
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2-Pyridyl substituents enhance the activity of palladium–phospha-adamantane catalysts for the methoxycarbonylation of phenylacetylene†
Timothy A.
Shuttleworth
,
Alexandra M.
Miles-Hobbs
,
Paul G.
Pringle
* and
Hazel A.
Sparkes
School of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: Paul.Pringle@bristol.ac.uk
First published on 6th December 2016
Abstract
The synthesis of a series of CgPAr ligands is reported, where CgP is the 6-phospha-2,4,8-trioxa-1,3,5,7-tetramethyladamant-6-yl moiety and Ar = 2-pyridyl (L2), 3-pyridyl (L3), 2-pyrimidyl (L4), 4-R-2-pyridyl [R = Me (L5a), CF3 (L6a), SiMe3 (L7a)] or 6-R-2-pyridyl [R = Me (L5b), CF3 (L6b), SiMe3 (L7b). Testing of these ligands in the Pd-catalysed methoxycarbonylation of phenylacetylene reveals that the activity and branched selectivity of the catalysts derived from these ligands varies as a function of the N-heterocycle, with the catalyst derived from L5b being the most active of those tested. This, together with the poor performance of catalysts derived from L3 supports the hypothesis that the catalysis proceeds by a “proton shuttling” mechanism, an idea that previously had only been applied to arylphosphines. Reaction of [PtCl2(cod)] with L where L = L2 or L4–7 yields a rac/meso mixture of the trans-[PtCl2(L)2] (1a–h) complexes, three of which are structurally characterised. 31P NMR spectroscopy shows that reaction of L3 with [PtCl2(cod)] gives a mixture of mononuclear and binuclear metal complexes in solution. The complex trans-[PdCl2(L2)2] (4) reacts with AgBF4 to give the [PdCl(κ1-L2)(κ2-L2)]BF4 (5) with spectroscopic and structural characterisation confirming the presence of a P,N-chelate. 1H and 31P NMR evidence supports the assignment of a pyridyl-protonated species being formed upon treatment of 4 with TsOH·H2O in CD2Cl2; both the protonated species and chelate 5 are observed when the reaction is carried out in MeOH.
Introduction
The palladium-catalysed methoxycarbonylation of alkynes (Scheme 1) is an atom-efficient way of producing branched or linear α,β-unsaturated esters from readily available feedstocks.1,2 | ||
| Scheme 1 | ||
The methoxycarbonylation of propyne (Scheme 1, R = Me) has been extensively studied owing to the industrial interest in the branched product, methyl methacrylate (MMA) which is a precursor to poly(methyl methacrylate).3 The methoxy-carbonylation of phenylacetylene (Scheme 1, R = Ph) produces methyl atropate (branched product), a precursor to ibuprofen2 or methyl cinnamate (linear product), which has applications in the perfume and flavouring industries.4
The activity, selectivity and stability of the palladium catalyst for methoxycarbonylation depend critically on the ligand. Drent et al. reported that changing the ligand from PPh3 to Ph2P(2-py) (L1) led to an increase in rate of three orders of magnitude for propyne and an increase in branched selectivity from 89% to 99%.3 It was postulated that a “non-classical” carbomethoxy mechanism involving P,N-chelates was in operation and the greater catalyst activity was a result of the Ph2P(2-py) acting as a “proton messenger”,3 by bringing the proton into close proximity to the metal and thereby promoting the protonolysis of the proposed vinyl-palladium intermediate as shown in Scheme 2.
 | ||
| Scheme 2 Drent's non-classical carbomethoxy mechanism.3 | ||
Recently, Bühl et al. reported a computational study of several possible pathways for methoxycarbonylation.5 Drent's original mechanism was challenged, as it was found that the postulated, selectivity-determining transition state would favour the formation of linear product (methyl crotonate) over the branched product (MMA). Instead, an alternative cycle involving labile P,N-chelates was proposed which proceeded by an in situ base mechanism (Scheme 3)5 closely related to that described by Scrivanti et al.6 This was suggested to be the most plausible mechanism due to it having surmountable barriers congruent with the high turnover and selectivity observed experimentally with L1. Common to the Drent and Bühl mechanisms (Schemes 2 and 3) is the chelation of the P,N ligand which acts to stabilise the catalyst. Catalysts derived from Ph2P(2-py) have been shown to be excellent for phenylacetylene methoxycarbonylation (Scheme 1, R = Ph).3,6
 | ||
| Scheme 3 Bühl's in situ base mechanism.5 | ||
It has also been observed that substituents on the 2-pyridyl ring have a pronounced effect upon the performance of the catalyst; moreover Ph2P(3-py) and Ph2P(4-py) give catalysts of much lower activity.3 Palladium complexes of other monophosphines with the potential to form heterodonor P,L-chelates (e.g. pyrimidylphosphines,7 iminophosphines,8 phosphinoquinilines,9 furylphosphines10 and so called TROPPs11) have been shown to be efficient methoxycarbonylation catalysts. It is notable that in all of these ligands an Ar2P moiety is present.12
Ligands incorporating the 6-phospha-2,4,8-trioxa-1,3,5,7-tetramethyladamant-6-yl (CgP) moiety are effective for a range of Pd-catalysed carbonylation reactions.13 CgPR and tBu2PR are comparable in terms of bulk but very different in σ/π-bonding characteristics: the donor properties of CgPR are comparable to a (PhO)2PR.14 It was therefore of interest to investigate CgP(2-py) (L2) and its derivatives as ligands for Pd-catalysed methoxycarbonylation of phenylacetylene. We show here that the Pd–L2 catalysts are active and that 4- and 6-substituents on the 2-pyridyl affect the performance of the catalyst. The platinum and palladium coordination chemistry of these ligands has been explored which has given some insight into the function of L2 and related ligands.

Results and discussion
Ligand synthesis
The synthesis of CgP(2-py) (L2) has previously been reported via a Pd-catalysed P-arylation of CgPH15 and we have extended the route to the preparation of cage phosphines containing 3-pyridyl (L3), 2-pyrimidyl (L4), 4-substituted-2-pyridyl (L5–7a) and 6-substituted-2-pyridyl (L5–7b) (Scheme 4). These air-stable ligands were purified by column chromatography and have been fully characterised. Crystals of all of the ligands L2–7 suitable for X-ray crystallography have been obtained. The structures are very similar to each other and so only L2, as a representative structure, is shown in Fig. 1 along with its main metrical parameters; the structures of L3–7 are given in the ESI.† | ||
| Scheme 4 | ||
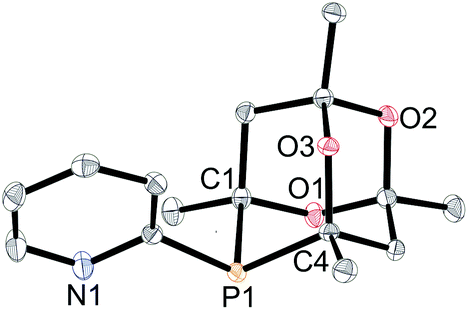 | ||
| Fig. 1 Crystal structure of L2. Hydrogen atoms omitted for clarity. Thermal ellipsoids at 50% probability. Selected bond lengths (Å) and bond angles (°): P1–C1 1.8857(12), P1–C4 1.8734(12), P1–C11 1.8395(12); C1–P1–C4 92.71(5), C1–P1–C11 101.83(5), C4–P1–C11 106.34(5). | ||
Methoxycarbonylation catalysis
In order to draw comparisons in performance among the ligands, the methoxycarbonylation of phenylacetylene (eqn (1)) has been carried out in the presence of L1–7, PPh3 and CgPPh under the same reaction conditions and the results are presented in Table 1.| Entry | ligand | % Conversionb | % Branchedc | |
|---|---|---|---|---|
| 4.5 h | 1 h | |||
| a Reaction conditions: 5.5 mmol of phenylacetylene, 5.5 × 10−3 mmol of Pd(OAc)2, 2.2 mmol of p-tolylsulfonic acid monohydrate, 1.1 mmol of ligand, 1.5 cm3 of MeOH, 45 bar of CO, 60 °C. b Conversion and selectivity determined by 1H NMR (see Experimental for details). Each result is an average of 2 or more runs. c The rest of the product was the linear isomer. | ||||
| 1 | PPh3 | 13 | — | 96 |
| 2 | CgPPh | 72 | — | 82 |
| 3 | L1 | 100 | 100 | 99 |
| 4 | L2 | 89 | 59 | 81 |
| 5 | L5a | 63 | 34 | 91 |
| 6 | L5b | 100 | 95 | 95 |
| 7 | L6a | 92 | 64 | 84 |
| 8 | L6b | 32 | 21 | 87 |
| 9 | L7a | 89 | 55 | 73 |
| 10 | L7b | 87 | 34 | 94 |
| 11 | L3 | 14 | — | 99 |
| 12 | L4 | 75 | 47 | 86 |
| 13 | — | <2 | — | — |
Under the conditions we used for the catalysis, the PPh3 catalyst produced low and inconsistent conversions (entry 1)
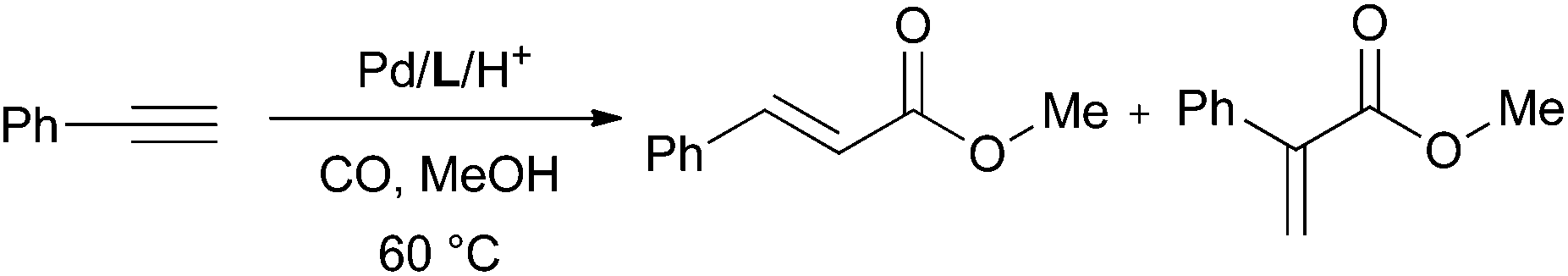 | (1) |
Ligand L2 (entry 4) produced a higher activity catalyst than CgPPh (entry 3) indicating that the 2-pyridyl group has a positive effect within the CgPAr series of ligands. This is reinforced by the low activity observed with the 3-pyridyl isomer L3 (entry 11). The pyrimidyl group in ligand L4 (entry 12) led to a catalyst with lower activity than L2.
The results for the catalysts derived from L5–7a and L5–7b show that substituents Me, CF3 and SiMe3 at the 4- and 6-positions in the 2-pyridyl ring can, in some cases, have a significant effect on the conversion compared to the unsubstituted L2. However, there appears to be no consistent trends associated with the stereoelectronic effect of the substituents. Relative to L2: (1) the 4-Me ligand L5a (entry 5) gives a significantly lower conversion while the 6-Me ligand L5b (entry 6) gives a significantly higher conversion; (2) the 4-CF3 and 4-SiMe3 ligands, L6a (entry 7) and L7a (entry 9), perform similarly to L2 while the 6-CF3 and 6-SiMe3 ligands, L6b (entry 8) and L7b (entry 10), produce catalysts of lower activity than L2. In terms of branched selectivity, there is some evidence that the 6-substituted 2-pyridyl ligands L5–7b give more branched-selective catalysts than their 4-substituted isomers L5–7a (entries 5–10). Others have reported similar observations with 4- and 6-substituted pyridyl derivatives of L1; that is, greater branched selectivity was obtained with 6-substituted than with 4-substituted pyridyl ligands.4
In order to elucidate the function of the 2-pyridyl in the Pd-catalysis, the coordination chemistry of L2–7 with Pt and Pd has been investigated.
Coordination chemistry
Ligands L2–7 have been reacted with [PtCl2(cod)] (cod = 1,5-cyclooctadiene) in CH2Cl2. With the exception of the reaction with L3 (see below), products of the type trans-[PtCl2(L)2] (1a–h) were obtained (Scheme 5) which have been fully characterised. The 31P NMR spectra of the products in each case showed two closely spaced singlets with 195Pt satellites (1JP–Pt values are typical of trans-[PtCl2(PR3)2] complexes) consistent with the presence of rac and meso diastereomers resulting from the chirality of the cage ligands.14 | ||
| Scheme 5 | ||
Crystals suitable for X-ray crystallography of 1c, 1d, 1e and 1g (meso isomers in each case) were grown by slow diffusion of hexane into a CH2Cl2 solution of a rac/meso mixture of the corresponding complex. As shown in Fig. 2–5, in each case, the ligands adopt an anti conformation, with the C(pyridyl)–P–P–C(pyridyl) torsion angle of 180°.
 | ||
| Fig. 2 Crystal structure of meso-1c showing the anti–trans conformation adopted by the ligands. Hydrogen atoms omitted for clarity. Atoms suffixed with a dash (′) are related by symmetry operation (−x, −y, −z). Thermal ellipsoids at 50% probability. Selected bond lengths (Å) and angles (°): Pt1–Cl1 2.3125(7), Pt1–P1 2.3259(7), P1–C1 1.884(3), P1–C4 1.879(3), P1–C11 1.837(3); C1–P1–C4 94.10(13), C1–P1–C11 109.47(13), C4–P1–C11 105.41(13). | ||
 | ||
| Fig. 3 Crystal structure of meso-1d, showing the anti–trans conformation adopted by the ligands. Hydrogen atoms omitted for clarity. Atoms suffixed with a dash (′) are related by symmetry operation (−x, 1−y, −z). Thermal ellipsoids at 50% probability. Selected bond lengths (Å) and angles (°): Pt1–Cl1 2.3119(6), Pt1–P1 2.3198(6), P1–C1 1.883(3), P1–C4 1.878(3), P1–C11 1.845(3); C1–P1–C4 93.99(11), C1–P1–C11 103.98(12), C4–P1–C11 111.54(12). | ||
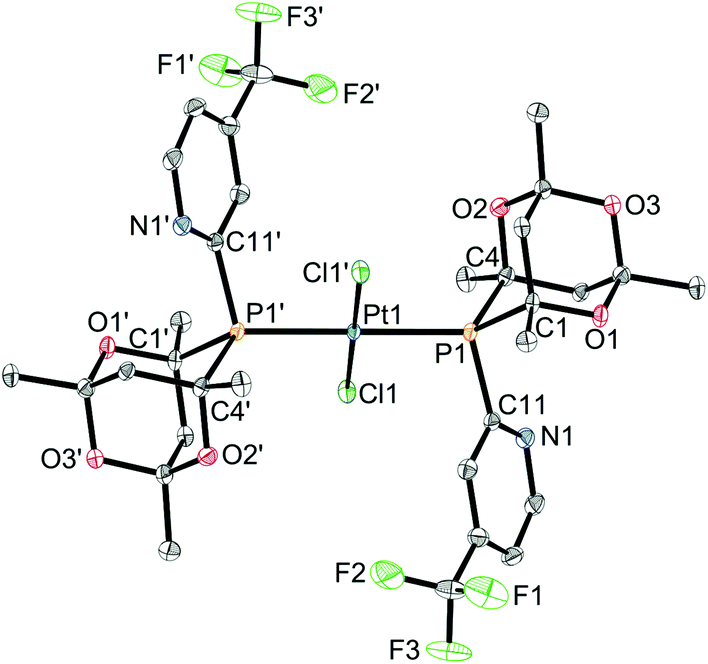 | ||
| Fig. 4 Crystal structure of meso-1e, showing the anti–trans conformation adopted by the ligands. Hydrogen atoms omitted for clarity. Atoms suffixed with a dash (′) are related by symmetry operation (−x, 1−y, −z). Thermal ellipsoids at 50% probability. Selected bond lengths (Å) and angles (°): Pt1–Cl1 2.3056(5), Pt1–P1 2.3227(5), P1–C1 1.877(2), P1–C4 1.886(3), P1–C11 1.842(2); C1–P1–C4 94.43(10), C1–P1–C11 105.04(13), C4–P1–C11 109.69(10). | ||
 | ||
| Fig. 5 Crystal structure of meso-1g, showing the anti–trans conformation adopted by the ligands. Disordered atoms in CgP moiety and hydrogen atoms omitted for clarity. Atoms suffixed with a dash (′) are related by symmetry operation (1−x, −y, −z). Thermal ellipsoids at 50% probability. Selected bond lengths (Å) and angles (°): Pt1–Cl1 2.3022(5), Pt1–P1 2.3193(5), P1–C1 1.877(2), P1–C4 1.875(2), P1–C11 1.833(2); C1–P1–C4 94.23(9), C1–P1–C11 103.66(8), C4–P1–C11 111.82(9). | ||
The reaction of [PtCl2(cod)] with the 3-pyridyl ligand L3 gave different results from those for all of the 2-pyridyl ligands L2, and L4–7. The 31P NMR spectrum of the products of the reaction of [PtCl2(cod)] with 2 equiv. of the 3-pyridyl ligand L3 showed four signals with 195Pt satellites, as well as free ligand (δP −29.5 ppm). Two of the signals were assigned to rac/meso-[PtCl2(L3)2] (1i) because of the similarity of their 31P NMR data (δP −2.6 ppm, 1JP,Pt = 2703 Hz; δP −2.8 ppm, 1JP,Pt = 2710 Hz) to those of the analogues 1a–h. The remaining two signals were assigned to binuclear complexes of the type [Pt2Cl2(L3)2(μ-Cl)2] on the basis of their large 1JP,Pt values (δP 8.3 ppm, 1JP,Pt = 4592 Hz; δP 7.9 ppm 1JP,Pt = 4608 Hz). Two types of isomerism (syn/anti and rac/meso) would be expected for [Pt2Cl2(L3)2(μ-Cl)2]; in view of the 0.4 ppm difference in their δP values, we tentatively assign the two observed 31P NMR signals to syn- and anti-2 (Scheme 6) with the signals expected for the rac and meso isomers unresolved. Mixtures of mononuclear and binuclear products were also reported in the reaction of [PtCl2(cod)] with 2 equiv. CgPPh14 and so the observation of exclusive formation of mononuclear complexes 1a–h appears to be an effect of the 2-pyridyl group. It is tempting to associate this with weak Pt⋯N interactions stabilising the mononuclear complexes although in the solid state at least, no such interactions were detected in complexes 1c–e and 1g (see Fig. 2–5).
 | ||
| Scheme 6 | ||
The ability of 2-pyridylphosphines to switch between P-monodentate and P,N-bidentate coordination has been invoked as part of the explanation for the high activity of Pd-complexes of 2-pyridylphosphines in methoxycarbonylation.3,5,6 It was therefore of interest to investigate if 2-pyridylphosphine L2 could form 4-membered P,N-chelates. Treatment of the trans complex 1a with 1 equiv. of AgBF4 gave a precipitate of AgCl and a solution whose 31P{1H} NMR spectrum was consistent with the formation of the monochelate 3[BF4] (Scheme 7). Two doublets were observed at −48.1 ppm (1JP,Pt = 2728 Hz) and +0.8 ppm (1JP,Pt = 3514 Hz) with a small 2JP,P of 11 Hz typical of cis coordinated phosphines. The signal at −48.1 ppm is assigned to the P,N-chelate since its high field shift is characteristic of a 4-membered chelate.16 The lower value of 1JP,Pt for the chelate-P is consistent with the ring strain being relieved by lengthened Pt–P bonds; the crystal structure of the Pd analogue 5[BF4] (see below) supports this inference.
 | ||
| Scheme 7 | ||
In a similar manner to that described above for the Pt analogues, reaction of [PdCl2(cod)] with 2 equiv. of L2 yielded trans-[PdCl2(L2)2] (4) which has been fully characterised. Treatment of 4 with 1 equiv. of AgBF4 gave a product assigned the structure 5[BF4], the Pd analogue of 3[BF4] (Scheme 8). Crystals of 5[BF4] suitable for X-ray crystallography were obtained and its crystal structure determined (Fig. 6) which confirmed the cis orientation of the two P-donors. The chelate ring strain is apparent from the acute P–Pd–N angle of 69.0°. The Pd–P length within the chelate is longer (by ca. 0.02 Å) than that for the monodentate ligand perhaps reflecting the ring strain.
 | ||
| Scheme 8 | ||
 | ||
| Fig. 6 Crystal structure of 5[BF4]. The [BF4]− counterion, hydrogen atoms and CH2Cl2 solvent molecule are omitted for clarity. Thermal ellipsoids at 50% probability. Selected bond lengths (Å) and angles (°): Pd1–Cl1 2.2989(10), Pd1–P1 2.3170(10), Pd1–P2 2.2779(10), Pd1–N1 2.073(3), P1–C11 1.829(4), P2–C26 1.819(4); P1–Pd1–P2 112.44(3), P1–Pd1–N1 68.95(9), P1–C11–N1 101.9(3), P2–Pd1–Cl1 86.17(4), P1–Pd1–Cl1 161.35(4), P2–Pd1–N1 176.87(9), C1–P1–C4 95.33(17), C16–P2–C19 94.48(16). | ||
The 31P{1H} NMR spectrum of 5[BF4] in CHCl2CHCl2 showed a slightly broadened (w1/2 ∼ 8 Hz) signal at 28.8 ppm, corresponding to the monodentate ligand, and a sharp doublet at −42.7 ppm (2JP,P = 3.2 Hz), assigned to the P,N-chelate. The signals remain distinct but broaden as the temperature is raised (e.g. w1/2 ∼ 70 Hz at 100 °C in CHCl2CHCl2) indicating that cation 5 is fluxional, which is associated with intramolecular interchange of chelating and non-chelating P,N ligands. Iggo et al. have shown that the complex [PdCl{κ1-Ph2P(2-py)}{κ2-Ph2P(2-py)}]BF4 (an analogue of 5[BF4]) is fluxional but with a coalescence temperature for the 31P NMR signals of −30 °C which is at least 150 °C lower than for cation 5.17 The high barrier to exchange in 5 can be rationalised in terms of steric hindrance to Pd–P bond rotation which may be required in order for the pyridyl-nitrogen in the κ1-L2 to coordinate. Energy barriers to M–P bond rotation in complexes containing cis-M(CgPH)2 moieties have previously been shown18 to be of the order of 50 kJ mol−1.
Complex 4 is only sparingly soluble in CD2Cl2 but addition of 1.1 equiv. of TsOH·H2O to a suspension of 4 in CD2Cl2 gave a homogenous yellow solution. The 31P NMR spectrum showed two broad peaks at 6.0 and 3.7 ppm (w1/2 ∼ 57 Hz and 53 Hz respectively), which are tentatively assigned to diastereoisomers of protonated species such as 6 (Scheme 9) with rapid proton exchange rendering the 31P NMR signals equivalent on the NMR timescale. In the 1H NMR spectrum of 6 a broad signal centred at 6.55 ppm is assigned to the exchanging N–H. When a suspension of 4 in MeOH was treated with TsOH, the 31P NMR spectrum of the resulting solution displayed two broad signals at 6.3 and 4.5 ppm (assigned to 6) but in addition, two signals at 28.8 and −42.2 ppm are present, suggesting that the P,N-chelate 5[OTs] is also generated (Scheme 9).
 | ||
| Scheme 9 | ||
The Pd and Pt coordination chemistry of L2 has shown that the ligand can be monodentate (κ1) or chelating (κ2) and the pyridyl-N can be protonated by TsOH. These properties are essential for the catalyst to be able to function in a manner similar to L1 shown in Schemes 1 and 2. Tellingly, the 31P{1H} NMR spectrum of the mixture obtained after catalysis runs involving Pd and L2 showed, amongst other peaks, a prominent signal at −37.8 ppm, reminiscent of the peak observed at −42.1 ppm for the cation 5, suggesting that a four-membered Pd chelate is present.
Conclusions
A series of substituted pyridyl-functionalised phospha-adamantyl ligands L2–7 have been made and fully characterised. It was found that, in some cases, substituents on the pyridyl ring had a marked effect on the rate of Pd-catalysed methoxycarbonylation of phenylacetylene. The 6-methyl substituted ligand L5b showed good activity and selectivity for the branched product albeit lower than that of the well-known Ph2P(2-py) (L1). The catalytic performance is a function of the substituents on the pyridine consistent with the idea that a mechanism involving P,N-chelation and “proton shuttling” may be operating.The Pt and Pd coordination chemistry of L2–7 has been probed and the ability of the ligands to form P,N-chelates has been established in solution from the characteristic 31P NMR spectra and in the solid state by the crystal structure of a palladium complex containing L2 ligands bound in a κ1- and κ2-mode. Moreover protonation of the pyridyl-N in a Pd-L2 complex has been observed in solution. In contrast to the CgP(2-py) ligands, the CgP(3-py) ligand L3 gives binuclear Pt complexes in a similar fashion to the CgPPh.
The methoxycarbonylation activity with catalysts derived from CgP(2-py) and its derivatives demonstrates that the 2-pyridyl effect is not restricted to Ph2P(2-py) and its derivatives. The CgP and Ph2P moieties has very different stereoelectronic effects and therefore the results presented hold out the prospect that other R2P(2-py) ligands may be useful for alkyne methoxycarbonylation catalysis.
Experimental
General procedures
All reactions were carried out under an atmosphere of dry nitrogen, unless otherwise stated, using standard Schlenk line techniques and oven dried (200 °C) glassware. CH2Cl2 and hexane were collected from a Grubbs type solvent purification system, and deoxygenated by bubbling with N2 for 30 minutes. Xylene and CD2Cl2 were dried over activated 4 Å molecular sieves for 72 hours and deoxygenated by successive freeze–pump–thaw cycles. MeOH was purchased as anhydrous, stored over 3 Å molecular sieves and deoxygenated by bubbling with N2 for 30 minutes. Other commercial reagents were used as supplied unless otherwise stated. [PtCl2(cod)],19 [PdCl2(cod)],20 1,3,5,7-tetramethyl-4,6,8-trioxa-2-phospha-adamantane (CgPH),21 CgPPh,14 Ph2P(2-py)22 (L1) 2-bromo-4-(trimethylsilyl)pyridine,23 and 2-bromo-6-(trimethylsilyl)pyridine24 were synthesised according to literature procedures. 1H, 11B, 13C, 19F and 31P NMR spectra were recorded at ambient temperature unless otherwise stated, on Jeol ECP (Eclipse) 300, Jeol ECS 300, Jeol ECS 400, Varian 400-MR, Varian VNMRS 500 spectrometers and a Bruker Avance III HD 500 spectrometer equipped with a 13C-observe (DCH) cryogenic probe. Chemical shifts δ are given in parts per million (ppm) and coupling constants J are in Hz. 1H and 13C chemical shifts were referenced to residual solvent peaks. 11B, 19F and 31P chemical shifts were referenced to BF3·OEt2, CFCl3 and 85% H3PO4 respectively. Mass Spectra were recorded by the University of Bristol Mass Spectrometry Service on VG Analytical Autospec (EI) or VG Analytical Quattro (ESI) spectrometers. Elemental Analysis was carried out by the Microanalytical Laboratory of the School of Chemistry, University of Bristol. X-ray crystallography was performed by the University of Bristol X-ray Analytical Service using Bruker AXS Microstar or Bruker Kappa Apex II diffractometers. Thin Layer Chromatography (TLC) was performed using Merck Kieselgel 60 F254 (Merck) aluminium backed plates (0.25 mm layer of silica). Flash column chromatography was performed using a Biotage Isolera Spektra One Chromatographic Isolation system.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :meso compounds observed in 3:2 ratio. 1H NMR (500 MHz, CD2Cl2): δH 8.76 (d, 3JH,H = 4.9 Hz) and 8.76 (d, 3JH,H = 4.9 Hz) (total 2H, ArH (H-6)), 8.04–8.01 (m, 2H, ArH (H-3)), 7.70–7.64 (m, 2H, ArH (H-4)), 7.32–7.26 (m, 2H, ArH (H-5)), 3.04 (dt, 2JH,H = 13.6 Hz, JH,P = 2.3 Hz) and 2.95 (dt, 2JH,H = 13.6 Hz, JH,P = 2.3 Hz) (total 2H, CgP CH2), 1.98–1.86 (m, 3H, CgP CH2), 1.85 (vir t, JH,P = 6.2 Hz) and 1.80 (vir t, JH,P = 6.2 Hz) (total 6H, CgP CH3), 1.74 (vir t, JH,P = 6.7 Hz) and 1.69 (vir t, JH,P = 6.7 Hz) (total 6H, CgP CH3), 1.66–1.59 (m, 1H, CgP CH2), 1.39 (s) and 1.34 (s) (total 6H, CgP CH3), 1.27 (s) and 1.26 (s) (total 6H, CgP CH3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 154.2 (vir t, JC,P = 154.1 Hz, ArC (C-2)), 149.6 (app q, JC,P = 8.4 Hz, ArC (C-6)), 134.7 (app q, JC,P = 3.3 Hz, ArC (C-4)), 130.7 (app q, JC,P = 8.3 Hz, ArC (C-3)), 123.9 (s, ArC (C-5)), 96.4–96.2 (m, CgP quat. C), 74.9 (vir t, JC,P = 14.1 Hz) and 74.7 (vir t, JC,P = 14.1 Hz) (CgP quat. C), 73.8 (vir t, JC,P = 11.1 Hz) and 73.7 (vir t, JC,P = 11.1 Hz) (CgP quat. C), 42.2 (vir t, JC,P = 3.8 Hz) and 42.1 (vir t, JC,P = 3.8 Hz) (CgP CH2), 41.9–41.8 (m, CgP CH2), 27.9 (s, CgP CH3), 27.7 (s, CgP CH3), 27.6 (br s) and 27.2 (br s) (CgP CH3), 26.0 (vir t, JC,P = 2.0 Hz) and 25.8 (vir t, JC,P = 2.5 Hz) (CgP CH3). 31P{1H} NMR (162 MHz, CD2Cl2): δP −0.6 (s, 1JP,Pt = 2740 Hz) and −1.1 (s, 1JP,Pt = 2727 Hz) (CgP). HRMS (ESI): m/z calc. for C30H40ClN2O6P2Pt [M − Cl]+ = 816.1695; obs. = 816.1690. Elem. Anal. found (calc. for C30H40Cl2N2O6P2Pt): C, 42.64 (42.26); H, 5.13 (4.73); N, 3.44 (3.29).
:meso compounds observed in 1:1 ratio. 1H NMR (500 MHz, CD2Cl2): δH 8.76 (d, 3JH,H = 4.9 Hz) and 8.75 (d, 3JH,H = 4.9 Hz) (total 4H, ArH (H-4 and H-6)), 7.25–7.22 (m, 2H, ArH (H-5)), 3.16 (d of vir t, 2JH,H = 13.7 Hz, JH,P = 1.8 Hz) and 3.13 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.3 Hz) (total 2H, CgP CH2), 2.29 (d, 3JH,H = 13.8 Hz) and 2.21 (d, 3JH,H = 13.8 Hz) (total 2H, CgP CH2), 1.92–1.83 (m, 12H, CgP CH3), 1.82–1.70 (m, 4H, CgP CH3), 1.37 (s) and 1.35 (s) (total 6H, CgP CH3), 1.20 (s, 6H, CgP CH3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 168.0 (vir t, JC,P = 46.1 Hz, ArC (C-2)), 156.2 (vir t, JC,P = 5.6 Hz) and 156.1 (vir t, JC,P = 5.6 Hz) (ArC (C-4 and C-6)), 121.3 (s, ArC (C-3)), 97.0 (s) and 96.96 (s) (CgP quat. C), 96.94 (s) and 96.92 (s) (CgP quat. C), 75.8–75.5 (m, CgP quat. C), 42.9–42.8 (m, CgP CH2), 42.6–42.4 (m, CgP CH2), 27.9 (s, CgP CH3), 27.7 (s) and 27.6 (s) (CgP CH3), 27.4 (vir t, JC,P = 2.4 Hz) and 27.3 (vir t, JC,P = 2.4 Hz) (CgP CH3), 26.7 (br s) and 26.6 (br s) (CgP CH3). 31P{1H} NMR (162 MHz, CD2Cl2): δP −0.8 (s, 1JP,Pt = 2707 Hz) and −0.9 (s, 1JP,Pt = 2706 Hz) (CgP). HRMS (ESI): m/z calc. for C28H38ClN4O6P2Pt [M − Cl]+ = 818.1600; obs. = 818.1606. Elem. Anal. found (calc. for C28H38Cl2N4O6P2Pt): C, 39.66 (39.35); H, 4.57 (4.48); N, 6.34 (6.56).
:meso compounds observed in 3:2 ratio. 1H NMR (500 MHz, CD2Cl2): δH 8.60 (d, 3JH,H = 5.0 Hz) and 8.58 (d, 3JH,H = 5.0 Hz) (total 2H, ArH (H-6)), 7.89 (br s) and 7.86 (br s) (total 2H, ArH (H-3)), 7.14 (d, 3JH,H = 4.7 Hz) and 7.10 (d, 3JH,H = 5.0 Hz) (total 2H, ArH (H-5)), 3.05 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.1 Hz) and 2.95 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.1 Hz) (total 2H, CgP CH2), 2.38 (s) and 2.35 (s) (total 6H, Ar–CH3), 2.05–2.02 (m, 1H, CgP CH2), 1.98–1.88 (m, 2H, CgP CH2), 1.84 (vir t, JH,P = 6.2 Hz) and 1.80 (vir t, JH,P = 6.2 Hz) (total 6H, CgP CH3), 1.73 (vir t, JH,P = 6.6 Hz) and 1.68 (vir t, JH,P = 6.6 Hz) (total 6H, CgP CH3), 1.65–1.56 (m, 3H, CgP CH2), 1.39 (s) and 1.34 (s) (total 6H, CgP CH3), 1.27 (s) and 1.26 (s) (total 6H, CgP CH3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 159.3 (vir t, JC,P = 34.6 Hz, ArC (C-2)), 149.8 (vir t, JC,P = 8.7 Hz) and 149.6 (vir t, JC,P = 8.7 Hz) (ArC (C-6)), 146.9–146.7 (br s, ArC (C-4)), 132.2 (vir t, JC,P = 8.7 Hz) and 132.0 (vir t, JC,P = 8.7 Hz) (ArC (C-3)), 125.6 (s, ArC (C-5)), 96.9 (s, CgP quat. C), 96.8 (s) and 96.7 (s) (CgP quat. C), 75.5 (vir t, JC,P = 14.0 Hz) and 75.3 (vir t, JC,P = 14.0 Hz) (CgP quat. C), 74.4 (vir t, JC,P = 11.1 Hz) and 74.3 (vir t, JC,P = 11.1 Hz) (CgP quat. C), 42.9 (app q, JC,P = 4.1 Hz, CgP CH2), 42.4 (s) and 42.3 (s) (CgP CH2), 27.8 (s, CgP CH3), 27.64 (s) and 27.56 (s) (CgP CH3), 27.5 (br s) and 27.4 (br s) (CgP CH3), 26.1 (vir t, JC,P = 2.2 Hz) and 25.9 (vir t, JC,P = 2.4 Hz) (CgP CH3), 21.7 (s) and 21.6 (s) (Ar–CH3). 31P{1H} NMR (162 MHz, CD2Cl2): δP −0.6 (s, 1JP,Pt = 2738 Hz) and −1.0 (s, 1JP,Pt = 2729 Hz) (CgP). HRMS (ESI): m/z calc. for C32H44ClN2O6P2Pt [M − Cl]+ = 844.2009; obs. = 844.1993. Elem. Anal. found (calc. for C32H44Cl2N2O6P2Pt): C, 43.47 (43.64); H, 5.07 (5.04); N, 3.19 (3.18).
:meso compounds observed in 3:2 ratio. 1H NMR (500 MHz, CD2Cl2): δH 7.87–7.84 (m, 2H, ArH (H-3)), 7.58–7.53 (m, 2H, ArH (H-4)), 7.16–7.12 (m, 2H, ArH (H-5)), 3.09 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.1 Hz) and 2.99 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.1 Hz) (total 2H, CgP CH2), 2.62 (s) and 2.57 (s) (total 6H, Ar–CH3), 2.17 (d, 2JH,H = 13.7 Hz, 1H, CgP CH2), 1.90–1.87 (m, 2H, CgP CH2) 1.85 1.70 (m, 12H, CgP CH3), 1.64–1.60 (m, 3H, CgP CH2), 1.37 (s) and 1.34 (s) (total 6H, CgP CH3), 1.25 (s, 6H, CgP CH3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 159.3 (vir t, JC,P = 8.6 Hz) and 159.0 (vir t, JC,P = 8.6 Hz) (ArC (C-6)), 153.7 (vir t, JC,P = 34.8 Hz) and 153.6 (vir t, JC,P = 35.6 Hz) (ArC (C-2)), 135.4–135.3 (m, ArC (C-4)), 128.7 (vir t, JC,P = 9.3 Hz) and 128.5 (vir t, JC,P = 8.2 Hz) (ArC (C-3)), 124.2 (s) and 124.1 (s) (ArC (C-5)), 97.0 (s) and 96.9 (s) (CgP quat. C), 96.8 (s) and 96.7 (s) (CgP quat. C), 75.6 (vir t, JC,P = 14.0 Hz) and 75.4 (vir t, JC,P = 14.0 Hz) (CgP quat. C), 74.5 (vir t, JC,P = 11.1 Hz) and 74.2 (vir t, JC,P = 11.1 Hz) (CgP quat. C), 42.9 (vir t, JC,P = 3.8 Hz) and 42.7 (vir t, JC,P = 3.8 Hz) (CgP CH2), 42.5 (s) and 42.4 (s) (CgP CH2), 27.9 (s, CgP CH3), 27.8 (s) and 27.6 (s) (CgP CH3), 27.7 (br s) and 27.4 (br s) (CgP CH3), 26.4 (vir t, JC,P = 2.4 Hz) and 26.0 (vir t, JC,P = 2.4 Hz) (CgP CH3), 24.8 (s) and 24.7 (s) (Ar–CH3). 31P{1H} NMR (162 MHz, CD2Cl2): δP −1.2 (s, 1JP,Pt = 2731 Hz) and −1.5 (s, 1JP,Pt = 2723 Hz) (CgP). HRMS (ESI): m/z calc. for C32H44ClN2O6P2Pt [M − Cl]+ = 844.2009; obs. = 844.2012. Elem. Anal. found (calc. for C32H44Cl2N2O6P2Pt): C, 43.73 (43.64); H, 5.12 (5.04); N, 3.24 (3.18).
:meso compounds observed in 3:1 ratio. 1H NMR (500 MHz, CD2Cl2): δH 8.96 (d, 3JH,H = 5.0 Hz) and 8.93 (d, 3JH,H = 5.0 Hz) (total 2H, ArH (H-6)), 8.29 (br s) and 8.26 (br s) (total 2H, ArH (H-3)), 7.53 (d, 3JH,H = 4.4 Hz) and 7.50 (d, 3JH,H = 4.7 Hz) (total 2H, ArH (H-5)), 3.02 (d of vir t, 2JH,H = 13.7 Hz, JH,P = 2.3 Hz) and 2.93 (d of vir t, 2JH,H = 13.7 Hz, JH,P = 2.4 Hz) (total 2H, CgP CH2), 1.98–1.86 (m, 4H, CgP CH2), 1.84–1.71 (m, 12H, CgP CH3), 1.69–1.67 (m, 2H, CgP CH2), 1.41 (s) and 1.36 (s) (total 6H, CgP CH3), 1.28 (s, 6H, CgP CH3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 156.9 (vir t, JC,P = 33.7 Hz, ArC (C-2)), 151.0 (vir t, JC,P = 8.2 Hz, ArC (C-6)), 137.3 (q of vir t, 2JC,F = 33.9 Hz, JC,P = 3.6 Hz, ArC (C-4)), 126.8–126.6 (m, ArC (C-3)), 123.2 (q, 1JC,F = 273.5 Hz, Ar–CF3), 118.4 (q, 3JC,F = 3.5 Hz, ArC (C-5)), 97.0–96.9 (m, CgP quat. C), 75.7 (vir t, JC,P = 14.0 Hz, CgP quat. C), 74.5 (vir t, JC,P = 11.0 Hz, CgP quat. C), 42.6 (vir t, JC,P = 4.0 Hz, CgP CH2), 42.5 (s, CgP CH2), 27.9 (s, CgP CH3), 27.7 (s) and 27.6 (s) (CgP CH3), 27.5 (br s) and 27.4 (br s) (CgP CH3), 26.0 (vir t, JC,P = 2.5 Hz) and 25.9 (vir t, JC,P = 2.5 Hz) (CgP CH3). 19F NMR (377 MHz, CD2Cl2): δF −65.13 (s) and −65.16 (s) (Ar–CF3). 31P{1H} NMR (162 MHz, CD2Cl2): δP 1.2 (s, 1JP,Pt = 2759 Hz) and 0.7 (s, 1JP,Pt = 2750 Hz) (CgP). HRMS (ESI): m/z calc. for C32H38ClF6N2O6P2Pt [M − Cl]+ = 952.1443; obs. = 952.1479. Elem. Anal. found (calc. for C32H38Cl2F6N2O6P2Pt): C, 38.66 (38.88); H, 3.89 (3.87); N, 2.85 (2.83).
:meso compounds observed in 2:1 ratio. 1H NMR (500 MHz, CD2Cl2): δH 8.30 (d, 3JH,H = 8.0 Hz) and 8.25 (d, 3JH,H = 8.0 Hz) (total 2H, ArH (H-3)), 7.90 (t, 3JH,H = 8.0 Hz) and 7.85 (d, 3JH,H = 8.0 Hz) (total 2H, ArH (H-4)), 7.68 (d, 3JH,H = 7.9 Hz) and 7.63 (d, 3JH,H = 7.9 Hz) (total 2H, ArH (H-5)), 3.08 (d of vir t, 2JH,H = 13.7 Hz, JH,P = 2.2 Hz) and 3.01 (d of vir t, 2JH,H = 13.8 Hz, JH,P = 2.2 Hz) (total 2H, CgP CH2), 2.16 (d, 2JH,H = 13.8 Hz) and 2.14 (d, 2JH,H = 13.8 Hz) (total 2H, CgP CH2), 1.95–1.84 (m, 2H, CgP CH2), 1.78–1.68 (m, 14H, CgP CH3 and CH2), 1.41 (s) and 1.36 (s) (6H, CgP CH3), 1.27 (br s, 6H, CgP CH3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 156.1 (vir t, JC,P = 32.0 Hz) and 156.0 (vir t, JC,P = 32.5 Hz) (ArC (C-2)), 148.1 (vir t, JC,P = 7.3 Hz) and 147.8 (vir t, JC,P = 7.8 Hz) (ArC (C-6)), 136.7 (vir t, JC,P = 5.6 Hz, ArC (C-4)), 134.2 (vir t, JC,P = 9.5 Hz, ArC (C-3)), 121.9 (q, 1JC,F = 274.0 Hz) and 121.8 (q, 1JC,F = 274.4 Hz) (Ar–CF3), 121.2 (br s) and 121.1 (br s) (ArC (C-5)), 97.1 (s) and 97.05 (s) (CgP quat. C), 97.03 (s) and 96.9 (s) (CgP quat. C), 75.7 (vir t, JC,P = 14.4 Hz) and 75.6 (vir t, JC,P = 14.4 Hz) (CgP quat. C), 74.8–74.5 (m, CgP quat. C), 42.7–42.6 (m, CgP CH2), 42.4 (s) and 42.3 (s) (CgP CH2), 27.8 (s, CgP CH3), 27.7 (s) and 27.6 (s) (CgP CH3), 27.2 (s) and 27.71 (s) (CgP CH3), 26.2 (br s, CgP CH3). 19F NMR (470 MHz, CD2Cl2): δF −68.4 (s) and −68.5 (s) (Ar–CF3). 31P{1H} NMR (202 MHz, CD2Cl2): δP 0.2 (s, 1JP,Pt = 2756 Hz, CgP). HRMS (ESI): m/z calc. for C32H38Cl2F6N2NaO6P2Pt [M + Na]+ = 1010.1029; obs. = 1010.1027. Elem. Anal. found (calc. for C32H38Cl2F6N2O6P2Pt): C, 38.92 (38.88); H, 4.08 (3.87); N, 2.75 (2.83).
:meso compounds observed in 1:1 ratio. 1H NMR (500 MHz, CD2Cl2): δH 8.69 (d, 3JH,H = 4.7 Hz) and 8.66 (d, 3JH,H = 4.7 Hz) (total 2H, ArH (H-3)), 8.19–8.17 (m) and 8.15–8.13 (m) (total 2H, ArH (H-6)), 7.40 (d, 3JH,H = 4.6 Hz) and 7.36 (d, 3JH,H = 4.7 Hz) (total 2H, ArH (H-5)), 3.07 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.3 Hz) and 2.97 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.3 Hz) (total 2H, CgP CH2), 2.04 (d, 2JH,H = 13.7 Hz) and 1.93 (d, 2JH,H = 13.5 Hz, 2H, CgP CH2), 1.92–1.85 (m, 2H, CgP CH2), 1.82 (vir t, JH,P = 6.2 Hz) and (vir t, JH,P = 6.1 Hz) (total 6H, CgP CH3), 1.72 (vir t, JH,P = 6.6 Hz) and 1.68 (vir t, JH,P = 6.6 Hz) (total 6H, CgP CH3), 1.74–1.67 (m, 6H, CgP CH3), 1.64–1.58 (m, 2H, CgP CH2), 1.40 (s) and 1.34 (s) (total 6H, CgP CH3), 1.26 (s) and 1.25 (s) (total 6H, CgP CH3), 0.30 (s) and 0.28 (s) (total 18H, Si(CH3)3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 153.5 (vir t, JC,P = 33.7 Hz, ArC (C-2)), 149.8–149.7 (m, ArC (C-4)), 149.0–148.8 (m, ArC (C-3)), 135.7–135.5 (m, ArC (C-6)), 128.9 (br s, ArC (C-5)), 96.9–96.7 (m, CgP quat. C), 75.5 (vir t, JC,P = 13.9 Hz) and 75.3 (vir t, JC,P = 13.9 Hz) (CgP quat. C), 74.5–74.2 (m, CgP quat. C), 42.9 (m, CgP CH2), 42.4 (m, CgP CH2), 28.0 (s, CgP CH3), 27.8 (s) and 27.7 (s) (CgP CH3), 27.6 (br s) and 27.5 (br s) (CgP CH3), 26.1 (br s) and 25.9 (br s) (CgP CH3), −1.59 (s) and −1.62 (s) (Si(CH3)3). 31P{1H} NMR (162 MHz, CD2Cl2): δP −0.8 (s, 1JP,Pt = 2728 Hz) and −1.3 (s, 1JP,Pt = 2647 Hz) (CgP). HRMS (ESI): m/z calc. for C36H56ClN2O6P2PtSi2 [M − Cl]+ = 960.2484; obs. = 960.2490. Elem. Anal. found (calc. for C36H56Cl2N2O6P2PtSi2): C, 43.61 (43.37); H, 5.80 (5.66); N, 2.95 (2.81).
:meso compounds observed in 1:1 ratio. 1H NMR (500 MHz, CD2Cl2): δH 7.96 (d, 3JH,H = 8.0 Hz) and 7.88 (d, 3JH,H = 8.0 Hz) (total 2H, ArH (H-3)), 7.60–7.52 (m, 2H, ArH (H-4)), 7.48 (d, 3JH,H = 7.4 Hz) and 7.44 (d, 3JH,H = 7.4 Hz) (total 2H, ArH (H-5)), 3.10–3.04 (m, 2H, CgP CH2), 2.29 (d, 2JH,H = 13.6 Hz) and 2.18 (d, 2JH,H = 13.6 Hz) (total 2H, CgP CH2), 1.87–1.58 (m, 16H, CgP CH3 CH2), 1.36 (s) and 1.33 (s) (total 6H, CgP CH3), 1.26 (s) and 1.25 (s) (total 6H, CgP CH3), 0.33 (s) and 0.32 (s) (total 18H, Si(CH3)3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 169.0 (br s) and 168.8 (br s) (ArC (C-6)), 155.1 (s) and 155.0 (s) (ArC (C-2)), 133.0–132.8 (m, ArC (C-4)), 130.4 (vir t, JC,P = 10.2 Hz) and 130.2 (vir t JC,P = 9.7 Hz) (ArC (C-3)), 128.9 (s) and 128.8 (s) (ArC (C-5)), 96.9–96.8 (m, CgP quat. C), 75.6–75.3 (m) and 74.7–74.3 (m) (CgP quat. C), 42.9 (vir t, JC,P = 3.8 Hz) and 42.8 (vir t, JC,P = 3.9 Hz) (CgP CH2), 42.4 (s) and 42.3 (s) (CgP CH2), 28.0 (s, CgP CH3), 27.8 (s) and 27.7 (s) (CgP CH3), 27.4 (br s) and 27.3 (br s) (CgP CH3), 26.4 (br s) and 26.3 (br s) (CgP CH3), −1.56 (s) and −1.57 (s) (Si(CH3)3). 31P{1H} NMR (202 MHz, CD2Cl2): δP −1.6 (s, 1JP,Pt = 2722 Hz) and −1.9 (s, 1JP,Pt = 2724 Hz) (CgP). HRMS (ESI): m/z calc. for C36H57Cl2N2O6P2PtSi2 [M + H]+ = 996.2251; obs. = 996.2247. Elem. Anal. found (calc. for C36H56Cl2N2O6P2PtSi2): C, 43.54 (43.37); H, 5.77 (5.66); N, 3.18 (2.81).
:meso compounds observed in 1:1 ratio. 1H NMR (500 MHz, CD2Cl2): δH 8.73 (d, 3JH,H = 4.3 Hz) and 8.71 (d, 3JH,H = 4.4 Hz) (total 2H, ArH (H-6)), 8.01 (d, 3JH,H = 4.4 Hz, 2H, ArH (H-3)), 7.70–7.66 (m, 2H, ArH (H-4)), 7.29 (appq, 3JH,H = 4.4 Hz, 2H, ArH (H-5)), 3.04 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.3 Hz) and 2.96 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.3 Hz) (total 2H, CgP CH2), 2.24 (s) and 2.35 (s) (total 1H, CgP CH2), 1.99–1.89 (m, 3H, CgP CH2), 1.84 (vir t, JH,P = 6.3 Hz) and 1.80 (vir t, JH,P = 6.3 Hz) (total 6H, CgP CH3), 1.73 (vir t, JH,P = 6.8 Hz) and 1.70 (vir t, JH,P = 6.6 Hz) (total 6H, CgP CH3), 1.59–1.54 (m, 2H, CgP CH2), 1.38 (s) and 1.35 (s) (total 6H, CgP CH3), 1.27 (s, 6H, CgP CH3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 155.6 (vir t, JC,P = 30.4 Hz) and 156.0 (vir t, JC,P = 29.7 Hz) (ArC (C-2)), 150.1 (app q, JC,P = 8.0 Hz, ArC (C-6)), 135.4 (br s, ArC (C-4)), 131.1 (vir t, JC,P = 8.7 Hz) and 130.1 (vir t, JC,P = 8.2 Hz) (ArC (C 3)), 124.5 (s, ArC (C-5)), 97.0–96.8 (m, CgP quat. C), 76.3 (vir t, JC,P = 10.9 Hz) and 76.2 (vir t, JC,P = 10.8 Hz) (CgP quat. C), 75.0 (vir t, JC,P = 7.1 Hz) and 74.8 (vir t, JC,P = 7.0 Hz) (CgP quat. C), 43.2–43.1 (m, CgP CH2), 42.5–42.4 (m, CgP CH2), 28.2 (br s) and 28.1 (br s) (CgP CH3), 27.8 (s, CgP CH3), 27.73 (s) and 27.68 (s) (CgP CH3), 26.8 (vir t, JC,P = 2.5 Hz) and 26.6 (vir t, JC,P = 2.7 Hz) (CgP CH3). 31P{1H} NMR (162 MHz, CD2Cl2): δP 4.4 (s) and 4.3 (s) (CgP). HRMS (ESI): m/z calc. for C30H40ClN2O6P2Pd [M − Cl]+ = 727.1088; obs. = 727.1103. Elem. Anal. found (calc. for C30H40Cl2N2O6P2Pd): C, 47.16 (47.17); H, 5.29 (5.28); N, 3.85 (3.67).
:meso compounds observed in 3:2 ratio. 1H NMR (500 MHz, CD2Cl2): δH 8.76 (d, 3JH,H = 4.9 Hz) and 8.76 (d, 3JH,H = 4.9 Hz) (total 2H, ArH (H-6)), 8.04–8.01 (m, 2H, ArH (H-3)), 7.70–7.64 (m, 2H, ArH (H-4)), 7.32–7.26 (m, 2H, ArH (H-5)), 3.04 (dt, 2JH,H = 13.6 Hz, JH,P = 2.3 Hz) and 2.95 (dt, 2JH,H = 13.6 Hz, JH,P = 2.3 Hz) (total 2H, CgP CH2), 1.98–1.86 (m, 3H, CgP CH2), 1.85 (vir t, JH,P = 6.2 Hz) and 1.80 (vir t, JH,P = 6.2 Hz) (total 6H, CgP CH3), 1.74 (vir t, JH,P = 6.7 Hz) and 1.69 (vir t, JH,P = 6.7 Hz) (total 6H, CgP CH3), 1.66–1.59 (m, 1H, CgP CH2), 1.39 (s) and 1.34 (s) (total 6H, CgP CH3), 1.27 (s) and 1.26 (s) (total 6H, CgP CH3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 154.2 (vir t, JC,P = 154.1 Hz, ArC (C-2)), 149.6 (app q, JC,P = 8.4 Hz, ArC (C-6)), 134.7 (app q, JC,P = 3.3 Hz, ArC (C-4)), 130.7 (app q, JC,P = 8.3 Hz, ArC (C-3)), 123.9 (s, ArC (C-5)), 96.4–96.2 (m, CgP quat. C), 74.9 (vir t, JC,P = 14.1 Hz) and 74.7 (vir t, JC,P = 14.1 Hz) (CgP quat. C), 73.8 (vir t, JC,P = 11.1 Hz) and 73.7 (vir t, JC,P = 11.1 Hz) (CgP quat. C), 42.2 (vir t, JC,P = 3.8 Hz) and 42.1 (vir t, JC,P = 3.8 Hz) (CgP CH2), 41.9–41.8 (m, CgP CH2), 27.9 (s, CgP CH3), 27.7 (s, CgP CH3), 27.6 (br s) and 27.2 (br s) (CgP CH3), 26.0 (vir t, JC,P = 2.0 Hz) and 25.8 (vir t, JC,P = 2.5 Hz) (CgP CH3). 31P{1H} NMR (162 MHz, CD2Cl2): δP −0.6 (s, 1JP,Pt = 2740 Hz) and −1.1 (s, 1JP,Pt = 2727 Hz) (CgP). HRMS (ESI): m/z calc. for C30H40ClN2O6P2Pt [M − Cl]+ = 816.1695; obs. = 816.1690. Elem. Anal. found (calc. for C30H40Cl2N2O6P2Pt): C, 42.64 (42.26); H, 5.13 (4.73); N, 3.44 (3.29).
:meso compounds observed in 1:1 ratio. 1H NMR (500 MHz, CD2Cl2): δH 8.76 (d, 3JH,H = 4.9 Hz) and 8.75 (d, 3JH,H = 4.9 Hz) (total 4H, ArH (H-4 and H-6)), 7.25–7.22 (m, 2H, ArH (H-5)), 3.16 (d of vir t, 2JH,H = 13.7 Hz, JH,P = 1.8 Hz) and 3.13 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.3 Hz) (total 2H, CgP CH2), 2.29 (d, 3JH,H = 13.8 Hz) and 2.21 (d, 3JH,H = 13.8 Hz) (total 2H, CgP CH2), 1.92–1.83 (m, 12H, CgP CH3), 1.82–1.70 (m, 4H, CgP CH3), 1.37 (s) and 1.35 (s) (total 6H, CgP CH3), 1.20 (s, 6H, CgP CH3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 168.0 (vir t, JC,P = 46.1 Hz, ArC (C-2)), 156.2 (vir t, JC,P = 5.6 Hz) and 156.1 (vir t, JC,P = 5.6 Hz) (ArC (C-4 and C-6)), 121.3 (s, ArC (C-3)), 97.0 (s) and 96.96 (s) (CgP quat. C), 96.94 (s) and 96.92 (s) (CgP quat. C), 75.8–75.5 (m, CgP quat. C), 42.9–42.8 (m, CgP CH2), 42.6–42.4 (m, CgP CH2), 27.9 (s, CgP CH3), 27.7 (s) and 27.6 (s) (CgP CH3), 27.4 (vir t, JC,P = 2.4 Hz) and 27.3 (vir t, JC,P = 2.4 Hz) (CgP CH3), 26.7 (br s) and 26.6 (br s) (CgP CH3). 31P{1H} NMR (162 MHz, CD2Cl2): δP −0.8 (s, 1JP,Pt = 2707 Hz) and −0.9 (s, 1JP,Pt = 2706 Hz) (CgP). HRMS (ESI): m/z calc. for C28H38ClN4O6P2Pt [M − Cl]+ = 818.1600; obs. = 818.1606. Elem. Anal. found (calc. for C28H38Cl2N4O6P2Pt): C, 39.66 (39.35); H, 4.57 (4.48); N, 6.34 (6.56).
:meso compounds observed in 3:2 ratio. 1H NMR (500 MHz, CD2Cl2): δH 8.60 (d, 3JH,H = 5.0 Hz) and 8.58 (d, 3JH,H = 5.0 Hz) (total 2H, ArH (H-6)), 7.89 (br s) and 7.86 (br s) (total 2H, ArH (H-3)), 7.14 (d, 3JH,H = 4.7 Hz) and 7.10 (d, 3JH,H = 5.0 Hz) (total 2H, ArH (H-5)), 3.05 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.1 Hz) and 2.95 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.1 Hz) (total 2H, CgP CH2), 2.38 (s) and 2.35 (s) (total 6H, Ar–CH3), 2.05–2.02 (m, 1H, CgP CH2), 1.98–1.88 (m, 2H, CgP CH2), 1.84 (vir t, JH,P = 6.2 Hz) and 1.80 (vir t, JH,P = 6.2 Hz) (total 6H, CgP CH3), 1.73 (vir t, JH,P = 6.6 Hz) and 1.68 (vir t, JH,P = 6.6 Hz) (total 6H, CgP CH3), 1.65–1.56 (m, 3H, CgP CH2), 1.39 (s) and 1.34 (s) (total 6H, CgP CH3), 1.27 (s) and 1.26 (s) (total 6H, CgP CH3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 159.3 (vir t, JC,P = 34.6 Hz, ArC (C-2)), 149.8 (vir t, JC,P = 8.7 Hz) and 149.6 (vir t, JC,P = 8.7 Hz) (ArC (C-6)), 146.9–146.7 (br s, ArC (C-4)), 132.2 (vir t, JC,P = 8.7 Hz) and 132.0 (vir t, JC,P = 8.7 Hz) (ArC (C-3)), 125.6 (s, ArC (C-5)), 96.9 (s, CgP quat. C), 96.8 (s) and 96.7 (s) (CgP quat. C), 75.5 (vir t, JC,P = 14.0 Hz) and 75.3 (vir t, JC,P = 14.0 Hz) (CgP quat. C), 74.4 (vir t, JC,P = 11.1 Hz) and 74.3 (vir t, JC,P = 11.1 Hz) (CgP quat. C), 42.9 (app q, JC,P = 4.1 Hz, CgP CH2), 42.4 (s) and 42.3 (s) (CgP CH2), 27.8 (s, CgP CH3), 27.64 (s) and 27.56 (s) (CgP CH3), 27.5 (br s) and 27.4 (br s) (CgP CH3), 26.1 (vir t, JC,P = 2.2 Hz) and 25.9 (vir t, JC,P = 2.4 Hz) (CgP CH3), 21.7 (s) and 21.6 (s) (Ar–CH3). 31P{1H} NMR (162 MHz, CD2Cl2): δP −0.6 (s, 1JP,Pt = 2738 Hz) and −1.0 (s, 1JP,Pt = 2729 Hz) (CgP). HRMS (ESI): m/z calc. for C32H44ClN2O6P2Pt [M − Cl]+ = 844.2009; obs. = 844.1993. Elem. Anal. found (calc. for C32H44Cl2N2O6P2Pt): C, 43.47 (43.64); H, 5.07 (5.04); N, 3.19 (3.18).
:meso compounds observed in 3:2 ratio. 1H NMR (500 MHz, CD2Cl2): δH 7.87–7.84 (m, 2H, ArH (H-3)), 7.58–7.53 (m, 2H, ArH (H-4)), 7.16–7.12 (m, 2H, ArH (H-5)), 3.09 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.1 Hz) and 2.99 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.1 Hz) (total 2H, CgP CH2), 2.62 (s) and 2.57 (s) (total 6H, Ar–CH3), 2.17 (d, 2JH,H = 13.7 Hz, 1H, CgP CH2), 1.90–1.87 (m, 2H, CgP CH2) 1.85 1.70 (m, 12H, CgP CH3), 1.64–1.60 (m, 3H, CgP CH2), 1.37 (s) and 1.34 (s) (total 6H, CgP CH3), 1.25 (s, 6H, CgP CH3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 159.3 (vir t, JC,P = 8.6 Hz) and 159.0 (vir t, JC,P = 8.6 Hz) (ArC (C-6)), 153.7 (vir t, JC,P = 34.8 Hz) and 153.6 (vir t, JC,P = 35.6 Hz) (ArC (C-2)), 135.4–135.3 (m, ArC (C-4)), 128.7 (vir t, JC,P = 9.3 Hz) and 128.5 (vir t, JC,P = 8.2 Hz) (ArC (C-3)), 124.2 (s) and 124.1 (s) (ArC (C-5)), 97.0 (s) and 96.9 (s) (CgP quat. C), 96.8 (s) and 96.7 (s) (CgP quat. C), 75.6 (vir t, JC,P = 14.0 Hz) and 75.4 (vir t, JC,P = 14.0 Hz) (CgP quat. C), 74.5 (vir t, JC,P = 11.1 Hz) and 74.2 (vir t, JC,P = 11.1 Hz) (CgP quat. C), 42.9 (vir t, JC,P = 3.8 Hz) and 42.7 (vir t, JC,P = 3.8 Hz) (CgP CH2), 42.5 (s) and 42.4 (s) (CgP CH2), 27.9 (s, CgP CH3), 27.8 (s) and 27.6 (s) (CgP CH3), 27.7 (br s) and 27.4 (br s) (CgP CH3), 26.4 (vir t, JC,P = 2.4 Hz) and 26.0 (vir t, JC,P = 2.4 Hz) (CgP CH3), 24.8 (s) and 24.7 (s) (Ar–CH3). 31P{1H} NMR (162 MHz, CD2Cl2): δP −1.2 (s, 1JP,Pt = 2731 Hz) and −1.5 (s, 1JP,Pt = 2723 Hz) (CgP). HRMS (ESI): m/z calc. for C32H44ClN2O6P2Pt [M − Cl]+ = 844.2009; obs. = 844.2012. Elem. Anal. found (calc. for C32H44Cl2N2O6P2Pt): C, 43.73 (43.64); H, 5.12 (5.04); N, 3.24 (3.18).
:meso compounds observed in 3:1 ratio. 1H NMR (500 MHz, CD2Cl2): δH 8.96 (d, 3JH,H = 5.0 Hz) and 8.93 (d, 3JH,H = 5.0 Hz) (total 2H, ArH (H-6)), 8.29 (br s) and 8.26 (br s) (total 2H, ArH (H-3)), 7.53 (d, 3JH,H = 4.4 Hz) and 7.50 (d, 3JH,H = 4.7 Hz) (total 2H, ArH (H-5)), 3.02 (d of vir t, 2JH,H = 13.7 Hz, JH,P = 2.3 Hz) and 2.93 (d of vir t, 2JH,H = 13.7 Hz, JH,P = 2.4 Hz) (total 2H, CgP CH2), 1.98–1.86 (m, 4H, CgP CH2), 1.84–1.71 (m, 12H, CgP CH3), 1.69–1.67 (m, 2H, CgP CH2), 1.41 (s) and 1.36 (s) (total 6H, CgP CH3), 1.28 (s, 6H, CgP CH3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 156.9 (vir t, JC,P = 33.7 Hz, ArC (C-2)), 151.0 (vir t, JC,P = 8.2 Hz, ArC (C-6)), 137.3 (q of vir t, 2JC,F = 33.9 Hz, JC,P = 3.6 Hz, ArC (C-4)), 126.8–126.6 (m, ArC (C-3)), 123.2 (q, 1JC,F = 273.5 Hz, Ar–CF3), 118.4 (q, 3JC,F = 3.5 Hz, ArC (C-5)), 97.0–96.9 (m, CgP quat. C), 75.7 (vir t, JC,P = 14.0 Hz, CgP quat. C), 74.5 (vir t, JC,P = 11.0 Hz, CgP quat. C), 42.6 (vir t, JC,P = 4.0 Hz, CgP CH2), 42.5 (s, CgP CH2), 27.9 (s, CgP CH3), 27.7 (s) and 27.6 (s) (CgP CH3), 27.5 (br s) and 27.4 (br s) (CgP CH3), 26.0 (vir t, JC,P = 2.5 Hz) and 25.9 (vir t, JC,P = 2.5 Hz) (CgP CH3). 19F NMR (377 MHz, CD2Cl2): δF −65.13 (s) and −65.16 (s) (Ar–CF3). 31P{1H} NMR (162 MHz, CD2Cl2): δP 1.2 (s, 1JP,Pt = 2759 Hz) and 0.7 (s, 1JP,Pt = 2750 Hz) (CgP). HRMS (ESI): m/z calc. for C32H38ClF6N2O6P2Pt [M − Cl]+ = 952.1443; obs. = 952.1479. Elem. Anal. found (calc. for C32H38Cl2F6N2O6P2Pt): C, 38.66 (38.88); H, 3.89 (3.87); N, 2.85 (2.83).
:meso compounds observed in 2:1 ratio. 1H NMR (500 MHz, CD2Cl2): δH 8.30 (d, 3JH,H = 8.0 Hz) and 8.25 (d, 3JH,H = 8.0 Hz) (total 2H, ArH (H-3)), 7.90 (t, 3JH,H = 8.0 Hz) and 7.85 (d, 3JH,H = 8.0 Hz) (total 2H, ArH (H-4)), 7.68 (d, 3JH,H = 7.9 Hz) and 7.63 (d, 3JH,H = 7.9 Hz) (total 2H, ArH (H-5)), 3.08 (d of vir t, 2JH,H = 13.7 Hz, JH,P = 2.2 Hz) and 3.01 (d of vir t, 2JH,H = 13.8 Hz, JH,P = 2.2 Hz) (total 2H, CgP CH2), 2.16 (d, 2JH,H = 13.8 Hz) and 2.14 (d, 2JH,H = 13.8 Hz) (total 2H, CgP CH2), 1.95–1.84 (m, 2H, CgP CH2), 1.78–1.68 (m, 14H, CgP CH3 and CH2), 1.41 (s) and 1.36 (s) (6H, CgP CH3), 1.27 (br s, 6H, CgP CH3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 156.1 (vir t, JC,P = 32.0 Hz) and 156.0 (vir t, JC,P = 32.5 Hz) (ArC (C-2)), 148.1 (vir t, JC,P = 7.3 Hz) and 147.8 (vir t, JC,P = 7.8 Hz) (ArC (C-6)), 136.7 (vir t, JC,P = 5.6 Hz, ArC (C-4)), 134.2 (vir t, JC,P = 9.5 Hz, ArC (C-3)), 121.9 (q, 1JC,F = 274.0 Hz) and 121.8 (q, 1JC,F = 274.4 Hz) (Ar–CF3), 121.2 (br s) and 121.1 (br s) (ArC (C-5)), 97.1 (s) and 97.05 (s) (CgP quat. C), 97.03 (s) and 96.9 (s) (CgP quat. C), 75.7 (vir t, JC,P = 14.4 Hz) and 75.6 (vir t, JC,P = 14.4 Hz) (CgP quat. C), 74.8–74.5 (m, CgP quat. C), 42.7–42.6 (m, CgP CH2), 42.4 (s) and 42.3 (s) (CgP CH2), 27.8 (s, CgP CH3), 27.7 (s) and 27.6 (s) (CgP CH3), 27.2 (s) and 27.71 (s) (CgP CH3), 26.2 (br s, CgP CH3). 19F NMR (470 MHz, CD2Cl2): δF −68.4 (s) and −68.5 (s) (Ar–CF3). 31P{1H} NMR (202 MHz, CD2Cl2): δP 0.2 (s, 1JP,Pt = 2756 Hz, CgP). HRMS (ESI): m/z calc. for C32H38Cl2F6N2NaO6P2Pt [M + Na]+ = 1010.1029; obs. = 1010.1027. Elem. Anal. found (calc. for C32H38Cl2F6N2O6P2Pt): C, 38.92 (38.88); H, 4.08 (3.87); N, 2.75 (2.83).
:meso compounds observed in 1:1 ratio. 1H NMR (500 MHz, CD2Cl2): δH 8.69 (d, 3JH,H = 4.7 Hz) and 8.66 (d, 3JH,H = 4.7 Hz) (total 2H, ArH (H-3)), 8.19–8.17 (m) and 8.15–8.13 (m) (total 2H, ArH (H-6)), 7.40 (d, 3JH,H = 4.6 Hz) and 7.36 (d, 3JH,H = 4.7 Hz) (total 2H, ArH (H-5)), 3.07 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.3 Hz) and 2.97 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.3 Hz) (total 2H, CgP CH2), 2.04 (d, 2JH,H = 13.7 Hz) and 1.93 (d, 2JH,H = 13.5 Hz, 2H, CgP CH2), 1.92–1.85 (m, 2H, CgP CH2), 1.82 (vir t, JH,P = 6.2 Hz) and (vir t, JH,P = 6.1 Hz) (total 6H, CgP CH3), 1.72 (vir t, JH,P = 6.6 Hz) and 1.68 (vir t, JH,P = 6.6 Hz) (total 6H, CgP CH3), 1.74–1.67 (m, 6H, CgP CH3), 1.64–1.58 (m, 2H, CgP CH2), 1.40 (s) and 1.34 (s) (total 6H, CgP CH3), 1.26 (s) and 1.25 (s) (total 6H, CgP CH3), 0.30 (s) and 0.28 (s) (total 18H, Si(CH3)3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 153.5 (vir t, JC,P = 33.7 Hz, ArC (C-2)), 149.8–149.7 (m, ArC (C-4)), 149.0–148.8 (m, ArC (C-3)), 135.7–135.5 (m, ArC (C-6)), 128.9 (br s, ArC (C-5)), 96.9–96.7 (m, CgP quat. C), 75.5 (vir t, JC,P = 13.9 Hz) and 75.3 (vir t, JC,P = 13.9 Hz) (CgP quat. C), 74.5–74.2 (m, CgP quat. C), 42.9 (m, CgP CH2), 42.4 (m, CgP CH2), 28.0 (s, CgP CH3), 27.8 (s) and 27.7 (s) (CgP CH3), 27.6 (br s) and 27.5 (br s) (CgP CH3), 26.1 (br s) and 25.9 (br s) (CgP CH3), −1.59 (s) and −1.62 (s) (Si(CH3)3). 31P{1H} NMR (162 MHz, CD2Cl2): δP −0.8 (s, 1JP,Pt = 2728 Hz) and −1.3 (s, 1JP,Pt = 2647 Hz) (CgP). HRMS (ESI): m/z calc. for C36H56ClN2O6P2PtSi2 [M − Cl]+ = 960.2484; obs. = 960.2490. Elem. Anal. found (calc. for C36H56Cl2N2O6P2PtSi2): C, 43.61 (43.37); H, 5.80 (5.66); N, 2.95 (2.81).
:meso compounds observed in 1:1 ratio. 1H NMR (500 MHz, CD2Cl2): δH 7.96 (d, 3JH,H = 8.0 Hz) and 7.88 (d, 3JH,H = 8.0 Hz) (total 2H, ArH (H-3)), 7.60–7.52 (m, 2H, ArH (H-4)), 7.48 (d, 3JH,H = 7.4 Hz) and 7.44 (d, 3JH,H = 7.4 Hz) (total 2H, ArH (H-5)), 3.10–3.04 (m, 2H, CgP CH2), 2.29 (d, 2JH,H = 13.6 Hz) and 2.18 (d, 2JH,H = 13.6 Hz) (total 2H, CgP CH2), 1.87–1.58 (m, 16H, CgP CH3 CH2), 1.36 (s) and 1.33 (s) (total 6H, CgP CH3), 1.26 (s) and 1.25 (s) (total 6H, CgP CH3), 0.33 (s) and 0.32 (s) (total 18H, Si(CH3)3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 169.0 (br s) and 168.8 (br s) (ArC (C-6)), 155.1 (s) and 155.0 (s) (ArC (C-2)), 133.0–132.8 (m, ArC (C-4)), 130.4 (vir t, JC,P = 10.2 Hz) and 130.2 (vir t JC,P = 9.7 Hz) (ArC (C-3)), 128.9 (s) and 128.8 (s) (ArC (C-5)), 96.9–96.8 (m, CgP quat. C), 75.6–75.3 (m) and 74.7–74.3 (m) (CgP quat. C), 42.9 (vir t, JC,P = 3.8 Hz) and 42.8 (vir t, JC,P = 3.9 Hz) (CgP CH2), 42.4 (s) and 42.3 (s) (CgP CH2), 28.0 (s, CgP CH3), 27.8 (s) and 27.7 (s) (CgP CH3), 27.4 (br s) and 27.3 (br s) (CgP CH3), 26.4 (br s) and 26.3 (br s) (CgP CH3), −1.56 (s) and −1.57 (s) (Si(CH3)3). 31P{1H} NMR (202 MHz, CD2Cl2): δP −1.6 (s, 1JP,Pt = 2722 Hz) and −1.9 (s, 1JP,Pt = 2724 Hz) (CgP). HRMS (ESI): m/z calc. for C36H57Cl2N2O6P2PtSi2 [M + H]+ = 996.2251; obs. = 996.2247. Elem. Anal. found (calc. for C36H56Cl2N2O6P2PtSi2): C, 43.54 (43.37); H, 5.77 (5.66); N, 3.18 (2.81).
:meso compounds observed in 1:1 ratio. 1H NMR (500 MHz, CD2Cl2): δH 8.73 (d, 3JH,H = 4.3 Hz) and 8.71 (d, 3JH,H = 4.4 Hz) (total 2H, ArH (H-6)), 8.01 (d, 3JH,H = 4.4 Hz, 2H, ArH (H-3)), 7.70–7.66 (m, 2H, ArH (H-4)), 7.29 (appq, 3JH,H = 4.4 Hz, 2H, ArH (H-5)), 3.04 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.3 Hz) and 2.96 (d of vir t, 2JH,H = 13.6 Hz, JH,P = 2.3 Hz) (total 2H, CgP CH2), 2.24 (s) and 2.35 (s) (total 1H, CgP CH2), 1.99–1.89 (m, 3H, CgP CH2), 1.84 (vir t, JH,P = 6.3 Hz) and 1.80 (vir t, JH,P = 6.3 Hz) (total 6H, CgP CH3), 1.73 (vir t, JH,P = 6.8 Hz) and 1.70 (vir t, JH,P = 6.6 Hz) (total 6H, CgP CH3), 1.59–1.54 (m, 2H, CgP CH2), 1.38 (s) and 1.35 (s) (total 6H, CgP CH3), 1.27 (s, 6H, CgP CH3). 13C{1H} NMR (126 MHz, CD2Cl2): δC 155.6 (vir t, JC,P = 30.4 Hz) and 156.0 (vir t, JC,P = 29.7 Hz) (ArC (C-2)), 150.1 (app q, JC,P = 8.0 Hz, ArC (C-6)), 135.4 (br s, ArC (C-4)), 131.1 (vir t, JC,P = 8.7 Hz) and 130.1 (vir t, JC,P = 8.2 Hz) (ArC (C 3)), 124.5 (s, ArC (C-5)), 97.0–96.8 (m, CgP quat. C), 76.3 (vir t, JC,P = 10.9 Hz) and 76.2 (vir t, JC,P = 10.8 Hz) (CgP quat. C), 75.0 (vir t, JC,P = 7.1 Hz) and 74.8 (vir t, JC,P = 7.0 Hz) (CgP quat. C), 43.2–43.1 (m, CgP CH2), 42.5–42.4 (m, CgP CH2), 28.2 (br s) and 28.1 (br s) (CgP CH3), 27.8 (s, CgP CH3), 27.73 (s) and 27.68 (s) (CgP CH3), 26.8 (vir t, JC,P = 2.5 Hz) and 26.6 (vir t, JC,P = 2.7 Hz) (CgP CH3). 31P{1H} NMR (162 MHz, CD2Cl2): δP 4.4 (s) and 4.3 (s) (CgP). HRMS (ESI): m/z calc. for C30H40ClN2O6P2Pd [M − Cl]+ = 727.1088; obs. = 727.1103. Elem. Anal. found (calc. for C30H40Cl2N2O6P2Pd): C, 47.16 (47.17); H, 5.29 (5.28); N, 3.85 (3.67).
Protonation studies
TsOH·H2O (0.003 g, 0.015 mmol) was added to a suspension of 4 (0.010 g, 0.013 mmol) in CD2Cl2 (0.7 cm3), upon which the solution became homogeneous, yielding species assigned to the protonated species 6. 31P{1H} NMR (121 MHz, CD2Cl2): δP 6.0 (br s, CgP) and 3.7 (br s, CgP). An analogous procedure in MeOH (0.7 cm3) also gave a homogenous solution, assigned to be a mixture of 6 and a P,N-chelate species related to 5[OTs]. 31P{1H} NMR (121 MHz, CD2Cl2): δP 28.8 (br s, monodentate CgP), 6.3 (br s, CgP) and 4.5 (br s, CgP), −42.2 (br s, chelate CgP).Phenylacetylene methoxycarbonylation
Adapted from previously reported procedure.9 Catalysis was performed using a Baskerville Multi-Cell autoclave. The ligand (0.11 mmol) was added to the autoclave and the system put under an atmosphere of N2. Solutions of Pd(OAc)2 (0.0055 mmol) in MeOH (0.5 cm3) and TsOH·H2O (0.22 mmol) in MeOH (0.5 cm3) were then added, followed by phenylacetylene (5.5 mmol). This was then washed in using MeOH (0.5 cm3) and the autoclave flushed with three cycles of CO (ca. 10 bar). The autoclave was then pressurised to 45 bar and heated to 60 °C. After 1 hour or 4.5 hours, the autoclave was transferred to an ice bath and once cooled, the system was vented. A small amount of each sample was dissolved in CDCl3 and analysed by 1H NMR spectroscopy. Conversion and selectivity was determined by integration of the phenylacetylene alkynyl proton (δH 3.10 ppm) and the methyl atropate (δH 6.38 and 5.90 ppm) and methyl cinnamate (δH 7.71 and 6.42 ppm) alkenyl protons.X-ray crystallography
All of the X-ray diffraction data were collected at 100 K on a Bruker Apex II diffractometer with CCD area detector using Mo-Kα radiation (λ = 0.71073 Å). Absorption corrections were carried out using SADABS.25 All of the structures were solved using Superflip26,27 and refined by full matrix least squares on F2 using ShelXL28,29 within Olex2.30 The structure of meso-1g displayed disorder in the cage, the occupancies of the disordered atoms were refined with the sum of the occupancies set to 1 before being fixed at the refined values. Restraints were applied to the bond lengths and the thermal parameters of pairs of disordered atoms on almost the same site were constrained to be equal. The structure of 5 was twinned and refined as a 2-component twin. In addition, the BF4− counterion displayed disorder in the fluorine positions, the sum of the occupancies were set to equal 1 and refined before being fixed at the refined values. Restraints were applied to maintain sensible thermal parameters and B–F distances. Crystal structure and refinement details are given in Tables S1–S3 in ESI.† Crystallographic data for the compounds have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication, CCDC 1497885–1497898.Acknowledgements
The Bristol Chemical Synthesis Centre for Doctoral Training (BCS CDT) funded by the Engineering and Physical Sciences Research Council (EPSRC) (EP/G036764/1) and the University of Bristol are thanked for a PhD studentship (to T. A. S.).References
- G. Kiss, Chem. Rev., 2001, 101, 3435–3456 CrossRef CAS PubMed.
- (a) A. Brennführer, H. Neumann and M. Beller, ChemCatChem, 2009, 1, 28–41 CrossRef and references therein; (b) P. Kalck and M. Urrutigoïty, Inorg. Chim. Acta, 2015, 431, 110–121 CrossRef CAS and references therein.
- (a) E. Drent, P. Arnoldy and P. H. M. Budzelaar, J. Organomet. Chem., 1993, 455, 247–253 CrossRef CAS; (b) E. Drent, P. Arnoldy and P. H. M. Budzelaar, J. Organomet. Chem., 1994, 475, 57–63 CrossRef CAS; (c) E. Drent and D. H. L. Pello, World Patent, WO95/03269, 1995 Search PubMed; (d) E. Drent, W. W. Jager and J. C. L. J. Suykerbuyk, World Patent, WO1995005357, 1995 Search PubMed; (e) E. Drent and W. W. Jager, US Patent, US5719313, 1998 Search PubMed.
- A. Viña and E. Murillo, J. Braz. Chem. Soc., 2003, 14, 744–749 CrossRef.
- (a) L. Crawford, D. J. Cole-Hamilton, E. Drent and M. Bühl, Chem. – Eur. J., 2014, 20, 13923–13926 CrossRef CAS PubMed; (b) L. Crawford, D. J. Cole-Hamilton and M. Bühl, Organometallics, 2015, 34, 438–449 CrossRef CAS.
- A. Scrivanti, V. Beghetto, E. Campagna, M. Zanato and U. Matteoli, Organometallics, 1998, 17, 630–635 CrossRef CAS.
- M. T. Reetz, R. Demuth and R. Goddard, Tetrahedron Lett., 1998, 39, 7089–7092 CrossRef CAS.
- A. Scrivanti, U. Matteoli, V. Beghetto, S. Antonaroli, R. Scarpelli and B. Crociani, J. Mol. Catal. A: Chem., 2001, 170, 51–56 CrossRef CAS.
- M. L. Clarke, D. J. Cole-Hamilton, D. F. Foster, A. M. Z. Slawin and J. D. Woollins, J. Chem. Soc., Dalton Trans., 2002, 1618–1624 RSC.
- (a) A. Scrivanti, V. Beghetto, M. Zanato and U. Matteoli, J. Mol. Catal. A: Chem., 2000, 160, 331–336 CrossRef CAS; (b) A. Scrivanti, V. Beghetto, E. Campagna and U. Matteoli, J. Mol. Catal. A: Chem., 2001, 168, 75–80 CrossRef CAS.
- L. Bettucci, C. Bianchini, W. Oberhauser, M. Vogt and H. Grützmacher, Dalton Trans., 2010, 39, 6509–6517 RSC.
- Propyne methoxycarbonylation catalysts based on RR′P(2-py) where R or R′ ≠ Ph have been claimed in patents: (a) E. Drent and P. H. M Budzelaar, Eur. Patent, EP0386834B1, 1993 Search PubMed; (b) E. Drent, US Patent, US005166411A, 1992 Search PubMed.
- (a) P. G. Pringle and M. B. Smith, in Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, John Wiley & Sons, Ltd, Chichester, UK, 2012 Search PubMed; (b) J. C. L. J. Suykerbuyk, E. Drent and P. G. Pringle, World Patent, WO9842717, 1998 Search PubMed; (c) R. I. Pugh, P. G. Pringle and E. Drent, Chem. Commun., 2001, 1476–1477 RSC; (d) G. R. Eastham, World Patent, WO2004024322, 2004 Search PubMed; (e) G. R. Eastham and N. Tindale, World Patent, WO2005079981, 2005 Search PubMed; (f) G. R. Eastham, World Patent, WO2005118519, 2005 Search PubMed; (g) M. Slany, M. Schäfer and M. Röper, German Patent, DE10228293, 2001 Search PubMed; (h) M. Schäfer, M. Slany, E. Zeller, M. Röper, M. Schultz and G. Grimm, World Patent, WO0226382, 2002 Search PubMed; (i) E. Drent, P. G. Pringle and R. I. Pugh, World Patent, WO0128972, 2001 Search PubMed.
- R. A. Baber, M. L. Clarke, K. M. Heslop, A. C. Marr, A. G. Orpen, P. G. Pringle, A. Ward and D. E. Zambrano-Williams, Dalton Trans., 2005, 1079–1085 RSC.
- T. Brenstrum, D. A. Gerristma, G. M. Adjabeng, C. S. Frampton, J. Britten, A. J. Robertson, J. McNulty and A. Capretta, J. Org. Chem., 2004, 69, 7635–7639 CrossRef CAS PubMed.
- V. K. Jain, V. S. Jakkal and R. Bohra, J. Organomet. Chem., 1990, 389, 417–426 CrossRef CAS.
- J. Liu, C. Jacob, K. J. Sheridan, F. Al-Mosule, B. T. Heaton, J. A. Iggo, M. Matthews, J. Pelletier, R. Whyman, J. F. Bickley and A. Steiner, Dalton Trans., 2010, 39, 7921–7935 RSC.
- P. Jankowski, C. L. McMullin, I. D. Gridnev, A. G. Orpen and P. G. Pringle, Tetrahedron: Asymmetry, 2010, 21, 1206–1209 CrossRef CAS.
- J. X. McDermott, J. F. White and G. M. Whitesides, J. Am. Chem. Soc., 1976, 98, 6521–6528 CrossRef CAS.
- C. T. Bailey and G. C. Lisensky, J. Chem. Educ., 1985, 62, 896 CrossRef CAS.
- M. Epstein and S. A. Buckler, J. Am. Chem. Soc., 1961, 83, 3279–3282 CrossRef CAS.
- H. Schmidbaur and Y. Inoguchi, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1980, 35, 1329–1334 Search PubMed.
- C. Y. Kim, D.-G. Ha, H. H. Kang, H.-J. Yun, S.-K. Kwon, J.-J. Kim and Y.-H. Kim, J. Mater. Chem., 2012, 22, 22721–22726 RSC.
- S. D. Bull, S. G. Davies, D. J. Fox, M. Gianotti, P. M. Kelly, C. Pierres, E. D. Savory and A. D. Smith, J. Chem. Soc., Perkin Trans. 1, 2002, 1858–1868 RSC.
- G. M. Sheldrick, SADABS V2012/1, University of Göttingen, Germany Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- L. Palatinus, S. J. Prathapa and S. van Smaalen, J. Appl. Crystallogr., 2012, 45, 575–580 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal structures of ligands L2–7 and crystal structure and refinement details. CCDC 1497885–1497898. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03983a |
| This journal is © The Royal Society of Chemistry 2017 |