33 |
| 7 | +1.3 | ±0.032–0.050 | 0.009 | 0.18–0.28 |
For the ferromagnetic compounds 6 and 7, which display a single broad band (Fig. 11), the anisotropy parameters could not be determined with accuracy and, consequently, the results are expressed as a range. As mentioned above, the DS=5 and ES=5 parameters are smaller than the respective DS<5 and ES<5 for the same DMn and EMn.48 Hence, the EPR spectra at low temperature of a ferromagnetic MnII2 compound, with S = 5 ground state, will be much less sensitive to the ZFS parameters of the MnII ions than the antiferromagnetic ones, where the first excited states, S = 1 and S = 2, are responsible for the splitting in the EPR signal. Indeed, while DS=5 ≈ 0.44DMn, DS=1 ≈ −6.4DMn and DS=2 ≈ −0.95DMn.48 The DMn values obtained from the fit of the experimental data at 4 K are rather small (≤0.05 cm−1). Aiming to see the accuracy of these results, the shape of the spectra with different DMn and EMn/DMn values was explored. As expected, no significant differences were found by changing the sign of DMn. For DMn = 0.12 cm−1, strong bands are observed at low fields. When DMn = 0.07 cm−1, the most intense features appear at g ≈ 2, but a simple band, like the one in the experimental spectra, is only found when DMn ≤ 0.5 cm−1 (Fig. S6†). Hence, in spite of the small effect of DMn on DS=5, we can confirm that these compounds possess small single-ion ZFS parameters, as indicated in Table 5.
For these ferromagnetic compounds (6 and 7), the shape of the spectra is similar at different temperatures, with an increase of 0.02 T in the linewidth from 77 K to 4 K. The simulation of the EPR spectra with the parameters obtained from the fit reproduces well the effect of temperature on the linewidth (Fig. S7†).
The spectra of compounds 1–5 at temperatures >10 K are rather similar; however, at 4 K significant differences can be observed (Fig. 12). These spectra can be separated into three regions: (a) below 0.2 T, where only 5 shows a significant band; (b) between 0.2–0.4 T, where compounds 1 and 2 show a unique broad band, while compounds 3–5 show two overlapped bands; and (c) above 0.4 T, where compounds 1, 2, and 5 show a well-defined band. Owing to the intrinsic complexity of these spectra, fitting these spectra was far more challenging. The assignment of a particular set of values to the parameters was not trivial either, since the simulations obtained from the fit are far from being perfect. However, after screening different DMn and EMn values within the expected range for MnII ions, the simulations presented in Fig. 12 correspond to those better representing the shape of the spectra.
Moreover, a surprising peculiarity was found: the sign of DMn was certainly relevant for the shape of the EPR spectra of compounds 3, 4, and 5. A good fit of the EPR spectra of these compounds can only be attained with DMn > 0. These latter facts suggest that one may likely establish the sign of DMn in some precise situations. In order to understand the effect of the sign of DMn on the resulting EPR spectra, several simulations were performed with g = 2.00 and 2J = −2.4 cm−1 and two different DMn values and four different EMn values. Fig. 13 shows the spectra for these hypothetical dinuclear compounds.
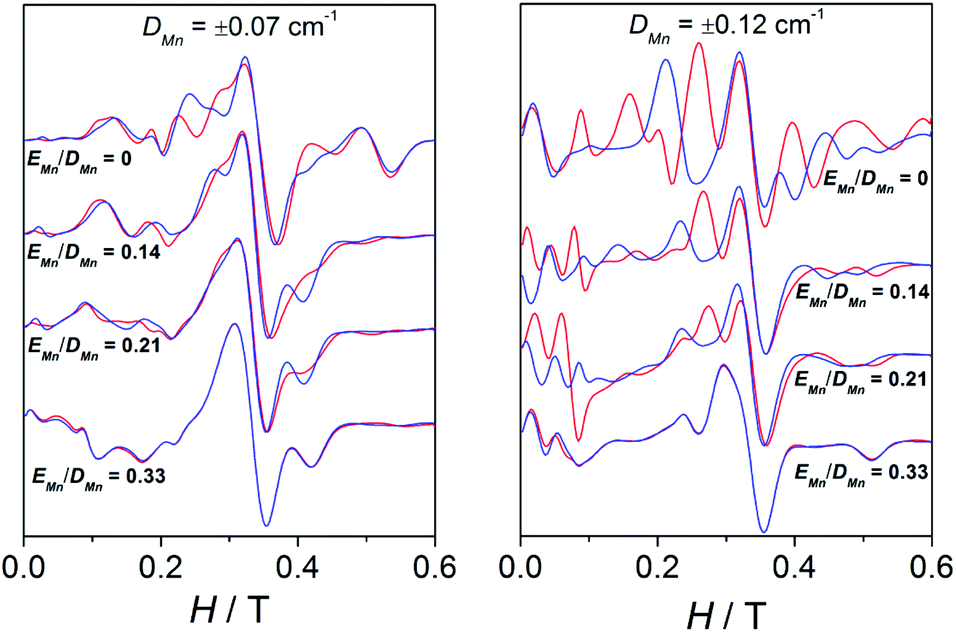 | ||
| Fig. 13 X-band EPR spectra for a hypothetical dinuclear compound with g = 2.00 and 2J = −2.4 cm−1 for two different values of DMn and four different |EMn|/|DMn| ratios. Red lines and blue lines correspond to the simulations performed with positive and negative DMn, respectively. | ||
As observed, for a “small” DMn value of 0.07 cm−1, the distinction between the spectra simulated with positive (red) or negative (blue) DMn appears very difficult to accomplish, since the bands are more or less situated in the same position and only the intensity differs. On the other hand, for a higher value (|DMn| = 0.12 cm−1), the differences between the spectra with positive or negative DMn become greater, as the bands do not only have different intensities but also appear at different fields. However, such variances become thinner as EMn increases, showing almost identical spectra when |EMn|/|DMn| = 0.33. The sign of DMn is indeed very difficult to determine upon approaching the rhombic limit (E/D = 0.33).49–51 To sum up, the degree of divergence between the spectra with positive and negative DMn depends on the magnitude of this parameter and the |EMn|/|DMn| ratio: (a) the higher the DMn becomes, the more different the spectra are; and (b) the higher the |EMn|/|DMn| ratio is, the less distinctive the spectra become.
To understand the cause of these latter points, a deeper analysis of the EPR spectra and Zeeman plots was performed. Firstly, it is worth recalling two important points: (a) in terms of populations the most relevant states are those with S = 0 (ground state, which display no EPR transitions), S = 1 (first excited state), and S = 2 (second excited state), whose populations at 4 K are ∼66, ∼28, and ∼5%, respectively; and (b) when the axial anisotropy (DMn) becomes relevant the Zeeman plot in the z magnetic field direction is rather different from those in the y and x ones (x = y ≠ z), so the resulting EPR spectra come from the addition of the permitted transitions of the three Zeeman plots.
Therefore, the Zeeman plots in the x/y and z magnetic field directions for a hypothetical MnII2 system with 2J = −2.4 cm−1 and DMn = ±0.12 cm−1 were simulated, and the expected transitions were marked according to the spectroscopic selection rules, ΔMS = ±1 and ΔS = 0.52 It is worth noting that no mixture of MS states coming from different S levels was observed for H < 1.0 T, so the EPR signals should come from the transitions between MS states within the same S level. To simplify, the Zeeman plots of the first and second excited states were extracted from that calculated for the entire system and presented in two separated figures (Fig. 14 and 15). Fig. 14 shows the Zeeman plots in the x/y and z magnetic field directions of the first excited state, S = 1, for a MnII2 system with positive and negative DMn. The pointed transitions correspond to those having ΔMS = 1 (at H = 0.5–0.6 T) and ΔMS = 2 (at H ≈ 0.2 T). These latter transitions should be forbidden according to the selection rules; however, when anisotropy is present, the MS states mix and that allows these transitions.48 As may be observed, the expected transitions should arise at the same field and should have the same intensity in both cases (for negative and positive DMn). Hence, the cause of the difference between these spectra should lie in the second excited state (S = 2).
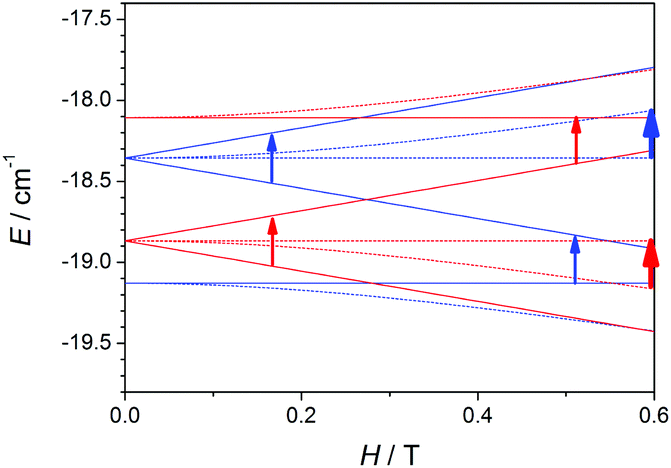 | ||
| Fig. 14 Zeeman plots in the x/y (dashed lines) and z (straight lines) magnetic field directions for the first excited state (S = 1), extracted from a complete Zeeman plot calculated for a MnII2 system with 2J = −2.4 cm−1 and positive (red) and negative (blue) ZFS (DMn = ±0.12 cm−1). The arrows point out the expected transitions in the X-band EPR spectra. The transitions coming from the x and y magnetic field directions are represented by thicker arrows than those from the z magnetic field direction because their contributions are double. | ||
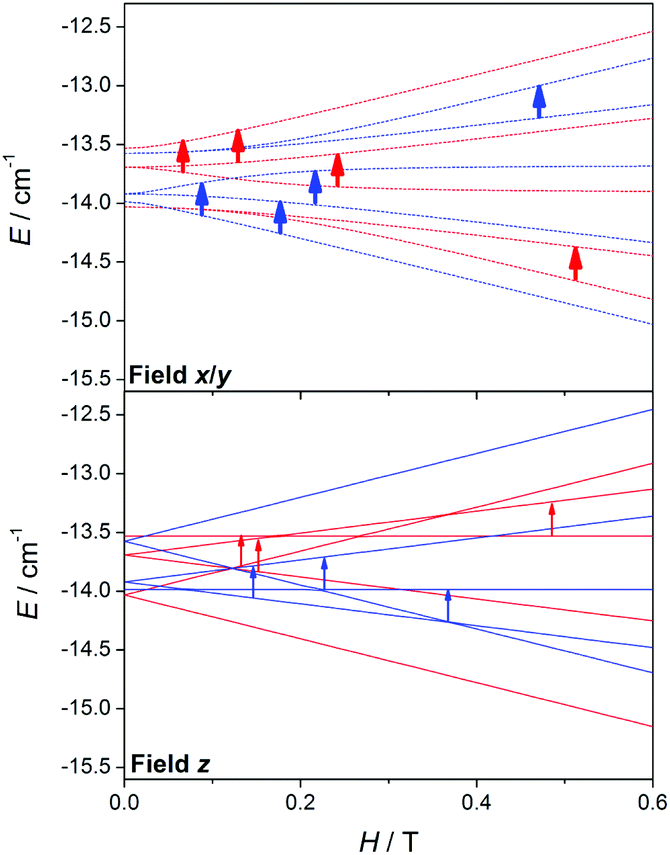 | ||
| Fig. 15 Zeeman plots in the x/y (dashed lines) and z (straight lines) magnetic field directions for the second excited state (S = 2), extracted from a complete Zeeman plot calculated for a MnII2 system with 2J = −2.4 cm−1 and positive (red) and negative (blue) ZFS (DMn = ±0.12 cm−1). The arrows point out the expected transitions in the X-band EPR spectra. The transitions coming from the x and y magnetic field directions are represented by thicker arrows than those from the z direction because their contributions are double. | ||
Fig. 15 shows the Zeeman plots in the x/y and z magnetic field directions of the second excited state, S = 2, for a MnII2 system with positive and negative DMn. Contrary to the S = 1, the expected transitions coming from the S = 2 are rather different in both position and intensity as a function of the sign of DMn. These latter facts indicate that the differences between the spectra with positive and negative DMn rely on the transitions coming from the second excited state, S = 2.
Another arising question is what dictates the sign of DMn in MnII ions. As far as we know, there are several studies in the literature concerning the ZFS of MnII ions;24,50,51,53,54 however, the factors determining the sign of DMn still remain unknown.
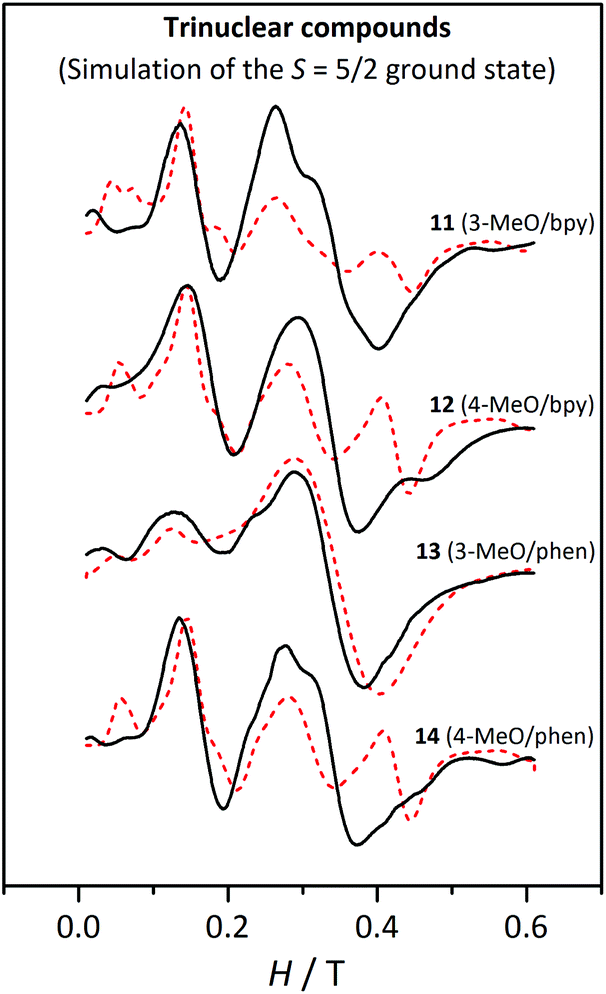 | ||
| Fig. 16 X-band EPR spectra for the trinuclear compounds 11, 12, 13 and 14 at 4 K. The dashed lines correspond to the best fit of the experimental data considering an isolated S = 5/2 ground state. | ||
Fitting the EPR spectra of the trinuclear compounds considering the whole system would be much more time-consuming, since each iteration takes about half an hour. So, in this case, the ZFS of the MnII ions were not determined. As seen with the magnetic measurements, these compounds show an S = 5/2 ground state. The first and second excited states, S = 3/2 and S = 7/2, respectively, are found in the 5.5–7.3 and 7.6–10 cm−1 ranges, respectively, depending on the 2J values. At 4 K, the population of these states is quite low, being 6.5–11.2% and 2.3–5.1% for the respective S = 3/2 and S = 7/2 states. Hence, at this temperature, the greatest contribution to the EPR spectra will be the ground state, whose population ranges from ∼80 to ∼90%.
Therefore, the spectra at 4 K were fitted using the PHI program47 considering an isolated S = 5/2 spin state with axial (D5/2) and rhombic (E5/2) ZFS, keeping g = 2.00. Before fitting the experimental data, several simulations were carried out in order to identify which were the best initial values. The results of these fits, listed in Table 6, are in agreement with those reported for analogous compounds, whose D5/2 and E5/2 are ∼0.16 and ∼0.05 cm−1, respectively.27 Compound 13 displays much smaller ZFS parameters; however, the lack of structural data for this compound does not allow us to reach any conclusion. As observed in Fig. 16, only the fit for 13 corresponds to a good simulation of the experimental spectra. For the rest of the compounds, the theoretical graphs reproduce the band at 0.15 T, but they show significant differences in the other regions of the spectra. Particularly, the intensity of the band at g ≈ 2 for compounds 11, 12, and 14 seems to be underestimated, since it is much weaker in the fitted spectra (red dashed lines) than in the experimental ones (black line). Nevertheless, one should remember that these fits have been performed considering just the ground state and the greater intensity of these bands may be due to the excited states.
| Compound | |D5/2|/cm−1 | |E5/2|/cm−1 | |E5/2|/|D5/2| |
|---|---|---|---|
| 11 | 0.14 | 0.040 | 0.28 |
| 12 | 0.14 | 0.046 | 0.33 |
| 13 | 0.045 | 0.015 | 0.33 |
| 14 | 0.14 | 0.046 | 0.33 |
To explain this, the Zeeman plot for this kind of trinuclear compound was simulated considering |DMn| = 0.1 cm−1 for the three Mn ions. The central ion, even though it is quite regular from the point of view of the angles (O6a–Mn2–O6, O4–Mn2–O4a, and O1–Mn2–O1a angles of 180°), shows a substantial elongation in the direction of the μ1,1-carboxylate ligand. From these calculations a relationship between the magnitudes of the ZFS parameters of each states could be found, where DMn ≈ 0.64 D5/2 and D5/2 ≈ 0.95 D3/2 ≈ 6.2 D7/2. So, while the two first states will have very similar D, the second excited state (S = 7/2) has a much smaller D parameter. To confirm the effect of the population of the S = 7/2 state on the shape of the graph, a new simulation was carried out for a system with an S = 7/2 spin state, whose ZFS parameters are D7/2 = 0.025 cm−1 and E7/2 = 0.0075 cm−1 (equivalent to the fitted D5/2 and E5/2 parameters divided by 6). This spectrum displays just a broad and intense signal at g ≈ 2. Hence, the higher intensity of this band in the experimental spectra of the trinuclear compounds may be due to a small contribution (population in the range 2.3–5.1%) of the second excited state (S = 7/2).
Simulations of the spectra of each compound were performed considering the entire molecule, including the magnetic coupling constant and the same DMn and EMn values for the three Mn(II) ions. DMn values were calculated from the D5/2 values obtained from the fit (listed in Table 6), with the relationship obtained with the analysis of the Zeeman plots (DMn = 0.64 D5/2) (see above). Moreover, we assumed that the EMn/DMn ratio is similar to the E5/2/D5/2 ratio. The parameters used for these simulations are listed in Table 7. They were performed for two different temperatures (4 and 60–80 K), considering the same linewidth and ZFS parameters. Fig. 17 shows these simulations at 4 K. To exemplify, Fig. S8† shows the experimental spectra and simulations at 4 and 60 K for compound 11.† From these simulations three remarkable conclusions could be drawn: (a) the simulation reproduces well the effect of temperature on the shape of the spectra; (b) no significant difference is observed by changing the sign of DMn; and (c) the intensity of the bands at g ≈ 2 for 11, 12, and 14 is well reproduced when the entire system is considered, confirming the contribution of the second excited state as suggested above.
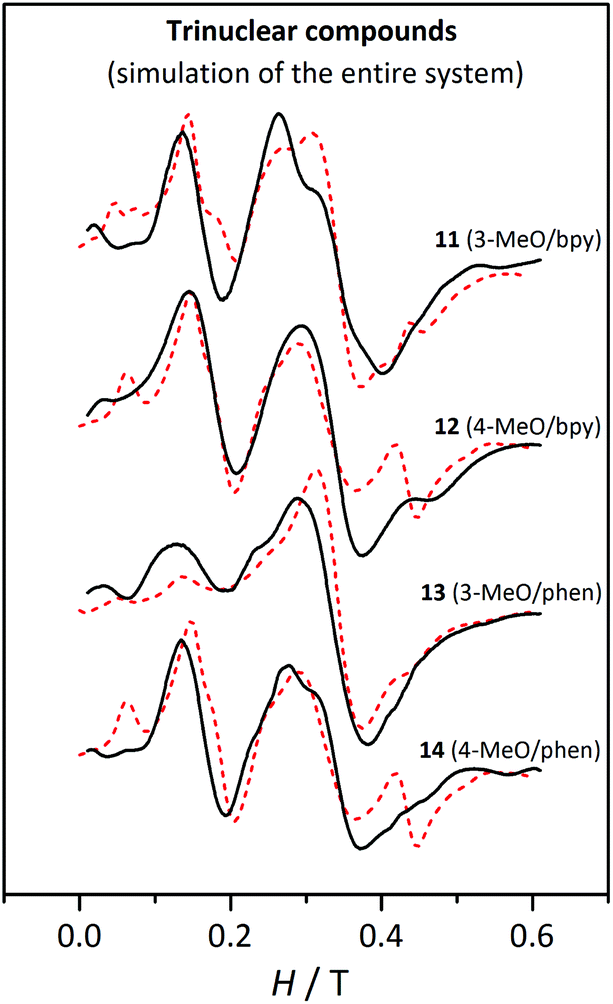 | ||
| Fig. 17 X-band EPR spectra for the trinuclear compounds 11, 12, 13 and 14 at 4 K. The dashed lines correspond to the simulation performed with the parameters listed in Table 7, considering the entire trinuclear system. | ||
As observed in Fig. 17, these simulations reproduce the experimental spectra much better. The differences between the spectra of 13 and the others (11, 12, and 14) are a consequence of the different DMn values, which are significantly smaller for 13.
Even though the experimental spectra are much better reproduced when the entire system is considered and in spite of our efforts, the simulations for 12 and 14 show two bands, at 0.06 T and at 0.45 T, that are not present in the experimental ones. Aiming to find an explanation, four new simulations were performed with 2J = −2.0 and −3.0 cm−1, and DMn (EMn) = 0.09 (0.03), and 0.03 (0.01) cm−1. From these simulations (presented in Fig. S9†) we concluded that: (a) the intensity of the bands at g ≈ 2 decreases when the magnetic interaction is more antiferromagnetic; (b) the band at 0.15 T is intense when D = 0.09 cm−1 and is independent of the 2J value; (c) the shoulder at 0.25 T is only detectable with D = 0.09 cm−1; (d) the band at 0.45 T is well defined when DMn = 0.09 cm−1 and EMn = 0.03 cm−1; and (e) when D = 0.03 cm−1 the effect of the magnitude of the magnetic interaction is negligible.
With the aim of clarifying why the band at 0.45 T in the experimental spectra was not observed in the simulation, the effect of EMn was also analysed by performing simulations (shown in Fig. S10†) with the same 2J = −3.0 cm−1 and DMn = 0.09 cm−1, and EMn = 0.02, 0.025, and 0.03 cm−1. As may be observed, a small decrease of EMn changes the shape of the spectra substantially.
Conclusions
The reaction between Mn(n-RC6H4COO)2, where n-R = 2-MeO, 3-MeO, 4-MeO, or 4-tBu, and the NN ligands bpy and phen led to the formation of seven ionic dinuclear, three mononuclear (ionic or neutral), and four trinuclear compounds. Each type of compound was formed depending on the presence or absence of ClO4− ions, the solvent used, and/or the presence of a small amount of water in the reaction medium. Regarding the dinuclear compounds, two different types of complexes were obtained: those with μ1,1-carboxylate bridges, which present ferromagnetic behaviour, and those having μ1,3-carboxylate bridges, which show an antiferromagnetic interaction. The formation of compounds with the μ1,1-carboxylate bridges occurs when the R group is in meta (n = 3) or para (n = 4) position and phen is the blocking ligand.26 However, the voluminous 4-tBu group apparently hinders the formation of these μ1,1-bridges.The EPR spectra of each type of compound give some peculiar features that allow its identification. All the EPR spectra presented in this work have been fitted in order to obtain the ZFS parameters, D and E. However, fitting these spectra is not as easy as we expected, since many simulations had to be performed in order to choose the best initial values of the parameters willing to determine. In spite of these complications, fitting the spectra helped us to easily adjust the theoretical curves to the experimental ones after obtaining the approximate values.
The spectra of the antiferromagnetic dinuclear compounds are much more complex than those of the ferromagnetic ones, very likely because the ZFS parameter values of the states (DS and ES) populated at low temperature are higher for the antiferromagnetic systems. Moreover, the splitting of the bands in the antiferromagnetic compounds is eventually very sensitive to the sign of DMn, particularly when DMn > 0.10 cm−1 and the |EMn|/|DMn| ratio is low. The analysis of the Zeeman plots revealed that the source of the difference between the spectra with positive and negative DMn lies in the second excited state (S = 2).
The shape of the EPR spectra at low temperature for the mononuclear and trinuclear complexes is different, in spite of showing the same S = 5/2 ground state. For the trinuclear compounds, the relative intensity of the bands depends on the population in the excited states. The influence of the population in the second excited state (S = 7/2) could be seen with the intensity of the band at g ≈ 2; this state has much smaller ZFS parameters than the ground and first excited states and contributes to the intensity of this g ≈ 2 band.
Contrary to the mono- and dinuclear compounds, the EPR spectra of the trinuclear ones were fitted just considering the ZFS of the ground state. A relationship between the ZFS parameters of the states and those of the MnII ion was then found from the analysis of the Zeeman plot of a trinuclear system. Therefore, the single-ion ZFS parameters could be estimated from those obtained just considering the ground state, giving a good simulation of the EPR spectra considering the entire system.
Acknowledgements
This work was supported by the Ministerio de Ciencia e Innovación of Spain (project no. CTQ2012-30662 and CTQ2015-63614-P). L. E. thanks the University of Barcelona for an APIF fellowship.References
- S. J. Lippard and J. M. Berg, Principles of Bioinorganic Chemistry, University Science Books, 1994 Search PubMed.
- V. L. Schramm, Manganese in Metabolism and Enzyme Function, Elsevier, 2012 Search PubMed.
- H. Sigel, Metal Ions in Biological Systems: Volume 37: Manganese and Its Role in Biological Processes, CRC Press, 2000 Search PubMed.
- A.-F. Miller, Curr. Opin. Chem. Biol., 2004, 8, 162–168 CrossRef CAS PubMed.
- T. A. Jackson and T. C. Brunold, Acc. Chem. Res., 2004, 37, 461–470 CrossRef CAS PubMed.
- C. Buy, G. Girault and J.-L. Zimmermann, Biochemistry, 1996, 35, 9880–9891 CrossRef CAS PubMed.
- R. Kappl, K. Ranguelova, B. Koch, C. Duboc and J. Hüttermann, Magn. Reson. Chem., 2005, 43, S65–S73 CrossRef CAS PubMed.
- A. Murphy, G. Dubois and T. D. P. Stack, J. Am. Chem. Soc., 2003, 125, 5250–5251 CrossRef CAS PubMed.
- A. Murphy, A. Pace and T. D. P. Stack, Org. Lett., 2004, 6, 3119–3122 CrossRef CAS PubMed.
- A. Murphy and T. D. P. Stack, J. Mol. Catal. A: Chem., 2006, 251, 78–88 CrossRef CAS.
- I. Garcia-Bosch, A. Company, X. Fontrodona, X. Ribas and M. Costas, Org. Lett., 2008, 10, 2095–2098 CrossRef CAS PubMed.
- D. Pijper, P. Saisaha, J. W. de Boer, R. Hoen, C. Smit, A. Meetsma, R. Hage, R. P. van Summeren, P. L. Alsters, B. L. Feringa and W. R. Browne, Dalton Trans., 2010, 39, 10375 RSC.
- D. M. L. Goodgame, H. E. Mkami, G. M. Smith, J. P. Zhao and E. J. L. McInnes, Dalton Trans., 2003, 34–35 RSC.
- R. Boča, Coord. Chem. Rev., 2004, 248, 757–815 CrossRef.
- R. B. Birdy and M. Goodgame, Inorg. Chim. Acta, 1981, 50, 183–187 CrossRef CAS.
- C. J. H. Jacobsen, E. Pedersen, J. Villadsen and H. Weihe, Inorg. Chem., 1993, 32, 1216–1221 CrossRef CAS.
- W. B. Lynch, R. S. Boorse and J. H. Freed, J. Am. Chem. Soc., 1993, 115, 10909–10915 CrossRef CAS.
- R. M. Wood, D. M. Stucker, L. M. Jones, W. B. Lynch, S. K. Misra and J. H. Freed, Inorg. Chem., 1999, 38, 5384–5388 CrossRef CAS.
- C. Mantel, C. Baffert, I. Romero, A. Deronzier, J. Pécaut, M.-N. Collomb and C. Duboc, Inorg. Chem., 2004, 43, 6455–6463 CrossRef CAS PubMed.
- C. Duboc, T. Phoeung, D. Jouvenot, A. G. Blackman, L. F. McClintock, J. Pécaut, M.-N. Collomb and A. Deronzier, Polyhedron, 2007, 26, 5243–5249 CrossRef CAS.
- C. Duboc, T. Phoeung, S. Zein, J. Pécaut, M.-N. Collomb and F. Neese, Inorg. Chem., 2007, 46, 4905–4916 CrossRef CAS PubMed.
- G. Berggren, P. Huang, L. Eriksson and M. F. Anderlund, Appl. Magn. Reson., 2009, 36, 9–24 CrossRef CAS.
- C. Mantel, C. Philouze, M.-N. Collomb and C. Duboc, Eur. J. Inorg. Chem., 2004, 2004, 3880–3886 CrossRef.
- C. Duboc, M.-N. Collomb, J. Pécaut, A. Deronzier and F. Neese, Chem. – Eur. J., 2008, 14, 6498–6509 CrossRef CAS PubMed.
- J. Rich, C. E. Castillo, I. Romero, M. Rodríguez, C. Duboc and M.-N. Collomb, Eur. J. Inorg. Chem., 2010, 2010, 3658–3665 CrossRef.
- V. Gómez, M. Corbella, M. Font-Bardia and T. Calvet, Dalton Trans., 2010, 39, 11664 RSC.
- V. Gómez and M. Corbella, Eur. J. Inorg. Chem., 2009, 2009, 4471–4482 CrossRef.
- SADABS, Version 2008/1, Sheldrick, Bruker AXS Inc., 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2007, 64, 112–122 CrossRef PubMed.
- G. Fernández, M. Corbella, J. Mahía and M. A. Maestro, Eur. J. Inorg. Chem., 2002, 2002, 2502–2510 CrossRef.
- B. Albela, M. Corbella, J. Ribas, I. Castro, J. Sletten and H. Stoeckli-Evans, Inorg. Chem., 1998, 37, 788–798 CrossRef CAS.
- H. Oshio, E. Ino, I. Mogi and T. Ito, Inorg. Chem., 1993, 32, 5697–5703 CrossRef CAS.
- C. Ma, W. Wang, X. Zhang, C. Chen, Q. Liu, H. Zhu, D. Liao and L. Li, Eur. J. Inorg. Chem., 2004, 2004, 3522–3532 CrossRef.
- Y.-S. Ma, X.-Y. Tang, F.-F. Xue, B. Chen, Y.-L. Dai, R.-X. Yuan and S. Roy, Eur. J. Inorg. Chem., 2012, 2012, 1243–1249 CrossRef CAS.
- S. Durot, C. Policar, G. Pelosi, F. Bisceglie, T. Mallah and J.-P. Mahy, Inorg. Chem., 2003, 42, 8072–8080 CrossRef CAS PubMed.
- D. Moon, J. Kim, M. Oh, B. J. Suh and M. S. Lah, Polyhedron, 2008, 27, 447–452 CrossRef CAS.
- S. G. Baca, I. L. Malaestean, T. D. Keene, H. Adams, M. D. Ward, J. Hauser, A. Neels and S. Decurtins, Inorg. Chem., 2008, 47, 11108–11119 CrossRef CAS PubMed.
- S. Menage, S. E. Vitols, P. Bergerat, E. Codjovi, O. Kahn, J. J. Girerd, M. Guillot, X. Solans and T. Calvet, Inorg. Chem., 1991, 30, 2666–2671 CrossRef CAS.
- V. Tangoulis, D. A. Malamatari, K. Soulti, V. Stergiou, C. P. Raptopoulou, A. Terzis, T. A. Kabanos and D. P. Kessissoglou, Inorg. Chem., 1996, 35, 4974–4983 CrossRef CAS PubMed.
- S. G. Baca, Y. Sevryugina, R. Clérac, I. Malaestean, N. Gerbeleu and M. A. Petrukhina, Inorg. Chem. Commun., 2005, 8, 474–478 CrossRef CAS.
- A. Escuer, B. Cordero, X. Solans, M. Font-Bardia and T. Calvet, Eur. J. Inorg. Chem., 2008, 2008, 5082–5087 CrossRef.
- C. J. Milios, T. C. Stamatatos, P. Kyritsis, A. Terzis, C. P. Raptopoulou, R. Vicente, A. Escuer and S. P. Perlepes, Eur. J. Inorg. Chem., 2004, 2004, 2885–2901 CrossRef.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- V. Gómez and M. Corbella, J. Chem. Crystallogr., 2011, 41, 843–846 CrossRef.
- V. Gómez, M. Corbella, F. A. Mautner, O. Roubeau, S. J. Teat, M. Font-Bardia and T. Calvet, Polyhedron, 2012, 45, 185–199 CrossRef.
- F. Nepveu, N. Gaultier, N. Korber, J. Jaud and P. Castan, J. Chem. Soc., Dalton Trans., 1995, 4005–4013 RSC.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- O. Kahn, Molecular magnetism, VCH, 1993 Search PubMed.
- A. Abragam and B. Bleaney, Electron Paramagnetic Resonance of Transition Ions, OUP, Oxford, 2012 Search PubMed.
- S. Zein, C. Duboc, W. Lubitz and F. Neese, Inorg. Chem., 2008, 47, 134–142 CrossRef CAS PubMed.
- R. Carmieli, T. M. Larsen, G. H. Reed, S. Zein, F. Neese and D. Goldfarb, J. Am. Chem. Soc., 2007, 129, 4240–4252 CrossRef CAS PubMed.
- J. R. Gispert, Coordination Chemistry, Wiley, 2008 Search PubMed.
- S. C. Hunter, A. A. Podlesnyak and Z.-L. Xue, Inorg. Chem., 2014, 53, 1955–1961 CrossRef CAS PubMed.
- T. D. Tzima, G. Sioros, C. Duboc, D. Kovala-Demertzi, V. S. Melissas and Y. Sanakis, Polyhedron, 2009, 28, 3257–3264 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables containing crystal data and structure refinement details, figure of the crystal structures of the cationic complexes of compounds 1–3, 5–9, and 12, tables with selected structural parameters for compounds 1–3, 5–9, and 12, χMT versus T and χMversus T plots for compounds 4 and 5, and several figures showing simulations of EPR spectra with diferent 2J, D, and E values and at different temperatures. X-ray crystallographic files in CIF format for the structure determination of compounds 1–3, 5–9, and 12. CCDC 1507565–1507573. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt04012h |
| This journal is © The Royal Society of Chemistry 2017 |