
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation of a new bis(carboxylate)triazole-based anchoring ligand for dye solar cell chromophore complexes†
Alessandro
Sinopoli
 a,
Fiona A.
Black
b,
Christopher J.
Wood
b,
Elizabeth A.
Gibson
*b and
Paul I. P.
Elliott
*a
a,
Fiona A.
Black
b,
Christopher J.
Wood
b,
Elizabeth A.
Gibson
*b and
Paul I. P.
Elliott
*a
aDepartment of Chemistry, University of Huddersfield, Queensgate, Huddersfield, HD1 3DH, UK. E-mail: p.i.elliott@hud.ac.uk
bSchool of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: elizabeth.gibson@ncl.ac.uk
First published on 5th January 2017
Abstract
A novel anchoring ligand for dye-sensitised solar cell chromophoric complexes, 1-(2,2′-bipyrid-4-yl)-1,2,3-triazole-4,5-dicarboxylic acid (dctzbpy), is described. The new dye complexes [Ru(bpy)2(dctzbpy)][PF6]2 (AS16), [Ir(ppy)2(dctzbpy)][PF6] (AS17) and [Re(dctzbpy)(CO)3Cl] (AS18) were prepared in a two stage procedure with intermediate isolation of their diester analogues, AS16-Et2, AS17-Et2 and AS18-Et2 respectively. Electrochemical analysis of AS16-Et2, AS17-Et2 and AS18-Et2 reveal reduction potentials in the range −1.50 to −1.59 V (vs. Fc+/Fc) which are cathodically shifted with respect to that of the model complex [Ru(bpy)2(dcbH2)]2+ (1) (Ered = −1.34 V, dcbH2 = 2,2′-bipyridyl-4,4′-dicarboxylic acid). This therefore demonstrates that the LUMO of the complex is correctly positioned for favourable electron transfer into the TiO2 conduction band upon photoexcitation. The higher energy LUMOs for AS16 to AS18 and a larger HOMO–LUMO gap result in blue-shifted absorption spectra and hence reduced light harvesting efficiency relative to their dcbH2 analogues. Preliminary tests on TiO2 n-type and NiO p-type DSSCs have been carried out. In the cases of the Ir(III) and Re(I) based dyes AS17 and AS18 these show inferior performance to their dcbH2 analogues. However, the Ru(II) dye AS16 (η = 0.61%) exhibits significantly greater efficiency than 1 (η = 0.1%). In a p-type cell AS16 shows the highest photovoltaic efficiency (η = 0.028%), almost three times that of cells incorporating the benchmark dye coumarin C343.
Introduction
The application of transition metal complexes, principally those of ruthenium(II), have attracted enormous attention for their application in n-type dye-sensitised solar cells (DSSC).1–4 The relatively long-lived triplet metal-to-ligand charge-transfer (3MLCT) states of these complexes5 allow efficient charge injection into the electrode on which the complex is adsorbed. Innumerable reports have appeared in the literature on the design and modification of the ligand set for these complexes in order to enhance the optical absorption cross-section to increase light harvesting efficiency and to optimise the ground state and excited state oxidation potentials for charge-injection and dye regeneration processes.6 A critical component of these dye complexes is therefore the ligand on which the 3MLCT state is localised and which anchors the complexes to the electrode. The energy of the lowest-unoccupied molecular orbital (LUMO) of the dye, localised on this ligand then largely determines the excited state oxidation potential which must be correctly positioned relative to the Fermi level of the electrode to favour efficient charge injection, and relative to the electrolyte redox couple to minimise short-circuit reactions. Whilst complexes of ruthenium dominate the literature dyes based on a variety of metals have been evaluated including osmium,7 platinum,8 rhenium,9 iron,10 and iridium11 as well as zinc porphyrins.12 Great strides have also been taken in the optimisation of metal free organic dyes.13,14Over thirty years ago, Goodenough and co-workers15 reported the use of 4,4′-dicarboxy-2,2′-bipyridine (dcbH2, Fig. 1) as an ambidentate ligand for coordination to Ru(II) and metal oxide semiconductors. Strong electronic coupling between the MLCT excited states of dcbH2-containing Ru compounds and TiO2 has been inferred from femtosecond transient absorption spectroscopy, and the timescales extracted from such data are on the order of <25 fs.16 To this day dcbH2 remains the most efficient and widely utilised anchoring ligand for applications in DSSCs, however, a number of other carboxylate-based variant ligands have been investigated. Heuer et al. reported the design of a new bipyridine based anchoring ligand (4,5-diazafluoren-9-ylidene)malonic acid (dfm, Fig. 1) together with its corresponding complex [Ru(bpy)2(dfm)]2+ (bpy = 2,2′-bipyridyl).6 Mishra et al. prepared the complex BCT-1, [Ru(dcthbpyH)2(NCS)2][NBu4]2, where the distance between the bpy core of the ligand and its anchoring carboxylate group has been extended by the introduction of a thiophene spacer (dcthbpyH2).17 This increases charge separation upon charge injection and reduces the rate of recombination but also leads to augmentation of the light absorption properties over the corresponding dcbH2 complex. This yielded an overall conversion efficiency of 6.1% compared with the 4.8% achieved using the archetypal DSSC dye N3 ([Ru(dcbH2)2(NCS)2]). In an attempt to minimise the rate constant for charge recombination, Abrahamsson et al. introduced a phenylenethynylene unit between a bpy ligand and the surface anchoring groups (dcpebpy, Fig. 1), preparing the corresponding [Ru(dcebpy)(bpy)(NCS)2] complex. This resulted in a recombination rate three times smaller compared to that for N3.18
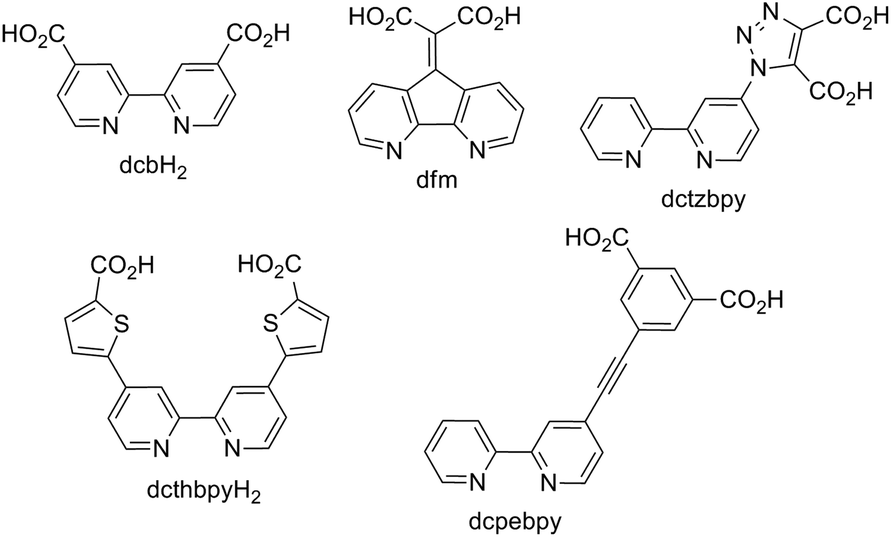 | ||
| Fig. 1 Structures of selected carboxylate-based DSSC anchoring ligands. | ||
Whilst carboxylates represent by far the most commonly encountered anchoring moiety DSSC dyes with other anchoring groups have also been investigated. These include phoshonic acid appended copper(I) and ruthenium(II) complexes19–21 and also boronic acid derivatised complexes.22
In contrast to the wealth of literature on transition metal-based dyes for n-type DSSCs, only a few examples have been reported for NiO p-type cells.23–30 Pellegrin et al. have reported ruthenium(II)-based sensitisers for p-type DSSC devices with phosphonic acid, thiocarboxylate and catechol derived anchoring groups.29 Ji et al. have reported cyclometalated ruthenium complexes of the type [Ru(N^N)2(C^N)]+ as sensitisers for p-type DSSCs.28 Addition of a rigid biphenylene spacer between the carboxylate anchor and the aryl ring of the cyclometalated ligand led to a marked increase in device efficiency (0.05% compared to 0.009% with no spacer). Recently Wood et al. reported bis(bidentate) ruthenium(II) based dye complexes bearing electron rich carboxylate derivatised triarylamine anchoring groups yielding efficiencies of up to 0.09%.27 Massin et al. have also reported carboxylate-appended triarylamine functionalised dyes with a [(Ph2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CRu(dppe)2)] core (dppe = 1,2-bis(diphenylphosphino)ethane).25
CRu(dppe)2)] core (dppe = 1,2-bis(diphenylphosphino)ethane).25
Through the use of triazole-based “click” chemistry31 we report here the design and synthesis of the novel anchoring ligand (2,2′-bipyrid-4-yl)-1,2,3-triazole-4,5-dicarboxylic acid (dctzbpy, Fig. 1). Triazole moieties have been utilised in conjugated polymers and results suggested that the linkage is somewhat electronically insulating.32,33 Whilst this might be expected to impair to some extent the rate of charge injection into TiO2 for an immobilised dye, this could significantly retard recombination reactions thereby enhancing overall photovoltaic efficiency. The designed anchor ligand also presents conformational freedom that would enable multiple possibilities for TiO2 surface coordination modes to be envisaged. For example, the various possibilities for coordination by one carboxylate group (monodentate (a), bidentate (b) or bridging (c) in Fig. 2), both carboxylate groups (bidentate (d), tridentate (e), tetradentate (f)) or anchoring through the 4-position carboxylate with additional surface coordination of the triazole-N3 atom (g) could result in highly favourable adsorption characteristics. Further anchoring modes through hydrogen bonding between carboxylic acid and surface oxygen atoms or carboxylate with surface hydroxyl groups can also be envisaged.
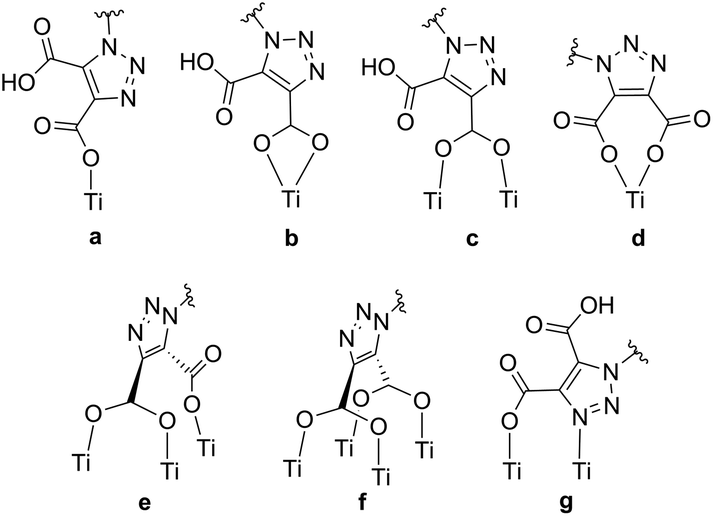 | ||
| Fig. 2 Selection of the potential TiO2 anchoring modes that could be adopted by the dctzbpy ligand. | ||
We also report the preparation of ruthenium(II), iridium(III) and rhenium(I) complexes AS16-Et2 to AS18-Et2 of the initial diethyl ester of the dctzbpy ligand and subsequent photophysical and electrochemical analyses. Further, we report the hydrolysis of these complexes to their corresponding diacids AS16 to AS18 (Fig. 3) and pilot studies on their utilisation in DSSC test devices with comparison to corresponding dcbH2 complexes 1 to 3.
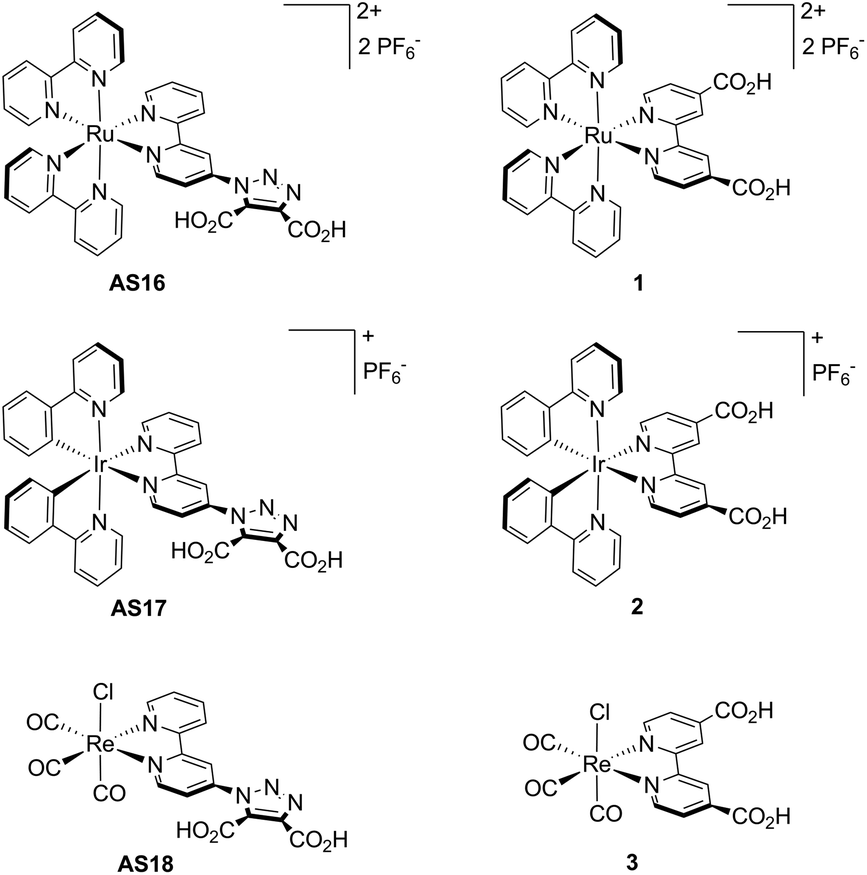 | ||
| Fig. 3 Structure of complexes AS16–18 and their dcbH2 analogues 1–3. | ||
Results & discussion
The above mentioned ligand, dctzbpy, was prepared by the route described in Scheme 1. We, and others, have previously reported the preparation and reactivity of the bpy derivative 4-azido-2,2′-bipyridyl.34,35 This azide precursor was subject to thermally driven cycloaddition with one equivalent of diethyl acetylenedicarboxylate in refluxing toluene to furnish the diester diethyl-1-(2,2′-bipyrid-4-yl)-1,2,3-triazole-4,5-dicarboxylate. Cycloaddition was confirmed by infrared spectroscopy with the disappearance of the azide stretch corresponding to the starting material (2115 cm−1) and the appearance of new CO stretching bands for the carboxylate substituents on the triazole ring (1735 cm−1). The ester groups then underwent hydrolysis to the corresponding carboxylic acids in refluxing dilute KOH in methanol before neutralising with HCl.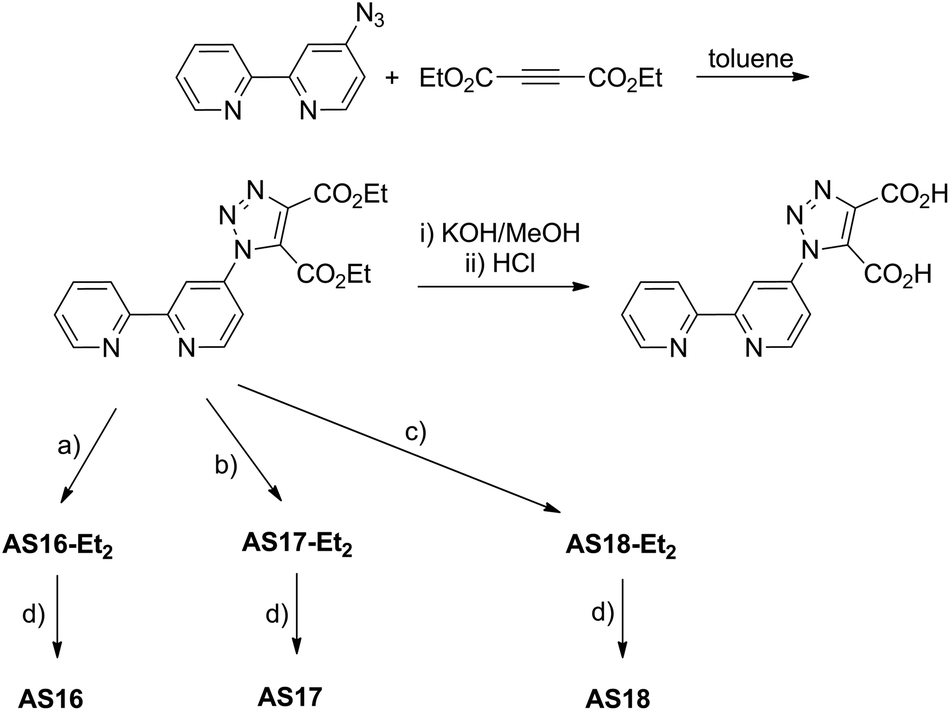 | ||
| Scheme 1 Synthesis of the target 1-(2,2′-bipyrid-4-yl)-1,2,3-triazole-4,5-dicarboxylic acid ligand and complexes AS16 to AS18: (a) (i) [Ru(bpy)2Cl2], EtOH, reflux, (ii) NH4PF6, MeOH; (b) (i) [Ir(ppy)2Cl]2, CH2Cl2/MeOH, reflux, (ii) NH4PF6, MeOH; (c) [Re(CO)5Cl], toluene, reflux; (d) (i) KOH/acetone, HCl, (ii) NH4PF6. | ||
The 1H NMR spectrum of the product shows seven unique environments (ESI†) for the protons on the bipyridyl fragment along with two sets of signals for the ethyl groups of the ester moieties. This is consistent with the cycloaddition of the acetylene to form the triazole and loss of magnetic equivalence of the two carboxylate groups. The resonances for the triazole-appended pyridine ring appear at δ 8.85, 8.67 and 7.61 and are shifted as a result of the cycloaddition relative to those of the azide starting material (δ 8.69, 8.15 and 7.34).35
Routes to ruthenium, iridium and rhenium dctzbpy complexes [Ru(bpy)2(dctzbpy)][PF6]2 (AS16), [Ir(ppy)2(dctzbpy)][PF6] (AS17, ppyH = 2-phenylpyridine) and [Re(CO)3(dctzbpy)Cl] (AS18) respectively were investigated involving direct reaction of the acid form of the ligand with precursor complexes, however, isolation of the corresponding complexes proved problematic. Isolation of complexes of the intermediate diester ligand, AS16-Et2, AS17-Et2 and AS18-Et2, followed by hydrolysis was deemed to give better results. Thus, two-step syntheses starting from [Ru(bpy)2Cl2], [Ir(ppy)2Cl]2 and [Re(CO)5Cl] were carried out. Firstly, the above mentioned starting metal precursor complexes were reacted together with detzbpy (Scheme 1) followed by anion metathesis with NH4PF6 to yield the desired diester complexes.
As a consequence of the asymmetry of the detzbpy ligand the [Ru(bpy)2] and [Ir(ppy)2] fragments in AS16-Et2 and AS17-Et2 do not possess the C2 symmetry present in 1 and 2. Hence, each bpy and ppy ligand in AS16-Et2 and AS17-Et2 respectively is magnetically unique resulting in complicated 1H NMR spectra due to the overlap of signals (ESI†). The 1H NMR spectrum of AS18-Et2 exhibits detzbpy signals shifted to higher field in comparison with the free diester ligand by ∼0.4 ppm with the resonances for the H-6 and H-6′ position appearing at δ 9.21 and 9.06 respectively.
The final dyes AS16 to AS18 can be readily accessed by hydrolysis with KOH in acetone and subsequent neutralisation. Due to solubility issues, however, and the fact that key properties are expected to differ little after hydrolysis electrochemical characterisation is reported for the more soluble esters AS16-Et2 to AS18-Et2.
Cyclic voltammetry traces and summarised electrochemical data are presented in Fig. 4 and Table 1 respectively. The complex AS16-Et2 presents a reversible Ru(II)/(III) oxidation at +1.00 V and an irreversible reduction peak at −1.50 V assigned to the detzbpy reduction. This is followed by two further reversible reduction peaks assigned to the bpy ligands. A quasi-reversible oxidation is observed at +0.90 V for AS17-Et2, corresponding to the one-electron Ir(III)/Ir(IV) couple. The first reduction presents as a quasi-reversible process at −1.59 V again assigned to be detzbpy ligand. Complex AS18-Et2 is characterised by a quasi-reversible oxidation peak at +1.00 V, in agreement with the potential of other Re(CO)3(dcb-Et2)X-based complexes (X = halide).36 It also presents a reversible detzbpy-based reduction at −1.56 V. The oxidation potentials for these complexes are similar to those of their corresponding dcbH2-based analogues consistent with the expectation of a largely metal-based HOMO.29,36,37 The detzbpy-based reduction potentials (−1.5 to −1.6 V) are more cathodically shifted than that reported for 1 (−1.34 V)29 consistent with a higher energy LUMO as indicated in the spectroscopic data.17–19
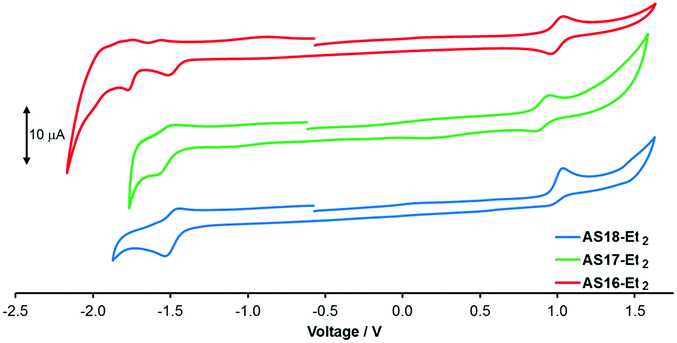 | ||
| Fig. 4 Cyclic voltammetry traces for complexes AS-16-Et2, AS17-Et2 and AS18-Et2vs. Fc+/Fc = 0.0 V. | ||
| Dye | E ox/V | E red/V |
|---|---|---|
| AS16-Et2 | 1.00 | −1.50 |
| AS17-Et2 | 0.90 | −1.59 |
| AS18-Et2 | 1.00 | −1.56 |
The isolated and purified complexes AS16-Et2 to AS18-Et2 were then refluxed in KOH/acetone and neutralised with HCl to yield the corresponding dctzbpy complexes. The analogous dcbH2 complexes [Ru(bpy)2(dcbH2)][PF6]2 (1),29 [Ir(ppy)2(dcbH2)][PF6] (2)20 and [Re(CO)3(dcbH2)(Cl)] (3)17 were also prepared for comparison. UV-Visible absorption spectra were recorded for all complexes in acetonitrile solutions at room temperature and are presented in Fig. 5 with summarised data listed in Table 2.
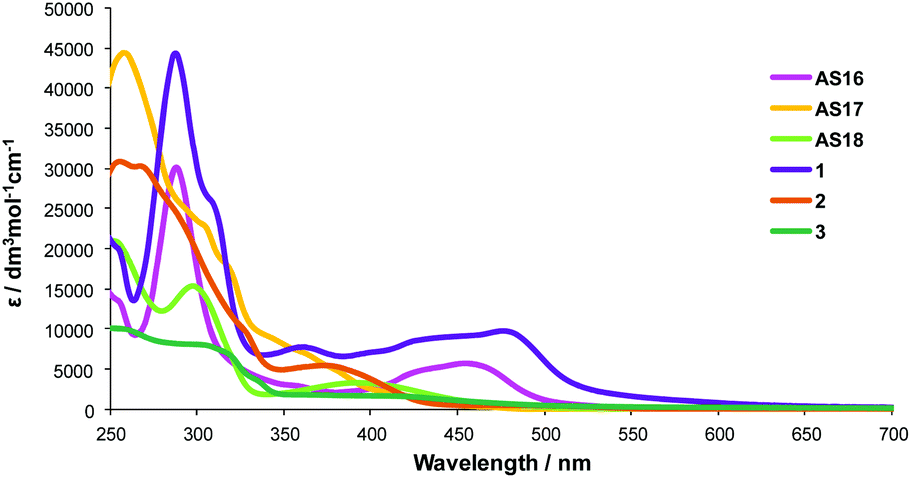 | ||
| Fig. 5 UV-visible absorption spectra for complexes AS16–18 and 1–3 in acetonitrile solutions. | ||
| Complex | λ abs/nm (ε/dm3 mol−1 cm−1) | λ emmax/nm | τ/ns |
|---|---|---|---|
| 1 | 245 (21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 923), 287 (44481), 308 (26079), 357 (7836), 430 (8807), 475 (9880) 923), 287 (44481), 308 (26079), 357 (7836), 430 (8807), 475 (9880) |
682 | 32 |
| 2 | 256 (30778), 289 (24016), 377 (6905) |
689 | 33 |
| 3 | 252 (10289), 303 (8202), 413 (1826) |
725 | 35 |
| AS16 | 242 (15624), 287 (30138), 424 (4642), 456 (5719) |
638 | 34 |
| AS17 | 255 (44311), 301 (23330), 364 (7771), 415 (1914) |
590 | 25 |
| AS18 | 254 (20204), 297 (15317), 397 (3815) |
553 | 38 |
The comparison between the dcbH2 complexes and their dctzbpy analogues shows similar absorption profiles but with a slight blue shift in the lower energy absorptions for complexes AS16–18. All complexes show a strong band at 250–300 nm attributed to π–π* 1LC transitions together with 1MLCT bands of modest intensity (ε ≈ 5000–10000 dm3 mol−1 cm−1) above 400 nm. The blue-shifted absorption bands in the spectra of the dctzbpy complexes suggests that the LUMO is higher in energy than those for the dcbH2 analogues and that the biscarboxytriazole group is less electron withdrawing than the two carboxylate groups bonded directly to the bipyridyl core in dcbH2. Nevertheless, LUMO is correctly positioned with reference to the TiO2 Fermi level for favourable charge injection when adsorbed on a photoanode.4 These similar absorption patterns will be expected to result in comparable photovoltaic performance for the dyes AS16–18 relative to 1–3.
The complexes AS16–18 exhibit broad emission bands between 550 and 650 nm which are similarly blue-shifted relative to their dcbH2 analogues, with similar life-times at about 32 ns. This again is indicative that the LUMO of the dctzbpy ligand, and thus that of its complexes, is higher in energy with respect to dcbH2 thereby leading to the observed destabilisation of 3MLCT T1 states in these complexes.
In order to gain a more complete understanding of the photophysical and electrochemical properties imparted by the new ligand dctzbpy we turned to density functional theory (DFT) calculations. These calculations were carried out on the free acid carboxylic acid complexes AS16–18, partly to reduce computational cost of the extra alkyl groups of the ester moieties, and due to the fact that the spectroscopic absorption properties of the free acid and ester complexes differ little suggesting that they are electronically very similar. Optimised singlet ground state geometries for the three new dyes AS16–18 and model complexes 1–3 were calculated at the B3LYP level of theory using Stuttgart–Dresden relativistic small-core effective core potentials and basis sets for the metallic elements and 6-311G* basis sets for all other atoms. Molecular orbital energies (Fig. 6) and localisations (Fig. 7) were then determined in single-point calculations using the COSMO solvation model (ε = 37.5 for acetonitrile). The HOMO has significant metallic d-orbital character in all cases with additional aryl π* character in the case of AS17 and CO π* and Cl p-orbital character for AS18. In all cases the LUMO is dominated by the dctzbpy ligand and is mostly localised over the bpy fragment. For AS16 there is also a minor contribution from the bpy ligands whilst for AS17 and AS18 there is an additional contribution from the dicarboxytriazole moiety.
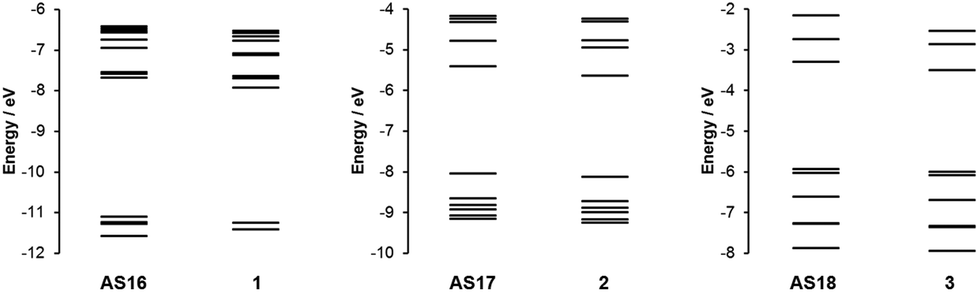 | ||
| Fig. 6 Calculated molecular orbital energy level diagram for complexes AS16–18 and 1–3. | ||
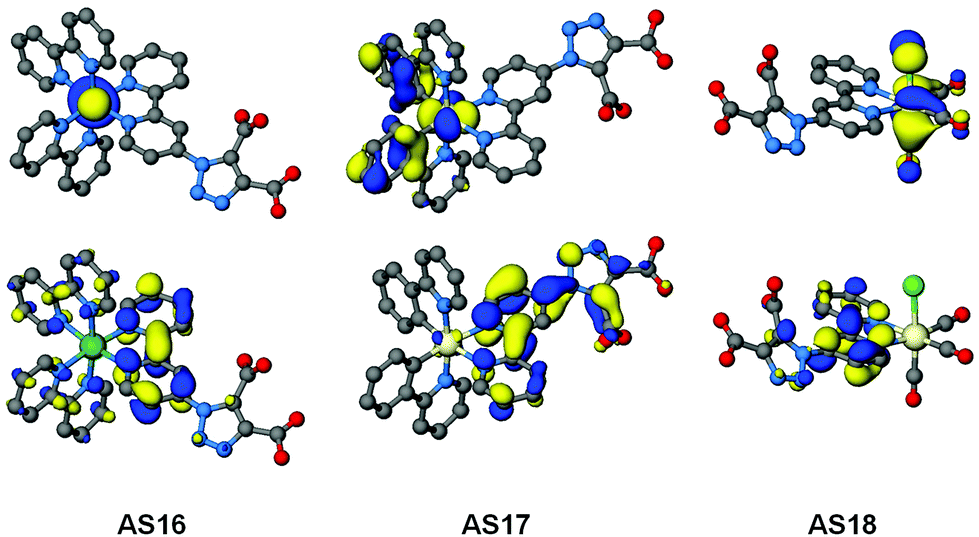 | ||
| Fig. 7 Optimised geometries and plots of HOMO (top) and LUMO (bottom) orbitals for complexes AS16 to AS18. | ||
Frontier molecular orbital energies are provided in Table 3. In agreement with UV-visible absorption and emission data there is a slight destabilization of both HOMO and LUMO orbitals, but to a greater extent for the latter, for AS16–18 relative to those of their dcbH2 analogues. Thus the HOMO–LUMO gaps for the dctzbpy complexes are an average of 0.12 eV larger than for 1–3 accounting for the experimental spectroscopic data. The ground state frontier molecular orbitals of AS16–18 are thus correctly localised to facilitate optimum charge-transfer directionality with respect to the carboxylate anchor for efficient charge injection when adsorbed onto a photoanode. Moreover, the relative energy of the dctzbpy ligand localised LUMO in complexes AS16–18, being slightly higher than those of the dcbH2 analogues 1 to 3, will inevitably be favourably positioned relative the TiO2 Fermi level in order to facilitate charge injection into the electrode.
| Dye | LUMO/eV | HOMO/eV | HOMO–LUMO/eV |
|---|---|---|---|
| 1 | −7.92 | −11.26 | 3.33 |
| AS16 | −7.68 | −11.10 | 3.42 |
| 2 | −5.65 | −8.13 | 2.48 |
| AS17 | −5.41 | −8.04 | 2.63 |
| 3 | −3.51 | −6.00 | 2.49 |
| AS18 | −3.30 | −5.94 | 2.64 |
Time-dependent DFT (TDDFT) calculations were carried out on the optimised ground state geometries of each complex in order to determine vertical excitation energies and the nature of the lowest energy singlet excited states. Simulated absorption spectra are overlaid with experimental spectra and shown in Fig. 8 and reveal that predicted transitions correlate well with the experimental spectra. The excitations to the S1 state of all complexes are primarily HOMO → LUMO in character, however, they are of low oscillator strength and will therefore contribute little to the observed absorption spectra. Consistent with the enlarged HOMO–LUMO gap in complexes AS16–18 relative to their respective dcbH2 analogues the S1 transitions occur at shorter wavelengths. The major transitions observed for all complexes between 350 and 550 nm are primarily of 1MLCT character. Higher energy intense absorptions (240 to 350 nm) are assigned as having predominantly 1LC π → π* character. The ruthenium complex AS16 presents two dominating transitions at 452 (S5) and 412 (S8) nm respectively, involving primarily HOMO−2 → LUMO+2 1MLCT character. The iridium complex AS17 exhibits one transition at 421 nm (S4) of mainly HOMO−1 → LUMO 1MLCT/1LLCT character and two transitions at 394 (S5) and 393 (S6) nm with 1MLCT/1ILCT and 1MLCT/1LLCT character respectively. The rhenium complex AS18 exhibits a strong transition at 443 nm (S2) with a predominant composition of HOMO−1 → LUMO 1MLCT character.
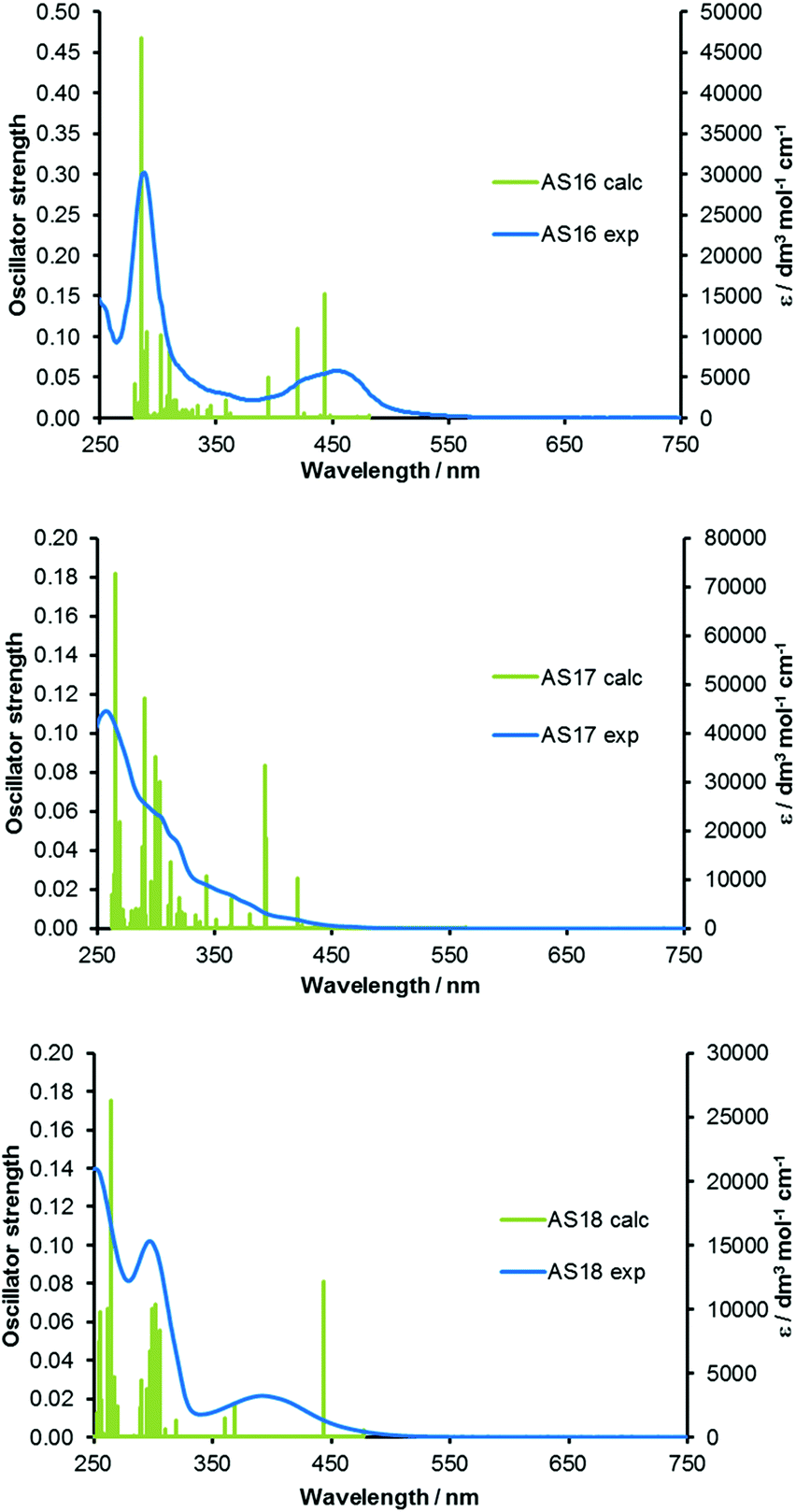 | ||
| Fig. 8 TDDFT calculated absorption spectra for complexes AS16–18 with experimental spectra overlaid. | ||
The relative positioning of the frontier orbitals in the dctzbpy complexes described would indeed seem to make them amenable to application in the photovoltaic sensitisation of n-type solar cells. We therefore prepared n-type TiO2-based test DSSC devices utilising complexes AS16 to AS18 along with those of the corresponding dcbH2 analogues for comparison.
The free dye AS16 and TiO2-immobilised AS16 were analysed by FTIR spectroscopy and microscopy in an attempt to gain insight into the anchoring mode of the dctzbpy ligand. Unfortunately the data do not enable any definitive conclusions to be drawn.
The principle photovoltaic parameters for the constructed DSSC devices are listed in Table 4. The overall conversion efficiencies η were derived from the equation η = JscVocFF, where Jsc is the short circuit current density, Voc is the open circuit voltage, and FF is the fill factor. Fig. 9 shows the photocurrent–voltage and IPCE traces of the n-type DSSCs based on the new dyes.
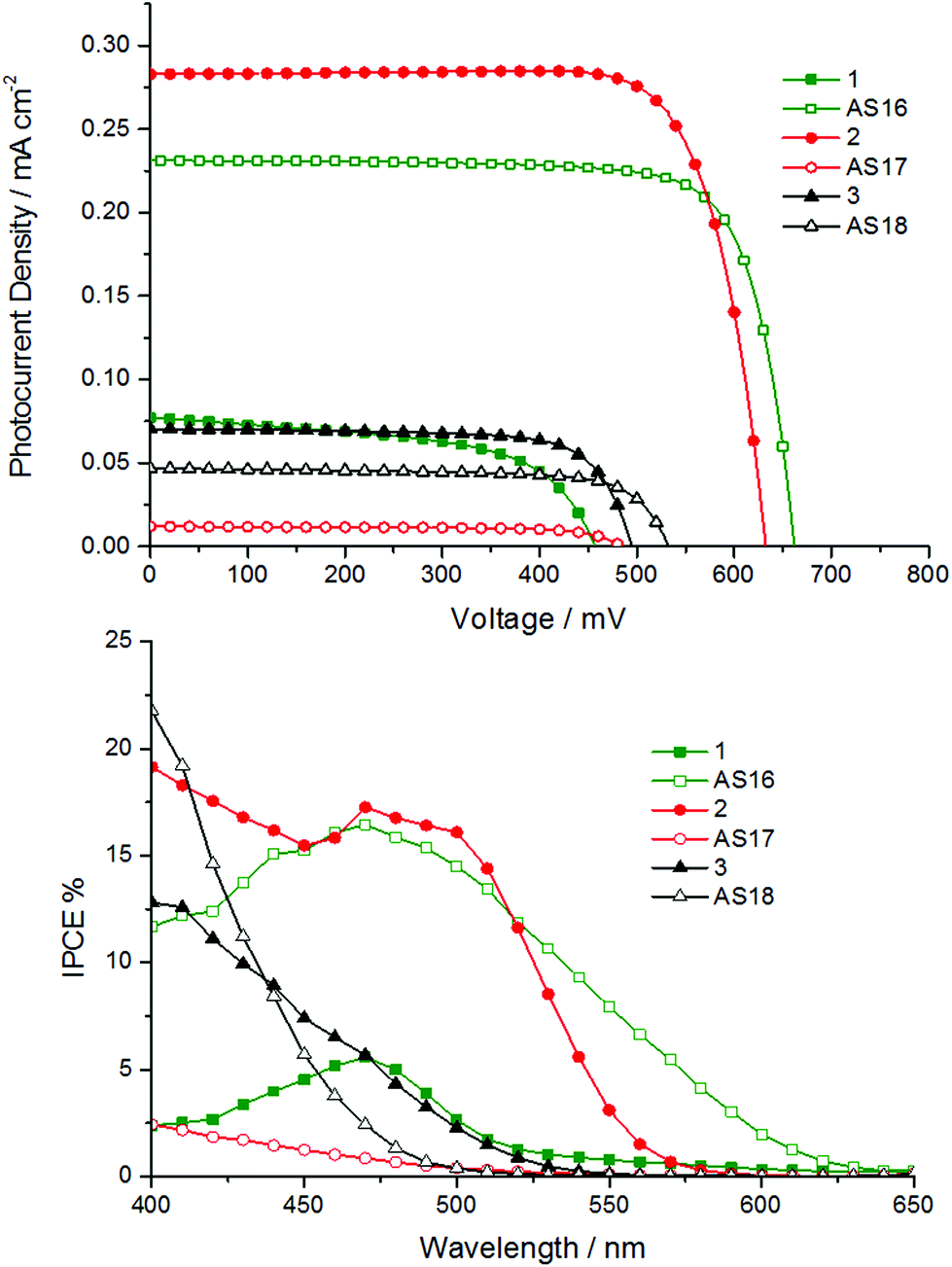 | ||
| Fig. 9 Current–voltage traces for pilot TiO2-based n-type DSSC devices utilising complexes AS16 to AS18 and model dyes 1 to 3 (top) along with IPCE traces for each cell (bottom). The electrolyte contained 0.6 M tetrabutylammonium iodide, 0.015 M I2, 0.1 M guanidinium thiocyanate and 0.5 M 4-tert-butylpyridine in MeCN. | ||
| Dye | J sc/mA cm−2 | V oc/mV | FF/% | η/% | IPCE/% |
|---|---|---|---|---|---|
| N719 | 10.7 | 779 | 69 | 5.76 | 65 |
| 1 | 0.39 | 457 | 57 | 0.1 | 6 |
| AS16 | 1.18 | 662 | 78 | 0.61 | 15 |
| 2 | 1.44 | 633 | 78 | 0.71 | 17 |
| AS17 | 0.06 | 485 | 72 | 0.02 | 3 |
| 3 | 0.36 | 495 | 73 | 0.13 | 6 |
| AS18 | 0.24 | 532 | 73 | 0.09 | 5 |
The IPCE values are generally below 10% over the visible region of the spectrum except for those of AS16 and 2 (Fig. 9). The IPCE profiles reflect the absorption profiles of the corresponding dyes; both AS16 and 1 exhibit IPCE maxima coincident with the region in which the complexes have a 1MLCT-based absorption band between 400–500 nm. All the other complexes have absorption maxima between 370 and 420 nm so their IPCE curves show only the tail of these bands into the visible region.
The obtained photovoltaic efficiencies for complexes AS17 and AS18 are lower than those for their dcbH2 analogues determined under identical conditions. This is not unexpected and attributable to their blue-shifted absorption profiles which would result in lower light harvesting efficiency. Further, whilst the LUMO energies for AS16–18 are expected to be in the correct position relative to the TiO2 Fermi level they are higher in energy than those of 1–3. This might imply less efficient overlap of the LUMO with the TiO2 conduction band and consequently lower electron injection efficiency. The best result for the new dyes is obtained for complex AS16 with an efficiency of 0.61% (compared to 0.1% for 1 and 5.76% for the benchmark dye N719 under the same conditions). AS16 achieved the highest open circuit voltage (0.66 V) which might suggest a longer excited state electron lifetime,38 hence a higher electron density on the TiO2 surface. The efficiency for AS17 was dramatically lower than that of its dcbH2 analogue 2. Indeed, the efficiency of 2 (0.71%) exceeds that of AS16 with a reasonably strong optical absorption shoulder when adsorbed on TiO2 (ESI†) that matches the band at 450–550 nm apparent in the IPCE trace. The rhenium complexes AS18 and 3, however, showed very similar Jsc, Voc and η values to each other.
Whilst AS16 indeed shows superior performance over its dcbH2 model in the cells test the performance of the other new dyes, especially in the case of AS17, is disappointing and likely stems from the elevated LUMO associated with the dctzbpy ligand. However, we reasoned that these dyes may yield greater sensitisation efficiency than their dcbH2 analogues in p-type devices for the very same reason.24 We therefore constructed and tested NiO-based p-type cells based on the new dyes and their dcbH2 analogues 1 to 3 along with coumarin C343 as a benchmark comparison. Current–voltage and IPCE plots are provided in Fig. 10 with photovoltaic parameters listed in Table 5.
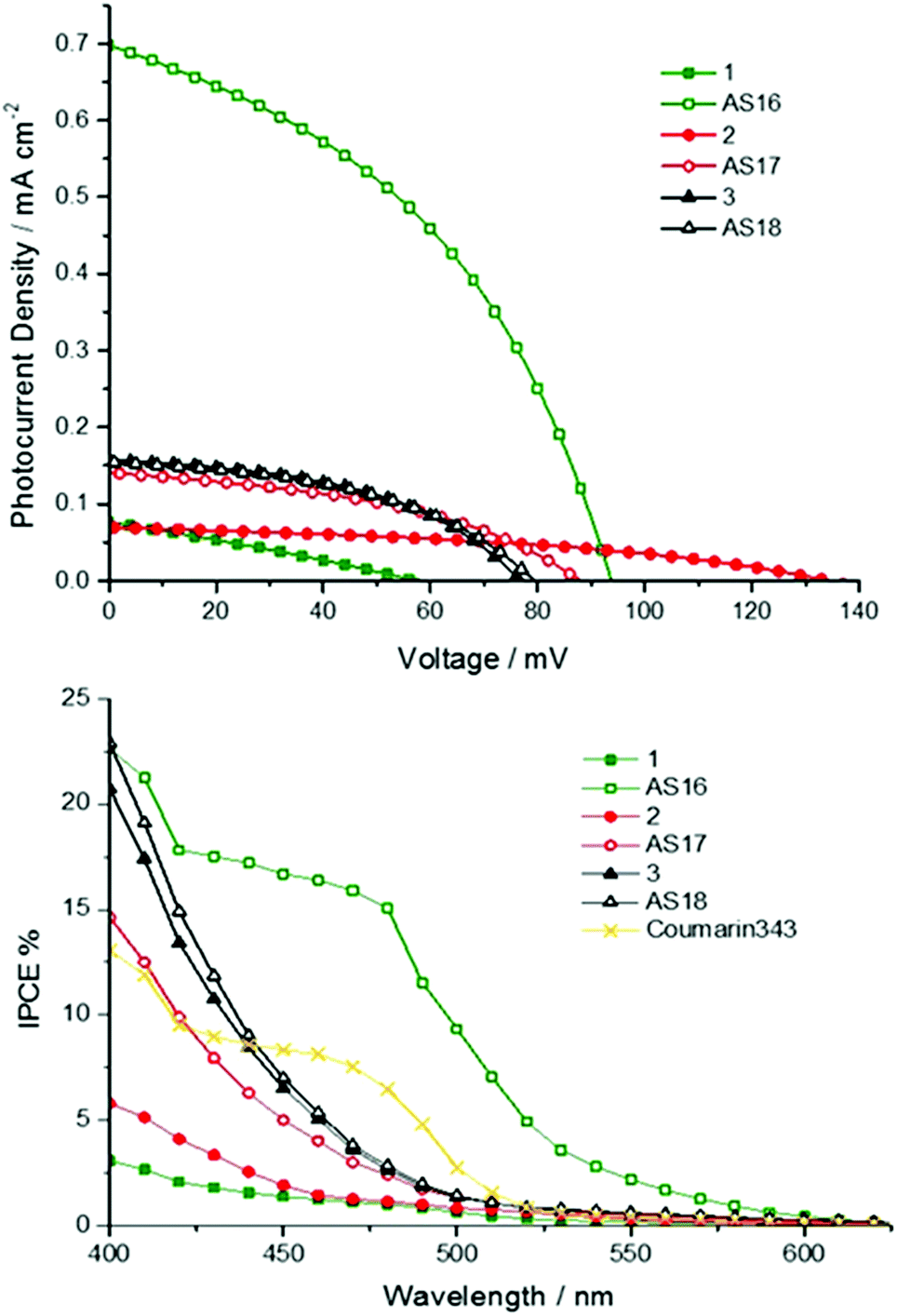 | ||
| Fig. 10 Current–voltage traces for pilot NiO-based p-type DSSC devices utilising complexes AS16 to AS18 and model dyes 1 to 3 (top) along with IPCE traces for each cell (bottom). The electrolyte contained 0.1 M I2 and 1 M LiI in MeCN. | ||
| Dye | J sc/mA cm−2 | V oc/mV | FF/% | η/% | IPCE/% |
|---|---|---|---|---|---|
| C343 | 0.26 | 105 | 37 | 0.01 | 8 |
| 1 | 0.076 | 58 | 27 | 0.0012 | 2 |
| AS16 | 0.69 | 94 | 42 | 0.028 | 17 |
| 2 | 0.069 | 134 | 40 | 0.0037 | 3 |
| AS17 | 0.14 | 89 | 42 | 0.0052 | 5 |
| 3 | 0.16 | 77 | 45 | 0.0056 | 6 |
| AS18 | 0.15 | 79 | 46 | 0.0055 | 6 |
The AS16 based cell exhibits the highest overall efficiency of all metal complex sensitised p-type cells (0.028% compared to 0.01% for C343) and the highest IPCE of 17%. As might be expected due to the dctzbpy anchoring ligand being less electron withdrawing than dcbH2 the performances of cells using AS16 and AS17 exceed those of 1 and 2 respectively. Indeed, 1 and 2 seem to be desensitisers of NiO in these cells with the current generated below 450 nm most likely stemming from photolysis of the I3− electrolyte as previously noted by Nattestad et al.39 Cells utilising AS18 and 3 yield near identical performance parameters.
The results for AS16 are generally encouraging in terms of dye development but are disappointing when considered in comparison to the benchmark N719. With further ancillary ligand development in combination with the new ligand it may be possible to arrive at much higher efficiency n-type sensitisers. Complexes of the new anchoring ligand dctzbpy may also find potential applications in p-type cells since the data show that the ruthenium(II)-based dye AS16 results in an efficiency three times that of the benchmark standard dye coumarin-C343. When combined with other ligands about a metal centre with greater electron withdrawing character this anchoring ligand provides potential for the development of far more efficient p-type sensitisers. Complexes based on the ligands described here may also find utility as photochemical solar fuel catalysts adsorbed on semiconductor nanoparticles.40 As the complexes here are luminescent and the carboxylate groups can be utilised for bioconjugation complexes bearing the dctzbpy core structure could also be used in biological imaging applications.41
Conclusions
We have designed and prepared a new anchoring ligand, 1-(2,2′-bipyridin-4-yl)-1,2,3-triazole-4,5-dicarboxylic acid, and its ruthenium, iridium and rhenium complexes. New ruthenium(II), iridium(III) and rhenium(I)-based dyes, AS16 to AS18 respectively, have been compared with their dcbH2 analogues. Spectroscopic and theoretical results suggest the LUMO of the dctzbpy ligand is higher in energy than that of dcbH2. Whilst correctly positioned for charge injection upon photoexcitation the resultant blue shift in optical absorption bands results in inferior light harvesting properties. Whilst improved photovoltaic performance is observed for AS16 this leads to a reduction in the photovoltaic efficiency for the other dyes in n-type DSSCs when compared to their dcbH2 analogues. In p-type cells the ruthenium(II) and iridium(III)-based dyes AS16 and AS17 display superior performance over their dcbH2 counterparts. Indeed, AS16 is shown to yield three times the efficiency of the benchmark dye coumarin C343. The work therefore offers insights into new anchor ligand systems. The newly reported ligand has potential for further development for dye complexes in NiO-based p-type DSSC devices especially in which a less electron withdrawing anchoring ligand is desirable, and in other applications including dye-sensitised electrochemical cells for solar catalysis and biological luminescent imaging.Experimental section
General methods
Chemicals were purchased from Aldrich and Acros, iridium and ruthenium were purchased from Precious Metal Online (Australia) and used as received. All complexation reactions were carried out under nitrogen. 4-Azido-2,2′-bipyridine,34 [Ru(bpy)2Cl2],24 [Ir(ppy)2Cl]2,20 [Ru(bpy)2(dcbH2)][PF6]2 (1),18 [Ir(ppy)2(dcbH2)][PF6] (2),20 [Re(CO)3(Cl)(dcbH2)] (3),17 were all prepared according to literature procedures. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance 400 MHz instrument. Mass spectrometry data were collected on a Bruker Micro Q-TOF instrument. UV-Visible absorption spectra were recorded on a Varian Cary 300 spectrophotometer and corrected emission spectra were recorded on a Horiba Fluoromax-4 spectrofluorometer. Luminescent lifetime measurements were carried out using an Edinburgh Instruments Mini-Tau spectrometer.Electrochemistry
For the cyclic voltammetry a EmStat3 electrochemical interface (Palm Sens, Netherlands) was employed, voltammograms were plotted on PSTrace software (Palm Sens, Netherlands). Cyclic voltammetry was carried out in a single compartment three electrode electrochemical cell under nitrogen atmosphere. Glassy carbon, platinum and Ag/AgCl were used respectively as working, counter and reference electrodes. The potential of the tested electrode was measured versus ferrocinium/ferrocene standard internal reference (FeCp2+/FeCp2). The electrolyte used for the cyclic voltammetry measurements was 0.1 M TBAPF6 in acetonitrile, prior to measurements all the solutions were degassed with nitrogen and during measurements a nitrogen blanket was maintained over the solution. The data were then plotted as current vs. voltage.Dye sensitised solar cell fabrication
FTO glass was used as current collector (TCO30-10, 3 mm thick glass substrate with a 10 Ω sq−1). For TiO2-based anodes cleaned and dried FTO electrodes were immersed into a 40 mM aqueous TiCl4 solution at 70 °C for 30 minutes and washed with pure water and ethanol and dried. A layer of nanocrystalline TiO2 paste (Solaronix Ti-Nanoxide SP-T) was coated on the FTO glass plates by doctor blade technique, using a round mask (5 mm diameter, 0.25 cm2 area) made by adhesive tape (3M Magic). The film was dried for 5 minutes on a hotplate and a second layer of TiO2 was deposited as before. Following this a light scattering layer (Solaronix Ti-Nanoxide R/SP) was applied in the same way and the films were sintered at 500 °C for 30 minutes. After a second treatment with TiCl4, the films were sintered for a final time as before. After cooling to 80 °C, the TiO2 electrodes were immersed into a 0.5 mM dye solution in acetonitrile overnight in the dark.The NiO films were prepared by using an F108-templated precursor solution containing NiCl2 (1 g), Pluronic® co-polymer F108 (1 g), distilled water (3 g) and ethanol (6 g) and the excess was removed by doctor blade. The film was sintered at 450 °C for 30 minutes and additional layers of precursor solution were applied and sintered until the film thickness was ca. 1.5 μm.
The Pt catalyst was deposited on the FTO glass, coating with 10 μL cm−2 of H2PtCl6 solution (5 mM ethanol solution), air dried and heated at 400 °C for 15 minutes. The dye-covered TiO2 or NiO electrodes and Pt-counter electrodes were assembled into a sandwich-type cell and sealed with a Surlyn hot-melt gasket of 60 μm thickness. A solution of 0.5 M TBP, 0.015 M I2, 0.6 M TBAI and 0.1 M GuSCN in acetonitrile was used as electrolyte in the TiO2-based n-type cells whereas an electrolyte containing 0.1 M I2 and 1 M LiI in acetonitrile was used for p-type cells.
Computational methods
DFT calculations were carried out using the NWChem 6.3 software package.42 B3LYP hybrid functional (20% Hartree–Fock) method has been used for calculations,43,44 Stuttgart relativistic small core ECP45 for transition metals and 6-311G*46 basis sets for all other atoms. Molecular geometries and molecular orbitals pictures were realised using the ccp1 graphical software. For all the studied complexes, the ground state geometries were first optimized and molecular orbital energies determined. TDDFT calculations47 on optimised structures in CH3CN by using the COSMO solvation model,48 built in NWChem software, were used to obtain the electronic spectra and molecular orbital energy levels.
Synthesis of diethyl-1-(2,2′-bipyrid-4-yl)-1,2,3-triazole-4,5-dicarboxylate
4-Azido-2,2′-bipyridine (0.1 g, 0.51 mmol) and diethyl acetylenedicarboxylate (0.16 g, 0.76 mmol) was dissolved in toluene (15 mL). The solution was allowed to stir at 70 °C for 24 h. The solvent was removed under vacuum and the product was recrystallised from DCM and hexane to yield a white solid. (175 mg, yield 93%).1H NMR (400 MHz, CDCl3) δ: 8.85 (d, 3J = 5.28 Hz, 1H, py-H); 8.67 (d, 3J = 2.08 Hz, 1H, py-H); 8.65 (d, 3J = 4.44 Hz, 1H, py-H); 8.45 (d, 3J = 7.92 Hz, 1H, py-H); 7.83 (td, 3J = 7.72, 4J = 1.72 Hz, 1H, py-H); 7.61 (dd, 3J = 5.28, 4J = 2.16 Hz, 1H, py-H); 7.43 (ddd, 1H, 3J = 7.52, 4J = 4.77, 5J = 1.00, py-H); 4.47 (q, 3J = 7.12, 2H, CH2–CH3); 4.46 (q, 3J = 7.12, 2H, CH2–CH3); 1.41 (t, 3J = 7.12 Hz, 3H, CH2–CH3); 1.35 (t, 3J = 7.16 Hz, 3H, CH2–CH3).
13C{1H} NMR (100 MHz, CDCl3) δ: 159.45, 158.90, 158.61, 154.21, 150.91, 149.38, 143.36, 139.38, 137.12, 132.45, 124.69, 121.24, 117.37, 114.59, 63.93, 62.09, 14.20, 13.77.
HRMS (ESI) m/z calcd for C18H17N5O4 367.1280, found 368.1359 (M + H)+.
Synthesis of 1-[2,2′-bipyridine-4-yl]triazole-4,5-dicarboxylic acid
1-[2,2′-Bipyridine-4-yl]triazole-4,5-ethyldicarboxylate (0.1 g, 0.51 mmol) was dissolved in 25 mL of NaOH 0.1 M and heated to reflux temperature for 4 h, after that the solution was neutralised with HCl 2 M. A white solid crushed out, collected by filtration and dried under reduced pressure. (134.84 mg, yield 85%).1H NMR (400 MHz, DMSO-d6) δ: 8.96 (d, 3J = 5.24, 1H, py-H); 8.77 (d, 3J = 4.72, 1H, py-H); 8.61 (d, 4J = 1.36, 1H, py-H); 8.54 (d, 3J = 7.96, 1H, py-H); 8.15 (t, 3J = 7.64, 1H, py-H); 7.81 (d, 3J = 5.20, 4J = 1.72, 1H, py-H); 7.64 (t, 3J = 5.48, 1H, py-H).
13C{1H} NMR (100 MHz, DMSO-d6) δ: 160.67, 158.62, 155.34, 152.77, 150.66, 148.55, 144.94, 139.97, 139.14, 134.14, 125.4, 121.45, 120.7, 116.84.
HRMS (ESI) m/z calcd for C14H9N5O4 311.0654, found 310.0582 (M − H)−.
Synthesis of [Ru(bpy)2(diethyl-1-(2,2′-bipyrid-4-yl)-1,2,3-triazole-4,5-dicarboxylate)](PF6)2AS16-Et2
100 mg of Ru(bpy)2Cl2 were dissolved in EtOH (20 mL) together with 113 mg of dectzbpy. The solution was heated to reflux temperature overnight, under N2 in the dark. The solvent was removed under reduced pressure and the resulting solid dissolved in MeOH, an excess of NH4PF6 was added and an orange crystalline solid was collected by filtration. The solid was columned on silica gel with MeCN:H2O:KNO3 7:1:0.5 as eluent. The main gloving fraction was then dried, redissolved in acetonitrile and filtered. The filtrate was dried and redissolved in a minimum amount of methanol, NH4PF6 was added, an orange solid crashed and it was collected by filtration. (168.1 mg, yield 76%).
1H NMR (400 MHz, CD3CN) δ: 8.79 (d, 3J = 2.02, 1H, bpy-H); 8.55–8.49 (m, 5H, bpy-H); 8.12–8.05 (m, 5H, bpy-H); 7.96 (d, 3J = 6.15, 1H, bpy-H); 7.83 (d, 3J = 5.32, 1H, bpy-H); 7.78 (d, 3J = 5.53, 1H, bpy-H); 7.76–7.69 (m, 3H, bpy-H); 7.55 (dd, 3J = 6.15, 3J = 2.20, 1H, bpy-H); 7.48–7.39 (m, 5H, bpy-H); 4.43 (q, 3J = 7.16, 2H, CH2–CH3); 4.31 (q, 3J = 7.07, 2H, CH2–CH3) 1.37 (t, 3J = 7.12, 3H, CH2–CH3); 1.11 (t, 3J = 7.03, 3H, CH2–CH3).
13C{1H} NMR (100 MHz, CD3CN) δ: 159.54; 159.10; 157.74; 156.97, 156.96; 156.93, 156.92; 156.88, 156.87; 156.83; 156.05; 153.52; 152.00; 151.94; 151.91; 151.83; 151.65; 143.10; 140.54, 138.19; 138.10; 138.09; 137.94, 128.41; 127.78; 127.74; 127.70; 127.62; 125.12; 125.08, 124.48; 124.43; 122.04; 119.67; 63.75; 62.25; 13.40; 13.04.
HRMS (ESI) m/z calcd for [C38H33N9O4Ru]2+ 390.5860, found 390.5864 (M)2+.
Synthesis of [Ru(bpy)2(1-[2,2′-bipyridine-4-yl]triazole-4,5-ethyldicarboxylic acid)]PF6AS16
50 mg of [Ru(bpy)2(detzbpy)](PF6)2 was dissolved in 8 mL 1 M KOH/acetone 1:1 and heated to reflux temperature for 12 hours. After cooling the acetone was removed under vacuum and the solution was neutralised with 1 M HCl. The solution was concentrated and a solid crashed out. The solid was collected by filtration, redissolved in a minimal amount of methanol and NH4PF6 was added, newly a red solid crashed and it was collected by filtration. (35.5 mg, yield 75%).
1H NMR (400 MHz, CD3CN) δ: 8.76 (d, 3J = 2.08, 1H, bpy-H); 8.55–8.46 (m, 5H, bpy-H); 8.13–8.03 (m, 5H, bpy-H); 7.92 (d, 3J = 6.12, 1H, bpy-H); 7.83–7.70 (m, 5H, bpy-H); 7.61 (d, 3J = 6.12, 4J = 2.2, 1H, bpy-H); 7.51–7.40 (m, 5H, bpy-H).
13C{1H} NMR (100 MHz, CD3CN) δ: 159.68, 159.63, 159.21, 157.86, 157.03, 156.99, 156.94, 156.90, 156.88, 156.46, 156.23, 154.69, 152.38, 151.89, 151.86, 151.84, 151.82, 151.81, 151.62, 143.42, 138.08, 138.03, 138.00, 137.96, 128.03, 127.73, 127.72, 127.66, 127.65, 124.75, 124.43, 124.40, 124.31, 121.72, 120.59.
Synthesis of [Ir(ppy)2(diethyl-1-(2,2′-bipyrid-4-yl)-1,2,3-triazole-4,5-dicarboxylate)]PF6AS17-Et2
107 mg of [Ir(ppy)2Cl]2 dimer were dissolved in DCM:MeOH 2:1 (12 mL) together with 77 mg of detzbpy and 53 mg of AgPF6. The solution was heated to reflux temperature overnight, under N2 in the dark. The solvent was removed under reduced pressure and the resulting solid dissolved in DCM and filtered on celite pad. The solvent was removed again and the solid recrystallised from acetonitrile/ether. A pale orange solid was collected by filtration and columned with 10% MeOH in DCM. (149.3 mg, yield 74%).
1H NMR (400 MHz, CD3CN) δ: 8.81 (d, 3J = 1.70, 1H, bpy-H); 8.54 (d, 3J = 8.04, 1H, bpy-H); 8.20–8.13 (m, 2H, bpy-H); 8.08 (d, 3J = 8.14, 2H, ppy-H); 8.02 (dd, 3J = 5.78, 4J = 0.78, 1H, bpy-H); 7.86 (td, 3J = 7.35, 4J = 0.84, 2H, ppy-H); 7.82 (d, 3J = 7.62, 2H, ppy-H); 7.72 (dd, 3J = 5.52, 3J = 0.81, 1H, ppy-H); 7.65 (dd, 3J = 6.09, 4J = 2.17, 1H, bpy-H); 7.63 (dd, 3J = 5.96, 4J = 0.81, 1H, bpy-H); 7.56 (t, 3J = 6.30, 1H, bpy-H); 7.09–7.02 (m, 4H, ppy-H); 6.96–6.90 (m, 2H, ppy-H); 6.29 (dd, 3J = 7.75, 4J = 0.88, 1H, ppy-H); 6.26 (dd, 3J = 7.63, 4J = 0.74, 1H, ppy-H); 4.42 (q, 3J = 6.95, 2H, CH2–CH3); 4.31 (qd, 3J = 7.10, 4J = 1.15, 2H, CH2–CH3); 1.36 (t, 3J = 7.10, 3H, CH2–CH3); 1.10 (t, 3J = 7.10, 3H, CH2–CH3).
13C{1H} NMR (100 MHz, CD3CN) δ: 167.35, 167.24, 159.48, 158.15, 157.72, 154.72, 152.48, 150.99, 149.52, 149.49, 149.43, 144.40, 144.09, 143.97, 140.50, 139.62, 138.78, 138.72, 131.80, 131.59, 131.44, 130.49, 130.44, 129.26, 125.37, 124.98, 124.95, 123.66, 123.54, 123.08, 122.82, 122.76, 120.10, 120.01, 119.98, 63.81, 62.24, 13.39, 12.97.
HRMS (ESI) m/z calcd for [C40H33N7O4Ir]+ 867.2230, found 867.2228 (M+).
Synthesis of [Ir(ppy)2(1-[2,2′-bipyridine-4-yl]triazole-4,5-ethyldicarboxylate)]PF6AS17
50 mg of AS17-Et2 was dissolved in 8 mL 1 M KOH/acetone 1:1 and heated to reflux temperature for 12 hours. After cooling the acetone was removed under vacuum and the solution was neutralised with 1 M HCl. The solution was concentrated and a solid crashed out. The solid was collected by filtration, redissolved in a minimal amount of methanol and NH4PF6 was added, newly a yellow solid crashed and it was collected by filtration. (32.1 mg, yield 68%).
1H NMR (400 MHz, CD3CN) δ: 8.46 (d, 3J = 6.56 Hz, 1H, bpy-H); 8.11 (td, 3J = 6.28, 4J = 1.24 Hz, 1H, bpy-H); 8.07 (t, 3J = 5.64 Hz, 2H, ppy-H); 8.00 (bs, 1H, bpy-H); 7.97 (dd, 3J = 4.3, 4J = 0.8 Hz, 1H, bpy-H); 7.89–7.84 (m, 2H, ppy-H); 7.81 (d, 3J = 6.08 Hz, 2H, ppy-H); 7.71 (d, 3J = 4.72 Hz, 1H, bpy-H); 7.65 (d, 3J = 4.6 Hz, 1H, ppy-H); 7.63 (d, 3J = 5.16 Hz, 1H, ppy-H); 7.47 (td, 3J = 5.24, 4J = 0.72 Hz, 1H, bpy-H); 7.09–7.02 (m, 5H, bpy-H&ppy-H); 6.97–6.9 (m, 3H, ppy-H); 6.31 (dd, 3J = 6.06, 4J = 0.6 Hz, 1H, ppy-H); 6.28 (dd, 3J = 6.04, 4J = 0.52 Hz, 1H, ppy-H).
13C{1H} NMR (100 MHz, CD3CN) δ: 167.63, 167.49, 157.16, 156.23, 151.26, 150.45, 149.14, 149.11, 144.28, 144.06, 139.99, 139.88, 139.86, 139.84, 139.21, 139.16, 138.36, 138.33, 131.91, 131.67, 131.65, 131.51, 130.31, 130.27, 130.23, 128.11, 124.82, 124.78, 124.33, 123.4, 123.37, 122.32, 122.3.
Synthesis of Re(CO)3(diethyl-1-(2,2′-bipyrid-4-yl)-1,2,3-triazole-4,5-dicarboxylate)Cl AS18-Et2
100 mg of [Re(CO)5Cl] and 113 mg of detzbpy were dissolved in 25 mL of toluene and heated at 70 °C for 12 hours. After cooling the volume was reduced under reduced pressure and a yellow solid crashed out, it was collected by filtration and dried with diethylether. (155.8 mg, yield 84%).1H NMR (400 MHz, CD3CN) δ: 9.21 (d, 3J = 5.96, 1H, bpy-H); 9.06 (dd, 3J = 5.38, 3J = 0.70, 1H, bpy-H); 8.70 (d, 3J = 2.10, 1H, bpy-H); 8.44 (d, 3J = 8.18, 1H, bpy-H); 8.23 (dt, 3J = 7.95 1.52, 1H, bpy-H); 7.78 (dd, 3J = 6.08, 3J = 2.34, 1H, bpy-H); 7.69 (dt, 3J = 6.49, 3J = 1.22, 1H, bpy-H); 4.44 (q, 3J = 7.13, 2H, CH2–CH3); 4.41 (q, 3J = 7.02, 2H, CH2–CH3); 1.38 (t, 3J = 7.13, 3H, CH2–CH3); 1.27 (t, 3J = 6.98, 3H, CH2–CH3).
13C{1H} NMR (100 MHz, CD3CN) δ: 197.63, 197.47, 189.26, 159.55, 157.96, 157.87, 154.73, 154.65, 153.25, 144.91, 140.59, 140.22, 131.79, 128.29, 124.65, 121.86, 119.36, 63.94, 62.24, 13.40, 13.07.
HRMS (ESI) m/z calcd for C21H17ClN5O7Re 671.0352, found 689.0694 (M + NH4)+.
Synthesis of Re(CO)3(1-[2,2′-bipyridine-4-yl]triazole-4,5-ethyldicarboxylate)Cl AS18
50 mg of [Re(CO)3(detzbpy)Cl] was dissolved in 8 mL 1 M KOH/acetone 1:1 and heated to reflux temperature for 12 hours. After cooling the acetone was removed under vacuum and the solution was neutralised with 1 M NaOH. The solution was concentrated and a yellow solid crashed out. The solid was collected by filtration. (30.7 mg, yield 67%).
1H NMR (400 MHz, DMSO-d6) δ: 8.94 (d, 3J = 5.44, 1H, bpy-H); 8.44 (d, 3J = 8.17, 1H, bpy-H); 8.22 (t, 3J = 7.78, 1H, bpy-H); 7.97 (d, 3J = 6.81, 1H, bpy-H); 7.63 (t, 3J = 6.61, 1H, bpy-H); 7.15 (d, 3J = 2.25, 1H, bpy-H); 6.14 (dd, 3J = 6.95, 4J = 2.64, 1H, bpy-H).
13C{1H} Low solubility, NMR not recorded.
Acknowledgements
The authors wish to thank the Leverhulme Trust and the University of Huddersfield for funding this research. As a member of the UK Materials Chemistry Consortium PIPE also thank the EPRSC (EP/L000202) and the UK HPC national resource, Archer, as well as the University of Huddersfield High Performance Computing Research Group for computational resources utilised in this project.References
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- M. K. Nazeeruddin, E. Baranoff and M. Grätzel, Sol. Energy, 2011, 85, 1172–1178 CrossRef CAS.
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef.
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2009, 38, 115–164 RSC.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. V. Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- W. B. Heuer, H.-L. Xia, W. Ward, Z. Zhou, W. H. Pearson, M. A. Siegler, A. A. Narducci Sarjeant, M. Abrahamsson and G. J. Meyer, Inorg. Chem., 2012, 51, 3981–3988 CrossRef CAS PubMed.
- S. Altobello, R. Argazzi, S. Caramori, C. Contado, S. Da Fré, P. Rubino, C. Choné, G. Larramona and C. A. Bignozzi, J. Am. Chem. Soc., 2005, 127, 15342–15343 CrossRef CAS PubMed.
- E. A. M. Geary, L. J. Yellowlees, L. A. Jack, I. D. H. Oswald, S. Parsons, N. Hirata, J. R. Durrant and N. Robertson, Inorg. Chem., 2005, 44, 242–250 CrossRef CAS PubMed.
- G. M. Hasselmann and G. J. Meyer, Z. Phys. Chem., 1999, 212, 39–44 CrossRef CAS.
- S. Ferrere, Chem. Mater., 2000, 12, 1083–1089 CrossRef CAS.
- E. Baranoff, J.-H. Yum, M. Graetzel and M. K. Nazeeruddin, J. Organomet. Chem., 2009, 694, 2661–2670 CrossRef CAS.
- A. Yella, H. W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W. G. Diau, C. Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- A. Mishra, M. K. R. Fischer and P. Büuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed.
- Z. Yao, M. Zhang, H. Wu, L. Yang, R. Li and P. Wang, J. Am. Chem. Soc., 2015, 137, 3799–3802 CrossRef CAS PubMed.
- J. B. Goodenough, J. Phys. Chem., 1982, 86, 3492–3492 CrossRef CAS.
- F. De Angelis, S. Fantacci, E. Mosconi, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. C, 2011, 115, 8825–8831 CAS.
- A. Mishra, N. Pootrakulchote, M. K. R. Fischer, C. Klein, M. K. Nazeeruddin, S. M. Zakeeruddin, P. Bauerle and M. Gratzel, Chem. Commun., 2009, 7146–7148 RSC.
- M. Abrahamsson, P. G. Johansson, S. Ardo, A. Kopecky, E. Galoppini and G. J. Meyer, J. Phys. Chem. Lett., 2010, 1725–1728 CrossRef CAS.
- P. Wang, C. Klein, J.-E. Moser, R. Humphry-Baker, N.-L. Cevey-Ha, R. Charvet, P. Comte, S. M. Zakeeruddin and M. Grätzel, J. Phys. Chem. B, 2004, 108, 17553–17559 CrossRef CAS.
- I. Gillaizeau-Gauthier, F. Odobel, M. Alebbi, R. Argazzi, E. Costa, C. A. Bignozzi, P. Qu and G. J. Meyer, Inorg. Chem., 2001, 40, 6073–6079 CrossRef CAS PubMed.
- A. Buttner, S. Y. Brauchli, R. Vogt, E. C. Constable and C. E. Housecroft, RSC Adv., 2016, 6, 5205–5213 RSC.
- S. Altobello, C. A. Bignozzi, S. Caramori, G. Larramona, S. Quici, G. Marzanni and R. Lakhmiri, J. Photochem. Photobiol., A, 2004, 166, 91–98 CrossRef CAS.
- F. Odobel, L. Le Pleux, Y. Pellegrin and E. Blart, Acc. Chem. Res., 2010, 43, 1063–1071 CrossRef CAS PubMed.
- F. Odobel, Y. Pellegrin, E. A. Gibson, A. Hagfeldt, A. L. Smeigh and L. Hammarström, Coord. Chem. Rev., 2012, 256, 2414–2423 CrossRef CAS.
- J. Massin, S. Lyu, M. Pavone, A. B. Munoz-Garcia, B. Kauffmann, T. Toupance, M. Chavarot-Kerlidou, V. Artero and C. Olivier, Dalton Trans., 2016, 45, 12539–12547 RSC.
- M. Gennari, F. Légalité, L. Zhang, Y. Pellegrin, E. Blart, J. Fortage, A. M. Brown, A. Deronzier, M.-N. Collomb, M. Boujtita, D. Jacquemin, L. Hammarström and F. Odobel, J. Phys. Chem. Lett., 2014, 5, 2254–2258 CrossRef CAS PubMed.
- C. J. Wood, K. C. D. Robson, P. I. P. Elliott, C. P. Berlinguette and E. A. Gibson, RSC Adv., 2014, 4, 5782–5791 RSC.
- Z. Ji, G. Natu, Z. Huang, O. Kokhan, X. Zhang and Y. Wu, J. Phys. Chem. C, 2012, 116, 16854–16863 CAS.
- Y. Pellegrin, L. Le Pleux, E. Blart, A. Renaud, B. Chavillon, N. Szuwarski, M. Boujtita, L. Cario, S. Jobic, D. Jacquemin and F. Odobel, J. Photochem. Photobiol., A, 2011, 219, 235–242 CrossRef CAS.
- A. Sinopoli, C. J. Wood, E. A. Gibson and P. I. P. Elliott, Eur. J. Inorg. Chem., 2016, 2887–2890 CrossRef CAS.
- T. P. Lodge, Macromol., 2009, 42, 3827–3829 CrossRef CAS.
- E. Lim, Int. J. Photoenergy, 2013, 2013, 607826, DOI:10.1155/2013/607826.
- D. Schweinfurth, K. I. Hardcastle and U. H. F. Bunz, Chem. Commun., 2008, 2203–2205 RSC.
- R. A. Fallahpour, Helv. Chim. Acta, 2000, 83, 384–393 CrossRef CAS.
- B. S. Uppal, A. Zahid and P. I. P. Elliott, Eur. J. Inorg. Chem., 2013, 2571–2579 CrossRef CAS.
- G. M. Hasselmann and G. J. Meyer, J. Phys. Chem. B, 1999, 103, 7671–7675 CrossRef CAS.
- E. I. Mayo, K. Kilsa, T. Tirrell, P. I. Djurovich, A. Tamayo, M. E. Thompson, N. S. Lewis and H. B. Gray, Photochem. Photobiol. Sci., 2006, 5, 871–873 CAS.
- T. N. Murakami, N. Koumura, M. Kimura and S. Mori, Langmuir, 2014, 30, 2274–2279 CrossRef CAS PubMed.
- A. Nattestad, M. Ferguson, R. Kerr, Y.-B. Cheng and U. Bach, Nanotechnology, 2008, 19, 295304 CrossRef PubMed.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef CAS PubMed.
- K. K.-W. Lo and S. P.-Y. Li, RSC Adv., 2014, 4, 10560–10585 RSC.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. V. Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. d. Jong, Comput. Phys. Commun., 2010, 181, 1477–1660 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1983, 98, 5648–5652 CrossRef.
- D. Andrae, U. Haussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- A. Klamt and G. Schuurmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6dt02905a |
| This journal is © The Royal Society of Chemistry 2017 |