
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAldose to ketose interconversion: galactose and arabinose isomerization over heterogeneous catalysts
Dmitry Yu.
Murzin
 *a,
Elena V.
Murzina
a,
Atte
Aho
a,
Mariya A.
Kazakova
bc,
Alexander G.
Selyutin
b,
David
Kubicka
d,
Vladimir L.
Kuznetsov
b and
Irina L.
Simakova
b
*a,
Elena V.
Murzina
a,
Atte
Aho
a,
Mariya A.
Kazakova
bc,
Alexander G.
Selyutin
b,
David
Kubicka
d,
Vladimir L.
Kuznetsov
b and
Irina L.
Simakova
b
aLaboratory of Industrial Chemistry and Reaction Engineering, Åbo Akademi University, Biskopsgatan 8, 20500, Turku, Finland. E-mail: dmurzin@abo.fi
bBoreskov Institute of Catalysis, akad. Lavrentieva 5, Novosibirsk, 630090, Russia
cNovosibirsk State University, Pirogova 2, Novosibirsk, 630090, Russia
dTechnopark Kralupy of University of Chemistry and Technology Prague, 278 01 Kralupy nad Vltavou, Czech Republic
First published on 16th May 2017
Abstract
Isomerization of glucose, galactose and arabinose to corresponding keto-sugars was studied in the present work over a range of heterogeneous catalysts. Magnesium aluminates with different ratios between oxides resulting in materials with a Mg/Al ratio from 0.2 to 0.9 were prepared, characterized and evaluated in terms of their catalytic behavior. The catalyst with a Mg/Al ratio close to hydrotalcites was the most efficient considering activity, selectivity and stability. The sugar structure was shown to have a minor influence on catalytic activity and selectivity.
Introduction
Interconversion of aldose to ketose has attracted a lot of attention recently as research efforts have been focused on valorization of carbohydrates derived from lignocellulosic biomass within the framework of the so-called biorefinery concept.1–3Formation of fructose from glucose in fact is a well-known reaction practiced on a million-tons-per-year scale in the production of high-fructose corn syrup (HFCS) using an immobilized enzymatic catalyst – glucose isomerase.4 This enzyme can be also used for interconversion of a C5 sugar – xylose to xylilose.4 The final equilibrium mixture of fructose and glucose contains ca. 55% of the former.
Chemical conversion of glucose to fructose can be also performed under alkaline conditions and at a temperature higher than that typically used (55–60 °C) for the enzymatic process. Transformations in an alkaline environment following the so-called Lobry de Bruyn–Alberda-van Ekenstein rearrangement5–7 are nonspecific leading to formation of side products. Conversion is typically limited resulting in fructose concentration of 40%.4 It should be mentioned, that higher fructose concentrations, i.e. 55% fructose, 41% glucose, and 4% mannose, have been recently reported using NaAlO2 in water.8In situ complexation of fructose under basic conditions by applying the concept of anionic extraction can increase the yield of fructose and as mentioned in ref. 7 could reach 72%. Aldose–ketose rearrangement is accompanied by epimerization which occurs in parallel to the main reaction. Other reactions, such as aldolization/retro-aldolization, β-elimination and benzylic rearrangement8–11 also contribute to formation of side products. The mechanism of isomerization reaction (Fig. 1) is thought12 to occur in the case of glucose via formation of glucose anions followed by generation of an ene–diol intermediate, which is then converted to fructose anions and finally into fructose.
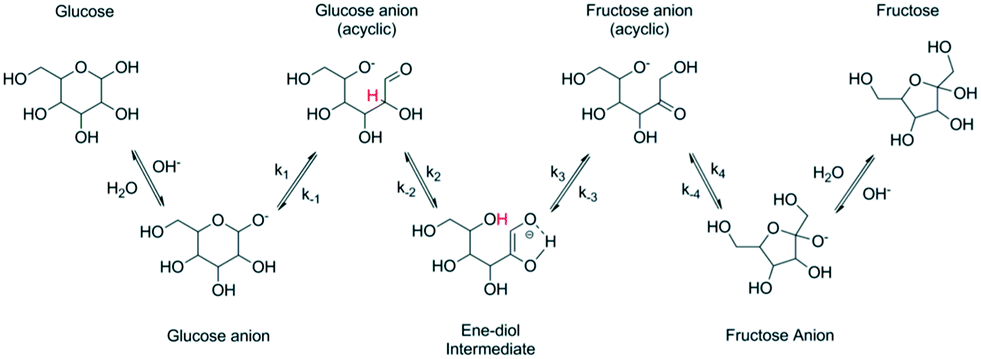 | ||
| Fig. 1 Base catalyzed isomerization of glucose to fructose (reproduced with permission from ref. 12). | ||
Besides homogeneous bases (alkali or alkaline earth hydroxides) heterogeneous base catalysts would be of high interest being devoid of the apparent drawbacks of their homogeneous counterparts, such as low selectivity, and challenges with separation.
In connection with heterogeneous catalysts, several types of them could be mentioned. Hydrotalcite (HT) with different Mg/Al molar ratios was studied in isomerization of glucose,13–18 while zeolites modified with basic metals were investigated for the same reaction. Commercial Mg–Al hydrotalcites prepared as hydroxide, carbonate and mixed hydroxide–carbonate were reported in ref. 14 to be prone to deactivation after 15% conversion. Hydrotalcite with the atomic ratio Mg/Al equal to 3 afforded a 25% yield of fructose at 42% glucose conversion.13 The same ratio between Mg and Al was used in calcined and rehydrated Mg–Al hydrotalcite in ref. 15.
In contrast to ref. 13, when water was used as a solvent, in ref. 15, application of dimethylformamide instead of an aqueous solution, resulted in a ca. 35% yield of fructose at ca. 50% conversion for the rehydrated sample. The same solvent was used in ref. 19. Nevertheless water seems to be the preferred solvent. It was applied in the recent work of Delidovich and Palkovits17 where a fructose yield of up to 30% with 89% selectivity was reached in glucose isomerization to fructose and the optimal molar content of aluminum was reported to be 0.23 < Al/(Mg + Al) < 0.30.17 Glucose conversion was seen to be linearly proportional to the basicity of hydrotalcite. The authors suggested that weak sites of hydrated hydrotalcites are more selective than the medium strong and strong sites. Moreover, HT in its carbonate or hydrated form was suggested to be catalytically active for glucose isomerization to fructose. As a consequence noncalcined HT was proposed to be more beneficial for the reaction than its calcined counterparts.17
Hydrotalcites were compared with other catalytic materials in a detailed review covering the applications of homogeneous and heterogeneous catalysts for isomerization of glucose.7 A viable alternative to hydrotalcites is magnesia. This implies that the presence of magnesium aluminate (MgAl2O4) or (M+AlO2−) formed during synthesis of hydrotalcites might not be needed to afford an active catalyst.
Commercially available MgO with increased basicity was reported as an efficient catalyst for glucose isomerization at 90 °C giving a yield of 33.3% fructose at a conversion of 44.1%.8 It should be noted that at the same time its selectivity was ca. 75% resulting not only in fructose but also in mannose, as well as oxidation products, such as glycolic, acetic, lactic and formic acids. Since the reaction conditions do not favor oxidation, formation of these products is most probably due to retro-aldol reactions.
The type of heterogeneous catalyst influences substantially selectivity. For instance non-acidic LiNbMoO6 and strongly acidic HNbMoO6 efficiently catalyze epimerization of glucose to mannose without any isomer formation.20 In contrast, glucose isomerization into fructose via an intramolecular 1,2-hydride shift mechanism with Lewis acid catalysts such as Sn-β zeolites can be very efficient giving a 55% glucose conversion and a 32% yield of fructose in water at 110 °C.21 Besides fructose, an epimer – mannose – was also formed. The same type of catalyst was used for isomerization of the C5 sugar xylose resulting in an 18% yield of xylulose at 85% xylose conversion at 110 °C in water.22
There is a limited amount of experimental data available for the isomerization of other sugars. Isomerization of lactose to lactulose using a range of homogeneous and heterogeneous catalysts was reported by Hajek et al.23 Commercial hydrotalcite with a Mg/Al ratio equal to 3 in its noncalcined form was inactive, while the calcination at a temperature in the range 643–923 K had a minor influence on either its activity or selectivity. For the same reaction Sn-MFI zeolites were reported in ref. 24.
Less is even known about isomerization of L-arabinose to L-ribulose (an intermediate for antiviral nucleosides) using heterogeneous catalysts, since the existing literature covers enzymatic and homogeneous catalysis. The same is valid for the isomerization of galactose (C4 epimer of glucose) to tagatose.
One of the very few reports on applications of hydrotalcites for isomerization of arabinose and galactose is related to one-pot synthesis of furanic compounds from arabinose and lactose (disaccharide of galactose and glucose) using a physical mixture of commercial HT with a Mg/Al ratio of 3 and acid catalysts of Amberlyst and Nafion types.25 No information was provided in ref. 25 about the influence of the basicity, Mg to Al ratio or calcination temperature on the activity and selectivity.
Besides homogeneous and heterogeneous catalysts containing magnesia, there are also reports on utilization of cobalt compounds as catalysts26 or co-factors in regard to homogeneous and enzymatic isomerization of glucose.27 Application of cobalt and magnesium ions as co-factors in enzymatic transformations of glucose with glucose isomerase27 is not necessarily related to their involvement in the binding of the reactant to the active site. However, according to ref. 26 Co(III) phthalocyanine species generated in situ from Co(II) phthalocyanines react with glucose giving a five membered ring chelate complex, which subsequently isomerizes to fructose. No reports are available on heterogeneous catalytic isomerization of carbohydrates with Co catalysts and it was therefore interesting to test if Co containing catalysts are active in this reaction.
The current work aims to explore isomerization of arabinose and galactose over heterogeneous catalysts, which contain MgO as such or as part of magnesium aluminates or CoMnAlMg mixed oxides. To this end besides commercial magnesia, several catalysts covering a broad range of magnesia to alumina ratios were synthesized, characterized, calcined at different temperatures and evaluated in the isomerization of carbohydrates. For the sake of comparison glucose isomerization was also performed with some catalysts.
A special emphasis was put on the evaluation of catalyst stability against leaching. The latter is especially important because catalyst deactivation due to its contact with aqueous reaction mixtures is often disregarded in the available literature.
Results and discussion
Catalyst characterization results
The elemental composition of monometallic cobalt catalysts was determined by ICP and corresponded well to the nominal loadings. The surface area (BET) decreased with increasing Co loading. For 10 wt% Co it decreased in the order Co/SBA-15 > Co–Al2O3 > Co/TiO2 and the BET values were 470, 215 and 98 m2 g−1.The elemental composition of the CoMnAlMg catalyst is given in Table 1, while characterization data for magnesium aluminates are presented in Fig. 2 and 3 and Tables 2–4.
| Mg | Ni | Co | Mn | O | N | C | Al |
|---|---|---|---|---|---|---|---|
| 15.6 | 0.3 | 4.2 | 5.0 | 59.5 | 0.8 | 4.7 | 9.9 |
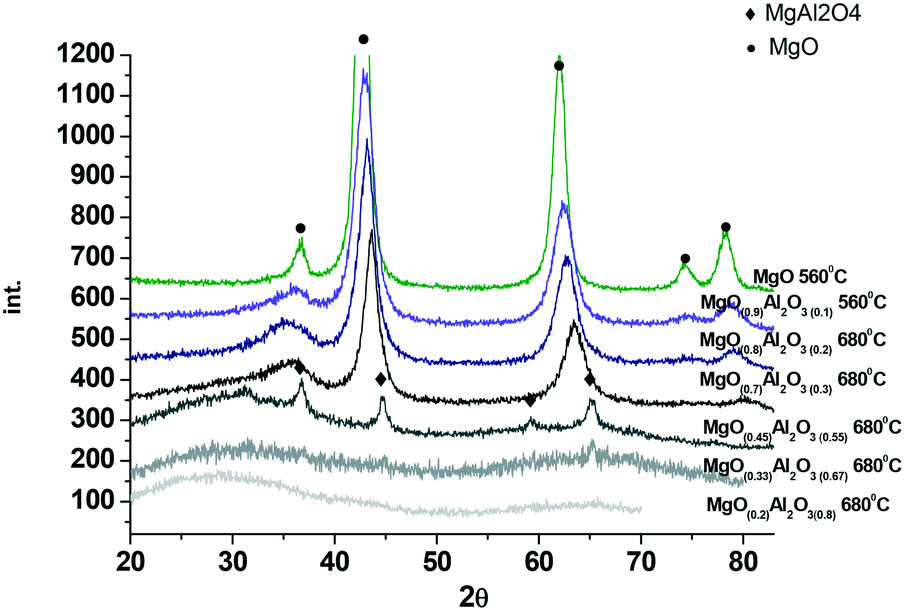 | ||
| Fig. 2 XRD of the (MgO)x(Al2O3)y samples after calcination at 560 °C or 680 °C. | ||
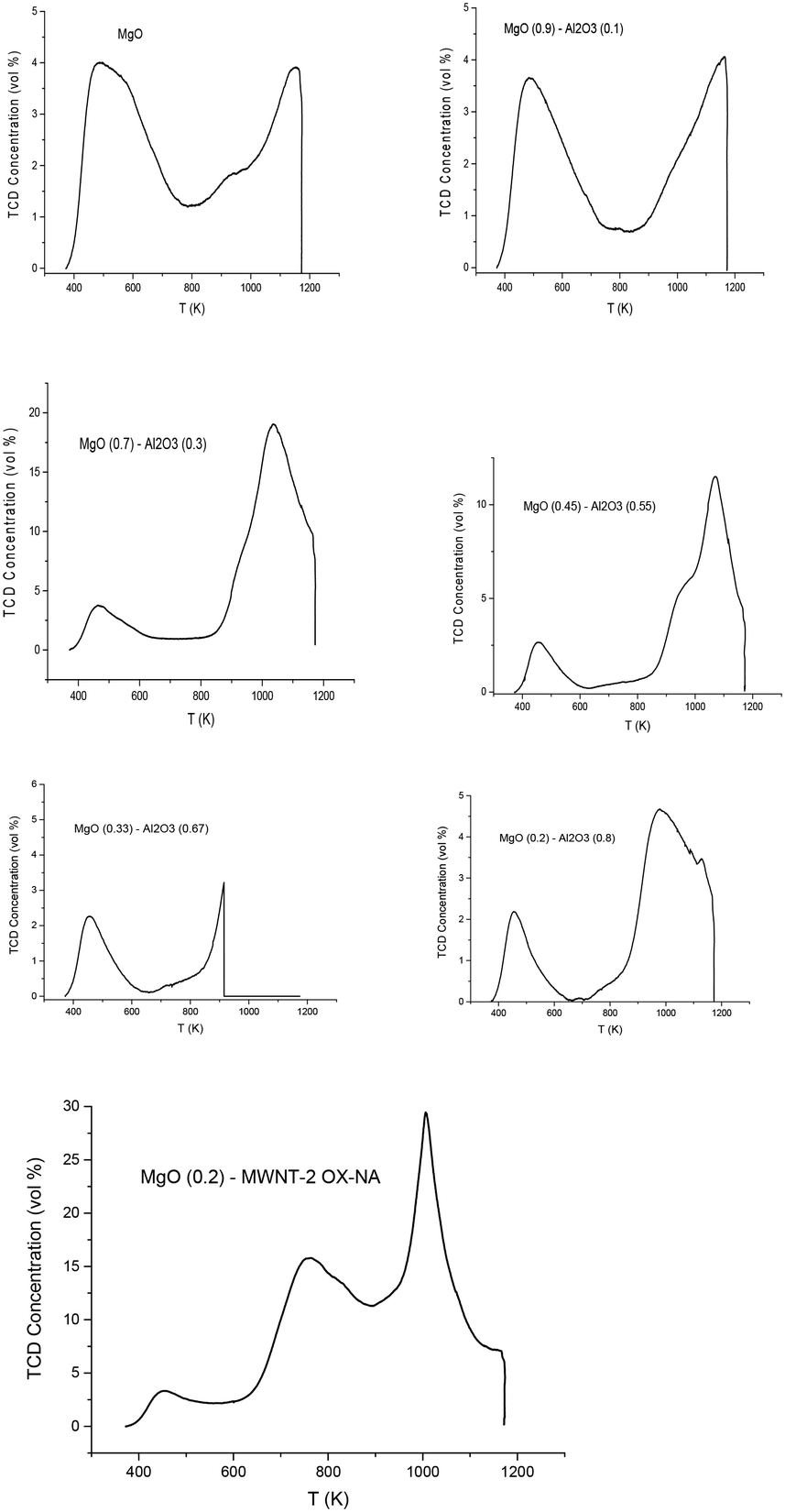 | ||
| Fig. 3 CO2 TPD for the catalysts in Table 3. | ||
| Sample | MgO, wt% | Al2O3, wt% | MgO/Al2O3 mass ratio (XRF) | MgO/Al2O3 mass ratio (XRF)a | MgO/Al2O3 mass ratio ICP-AES | MgO/Al2O3 mole ratio (XRF) | S BET, m2 g−1 | |
|---|---|---|---|---|---|---|---|---|
| a After hydrothermal stability tests at 110 °C; nm – not measured. | ||||||||
| MgO | 560 °C | 99.2 | 176 | |||||
| (MgO)0.2(Al2O3)0.8 | T = 560 °C | 11.0 | 87.2 | 0.126 | 0.319 | 165 | ||
| T = 680 °C | 0.116 | 0.091 | ||||||
| (MgO)0.33(Al2O3)0.67 | T = 560 °C | 19.7 | 78.5 | 0.251 | 0.634 | 97 | ||
| T = 680 °C | 0.233 | nm | ||||||
| (MgO)0.45(Al2O3)0.55 | T = 560 °C | 29.3 | 69.0 | 0.425 | 1.075 | 130 | ||
| T = 680 °C | 0.397 | 0.318 | ||||||
| (MgO)0.7(Al2O3)0.3 | T = 560 °C | 54.0 | 43.8 | 1.233 | 3.118 | 132 | ||
| T = 680 °C | 1.215 | |||||||
| (MgO)0.8(Al2O3)0.2 | T = 560 °C | 69.7 | 29.6 | 2.354 | 5.956 | 205 | ||
| T = 680 °C | 68.5 | 30.9 | 2.217 | 1.55 | 5.607 | 272 | ||
| (MgO)0.9(Al2O3)0.1 | T = 560 °C | 82.9 | 16.4 | 5.055 | 12.784 | 276 | ||
| T = 680 °C | 3.59 | nm | ||||||
Fig. 2 displays the XRD data for the synthesized magnesium aluminate materials. As can be seen from Fig. 2 the samples with a higher content of alumina are X-ray amorphous, while an increase of MgO content affords crystalline materials. Already the (MgO)0.45(Al2O3)0.55 material calcined at 680 °C contains reflections typical for MgAl2O4 spinels which is in agreement with the phase diagram. Further increase of MgO content in magnesium aluminates ((MgO)0.7(Al2O3)0.3 and (MgO)0.9(Al2O3)0.1 calcined at 680 °C) results in reflections, which can be attributed to the cubic structure of MgO, which somewhat shifted in terms of 2θ values compared to neat magnesia. The reason for the shift can be attributed to replacement of some Mg by Al. Reflections typical for the basal planes of double layer hydroxide (hydrotalcite) at 2θ values of 11.4°, 22.9° and 34.7° were not observed not only in the samples with a low Mg content, but also in materials with a high Mg content. According to XRD analysis there were no significant differences in the values of the coherent scattering region (CSR) for the studied samples. The CSR values obtained by the Rietveld method varied within 3–6 nm. The range of CSR values is within the error of the method (significant deviation of ∼5 nm).
The MgO and Al2O3 composition in the synthesized materials was evaluated by XRF. The results presented in Table 2 clearly demonstrate that calcination did not influence the composition of magnesia–alumina materials.
The measured mass ratios displayed some deviations from the theoretical ones which can be at least partially explained by the presence of some organic leftovers in the catalysts even after calcination and inherent errors of the applied methods associated with for example high concentration of elements (ICP-AES).
The values of surface areas for the tested magnesium aluminates given also in Table 2 are in line with the characterization data typical for these types of materials.16
Basicity data presented in Table 3 and Fig. 3 clearly show that the basicity of magnesium aluminates increases by increasing the content of MgO. All catalysts displayed well-defined CO2 TPD peaks with the peak maxima at ca. 450 K. This could be assigned to weak and medium basic sites, while some additional peaks present at the high temperature domain could be related to strong basic sites. In ref. 16 the activity for isomerization was related to the weak base sites; therefore for calculations of weak and medium basic sites, the cut-off temperatures mentioned in Table 3 were applied. The strong sites present in the catalyst might lead to the formation of degradation products as mentioned in ref. 16. A special case was 20 wt% MgO/MWNT displaying a more complicated TPD pattern, which could be related to the properties of the support only, in particular, the decomposition of various functional groups present in MWNTs during TPD. In particular, decomposition of carboxylic (–COOH) and more stable keto groups (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) occurs respectively at 250–300 °C and 680–720 °C.
O) occurs respectively at 250–300 °C and 680–720 °C.
| Catalyst | Cut off T, K | Weak and medium basic sites, μmol g−1 |
|---|---|---|
| 20 wt% MgO/MWNT | 600 | 218 |
| (MgO)0.2(Al2O3)0.8 | 700 | 118 |
| (MgO)0.33(Al2O3)0.67 | 650 | 125 |
| (MgO)0.45(Al2O3)0.55 | 650 | 140 |
| (MgO)0.7(Al2O3)0.3 | 650 | 268 |
| (MgO)0.9(Al2O3)0.1 | 750 | 350 |
| MgO | 750 | 507 |
Catalytic results: screening
None of the monometallic cobalt catalysts displayed any meaningful activity in the isomerization of galactose at 110 °C and 5 bar of nitrogen with a catalyst loading of 0.2 g.The results for the activity tests with commercial hydrotalcite and MgO are presented in Table 4. As follows from Table 4 besides the desired ketoses, some by-products, such as oligomers, epimers and degradation products were also observed with different selectivities depending on the catalyst.
| Catalyst | Substrate | T, °C | m × t, (g × min) | C aldose | C ketose | Conv.% | Selec% |
|---|---|---|---|---|---|---|---|
| a m cat = 0.2 g. b The rest of the oligomers, epimers and degradation products. c C initial = 0.11 M. d C initial = 0.13 M. Gal, Glu and Ara stand for galactose, glucose and arabinose, respectively. Concentrations are given in M. | |||||||
| MgO | Galc | 110 | 12.0 | 0.05 | 0.03 | 55 | 50 |
| MgO | Gal | 110 | 48 | 0.03 | 0.02 | 73 | 25 |
| MgO | Gal | 90 | 12.0 | 0.07 | 0.03 | 36 | 75 |
| MgO | Gal | 90 | 48.0 | 0.05 | 0.03 | 45 | 50 |
| HT, noncalcined | Gal | 110 | 48.0 | 0.10 | 0.00 | 9 | 0 |
| HT, calcined at 500 °C | Gal | 110 | 24.0 | 0.07 | 0.02 | 36 | 50 |
| HT, calcined at 500 °C | Gal | 110 | 48.0 | 0.07 | 0.02 | 36 | 50 |
| HT, calcined at 500 °C | Gluc | 120 | 48.0 | 0.07 | 0.03 | 36 | 75 |
| HT, noncalcined | Arad | 110 | 48.0 | 0.12 | 0.00 | 8 | 0 |
| HT, calcined at 500 °C | Ara | 110 | 6.0 | 0.08 | 0.01 | 38 | 20 |
| HT, calcined at 500 °C | Ara | 110 | 48.0 | 0.06 | 0.01 | 54 | 14 |
It follows from Table 4 that commercial hydrotalcite with a Mg/Al ratio equal to 3 in its noncalcined form was rather inactive in isomerization independent of the type of substrate, while calcination at 500 °C was sufficient to boost the catalytic activity. Although the results in terms of conversion are like those for glucose to fructose isomerization at the same temperature reported in ref. 17, the influence of calcination seems to be different. In ref. 17 noncalcined HT was proposed to be more beneficial for the reaction than the calcined counterparts based on a suggestion that hydrotalcite in its carbonate or hydrated forms are the catalytically active species. In the current work as well as in the previous study on lactose isomerization23 calcination was found to be beneficial for catalytic activity. Inactivity of noncalcined HT compared to that of the calcined counterpart in general could be related to basicity. As it is challenging to perform TPD–CO2 for samples when carbonates are decomposed at ca. 200–300 °C, a comparison of the pH value of water suspensions with HT prior to and after calcination was performed. It was observed that the pH value of the water suspension with the noncalcined hydrotalcite being equal to 8.95 was slightly elevated when hydrotalcite was calcined for 4 h at 680 °C reaching 9.55. Apart from these small differences which probably in itself cannot be responsible for the differences in the catalytic behaviour, it should be noted that the noncalcined sample was clearly hydrophobic in contrast to its calcined counterpart. This is probably the reason for their different performances in isomerization of sugars occurring in aqueous solutions.
Similar to ref. 8 where glucose to fructose isomerization with commercially available MgO was studied, in the current work isomerization of galactose was investigated. The catalyst is also active in isomerization allowing high conversion exceeding 50%. At the same time selectivity to ketose clearly decreased with conversion as the reaction resulted not only in tagatose, but also in oligomers and degradation products (Fig. 4). Another serious issue is catalyst deactivation through leaching. In a separate experiment on leaching of magnesia in galactose solution at 110 °C severe dissolution of MgO in the solution was observed.
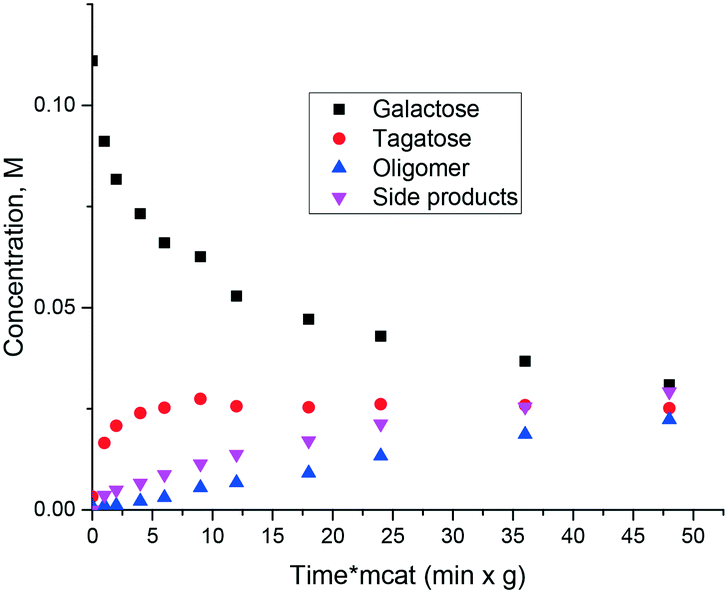 | ||
| Fig. 4 Isomerization of galactose with commercial MgO at 90 °C. | ||
Thus, even if magnesia is superficially active in the isomerization of sugars there is still a need for obtaining an active and at the same time stable and selective catalyst which, on the one hand, would be stable against leaching and on the other would avoid or at least minimize formation of oligomers or degradation products. It was thus interesting to test catalysts where MgO was deposited on an inert support: multiwall carbon nanotubes. Fig. 5 displays the results illustrating that application of supported MgO resulted in limited conversion, thereafter the reaction stopped completely. The hydrothermal stability was very moderate and will be discussed below.
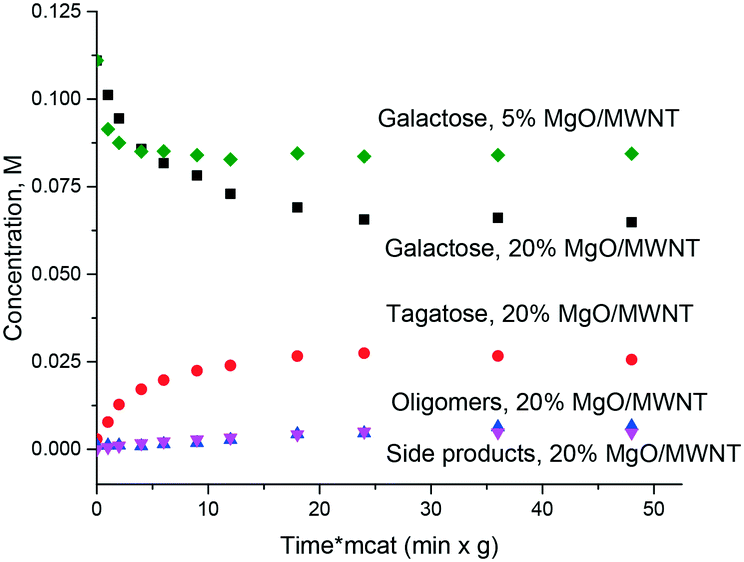 | ||
| Fig. 5 Isomerization of galactose with MgO supported on MWNTs at 110 °C. | ||
Synthesis of carbon nanotubes is typically performed using a suitable growth catalyst. One such catalyst is the CoMnAlMg mixed oxide which was synthesized following ref. 28. The content of magnesium oxide in this catalyst was enough to expect non-negligible catalytic activity. Experiments were conducted with three sugars (arabinose, galactose and glucose) at three temperatures allowing a straightforward comparison between different substrates and calculations of activation energies. The results are presented in Fig. 6. Due to a narrow temperature interval it was challenging to establish very precisely the temperature dependence. For example, while some of the data points for glucose might coincide at 100 and 110 °C there was still a difference. For instance, a concentration of 0.09 M is reached within 50 min at a higher temperature, while at 100 °C the same concentration is obtained after 100 min. There is also a clear difference for arabinose. In order to have a more reliable evaluation of the temperature dependence the kinetic constants for first order reactions were calculated by numerical data fitting for respective kinetic curves across the whole experimental duration (Fig. 6d). The apparent activation energy for all sugars can be in fact considered the same being equal to 99 ± 8 kJ mol−1. Information about the activation energy of sugar isomerization on magnesium aluminate, available in the literature, is limited. In ref. 16 data of the final conversion after 5 h in glucose to fructose isomerization are presented as a function of temperature. By using the conversion values in the temperature range 80–100 °C reported in ref. 16 the value of activation energy equal to 100 kJ mol−1 can be calculated.
 | ||
| Fig. 6 Isomerization of sugars over the CoMnAlMg mixed oxide catalyst: a) arabinose, b) galactose, c) glucose, d) Arrhenius plot. | ||
The conclusion that different sugars display almost the same reactivity was further confirmed by studying the mixtures of arabinose and galactose (Fig. 7a) and glucose and galactose (Fig. 7b). In both cases, there was a non-negligible formation of oligomers, which eventually deactivates the catalyst.
 | ||
| Fig. 7 Isomerization of the mixtures of sugars over the CoMnAlMg mixed oxide catalyst at 110 °C: a) arabinose–galactose, b) glucos–galactose. | ||
Catalytic results: Mg/Al ratio
The influence of the Mg/Al ratio on catalytic activity in isomerization of glucose to fructose was studied in ref. 17 for hydrotalcites. The latter materials being double layer hydroxides have a limited composition range. Namely in ref. 17, the content of Mg was varied between 0.78 and 0.89. In the current work a much broader range of Mg/Al was tested. In the prepared magnesium aluminates the hydrotalcite phases were not present, thus it can be suggested that the layered structure of hydrotalcite is not necessarily needed for catalytic activity.The experimental data of galactose isomerization are displayed in Fig. 8 for materials calcined at either 560 or 680 °C. As can be seen from this figure an increase in the concentration of MgO brought along an increase of activity. Elevation of the calcination temperature did not influence substantially the catalytic activity.
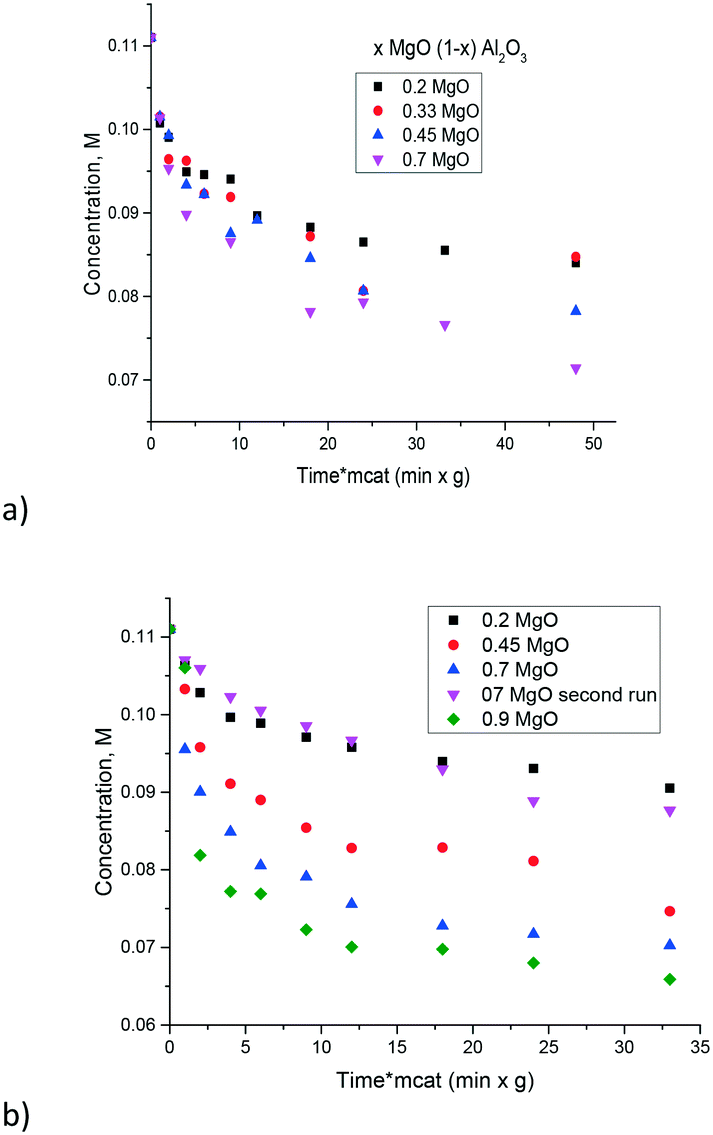 | ||
| Fig. 8 Isomerization of galactose over the magnesium aluminates with different Mg/Al ratios at 110 °C and different calcination temperatures, a) 560 °C and b) 680 °C. | ||
A simple recycling test with magnesium aluminate having the composition 0.7 MgO 0.3 Al2O3 was performed by just filtering the catalyst after the reaction, washing with water and subsequent drying at 110 °C for 24 h and demonstrated a substantial decrease of catalytic activity (Fig. 8b). Therefore, catalyst regeneration should involve also burning of organic compounds by calcination similar to the procedure described in ref. 17. In particular previously performed recycling tests with hydrotalcites having a similar MgO content to that in the recycling experiment in the present study, were conducted by first regenerating the catalyst by calcination followed by rehydration in a sodium carbonate solution.17 While the molar ratio of Mg to Al changed from 3.34 to 2.03 reflecting a substantial loss of Mg during the recycling tests,17 the conversion did not change much in recycling experiments, being around 18–20%.
The TOF values reported in ref. 17 were in the range of 18–23 h−1. Direct comparison of the TOF values is not straightforward as this parameter depends on conversion for first order reactions. Nevertheless, calculation of the TOF for the (MgO)0.7(Al2O3)0.3 catalyst using XRF values from Table 3 and conversion after 30 min gave a TOF equal to 22 h−1, which is the same as the values reported in ref. 17. To avoid uncertainties in the determination of TOF values and rationalize the influence of catalyst composition on the reactivity the first order kinetic constants have been plotted against magnesia content and overall basicity (Fig. 9). The errors in the determination of the rate constants were 15 ÷ 20% depending on the Mg/Al ratio.
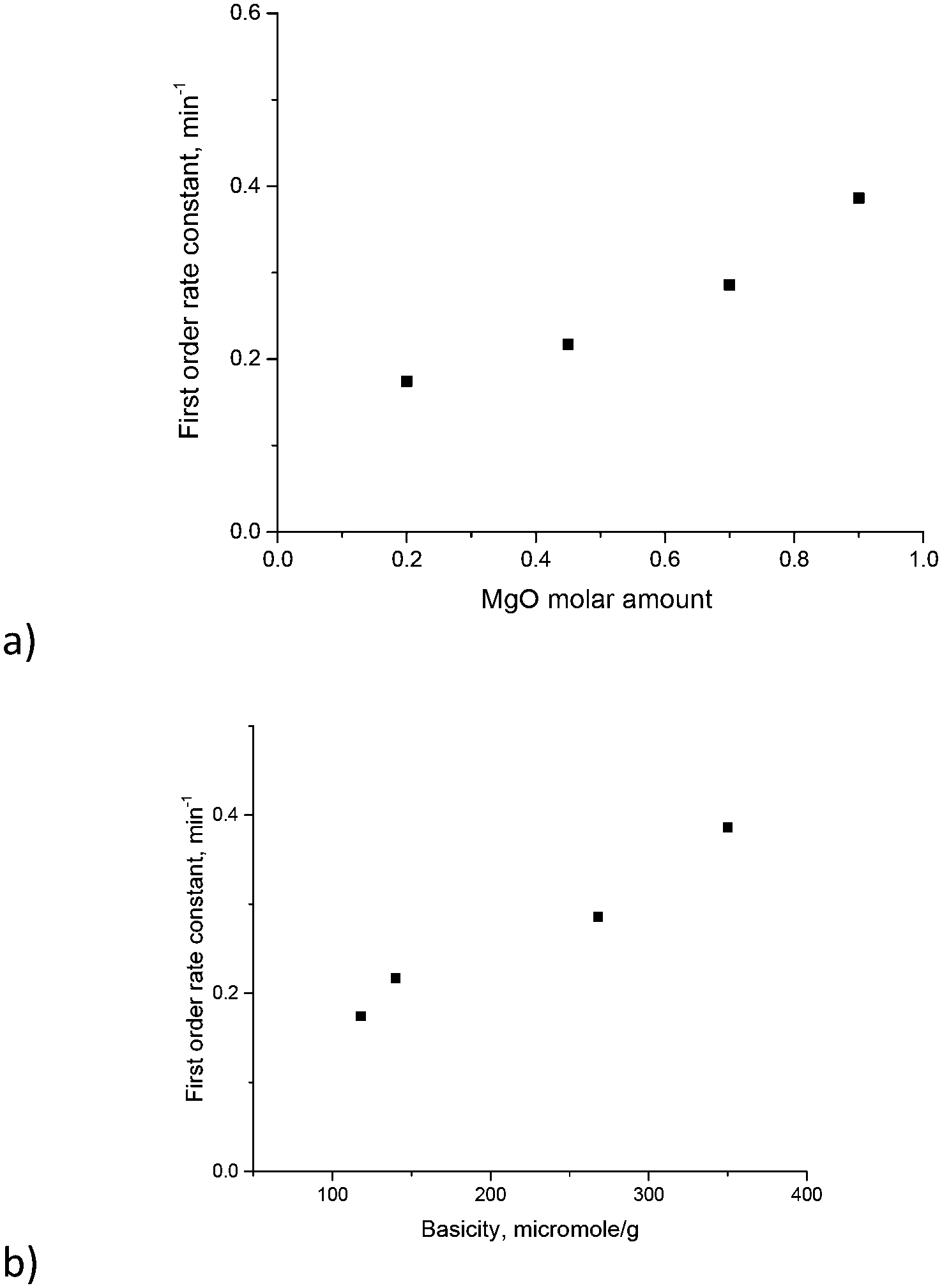 | ||
| Fig. 9 Galactose isomerization first order rate constant vs. a) molar Mg content and b) basicity of magnesium aluminates. Reaction conditions: 110 °C, mcat = 9.2 g, 5 bar, 1000 rpm and calcination temperature = 680 °C. | ||
As can be seen from Fig. 9 there is a clear linear correlation between the first order rate constant and the molar content of MgO or basicity as the latter parameters are related to each other. Previously glucose conversion in isomerization was related to the basicity of hydrotalcites with a rather narrow range of Mg/Al ratios.17 The same conclusion can be also applied for another sugar – galactose – and can be extended to a broader range of Mg/Al ratios.
The influence of the sugar structure for magnesium aluminates was tested by conducting experiments with galactose–glucose and galactose–arabinose mixtures using the (MgO)0.8(Al2O3)0.2 catalyst calcined at 680 °C. Fig. 10 clearly illustrates that the kinetic behavior was independent of the sugar structure.
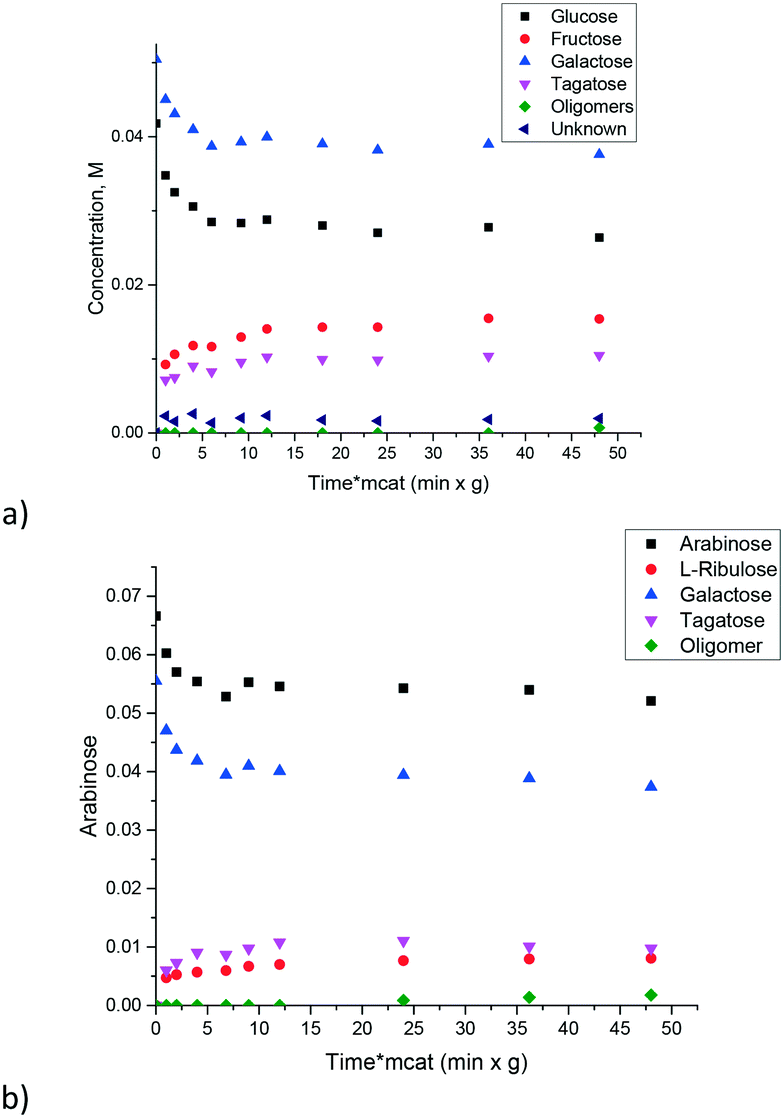 | ||
| Fig. 10 Isomerization of sugar mixtures over the (MgO)0.8(Al2O3)0.2 catalyst for a) galactose–glucose and b) galactose–arabinose mixtures. Reaction conditions: temperature = 110 °C, calcination temperature = 680 °C, mcat = 0.2 g, 5 bar and 1000 rpm. | ||
Hydrothermal stability tests
Literature data along with the catalytic results presented above clearly indicate that magnesium based materials (MgO, hydrotalcite and magnesium aluminate) could be promising catalysts for isomerization of sugars. A serious issue with this type of catalyst is leaching of the active phase into the solution. To address this issue hydrothermal activity tests were conducted at 110 °C for 2.5 h using 0.215 wt% catalyst loading in 2 wt% galactose solutions. Analysis of the liquid phase by AAS for several catalyst samples is shown in Table 5.| Catalyst | Mg content, mg ml−1 | Al content, mg ml−1 |
|---|---|---|
| a Hydrothermal treatment was performed in the absence of galactose. | ||
| (MgO)0.2(Al2O3)0.8a | 0.0015 | 0.00002 |
| (MgO)0.33(Al2O3)0.67a | 0.0006 | 0.00002 |
| (MgO)0.33(Al2O3)0.67 | 0.008 | 0.00002 |
| (MgO)0.45(Al2O3)0.55a | 0.0017 | 0.00002 |
| (MgO)0.7(Al2O3)0.3a | 0.0007 | 0.0014 |
| (MgO)0.8(Al2O3)0.2 | 0.213 | 0.0013 |
| (MgO)0.9(Al2O3)0.1 | 0.318 | 0.0013 |
| MgO | 0.750 | — |
| 5% MgO/MWNT | 0.0825 | — |
| 20% MgO/MWNT | 0.1967 | — |
The sample containing only MgO was completely dissolved during hydrothermal treatment giving the brownish color of the resulting solution. 20 wt% MgO/MWNT was also prone to leaching resulting in a relatively high concentration of magnesium ions after hydrothermal stability tests in a galactose solution at 110 °C (0.1967 mg ml−1). The color of the solution after hydrothermal stability tests was yellowish. XRD of the hydrothermally treated 20 wt% MgO/MWNT (not shown) displayed only the MgO phase with lower intensity, which can be attributed to the leaching of magnesia from the catalyst.
Interestingly enough, leaching in the presence of galactose was much more severe compared to that of the treatment in neat water at the same temperature (Table 5). XRD data for the (MgO)0.7(Al2O3)0.3 catalyst calcined at 680 °C and hydrothermally treated at 110 °C for 2.5 h are presented in Fig. 11.
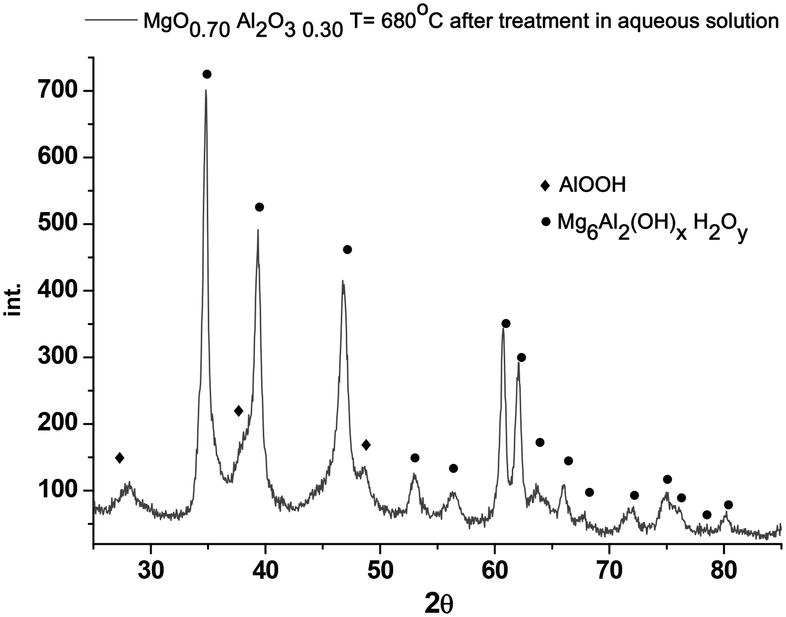 | ||
| Fig. 11 XRD data for the (MgO)0.7(Al2O3)0.3 catalyst calcined at 680 °C and hydrothermally treated at 110 °C for 2.5 h in galactose solution. | ||
Comparison of Fig. 11 with XRD data for non-treated catalyst (Fig. 2) clearly shows that the structure of the material changed significantly after hydrothermal treatment resulting in a structure close to hydrotalcite. This is an interesting observation showing that rehydration of magnesium aluminate results in double layer hydroxide. The activity of calcined hydrotalcite (Table 5) in galactose isomerization was similar to that of (MgO)0.7(Al2O3)0.3 (Fig. 8b). This and the absence of any induction period or inflection points can indicate that if solid phase transformations are slow, different phases possess similar activities. Therefore catalytic activity can be attributed to the same MgO content in both samples, rather than to the presence of a HT phase as there was a linear increase of activity as a function of MgO content and even catalysts with low MgO loading not possessing a hydrotalcite structure were moderately active. Transformation of magnesium aluminate to hydrotalcite could have happened during the initial heating to the reaction temperature of the catalyst in a water solution, as well as during the reaction.
It was interesting to check if another magnesium aluminate with a much lower Mg content would undergo structural changes during hydrothermal treatment at 110 °C in galactose solution. For initially amorphous (MgO)0.33(Al2O3)0.67 calcined at 680 °C, hydrothermal treatment also resulted in the formation of a crystal structure, which is visible by the appearance of reflexes attributed to MgAl2O4 and AlO(OH) (Fig. 12).
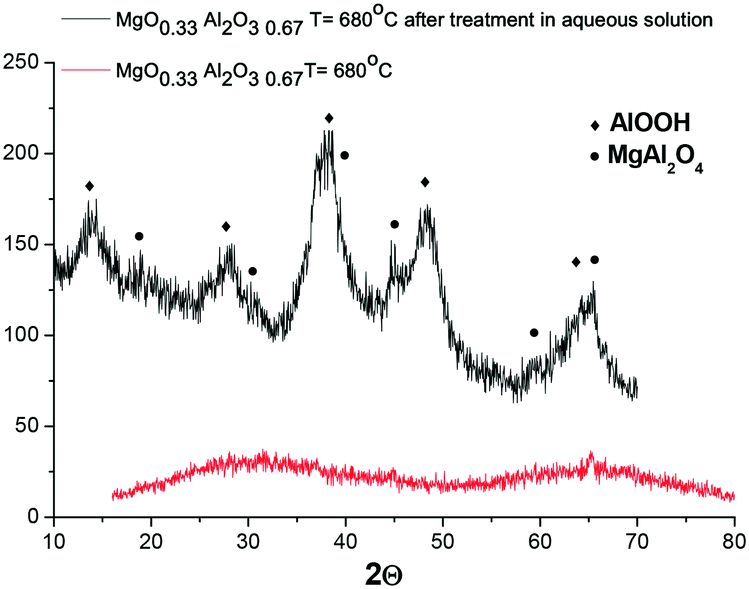 | ||
| Fig. 12 XRD diffractogram of (MgO)0.33(Al2O3)0.67 calcined at 680 °C prior to and after hydrothermal treatment at 110 °C for 2.5 h in galactose solution. | ||
Investigation of the Mg/Al ratio by XRF (Table 2) shows that the ratio between Mg and Al does not change substantially after hydrothermal treatment. At the same time the overall content of Mg and Al oxides decreased from 98 wt% to 87 wt%, which can be related to the appearance of water in the crystal structure as well as the existence of complex oxy- and hydroxy-mixed compounds containing Mg and Al.
For the (MgO)0.7(Al2O3)0.3 catalyst the leached amount of MgO was very minor in contrast to the catalysts with higher MgO content, such as (MgO)0.9(Al2O3)0.1 and (MgO)0.8(Al2O3)0.2, although the latter catalysts were more active in isomerization of aldoses that have resulted in a larger amount of side-products and displayed significant leaching with concentration of magnesium ions being 0.318 and 0.213 mg ml−1, respectively. It can be thus concluded that the (MgO)0.7(Al2O3)0.3 catalyst exhibited a Mg/Al ratio close to the hydrotalcites often reported as efficient base catalysts for isomerization of aldoses. The TEM images for this material are illustrated in Fig. 13.
 | ||
| Fig. 13 TEM of the (MgO)0.7(Al2O3)0.3 catalyst. | ||
As can be seen from the micrographs the catalyst is rather heterogeneous featuring particles ranging from 3 to 200 nm. Large (transparent) particles are spinel species with a large number of defects and in part are not well crystallized. No special phase of MgO is visible which can be also related to the poor leaching behavior of this catalyst as magnesia is present in the catalyst in a bound form. Smaller particles could tentatively be assigned to alumina even if the corresponding reflexes were not pronounced in the XRD patterns.
Conclusions
Isomerization of several aldoses (arabinose, galactose, glucose) was performed at 100–120 °C using a range of base catalysts including magnesia, hydrotalcites, magnesium aluminates with varying Mg/Al ratios and mixed oxides containing magnesia and alumina.Experiments with different aldoses showed that the sugar structure did not influence the catalytic properties. The apparent activation energy for all sugars obtained for a mixed oxide catalyst containing, in addition to magnesia and alumina, cobalt and manganese oxide was close to 99 kJ mol−1.
There was a linear correlation between the catalytic activity in galactose isomerization and the molar content of MgO in magnesium aluminates covering a broad range of Mg/Al ratios. The amount of MgO in the catalysts was closely related to their basicity.
Even if magnesium based materials (MgO, hydrotalcite and magnesium aluminate) could be applied for isomerization of sugars a serious issue is catalyst leaching. Hydrothermal stability tests with galactose solutions demonstrated that while a sample containing only MgO was dissolved completely, reasonable hydrothermal stability was displayed by the (MgO)0.7(Al2O3)0.3 catalyst calcined at 680 °C. The latter catalyst was active in isomerization of galactose exhibiting a TOF equal to 22 h−1.
Experimental section
Catalysts
Commercial samples of MgO (Fluka, 63093) and hydrotalcite (Mg/Al ratio of 3, Aldrich, 652288) were used as-received and after calcination. The CoMnAlMg mixed oxide catalyst (Mn:Co:Al2O3:MgO) was synthesized from the corresponding nitrates following the procedure described in ref. 28. This catalyst, as well as several supported cobalt catalysts, was applied to assess a potential role of cobalt and in combination with magnesia and alumina. The metal loading was 10 wt% and as supports alumina, silica, titania and SBA-15 were applied.Magnesium aluminate (Mg–Al–O) catalysts were prepared using the polymeric precursor or Pechini method.29 Predetermined amounts of magnesium nitrate (Mg(NO3)2·6H2O, 98%, Vekton) and aluminum nitrate (Al(NO3)3·9H2O, 98%, Vekton) were mixed and stirred at room temperature in distilled water. Thereafter, appropriate amounts of citric acid (99%) and ethylene glycol (>99%) (Sigma Aldrich) were added to this solution at room temperature. The polymerization reaction was conducted at 120 °C until no gas evolution was visible. The dark solid was calcined at 560 °C for 4 h under air with a heating rate of 10 °C min−1 to remove all organic components. Incomplete removal of organic compounds was observed at 560 °C judging by the color of the obtained samples; therefore, some samples were calcined at 680 °C for 24 h. It was decided to synthesize catalysts with the molar ratio MgO/(MgO + Al2O3) equal 0.2; 0.33; 0.45; 0.7; 0.8 and 0.9 which corresponds to different phases in the phase diagram (Fig. 14).
 | ||
| Fig. 14 Phase diagram of magnesium aluminates. | ||
Multiwall nanotubes (MWNTs) were synthesized by a CVD method of ethylene decomposition over bimetallic Fe–Co catalysts at 680 °C (surface area of 305 m2 g−1, average particle diameter of 9.4 nm).30 The functionalized MWCNTs containing surface carboxylic groups (0.76 groups per 1 nm2) were produced by treatment with boiling in concentrated nitric acid (denoted as MWNT–Ox–NA).31 Mg-containing samples were prepared by impregnation of functionalized MWNTs with the aqueous solutions of magnesium nitrate (Mg(NO3)2·6H2O, 98%, Vekton).32 After impregnation for 6 h the samples were dried at 110 °C for 4 hours followed by calcination at 350 °C for 4 hours under argon. By varying the concentration of the magnesium nitrate solutions, samples of 5 and 20 wt% Mg loadings were obtained. The cobalt catalysts were prepared by an impregnation method using Co(NO3)2·6H2O (Lach-ner, s.r.o.) described previously.33
Catalyst characterization
The metal content in the catalysts was determined by X-ray fluorescence analysis (XRF) on an ARL Perform'X analyzer with a Rh-anode X-ray tube. The element content was assessed by the UniQuant program for standard-less analysis. Prior to analysis, the samples were ground in an agate mortar, and then mixed with cellulose in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 (wt) in order to obtain the required volume for filling the cuvette. The sample mixed with cellulose was pressed into a tablet and placed in a cassette for analysis. Thereafter, the cassette was loaded into the vacuum chamber of the spectrometer and the measurements were carried out. To cover a range of elements from F to U, 5 scans were conducted for each sample.
:5 (wt) in order to obtain the required volume for filling the cuvette. The sample mixed with cellulose was pressed into a tablet and placed in a cassette for analysis. Thereafter, the cassette was loaded into the vacuum chamber of the spectrometer and the measurements were carried out. To cover a range of elements from F to U, 5 scans were conducted for each sample.
Reactor set-up
Isomerization of D(+) galactose (Sigma-Aldrich, ≥99% purity), L(+)-arabinose (Sigma, ≥99% purity) and D(+) glucose (Fluka, ≥98% purity) over different catalysts was investigated using a Parr 4561 autoclave. The liquid load was typically 100 ml of the overall volume of 300 ml. The sugar concentration in aqueous solution was equal to 2 wt% corresponding to 0.11 (galactose and glucose) and 0.13 M (arabinose). The autoclave was equipped with a gas entrainment impeller, baffles, heating jacket and cooling coil, sampling line, pressure, temperature and stirring rate controllers. The sugar solution was pre-heated under a nitrogen flow in a separate chamber. The catalyst (0.2–0.4 g) was put in the reactor which was flushed with nitrogen before heating. The reaction temperature was in the range 90–120 °C with the major part of experiments conducted at 110 °C. The stirring rate was 1000 rpm to overcome potential influence of external mass transfer, while application of fine catalyst powder (below 63 μm) ensured negligible influence of internal mass transfer. The catalytic experiments were performed under nitrogen with the overpressure 5 bar. Samples (1–2 ml) were periodically withdrawn through a 0.5 μm sinter during the semi-batch experiments. The experiments were carried out typically for 240 minutes.The pH of sugar and the catalyst slurry was measured for some cases after the reaction. As mentioned in the Introduction section, isomerization of glucose to fructose can occur at high alkaline pH (ca. 12–13). Much milder conditions in the present work rule out contribution of the homogeneous base catalysts to the experimental data.
HPLC analysis
The concentrations of reactants and products were determined by high-performance liquid chromatography (HPLC) (HITACHI Chromaster HPLC) equipped with an RI detector. A Biorad HPX-87C carbohydrate column was used and the mobile phase was 1.2 mM CaSO4. The temperature of the column was 70 °C, the flow rate of the mobile phase was 0.5 ml min−1 and the detector was at 40 °C. Calibrations were made for all reacting components as well as potential side products.Acknowledgements
The SusFuelCat project has received funding from the European Union's Seventh Framework Programme for research, technological development and demonstration under grant agreement no. 310490 (www.susfuelcat.eu). The work is a part of the Process Chemistry Centre (PCC), a centre of excellence financed by the Åbo Akademi University. M.A. Kazakova is grateful to the Novosibirsk regional government for the financial support (Scholarship for training of young scientists 2017).Notes and references
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539 RSC.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493 RSC.
- D. Yu. Murzin and I. L. Simakova, in Comprehensive Inorganic Chemistry II, vol. 7, From Elements to Applications, ed. R. Schlögl and J. W. Niemantsverdriet, 2013, vol. 7, p. 559 Search PubMed.
- S. H. Bhosale, M. B. Rao and V. V. Deshpande, Microbiol. Rev., 1996, 60, 280 CAS.
- C. A. Lobry de Bruyn and W. Alberda van Ekenstein, Recl. Trav. Chim. Pays-Bas, 1895, 14, 201–206 CAS.
- S. J. Angyal, Carbohydr. Res., 1997, 300, 279–281 CrossRef CAS.
- I. Delidovich and R. Palkovits, ChemSusChem, 2016, 9, 547 CrossRef CAS PubMed.
- A. A. Marianou, C. M. Michailof, A. Pineda, E. F. Iliopoulou, K. S. Triantafyllidis and A. A. Lappas, ChemCatChem, 2016, 8, 1100 CrossRef CAS.
- C. Kooyman, K. Vellenga and H. de Wilt, Carbohydr. Res., 1977, 54, 33 CrossRef CAS.
- B. Y. Yang and R. Montgomery, Carbohydr. Res., 1996, 280, 27 CrossRef CAS.
- C. J. Knill and J. F. Kennedy, Carbohydr. Polym., 2003, 51, 281 CrossRef CAS.
- J. M. Carraher, C. N. Fleitman and J.-P. Tessonnier, ACS Catal., 2015, 5, 3162 CrossRef CAS.
- C. Moreau, R. Durand, A. Roux and D. Tichit, Appl. Catal., A, 2000, 193, 257 CrossRef CAS.
- J. Lecomte, A. Finiels and C. Moreau, Starch/Staerke, 2002, 54, 75 CrossRef CAS.
- C. Moreau, J. Lecomte and A. Roux, Catal. Commun., 2006, 7, 941 CrossRef CAS.
- S. Yu, E. Kim, S. Park, I. K. Song and J. C. Jung, Catal. Commun., 2012, 29, 63 CrossRef CAS.
- I. Delidovich and R. Palkovits, J. Catal., 2015, 327, 1 CrossRef CAS.
- I. Delidovich and R. Palkovits, Catal. Sci. Technol., 2014, 4, 4322 CAS.
- A. Takagaki, M. Ohara, S. Nishimura and K. Ebitani, Chem. Commun., 2009, 6276 RSC.
- A. Takagaki, S. Furusato, R. Kikuchi and S. T. Oyama, ChemSusChem, 2015, 8, 3769 CrossRef CAS PubMed.
- M. Moliner, Y. Roman-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164 CrossRef CAS PubMed.
- V. Choudhary, A. B. Pinar, S. I. Sandler, D. G. Vlachos and R. F. Lobo, ACS Catal., 2011, 1, 1724 CrossRef CAS.
- J. Hajek, D. Yu. Murzin, T. Salmi and J.-P. Mikkola, Top. Catal., 2013, 56, 839 CrossRef CAS.
- L. Ren, Q. Guo, P. Kumar, M. Orazov, D. Xu, S. M. Alhassan, K. A. Mkhoyan, M. E. Davis and M. Tsapatsis, Angew. Chem., Int. Ed., 2015, 54, 10848 CrossRef CAS PubMed.
- J. Tuteja, S. Nishimura and K. Ebitani, Bull. Chem. Soc. Jpn., 2012, 85, 275 CrossRef CAS.
- K. K. Yadav, S. Ahmad and S. M. S. Chauhan, J. Mol. Catal. A: Chem., 2014, 394, 170 CrossRef CAS.
- J. A. Moulijn, M. Makee and A. E. van Diepen, Chemical Process Technology, Wiley, Chichester, 2013 Search PubMed.
- S. Buchholz, V. Michele, L. Mleczko, C. Muennich, R. Rudolf and A. Wolf, Patent WO2007093337 to Bayer Material Science AG, 2007.
- J. Rufner, D. Anderson, K. van Benthem and H. R. Castro, J. Am. Ceram. Soc., 2013, 96, 2077 CrossRef CAS.
- V. L. Kuznetsov, S. N. Bokova-Sirosh, S. I. Moseenkov, A. V. Ishchenko, D. V. Krasnikov, M. A. Kazakova, A. I. Romanenko, E. N. Tkachev and E. D. Obraztsova, Phys. Status Solidi B, 2014, 251, 2444 CrossRef CAS.
- I. Mazov, V. Kuznetsov, I. Simonova, A. Stadnichenko, A. Ishchenko, A. Romanenko, E. Tkachev and O. Anikeeva, Appl. Surf. Sci., 2012, 258, 6272 CrossRef CAS.
- A. S. Andreev, M. A. Kazakova, A. V. Ishchenko, A. G. Selyutin, O. B. Lapina, V. L. Kuznetsov and J.-B. d'Espinose de Lacaillerie, Carbon, 2017, 114, 39 CrossRef CAS.
- D. Kubička and R. Černý, Ind. Eng. Chem. Res., 2011, 51, 8849 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |