
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assessing the stereoselectivity of Serratia marcescens CECT 977 2,3-butanediol dehydrogenase†‡
Rosario
Médici
,
Hanna
Stammes
,
Stender
Kwakernaak
,
Linda G.
Otten
and
Ulf
Hanefeld
 *
*
Biokatalyse, Afdeling Biotechnologie, Technische Universiteit Delft, van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: U.Hanefeld@tudelft.nl; Tel: +31 15 2789304
First published on 22nd February 2017
Abstract
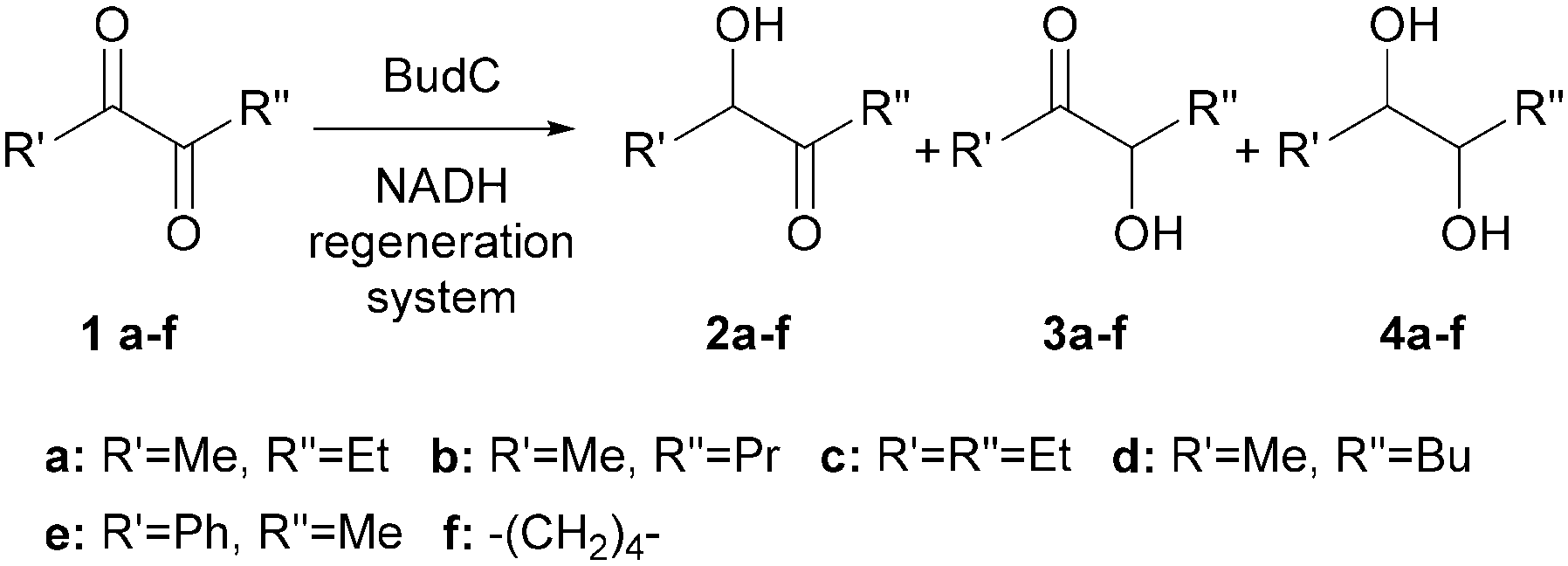
α-Hydroxy ketones and vicinal diols constitute well-known building blocks in organic synthesis. Here we describe one enzyme that enables the enantioselective synthesis of both building blocks starting from diketones. The enzyme 2,3-butanediol dehydrogenase (BudC) from S. marcescens CECT 977 belongs to the NADH-dependent metal-independent short-chain dehydrogenases/reductases family (SDR) and catalyses the selective asymmetric reductions of prochiral α-diketones to the corresponding α-hydroxy ketones and diols. BudC is highly active towards structurally diverse diketones in combination with nicotinamide cofactor regeneration systems. Aliphatic diketones, cyclic diketones and alkyl phenyl diketones are well accepted, whereas their derivatives possessing two bulky groups are not converted. In the reverse reaction vicinal diols are preferred over other substrates with hydroxy/keto groups in non-vicinal positions.
1. Introduction
Enantiomerically pure α-hydroxy ketones and vicinal diols constitute a valuable group of molecules useful directly as fine chemicals or as building blocks for asymmetric synthesis of bioactive compounds and agrochemicals.1,2 Numerous chemical and enzymatic strategies have been devised to address this challenge.2 To date all chemical approaches3–5 and most of the biochemical approaches6–8 only allow the formation of one of the two chiral products: either the α-hydroxy ketones or vicinal diols. A particular challenge is the development of methodologies allowing for the targeted selective synthesis of both α-hydroxy ketones and vicinal diols, in particular with aliphatic side chains (Scheme 1).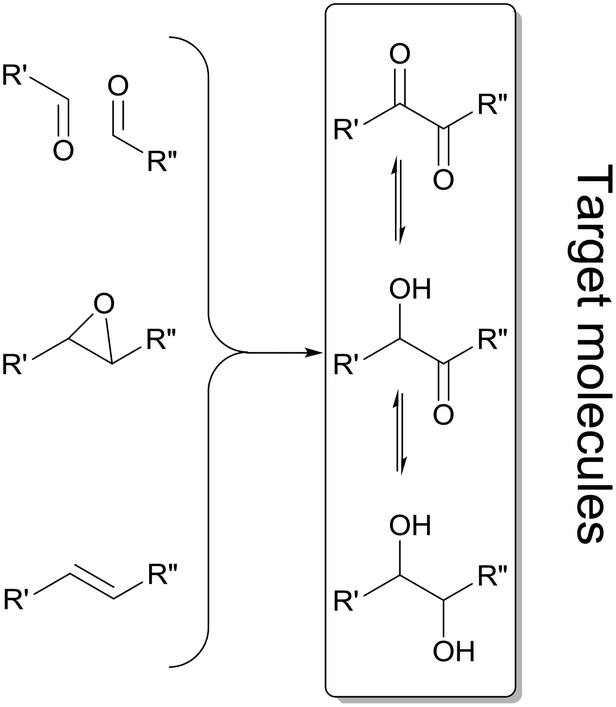 | ||
| Scheme 1 Summary of enzymatic methods applied for the synthesis of α-hydroxy ketones and vicinal diols, employing oxidoreductases,7,12–14 lyases,15,16 hydrolases17 and epoxide hydrolases.18 Double reduction of α-diketones is achieved by a few oxidoreductases19–22 and enzymatic cascade strategies involving two enzymatic steps.8,23,24 | ||
Acetoin reductases/2,3-butanediol dehydrogenases (EC 1.1.1.4 and 1.1.1.76) constitute a less explored group of enzymes. They belong to the family of NADH-dependent metal-independent short-chain dehydrogenases/reductases and are responsible for the accumulation of 2,3-butanediols in high titers (>100 g L−1) during the cultivations of species such as Klebsiella, Enterobacter, Serratia and Bacillus.9 In general, 2,3-butanediol is not produced as a single isomer, but as a mixture of meso-2,3-butanediol with (S,S)-butanediol (L-(+)-form) or (R,R)-2,3-butanediol (D-(−)-form). Microorganisms that do not produce the meso diol, generate optically enriched (R,R)-butanediol. The variations in optical purity might be due to a single unselective enzyme or due to mixtures of selective enzymes. In particular, the Bacillus stearothermophilus10 enzyme (2S,3S)-2,3-butanediol dehydrogenase (BDH, EC 1.1.1.76) gave excellent enantioselectivities for a range of substrates towards the S,S-diols. R,R-Diols could be obtained employing for instance Saccharomyces cerevisiae BDH.11 This indicates that more enzymes with similarly high specificities for other substrates and isomers including the meso isomer should be available. The oxidation of the meso isomer would be of particular interest. In principle, this enzymatic transformation could afford enantio-enriched α-hydroxy ketones with up to 100% yield rather than the 50% obtained through a kinetic resolution approach.
Here we describe the investigation of meso-2,3-butanediol dehydrogenase (BudC) from Serratia marcescens CECT 977, including its stereoselectivity and substrate range.
2. Results and discussion
2.1 Cloning and heterologous expression of BudC
In the present work, the budC gene encoding to a 2,3-BDH from S. marcescens CECT 977 (756 bp) was amplified from genomic DNA and cloned into pET-28a in frame with the N-terminal His6-tag to facilitate purification of the recombinant enzyme. The DNA sequence exhibited a 91.8% identity with S. marcescens H30 strain25 and both proteins were identical at protein level. This means that the production and purification described earlier25 is also valid for the enzyme described here. We therefore concentrated on substrate range and enantioselectivity.2.2 Enzyme kinetics
The budC gene was successfully expressed in E. coli BL21 (DE3) star cells and the enzyme was purified from E. coli cell-free extracts by Ni-NTA affinity chromatography. Activity of crude extracts reached 14.4 U mg−1 using rac-acetoin (50 mM) as substrate at pH 7.0 in a reductive assay (ESI‡ Fig. S1). SDS-PAGE gels revealed the production of a protein of approximately 28 kDa in agreement with the calculated molecular weight (28.4 kDa). Native gels confirmed the tetrameric oligomerization state, yielding a band between 120–140 KDa (ESI‡ Fig. S2) in line with the earlier results.25BudC catalyses the reduction of rac-acetoin preferably at weakly acidic pH 5.0 rather than neutral pH (975 U mg−1vs. 250 U mg−1). However, kinetic parameters at pH 5.0 were difficult to determine, yielding inconsistent data due to the low stability of the enzyme at this pH (t1/2: 2.5 h, Fig. 1).
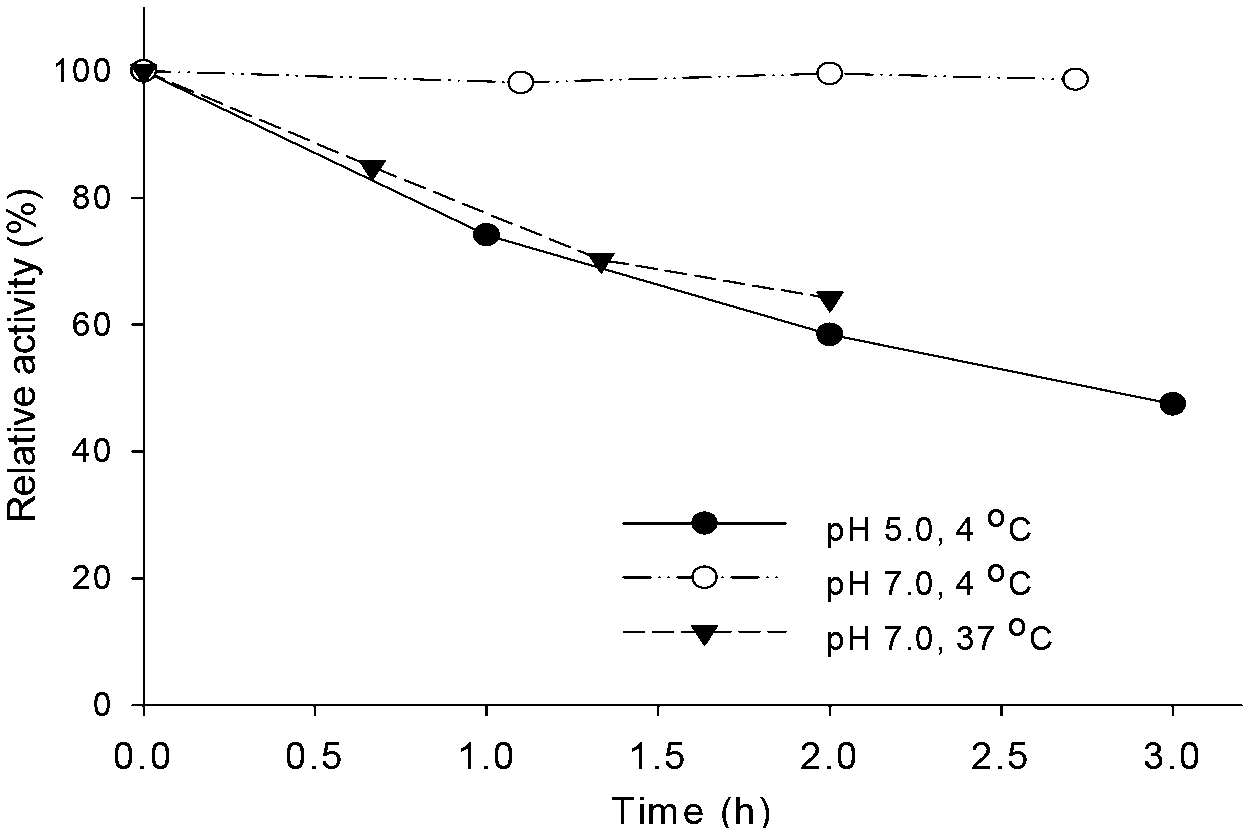 | ||
| Fig. 1 Effect of the temperature and pH on BudC activity. Reaction conditions: 0.16 mM NADH, 0.27 nM BudC, 50 mM rac-acetoin in potassium phosphate buffer (50 mM, pH 7.0) or potassium acetate buffer (50 mM, pH 5.0) at the corresponding temperatures. | ||
In line with these observations, only kinetic parameters at pH 7.0 were determined (Table 1, ESI‡ Fig. S4, A–D). In the reductive reaction diacetyl is the preferred substrate of BudC over acetoin, while only meso-2,3-butanediol oxidation was catalysed by the enzyme under the conditions assessed. Despite its protein sequence identity with the enzyme from S. marcescens H30, the specific activities obtained in our study with BudC were significant higher than those reported earlier (up to 4.5-fold at pH 5.0)25 under identical reaction conditions.
| Substrate | V max (U mg−1) | K m (mM) | k cat (s−1) | k cat/Km (M−1 s−1) |
|---|---|---|---|---|
| a Reaction conditions: 0.16 mM/0.32 mM NADH/NAD+, 0.27 nM BudC, substrate concentration 100 μM to 150 mM in potassium phosphate buffer (50 mM, pH 7.0) at 37 °C. Turnover numbers (kcat) are expressed per monomer of BudC. b Oxidation reaction. c Reduction reaction. | ||||
| meso-2,3-BDOb | 140.7 ± 5.2 | 6.9 ± 1 | 66.6 | 9.6 × 103 |
| Diacetylc | 412 ± 11.0 | 1.7 ± 0.2 | 195.0 | 1.1 × 105 |
| (±)-Acetoinc | 250.8 ± 3.1 | 3.1 ± 0.3 | 118.7 | 3.8 × 104 |
| (2S,3S)-2,3-BDOb | — | — | — | — |
| (2R,3R)-2,3-BDOb | — | — | — | — |
2.3 Bioconversions
When reduction reactions containing stoichiometric amounts of substrate (diacetyl or acetoin), NADH (5 mM) and BudC (0.5 μM) were performed, no product formation was observed in any case. Variation of the NADH concentration revealed this to be due to NADH inhibition. Although not previously reported for this enzyme, NADH inhibition was observed at concentrations higher than 0.3 mM; 1 mM being sufficient for complete inhibition (ESI‡ Fig. S5).Consistent with the previous results, nicotinamide cofactor regeneration strategies are essential to achieve significant product yields. Ethanol, 2-propanol, and acetone were tested as co-substrates for substrate-coupled cofactor regeneration. However, none of them was accepted by BudC (Table 2). An enzyme-coupled approach was accomplished employing formate dehydrogenase and glucose dehydrogenase (both as crude cell extracts) to recycle NADH, and NADH oxidase (as crude cell extracts) to regenerate NAD+. With these systems at hand, both the BudC catalysed oxidation and reduction could be investigated.
| Compound | U mg−1 |
|---|---|
| a Reaction conditions: 50 mM substrate, 0.16 mM/0.32 mM NADH/NAD+, 0.27 nM BudC in potassium phosphate buffer (50 mM, pH 7.0) at 37 °C. Experiments were performed in triplicate and the data are presented as means with standard deviations. b n.d.: not detected. | |
| 1a 2,3-pentanedione | 249.2 ± 11.4 |
| 1b 2,3-hexanedione | 220.5 ± 12.6 |
| 1c 3,4-hexanedione | 88.5 ± 2.1 |
| 1d 2,3-heptanedione | 19.4.6 ± 0.7 |
| 1e 1-phenyl-1,2-propanedione | 23.4 ± 3.4 |
| 1f 1,2-cyclohexanedione | 6.2 ± 0.5 |
| R-Benzoin | n.d.b |
| rac-Benzoin | n.d. |
| Benzil | n.d. |
| Acetone | n.d. |
| 2,4-Pentanediol | <1.0 |
| 1,3-Butanediol | <1.0 |
| Ethanol | n.d. |
| 2-Propanol | n.d. |
Whereas BudC was S-selective for the reduction of diacetyl yielding (S,S)-2,3-butanediol ((S,S)-2,3-BDO), rac-acetoin was reduced to both meso-2,3-BDO and (S,S)-2,3-BDO. Here (R)-acetoin was the preferred substrate and 15% (S)-acetoin remained unconverted after 24 h. Thus, BudC showed a stereo-preference consistent with meso-2,3-butanediol dehydrogenases with respect to acetoin.
In the oxidation reaction meso-2,3-BDO led to a complex product mixture. Starting with the initial formation of (R)-acetoin BudC catalysed its racemization within 24 h, concomitant with oxidation and reduction reactions, yielded small amounts of (2S,3S)-2,3-BDO as a side product.
In agreement with the kinetic measurements (Table 1), (2S,3S)-2,3-BDO was poorly converted to (S)-acetoin (<5% yield). No measurable amounts of diacetyl were detected.
Overall, BudC is S-selective for the reduction reaction of diacetyl to (2S,3S)-2,3-BDO but displays a preference for (R)-acetoin as a substrate for the second reduction step, which is also reduced with S-selectivity. In the reverse oxidation reaction meso-2,3-BDO was converted under the tested conditions, while the other diols and acetoin are poor substrates for the oxidation reaction (Scheme 2).
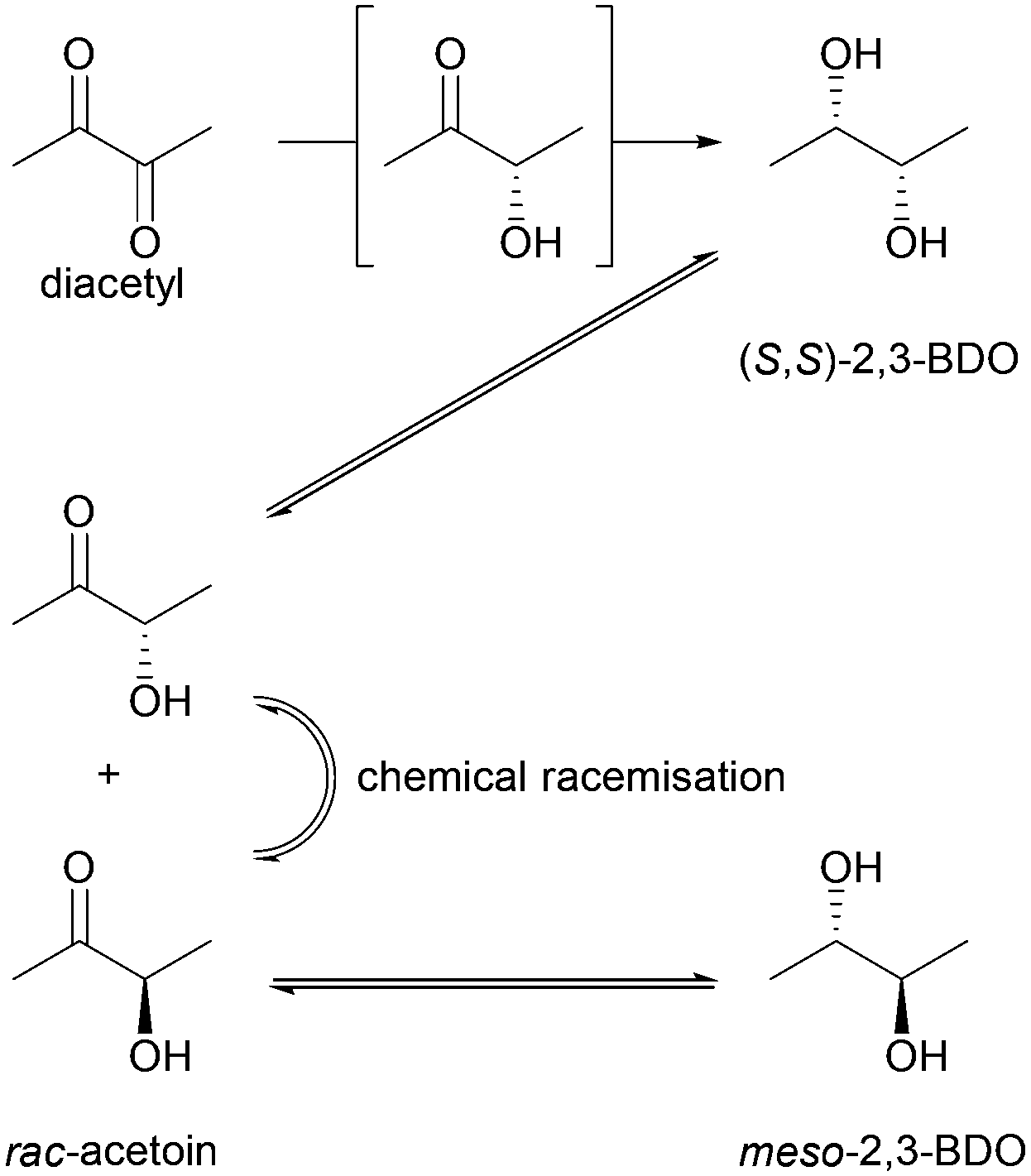 | ||
| Scheme 2 Specificity of BudC in the reduction and oxidation of 2,3-dioxygenated butane. | ||
2.4 Substrate scope
In order to broaden the applicability of BudC, we extended our investigation to structurally diverse α-diketones and vic-diols. Enzymatic activity was determined spectrophotometrically and the results are presented in Table 2. In the reduction reaction 2,3-diketones were good substrates of BudC, while the enzyme activity decreases with the increase of alkyl chain length. Sterically more demanding ketone moieties, such as 1,2-cyclohexanedione and 1-phenyl-1,2-propanedione were also converted by BudC, while bulky-bulky α-diketones, such as benzil, or α-hydroxy ketones (i.e. benzoin) were not converted. Thus, BudC catalyses the reduction of aliphatic symmetric and non-symmetric α-diketones, while its activity is severely affected by the presence of bulky moieties next to the carbonyl groups either due to steric hindrance (i.e.1c, 1d and 1e) or conformational effects (1c and 1f).In the oxidation reaction non-vicinal diols such as 2,4-pentanediol and 1,3-butanediol were no substrates of BudC. These results are in agreement with those previously reported by Zhang et al.25 indicating that vicinal diols (1,2-, 2,3- or 3,4) are required for catalysis.
2.5 Analysis of BudC stereoselectivity
The reduction of compounds 1a–1f was further investigated employing both NADH regeneration systems using formate dehydrogenase or glucose dehydrogenase, depending the reaction scale (Fig. 2 and Scheme 3).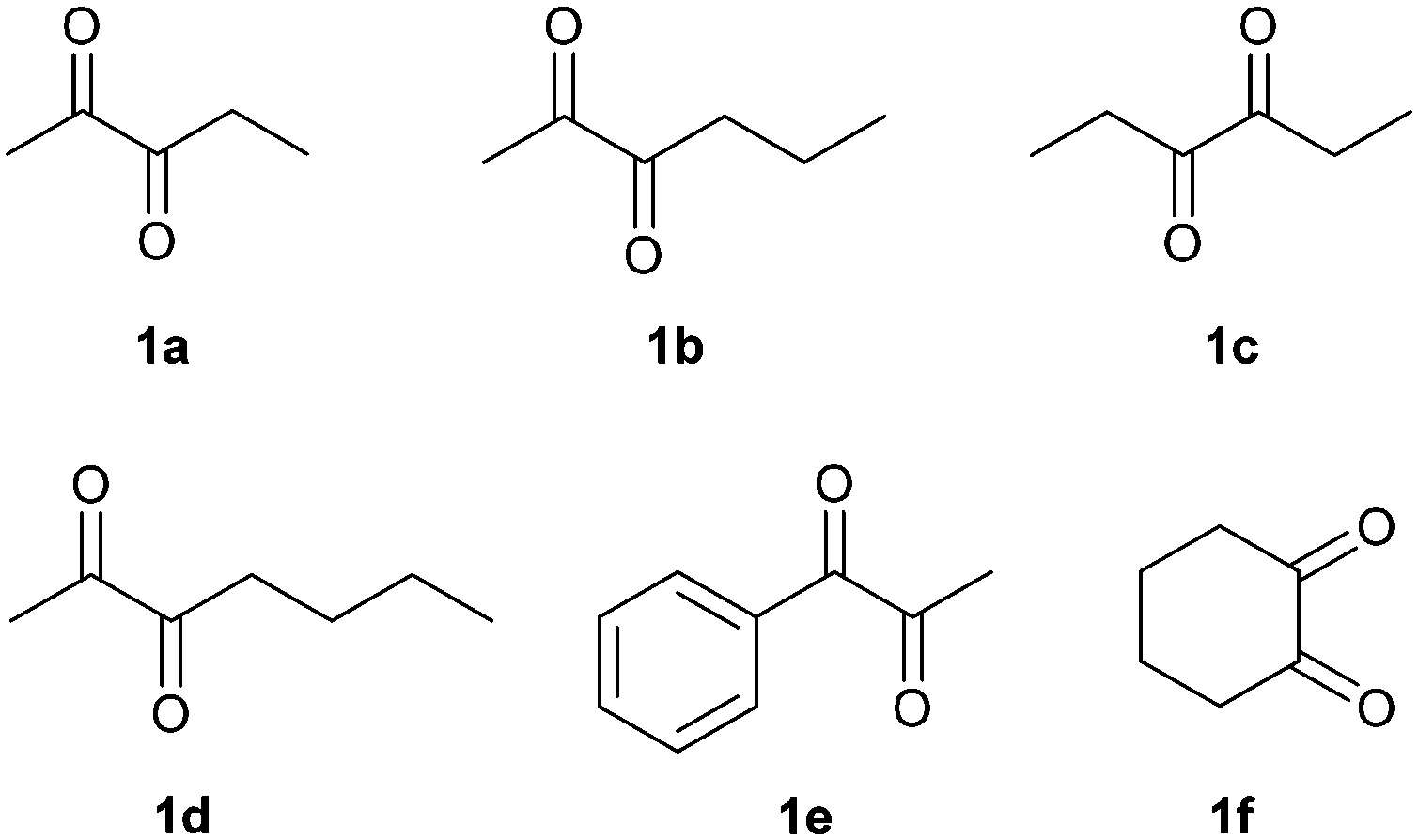 | ||
| Fig. 2 α-Diketones reduced by BudC. | ||
 | ||
| Scheme 3 Stereoselective reduction of 1,2-diketones using BudC from S. marcescens CECT 977. | ||
Reaction products were isolated by column chromatography and characterised by 1H and 13C NMR, optical rotation, and by comparison with the products obtained under the same conditions with a well characterised commercial enzyme ADH-380 (Evoxx, Düsseldorf, Germany), which is known to yield the corresponding S-stereoisomers. Moreover, ADH-380 catalysed reactions were used as positive control.26 Slight differences in reaction yields and stereoselectivities were observed depending on the regeneration system employed (crude extracts of formate or glucose dehydrogenase).
2,3-Pentanedione (1a) was reduced to the diol (2S,3S)-4a in good yield and selectivity after 1 h. After 24 h the yield was improved further but a slight loss of selectivity was observed (Table 3, entry 1–2, ESI‡ Fig. S7 and S11). The results are in line with those for diacetyl (Scheme 2).
| Entry | Substrate | Conc. [mM] | Enzyme | Reg. syst. | 2 [%] | eeb [%] | 3 [%] | ee [%] | 4 [%] | de [%] | ee [%] | Time (h) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ND stands for not determined. a Reaction conditions: the corresponding diketone (1a–1f, 10 or 50 mM) was added to 50 mM potassium phosphate buffer pH 7.0 with the nicotinamide cofactor (0.16 mM), BudC (2 U ml−1) and regeneration system (FDH or GDH, 2 U mL−1). b Yields, ees and des were determined by chiral GC analysis (when possible). c Small peaks could not be assigned due to missing standards, therefore it was impossible to distinguish between ee and de. | ||||||||||||
| 1 | 1a | 10 | BudC | FDH | — | — | — | — | 71 | (2S,3S) | 96c | 1 |
| 2 | 1a | 10 | BudC | FDH | — | — | — | — | 90 | (2S,3S) | 94c | 24 |
| 3 | 1a | 10 | ADH-380 | FDH | — | — | — | — | 92 | 99 (2S,3S) | >99 | 24 |
| 4 | 1b | 10 | BudC | FDH | <5 | ND | 80 | 99 | <5 | ND | — | 24 |
| 5 | 1b | 50 | BudC | GDH | — | — | 90 | 96 | <5 | ND | — | 48 |
| 6 | 1c | 10 | BudC | FDH | 15 | 80 (S) | — | — | 79 | (2S,3S) | 92c | 24 |
| 7 | 1c | 50 | BudC | GDH | 92 | 93 (S) | — | — | <5 | ND | — | 1 |
| 8 | 1c | 10 | ADH-380 | FDH | — | — | — | — | 99 | 99 (2S,3S) | 99 | 24 |
| 9 | 1d | 10 | BudC | FDH | 31 | 91 (S) | 57 | 99 (ND) | <5 | ND | — | 24 |
| 10 | 1d | 10 | BudC | GDH | 46 | 97 (S) | 40 | 99 (ND) | <5 | ND | 24 | |
| 11 | 1d | 50 | BudC | GDH | 20 | 89 (S) | 68 | 99 (ND) | <5 | ND | 48 | |
| 12 | 1d | 10 | ADH-380 | FDH | 29 | 99 (S) | — | — | 65 | (2S,3R) | 94c | 24 |
| 13 | 1d | 10 | ADH-380 | GDH | <5 | ND | — | — | 95 | 99 (2S,3R) | 99 | 24 |
| 14 | 1e | 10 | BudC | FDH | 25 | 85 (S) | 55 | 89 (S) | 9 | 59 (1S,2S) | 99 | 2 |
| 15 | 1e | 10 | BudC | FDH | 15 | 85 (S) | 40 | 83 (S) | 30 | 5 (1S,2S) | 99 | 24 |
| 16 | 1e | 10 | ADH-380 | FDH | <5 | ND | 65 | 99 (S) | 36 | 69 (1S,2S) | 99 | 24 |
| 17 | 1f | 10 | BudC | FDH | — | — | — | — | 66 | 67 (1S,2S) | 99 (1S,2S) | 2 |
| 18 | 1f | 10 | BudC | FDH | — | — | — | — | 66 | 67 (1S,2S) | 99 | 24 |
| 19 | 1f | 10 | ADH-380 | FDH | — | — | — | — | 81 | 99 (1S,2S) | 99 | 24 |
2,3-Hexanedione (1b) was also rapidly reduced to the corresponding α-hydroxy ketone, with (S)-3-hydroxy-2-hexanone (3b) as the main reaction product (80% yield, 99% ee, Table 3, entry 4), while 2,3-hexanediol was produced only in low yields. High yields of 3b were also achieved at semi-preparative scale with a minor loss of enantioselectivity (Table 3, entry 5, ESI‡ Fig. S8 and S12). It should be noted that the less accessible 3-ketogroup was selectively reduced and the reaction almost stopped completely after a single reduction. The second reduction step was much slower, allowing to selective production of the acyloin.
A modified reaction profile was observed with 2,3-heptanedione (1d). A mixture of 2-hydroxy-3-heptanone (2d) and 3-hydroxy-2-heptanone (3d) was produced, the latter again as the main reaction product (Table 3, entries 9–10). As with 1b the less accessible 3-ketogroup was thus reduced preferably. Despite extensive efforts, the regio-isomers could not be separated by column chromatography; therefore the absolute stereochemistry of 3d could not be determined (ESI‡ Fig. S10 and S15–S17). Based on the other results it can tentatively be assigned as (S). (2S,3R)-2,3-Heptanediol (4d) was obtained in low yields (<5%). Neither longer reaction times nor fresh pulses of the enzyme improved the yield of the diol.
In contrast to other dehydrogenases (i.e., ADH-A from Rhodococcus ruber or ADH-T from Thermoanaerobacter sp.),19,20 where the extension of α-diketone alkyl chain (n = 1 to n = 3) increased the selectivity of the enzymes, a lower regio-selectivity was observed in case of budC, indicating its preference for small substrates.
The reduction of 3,4-hexanedione (1c) proceeded with the same stereoselectivity as for diacetyl, (S)-acetoin and 1a yielding (3S,4S)-3,4-hexanediol (4c) (79% yield, 92% stereoselectivity). When higher substrate concentrations were employed and the reaction time shortened (Table 3, entries 7), (S)-4-hydroxy-3-hexanone (2c) was produced with excellent yields (92%) and stereoselectivity (93% ee). This supports the stereochemical assignment of 3d (ESI‡ Fig. S8, S13 and S14).
1-Phenyl-1,2-propanedione reduction yielded (S)-2-hydroxy-1-phenylpropan-1-one (3e) as the main product (55% yield, 89% ee), (S)-1-hydroxy-1-phenylpropan-2-one (2e, 25% yield, 85% ee) and (S,S)-1-phenyl-1,2-propanediol (4e) (9% yield, 99% ee, 59% de) (Table 3, entry 14, ESI‡ Fig. S6). Longer incubation times increased the yield of 4e up to 30%, but reduced the stereoselectivity (Table 3, entry 15). While the S-selectivity was maintained BudC surprisingly cannot differentiate between a methyl and a phenyl-group.
1,2-Cyclohexanedione was completely reduced to (S,S)-1,2-cyclohexanediol (4f) in good yields (66%) and with excellent ee (99%) although poorer de (67%), as cis-1,2-cyclohexanediol was also detected as a reaction product. It could not be distinguished whether the low de was due to poor selectivity in the first or second reduction step.
2.6 Homology modelling of BudC and substrate docking
An homology model of BudC was made based solely on the structure of the meso-2,3-BDH from Klebsiella pneumoniae (KpBDH, PDB 1GEG).27 This resulted in a model in which the positioning of several active site residues is in line with another meso-BDH, which seems important for the enantioselectivity of the enzyme.28 The model shows the enzyme as a tetramer, which is in accordance with the results from the native PAGE and other SDR family members (see ESI‡ Fig. S18 and S19). The important active site residues for SDRs (Asn-Ser-Tyr-Lys) are in the exact same position as in other BDHs, the only difference is Asp111 in the place were normally an Asn is located. This residue plays an essential role in keeping the conformation of the active site and allowing a proton relay system,29 which is still there involving the Asp residue.Docking different substrates and products did not result in good binding poses. This is probably due to the small nature of the substrates. This was confirmed by the fact that docking these small compounds into the crystal structure of the KpBDH also did not result in proper binding modes. Furthermore, the loop just outside the binding pocket is probably moving upon binding of the substrate which cannot be mimicked by the docking in silico.
Normally the reduction of the proximal keto group results in the S-enantiomer of the corresponding α-hydroxy ketone. The pro-R NADH hydride transfer from NADH to the re-face of diketones yields (S)-hydroxy ketones, indicating that the enzyme obeys canonical Prelog addition (Fig. 3). Small diketones are most probably fixed at the correct distance to NADH with hydrogen bonds, similar to KpBDH. Since the active site easily can accommodate larger substrates, the larger and more aliphatic penta-, hexa- and heptadione can bind into the active site in an alternative orientation. In this way the 3-keto group may be positioned properly towards the NADH, resulting in the respective 3S-hydroxyketone. Reduced affinity of this alternative binding mode can explain the decreased activity for the larger substrates.
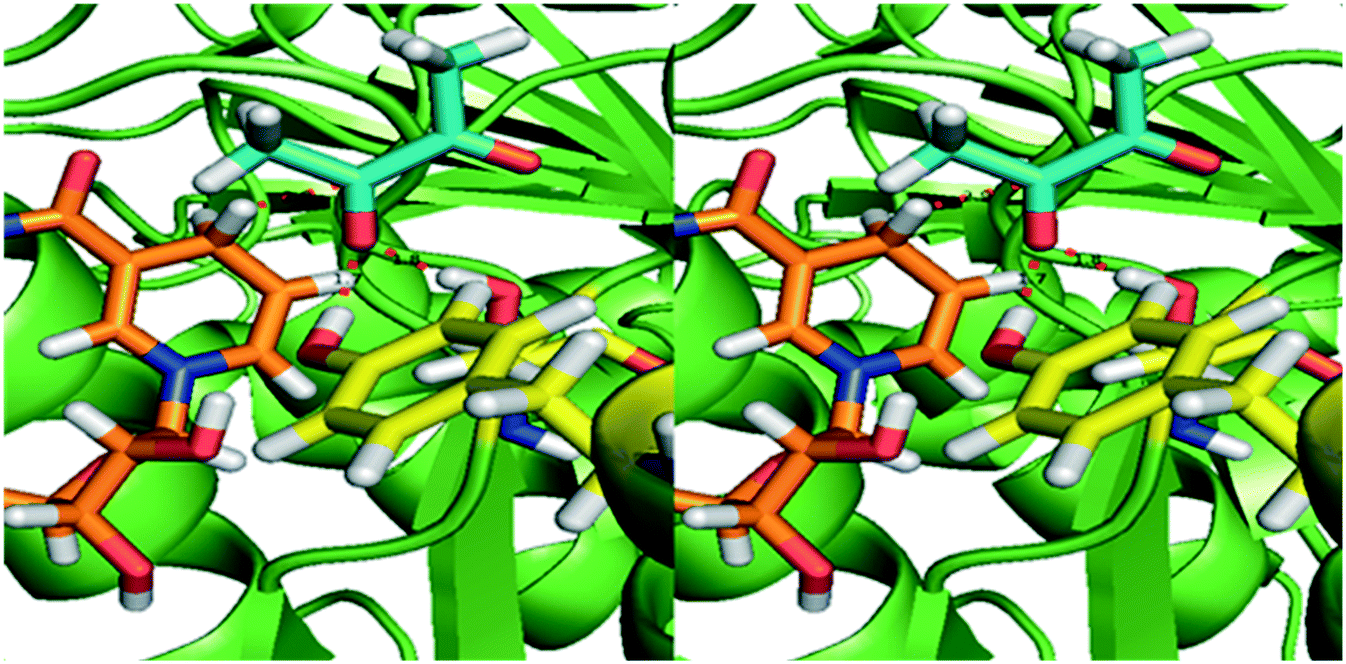 | ||
| Fig. 3 Homology model of BudC based on meso-2,3-butanediol dehydrogenase from K. pneumoniae (1GEG). The substrate diacetyl (blue sticks) was modelled based on the co-crystalised beta-mercaptoethanol in 1GEG. Tyr155 and Ser138 (yellow) are both hydrogen bonded to the proximal keto-group. The Tyr donates a hydride to the oxygen of the substrate, while the pro-R hydride from NADH (orange) transfers to the carbon, resulting in the S-configuration of the hydroxyl group. | ||
3. Conclusions
The BudC gene was successfully cloned and recombinantly expressed in E. coli. The enzyme is (S)-selective for all tested substrates and has a broad substrate scope. Unlike other ADHs it displays a better stereoselectivity for smaller substrates. Although the enzyme is strictly S-specific for all small substrates, meso-2,3-BDO is produced with excellent selectivity using Serratia marcescens strains. The production of this compound can be explained by the accumulation of R-acetoin in these strains, which is a very good substrate for the S-specific BudC.By varying reaction conditions, single or double reductions as well as oxidations could be achieved in one step. This makes this enzyme an outstanding biocatalyst for the synthesis of different chiral compounds from prochiral diketones and diols.
4. Experimental
4.1 Strains, plasmids and culture conditions
All standard recombinant DNA procedures were performed as described by Sambrook et al.30 The host strains Escherichia coli DH5α (Thermo Fisher Scientific, Breda, The Netherlands) and Escherichia coli BL21 star (DE3) (Thermo Fisher Scientific) were used in sub-cloning and expression experiments, respectively.Serratia marcescens CECT 977 was supplied by the Spanish Type Culture Collection (CECT, University of Valencia, Spain) and as DNA source for budC cloning. pGEMT-easy (Promega, Promega Benelux B.V., Leiden, The Netherlands) was used as cloning vector and pET-28a (Merck Millipore, Amsterdam, The Netherlands) as expression vector.
Bacteria were routinely grown at 37 °C in liquid Luria-Bertani broth medium (NaCl, 5 g L−1; tryptone, 10 g L−1; yeast extract, 5 g L−1) containing 100 μg ml−1 ampicillin or 50 μg ml−1 kanamycin when required.
4.2 Construction of pET-28a budC expression vector
S. marcescens genomic DNA was extracted following a standard phenol/chloroform procedure as reported by Sambrook et al.30 A budC consensus sequence was generated based on the alignment of homologous sequences from five S. marcescens strains (S. marcescens SM39, accession number: AP013063; S. marcescens MG1, accession number: JF519738; S. marcescens subsp. marcescens Db11, accession number HG326223; S. marcescens G12, accession number: KF547938; S. marcescens WW4, accession number: CP003959) with budC gene sequence of S. marcescens H30 (accession number: JQ63959) reported by Zhang et al.,25 as template.The budC gene was PCR amplified from S. marcescens CECT 977 genomic DNA using the forward 5′- GGAATTCCATATGCGTTTTGACAATAAAGTCGTGGTTATC -3′ (NdeI site in bold) and reverse primer 5′- CGCTCGAGTTAGACGATCTTCGGTTGGCCGTCCGA-3′ (XhoI site in bold). The PCR was performed using Accuprime Pfx DNA polymerase (Thermo Fisher Scientific) with the following conditions: 0.1 μg of chromosomal DNA, 0.3 μM of each primer, 5 μl of 10× PFx reaction mix and 2.5 units of Pfx DNA polymerase in a final volume of 50 μl. A hot start of 2 minutes at 95 °C was followed by 30 cycles of denaturation (15 s at 95 °C), annealing (30 s at 56 °C), and extension (1 min at 68 °C). The PCR product was first purified using a Gel purification kit (Qiagen, Venlo, The Netherlands), followed by an A-tailing procedure with Taq DNA polymerase (Qiagen).
BudC gene was ligated into pGEM® T-Easy vector system (Promega, Leiden, The Netherlands), and the resulting plasmids were produced in E. coli DH5α. The resulting constructs were confirmed by DNA sequencing, restricted with NdeI and XhoI, and the coding gene was ligated into the corresponding sites of pET-28a vector (Merck Millipore, Amsterdam, The Netherlands), introducing a (His)6-tag at the N-terminus of budC gene product.
Recombinant expression of budC was performed using E. coli BL21 star (DE3) cells transformed with pET-28a-budC in autoinduction media (ZYM-5052)31 supplemented with 100 μg ml−1 of kanamycin at 22 °C and 150 rpm during 24 h. Cells were harvested by centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 × g for 15 minutes at 4 °C, washed twice with potassium phosphate buffer (50 mM, pH 7.0), re-centrifuged stored at −80 °C until further use.
500 × g for 15 minutes at 4 °C, washed twice with potassium phosphate buffer (50 mM, pH 7.0), re-centrifuged stored at −80 °C until further use.
4.3 BudC purification
Cell-free extracts (CFE) were obtained incubating first the cells in binding buffer (sodium phosphate 50 mM, 300 mM NaCl, 20 mM imidazole, pH 8.0) containing egg white lysozyme (1 mg ml−1) and DNAse I (0.1 mg ml−1) for 1 h at 4 °C, followed by 2 cycles of cell disruption (French Press, 1.5 kBar) and finally centrifuged at 12500 × g and 4 °C for 45 min. CFE was filtered through 0.45 μm filter and applied onto a 5 ml Ni-NTA HisTrap HP column (GE Healthcare Life Sciences, Eindhoven, The Netherlands) previously equilibrated with binding buffer. An imidazole gradient was then applied (20 mM–500 mM imidazole, 3 ml min−1, 60 minutes) and BudC was eluted with about 350 mM of imidazole.
Enzyme fractions were analysed by SDS-PAGE (12% Bis-Tris gels, Bio-Rad, Munich, Germany). Fractions containing BudC were combined, concentrated by centrifugation using an Amicon filter (10 kDa, Merck Millipore) and applied onto a PD-10 desalting column (Thermo Fischer Scientific) previously equilibrated with 50 mM potassium phosphate, pH 7.0. Enzyme aliquots were frozen in liquid nitrogen and stored at −80 °C. Protein concentration was determined by the bicinchoninic acid assay (Interchim Uptima BC assay, Interchim, Montluçon France) using bovine serum albumin (Bio-Rad, Veenendaal, The Netherlands) as standard.
4.4 Enzyme kinetics
Enzymatic activity was determined spectrophotometrically by monitoring NADH consumption (oxidation) or formation (reduction) at 340 nm and 37 °C using an extinction coefficient of 6.22 mM−1 cm−1. One unit of activity was defined as the amount of enzyme catalysing the oxidation/reduction of 1 μmol of nicotinamide cofactor per minute under standard conditions (pH 7.0, 37 °C).Specific activities were measured in potassium phosphate buffer (50 mM, pH 7.0, 1 ml) in presence of 50 mM of substrate and 0.16 mM NADH or 0.32 mM NAD+. DMSO (5% (v/v)) was added as a co-solvent to improve the solubility of benzil and benzoin. Reactions were started by addition of the enzyme solution and measured for 3 min. Activities of the CFE was determined using E. coli BL21 (DE3) star pET28(a)-buC or E. coli BL21 (DE3) star pET28(a) as negative control.
Kinetics parameters were investigated using 0.27 nM enzyme in presence of varying substrate (0.01–200 mM) or nicotinamide cofactor concentrations (12.5 μM to 5 mM) at 37 °C. Data were fitted to the Michaelis–Menten equation (non-linear regression, SigmaPlot 8.0) to estimates of Km and kcat.
4.5 Standard reaction conditions
The standard reduction mixture consisted of 10 mM diketones, 0.16 mM of NADH, 2 U ml−1 of BudC in potassium phosphate buffer (50 mM, pH 7.0) and NADH-regeneration system containing 100 mM ammonium formate and 2 U ml−1 formate dehydrogenase (Prozomix, Northumberland, UK). Oxidation reactions comprised: 10 mM diols, 0.32 mM NAD+, 2 U ml−1 of BudC in potassium phosphate buffer (50 mM, pH 7.0) and NAD+-regeneration system containing 4 U ml−1 of NADH-oxidase (Prozomix).Reactions were incubated at 37 °C and 600 rpm during 24 h. Aliquots (1 ml) were taken at different reaction times, centrifuged at 13000 rpm for 2 minutes and supernatants were first saturated with a NaCl saturated solution (0.1 ml) followed by extraction with ethyl acetate (containing isoamyl alcohol as internal standard) (0.5 mL × 2). The combined organic layer was dried over Na2SO4 and analysed with chiral GC with respect to yield, chemoselectivity and ee (see ESI‡).
Aliphatic α-hydroxy ketones and diol standards were obtained carrying out the reduction reactions at 50 ml scale. The reaction mixture contained 50 mM substrate (200 mg 2,3-pentanedione, 285 mg 2,3-hexanedione, 285 mg 3,4-hexanedione, 320 mg 2,3-heptanedione), 7.1 mg NADH, 1.5 g glucose-1-monohydrate, 0.75 g calcium carbonate, 2 mg glucose dehydrogenase and 2 mg ADH-380 (Evoxx) or 1.4 mg of BudC in potassium phosphate buffer (50 mM, pH 7.0). Reaction vessels were incubated at 37 °C and 100 rpm during 24 or 48 h. α-Hydroxy ketones and diols were purified by column chromatography as detailed in ESI‡ (sections 3 and 4), characterised by their optical rotation, 1H and 13C NMR spectra and compared with authentic standards or literature data when available (see ESI‡ Fig. S6 to S11).
4.6 Homology modelling of BudC and docking of substrates
An homology model of BudC was build using the standard homology parameters of Yasara.32 Only meso-2,3-BDH (1GEG) was used as a template in order to preserve the position of catalytically important side chains.Docking was done in the Yasara program with 100 docking runs using VINA and a minimum ligand RMSD of 1.5 Å. All residues were fixed except the important residues lining the active site, namely Ser138, Val139, Trp146, Tyr151, Lys155, Trp192 and the nicotinamide part of NADH. All docked compounds were visually inspected to determine the most probable catalytically active position. This binding mode was minimised with the NOVA force field to improve binding interactions with the protein.
Acknowledgements
This study was financially supported by The Netherlands Organization for Scientific Research (NWO) under the framework of Technology Area TA-Biomass. We thank Martina Pohl and Dörte Rother (Forschungszentrum Jülich GmBH, IBG-1: Biotechnology, Jülich, Germany) for providing 1-phenyl-1,2-propanediol and 2-hydroxy-1-phenylpropan-1-one isomers and the enzymes for the synthesis of 1-hydroxy-1-phenylpropan-2-ones and for revising the manuscript.References
- C. Palomo, M. Oiarbide and J. M. Garcia, Chem. Soc. Rev., 2012, 41, 4150–4164 RSC.
- P. Hoyos, J. V. Sinisterra, F. Molinari, A. R. Alcantara and P. Dominguez de Maria, Acc. Chem. Res., 2010, 43, 288–299 CrossRef CAS PubMed.
- C. R. Unelius, B. Bohman, M. G. Lorenzo, A. Tröger, S. Franke and W. Francke, Org. Lett., 2010, 12, 5601–5603 CrossRef CAS PubMed.
- B. Plietker, Tetrahedron: Asymmetry, 2005, 16, 3453–3459 CrossRef CAS.
- S. E. Schaus, B. D. Brandes, J. F. Larrow, M. Tokunaga, K. B. Hansen, A. E. Gould, M. E. Furrow and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 1307–1315 CrossRef CAS PubMed.
- P. P. Giovannini, O. Bortolini and A. Massi, Eur. J. Org. Chem., 2016, 2016, 4441–4459 CrossRef CAS.
- J. Kulig, R. C. Simon, C. A. Rose, S. M. Husain, M. Häckh, S. Lüdeke, K. Zeitler, W. Kroutil, M. Pohl and D. Rother, Catal. Sci. Technol., 2012, 2, 1580 CAS.
- D. Kihumbu, T. Stillger, W. Hummel and A. Liese, Tetrahedron: Asymmetry, 2002, 13, 1069–1072 CrossRef CAS.
- E. Celinska and W. Grajek, Biotechnol. Adv., 2009, 27, 715–725 CrossRef CAS PubMed.
- O. Bortolini, G. Fantin, M. Fogagnolo, P. P. Giovannini, A. Guerrini and A. Medici, J. Org. Chem., 1997, 62, 1854–1856 CrossRef CAS.
- E. Calam, E. Gonzalez-Roca, M. R. Fernandez, S. Dequin, X. Pares, A. Virgili and J. A. Biosca, Appl. Environ. Microbiol., 2016, 82, 1706–1721 CrossRef CAS PubMed.
- E. Calam, S. Porte, M. R. Fernandez, J. Farres, X. Pares and J. A. Biosca, Chem.-Biol. Interact., 2013, 202, 195–203 CrossRef CAS PubMed.
- C. Loderer and M. B. Ansorge-Schumacher, RSC Adv., 2015, 5, 38271–38276 RSC.
- C. E. Paul, I. Lavandera, V. Gotor-Fernández, W. Kroutil and V. Gotor, ChemCatChem, 2013, 5, 3875–3881 CrossRef CAS.
- M. Müller, D. Gocke and M. Pohl, FEBS J., 2009, 276, 2894–2904 CrossRef PubMed.
- P. Domínguez de María, M. Pohl, D. Gocke, H. Gröger, H. Trauthwein, T. Stillger, L. Walter and M. Müller, Eur. J. Org. Chem., 2007, 2007, 2940–2944 CrossRef.
- A. Petrenz, P. Domínguez de María, A. Ramanathan, U. Hanefeld, M. B. Ansorge-Schumacher and S. Kara, J. Mol. Catal. B: Enzym., 2015, 114, 42–49 CrossRef CAS.
- M. Kotik, A. Archelas and R. Wohlgemuth, Curr. Org. Chem., 2012, 16, 451–482 CrossRef CAS.
- K. Edegger, W. Stampfer, B. Seisser, K. Faber, S. F. Mayer, R. Oehrlein, A. Hafner and W. Kroutil, Eur. J. Org. Chem., 2006, 2006, 1904–1909 CrossRef.
- M. Kurina-Sanz, F. R. Bisogno, I. Lavandera, A. A. Orden and V. Gotor, Adv. Synth. Catal., 2009, 351, 1842–1848 CrossRef CAS.
- P. Besse, J. Bolte, A. Fauve and H. Veschambre, Bioorg. Chem., 1993, 21, 342–353 CrossRef CAS.
- L. N. Monsalve, P. Cerrutti, M. A. Galvagno and A. Baldessari, Biocatal. Biotransform., 2010, 28, 137–143 CrossRef CAS.
- J. Zhang, S. Wu, J. Wu and Z. Li, ACS Catal., 2015, 5, 51–58 CrossRef CAS.
- A. Jakoblinnert and D. Rother, Green Chem., 2014, 16, 3472–3482 RSC.
- L. Zhang, Q. Xu, S. Zhan, Y. Li, H. Lin, S. Sun, L. Sha, K. Hu, X. Guan and Y. Shen, Appl. Microbiol. Biotechnol., 2014, 98, 1175–1184 CrossRef CAS PubMed.
- Evocatal GmbH, http://www.biocatal.com/download.php?dl=adh380, (accessed 16-12-2016, 2016).
- M. Otagiri, G. Kurisu, S. Ui, Y. Takusagawa, M. Ohkuma, T. Kudo and M. Kusunoki, J. Biochem., 2001, 129, 205–208 CrossRef CAS PubMed.
- M. Otagiri, S. Ui, Y. Takusagawa, T. Ohtsuki, G. Kurisu and M. Kusunoki, FEBS Lett., 2010, 584, 219–223 CrossRef CAS PubMed.
- C. Filling, K. D. Berndt, J. Benach, S. Knapp, T. Prozorovski, E. Nordling, R. Ladenstein, H. Jörnvall and U. Oppermann, J. Biol. Chem., 2002, 277, 25677–25684 CrossRef CAS PubMed.
- J. Sambrook, E. F. Fritsch and T. Maniatis, Molecular cloning: a laboratory manual, Cold Spring Harbor Laboratory Press, New York, 3rd edn, 2001 Search PubMed.
- F. W. Studier, Protein Expression Purif., 2005, 41, 207–234 CrossRef CAS PubMed.
- E. Krieger and G. Vriend, Bioinformatics, 2014, 30, 2981–2982 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to W. R. “Fred” Hagen on the occasion of his retirement. |
| ‡ Electronic supplementary information (ESI) available: Chemicals, strains and plasmids, analytical methods; and additional figures. See DOI: 10.1039/c7cy00169j |
| This journal is © The Royal Society of Chemistry 2017 |