
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEpoxidation of propene using Au/TiO2: on the difference between H2 and CO as a co-reactant†
Shamayita
Kanungo‡
a,
Yaqiong
Su‡
b,
M. F.
Neira d'Angelo
a,
Jaap C.
Schouten
a and
Emiel J. M.
Hensen
 *b
*b
aLaboratory of Chemical Reactor Engineering, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
bLaboratory of Inorganic Materials Chemistry, Schuit Institute of Catalysis, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: e.j.m.hensen@tue.nl
First published on 21st April 2017
Abstract
The role of the reducing gas in the direct epoxidation of propene to propene oxide (PO) using O2 over a Au/TiO2 catalyst was studied through experiments and density functional theory calculations. It was found that PO can be obtained using both H2 and CO as co-reactants. The yield of PO was much lower with CO than that with H2. The role of the oxygen atoms of the titania support was studied by quantum-chemical investigations, which show that the mechanism involving CO as a co-reactant should proceed via surface oxygen vacancies, whereas with H2 the well-accepted pathway involving OOH is favored. Steady-state isotopic transient kinetic analysis experiments demonstrate that support oxygen atoms are involved in PO formation when CO is used as the co-reactant.
Introduction
Propene oxide (PO) is an important bulk chemical intermediate, which finds application in the production of a wide range of commercial commodities.1 The demand for PO is steadily growing. Unlike its homologue ethylene oxide, PO cannot be obtained directly by oxidation of the olefin with oxygen. Accordingly, alternative approaches have been developed for producing this valuable compound. Several of these processes are carried out on an industrial scale, despite some shortcomings such as the production of hazardous wastes (chlorohydrin process), the dependence on the economics of a co-product (SMPO and MTBE/PO processes), the requirement of multiple processing steps (chlorohydrin, SMPO, HPPO and cumene recycling process) and the use of an expensive reactant (HPPO process). Therefore, the one-step synthesis of PO directly from propene and O2 remains the most coveted route.2Despite the development of several catalysts for direct epoxidation of propene, the reported conversions and selectivities are poor.3 One of the most promising approaches is to use gold nanoparticle catalysts in combination with Ti, either in the form of titania or as isolated Ti species dispersed on silica, in combination with a sacrificial reducing gas such as H2.4 The use of a reducing co-reactant has been found to be indispensable for obtaining reasonable conversion of propene (5–10%), while maintaining high PO selectivity (>85%). Since this discovery by Haruta et al.,4 significant research efforts have been made to improve the catalyst systems. Some examples of active catalysts are gold dispersed on TS-1, Ti-SiO2, Ti-MCM-41 and Ti-SBA-15.5–10 An inherent drawback of this approach is the formation of an excessive amount of water by oxidation of hydrogen. Overall, the low H2 efficiency constitutes a major economic hurdle. It has been reported that water can replace hydrogen, although propene conversion and PO selectivity then become very low.11,12
It is generally assumed that propene oxidation takes place at the interface between Au nanoparticles and Ti sites. Radical ˙OOH (hydroperoxo) species formed on gold by the interaction of O2 and H species are thought to be the oxidizing species that convert propene to PO.13 Theoretical studies have contributed to this discussion by exploring different reaction pathways and reaction intermediates. Quantum-chemical calculations performed by Molina et al.14 used a one-dimensional gold rod on an anatase-TiO2 (101) slab to model Au/TiO2. This study indicated that propene can be adsorbed with one end on the titania surface and with the other on Au. Reactive ˙OOH radicals were argued to be essential for converting propene to a metallacycle C3H6O intermediate. A theoretical study of Delgass and co-workers on Au/TS-1 emphasized the role of gold clusters in forming H2O2 and the role of Ti defect sites in their reaction with propene and H2O2 to form the desired product.15 In a later work, the influence of Au–Ti proximity on the reaction mechanism was studied. Based on the finding that the formation of Ti–OOH on a Au3/Ti-defect site model was unfavorable, a concerted mechanism was proposed in which propene is adsorbed on the Au–Ti interface rather than on a Ti site.16 Haruta et al.17 proposed that electron transfer from Au to TiO2 may lead to the formation of Ti3+–O–Au+ species. These Ti3+ surface sites can activate molecular oxygen with subsequent formation of a hydroperoxo species, which reacts with propene adsorbed on Au to form PO. In summary, although the exact mechanism remains debated, it is clear that ˙OOH radicals and the Au–Ti interface play significant roles in the pathway towards PO formation.
A recent study by Sobolev and co-workers demonstrated that PO can also be formed when H2 is replaced by CO, using a Au/TiO2 catalyst.18 An advantage of replacing H2 with CO is that it largely suppresses the formation of propane,19 which is an undesirable side reaction observed.8,19,20 These authors proposed that the reaction propagates via oxygen vacancy formation on the titania support. They also argued that this mechanism may also be operative in the propene/oxygen/hydrogen case, which is in discord with the well-accepted mechanism involving surface hydrogen peroxide/hydroperoxo (HOOH/˙OOH) species.18,21 This discrepancy gives rise to a number of important research questions that have not been addressed yet, namely (a) what is the role of support oxygen atoms in the epoxidation of propene, (b) can one indeed replace H2 with other reducing gases such as CO, and if so (c) what is the underlying mechanism and what role does the support play in such a mechanism. Earlier investigations into the role of the support oxygen atoms in propene epoxidation were inconclusive.22
In this work, we compared the direct epoxidation of propene using a Au/TiO2 catalyst in the presence of H2 or CO as co-reactants. Steady-state isotopic transient kinetic analysis (SSITKA) was employed to determine the role of support oxygen atoms. The experimental part of this study was complemented by density functional theory (DFT) calculations of the reaction mechanism using a model consisting of a small Au cluster supported on titania. The catalytic activity measurements show that the PO yield with CO as the co-reactant is much lower than that obtained using H2 as the co-reactant. Similar observations were made for other catalyst compositions and under different reaction conditions. The DFT calculations explain this difference by identifying different favorable routes for the formation of PO with CO and H2. Significantly, these data show that the O atoms of titania are involved in the catalytic cycle, a prediction corroborated by SSITKA experiments. The combined experimental and theoretical data provide new insight into the role of the co-reactant in the selective oxidation of propene to PO.
Experimental methods
Catalyst preparation
Au/TiO2 was prepared by two different preparation methods. A catalyst denoted Au/TiO2-NM was prepared as outlined by Nijhuis et al.,22i.e., by deposition–precipitation of Au on titania. In a typical synthesis, 1 g of TiO2 (Degussa P-25) was suspended in 50 ml of demineralized water. The pH was adjusted to ∼9.5 using a 2.5 wt% ammonia solution. Then, 35.5 μl of HAuCl4 (Aldrich, 17 wt% Au), diluted in 20 ml of water, was added dropwise (over 15 min) using a burette. The slurry was kept stirring for 1 h, while maintaining the pH at 9.4–9.5 by adding ammonia. Next, the solid was collected by filtration and washed three times using demineralized water. The catalyst was dried overnight at 80 °C and calcined in static air at 120 °C (5 °C min−1, isothermal period = 2 h) and 400 °C (10 °C min−1, isothermal period = 4 h). The resulting catalyst had an intense purple blue color.Another catalyst, denoted Au/TiO2-SM, was made as outlined by Sobolev and Koltunov.18,21 A gold precursor solution was made by dissolving 71 μl of HAuCl4 (Aldrich, 17 wt% Au) in 100 ml of demineralized water and heated to 70 °C. Then, 1 g of TiO2 (Degussa P-25) was added to the solution and stirring was continued for 1 h. After collecting the solid by centrifugation, it was suspended in 50 ml of NH4OH (4 M) and the mixture was stirred at 70 °C for 1 h. The suspension was then filtered and washed with 100 ml of demineralized water. Calcination was carried out in static air at 80 °C for 10 h, followed by 300 °C for 4 h. The catalyst had a similar appearance to Au/TiO2-NM, but was slightly lighter in color suggestive of a lower Au uptake.
The catalyst Au/Ti–SiO2 was made in the same way as Au/TiO2-NM. The support was Ti grafted on commercial SiO2 (Ti–SiO2). The detailed support preparation can be found in the literature.8,9
Catalyst characterization
Transmission Electron Microscopy (TEM) was used to determine the size distribution of the Au particles in both catalysts. The TEM images were recorded using a FEI Tecnai G2 Sphera transmission electron microscope at an accelerating voltage of 200 kV. Sample preparation involved sonication of the finely ground catalyst in ethanol and application of a few drops of the suspension onto a TEM grid. The size distribution was determined by counting all visible gold particles from approximately 20 images (at least 150 gold particles in total).The gold loading in the catalysts was analyzed by inductively coupled plasma optical emission spectroscopy (ICP-OES) using a SpectroCiros CCD spectrometer. First, aqua regia was added to the samples, and then these mixtures were heated under stirring for 30 min. The solutions were cooled and then dilute HF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 by volume in water) was added, followed by filtration.
:15 by volume in water) was added, followed by filtration.
Catalytic activity measurements
Catalytic activity measurements were performed in a flow set-up equipped with an Interscience gas chromatography system (analysis time of ∼5 min), containing two analysis channels with Porabond Q and Molsieve 5A columns and thermal conductivity detectors. The catalyst was loaded into a quartz reactor tube (inner diameter = 6 mm) which was placed in a tubular oven. A typical reaction cycle was 2 h long with a regeneration step in between at 300 °C with 10 vol% O2 in He for 1 h. In a typical test, 150 mg of catalyst was loaded into the reactor, and the total flow was adjusted to 25 ml min−1 (GHSV = 10000 ml gcat−1 h−1). The catalysts were tested under two conditions, namely at X/O2/C3H6/He = 1:1:1:7 and X/O2/C3H6/He = 5:10:5:80, where X is H2 or CO. The propylene conversion and selectivity to PO are expressed as: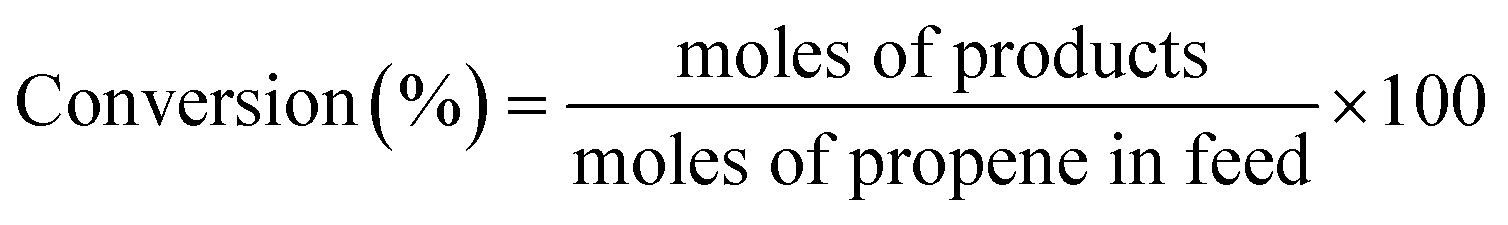 | (1) |
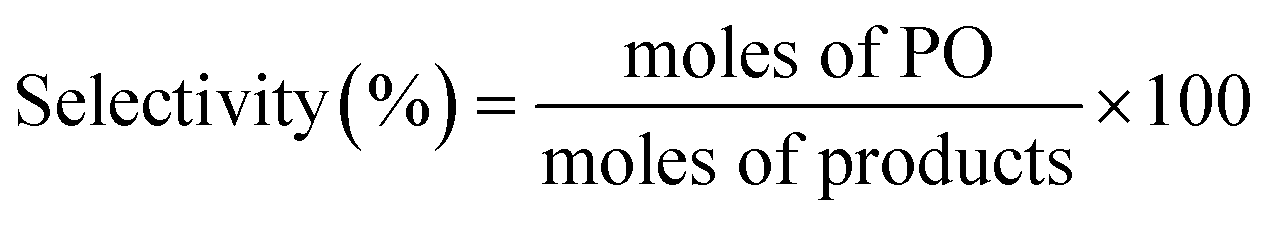 | (2) |
Steady-state isotopic transient kinetic analysis (SSITKA)
SSITKA experiments were performed in a similar way as described in the literature.22 In brief, 0.3 g of catalyst was loaded into the reactor after dilution with SiC to maintain a uniform reaction temperature. The reactor feed was then switched to a mixture of 30 ml min−1 Ar, 5 ml min−1 propene, 5 ml min−1 16O2 and an additional 5 ml min−1 Ar. After reaching a steady state, the latter two flows (5 ml min−1 16O2 and 5 ml min−1 Ar) were replaced by 5 ml min−1 18O2 and 5 ml min−1 Ne. The reaction was then continued for 20 min using 18O2. Product analysis was performed using a mass spectrometer (MS) and a gas chromatography–mass spectrometry (GC–MS) system.Computational methods
The mechanism of propene oxidation was investigated by spin-polarized periodic DFT calculations using the Vienna Ab Initio Simulation Package (VASP).23 The ion–electron interactions were represented by the projector-augmented wave (PAW) method,24 and the electron exchange–correlation by the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional.25 A plane-wave basis set with a cut-off energy of 400 eV was used. The Brillouin zone integration was carried out in the Γ-point for a Au6 cluster placed on a 5 × 2 supercell of a rutile-TiO2(110) surface containing three O–Ti–O layers, with a vacuum space of 20 Å to avoid spurious self-interactions.26 The (110) termination is the most stable among the rutile low-index surfaces. The small 6-atom gold cluster was chosen as a model for gold nanoparticles in their interaction with the titania support. The convergence criteria for the force and the energy were set at 50 meV Å−1 and 0.1 meV, respectively. During geometry optimization, the bottom layer was frozen, while the upper two layers were fully relaxed. All adsorbates were fully relaxed during all calculations. The DFT+U methodology (U is a Hubbard-like term describing the on-site Coulomb interactions) was used. Ueff was set to 4.0 eV for the Ti 3d orbital.27 The climbing image nudged-elastic band (CI-NEB) algorithm was used to search for the minimum energy pathways.28,29Results and discussion
Characterization
Fig. 1 shows representative TEM images of Au/TiO2-NM and Au/TiO2-SM. These images clearly show the presence of gold nanoparticles homogeneously distributed over the support. Statistical analysis shows that the average particle size and the distribution are comparable between the two catalysts. The average gold particle size is 3.4 nm for Au/TiO2-NM, in close agreement with values reported for gold catalysts synthesized using this method.22 The average particle size is 3.1 nm for Au/TiO2-SM, which is slightly higher than the Au particle size reported by Sobolev and Koltunov.21Table 1 lists the targeted and analyzed Au content as determined by ICP-OES. For the TiO2-NM support, the actual Au loading is close to the targeted loading. For Au/TiO2-SM, the actual Au loading is lower than the targeted loading.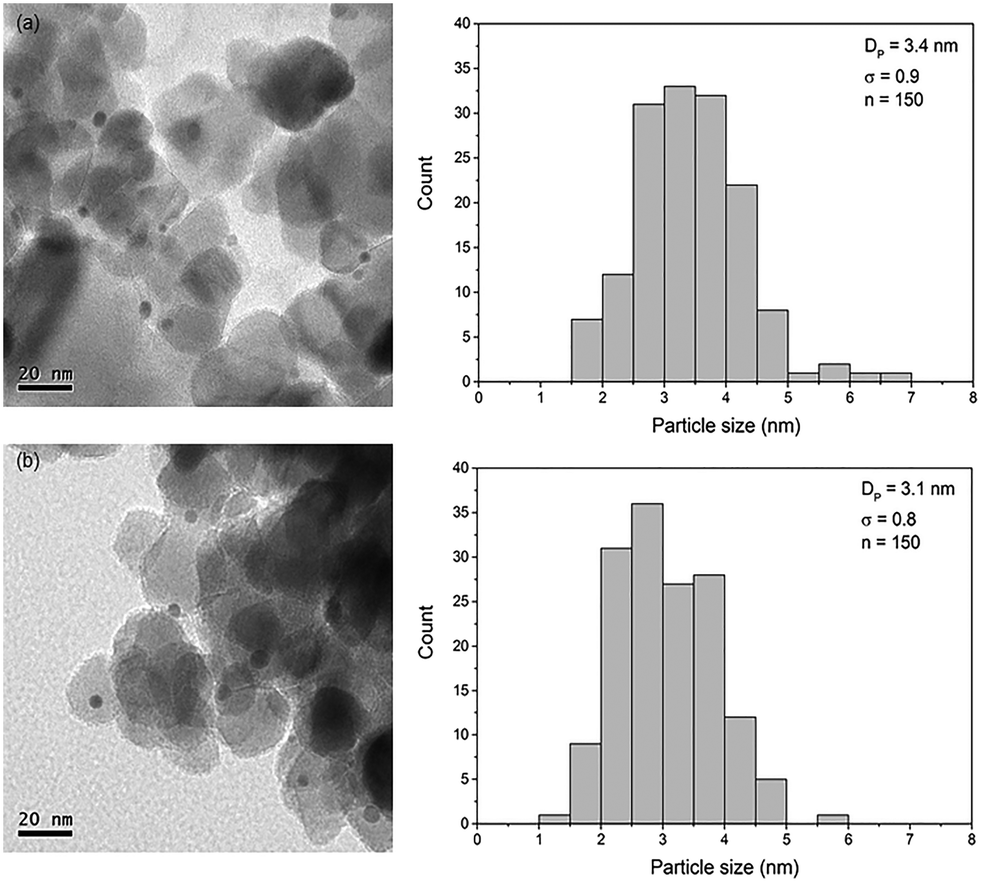 | ||
| Fig. 1 TEM images of (a) Au/TiO2-NM and (b) Au/TiO2-SM along with the corresponding particle size distribution, showing the average particle size with standard deviation and the total number of particles counted. | ||
Catalytic activity measurements
The performance of Au/TiO2-NM and Au/TiO2-SM in propene epoxidation was evaluated using a gas feed composed of either H2/O2/C3H6 or CO/O2/C3H6. The reaction conditions were a temperature of 50 °C, a GHSV of 10000 mL gcat−1 h−1 and a volumetric feed composition of H2 (or CO)/O2/C3H6/He = 1/1/1/7. The catalyst prepared as outlined by Sobolev and Koltunov21 was also tested at 70 °C, a GHSV of 6000 mL gcat−1 h−1, and a volumetric feed composition of H2 (or CO)/O2/C3H6/He = 5:10:5:80 for reasons of comparison to literature data.18,21
Fig. 2 and 3 show the performance in terms of the rate of formation of PO with time on stream of Au/TiO2-NM and Au/TiO2-SM, respectively. For both catalysts, the rate of PO formation is higher when H2 is used as the reducing gas in comparison with CO. With time on stream, the catalysts display initially increasing activity followed by a sharp decline, which is typical for titania-supported gold catalysts used in propene oxidation.30 The maximum PO yield using H2 for Au/TiO2-NM is 1.6% (∼20 gPO kgcat−1 h−1) and the selectivity to PO is higher than 99%, which is in good accordance with the literature.4 When CO is used in place of H2, the maximum yield obtained is 0.6% (∼14 gPO kgcat−1 h−1) with similarly high PO selectivity. It should be mentioned here that CO2 was excluded from the conversion and selectivity data when CO was the co-reactant. This is based on the assumption that CO2 is the product of CO oxidation rather than a full combustion product from propene. Water is the undesired by-product of combustion of the H2 co-reactant. The rates of H2O and CO2 formation for both co-reactants are shown in Fig. 4. These data trend with time on stream in the same way as the PO formation rate, suggesting that the active sites for PO formation and co-reactant combustion are similar. Water is primarily formed by the undesired oxidation of hydrogen at Au sites.2,4,8 Au/TiO2 is a known catalyst for CO oxidation to CO2, so it is reasonable to argue that the majority of the CO2 product stems from the oxidation of CO at Au sites.26,31,32 There are strong indications that the Au–TiO2 interface plays an important role in catalyzing CO oxidation.26,33 In both cases, the co-reactant efficiency, defined as the ratio of PO to CO2 or H2O, is around 20–25%.
 | ||
| Fig. 2 Time-on-stream formation rate of PO during a 2 h catalytic test over Au/TiO2-NM at X/O2/C3H6/He = 1:1:1:7, where X = CO (black box) and H2 (red circles), at 50 °C and GHSV = 10000 mL gcat−1 h−1 (N.B.: 1 × 10−7 mol gcat−1 s−1 corresponds to 20.9 gPO kgcat−1 h−1; solid lines are drawn to guide the eye). | ||
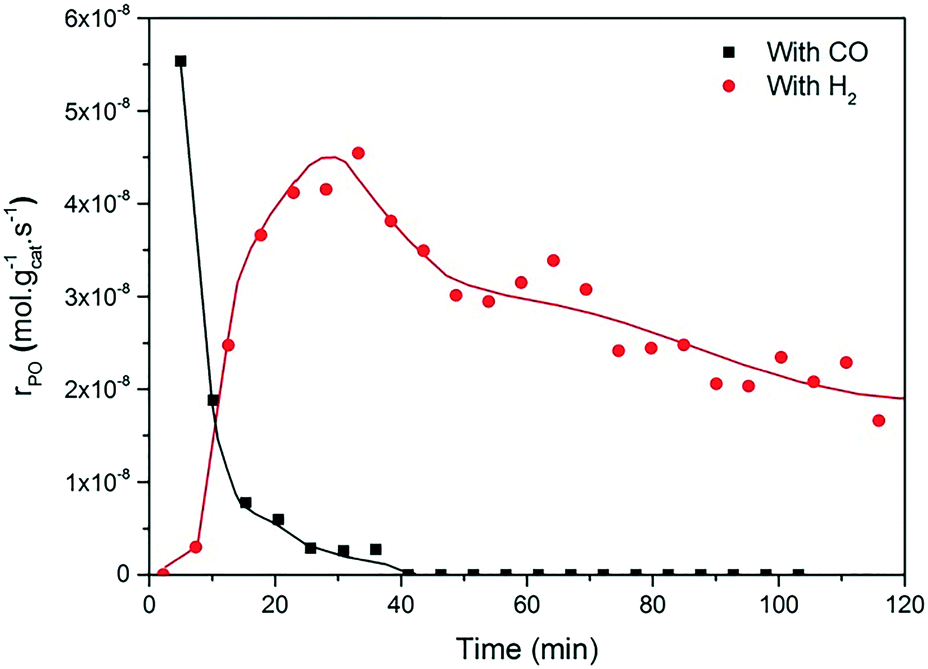 | ||
| Fig. 3 Time-on-stream formation rate of PO during a 2 h catalytic test over Au/TiO2-SM at X/O2/C3H6/He = 1:1:1:7, where X = CO (black box) and H2 (red circles), at 50 °C and GHSV = 10000 mL gcat−1 h−1. | ||
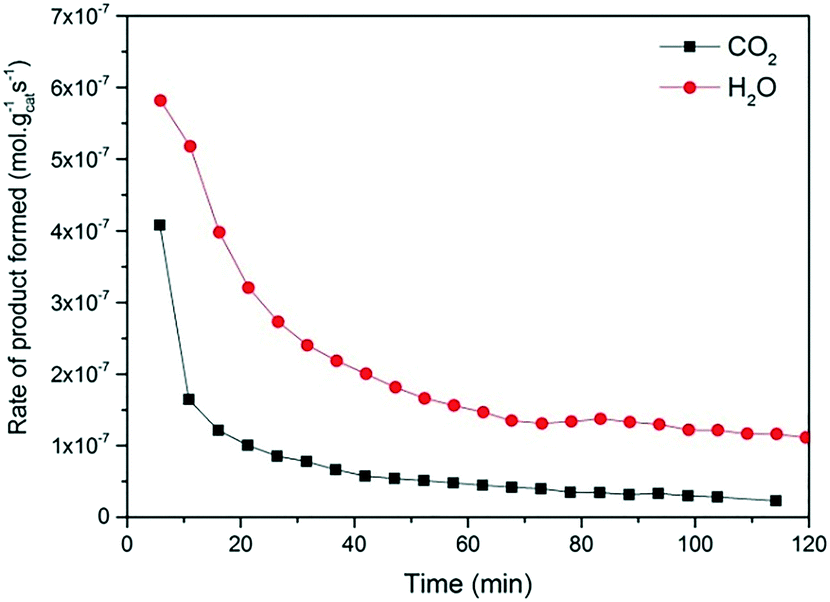 | ||
| Fig. 4 Time-on-stream formation rates of CO2 and water during a 2 h catalytic test over Au/TiO2-NM at X/O2/C3H6/He = 1:1:1:7, where X = CO (black box) and H2 (red circles); temp = 50 °C and GHSV = 10000 mL gcat−1 h−1. | ||
The strong deactivation observed for both catalysts has been extensively studied in the past and is attributed to the strong adsorption of reaction products on the catalyst surface, possibly involving subsequent oligomerization.30,34 The activity can be fully recovered by treating the catalysts in 10 vol% O2/He at 300 °C for 1 h. This regeneration is effective for catalysts operated with either H2 or CO as a co-reactant. This may indicate that the deactivation mechanism for both cases is the same.
The present results are at odds with those of Sobolev and Koltunov,18,21 who reported higher PO yields when CO was the co-reactant. The present data show that PO is formed when CO is the co-reactant, but the activity is substantially lower in comparison with that obtained using H2 as the co-reactant. To ensure that the slightly different reaction conditions do not cause this difference, we evaluated the catalytic performance of Au/TiO2-SM, the same catalyst explored by Sobolev and Koltunov, under the same conditions. The results shown in Fig. 5 confirm that the PO formation rates are lower with CO than those with H2. Deactivation is more pronounced in this case and the PO formation rate drops to negligible values after about 1 h.
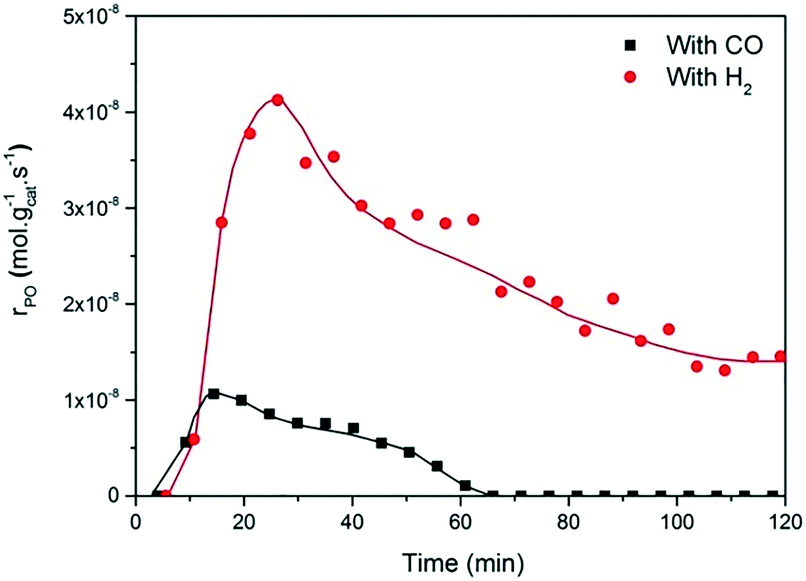 | ||
| Fig. 5 Time-on-stream formation rate of PO during a 2 h catalytic test over Au/TiO2-SM at X/O2/C3H6/He = 5:10:5:80, where X = CO (black box) and H2 (red circles), at 70 °C and GHSV = 6000 mL gcat−1 h−1, the same conditions used by Sobolev and Koltunov.18 | ||
We noted that the Au loading in our case was lower than that reported in the literature.21 Therefore, we prepared a 2 wt% Au-loaded catalyst (Au/TiO2-NM) by deposition–precipitation as this method ensures complete uptake of the gold in the solution. The resulting performance of this catalyst with CO and H2 (ESI†) confirmed that H2 is the preferred co-reactant. Finally, as it is also known that catalysts based on Ti sites dispersed on silica as the support for gold exhibit more stable and active catalytic performance, we compared the performance of 0.1% Au/Ti–SiO2 in both reactions (Fig. 6). We found that the PO formation rates were stable and significantly higher with H2 as the co-reactant.
 | ||
| Fig. 6 Time-on-stream formation rate of PO during a 2 h catalytic test over 0.1% Au/Ti–SiO2 at X/O2/C3H6/He = 1:1:1:7, where X = CO (black box) and H2 (red circles), at 200 °C and GHSV = 10000 mL gcat−1 h−1. | ||
Computational modeling
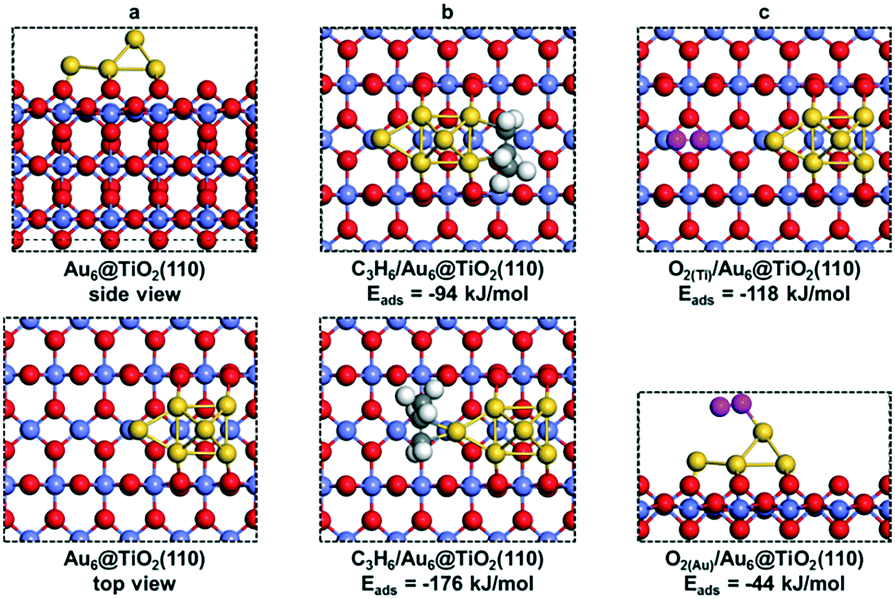 | ||
| Fig. 7 (a) Side (above) and top (below) views of Au6@TiO2(110). (b) C3H6 adsorbed on dual Au atoms (above) and single Au atom (below) at the Au6@TiO2(110) interface. (c) O2 adsorbed on exposed Ti atoms (above) and supported Au6 cluster (below) at the Au6@TiO2(110) interface. Color code: red, titania surface oxygen; pink, oxygen of adsorbed O2; grey, carbon; yellow, Au; blue, Ti; white, hydrogen. | ||
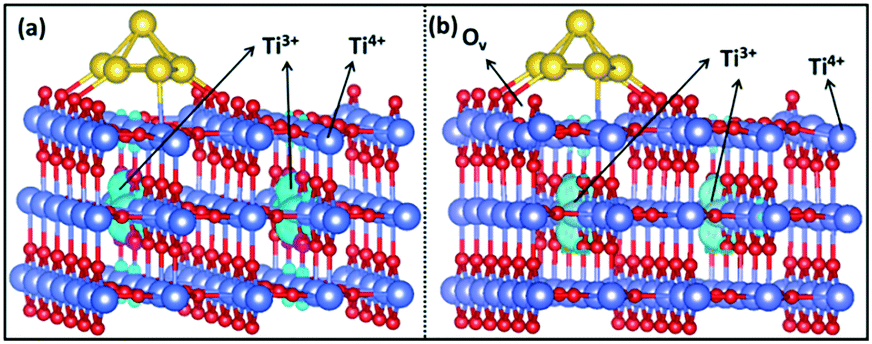 | ||
| Fig. 8 The spin-density iso-surface (in light blue) of Ti3+ cations in (a) intact Au6@TiO2(110) and (b) defective Au6@TiO2(110) with one oxygen vacancy. | ||
 | ||
| Fig. 9 The reaction energy diagram with elementary reaction steps for the epoxidation of propylene to propylene oxide (PO) using an O2 molecule with or without H2 assistance. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of the adsorbed propene, involving a barrier of 63 kJ mol−1 (TS2), resulting in the formation of an OH group on the surface. Desorption of PO costs 137 kJ mol−1 and the adsorbed OH intermediate reacts with the acidic OH group to form water (barrier of 14 kJ mol−1). The reaction cycle ends by the desorption of water (ΔE = 112 kJ mol−1).
C double bond of the adsorbed propene, involving a barrier of 63 kJ mol−1 (TS2), resulting in the formation of an OH group on the surface. Desorption of PO costs 137 kJ mol−1 and the adsorbed OH intermediate reacts with the acidic OH group to form water (barrier of 14 kJ mol−1). The reaction cycle ends by the desorption of water (ΔE = 112 kJ mol−1).
It is interesting to compare the transition states for PO formation involving OOH and O2 (see the ESI†). In the former case, the C1–O bond is substantially longer and the C1–Au bond is shorter than those in the O2-assisted pathway. In the transition state TS2, the O–O and C1–O bond lengths are 1.74 and 2.00 Å, and the Au–C1 and Au–C2 bond lengths are 2.47 and 2.20 Å. In the transition state TS1′ for PO formation, the O–O and C1–O bond lengths are 1.74 Å and 1.83 Å, respectively; those between Au and C1 and C2 are 2.67 Å and 2.18 Å, respectively. The O–O bond length of ˙OOH is 1.46 Å, which is longer than that of adsorbed O2 (1.41 Å), demonstrating that the O–O bond becomes weakened after hydrogenation. Moreover, one OH˙ species is left after the reaction of ˙OOH with propene, while one isolated oxygen is left on the surface without H2. The presence of hydrogen stabilizes the isolated oxygen atom.
As mentioned earlier, two Ti3+ ions are present in the Au6/TiO2(110) model (Fig. 8a). When O2 is adsorbed, the Ti3+ ions are reoxidized to Ti4+ and the adsorbed O2 molecule becomes negatively charged, obtaining a superoxide character. H2 adsorption and dissociation will reduce two Ti4+ into Ti3+ ions again and generate the OOH− anion involved in propylene epoxidation. Obviously, the Ti3+ cation plays an important role in the overall reaction, which increases the reducibility of the support surface and catalyzes the O22−/OOH− anion formation for subsequent propene epoxidation.
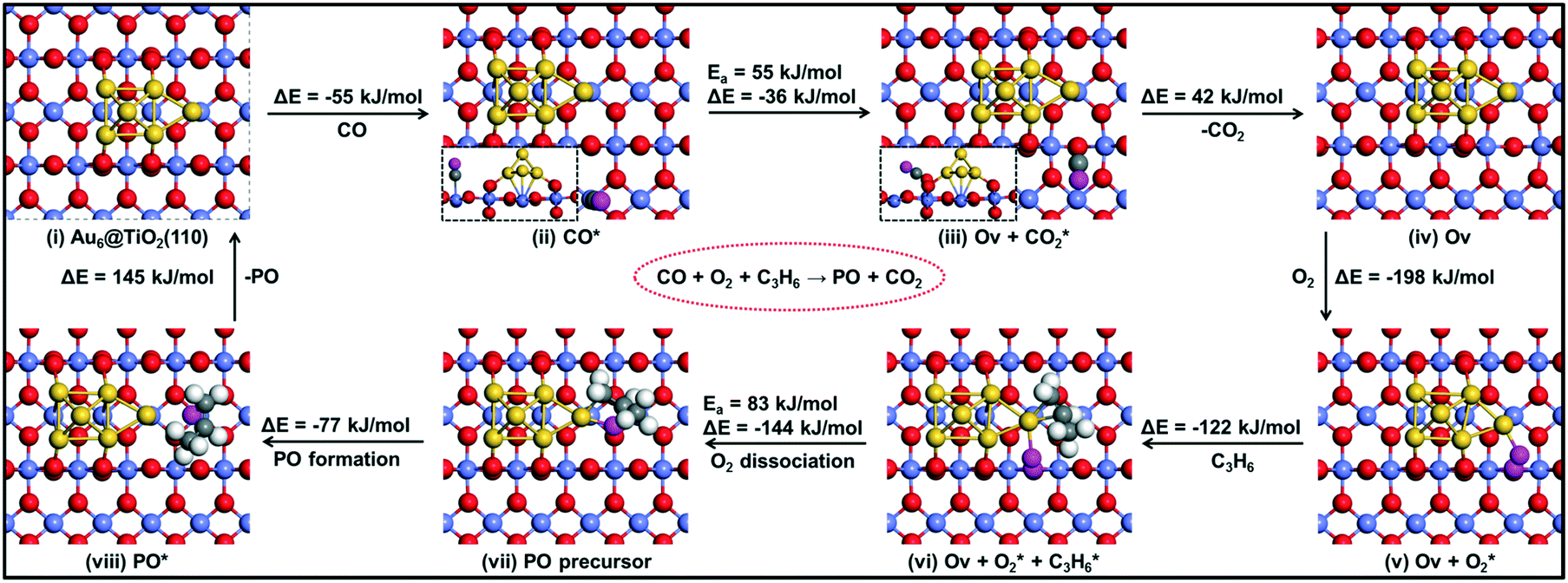 | ||
| Fig. 10 The reaction energy diagram with elementary reaction steps for the epoxidation of propylene to propylene oxide (PO) using a CO molecule. | ||
It is useful to compare the energy barriers to alternative mechanisms involving only titania or gold. Molina et al. reported a barrier of ∼150 kJ mol−1 for PO formation on titania.43 Another study focusing on gold itself showed that adsorption of molecular oxygen is weak (Eads > −20 kJ mol−1) and the barrier for PO formation via the ˙OOH radical is ∼95 kJ mol−1.44 Both of these mechanisms are more difficult than that of the reaction explored by our group at the Au–TiO2 interface. Accordingly, the present results emphasize the important role of the Au–Ti interface, in line with experimental data.
We summarize the DFT calculations by comparing the different pathways without and with co-reactants. In the absence of a co-reactant, the activation barrier for PO formation is 107 kJ mol−1, while the involvement of OOH˙ due to the presence of H2 lowers this activation energy to 63 kJ mol−1. When CO is used as the co-reactant, the mechanism proceeds via a surface oxygen vacancy and the corresponding activation energy is found to be 83 kJ mol−1. We observe that these trends are in good agreement with the experimental findings, where PO is not reported to be formed in the absence of a sacrificial agent on Au–Ti catalysts,1–3 and the PO yield in the CO-assisted epoxidation of propene is much lower than that in its hydro-epoxidation, as also revealed by the catalytic performance data in the present study.
SSITKA
From the catalytic experiments, we established that CO can be used as an alternative co-reactant in the direct epoxidation of propene, while DFT calculations for this reaction revealed that support oxygen plays a crucial role in the reaction mechanism towards PO formation. To explore the role of support oxygen experimentally, SSITKA studies were carried out by switching 16O2 with 18O2 under steady-state reaction conditions.Scheme 1 shows the mechanism of PO formation with CO as the co-reactant, as proposed by DFT calculations and the most likely surface species present in the SSITKA experiment. The product stream is expected to predominantly consist of C3H618O (PO18), C16O18O and C16O2. C18O2 is expected as a combustion product of PO18 and to be present only in small amounts given the low PO yield.
 | ||
| Scheme 1 Surface species expected, before and after isotopic switch, in the direct epoxidation of propene with CO. | ||
Fig. 11 shows the transients for CO2 isotopomers during epoxidation with CO, after switching from 16O2 to 18O2. All three CO2 isotopomers are observed and their relative concentrations at the (pseudo) steady state are C16O2:C16O18O:C18O2 ∼ 9:6:1. That is to say, the predominant product is C16O2 and it is still seen 15 min after the isotopic switch, indicating the contribution of the support surface oxygen atoms to the reaction mechanism. If the O atoms of the support were not involved, we would expect the predominant product to be C16O18O, with the C16O2 concentration dropping to negligible levels within a very short time after the switch. Nevertheless, it can be argued that some of the formed C16O18O exchanges with surface oxygen to give C16O2. This was for instance observed in a SSITKA study of CO oxidation on Au/TiO2, but the relative amount of C16O2 was much lower with a C16O2:C16O18O:C18O2 ratio of ∼1:2:1.31,32 Hence, even upon considering some exchange with support oxygen, it is highly unlikely that C16O2 would be the predominant product. Based on these observations, we infer that the steady release of C16O2 is due to the reaction of C16O with support oxygen, (16O)s as described in eqn (3.2) (Scheme 1). It must be noted that the amount of PO was too low to be quantified accurately in the SSITKA experiments and therefore is not shown here. Qualitatively, however, it can be said that PO18 was observed as soon as the switch was made, which is in agreement with Scheme 1.
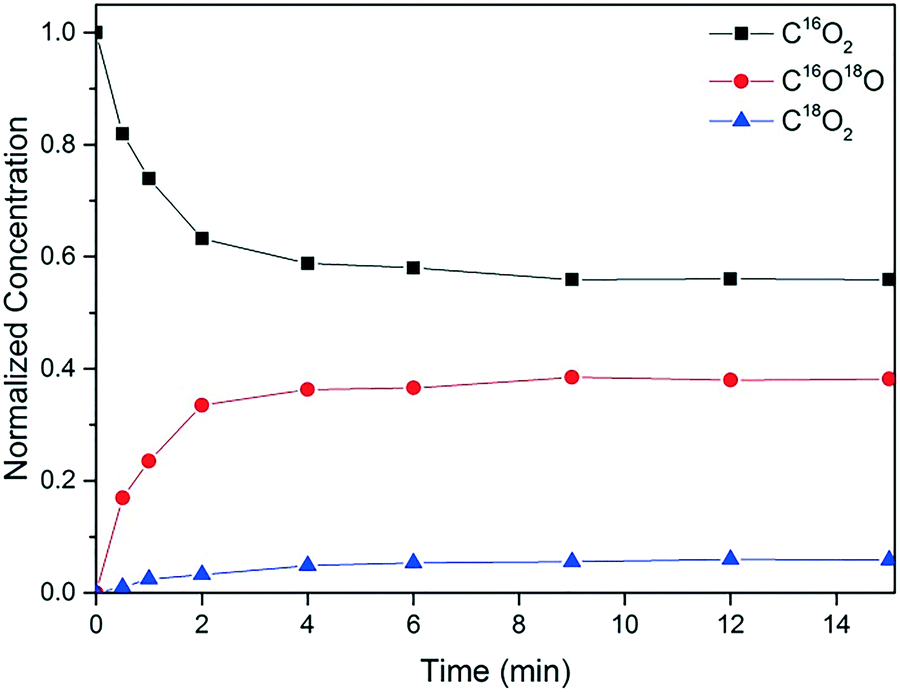 | ||
| Fig. 11 SSITKA transients for the switch from 16O2 to 18O2 (at t = 0) after performing propene epoxidation over the Au/TiO2-NM catalyst at 60 °C for 10 min (GHSV = 10000 mL gcat−1 h−1, 10 vol% CO, O2 and propene in Ar/Ne). Isotopic fractions for the products are given normalized to the amount of all isotopic varieties of the product produced at that moment. | ||
Conclusions
The present study explores the role of co-reactants in the direct epoxidation of propene to PO on Au/TiO2 catalysts, using experimental and theoretical tools. When H2 was substituted by CO as a co-reactant, PO formation was observed but with lower PO yields. PO selectivity, on the other hand, was >99% in both cases and catalyst deactivation, which is a common feature on Au/TiO2, was also observed in the case of CO, with time on stream.DFT calculations were employed to explore the probable reaction mechanism for CO-assisted epoxidation and to obtain further insight into the role of support oxygen en route to PO formation. These calculations revealed that formation of oxygen vacancies on the support is energetically more feasible using CO, as compared to H2. Hence, PO formation with CO as the co-reactant was calculated via this route and the resulting activation energy was 83 kJ mol−1. It was also found that epoxidation without the use of any sacrificial agent on Au/TiO2 is energetically highly demanding (107 kJ mol−1) and hence improbable. The use of H2 was found to redirect the mechanism through OOH radical formation, which subsequently lowered the activation energy to 63 kJ mol−1 – the lowest among the three pathways explored. Accordingly, the theoretical findings are in agreement with the experimental observations where the PO yield was observed to be the highest in hydro-epoxidation, followed by CO-assisted epoxidation, and negligible in the absence of any reducing gas. Furthermore, isotopic (SSITKA) measurements experimentally revealed the role of support oxygen in the case of CO epoxidation. This study provides valuable fundamental insight into the important role of a co-reactant in the direct epoxidation of propene and the corresponding mechanisms leading to PO formation on Au–Ti catalysts.
Acknowledgements
We acknowledge financial support for the research from The Netherlands Organization for Scientific Research (NWO) through project number 10017587, a VICI grant and Nuffic funding. Supercomputing facilities were funded by the NWO.References
- T. A. Nijhuis, M. Makkee, J. A. Moulijn and B. M. Weckhuysen, Ind. Eng. Chem. Res., 2006, 45(10), 3447–3459 CrossRef CAS.
- J. Huang and M. Haruta, Res. Chem. Intermed., 2012, 38(1), 1–24 CrossRef CAS.
- J. R. Monnier, Appl. Catal., 2001, 221, 73–91 CrossRef CAS.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178(2), 566–575 CrossRef CAS.
- X. Feng, N. Sheng, Y. Liu, X. Chen, D. Chen, C. Yang and X. Zhou, ACS Catal., 2017, 7(4), 2668–2675 CrossRef CAS.
- X. Feng, Y. Liu, Y. Li, C. Yang and D. Chen, AIChE J., 2016, 62(11), 3963–3972 CrossRef CAS.
- W. S. Lee, M. Cem Akatay, E. A. Stach, F. H. Ribeiro and W. Nicholas Delgass, J. Catal., 2012, 287, 178–189 CrossRef CAS.
- S. Kanungo, D. M. Perez Ferrandez, F. Neira d'Angelo, J. C. Schouten and T. A. Nijhuis, J. Catal., 2016, 338, 284–294 CrossRef CAS.
- S. Kanungo, K. S. Keshri, A. J. F. van Hoof, M. F. N. d'Angelo, J. C. Schouten, T. A. Nijhuis, E. J. M. Hensen and B. Chowdhury, J. Catal., 2016, 344, 434–444 CrossRef CAS.
- C. H. Liu, Y. Guan, E. J. M. Hensen, J. F. Lee and C. M. Yang, J. Catal., 2011, 282(1), 94–102 CrossRef CAS.
- M. Ojeda and E. Iglesia, Chem. Commun., 2009, 352–354 RSC.
- J. Huang, T. Akita, J. Faye, T. Fujitani, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2009, 48(42), 7862–7866 CrossRef CAS PubMed.
- C. Sivadinarayana, T. V. Choudhary, L. L. Daemen, J. Eckert and D. W. Goodman, J. Am. Chem. Soc., 2004, 126(1), 38–39 CrossRef CAS PubMed.
- S. Lee, L. M. Molina, M. J. López, J. A. Alonso, B. Hammer, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, M. J. Pellin and S. Vajda, Angew. Chem., Int. Ed., 2009, 48(8), 1467–1471 CrossRef CAS PubMed.
- D. H. Wells, W. N. Delgass and K. T. Thomson, J. Am. Chem. Soc., 2004, 126(9), 2956–2962 CrossRef CAS PubMed.
- A. M. Joshi, W. N. Delgass and K. T. Thomson, J. Phys. Chem. C, 2007, 765, 7841–7844 Search PubMed.
- A. K. Sinha, S. Seelan, S. Tsubota and M. Haruta, Top. Catal., 2004, 29(3/4), 95–102 CrossRef CAS.
- V. I. Sobolev and K. Y. Koltunov, Catal. Commun., 2013, 40, 103–105 CrossRef CAS.
- J. Chen, S. J. A. Halin, D. M. Perez Ferrandez, J. C. Schouten and T. A. Nijhuis, J. Catal., 2012, 285(1), 324–327 CrossRef CAS.
- C. Qi, J. Huang, S. Bao, H. Su, T. Akita and M. Haruta, J. Catal., 2011, 281, 12–20 CrossRef CAS.
- V. I. Sobolev and K. Y. Koltunov, Appl. Catal., A, 2014, 476, 197–203 CrossRef CAS.
- T. A. Nijhuis, E. Sacaliuc-Parvulescu, N. S. Govender, J. C. Schouten and B. M. Weckhuysen, J. Catal., 2009, 265(2), 161–169 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49(20), 14251–14269 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- L. M. Molina, M. D. Rasmussen and B. Hammer, J. Chem. Phys., 2004, 120(16), 7673–7680 CrossRef CAS PubMed.
- K. Liu, A. Litke, Y. Su, B. G. van Campenhout, E. A. Pidko and E. J. M. Hensen, Chem. Commun., 2016, 1(Table 1), 2–5 Search PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113(22), 9978–9985 CrossRef CAS.
- D. Sheppard, R. Terrell and G. Henkelman, J. Chem. Phys., 2008, 128(13), 134106 CrossRef PubMed.
- T. A. Nijhuis, T. Q. Gardner and B. M. Weckhuysen, J. Catal., 2005, 236(1), 153–163 CrossRef CAS.
- H. Liu, A. I. Kozlov, A. P. Kozlova, T. Shido, K. Asakura and Y. Iwasawa, J. Catal., 1999, 185(3), 252–264 CrossRef CAS.
- J. T. Calla and R. J. Davis, J. Catal., 2006, 241(2), 407–416 CrossRef CAS.
- I. X. Green, W. Tang, M. Neurock and J. T. Yates, Science, 2011, 333, 736–739 CrossRef CAS PubMed.
- A. Ruiz, B. van der Linden, M. Makkee and G. Mul, J. Catal., 2009, 266(2), 286–290 CrossRef CAS.
- M. F. Camellone, J. Zhao, L. Jin, Y. Wang, M. Muhler and D. Marx, Angew. Chem., Int. Ed., 2013, 52(22), 5780–5784 CrossRef CAS PubMed.
- N. A. Deskins, R. Rousseau and M. Dupuis, J. Phys. Chem. C, 2011, 115(15), 7562–7572 CAS.
- S. Chrétien, M. S. Gordon and H. Metiu, J. Chem. Phys., 2004, 121(8), 3756–3766 CrossRef PubMed.
- H. Li, H. Wang, X. Gong, Y. Guo, Y. Guo, G. Lu and P. Hu, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 193401 CrossRef.
- M. Huang and S. Fabris, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 081404(R) CrossRef.
- A. Linsebigler, G. Lu and J. T. Yates, J. Chem. Phys., 1995, 103(21), 9438–9443 CrossRef CAS.
- S. Derrouiche, P. Gravejat and D. Bianchi, J. Am. Chem. Soc., 2004, 126(40), 13010–13015 CrossRef CAS PubMed.
- Y. Wang, Y. Yoon, V. Glezakou, J. Li and R. Rousseau, J. Am. Chem. Soc., 2013, 135(29), 10673–10683 CrossRef CAS PubMed.
- S. Lee, L. M. Molina, M. J. López, J. A. Alonso, B. Hammer, B. Lee, S. Seifert, R. E. Winans, J. W. Elam and M. J. Pellin, Angew. Chem., 2009, 121, 1495–1499 CrossRef.
- C. Chang, Y. Wang and J. Li, Nano Res., 2011, 4, 131–142 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cy00525c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |