
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multifunctional supported bimetallic catalysts for a cascade reaction with hydrogen auto transfer: synthesis of 4-phenylbutan-2-ones from 4-methoxybenzyl alcohols†
Moataz
Morad
ab,
Ewa
Nowicka
a,
Mark
Douthwaite
a,
Sarwat
Iqbal
a,
Peter
Miedziak
a,
Jennifer K.
Edwards
a,
Gemma L.
Brett
a,
Qian
He
a,
David
Morgan
a,
Hamed
Alshammari
ac,
Donald
Bethell
a,
David W.
Knight
a,
Meenakshisundaram
Sankar
*a and
Graham J.
Hutchings
 *a
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, CF10 3AT Cardiff, UK. E-mail: sankar@cardiff.ac.uk; hutch@cardiff.ac.uk; Fax: +44 (0)2920 874 030
bChemistry Department Faculty of Science, Umm Al-Qura University, PO Box 21955, 9264 Makkah, Saudi Arabia
cChemistry Department, Faculty of Science, Ha'il University, P. Box 2440, 81451 Ha'il, Saudi Arabia
First published on 28th March 2017
Abstract
We report the one-pot tandem synthesis of 4-(4-methoxyphenyl)butan-2-one directly from 4-methoxybenzyl alcohol and acetone using a multifunctional supported AuPd nanoalloy catalyst. This one-pot synthesis involves dehydrogenation, aldol condensation and hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C. In this supported AuPd catalyst, the bimetallic sites catalyse the dehydrogenation and hydrogenation steps and, in combination with the support, catalyse the C–C coupling (aldol) process. This supported bimetallic catalyst is also effective in utilizing hydrogen from the dehydrogenation reaction for the hydrogenation of 4-(4-methoxyphenyl)but-3-en-2-one to 4-(4-methoxyphenyl)butane-2-one via a hydrogen auto transfer route. These multifunctional catalysts were characterised using transmission electron microscopy, X-ray diffraction and X-ray photoelectron spectroscopy.
C. In this supported AuPd catalyst, the bimetallic sites catalyse the dehydrogenation and hydrogenation steps and, in combination with the support, catalyse the C–C coupling (aldol) process. This supported bimetallic catalyst is also effective in utilizing hydrogen from the dehydrogenation reaction for the hydrogenation of 4-(4-methoxyphenyl)but-3-en-2-one to 4-(4-methoxyphenyl)butane-2-one via a hydrogen auto transfer route. These multifunctional catalysts were characterised using transmission electron microscopy, X-ray diffraction and X-ray photoelectron spectroscopy.
Introduction
4-(4-Methoxyphenyl)butan-2-one (4), also known as “raspberry ketone methyl ether”, is an important fine chemical and is used extensively in several consumer products such as (a) raspberry scent, (b) food additives, (c) insect attractants as well as (d) in the preparation of melanin formation inhibitors.1,2 However, the quantities available from natural sources are exceedingly low (ca. 1–4 mg per kg of raspberries) and hence an efficient synthetic route leading to 4 is in high demand. Commercially, ketone 4-(4-methoxyphenyl)butan-2-one is prepared by Friedel–Crafts alkylation of anisole with 4-hydroxybutan-2-one or methyl vinyl ketone (MVK) using homogeneous or heterogeneous Lewis or Brønsted acid catalysts.3 This method produces many side products (as a result of transalkylation, isomerization, polyalkylation and polymerisation reactions) besides significant quantities of waste. Corma et al. proposed another route for the synthesis of 4 using the Heck reaction between 4-methoxyiodobenzene and methyl vinyl ketone.4 Although this is an efficient method, it is not environmentally benign, especially as iodide is a by-product. Recently, the same group reported a one-pot synthesis of 4-(4-methoxyphenyl)butan-2-ol (5) from 4-methoxybenzaldehyde (2) (R1 = OCH3) and acetone by combining aldol coupling and hydrogenation reactions in one pot (2 to 5 in Scheme 1). They compared the three processes and deduced that the tandem synthesis, employing a multifunctional catalyst, has the smallest E factor.5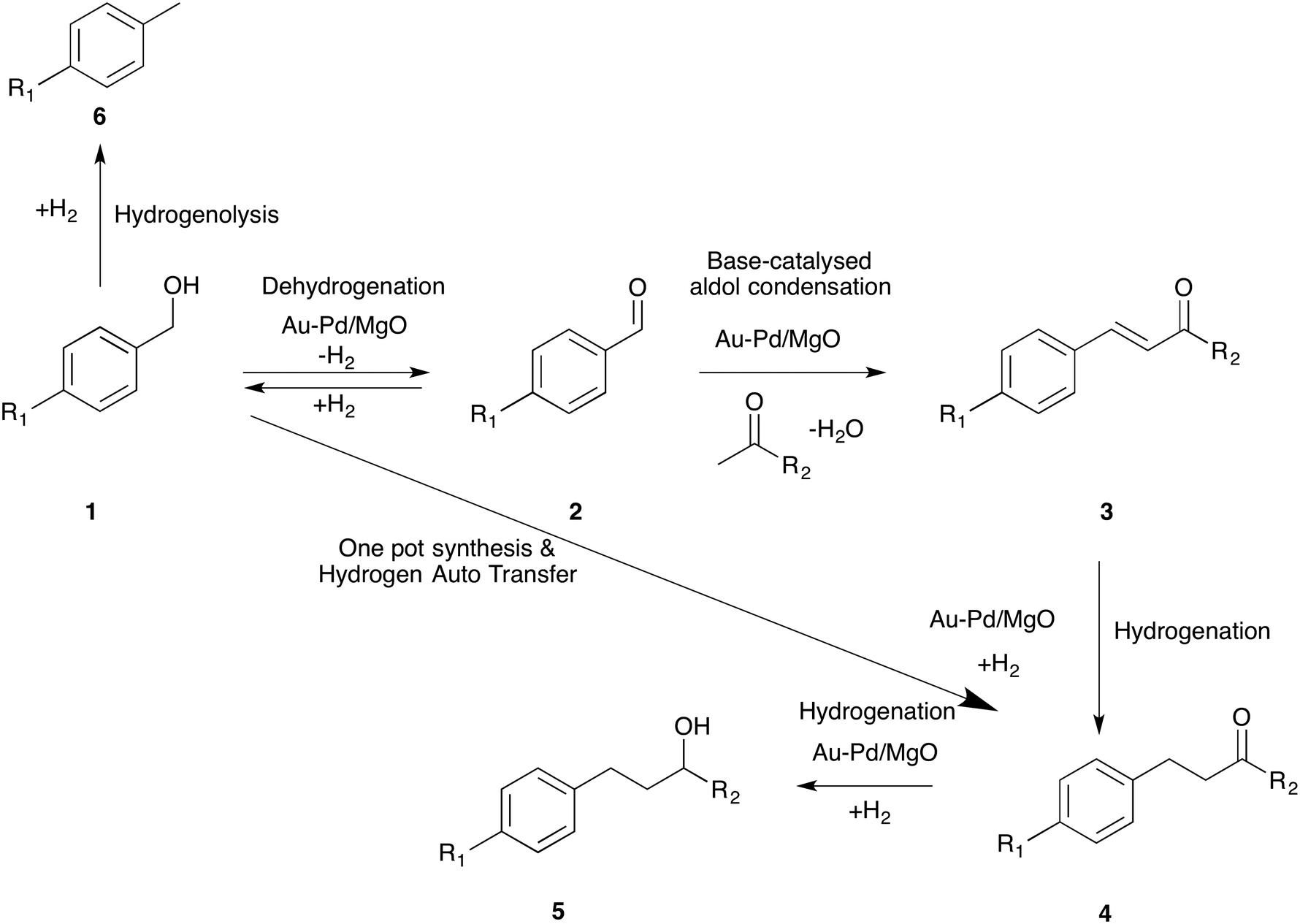 | ||
| Scheme 1 General scheme for the tandem synthesis of aldols from alcohol and ketone by combining dehydrogenation, coupling and hydrogenation reactions. | ||
One of the grand challenges in chemistry is to develop alternative technologies to produce chemicals in a cleaner, safer, and environmentally benign manner.6 Multi-enzymatic systems that perform multi-step reactions in nature have inspired catalytic chemists to develop multifunctional catalysts that can catalyse several chemical transformations in a single reactor, known as tandem, domino or cascade reactions.7–9 This strategy helps to improve the atom economy or the E factor (kgwaste/kgproduct) by decreasing the number of energy-intensive separation and purification steps of the intermediates.10 Designing a multi-functional catalyst is a challenging task because each transformation requires different, often incompatible, catalytic active sites (e.g. acidic and basic sites) and/or reaction conditions.11,12 There are examples of bi-functional catalysts, including the widely used alkane hydroisomerization/hydrocracking catalyst; in this case the metal (Pt) centre catalyses the dehydrogenation of an n-alkane to form the corresponding olefin and H2 whilst the acidic support (alumina or zeolite) catalyses the isomerization or cracking reactions.13 The metal (Pt) site subsequently catalyses the hydrogenation of the branched olefins.
Following initial reports by Sinfelt on the use of bimetallic catalysts, we have reported the application of supported gold–palladium bimetallic catalysts for the aerobic oxidative dehydrogenation of primary alcohols and the hydrogenation of levulinic acid (LA) to γ-valerolactone (GVL).14–16 In these examples, we found that alloying two metals substantially increased the activity and/or selectivity compared to their monometallic counterparts. A number of other bimetallic catalysts have also been reported to be exceptionally active and/or selective for oxidation, hydrogenation, hydrochlorination, reforming reactions and biomass conversion.17,18 Recently we found that for the aerobic oxidative dehydrogenation of benzyl alcohol, supporting the gold–palladium nanoalloys on MgO, the disproportionation pathway, which leads to the undesired formation of toluene, is completely inhibited, thereby increasing the selectivity for the desired benzaldehyde to >99%.19 We further exploited the basicity of MgO in this catalyst for the (otherwise) base-free oxidation of glycerol to glyceric acid under mild reaction conditions.20,21
In this work, we report an efficient multi-functional catalyst for the tandem synthesis of 4-(4-methoxyphenyl)butan-2-one (4) starting from 4-methoxybenzyl alcohol (1) instead of 4-methoxybenzaldehyde (2) (Scheme 1). Adding an initial dehydrogenation reaction is advantageous because alcohols are generally more readily available starting materials than aldehydes and inherently they are more stable. Importantly, we further report that the hydrogen generated from this dehydrogenation reaction is effectively used in the hydrogenation reaction (3 to 4) using a hydrogen auto-transfer strategy. Moreover, we demonstrate, using different derivatives of benzyl alcohol, that this one-pot reaction proceeds in a similar way to that of 4-methoxybenzyl alcohol.
Experimental
Catalyst preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 80% hydrolyzed) was added to the solution and stirred with HAuCl4·3H2O for 15 min. 0.1 M NaBH4 (>96% Aldrich, NaBH4/Au (mol/mol = 5)) was freshly prepared and then added to the solution to form a dark brown sol. The mixture was stirred for 30 min and adjusted to pH 1 by the drop-wise addition of sulphuric acid. The supports were added to the mixture and the slurry was then stirred for 1 h. Following this, the catalyst was recovered by filtration, washed with 2 L distilled water and dried at 383 K overnight. The 0.5% Au–Pd/Mg(OH)2 catalyst was then treated at 723 K for 5 h under N2 (ramp rate 20 K min−1) to form 0.5% Au–Pd/MgO. All these catalysts were used without any further treatment.
000, 80% hydrolyzed) was added to the solution and stirred with HAuCl4·3H2O for 15 min. 0.1 M NaBH4 (>96% Aldrich, NaBH4/Au (mol/mol = 5)) was freshly prepared and then added to the solution to form a dark brown sol. The mixture was stirred for 30 min and adjusted to pH 1 by the drop-wise addition of sulphuric acid. The supports were added to the mixture and the slurry was then stirred for 1 h. Following this, the catalyst was recovered by filtration, washed with 2 L distilled water and dried at 383 K overnight. The 0.5% Au–Pd/Mg(OH)2 catalyst was then treated at 723 K for 5 h under N2 (ramp rate 20 K min−1) to form 0.5% Au–Pd/MgO. All these catalysts were used without any further treatment.
| X = (Sin − Sout)/(Sin) × 100% |
Selectivities were calculated taking into account only identified products. Unidentified products accounted for 20% to 30% of all products (depending on the reaction). These were not taken into account during the analysis, resulting in a carbon mass balance of 70–80%. Selectivity was calculated based on the following equation:
| SP = (P/∑Pidentified) × 100% |
Catalyst characterisation
Results and discussion
Initially we used aldehyde 2 (R1 = OCH3) as the starting material to identify a suitable active support for the aldol condensation step. Basic metal oxides such as MgO (BDH), ZnO (Sigma Aldrich) and La2O3 (Sigma Aldrich) were tested as received; for comparison TiO2 (Degussa P-25) and Al2O3 (Sigma Aldrich) were also tested (see the ESI† Table S1). Surprisingly, the basic metal oxides are not effective for this transformation. This is not due to poisoning of the basic sites by CO2 as even after calcination at 673 K there was no improvement in their catalytic activities. Al2O3 and TiO2, however, show appreciable ability to catalyse this coupling, possibly via acid catalysed aldol condensation reaction. In the next stage, we supported AuPd nanoalloy particles, which were between 1 and 7 nm, on all these supports using a sol-immobilization technique.19 In addition to these supports, a nano MgO, produced by the calcination of commercial nano Mg(OH)2 at 673 K, was also used to support these AuPd nanoalloys.23 All supported AuPd catalysts were tested for the conversion of 4-methoxybenzaldehyde (2) to 4-(4-methoxyphenyl)butan-2-one (4). Here we sought to combine the coupling reaction with hydrogenation of the CC bond in the resulting α,β unsaturated ketone 3. For comparison, we also prepared monometallic Au and Pd catalysts and tested these in this one-pot synthesis (see the ESI† Table S2). The results indicate that the nature of the metallic site and the metal oxide support play crucial roles in determining the reaction pathways and hence the selectivity towards the products. Monometallic Pd supported on TiO2 (5% Pd/TiO2) is extremely active and selective for the transformation of 2 into 4-methylanisole (6) with 100% conversion and 99% selectivity, presumably by sequential reduction to the corresponding benzyl alcohol 1 and its hydrogenolysis. This is a totally different pathway compared to the formation of 3 and 4. A different 1% Pd/TiO2 catalyst was prepared by a conventional wet-impregnation method and was used for this reaction. This catalyst is less active than the previous catalyst (5% Pd/TiO2); however, it leads predominantly to formation of ketone 4 with no trace of anisole 6. This change in reaction pathway between 1% Pd/TiO2 and 5% Pd/TiO2 could be because of the difference in the acidity of these two catalysts. The Pd precursor solution is made by dissolving solid PdCl2 in 0.58 M HCl solution to form palladate because solid PdCl2 is sparingly soluble in cold water, whereas palladate is. This means that a higher Pd loading increases the HCl concentration in the catalyst synthesis medium. This could have resulted in an increased acidic surface of the catalyst for higher Pd loading which results in the hydrogenolysis pathway rather than the aldol condensation pathway. Another explanation is that metal catalysed reactions (hydrogenation and hydrogenolysis) dominate in the competition for 2 when catalysts with higher metal loading are used. Metal oxide catalysed aldol condensation dominates when the catalysts with lower metal loading are used. However, the role of the nature of the metal particles (specifically size and structure) in determining the reaction pathways cannot be ruled out at this stage. A further in-depth investigation to study this interesting behaviour is currently in progress. All other observations are understandable in terms of the flow chart in Scheme 1.
When bimetallic 1% AuPd/TiO2 was used for this reaction, the conversion increased to 63% compared to 16% for the monometallic Pd catalyst prepared by the same method. However, the selectivity to 4 dropped dramatically to 16% with reduction to alcohol 1 becoming the major pathway (62%). We also used Al2O3 as the support for the AuPd nanoalloys since it was found to be active for the transformation of 2 into 3 in the absence of a metal (Table 1). Surprisingly, this catalyst is less active, with only 7% conversion and a higher selectivity for reduction to alcohol 1. However, when 1% AuPd/MgO was used as the catalyst, the selectivity to ketone 4 increased markedly to 58% with 100% conversion. This indicates that MgO-supported catalysts are preferred for this reaction and validates the initial hypothesis of this work. We then employed nano-MgO23 as support for the AuPd nanoalloys and decreased the metal content by 50% to 0.5 wt%. As shown in Fig. S2,† XRD confirmed that the final phase of the support was in fact MgO. The final heat treatment conducted under flowing N2 appears to result in a complete phase change from Mg(OH)2 after the immobilisation of Au and Pd to MgO. Both catalysts displayed 100% conversion but for this new catalyst there was a marginal increase in the selectivity to the desired product 4. For comparison we used the monometallic counterparts of this catalyst [0.5% Au/MgO and 0.5% Pd/MgO] for this reaction and found 0.5% Pd/MgO to be the most effective (see the ESI† Table S2). The most interesting observation is that MgO alone does not catalyse the aldol coupling reaction (see the ESI† Table S1); however, when combined with metal nanoparticles, it is highly effective. This is completely reversed in the case of Al2O3, where the metal oxide alone is active for the coupling reaction, whereas AuPd supported on Al2O3 is not. These two examples indicate that the active sites for these two reactions (coupling and hydrogenation) are interdependent: the metal sites appear to promote the catalytic activities of metal oxide sites for the coupling reactions.
| Catalyst | Conv (%) | Product selectivitya (%) | ||||
|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 6 | ||
| Reaction conditions: 4-methoxybenzyl alcohol: 1.3 g; acetone: 6.3 g; mesitylene (internal standard): 1 g; catalyst: 0.5 g; pH2: 5 bar; T: 348 K; time: 17 h. a Selectivity is calculated based on known and identified products and we have used a normalisation method based on these products. The C balance for these reactions was calculated to be between 70–80%. Remaining products are unidentified (20–30%). b Oxidative dehydrogenation for 3 h followed by reduction for 2 h. c Oxidation for 3 h followed by coupling reaction for 17 h and hydrogenation for 2 h. d Oxidative dehydrogenation for 3 h followed by coupling reaction for 17 h and hydrogenation for 3 h. | ||||||
| 1% AuPd/TiO2b | 100 | 0 | <1 | 27 | 14 | 57 |
| 1% AuPd/TiO2c | 100 | 0 | 0 | 55 | 21 | 24 |
| 1% Pd/TiO2d | 100 | 0 | 0 | 55 | 11 | 34 |
| 1% AuPd/MgO | 57 | 0 | 44 | 32 | 1 | <1 |
Inspired by our discovery that supported AuPd nanoalloys are extremely active for the selective oxidative dehydrogenation of benzyl alcohol to benzaldehyde,15 we then investigated the tandem synthesis of ketone 4 (R2 = CH3) directly from alcohol 1 (R1 = OCH3). The results are presented in Table 1. Initially we used 1% AuPd/TiO2 and performed the oxidative dehydrogenation reaction (step 1) under O2 pressure at 398 K for 3 h, resulting in 100% conversion with selectivities of 64% and 34% for 2 (aldehyde) and 3 (enone), respectively. The reaction mixture was then hydrogenated (step 3) under a hydrogen atmosphere (after purging the reaction mixture with N2 thoroughly) at 398 K for 2 h. The product mixture contained mostly anisole 6 (57%) together with small amounts of ketone 4 (27%) and alcohol 5 (14%). This catalyst, although very suitable for the oxidative dehydrogenation reaction, is not effective for either the coupling or the hydrogenation reaction. Instead, it catalyses the hydrogenation of 2 to 1 which further undergoes hydrogenolysis to 6. To force 2 to undergo the coupling reaction instead of hydrogenation, we introduced another step (step 2) between steps 1 and 3 (Scheme 1), where N2 replaced the oxygen atmosphere in a reaction carried out for 17 h at 398 K. At the end of this, the product mixture was mostly enone 3 (76%) together with some residual aldehyde 2 (21%). This mixture was further hydrogenated at 348 K for 2 h. This additional step increased the selectivity of 4 to 55% with selectivities of 24% and 21% for 6 and 5, respectively. When monometallic Pd (1% Pd/TiO2) was used for this reaction, the selectivity to ketone 4 increased slightly to 61%.
Finally, we used 1% AuPd/MgO for this one pot synthesis because MgO does not catalyse the hydrogenolysis reaction. The resulting reaction mixture had a substantially lower amount of anisole 6 (<1%), however with a lower conversion (57%), the desired products 3 and 4 were formed with selectivities 44% and 32% respectively. The lower conversion was deemed to be due to the hydrogenation of 2 back to 1. Instead of this three-step strategy, we decided to perform an experiment in a hydrogen atmosphere, which resulted in better activity. After a 22 h reaction at 398 K, we achieved 100% conversion with 85% selectivity for the formation of ketone 4 (Table 2). The reaction was performed in an inert atmosphere (N2) and the results, presented in Table 2 (last entry), clearly indicate that the hydrogen from the dehydrogenation of alcohol is utilized for the hydrogenation of 3 to 4. This is evidently the hydrogen auto transfer methodology (or hydrogen borrowing) proposed by Williams and co-workers.24 This heterogeneous catalyst is one of the very few that are active for this hydrogen auto transfer under mild reaction conditions.25,26 This hydrogen auto transfer route has potentially many applications, especially in the synthesis of fine chemicals.27,28 In order to test the substrate tolerance of this catalyst, in one reaction, benzyl alcohol was coupled with acetone and in a different reaction, 4-methoxybenzyl alcohol and 4-tert-butylbenzyl alcohol were coupled with 2-butanone (Table 2). In this hydrogen auto transfer reaction, we have combined dehydrogenation, coupling and hydrogenation reactions in one-pot for the sequential synthesis of an industrially very important compound (4). It is interesting to note that the first step of this synthesis (dehydrogenation of 1 to 2) proceeds without any oxidant and even in the presence of H2. Further detailed investigation is needed to understand this observation. To the best of our knowledge, this is the first time that these three reactions (dehydrogenation, coupling and hydrogenation) have been combined in one pot for the selective synthesis of this industrially very important ketone (4).
| R1 | R2 | Atm | Conv (%) | Product selectivitya (%) | ||||
|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 6 | ||||
| Reaction conditions: 4-methoxybenzyl alcohol: 1.3 g; benzyl alcohol: 1.1 g, 4-tert-butyl benzyl alcohol: 1.54 g, 2-butanone: 7.7 g acetone: 6.3 g; mesitylene (internal standard): 1 g; catalyst: 0.5 g; p: 5 bar; T: 398 K; time: 22 h. a Selectivity is calculated based on known and identified products and we have used a normalisation method based on these products. The C balance for these reactions was calculated to be between 70% and 80%. The remaining products are unidentified (20–30%). | ||||||||
| OCH3 | CH3 | N2 | 96 | 1 | 55 | 43 | 0 | 4 |
| H2 | 100 | 0 | 0 | 85 | 10 | 5 | ||
| OCH3 | C2H5 | N2 | 79 | 14 | 73 | 12 | 2 | 0 |
| H2 | 97 | 0 | 4 | 78 | 13 | 5 | ||
| H | CH3 | N2 | 65 | 0 | 74 | 17 | 6 | 3 |
| H2 | 65 | 0 | 11 | 6 | 2 | 81 | ||
| C(CH3) | C2H5 | N2 | 34 | 6 | 88 | 6 | 0 | 0 |
| H2 | 90 | 0 | 0 | 95 | 2 | 3 | ||
The conversion of 1 to 4 is a sequence of reactions and the mechanism of formation of all these products on a multi-functional catalyst is complex. However, based on the currently available experimental evidence, we can say that the dehydrogenation of the alcohol 1, presumably reversibly, gives aldehyde 2 + 2Cat-H (potential side reaction → Cat + H2). The aldol reaction of 2 with acetone gives unsaturated ketone 3 + water. Reaction of 3 with Cat-H, gives 4, followed by hydrogenation of the carbonyl group to give 5, these steps being limited by the amount of Cat-H remaining after the first step, in reactions not conducted in a hydrogen atmosphere. With a hydrogen atmosphere, another source of Cat-H is available and the hydrogenation steps 3 → 4 → 5 progress further; however, the probability of hydrogenolysis of the alcohol, giving toluene 6 + water, in competition with dehydrogenation, is now increased.
To study the effect of the electronic nature of the substituents on the reactivity and the versatility of this catalyst, we tested 1% AuPd/MgO using different substituted benzyl alcohols and ketones and the results are presented in Table 2. The electron donating methoxy group (−OCH3) in the phenyl ring displays the maximum reactivity, whereas a tertiary butyl group suppresses the activity for both the one-pot synthesis and the hydrogen auto transfer reaction. Acetone is a better carbonyl compared to butanone for this one-pot synthesis.
We assessed the stability of these catalysts by reusing them for the reactions conducted under both H2 and N2 and the results are presented in Table 3. A significant decrease in catalytic performance was observed for the catalysts recovered from the reactions under both H2 and N2. A drop in conversion from 100% to 46% was observed for the reaction conducted under H2. The hydrogenation of 3 to 4 appears to have a reduced rate, which suggests that this pathway may also be affected by the deactivation of the catalyst. Interestingly, the selectivity to product 6 is significantly reduced during the re-use study under H2, which indicates that the used catalyst is more selective to our desired reaction pathway. For the re-use study under N2 no conversion was observed, suggesting that the catalyst was fully deactivated.
| Atm | Conv (%) | Product selectivitya (%) | ||||
|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 6 | ||
| Reaction conditions: 4-methoxybenzyl alcohol: 1.3 g, acetone: 6.3 g; mesitylene (internal standard): 1 g; catalyst: 0.5 g; p: 5 bar; T: 398 K; time: 22 h. a Selectivity is calculated based on known and identified products and we have used a normalisation method based on these products. The C balance for these reactions was calculated to be between 70% and 80%. The remaining products are unidentified (20–30%). | ||||||
| N2 | 96 | 1 | 55 | 43 | 0 | 4 |
| — | — | — | — | — | — | |
| H2 | 100 | 0 | 0 | 85 | 10 | 5 |
| 46 | 0 | 6 | 77 | 16 | 0.3 | |
In order to understand the reason for the catalyst deactivation, the fresh and used catalysts were studied. Representative TEM images of fresh and used AuPd/MgO catalysts prepared from nano-Mg(OH)2 are shown in Fig. 1 and 2. From Fig. 1, it is clearly seen that small Au–Pd nanoparticles can be found in the fresh catalyst (Fig. 1(a)) and are still present in the used catalysts (Fig. 1(b and c)), suggesting that particle agglomeration is not likely to be responsible for deactivation. However, the support material appears to have undergone some changes after the reaction. In the fresh catalyst, well crystallised MgO particles can be found (Fig. 2(a)), while in the catalyst used in a H2 environment, a mixture of MgO (Fig. 2(b)) and brucite Mg(OH)2 phase (Fig. 2(c)) can be found. The difference in the electron diffraction can be clearly seen in the rotational averaged profile shown in Fig. 2(d). This also agrees with the XRD data shown in Fig. S3,† which indicates that there is a clear change in the support during the reactions. The support in the used catalyst appears to be partially hydroxylated, whereas the fresh sample is clearly an oxide. It is known that MgO readily converts to Mg(OH)2 in the presence of H2O,20 so it is likely that the production of H2O as a result of the aldol condensation from 3 to 4 is responsible for this change in phase from MgO to Mg(OH)2. This subtle change in the support phase may also contribute to the clear deactivation of the catalyst. Another possible source of deactivation could be the substantial quantity of organics on the surface. These organic materials, covering some of the metallic sites, could explain the reduced reduction rate for the used catalyst. TGA experiments revealed a large loss in mass at approximately 630 K (Fig. S1†), which is not observed in the corresponding fresh catalytic sample. ICP analysis confirmed that metal leaching was not responsible for the deactivation. The concentrations of Au and Pd observed in the post reaction effluents of the H2 and N2 reactions were comparable with an acetone background blank, suggesting that no Au or Pd was leached from the support during the reactions (see the ESI† Table S3).
 | ||
| Fig. 1 Representative TEM images of Au–Pd/MgO catalysts prepared using nano-Mg(OH)2 showing the Au–Pd nanoparticles on (a) the catalyst prior to reaction, (b) the used catalyst after the reaction with 4-methoxybenzyl alcohol under H2 and (c) the used catalyst after the reaction with 4-methoxybenzyl alcohol under N2. | ||
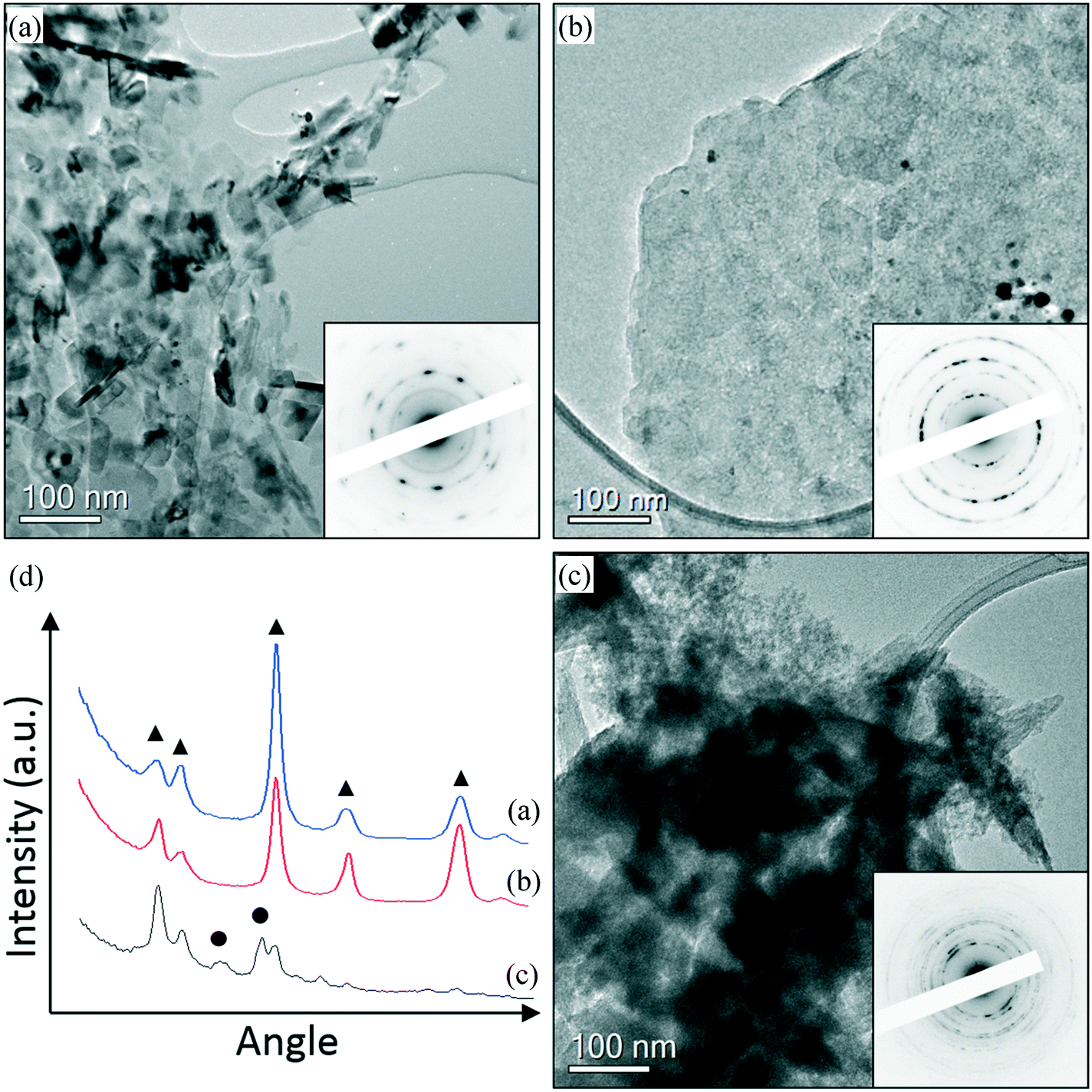 | ||
| Fig. 2 Representative TEM images of Au–Pd/MgO catalysts prepared using nano-Mg(OH)2 showing the support structure change on the (a) AuPd/MgO catalyst prior to reaction and (b and c) the used AuPd/MgO catalyst after the reaction with 4-methoxybenzyl alcohol under H2. The insets are corresponding select area electron diffractions. (d) Shows the rotational average of the diffraction pattern shown in (a)–(c). ●: Mg(OH)2 brucite phase; ▲: MgO phase. | ||
XPS analysis of low loading Au and Pd supported materials on MgO is complicated by the overlap of the Mg(2s) and Mg(KLL) Auger peaks, respectively.29 However, careful fitting of peak components and also application of “vector” techniques for determination of peak positions and lineshapes30 can greatly enhance the information which can be elucidated from such complicated data. For the fresh catalyst, Au is found in two states with binding energies of 83.8 and 85.5 eV, which we attribute to metallic Au and ionic Au, potentially anchored to defect sites such as F-centres. For the fresh sample, we have been unable to determine the Pd content with significant confidence. Fig. 3 shows the Au(4f)/Mg(2s) and Pd(3d)/Mg(Auger) fitted spectra.
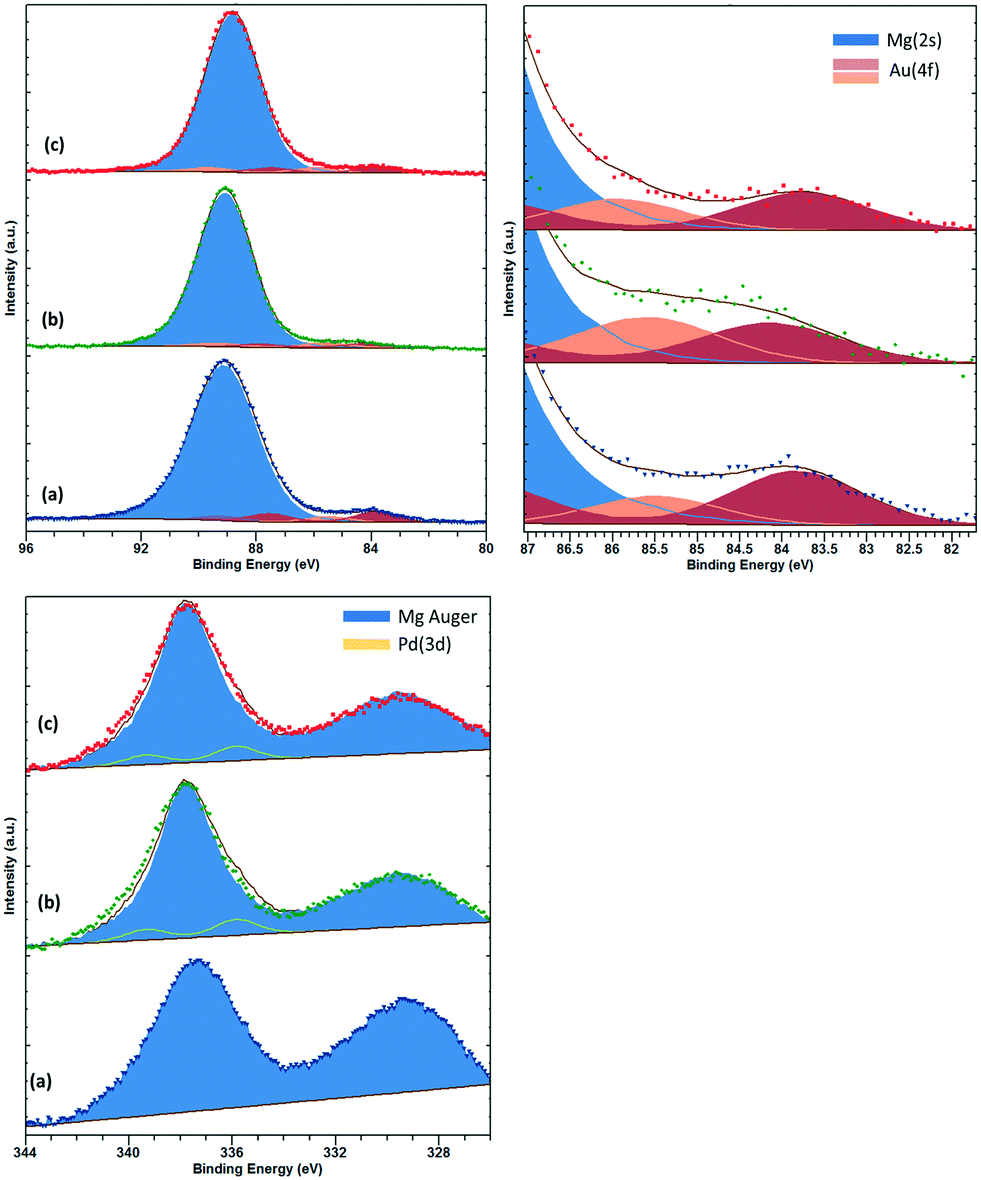 | ||
| Fig. 3 Top: Au(4f)/Mg(2s), with close up for Au(4f) region and bottom: Pd(3d)/Mg(KLL) Auger photoemission spectra for (a) AuPd/MgO fresh, (b) AuPd/MgO–N2 and (c) AuPd/MgO–H2. | ||
For the H2 sample, the Au is found to shift upward slightly to 84.1 and 86.3 eV, respectively, and may be due to increased stabilisation of the Au by hydroxide groups.31 The Pd content in this sample can be determined by the vector method and is found to have a binding energy of 335.8 eV (±0.2 eV) and attributed to partially oxidised Pd or binding energy shifts due to reduced final state screening effects.32 However, with the small metal loadings employed in this study, we are unable to perform analysis of the Pd(LMM) Auger signal to elucidate this shift any further. For the N2 sample, similar Au and Pd species remain; however, the intensity of the Au species is diminished, whilst the high binding energy Au species is downshifted ca. 0.4 eV, potentially indicating an increase in particle size.
XPS was also used to determine the estimated surface content of Au, Pd, C, O and Mg which is displayed in Table 4. Although it was not possible to accurately determine the quantity of Pd in the fresh AuPd/MgO sample, near stoichiometric quantities of Au and Pd were observed in the used catalysts after reaction under H2 and N2. Interestingly, a significantly larger quantity of oxygen is observed on the surface of the used catalysts, which provides further evidence that the MgO undergoes partial hydroxylation during the reaction. It was previously suggested that this is a result of H2O from the aldol condensation reaction taking place and is supported by the diffraction data of the used catalysts displayed in Fig. S3.† Perhaps the most significant finding from this technique is the larger quantities of carbon observed on the surface of the used catalysts. This further supports the suggestion that the catalytic deactivation observed could be the result of coking on the surface of the catalysts during the reactions. The deactivation mechanism of the catalyst still remains unclear, although there is evidence to suggest that the deactivation may be a consequence of particle agglomeration and/or substrate coking on the catalyst surface. Enhancements and modifications in the design of the catalyst that could alleviate these deactivation issues and allow for greater potential for industrialisation of this process are needed.
| Catalyst | Atomic % | ||||
|---|---|---|---|---|---|
| C | Mg | O | Au | Pd | |
| AuPd MgO fresh | 5.84 | 46.58 | 47.4 | 0.19 | n.d. |
| AuPd MgO used H2 | 9.65 | 31.54 | 58.59 | 0.12 | 0.10 |
| AuPd MgO used N2 | 9.63 | 32.53 | 57.62 | 0.10 | 0.12 |
Conclusion
We report that AuPd nanoalloys supported on MgO and TiO2 are effective multi-functional catalysts for the one-pot synthesis of 4-(4-methoxyphenyl)butan-2-one (4) from 4-methoxybenzyl alcohol (1) and acetone. This one-pot synthesis comprises dehydrogenation, condensation and hydrogenation reactions. This is the first report of combining all these reactions in one-pot. Throughout, we have portrayed this chemistry as a sequential dehydrogenation–aldol–hydrogenation sequence. However, it can be viewed in another, very general way as the overall alkylation of a ketone (acetone) by a benzyl alcohol 1. This places the foregoing methodology in the same area as the recently developed protocols for achieving the same overall transformations.28,33,34 Furthermore we have shown that the MgO supported material is very active, as a heterogeneous catalyst, for the hydrogen auto transfer reaction.Author contributions
MM carried out initial catalytic experiments including catalyst preparation and testing under the supervision of MS and this was followed by EN, MD, SI and HA under the supervision of PM, GB and JKE. QH carried out the electron microscopy studies of the catalysts. DM was involved in the XPS data acquisition and analysis. DWK and DB helped in the mechanistic studies and data interpretation. MS initiated and supervised this project and GJH directed this research program. All authors contributed to the manuscript preparation. PM designed the graphical abstract.Acknowledgements
M. M. thanks the Umm Al-Qura University, Saudi Arabia, for his Ph.D. studentship. MS, QH and JKE thank Cardiff University for their University Research Fellowships. The authors thank the UK Catalysis Hub for resources and support provided via their membership in the UK Catalysis Hub Consortium funded by EPSRC (grants EP/K014706/2, EP/K014668/1, EP/K014854/1, EP/K014714/1 and EP/M013219/1). The authors further thank the EPSRC for providing the ICP and MS equipment in the School of Chemistry, Cardiff University (grant EP/L027240/1). Information on the data supporting the results presented here, including how to access them, can be found in the Cardiff University data catalogue at http://doi.org/10.17035/d.2017.0033515199References
- W. Borejsza-Wysocki and G. Hrazdina, Phytochemistry, 1994, 35, 623–628 CrossRef CAS.
- C. Morimoto, Y. Satoh, M. Hara, S. Inoue, T. Tsujita and H. Okuda, Life Sci., 2005, 77, 194–204 CrossRef CAS PubMed.
- J. I. Tateiwa, H. Horiuchi, K. Hashimoto, T. Yamauchi and S. Uemura, J. Org. Chem., 1994, 59, 5901–5904 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra and M. Mifsud, Adv. Synth. Catal., 2007, 349, 1949–1954 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra, M. Mifsud and A. Velty, Green Chem., 2010, 12, 99–107 RSC.
- I. T. Horvath and P. T. Anastas, Chem. Rev., 2007, 107, 2169–2173 CrossRef CAS PubMed.
- C. Khosla, Chem. Rev., 1997, 97, 2577–2590 CrossRef CAS PubMed.
- M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072–1133 CrossRef CAS PubMed.
- M. J. Climent, A. Corma, S. Iborra and M. J. Sabater, ACS Catal., 2014, 4, 870–891 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- E. Merino, E. Verde-Sesto, E. M. Maya, M. Iglesias, F. Sanchez and A. Corma, Chem. Mater., 2013, 25, 981–988 CrossRef CAS.
- S. Shylesh and W. R. Thiel, ChemCatChem, 2011, 3, 278–287 CrossRef CAS.
- V. Calemma, S. Peratello and C. Perego, Appl. Catal., A, 2000, 190, 207–218 CrossRef CAS.
- J. H. Sinfelt, Acc. Chem. Res., 1977, 10, 15–20 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS PubMed.
- W. H. Luo, M. Sankar, A. M. Beale, Q. He, C. J. Kiely, P. C. A. Bruijnincx and B. M. Weckhuysen, Nat. Commun., 2015, 6, 6540 CrossRef PubMed.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- M. Sankar, E. Nowicka, R. Tiruvalam, Q. He, S. H. Taylor, C. J. Kiely, D. Bethell, D. W. Knight and G. J. Hutchings, Chem. – Eur. J., 2011, 17, 6524–6532 CrossRef CAS PubMed.
- G. L. Brett, Q. He, C. Hammond, P. J. Miedziak, N. Dimitratos, M. Sankar, A. A. Herzing, M. Conte, J. A. Lopez-Sanchez, C. J. Kiely, D. W. Knight, S. H. Taylor and G. J. Hutchings, Angew. Chem., Int. Ed., 2011, 50, 10136–10139 CrossRef CAS PubMed.
- S. A. Kondrat, P. J. Miedziak, M. Douthwaite, G. L. Brett, T. E. Davies, D. J. Morgan, J. K. Edwards, D. W. Knight, C. J. Kiely, S. H. Taylor and G. J. Hutchings, ChemSusChem, 2014, 7, 1326–1334 CrossRef CAS PubMed.
- V. A. Naumkin, A. Kraut-Vass, S. W. Gaarenstroom and C. J. Powell, Version 4.1 National Institute of Standards and Technology Search PubMed.
- C.-J. Jia, Y. Liu, H. Bongard and F. Schuth, J. Am. Chem. Soc., 2010, 132, 1520–1522 CrossRef CAS PubMed.
- A. J. A. Watson and J. M. J. Williams, Science, 2010, 329, 635–636 CrossRef CAS PubMed.
- G. Guillena, D. J. Ramon and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed.
- M. Sankar, Q. He, S. Dawson, E. Nowicka, L. Lu, P. C. A. Bruijnincx, A. M. Beale, C. J. Kiely and B. M. Weckhuysen, Catal. Sci. Technol., 2016, 6, 5473–5482 CAS.
- C. Gunanathan and D. Milstein, Science, 2013, 341(6143), 1229712 CrossRef PubMed.
- L. K. M. Chan, D. L. Poole, D. Shen, M. P. Healy and T. J. Donohoe, Angew. Chem., Int. Ed., 2014, 53, 761–765 CrossRef CAS PubMed.
- M. A. Brown, F. Ringleb, Y. Fujimori, M. Sterrer, H. J. Freund, G. Preda and G. Pacchioni, J. Phys. Chem. C, 2011, 115, 10114–10124 CAS.
- J. Baltrusaitis, B. Mendoza-Sanchez, V. Fernandez, R. Veenstra, N. Dukstiene, A. Roberts and N. Fairley, Appl. Surf. Sci., 2015, 326, 151–161 CrossRef CAS.
- M. Sterrer and H. J. Freund, Catal. Lett., 2013, 143, 375–385 CrossRef CAS.
- F. Ringleb, M. Sterrer and H. J. Freund, Appl. Catal., A, 2014, 474, 186–193 CrossRef CAS.
- D. Shen, D. L. Poole, C. C. Shotton, A. F. Kornahrens, M. P. Healy and T. J. Donohoe, Angew. Chem., Int. Ed., 2015, 54, 1642–1645 CrossRef CAS PubMed.
- L. Guo, X. C. Ma, H. Q. Fang, X. Q. Jia and Z. Huang, Angew. Chem., Int. Ed., 2015, 54, 4023–4027 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cy00184c |
| This journal is © The Royal Society of Chemistry 2017 |