
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deoxydehydration of vicinal diols and polyols catalyzed by pyridinium perrhenate salts†
Danny S.
Morris‡
a,
Karlotta
van Rees‡
ab,
Massimiliano
Curcio‡
a,
Mirza
Cokoja
c,
Fritz E.
Kühn
 d,
Fernanda
Duarte
a and
Jason B.
Love
*a
d,
Fernanda
Duarte
a and
Jason B.
Love
*a
aEaStCHEM School of Chemistry, University of Edinburgh, David Brewster Road, Edinburgh EH9 3FJ, UK. E-mail: jason.love@ed.ac.uk; Tel: +44 (0)131 650 4762
bVan't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands
cChair of Inorganic and Metal Organic Chemistry, Faculty of Chemistry, Technical University Munich, Lichtenbergstr. 4, 85748 Garching, Germany
dMolecular Catalysis, Faculty of Chemistry and Catalysis Research Center, Technical University Munich, Lichtenbergstr. 4, Garching, 85748 Germany
First published on 25th October 2017
Abstract
Simple ammonium and pyridinium perrhenate salts were evaluated as catalysts for the deoxydehydration (DODH) of diols into alkenes. Pyridinium perrhenates were found to be effective catalysts at much lower temperatures than those in previous reports, outperforming primary, secondary, and tertiary ammonium salts, while quaternary ammonium salts are effectively inactive. The mechanism of reaction was studied computationally using DFT calculations which indicate that proton shuttling between the ion pair is intrinsic to the mechanism and that the reduction of rhenium by the phosphine occurs before the diol condensation.
Introduction
The deoxygenation of oxygen-rich, biomass-derived materials such as glycerides and carbohydrates could potentially provide renewable routes to platform chemicals and transportation fuels, so delivering alternatives to the use of fossil-based resources.1 One strategy towards this target is the use of catalytic deoxydehydration (DODH),2 a reaction that transforms vicinal diols, polyols, and carbohydrates into alkenes in the presence of a reducing agent (eqn (1)).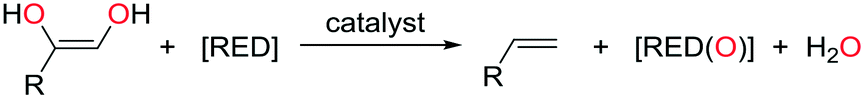 | (1) |
Although some oxo complexes of V and Mo have been exploited as catalysts for DODH,3 the majority make use of the redox activity of rhenium. These include low oxidation-state complexes such as Re2(CO)10 and BrRe(CO)5,4 but more commonly ReVII/ReV oxo complexes such as MeReO3, Cp*/CptttReO3 (Cpttt = But3C5H2), ReO2I(PPh3)2, and ReOCl3(PPh3)2.4a,b,5 The simple perrhenate salts HReO4 (and Re2O7), NaReO4, (NH4)ReO4, and (Bun4N)ReO4 act as molecular or supported catalysts for DODH reactions,5d,6 but are significantly less active than, for example MeReO3 which has been ascribed to the poor solubility of the former complexes.7 The majority of these Re catalysts need elevated temperatures (130–250 °C) for turnover and, with the exception of perrhenate, are toxic8 and not easily recycled.
In 2015, we reported that the toluene-soluble perrhenate salt of the cation 1 (Chart 1) acts as a catalyst for the epoxidation of alkenes by H2O2 under biphasic conditions.9 Through microscopic reversibility and the literature precedent above, we envisaged that 1 should also be able to act as a catalyst for reductive deoxygenation reactions. As such, with a view to understanding the use of perrhenate in DODH and the effect of its attendant cation, we show here that pyridinium perrhenates such as 1 are effective and selective catalysts for the DODH of diols. These catalysts operate at significantly lower temperatures than for previously reported oxo-metal catalysts and, furthermore, the identity of the cation and its ability to act as a Brønsted acid is important to the efficacy of this catalyst system.
 | ||
| Chart 1 Cation variation in perrhenate salts (Z)ReO4 (Z = cation) used as catalysts for the deoxydehydration of vicinal diols. | ||
Results and discussion
Synthesis and evaluation of perrhenate salts as DODH catalysts
The pyridinium (1–7) and ammonium (8–13) salts of perrhenate were prepared by liquid–liquid extraction of perrhenic acid into a solution of the base in toluene. The solid-state structures of 1,92, 3, 4, 6, 9 and 10 were determined by single-crystal X-ray diffraction and, with the exception of 1 and 2 show formation of an ion pair with a strong hydrogen-bonding interaction between the cation and a single Re oxo (Fig. 1 and ESI†); in contrast, a C–H–O hydrogen bond is seen in 2 due to the sterically hindering tert-butyl groups that flank the pyridinium nitrogen atom, and the diamido motif in the cation of 1 promotes diffuse N–H and C–H hydrogen bonding to the perrhenate anion.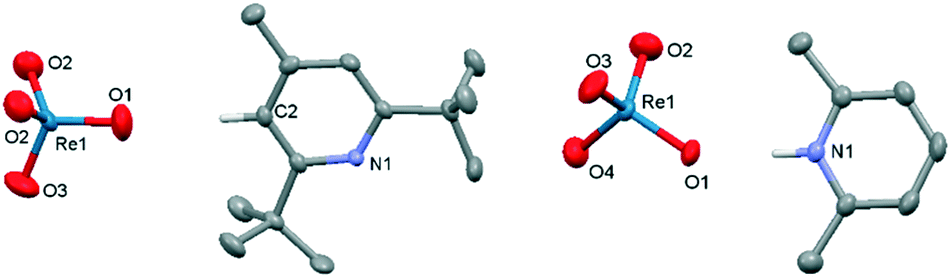 | ||
| Fig. 1 Solid-state structures of 2 (left) and 3 (right). For clarity, all hydrogen atoms, except those involved in hydrogen-bonding, are omitted (displacement ellipsoids are drawn at 50% probability). Selected bond lengths (Å): 2: Re1–O1 1.703(4); Re1–O2 1.723(4); Re1–O3 1.703(4); C2–O1 3.312. 3: Re1–O1 1.736(2); Re1–O2 1.714(2); Re1–O3 1.710(2); Re1–O4 1.716(2); N1–O1 2.754. | ||
Along with the commercially available 14 and 15, compounds 1 to 13 were evaluated as catalysts for the DODH of styrene diol in CDCl3 at 80 °C (Table 1). Blank tests displayed that DODH does not take place in the absence of catalyst and that the substrate slowly acts as a reducing agent (i.e. in the absence of added reducing agent such as PPh3). Disappointingly, the quaternary ammonium salts 13 to 15 were ineffective as catalysts (<5% conversion), whereas the 1°, 2°, and 3° ammonium salts 8 to 12 gave only moderate conversion (30–65%); in contrast, the pyridinium salts 1 to 7 afforded high conversion. While 2 results in the highest conversion after 4 h at 80 °C, it displayed poor solubility in CDCl3 so its concentration during the reaction is likely imprecise. As a result, the reaction conditions were optimized using 3 (Table 2). Although the TOFs at 80 °C for 1 to 7 are low (1.2 to 3.2 h−1), they are comparable to previous reports and 2 to 3× greater than the other perrhenate salts reported here.
| Entry | Catalyst | Load (mol%) | Time (h) | Conv.a (%) | TOF (h−1) | pKa |
|---|---|---|---|---|---|---|
| a Conversion was determined by 1H-NMR spectroscopy using Ph3CH as an internal standard. | ||||||
| 1 | 1 | 5.0 | 16 | >99 | 1.3 | — |
| 2 | 2 | 5.0 | 16 | >99 | 1.3 | 4.4 |
| 3 | 3 | 5.0 | 16 | >99 | 1.3 | 6.7 |
| 4 | 4 | 5.0 | 16 | >99 | 1.3 | 0.5 |
| 5 | 10 | 5.0 | 16 | 37 | 0.5 | 10.6 |
| 6 | 11 | 5.0 | 16 | 39 | 0.5 | 9.3 |
| 7 | 12 | 5.0 | 16 | 64 | 0.8 | 8.5 |
| 8 | 13 | 5.0 | 16 | 21 | 0.3 | — |
| 9 | 14 | 5.0 | 16 | 21 | 0.3 | — |
| 10 | 15 | 5.0 | 16 | <5 | — | 9.2 |
| 11 | 2 | 5.0 | 1/2/4 | 21/39/63 | 3.2 | 4.4 |
| 12 | 3 | 5.0 | 1/2/4 | 12/20/29 | 1.5 | 6.7 |
| 13 | 4 | 5.0 | 1/2/4 | 17/26/38 | 2.0 | 0.5 |
| 14 | 5 | 5.0 | 1/2/4 | 18/30/50 | 2.5 | 3.3 |
| 15 | 6 | 5.0 | 1/2/4 | 10/15/24 | 1.2 | 2.8 |
| 16 | 7 | 5.0 | 1/2/4 | 22/36/59 | 3.0 | −0.4 |
| 17 | 8 | 5.0 | 1/2/4 | 5/10/16 | 1.0 | 10.1 |
| 18 | 9 | 5.0 | 1/2/4 | 18/27/41 | 2.0 | 5.1 |
| Entry | Reducing agent | Solvent | T [°C] | Time (h) | Cat. (mol%) | Conv.a (%) |
|---|---|---|---|---|---|---|
| Reaction conditions: 0.560 M styrene glycol, 0.028 M 3 (5 mol%), 0.610 M reducing agent. a Conversion was determined by 1H-NMR spectroscopy using Ph3CH as an internal standard. b Addition of 0.028 M Ph3PO. c Addition of 0.028 M styrene. d Addition of 0.028 M water. | ||||||
| 1 | Ph3P | C6H6 | 80 | 16 | 5 | >99 |
| 2 | Ph3P | THF | 80 | 16 | 5 | 65 |
| 3 | Ph3P | CH3CN | 80 | 24 | 5 | <5 |
| 4 | Ph3P | Pyridine | 80 | 16 | 5 | 0 |
| 5 | Ph3P | CHCl3 | 90 | 16 | 5 | >99 |
| 6 | Ph3P | CHCl3 | 80 | 16 | 5 | >99 |
| 7 | Ph3P | CHCl3 | 60 | 16 | 5 | 23 |
| 8 | Ph3P | CHCl3 | 40 | 16 | 5 | <5 |
| 9 | H2, 1 bar | CHCl3 | 80 | 16 | 5 | 10 |
| 10 | CO, 1 bar | CHCl3 | 80 | 24 | 5 | 40 |
| 11 | 1-Phenyl ethanol | CHCl3 | 80 | 16 | 5 | 19 |
| 12 | 1,2,3,4-C10H12 | CHCl3 | 80 | 16 | 5 | 15 |
| 13 | Ph3P | CHCl3 | 80 | 4 | 10 | 39 |
| 14 | Ph3P | CHCl3 | 80 | 4 | 5 | 33 |
| 15 | Ph3P | CHCl3 | 80 | 4 | 2.5 | 28 |
| 16b | Ph3P | CHCl3 | 80 | 4 | 5 | 32 |
| 17c | Ph3P | CHCl3 | 80 | 4 | 5 | 32 |
| 18d | Ph3P | CHCl3 | 80 | 4 | 5 | 22 |
The use of different solvents has a significant impact on reaction yield. Reactions conducted in benzene and chloroform result in complete conversion of the diol (entries 1 and 6) whereas more polar tetrahydrofuran, acetonitrile, and pyridine result in moderate to low conversion (Table 2, entries 2–4). It is likely that these latter solvents coordinate to rhenium to inhibit reaction, as observed for CptttReO3.5c It is possible to decrease the catalyst loading from 10 to 2.5 mol% without significant decrease of yield (Table 2, entries 13–15), therefore a loading of 5 mol% was adopted to compare this system with previously reported oxo-rhenium catalysts. Experiments were also carried out to investigate deactivation by the starting materials and/or products (Table 2, entries 16–18). While the addition of 5 mol% of Ph3PO or styrene does not affect the profile of the reaction, water is an inhibitor, with a 10% drop in conversion after 4 h. It should be noted that the inhibition by water, the low conversions seen in more polar solvents, and the toxicities of the benzene and chloroform solvents used to ensure best activity provides a significant challenge to up-scaling this catalyst system.
In contrast with previous DODH studies which operate at high reaction temperatures (≫100 °C), these pyridinium perrhenate catalysts are effective at 80 °C. Significantly, when the reactions are stirred (in contrast to NMR tube reactions), complete conversion is seen in under 4 h at 80 °C. However, reactions carried out below this temperature display a net decrease in activity with conversions of 23 and <5% at 60 and 40 °C respectively (Table 2, entries 7 and 8), while increasing the temperature to 90 °C retains full conversion (Table 2, entry 5).
Alternative reducing agents were evaluated, with H2 and CO resulting in 10% and 40% conversion, respectively (entries 9 and 10). In the latter case, styrene glycol also acts as the reducing agent, which was evident by the presence of oxidized styrene glycol species. Organic reducing agents such as 1-phenylethanol or 1,2,3,4-tetrahydronapthalene were also investigated, but result in poor conversions of 19 and 15%, respectively (Table 2, entries 11 and 12). The lack of effective alternative reducing agents to phosphines as a stoichiometric reductant in this reaction is clearly a hindrance to its large-scale use.
Catalyst scope
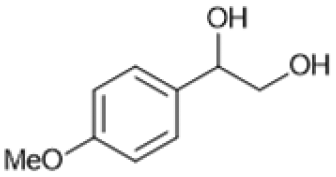
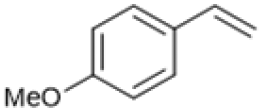
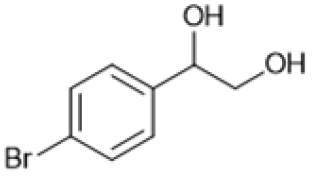
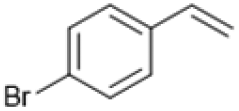
Following these optimization studies, the scope of the reaction was investigated (Table 3). The aromatic vicinal diol styrene glycol (s1) produces a quantitative yield of styrene (p1) without polymerization of the product (Table 3, entry 1), whereas a strongly electron-donating methoxy group (s2), or strongly electron-withdrawing nitro group (s4) decreases both activity and selectivity towards alkene formation (Table 3, entries 2 and 4); the effect is more pronounced for the nitro group. In contrast, using bromide (s3) as a weakly deactivating group provides a quantitative yield of styrene (Table 3, entry 3). No change in activity and selectivity for alkene is seen when the size of the conjugated system is increased by substituting phenyl for naphthalene (s5) (Table 3, entry 5). Although the same activity and selectivity for alkene is seen for meso-hydrobenzoin (s6), this substrate also forms trace benzaldehyde due to C–C bond cleavage (Table 3, entry 6); similar oxidative cleavage has been observed to a higher degree using MeReO3 as a catalyst.10 The functionalized ester, (+)-diisopropyl L-tartrate (s7) is converted with excellent yield (Table 3, entry 7). The alkyl ether, 1,4-anhydroerythritol (s8), requires elevated temperature for the reaction to proceed and results in 51% yield of 2,5-dihydrofuran (p8). The linear aliphatic terminal diols (s9 and s10) also require elevated temperature to give moderate conversions comparable to literature (Table 3, entries 9 and 10).| Entry | Substrate | Product | Time [h] | Conv. [%] | TOF [h−1] | Yielda [%] | ||
|---|---|---|---|---|---|---|---|---|
| Reaction conditions: 0.560 M styrene glycol, 0.028 M 3 (5 mol%), 0.610 M Ph3P, 0.5 mL chloroform solvent, 90 °C. a Conversion and NMR yield were determined by 1H NMR spectroscopy using Ph3CH as an internal standard. b Isolated yield. c Substrate not miscible. d At 140 °C. e Conversion was not measured due to poor substrate solubility. | ||||||||
| 1 |
|
S1 |
|
P1 | 4 | >99 | 5.0 | >99(74)b |
| 2 |
|
S2 |
|
P2 | 4 | 80 | 4.0 | 50 |
| 3 |
|
S3 |
|
P3 | 4 | >99 | 5.0 | >99 |
| 4 |

|
S4 |
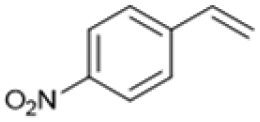
|
P4 | 16 | 55 | 0.7 | 22 |
| 5 |
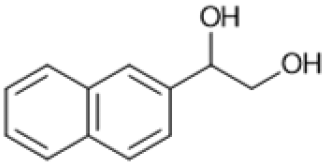
|
S5 |
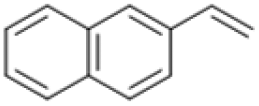
|
P5 | 4 | >99 | 5.0 | >99 |
| 6 |
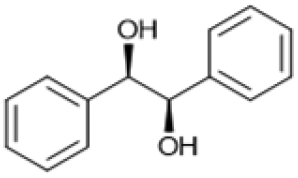
|
S6 |

|
P6 | 4 | —c | 4.9 | 98 |
| 7 |
|
S7 |
|
P7 | 16 | >99 | 1.3 | >99 |
| 8d |
|
S8 |
|
P8 | 16 | 77 | 1.0 | 51 |
| 9d |
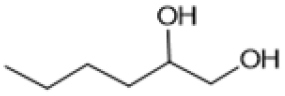
|
S9 |

|
P9 | 16 | 41 | 0.5 | 28 |
| 10d |
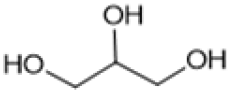
|
S10 |

|
P10 | 16 | —e | 0.3 | 21 |
Evaluation of the mechanism of DODH by perrhenate salts
The relationship between the nature of the cation and catalyst activity under the optimized conditions was probed. The clear requirement of a protic cation led us to study the effect of its pKa and related electronic and steric effects. While the range of pyridinium cations 2 to 7 spans pKa values between −0.4 and 6.7, no clear relationship with their activity is seen (Fig. S26 and S27†). For the ortho-substituted pyridinium ions, there is an increase in catalytic activity going from Me < OMe < F which is allied to an increase in Hammett sigma constant.11 This possible electronic correlation with conversion led us to compare the observed conversions with nucleophilicity (N)12 and Lever electronic parameters (LEP)13 of the neutral amine/pyridine. While no correlation is seen with N, good correlation between LEP and close analogues of the pyridines 3 and 4 and amines 10 and 15 is seen (Fig. S28†). This suggests that the nature of the neutral amine or pyridine has mechanistic consequence, e.g. through coordination to an intermediate (see later). Similar effects were seen in DODH catalysis using MeReO3 with the addition of pyridine or bipy resulting in lower product yields,14 but contrasts to previous evidence that simple ammonium perrhenates are the most active, yet least selective, for alkene formation.14 Besides this apparent electronic parameter effect, there is also a decrease in activity going from o-tBu-substituted 2 to o-Me-substituted 3 (Table 1). Water inhibition could also play a role, as all of these perrhenate salts have some solubility in water, but as no direct relationship is seen a combination of these factors may be operating.The general mechanism of the DODH of diols by oxorhenium complexes has been examined,2c,4b,6d,10,15 and consensus exists in that it proceeds through three steps: (1) reduction of rhenium(VII) to rhenium(V); (2) condensation of the vicinal diol and (3) oxidative extrusion of the alkene and catalyst regeneration. To elucidate the mechanism of this process stoichiometric reactions were targeted. No reaction between 3 and styrene glycol occurs and instead heating triggers substrate oxidation to phenylglyoxal and 2-hydroxy-1-phenylethanone along with the production of styrene. Interestingly, styrene formation is not concomitant with diol oxidation, but delayed (Fig. S30†), which suggests that an initial reduction to a catalytically active rhenium species occurs. To support this, it is found that heating 3 with PPh3 results in phosphine consumption and the slow formation of Ph3PO along with a black, unidentifiable material which is ascribed to excessive rhenium reduction.2c,6e,10
The reaction mechanism was investigated computationally using density functional theory (DFT) at the SMD-B3LYP-D3(BJ)/Def2QZVP//B3LYP-D3(BJ)/6-31G+(d) level of theory (see ESI† for computational details). The preferred pathway proceeds via reduction of Re(VII) to Re(V) followed by diol condensation (path A, Fig. 2) with the route involving initial coordination of the diol to perrhenate (path B, Fig. S32†) and its subsequent reduction much higher in energy. In both cases, reduction of Re(VII) to Re(V) is the rate-determining step. In path A, the first step corresponds to reduction of the metal and has an activation energy of 30.1 kcal mol−1 relative to the independent species. The transition state (TS) structure for this step demonstrates that proton shuttling between the cation and the anionic rhenium species is important. Following this step, dissociation of OPPh3 affords the trigonal planar ReO3− and subsequent addition of the diol to Re(V) viaTS2 (22.6 kcal mol−1) results in tetra-coordinated C; the associated barrier for this step from the intermediate B is similar to that computed for the activation of ethylene glycol by MeReO3 (29.3 kcal mol−1).15 Elimination of water occurs viaTS3 and is the first exothermic process along the path (−13.3 kcal mol−1). Here the hydrogen atom being transferred is located almost halfway between the hydroxide and the oxygen atom of the phenyl glycol, which is stabilized by the lutidinium cation by hydrogen bonding. Finally styrene extrusion further drives the reaction downhill allowing regeneration of the catalyst.
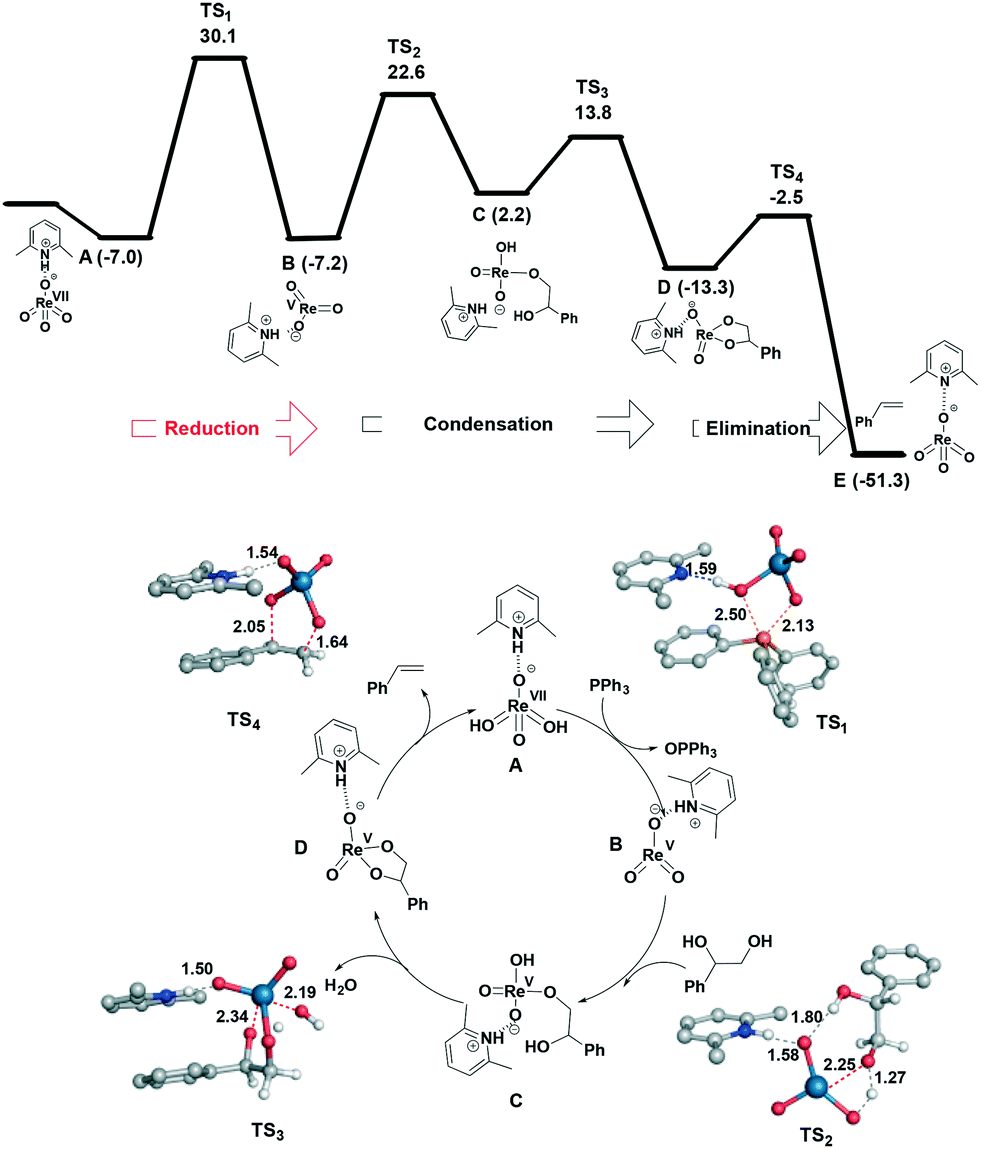 | ||
| Fig. 2 Structures and relative free energies (ΔG, in kcal mol−1; selected distances in Å) of stationary points involved in the catalytic cycle for the DODH reaction of styrene diol by 3. Energy values were calculated at the SMD(CHCl3)B3LYP-D3(BJ)/Def2QZVP//B3LYP-D3(BJ)/6-31G+(d) level of theory relative to the separated species. | ||
Despite the differences observed experimentally for ammonium (15) and lutidinium (3) catalysts, similar energetics were found for both systems and suggests that electronic effects are not the origin of the differences in activity. To probe this further, the coordination of the neutral amine/pyridine to intermediate D (as a consequence of proton transfer, PSoff-cycle) was evaluated (Fig. 3). Modelling this pathway shows an activation energy very similar to the canonical pathway but with a significant decrease in free energy when forming the neutral complex, −28.4 kcal mol−1 for ammonium and −16.6 kcal mol−1 for lutidinium. While this potential off-cycle correlates with the LEP effects seen above, in that the stability of the neutral product Re(OH)(O)(diol)(L) (L = amine or pyridine) decreases with increasing steric bulk, i.e. NH4 > 2,6-Me2py > 2,6-tBu2py, the products of elimination E from the catalytic pathway are of significantly lower energy, indicating that access to this off-cycle should be limited (Fig. S32†). As such, we attribute the differences seen in the catalytic reactivity to the variation in solubility of these complexes which, in the case of the ammonium salt 15, would limit the availability of the perrhenate salt in solution.
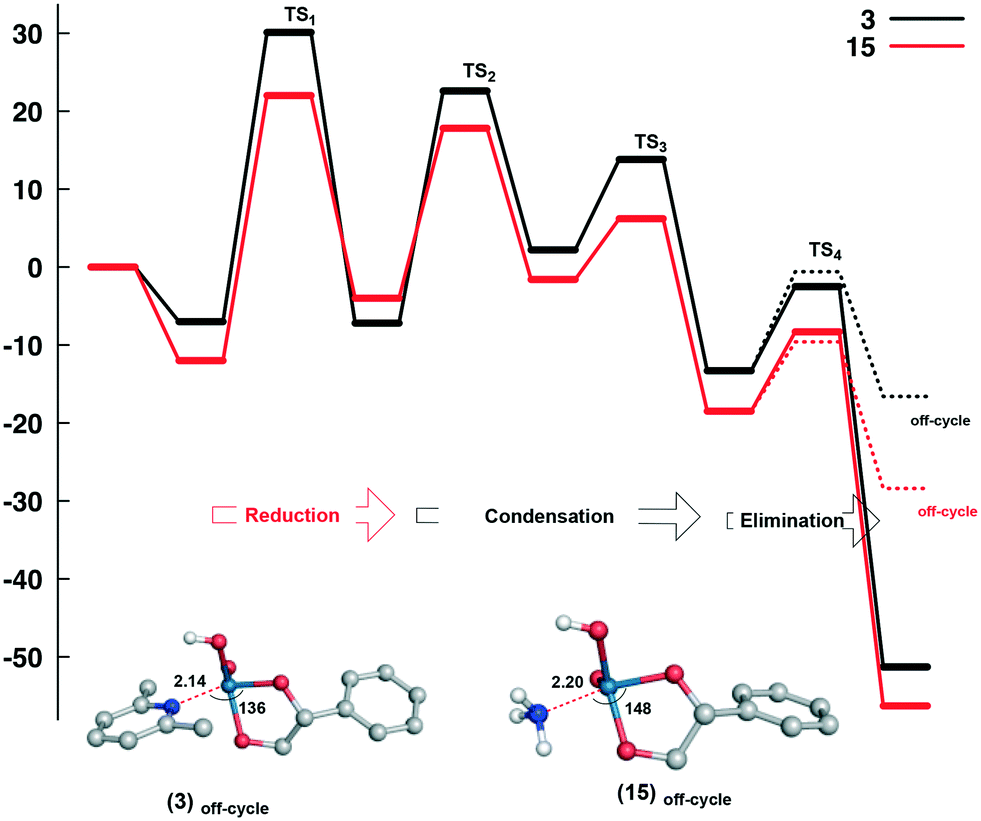 | ||
| Fig. 3 Comparison of the structures and relative free energies (ΔG, in kcal mol−1) for the catalytic DODH reaction of styrene diol by 3 (black) and 15 (red) proceeding via reduction, condensation and elimination (path A). Energy values were calculated at the SMD(CHCl3)B3LYP-D3(BJ)/Def2QZVP//B3LYP-D3(BJ)/6-31G+(d) level of theory relative to the separated species. The production of off-cycle products as a result of proton-shuttling and base coordination is shown. | ||
Conclusions
We have shown that pyridinium perrhenates act as catalysts for the deoxydehydrogenation of diols to alkenes at significantly lower temperatures than other perrhenate salts and rhenium oxo complexes; in the latter cases, these complexes often decompose to poorly active perrhenate salts during the reaction. The supramolecular interactions between the cation and the perrhenate anion are important, not only in stabilizing reaction intermediates and transition states but also in the ability of the cation to act as a Brønsted acid in a proton-shuttling mechanism. Furthermore, enhancing the solubility of the catalyst in hydrophobic solvents is important to its efficacy. It will be interesting to see if this supramolecular ion pair strategy can be applied to other oxo-anions and their chemistry, especially for compounds of earth-abundant metals. Whilst the DODH reactions catalysed by pyridinium perrhenates occur at a lower temperature than previous examples, the use of non-polar, toxic solvents and expensive reducing agents means that alternatives must be sought before these catalysts are applicable to large-scale reactions. Even so, the simplicity in modifying the pyridinium perrhenates described here along with the potential for their heterogenisation on suitable anion-exchange resins could overcome some of these challenges.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Edinburgh and the EPSRC CRITICAT Centre for Doctoral Training (Ph.D. studentship to M.C.; Grant code EP/L016419/1) for financial support.Notes and references
- (a) R. G. Harms, I. I. E. Markovits, M. Drees, W. A. Herrmann, M. Cokoja and F. E. Kühn, ChemSusChem, 2014, 7, 429–434 CrossRef CAS PubMed; (b) S. Dutta, ChemSusChem, 2012, 5, 2125–2127 CrossRef CAS PubMed; (c) C. Wang, Z. Tian, L. Wang, R. Xu, Q. Liu, W. Qu, H. Ma and B. Wang, ChemSusChem, 2012, 5, 1974–1983 CrossRef CAS PubMed.
- (a) S. Raju, M.-E. Moret and R. J. M. Klein Gebbink, ACS Catal., 2015, 5, 281–300 CrossRef CAS; (b) R. G. Harms, W. A. Herrmann and F. E. Kühn, Coord. Chem. Rev., 2015, 296, 1–23 CrossRef CAS; (c) J. R. Dethlefsen and P. Fristrup, ChemSusChem, 2015, 8, 767–775 CrossRef CAS PubMed; (d) C. Boucher-Jacobs and K. M. Nicholas, Top. Curr. Chem., 2014, 353, 163–184 CrossRef CAS PubMed; (e) S. C. A. Sousa, I. Cabrita and A. C. Fernandes, Chem. Soc. Rev., 2012, 41, 5641–5653 RSC.
- (a) D. Lupp, N. J. Christensen, J. R. Dethlefsen and P. Fristrup, Chem. – Eur. J., 2015, 21, 3435–3442 CrossRef CAS PubMed; (b) J. R. Dethlefsen, D. Lupp, A. Teshome, L. B. Nielsen and P. Fristrup, ACS Catal., 2015, 5, 3638–3647 CrossRef CAS; (c) G. Chapman and K. M. Nicholas, Chem. Commun., 2013, 49, 8199–8201 RSC.
- (a) V. Canale, L. Tonucci, M. Bressan and N. d'Alessandra, Catal. Sci. Technol., 2014, 4, 3697–3704 RSC; (b) M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2012, 51, 8082–8086 CrossRef CAS PubMed; (c) E. Arceo, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2010, 132, 11408–11409 CrossRef CAS PubMed.
- (a) S. Raju, J. T. B. H. Jastrzebski, M. Lutz, L. Witteman, J. R. Dethlefsen, P. Fristrup, M.-E. Moret and R. J. M. Klein Gebbink, Inorg. Chem., 2015, 54, 11031–11036 CrossRef CAS PubMed; (b) J. Davis and R. S. Srivastava, Tetrahedron Lett., 2014, 55, 4178–4180 CrossRef CAS; (c) S. Raju, J. T. B. H. Jastrzebski, M. Lutz and R. J. M. Klein Gebbink, ChemSusChem, 2013, 6, 1673–1680 CrossRef CAS PubMed; (d) J. Yi, S. Liu and M. M. Abu-Omar, ChemSusChem, 2012, 5, 1401–1404 CrossRef CAS PubMed; (e) S. C. A. Sousa and A. C. Fernandes, Tetrahedron Lett., 2011, 52, 6960–6962 CrossRef CAS; (f) G. K. Cook and M. A. Andrews, J. Am. Chem. Soc., 1996, 118, 9448–9449 CrossRef CAS.
- (a) L. Sandbrink, E. Klindtworth, H.-U. Islam, A. M. Beale and R. Palkovits, ACS Catal., 2016, 6, 677–680 CrossRef CAS; (b) H. Sun, C. Hu, Z. Hao, Y. Zuo, T. Wang and C. Zhong, Chin. J. Org. Chem., 2015, 35, 1904–1909 CrossRef CAS; (c) M. J. McClain and K. M. Nicholas, ACS Catal., 2014, 4, 2109–2112 CrossRef; (d) X. Li, D. Wu, T. Lu, G. Yi, H. Su and Y. Zhang, Angew. Chem., Int. Ed., 2014, 53, 4200–4204 CrossRef CAS PubMed; (e) M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2013, 52, 12905–12909 CrossRef CAS PubMed; (f) C. Boucher-Jacobs and K. M. Nicholas, ChemSusChem, 2013, 6, 597–599 CrossRef CAS PubMed; (g) S. Vkuturi, G. Chapman, I. Ahmad and K. M. Nicholas, Inorg. Chem., 2010, 49, 4744–4746 CrossRef CAS PubMed.
- I. Ahmad, G. Chapman and K. M. Nicholas, Organometallics, 2011, 30, 2810–2818 CrossRef CAS.
- S. Stolte, H. T. T. Bui, S. Steudte, V. Korinth, J. Arning, A. Białk-Bielińska, U. Bottin-Weber, M. Cokoja, A. Hahlbrock, V. Fetz, R. Stauber, B. Jastorff, C. Hartmann, R. W. Fischer and F. E. Kühn, Green Chem., 2015, 17, 1136–1144 RSC.
- M. Cokoja, I. I. E. Markovits, M. H. Anthofer, S. Poplata, A. Pöthig, D. S. Morris, P. A. Tasker, W. A. Herrmann, F. E. Kühn and J. B. Love, Chem. Commun., 2015, 51, 3399–3402 RSC.
- S. Liu, A. Senocak, J. L. Smeltz, L. Yang, B. Wegenhart, J. Yi, H. I. Kenttämaa, E. A. Ison and M. M. Abu-Omar, Organometallics, 2013, 32, 3210–3219 CrossRef CAS.
- C. Hansch, S. D. Rockwell, P. Y. C. Jow, A. Leo and E. E. Steller, J. Med. Chem., 1977, 20, 304–306 CrossRef CAS PubMed.
- H. Mayr and A. R. Ofial, J. Phys. Org. Chem., 2008, 21, 584–595 CrossRef CAS.
- A. B. P. Lever, Inorg. Chem., 1990, 29, 1271–1285 CrossRef CAS.
- X. Li and Y. Zhang, ChemSusChem, 2016, 9, 2774–2778 CrossRef CAS PubMed.
- (a) D. Wu, Y. Zhang and H. Su, Chem. – Asian J., 2016, 11, 1565–1571 CrossRef CAS PubMed; (b) C. Boucher-Jacobs and K. M. Nicholas, Organometallics, 2015, 34, 1985–1990 CrossRef CAS; (c) P. Liu and K. M. Nicholas, Organometallics, 2013, 32, 1821–1831 CrossRef CAS; (d) S. Qu, Y. Dang, M. Wen and Z.-X. Wang, Chem. – Eur. J., 2013, 19, 3827–3832 CrossRef CAS PubMed; (e) S. Bi, J. Wang, L. Liu, P. Li and Z. Lin, Organometallics, 2012, 31, 6139–6147 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental, crystallographic, and computational details. CCDC 1568774–1568780. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cy01728f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |