
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the Mn promoted synthesis of higher alcohols over Cu derived ternary catalysts†
Eleni T.
Liakakou
ab,
Mark A.
Isaacs
d,
Karen
Wilson
 d,
Adam F.
Lee
d and
Eleni
Heracleous
*ac
d,
Adam F.
Lee
d and
Eleni
Heracleous
*ac
aChemical Process & Energy Resources Institute (CPERI), Centre for Research and Technology Hellas (CERTH), 6th km Charilaou - Thermi Road, P.O. Box 361, 57001 Thessaloniki, Greece. E-mail: eheracle@cperi.certh.gr; Fax: +30 2310 498380; Tel: +30 2310 498345
bDepartment of Chemical Engineering, Aristotle University of Thessaloniki, POBox 1517, University Campus, 54006 Thessaloniki, Greece
cSchool of Science & Technology, International Hellenic University (IHU), 14th km Thessaloniki – Moudania, 57001 Thessaloniki, Greece
dEuropean Bioenergy Research Institute, Aston University, Birmingham B4 7ET, UK
First published on 23rd January 2017
Abstract
This work provides insight into the promotional effect of Mn on the synthesis of higher alcohols over Cu-based ternary catalysts through XPS and in situ DRIFTS and powder XRD. These revealed that the surface of K-CuZnAl, an active catalyst for CO hydrogenation to methanol, possesses Cu+ sites able to adsorb CO associatively and Cu0 sites for H2 dissociation. Here we show that exchanging Zn with Mn induces a strong interaction between Cu and Mn that decreases the overall copper surface area and increases the Cu+/Cu0 ratio. In situ DRIFTS showed that electronic modification of Cu+ sites by proximate Mn favors dissociative CO chemisorption, resulting in the formation of C and O adspecies that are precursors to higher alcohol formation. The decrease in metallic copper limits available sites for H2 dissociation, and hence retards the hydrogenation of oxygen-containing intermediates, thereby further promoting carbon-chain growth. Mn also increases the dispersion of the K promoter over the catalyst surface, providing abundant basic sites for aldol-type condensation reactions to branched oxygenates.
1. Introduction
The conversion of synthesis gas to oxygenates represents an attractive route for the production of drop-in fuel components. The process is versatile in terms of feedstock since both renewable and non-renewable carbon-containing feedstocks can be employed, while modification of the process parameters and the catalyst yields a wide range of products, spanning from methanol and higher alcohols to ketones, aldehydes and esters. Alcohols with a carbon number higher than two demonstrate more favorable properties for fuel use than ethanol, such as higher octane number, improved fuel blend water tolerance, better volatility characteristics and material compatibility.1 More importantly, higher alcohols have been identified as a suitable blending component for aviation,2 a transport sector with limited alternative fuel options due to the extremely stringent fuel specifications. Mixing 10% C2–C6 alcohols in jet fuel was shown to meet current jet fuel specifications. The use of the fuel blend in aircraft engines led to reduced emissions, with no compromise in the engine performance.2Although the conversion of syngas to methanol over Cu/ZnO/Al2O3 catalysts is an established, large-scale industrial process, CO hydrogenation to higher alcohols still remains challenging. Despite the substantial amount of research work, the commercialization of the higher alcohol synthesis (HAS) process is still hindered by low yields and poor catalyst selectivity. Promotion of Cu-based methanol synthesis catalysts with alkalis, such as Li, Na, K and Cs, shifts the synthesis to higher molecular weight products,3–8 however almost always at the expense of CO conversion.9 The addition of transition metals has also been reported to act beneficially towards higher alcohol formation. Among others, the IFP-developed Cu–Co catalysts and Fe–Cu and Ni–Cu catalysts have been shown to be active in the higher alcohol synthesis reaction.10 The addition of Mn,11–14 Cr,11 Th,11,15,16 and Ce (ref. 11, 12, 15 and 16) together with alkali compounds to Cu-based ZnO or Cr2O3 catalysts improved selectivity to higher alcohols, especially to isobutanol.
The mechanism of higher alcohol formation and the nature of the active site(s) still remain unclear. Several different mechanistic routes and reactive intermediates have been proposed in the literature and these have been nicely summarized in several older and more recent reviews.3–5,9,17–21 It is generally accepted that the reaction mechanism depends on the type of catalyst employed. Over noble metals and modified Fischer–Tropsch and Mo-containing catalysts, the reaction yields mostly linear alcohols, produced via the insertion of non-dissociated CO in (CHx)ad species formed from the hydrogenation of dissociative CO. This reaction mechanism is often described as a “dual-site” mechanism, where one active site catalyzes CO dissociation and chain propagation, while another site has functionalities for CO non-dissociative activation and insertion.22 The mechanism is much more complex on Cu-based catalysts and comprises several reaction steps, depending on the metals and promoters used: CO adsorption (associative/dissociative), hydrogenation of the adsorbed CO to formyl species, carbon chain growth via aldol-type condensation of formyl species to acetyl and higher species or CO insertion to form a C–C bond followed by hydrogenation of the intermediate species to produce a complex product mixture consisting of linear and branched alcohols, other oxygenates and hydrocarbons.4 In this context, the C–C growth over alkali-promoted Cu catalysts has been ascribed to the aldol condensation reactions over basic sites provided by the alkalis.
Most studies point to a common intermediate for the synthesis of methanol and higher alcohols, with syngas or methanol forming a C1 surface species that further reacts, leading to carbon chain growth. On the basis of isotopic studies with the addition of methanol and ethanol with 13C in the syngas feed, Nunan et al.23 showed that lower alcohols were incorporated into the synthesis to form higher alcohols on Cs-doped Cu/ZnO catalysts. Iglesia et al.,24 Baysar and Schrader,25 Kiennemann et al.26 and Elliott et al.27–29 reached similar conclusions for alkali promoted and unpromoted Cu/ZnO/Al2O3 catalysts by isotopic studies and addition of probe molecules, such as ethanol and 1-propanol, to the H2/CO feed. Therefore, there is a consensus that the synthesis of methanol and higher alcohols occurs via the same C1 intermediate, but it is still not clear if the two syntheses share the same catalytic site and what the exact nature of this site is. In fact, despite numerous investigations, the precise type of active center for methanol synthesis is still uncertain. Most studies agree on the contribution of ZnO to the active site, but several different models have been proposed, such as Cu metal supported on ZnO, Cu+ ions in the ZnO matrix, ZnO segregated on Cu+, electron-rich Cu at the Cu–ZnO heterojunction, CuZn surface alloy formation etc. [ref. 30 and references therein]. Regarding higher alcohol synthesis, earlier works of Hofstadt et al.16 over Cu catalysts promoted with alkalis and different transition metals suggest that oxygen-containing intermediates form on Cu+, while Cu0 favors the formation of CHx structures. The authors argue that both are needed to form ethanol via addition of the oxygen-containing intermediates in the CHx species. More recently, a series of publications from Wang et al.31–33 showed via experimental and molecular-level theoretical studies that both Cu+ and Cu0 sites are needed for ethanol formation on Cu/ZnO/Al2O3 catalysts. XPS results showed that ethanol synthesis requires a higher Cu+ content and higher Cu/Zn ratio on the catalyst surface.32
In a previous study, we investigated a series of K-promoted CuZnAl, CuZnMn, CuZnCr and CuMnAl catalysts with constant composition for the production of higher alcohols from syngas.34 We found that Mn favors the formation of alcohols with a carbon chain length over two. The most appreciable changes were recorded for the K-CuMnAl catalyst that exhibited much higher alcohol selectivity than K-CuZnAl at the expense of methanol formation. This was tentatively ascribed to strong interaction between Cu and Mn and the reduction of catalyst acidity that enhances aldol-type condensation reactions leading to higher alcohols. In this paper, we obtain further insight on the nature of the active sites over these catalysts and in particular on the promotional effect of Mn by monitoring the working state of the most active, K-CuZnAl, and the most selective, K-CuMnAl, catalyst under reaction conditions using in situ techniques. Moreover, the interaction of CO in the absence and presence of H2 is followed with time-resolved in situ DRIFTS to probe the active sites and intermediates formed on the surface.
2. Experimental section
2.1 Catalytic materials
The study was conducted over K-CuZnAl and K-CuMnAl catalysts synthesized via co-precipitation with a constant Cu/(Zn or Mn)/Al molar composition of 45/45/10. Details on the synthesis, characterization and testing of the materials in CO hydrogenation to higher alcohols have been previously reported.34 Briefly, co-precipitation was performed by adding an aqueous 0.1 M solution of Na2CO3 (Merck) in a dropwise manner to an aqueous solution of the corresponding nitrate precursors in appropriate stoichiometric amounts at 70 °C and pH of 6–7. The precursors employed were Cu(NO3)2·3H2O (Merck), Zn(NO3)2·6H2O (Merck), Al(NO3)3·9H2O (Fluka) and Mn(NO3)2·4H2O (Merck). The precipitate was washed and dried for 24 h at 100 °C and calcined under air flow for 4 h at 350 °C. The potassium promoter (0.5 wt% K) was added on the resulting powders by dry impregnation with an aqueous solution of K2CO3 (Aldrich), followed by calcination in synthetic air for 4 h at 350 °C. The catalysts were characterized by ICP-AES, BET analysis, XRD, SEM, TPR-H2 and NH3-TPD. The key physicochemical properties of the materials are repeated in Table 1. The catalytic performance in CO hydrogenation was measured in a high pressure small-scale test unit, equipped with a stainless steel fixed bed reactor (ID: 9.3 mm). Prior to the measurements, the catalysts were reduced in situ under pure hydrogen at 350 °C for 3 h and atmospheric pressure. The higher alcohol synthesis reaction was investigated in the temperature range 280–320 °C at 40 bar, W/F ratio of 0.74 g s cm−3 and inlet feed composition H2/CO = 2. The steady-state activity measurements were taken after at least 24 h time-on-stream. A summary of the catalytic performance in terms of CO conversion, C-based selectivity and space time yield is shown in Table 1.| Samples | SA, m2 g−1 | Crystallite sizeab, nm | CO conversionc, % | ROH STY, mg gcat−1 h | Carbon selectivity, C mol% | |||
|---|---|---|---|---|---|---|---|---|
| MeOH | C2+OH | HC | CO2 | |||||
| a Calculated from XRD patterns using the Scherrer equation. b The crystallite size refers to the CuO phase in K-CuZnAl and the Cu1.5Mn1.5O4 spinel phase in K-CuMnAl. c Reaction conditions: T = 300 °C, P = 40 bar, W/F = 0.74 g s cm−3, H2/CO = 2. | ||||||||
| K-CuZnAl | 33 | 8.2 | 21.5 | 449.6 | 75.8 | 10.8 | 2.8 | 10.6 |
| K-CuMnAl | 48 | 27.4 | 9.1 | 172.4 | 65.4 | 14.7 | 5.4 | 14.5 |
2.2 In situ XRD
In situ X-ray diffraction was performed at 7 bar pressure under H2 and syngas (ratio H2/CO = 2) at a temperature range of 25 to 400 °C in 50 °C intervals. The measurements were performed on a Bruker d8 advance XRD instrument with a Cu Kα source (λ = 1.54 Å) and a nickel Kβ filter, fitted with an Anton Parr XRK900 cell with Bronkhorst EL-Flow BASE mass flow controllers and an EL-Flow Select pressure regulator.2.3 Ex situ XPS
X-ray photoelectron spectra were acquired on a Kratos AXIS HSi spectrometer equipped with a charge neutraliser and monochromated Al Kα excitation source (1486.7 eV). Binding energy (BE) referencing was employed using the adventitious carbon peak at 284.6 eV. Survey scans were recorded for surface elemental analysis (pass energy 160 eV), with high resolution spectra recorded at 40 eV pass energy. Spectral fitting was performed using CasaXPS Version 2.3.15. Standard spectra of the samples were collected in an ultrahigh-vacuum system (10−8 torr) at ambient temperature in fresh form and after ex situ reduction at 350 °C in H2 under atmospheric pressure. Following reduction, samples were transferred to the instrument in a glove bag to avoid exposure to air.2.4 In situ DRIFTS studies
In situ DRIFTS spectra were collected on a Bruker Tensor 27 FT-IR spectrometer. The powdered samples were placed in a Zn/Se window chamber (Specac) controlled by a Specac temperature controller. The samples were first reduced in situ in H2 at 350 °C for 30 min. The chamber was then cooled to 50 °C in He flow. After the acquisition of stable reference spectra under He flow at 50 °C, the reactant gas was introduced to the chamber and the infrared spectra were recorded at 4 cm−1 resolution with 68 scans. CO adsorption was carried out under a 3% CO/He gas mixture at 50 °C, followed by purging with He or 6% H2/He flow. Spectra were also collected under a simultaneous 3% CO/6% H2/He gas atmosphere at 50 °C, followed by purging with He.3. Results
3.1 Catalytic performance in higher alcohol synthesis
The catalytic performance of the K-promoted Cu catalysts was evaluated in CO hydrogenation tests performed at 300 and 320 °C at 40 bar with a CO/H2 molar ratio of two in a high pressure fixed bed reactor unit. These results have been presented and discussed in detail in a previous publication.34 The main observations are briefly reported here to aid with the interpretation of the in situ studies presented in the following sections. Table 1 presents the catalytic performance at 300 °C in terms of CO conversion, carbon-based selectivity to the different products and space time yield to alcohols. K-CuZnAl demonstrated the highest activity, with 20% CO conversion. The introduction of Mn in the position of Zn in the material reduced the activity to ∼10%. The decrease was tentatively ascribed to the larger Cu-containing crystallites in K-CuMnAl, as calculated from XRD, and the lower accessibility of copper in the Cu–Mn spinel complex formed. The main reaction product over both catalysts was alcohols, with methanol being the major carbon oxygenated compound, along with CO2 and small concentrations of hydrocarbons (mainly methane and ethane). The detailed distribution of the alcohols presented in Fig. S1† shows that the synthesis yields mainly primary linear (ethanol and 1-propanol) and branched alcohols (2-methyl-1-propanol), consistent with the aldol-type condensation mechanism that has been proposed for Cu-based catalysts.3 Substitution of Zn by Mn in the catalyst formulation was found to favor the synthesis of higher alcohols, with K-CuMnAl exhibiting higher selectivity compared to the reference K-CuZnAl. The Mn-containing catalyst produced the longest chain alcohols, with propanol and methyl-propanols being in highest concentration in the higher alcohol mixture, as shown in Fig. S1.†3.2 In situ XRD under H2 and H2/CO flow
In order to elucidate the evolution of the catalyst precursor and the promotional effect of Mn on higher alcohol selectivity, the structural state of the catalysts during reduction and under working conditions was investigated by in situ XRD under reductive and reactive conditions similar to those employed in the catalytic reaction tests. In situ XRD diffractograms were recorded at 7 bar from room temperature to 400 °C in 50 °C temperature intervals under both H2 and H2/CO flow over the most active K-CuZnAl and the most selective K-CuMnAl catalyst. The Scherrer equation was used to calculate the crystal size of the different phases from the X-ray patterns.Fig. 1A and B show the in situ XRD patterns and the respective crystallite sizes as a function of temperature for K-CuZnal and K-CuMnAl. In accordance with previous ex situ XRD characterization results,34 the reference K-CuZnAl catalyst exhibits, at room temperature, highly disordered CuO (JCPDS entry 5-661) and ZnO (JCPDS entry 36-1451) crystal phases, with crystallites sizes ∼10 nm and 7–8 nm, respectively. At 100 °C, the reflections corresponding to CuO start decreasing with the simultaneous appearance of peaks corresponding to metallic Cu, signifying the onset of the catalyst reduction. The XRD pattern at 150 °C exhibits only metallic Cu reflections (JCPDS entry 4-836), indicating that reduction is completed at low temperatures with a fast rate. The Cu XRD peaks increase with temperature due to increased crystallinity. This is also reflected in the size of the Cu crystallites that become larger, growing linearly with temperature and almost doubling in size (∼17 nm) at 400 °C. No changes in the ZnO phase are observed during the entire investigated temperature range, with its reflections, and therefore its size, remaining practically constant with temperature.
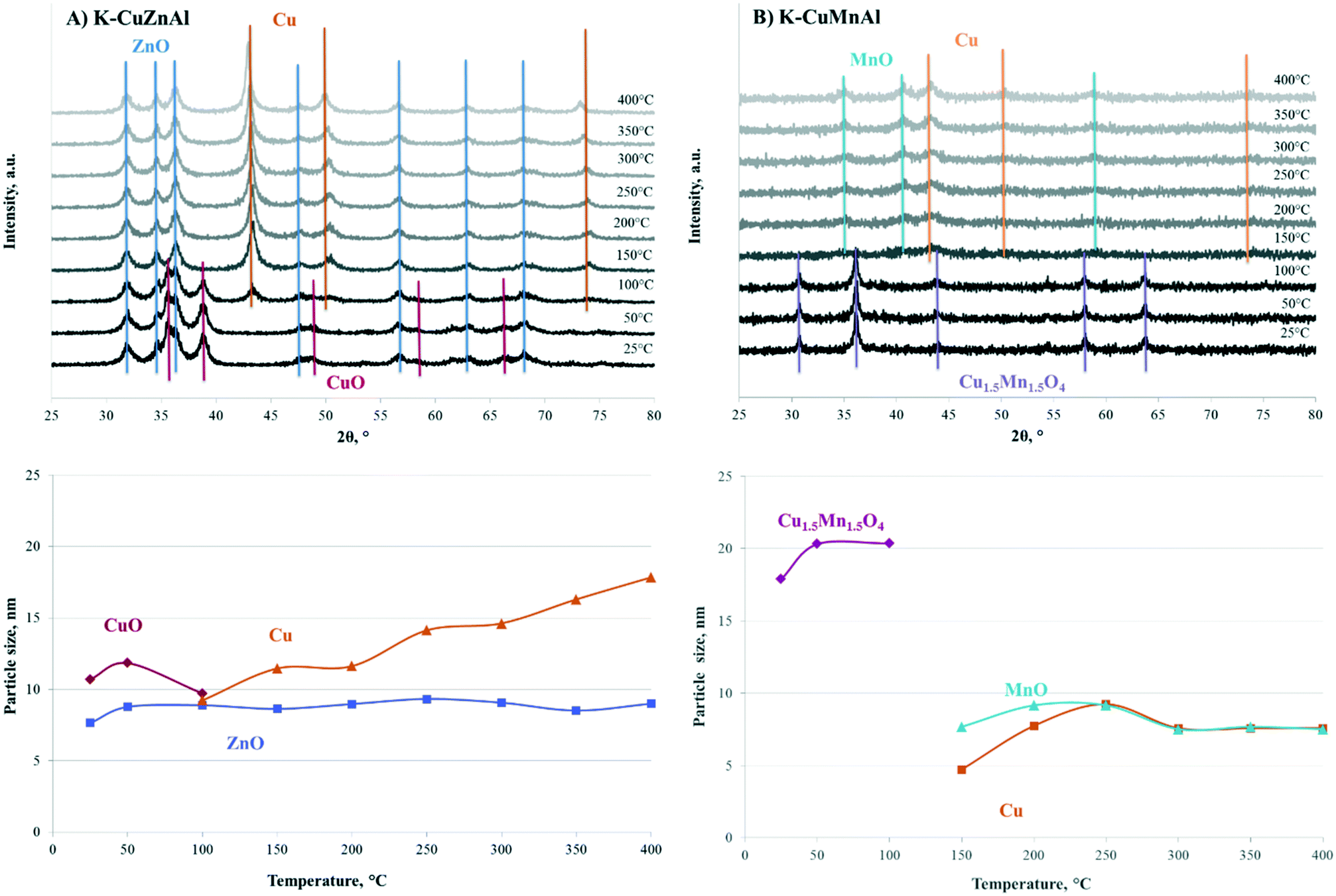 | ||
| Fig. 1 In situ XRD patterns and crystallite size as a function of temperature in H2 flow at 7 bar for (A) K-CuZnAl and (B) K-CuMnAl. | ||
The K-CuMnAl sample exhibits a different structure, with a single well-defined crystalline phase at room temperature, attributed to the Cu1.5Mn1.5O4 spinel (JCPDS entry 35-1171), with a crystallite size of ∼20 nm. No CuO reflections are detected, indicating strong interaction between Cu and Mn and incorporation of copper ions into the spinel lattice in accordance with our own34 and others'35 previous results. The XRD patterns remain unchanged up to 100 °C. Further increase to 150 °C leads to reduction of the spinel phase and formation of metallic Cu (JCPDS entry 4-836) and MnO (JCPDS entry 7-230). The intensity of the XRD peaks increases slightly with temperature up to 250 °C, with no further changes up to 400 °C. In contrast to the K-CuZnAl catalyst, the size of the Cu crystallites ranges around 5–8 nm and does not change significantly with increasing temperature. These findings confirm the stronger interaction between Cu and Mn that hinders Cu reducibility and potentially maintains Cu in a partially oxidized state. Promotion of Cu/ZrO2 catalysts with Mn was also shown to shift the reduction temperature of Cu to a higher temperature.36 The maximum temperature of reduction was found to inversely correlate with activity in CO hydrogenation over a series of CuZnAl catalysts modified with different promoters (Fe, Co, Ru, Zr, Mo, Mg, Mn, and Cr).37 The lower activity of the K-CuMnAl can be thus ascribed to the harder-to-reduce Cu species. In situ XRD patterns for samples reduced in situ at 350 °C resemble those obtained following ex situ reduction in H2 (Fig. S2†), suggesting that the trends in catalyst evolution observed from 300 to 400 °C are representative of those observed under operational conditions.
The in situ XRD study of the two catalytic systems was repeated at the same temperature range and pressure, but in syngas flow with a H2/CO ratio of 2, to detect possible structural changes under reaction conditions. The XRD patterns and the crystallite sizes as a function of temperature for K-CuZnAl and K-CuMnAl are presented in Fig. 2A and B, respectively. Exposure to synthesis gas also reduces the catalytic materials, forming the same structural phases as those formed in the presence of only hydrogen, i.e. Cu and ZnO for K-CuZnAl and Cu and MnO for K-CuMnAl. This indicates that the reaction can be run on the fresh catalysts, omitting the need for pre-reduction in H2 prior to the reaction. The main differences observed lie in the temperature of reduction when CO is co-fed with H2. In the case of the reference K-CuZnAl, reduction is completed at temperatures as low as 100 °C, compared to 150 °C required only in hydrogen flow. The sizes of the crystallites remain at similar levels, with the same trend of Cu sintering with increasing temperature. On the contrary, over the K-CuMnAl sample, reduction is retarded and occurs at 200 °C, a temperature higher than that observed in hydrogen flow. This further supports the hypothesis that Cu is retained at a partially oxidized state over this material due to the strong Cu–Mn interaction. As discussed later in the manuscript, the presence of partially oxidized Cu species at an appropriate ratio has important implications on the selectivity to higher alcohols.
 | ||
| Fig. 2 In situ XRD patterns and crystallite size as a function of temperature in syngas flow (H2/CO ratio 2) at 7 bar for (A) K-CuZnAl and (B) K-CuMnAl. | ||
3.3 XPS studies
The oxidation state of copper and the surface composition of K-CuZnAl and K-CuMnAl were investigated with X-ray photoelectron spectroscopy. Initially, XPS spectra were recorded for the freshly calcined materials in ultra-high vacuum at ambient temperature. The catalysts were then reduced ex situ under hydrogen flow at 350 °C and 1 bar and were transferred to the XPS instrument in a glove bag to minimize exposure to air. Fig. 3A and B present the Cu 2p XP spectra of the catalysts in oxidized and reduced form, respectively. The Cu 2p3/2 binding energy values in the oxidized catalysts are in the range of 932.6–933.8 eV with Cu 2p3/2–2p1/2 spin–orbit splitting of 20.0 ± 0.1 eV, characteristic of CuO.38–41 The shake-up satellites at ∼942 eV and ∼962 eV are attributed to shake-up transitions by ligand-to-metal 3d charge transfer41 that can occur only for copper in the +2 oxidation state, confirming the presence of Cu2+ in both catalysts. The substitution of Zn by Mn induces a clear shift of the Cu 2p3/2 binding energy to 933.8 eV in K-CuMnAl, compared to 932.6 eV in K-CuZnAl, associated with the different coordination of copper in the two catalysts. While CuO consists of tetrahedrally coordinated Cu2+ cations with BE in the range of 932.7–933.6 eV,38–42 Cu1.5Mn1.5O4 contains Cu both in tetrahedral and octahedral coordination.43,44 According to the literature, higher Cu 2p binding energies (in the range of 933.4–934.5 eV) are associated with a higher percentage of copper in octahedral coordination,45,46 while lower values show a higher percentage of tetrahedrally coordinated copper cations.47 Therefore, the shift of the binding energy of Cu2+ to higher values in the K-CuMnAl catalyst can be related to both the coordination of copper in the Cu1.5Mn1.5O4 spinel and the stronger interaction between Cu and Mn and the formation of Cu–O–Mn bonds.44,48 In addition to the main peak, the Cu 2p spectrum of the K-CuMnAl catalyst features an additional shoulder at lower binding energy. This peak signifies the presence of Cu+ in Cu1.5Mn1.5O4, as previously reported.49 The presence of Cu+ in K-CuMnAl is also suggested by the observation that the area ratio of the satellite 2p3/2 is 0.37, notably lower than the 0.51 value observed for K-CuZnAl. | ||
| Fig. 3 Cu 2p XP spectra of the K-CuZnAl and the K-CuMnAl catalysts (A) fresh and (B) reduced at 350 °C under H2. | ||
As shown in Fig. 3B, after catalyst reduction at 350 °C, the binding energy of Cu 2p3/2 shifts down to 931.5–932.5 eV, which is characteristic of copper species in lower oxidation states.22,50 The absence of the characteristic shake-up satellites signifies the reduction of Cu2+ to Cuδ+ (0 ≤ δ ≤ 1) species.22,40,41 The shift of the Cu binding energy to higher values in K-CuMnAl (932.5 eV) compared to K-CuZnAl (931.7 eV) is retained, similarly to the oxidized samples, indicating that the stronger interaction between Cu and Mn is maintained even in the reduced form.47 Chemical state differentiation between Cu+ and Cu0 is difficult from the 2p XP region, since the XP binding energies are practically the same.50,51 Although bigger chemical shifts are in principle observed in the LMM Auger features of Cu (KE = 917 eV), these strongly overlap with the Zn LMM Auger features (KE = 914 eV). N(E) XP derived Auger spectra are inherently broad; calculation of the Cu Auger parameter using the differential Auger spectra should however prove to be indicative of the oxidation state of Cu. Fig. 4A and B show the differentiated Cu LMM Auger transition for the fresh and reduced K-CuZnAl and K-CuMnAl, respectively. Both fresh samples show a broad peak (α) in the differential signal centered at kinetic energies of 919–920 eV, which gives rise to an Auger parameter of 1852.3–1852.5 consistent with Cu2+ species. Following reduction at 350 °C in H2, there is a slight increase in the kinetic energy of the Cu LMM α state, and a new state (β) evolves at 916.5 eV which has a corresponding Auger parameter of 1848.6–1849 eV. The new β peak is most prominent in the K-CuMnAl sample, accounting for 54% of the Cu signal, compared to only 27% in K-CuZnAl. Table 2 summarizes the measured binding and kinetic energies for the Cu 2p3/2, LMM Auger transitions and calculated Auger parameters.
 | ||
| Fig. 4 Cu and Zn LMM Auger spectra of the (A) K-CuZnAl and (B) the K-CuMnAl fresh and reduced catalysts at 350 °C under H2. | ||
| Sample | Cu 2p3/2 BE/eV | Auger KE/eV peak αa | Auger KE/eV peak βa | Auger parameter/eV peak α | Auger parameter/eV peak β |
|---|---|---|---|---|---|
| a Values in brackets show the % of Cu present in this state. | |||||
| CuZnAl fresh | 932.7 | 919.6 | 1852.3 | ||
| CuZnAl reduced | 932.1 | 919.8 (73%) | 916.5 (27%) | 1851.9 | 1848.6 |
| CuMnAl fresh | 933.5 | 919 | 1852.5 | ||
| CuMnAl reduced | 932.5 | 919.2 (46%) | 916.5 (54%) | 1851.7 | 1849.0 |
Literature values report the Auger parameters for Cu0, Cu+ and Cu2+ species as 1851.24 eV, 1849.17 and 1851.33 eV, respectively,52 with the absence of the satellite species in the Cu 2p region allowing Cu0 and Cu2+ to be distinguished. Based on the observations from Fig. 3 and 4, it can be concluded that the fresh samples are composed mainly of Cu2+ species as previously discussed, with complete loss of the satellite feature on reduction, evidencing the formation of both Cu+ and Cu0 species. The two Auger parameters calculated for the α′ and β states of reduced K-CuMnAl and K-CuZnAl are thus consistent with Cu0 and Cu+, respectively, with the former sample containing a higher proportion of Cu+, owing to its spinel structure and the strong Cu–Mn interaction.
Apart from determining the oxidation state, XPS studies also provide important information about the elemental composition of the catalysts on the surface. Integration of the XPS peaks allowed the calculation of the surface composition in the two materials in fresh (oxidized) and reduced form, presented in Fig. 5A and B for K-CuZnAl and K-CuMnAl, respectively. The bulk values for the fresh catalysts determined by ICP (ref. 34) are also included for comparison. In both materials, the amount of Cu on the surface is much lower than that in the bulk, with the surface being strongly enriched in Al. Although no bulk Al2O3 phase was detected by XRD, Al-containing compounds exhibit a strong affinity to accumulate on the surface in accordance with previous studies.53 The surface Cu exposure increases considerably after reduction in hydrogen. K-CuZnAl demonstrates a higher Cu content on the reduced surface, consistent with its higher reactivity compared to K-CuMnAl. A distinct difference between the two materials is the much higher accumulation of Mn (compared to Zn) and K on the reduced surface. It is interesting to note that the surface K content increases significantly on reduction of the K-CuMnAl. This can be directly related to the increased selectivity of the K-CuMnAl catalyst towards higher alcohols.
 | ||
| Fig. 5 Bulk (ICP) and surface (XPS) elemental analyses of (A) K-CuZnAl and (B) K-CuMnAl fresh and reduced catalysts. | ||
3.4 In situ DRIFTS studies
To obtain information on the nature and the thermal stability of the carbonyl species formed on the catalytic surface, the pre-reduced catalysts were exposed to 3% CO/He flow at 50 °C for 15 min. The peaks reached steady state in the first 2–3 min of CO exposure, indicating a fast saturation of the surface with carbonyl adsorbates. The flow was then switched to He or 6% H2/He flow and time-resolved DRIFTS spectra were recorded to monitor and quantify the extinction coefficient of the carbonyls in inert and reductive atmosphere. Fig. 6 shows the IR spectra recorded as a function of flushing time in He for K-CuZnAl and K-CuMnAl, respectively, while the same information with flushing performed in hydrogen is shown in Fig. 7. It should be noted that the detection of carboxylic or alkoxy species in the 1300–1650 cm−1 range was not possible due to the dark color of the samples and the high noise-to-signal ratio. The peak centered on 2175 cm−1 is due to vibrations of gaseous CO,54 while the band extending from 2150 to 2050 cm−1 corresponds to different linearly adsorbed CO species.54–58 It is commonly acknowledged that bands located around 2080–2090 cm−1 are due to carbonyl species adsorbed on metallic copper sites, while higher frequency bands with wavenumbers greater than ∼2110 cm−1 are related to imperfectly reduced catalysts and the presence of linear Cu+–CO species.59 The former carbonyls are very unstable, while the latter exhibit a strong resistance to flushing/outgassing. Based on the above, the carbonyl species located at ∼2120–2130 cm−1 in the K-promoted copper catalysts can be assigned to linearly adsorbed CO species on Cu+, without excluding a minor contribution from CO species on metallic copper sites. In addition, a weak shoulder at 2115 cm−1, detectable on both catalysts, can be tentatively assigned to the symmetric vibration of Cu+–(CO)2 dicarbonyl species.60 The vibration of linear Cu+–CO species is red shifted from 2129 cm−1 to 2120 cm−1 on K-CuMnAl, signifying stronger binding of CO to the Cu surface catalyst (Fig. S3†). These observations pertain also in the case of exposure of the surface to synthesis gas (CO/H2) with a molar ratio of 2, as shown in Fig. S4.† | ||
| Fig. 6 In situ DRIFTS spectra of 3% CO/He adsorption followed by flushing in He over (A) K-CuZnAl and (B) K-CuMnAl. | ||
 | ||
| Fig. 7 In situ DRIFTS spectra of 3% CO/He adsorption followed by flushing in 6% H2/He over (A) K-CuZnAl and (B) K-CuMnAl. | ||
The intensity of the DRIFTS bands suggests that the CO uptake of the Mn-substituted catalyst is higher than that of the reference material, in agreement with the XPS results that clearly showed a higher concentration of Cu+ species on K-CuMnAl. However, the higher CO uptake is inconsistent with the lower reactivity of the K-CuMnAl in the CO hydrogenation reaction, which demonstrated almost half CO conversion compared to the reference material.34 Considering that the main species detected by IR are Cu+–CO complexes, this suggests that the primary activation of the carbon oxide molecule to methanol and/or higher alcohols occurs on both Cu0 and Cu+ sites on the surface, as discussed in detail in the Discussion section. It is interesting to note that the CO uptake reduces considerably in the co-presence of H2 (Fig. S4†), especially for the K-CuMnAl catalyst. The much higher intensity loss over this catalyst is probably related to the competition of H2 and CO for the same sites,61 due to the higher Cu+/Cu0 ratio, as evidenced by XPS, and therefore the reduced concentration of metallic Cu sites which catalyze H2 dissociation.
When the flow was switched to either helium or hydrogen, the peak attributed to gaseous CO was quickly removed, while the bands attributed to carbonyl complexes were retained even after 14 min of flushing, indicating that these species are indeed chemisorbed on the surface. The time-resolved spectra shown in both Fig. 6 and 7 point out that the intensity of the adsorbates decreases more quickly over the Mn-containing catalyst, especially in the case of H2 flushing, suggesting that the adsorbed CO undergoes reaction with hydrogen.
In order to quantify the above observations, we integrated the area corresponding to chemisorbed CO in the time resolved in situ DRIFTS spectra of CO desorption in helium and in hydrogen over the two catalysts and normalized the values to the surface area of each catalyst. The quantified results are presented in Fig. 8 as a function of time. As already discussed, CO uptake is almost double on K-CuMnAl due to the higher amount of Cu+ sites. The rate of CO removal is also much faster on K-CuMnAl for the first minutes of flushing, especially in the presence of H2, but slows down after about ∼6 min and approaches that of K-CuZnAl. On the contrary, the CO desorption rates in the presence and in the absence of H2 are very similar on the reference catalyst.
 | ||
| Fig. 8 Amount of surface CO species as a function of time after 3% CO/He adsorption followed by flushing in He (solid lines) or 6% H2/He (dotted lines) over K-CuZnAl and K-CuMnAl. | ||
Assuming first order desorption of CO from the surface, we calculated the apparent kinetic constants for desorption from the slope of the logarithm of CO concentration as a function of time, shown in Fig. 9A and B for desorption in He and H2, respectively. Whereas a straight line describes kinetically the desorption of CO from K-CuZnAl, in the case of K-CuMnAl there is a break point in the case of both He and H2 flushing, suggesting contributions from two different CO adsorption sites with different desorption kinetic parameters. The first type of carbonyl species is very unstable/reactive and is characterized by a high apparent extinction coefficient equal to 0.24 min−1 in He. This value increases to 0.61 min−1 when flushing is performed in H2, indicating that the higher removal rate of CO from these sites is not due to thermal instability but to further reaction, which is enhanced with hydrogen. We tentatively ascribe this species to CO adsorbed on Cu+ sites that are created/modified in the presence of Mn and bind CO more strongly, as indicated by the shift of the CO vibration to lower wavenumbers. Chemisorbed CO is more likely to undergo fast dissociation to C and O species due to the weakening of the C–O bond, with the rate increasing in the presence of hydrogen due to hydrogen-assisted dissociation. The second type of CO species is removed much slower, with a similar rate in both hydrogen and helium. The kinetic constant for desorption is an order of magnitude lower and equal to ∼0.06 min−1. This value coincides with the extinction coefficient calculated for K-CuZnAl in both helium and hydrogen, suggesting that these species correspond to CO adsorbed on unmodified Cu+ sites that exist on both materials, with weaker metal–CO bonding and high thermal stability, which adsorb CO associatively.
 | ||
| Fig. 9 Logarithm of CO concentration as a function of time after 3% CO/He adsorption followed by flushing in (A) He and (B) 6% H2/He flow. | ||
4. Discussion
Detailed characterization of K-CuZnAl and K-CuMnAl provided important insight on the effect of Mn in the hydrogenation of CO to alcohols higher than methanol. In situ XRD and XPS clearly showed the existence of stronger interaction between Cu and Mn, compared to Zn, leading to the incorporation of copper ions in a Cu–Mn spinel lattice. This in turn hinders reducibility and stabilizes copper in the Cu+ state. Several studies on Cu/Zn/Al catalysts for methanol synthesis argue that stabilization of Cu+ can be attributed to its incorporation into the ZnO lattice due to a strong metal–support interaction.62 It is very likely that the role of Mn is similar in the K-CuMnAl catalyst. The increased population of Cu+ species can also be related with the considerably higher dispersion of K on the surface of K-CuMnAl, as evidenced by XPS. Earlier works63,64 showed that the primary role of alkali promoters, even in impurity levels, is to stabilize Cu+ on the surface of Cu metal, creating in this way active centers, which are supplied by Hads from the Cu–metal surface.The stronger Cu–Mn interaction changes significantly the population and type of carbonyl species formed on the surface and consequently the activity and selectivity of the catalyst in the CO hydrogenation reaction. Linearly adsorbed CO species on Cu+ sites were detected on the surface of both catalysts by DRIFTS, with the CO uptake being much higher on K-CuMnAl than K-CuZnAl, in contrast to its lower reactivity in the reaction. Considering that the main species detected by IR are Cu+–CO complexes, this suggests that the primary activation of the carbon oxide molecule to methanol and/or higher alcohols cannot be ascribed exclusively to the partially oxidized Cu sites. As discussed in the Introduction, the active site for methanol synthesis has been debated for decades and opposing results appear in the literature with some studies linearly correlating methanol activity to the amount of metallic Cu65 and others to the surface Cu+ species.64,66,67 Our results possibly indicate that CO is activated on both Cu0 and Cu+ sites on the surface. This has also been demonstrated recently in a detailed study by Pérez-Ramírez's group,53 who investigated simultaneously CO hydrogenation, CO2 hydrogenation, and water-gas shift (WGS) reactions over industrial Cu/Zn/Al catalysts and concluded that CO is converted to methanol over both metallic and partially oxidized copper. It was suggested that monovalent copper species might only account for the WGS reaction. Considering the selectivity of the two catalysts under investigation in the present study (Table 1), selectivity to CO2, a product of the WGS reaction, is 37% higher on the Mn-promoted catalysts compared to K-CuZnAl, supporting the hypothesis that Cu+ plays a determining role in catalyzing CO conversion to CO2via the WGS route.
DRIFTS also evidenced that CO species are more strongly bonded to copper in the K-CuMnAl catalyst. It can be postulated that the strong interaction evidenced between Cu and Mn increases the electron density of surface Cu sites and hence increases π* back-donation to the adsorbed CO, strengthening the carbon–metal bond and weakening the carbon–oxygen bond in CO. Similar results were also reported by Cai et al. for CO adsorption on mixed CuMnOx oxides.68 The weakening of the carbonyl bonds implies that the CO species on K-CuMnAl would be more readily dissociated to C and O than the carbonyl intermediates on K-CuZnAl.69 Calculation of the extinction coefficients of the carbonyl species in He and H2 by time resolved in situ DRIFTS confirmed the above. Whereas CO adsorbed linearly and associatively on unmodified Cu+ sites was found on both K-CuZnAl and K-CuMnAl, an additional, highly reactive type of carbonyl was detected on K-CuMnAl, attributed to CO adsorbed on Cu+ sites that are created/modified in the presence of Mn and bind CO more strongly. These species undergo fast dissociation to C and O. This observation is also validated by the results of the catalytic performance measurements that showed that the selectivity to hydrocarbons, which are produced via dissociative adsorption of CO, is double on K-CuMnAl compared to K-CuZnAl. Time-resolved DRIFTS also showed that the CO dissociation rate increases in the presence of hydrogen. Hydrogen has been found to have an important effect on CO dissociation. Based on time resolved IR spectra, the CO dissociation rate on Ru was found to be two orders of magnitude higher, affected by hydrogen.70 Moreover, kinetic studies clearly showed the positive effect of hydrogen in accelerating the CO dissociation rate on metal surfaces.71,72 Considering the above, the faster reduction of the CO peaks in the presence of hydrogen over K-CuMnAl can be attributed to the hydrogen-assisted dissociation of the strongly bonded CO species on the surface to C and O atoms.
The higher selectivity of the K-CuMnAl can thus be ascribed to the existence of sites for both dissociative and associative CO adsorption on K-CuMnAl, with higher alcohols forming via the insertion of molecular CO to CHx species that form from CO dissociation to C and O and hydrogenation of the former. Formation of longer carbon chain alcohols is also favored by the higher Cu+/Cu0 ratio and K dispersion in K-CuMnAl compared to K-CuZnAl. On the basis of mean-field microkinetic models for the methanol synthesis and WGS reactions, Grabow and Mavrikakis73 showed that the hydrogenation of methoxy species is the slow step for the CO methanol synthesis route. Since the concentration of metallic copper on the surface (which catalyzes H2 dissociation) is lower over K-CuMnAl, the availability of hydrogen on the surface is probably lower, thus limiting the formation of methanol and increasing the lifetime of methoxy species that could undergo CO insertion to produce higher alcohols. Regarding K dispersion, alkalis have been known to provide basic sites for the aldol-type condensation of lower to higher alcohols.3 In our previous work,34 we established a linear inverse correlation between acidity and higher alcohol selectivity and attributed the increased selectivity of K-CuMnAl to its lower acidity and higher basicity that catalyzes the formation of various C–C and C–O bonds. Based on the XPS results of this work, we can now confirm the higher concentration of basic sites on the surface and their positive role in the production of higher alcohols over the K-CuMnAl catalyst.
5. Conclusions
The K-promoted CuZnAl catalyst, with a Cu/Zn/Al ratio of 45/45/10, was previously identified as an active catalyst for the hydrogenation of CO to CH3OH at 300 °C and 40 bar. Substitution of Zn by Mn at constant chemical composition shifted the products of the reaction to higher molecular weight alcohols (mainly propanol and methyl-propanol), with a substantial increase in selectivity compared to K-CuZnAl.34 In this study, we performed detailed characterization of the two materials, K-CuZnAl and K-CuMnAl, to understand the promotional effect of Mn in higher alcohol synthesis. Our results showed that K-CuZnAl, which is active towards methanol formation, adsorbs CO on Cu+ surface species via the formation of weak carbon–metal bonds. The carbon–oxygen bond in the carbonyl species remains strong and CO does not dissociate on the surface. H2 is dissociated on the metallic Cu sites and the H atoms hydrogenate the molecularly adsorbed CO to methanol. In the K-CuMnAl catalysts, the strong interaction between Cu and Mn increases the percentage of Cu+ species on the surface and modifies copper sites in close vicinity to Mn. These modified Cu+ sites bind CO more strongly, leading to dissociation of CO to C and O species. The C species are hydrogenated and higher alcohols are produced via the addition of non-dissociated CO in the CHx species, accounting for the higher selectivity of K-CuMnAl. The higher Cu+/Cu0 ratio also limits the population of hydrogen on the surface (due to decreased concentration of metallic copper sites that catalyze H2 dissociation) and retards the fast hydrogenation of the oxygen-containing intermediates to methanol, thus increasing the probability of carbon-chain growth. Once ethanol precursors form on the surface, higher alcohols are produced via aldol-type condensation reactions on the basic sites of the surface, that are more abundant on K-CuMnAl compared to K-CuZnAl.Acknowledgements
This work received funding from the “European Biofuels Research Infrastructure for Sharing Knowledge (BRISK)” (www.briskeu.com) project (contract no. 284498) in the framework of the funding of transnational activities. Karen Wilson acknowledges the Royal Society for the award of an Industry Fellowship, and Adam F. Lee acknowledges the EPSRC for funding under grant EP/N009924/1. Supporting data can be found at http://doi.org/10.17036/researchdata.aston.ac.uk.00000189.References
- Y. Yacoub, R. Bata, M. Gautam and D. Martin, Proc. Inst. Mech. Eng., Part A, 1998, 212, 363–379 CrossRef.
- Eurobioref Public Booklet, Available from: http://eurobioref.org/images/Eurobioref_livret_resultats_total_v4.pdf (accessed July 2016).
- J. J. Spivey and A. Egbebi, Chem. Soc. Rev., 2007, 36, 1514–1528 RSC.
- V. Subramani and S. K. Gangwal, Energy Fuels, 2008, 22, 814–839 CrossRef CAS.
- M. Gupta, M. L. Smith and J. J. Spivey, ACS Catal., 2011, 1, 641–656 CrossRef CAS.
- K. J. Smith and R. B. Anderson, Can. J. Chem. Eng., 1983, 61, 40–45 CrossRef CAS.
- J. J. F. Scholten, X. Xiaoding, C.-B. von der Decken, B. Höhlein, D. Mausbeck, J. R. H. Ross, J. Slaa, U. Hoffmann, U. Kunz and H. Stölting, Int. J. Energy Res., 1994, 18, 185–196 CrossRef CAS.
- K. Klier, R. G. Herman, J. G. Nunan, K. J. Smith, C. E. Bogdan, C.-W. Young and J. G. Santiesteban, in Methane Conversion, ed. D. M. Bibby, C. D. Chang, R. F. Howe and S. Yurchak, Elsevier, Amsterdam, 1988, p. 109 Search PubMed.
- J. C. Slaa, J. G. Van Ommen and J. R. H. Ross, Catal. Today, 1992, 15, 129–148 CrossRef CAS.
- A. Egbebi and J. J. Spivey, in Thermochemical Conversion of Biomass to Liquid Fuels and Chemicals, ed. M. Crocker, RSC publishing, 2010, pp. 125–145 Search PubMed.
- G. Natta, I.-I. Colombo and I. Pasquon, in Catalysis, ed. P. H. Emmett, Reinhold Publ. Co., New York, 1957, vol. V, p. 131 Search PubMed.
- W. Falter, Isobutanol aus Synthesegas Oktanzahlverbesserer der Zukunft?, Ph.D. Thesis, T. H. Aachen, Germany, 1988 Search PubMed.
- W. Kotowski, J. Klimiec, J. Tomaszewska and W. Czelakowskii, Chem. Ing. Tech., 1983, 55, 871 CrossRef CAS.
- J. C. Slaa, J. G. van Ommen and J. R. H. Ross, Top. Catal., 1995, 2, 79–89 CrossRef CAS.
- C. E. Hofstadt, K. Kochloefl and O. Bock (Siid Chemie A.G.), EP 0.034.338-A2 (1981).
- C. E. Hofstadt, M. Schneider, O. Bock and K. Kochloefl, in Preparation of Catalysts III, ed. G. Poncelet, P. Grange and P. A. Jacobs, Elsevier, Amsterdam, 1983, p. 709 Search PubMed.
- R. G. Herman, Catal. Today, 2000, 55, 233–245 CrossRef CAS.
- P. Forzatti, E. Tronconi and I. Pasquon, Catal. Rev.: Sci. Eng., 1991, 33(1&2), 109–168 CAS.
- K. Fang, D. Li, M. Lin, M. Xiang, W. Wei and Y. Sun, Catal. Today, 2009, 147, 133–138 CrossRef CAS.
- V. R. Surisetty, A. K. Dalai and J. Kozinski, Appl. Catal., A, 2011, 404, 1–11 CAS.
- K. A. N. Verkerk, B. Jaeger, C.-H. Finkeldei and W. Keim, Appl. Catal., A, 1999, 186, 407–431 CrossRef CAS.
- Y. Yang, L. Wang, K. Xiao, T. Zhao, H. Wang, L. Zhong and Y. Sun, Catal. Sci. Technol., 2015, 5, 4224–4232 CAS.
- J. G. Nunan, C. E. Bogdan, K. Klier, K. J. Smith, C.-W. Young and R. G. Herman, J. Catal., 1989, 116, 195–221 CrossRef CAS.
- A.-M. Hilmen, M. Xu, M. J. L. Gines and E. Iglesia, Appl. Catal., A, 1998, 169, 355–372 CrossRef CAS.
- A. Baysar and G. L. Schrader, Turk. J. Chem., 1994, 18, 223–241 CAS.
- A. Kiennemann, H. Idriss, R. Kieffer, P. Chaumette and D. Durand, Ind. Eng. Chem. Res., 1991, 30, 1130–1138 CrossRef CAS.
- D. J. Elliott, J. Catal., 1988, 111, 445–449 CrossRef CAS.
- D. J. Elliott and F. J. Pennella, J. Catal., 1988, 114, 90–99 CrossRef CAS.
- D. J. Elliott and F. J. Pennella, J. Catal., 1986, 102, 464–466 CrossRef CAS.
- E. Kunkes and M. Behrens, in Methanol Chemistry, ed. R. Schlögl, Chemical Energy Storage, De Gruyter, Berlin, 2013, pp. 413–442 Search PubMed.
- Z.-J. Zuo, L. Wang, L.-M. Yu, P.-D. Han and W. Huang, J. Phys. Chem. C, 2014, 118, 12890–12898 CAS.
- Z.-J. Zuo, L. Wang, Y.-J. Liu and W. Huang, Catal. Commun., 2013, 34, 69–72 CrossRef CAS.
- X. Sun, R. Zhang and B. Wang, Appl. Surf. Sci., 2013, 265, 720–730 CrossRef CAS.
- E. Heracleous, E. T. Liakakou, A. A. Lappas and A. A. Lemonidou, Appl. Catal., A, 2013, 455, 145–154 CrossRef CAS.
- X. Du, Z. Yuan, L. Cao, C. Zhang and S. Wang, Fuel Process. Technol., 2008, 89, 131–138 CrossRef CAS.
- R. Xu, Z. Ma, C. Yang, W. Wei and Y. Sun, Prepr. Symp. - Am. Chem. Soc., Div. Fuel Chem., 2003, 48(2), 927–928 CAS.
- J. M. Beiramar, A. Griboval-Constant and A. Y. Khodakov, ChemCatChem, 2014, 6(6), 1788–1793 CrossRef CAS.
- S. Poulston, P. M. Parlett, P. Stone and M. Bowker, Surf. Interface Anal., 1996, 24, 811–820 CrossRef CAS.
- J. A. Rodriguez and J. Hrbek, J. Vac. Sci. Technol., A, 1994, 12, 2140–2144 CAS.
- J. A. Rodriguez, J. Y. Kim, J. C. Hanson, M. Perez and A. I. Frenkel, Catal. Lett., 2003, 85(3–4), 247–254 CrossRef CAS.
- Y. Lu, F. Yu, J. Hu and J. Liu, Appl. Catal., A, 2012, 429–430, 48–58 CrossRef CAS.
- G. Moretti, G. Fierro, M. Lo Jacono and P. Porta, Surf. Interface Anal., 1989, 14, 325–326 CrossRef.
- M. Lenglet, P. Foulatier, J. Düeb and J. Arséne, Phys. Status Solidi A, 1986, 94, 461–466 CrossRef CAS.
- F. Li, L. Zhang, D. G. Evans and X. Duan, Colloids Surf., A, 2004, 244, 169–177 CrossRef CAS.
- B. Gillot, S. Buquet and E. Kester, J. Mater. Chem., 1997, 7, 2513–2517 RSC.
- V. J. Rico, J. L. Hueso, J. Cotrino, V. Gallardo, B. Sarmiento, J. J. Brey and A. R. Gonzalez-Elipe, Chem. Commun., 2009, 6192–6194 RSC.
- X. Han, K. Fang and Y. Sun, RSC Adv., 2015, 5, 51868–51874 RSC.
- P. Decyk, A. B. Więckowski, L. Najder-Kozdrowska and I. Bilkova, Nukleonika, 2015, 60(3), 423–428 CrossRef CAS.
- J. Quan, L. Mei, Z. Ma, J. Huang and D. Li, RSC Adv., 2016, 6, 55786–55791 RSC.
- J. Sun, S. Wan, F. Wang, J. Lin and Y. Wang, Ind. Eng. Chem. Res., 2015, 54(32), 7841–7851 CrossRef CAS.
- J. M. Campos-Martin, J. L. G. Fierro, A. Guerrero-Ruiz, R. G. Herman and K. Klier, J. Catal., 1996, 163, 418–428 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. St. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- O. Martin and J. Pérez-Ramírez, Catal. Sci. Technol., 2013, 3, 3343–3352 CAS.
- N. D. Subramanian, C. S. S. R. Kumar, K. Watanabe, P. Fischer, R. Tanaka and J. J. Spivey, Catal. Sci. Technol., 2012, 2, 621–631 CAS.
- M. L. Smith, N. Kumar and J. J. Spivey, J. Phys. Chem. B, 2012, 116, 7931–7939 CrossRef CAS PubMed.
- K. Hadjiivanov and H. Knozinger, Phys. Chem. Chem. Phys., 2001, 3(6), 1132–1137 RSC.
- K. Hadjiivanov, T. Venkov and H. Knozinger, Catal. Lett., 2001, 75(1–2), 55–59 CrossRef CAS.
- H. Z. Guo, Y. Z. Chen, X. Z. Chen, R. T. Wen, G. H. Yue and D. L. Peng, Nanotechnology, 2011, 22(19), 195604–195608 CrossRef PubMed.
- P. Hollins, Surf. Sci. Rep., 1992, 16, 51–94 CrossRef CAS.
- D. Scarano, S. Bordiga, C. Lamberti, G. Spoto, G. Ricchiardi, A. Zecchina and C. O. Arean, Surf. Sci., 1998, 411(3), 272–285 CrossRef CAS.
- N.-Y. Topsoe and H. Topsoe, J. Mol. Catal. A: Chem., 1999, 141, 95–105 CrossRef CAS.
- M. Santiago, K. Barbera, C. Ferreira, D. Curulla-Ferré, P. Kolb and J. Pérez-Ramírez, Catal. Commun., 2012, 21, 63–67 CrossRef CAS.
- L. E. Y. Nonneman and V. Ponec, Catal. Lett., 1990, 7, 213–218 CrossRef CAS.
- G. R. Sheffer and T. S. King, J. Catal., 1989, 115, 376–387 CrossRef CAS.
- G. C. Chinchen, M. S. Spencer, K. W. Waugh and D. A. Whan, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 2193 RSC.
- J. Szanyi and D. Wayne Goodman, Catal. Lett., 1991, 10(5), 383–390 CrossRef CAS.
- G. R. Apai, J. R. Monnier and M. J. Hanrahan, J. Chem. Soc. Chem. Commun., 1984, 212–213 RSC.
- L. Cai, Z. Hu, P. Branton and W. Li, Chin. J. Catal., 2014, 35, 159–167 CrossRef CAS.
- G. R. Johnson, S. Werner and A. T. Bell, ACS Catal., 2015, 5, 5888–5903 CrossRef CAS.
- F. M. Hoffmann and J. L. Robbins, J. Electron Spectrosc. Relat. Phenom., 1987, 45, 421 CrossRef CAS.
- D. Bianchi and C. O. Bennett, J. Catal., 1984, 86, 433 CrossRef CAS.
- G. Blyholder and M. Lawless, Langmuir, 1991, 7, 140–141 CrossRef CAS.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cy00018a |
| This journal is © The Royal Society of Chemistry 2017 |