
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic hydrogenation of CO2 to methane over supported Pd, Rh and Ni catalysts†
Natalia M.
Martin
 *a,
Peter
Velin
a,
Magnus
Skoglundh
a,
Matthias
Bauer
b and
Per-Anders
Carlsson
a
*a,
Peter
Velin
a,
Magnus
Skoglundh
a,
Matthias
Bauer
b and
Per-Anders
Carlsson
a
aCompetence Centre for Catalysis, Chalmers University of Technology, Gothenburgh, 412 96, Sweden. E-mail: Natalia.Martin@chalmers.se; Fax: +46 31 160062; Tel: +46 31 772 29 04
bDepartment of Chemistry, Paderborn University, 33098 Paderborn, Germany
First published on 26th January 2017
Abstract
As a step in production of so-called electrofuels, ambient pressure CO2 hydrogenation has been investigated over different catalytic model systems based on metal particles (Pd, Rh and Ni) supported on various metal oxides (SiO2, Al2O3 and CeO2) and aluminosilicates (ZSM-5 and MCM-41) at different specific reactant ratios and temperatures between 150 and 450 °C. Catalytic activity and selectivity measurements in a flow reactor show that the highest CO2 conversion towards methane is obtained for the Rh/Al2O3 and Rh/CeO2 catalysts, followed by Ni/CeO2. Generally, the results suggest that both the support material and reaction conditions play an important role in the hydrogenation process. Further, in situ diffusive reflectance infrared Fourier transform spectroscopy reveals the intermediate species during transient CO2 hydrogenation over the Rh and Ni containing catalysts. Adsorption and dissociation of CO2 occurs over the Rh/Al2O3 catalyst in the presence of H2, resulting in the formation of linear Rh–CO species, while formates and carbonates are formed over the Rh/CeO2 and Ni/CeO2 catalysts, likely at the metal–support interface.
1. Introduction
Because of the high demand on the limited fossil fuel resources and the environmental problems associated with the increased anthropogenic CO2 emissions, significant attention is paid to exploring means to utilise CO2 as a renewable source of energy. A possible viable route for recycling of CO2 is through catalytic hydrogenation preferably directly to a usable hydrocarbon-based fuel, e.g., methane, methanol or dimethyl ether.1 Although some progress has been made in this direction,2,3 development of efficient routes for CO2 hydrogenation to produce such green fuels remains an important topic in catalysis. In particular, the promising progress in sustainable production of H2 by wind and/or solar powered electrolysis of water or by photocatalytic water splitting4 stimulates studies on catalytic CO2 hydrogenation at ambient pressures as a realistic technology for producing so-called electrofuels or sunfuels through CO2 recycling not only at large scale biorefineries but also at smaller scale remote plants.5To catalyse the reaction between CO2 and H2, surface sites that bind and activate CO2 need to co-exist and cooperate with sites for dissociation of H2. Activation of CO2 by heterogeneous catalysis is often carried out using basic or amphoteric reducible oxides as for example ceria, zirconia or titania, while metals are most frequently used to dissociate H2. The transformation of CO2, however, is still an issue due to the difficulties in activating the thermodynamically stable CO2 molecule, a process that is highly energy-intensive.6 In the present study, the conversion of CO2 into methane and/or methanol is the prime target. There are different competing reactions during CO2 hydrogenation, the ones relevant for this study being enumerated in eqn (1)–(3) below.
| CO2 + H2 ⇌ CO + H2O, ΔH298 = 41.2 kJ mol−1 | (1) |
| CO2 + 4H2 ⇌ CH4 + 2H2O, ΔH298 = −252.9 kJ mol−1 | (2) |
| CO2 + 3H2 ⇌ CH3OH + H2O, ΔH298 = −49.5 kJ mol−1 | (3) |
The formation of by-products and particularly the formation of carbon monoxide and water through the strongly competing reverse water-gas shift reaction (rWGS, eqn (1)), consumes CO2 and H2 such that the desired product formation and thereby the selectivity become low. Therefore, the sole formation of the desired product is not an easy task. For several decades, methanol has been produced industrially over Cu–ZnO/Al2O3 catalysts at 200–300 °C and 50–100 bar using syngas (CO/H2/CO2).7,8 Using the industrial Cu–ZnO/Al2O3 catalyst at low pressures methanol synthesis leads to significant CO production, lowering the desired product formation, suggesting that a low-pressure CO2 reduction process may require a different catalyst.9 An important challenge for atmospheric pressure CO2 hydrogenation is to design catalysts that efficiently can suppress the rWGS reaction as well as formation of other by-products in favour of methanol and/or methane formation.
Hydrogenation of CO2 to methane (eqn (2)) is the most advantageous reaction with respect to thermodynamics (ΔG298 = −130.8 kJ mol−1) among the CO2 conversion reactions. Previous work has reported different transition metals such as Ru, Rh, Ni and Pd to be highly active and selective for the methane formation by CO2 hydrogenation, especially at low temperatures.10–12 Among these, the supported Ni catalysts were reported to have the highest selectivity to form methane.13
In the present work we have synthesised and studied industrially relevant polycrystalline catalysts that can bind CO2 and facilitate H2 dissociation and further reaction with activated CO2 to form methane at atmospheric pressure conditions. We also consider possible methanol formation, even though no methanol has been observed to form in the present study, under the investigated conditions. A series of metal oxide supported catalysts containing Pd, Ni and Rh have been prepared with impregnation and ion-exchange methods, characterised and evaluated in terms of activity and selectivity for CO2 hydrogenation. The kinetic study showed an increased conversion of CO2 to methane and CO at elevated temperature, with varying selectivity depending on the active phase. The Rh/Al2O3 and Rh/CeO2 catalysts were singled out as the ones giving the highest CO2 conversion and lowest selectivity for the rWGS reaction in the methanation reaction, followed by the Ni/CeO2 catalyst. Furthermore, ceria is concluded to be the most active support for hydrogenation of CO2, probably due to a participating role in CO2 adsorption and a highly dispersed active phase. In addition, the CO2 methanation reaction mechanism was investigated by in situ diffusive reflectance Fourier transform infrared spectroscopy and the results revel clear differences between Rh/Al2O3 and Rh/CeO2 catalysts.
2. Experimental section
2.1 Catalyst preparation and ex situ characterization
In this study, 13 different catalysts were prepared with Pd, Ni and Rh supported on silica, alumina, ceria, MCM-41 and ZSM-5. The samples with silica, alumina, ceria and MCM-41 were prepared by incipient wetness impregnation, while the Ni/ZSM-5 sample was prepared by ion-exchange as described below. Powders of silica (Kromasil Silica KR-300-10, Akzo Nobel Eka Chemicals), alumina (Puralox SBa200, Sasol) and ceria (99.5 H.S.A. 514, Rhône-Poulenc) were calcined in air at 600 °C for 2 h starting from room temperature with a heating rate of 5 °C min−1 to remove carbonaceous impurities and stabilise the structure of the support materials. Since MCM-41 (Sigma-Aldrich) is more fragile, a lower calcination temperature (450 °C) was used for this material. Precursor solutions of nickel and rhodium were prepared by dissolving Ni(NO3)2 × 6H2O (Alfa Aesar) and Rh(NO3)3 × 2H2O (Alfa Aesar) salts in Milli-Q water (18 MΩ cm−1). To increase the solubility of the rhodium salt, 25 droplets of 70% HNO3 were added. The precursor for palladium was an aqueous solution of the complex (NH3)4Pd(NO3)2 (Alfa Aesar). The specific amount of precursor solution to obtain 3 wt% of the metal was added to 3 g of each support. The impregnated silica, alumina and ceria samples were instantly frozen with liquid nitrogen for 24 h and finally calcined in air at 550 °C for 1 h. After freeze-drying, the impregnated MCM-41 sample was calcined in air at 450 °C for 1 h.For the Ni/ZSM-5 catalyst, 3 g ZSM-5 (SAR 27, Akzo Nobel) was added to a beaker together with 1.5 liter 0.025 molar nickel nitrate solution. After 24 h of stirring at room temperature the solution was filtered and washed with Milli-Q water. The aqueous ion-exchanged zeolite was then dried at 110 °C for 24 h before calcination in air at 450 °C for 1 h starting from room temperature with a heating rate of 5 °C min−1. Table 1 gives an overview of the prepared catalysts.
| Sample | Metal loading (wt%) | Calcination temperature (°C) | Specific surface area (m2 g−1) | ||
|---|---|---|---|---|---|
| Pd | Ni | Rh | |||
| Pd/SiO2 | 3.0 | — | — | 550 | 315 |
| Pd/Al2O3 | 3.0 | — | — | 550 | 187 |
| Pd/CeO2 | 3.0 | — | — | 550 | 131 |
| Pd/MCM-41 | 3.0 | — | — | 450 | — |
| Ni/SiO2 | — | 3.0 | — | 550 | 311 |
| Ni/Al2O3 | — | 3.0 | — | 550 | 182 |
| Ni/CeO2 | — | 3.0 | — | 550 | 128 |
| Ni/MCM-41 | — | 3.0 | — | 450 | — |
| Ni/ZSM-5 | — | 1.5 | — | 450 | — |
| Rh/SiO2 | — | — | 3.0 | 550 | 310 |
| Rh/Al2O3 | — | — | 3.0 | 550 | 180 |
| Rh/CeO2 | — | — | 3.0 | 550 | 125 |
| Rh/MCM-41 | — | — | 3.0 | 450 | — |
Monolith samples were prepared by coating cordierite monoliths with approximately 200 mg of the powder samples through dip coating. Colloidal alumina sol (Disperal, Sasol) was used as binder for all samples. The monolith samples were thereafter dried at 90 °C in air until all water evaporated and finally calcined at 600 °C for 5 min.
The crystal structure of the as-prepared samples was studied by X-ray diffraction (XRD) under ambient atmosphere using a Bruker AXS D8 ADVANCE diffractometer with Cu-Kα radiation equivalent to 0.15418 nm. The diffraction data was recorded in the 2θ range of 20–66° with incremental steps of 0.03° for 28 minutes.
The specific surface area of the catalysts was determined by nitrogen sorption at 77 K (Micrometrics Tristar 3000) using the Brunauer Emmet and Teller (BET) method.14 The samples were dried in vacuum at 230 °C for 3 h before the measurement.
2.2 Flow-reactor experiments
The catalytic performance of the catalysts was analysed in a flow reactor. An FTIR instrument (MultiGas 2030, MKS Instruments) was used to measure the composition of the effluent gas. The flow reactor, described elsewhere,15 consists of a quartz tube wherein the sample was positioned. Heating of the inlet gas and the sample occurred via resistive heating of a metal coil surrounding the reactor tube. Both the inlet gas and the catalyst temperature were measured by separate thermocouples. Feed gases were mixed from H2, CO2 and Ar and introduced into the reactor via individual mass flow controllers, providing a total flow of 2000 ml min−1, corresponding to a gas hourly space velocity (GHSV) of about 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. Prior to each experiment the samples were pre-treated at 450 °C under a flow of 5% oxygen followed by a flow of 5% hydrogen, each for 10 minutes. The catalytic performance of the catalysts was measured under a flow of 1% CO2 and 5% H2, corresponding to a molar ratio of 1:5, for 10 minutes at four different temperatures (450, 350, 250, and 150 °C).
000 h−1. Prior to each experiment the samples were pre-treated at 450 °C under a flow of 5% oxygen followed by a flow of 5% hydrogen, each for 10 minutes. The catalytic performance of the catalysts was measured under a flow of 1% CO2 and 5% H2, corresponding to a molar ratio of 1:5, for 10 minutes at four different temperatures (450, 350, 250, and 150 °C).
2.3 In situ FTIR spectroscopy measurements
The in situ FTIR spectroscopy experiments were performed in diffusive reflectance (DRIFT) mode with a BRUKER Vertex 70 spectrometer equipped with a nitrogen cooled MCT detector and a high-temperature stainless steel reaction cell (Harrick Praying Mantis™ high temperature reaction chamber) with KBr windows. The temperature of the sample holder was measured by a thermocouple (type K) and controlled by a PID regulator (Eurotherm). Feed gases were introduced into the reaction cell via individual mass flow controllers, providing a total flow of 100 ml min−1 in all experiments. Moreover, the H2 feed was introduced via a high-speed gas valve (Valco, VICI) in order to provide precise transients. Prior to each experiment the samples were pre-treated at 350 °C with 5% O2 in Ar for 10 min and 0.8% H2 in Ar for 10 min and then cooled in Ar to the desired temperature where a background spectrum was collected. The experiment started by introducing a flow of 0.2% CO2 and 0.8% H2 to the reaction cell. After 10 min the H2 flow was turned off for 10 min and reintroduced again for 10 minutes. The measurements were repeated five times giving a total length of the experiments of 100 minutes. The hydrogen pulse-response measurements were performed at different temperatures (400, 350, 300, and 250 °C). The region between 790–3800 cm−1 was investigated with a spectral resolution of 8 cm−1. The product stream was continuously analysed with mass spectrometry (Hiden Analytical, HPR-20 QIC) following the m/z ratios 2 (H2), 15 and 16 (CH4), 18 (H2O), 28 (CO), 40 (Ar), and 44 (CO2).3. Results and discussion
3.1 Ex situ characterization of as-prepared samples
The specific surface areas and the X-ray diffraction patterns of the as-prepared powder catalyst samples are presented in Table 1 and Fig. 1, respectively. The surface area measurements reveal that all samples initially have high specific surface areas. The silica supported catalysts had the highest surface area (310–315 m2 g−1), followed by the alumina (180–187 m2 g−1) and the ceria (125–131 m2 g−1) supported catalysts. The surface area measurements of MCM-41 and ZSM-5 were cancelled due to the long measurement times, which indicates that the high surface areas are preserved after metal impregnation and calcination. Fig. 1 shows the XRD patterns for the as-prepared Pd-, Ni- and Rh-based catalysts. It can be observed that the pure metal oxide patterns are unaffected by calcination at 600 °C and impregnation with Pd, Ni and Rh, suggesting that the metals are well dispersed over the oxide supports. Additional peaks are observed for some of the samples, which were found to correspond to the diffraction patterns of PdO, NiO and Rh2O3, seen as coloured marks above the diffraction patterns (Fig. 1a), c) and d)). The XRD results indicate that the metal particle sizes are different for the metals supported on each oxide. The metals supported by cerium oxide nearly have diffraction peaks, which might indicate that Pd, Ni, and Rh are in amorphous phase or in extreme small crystalline size (below 3 nm), while the metals supported on silica show diffraction peaks for PdO, NiO and Rh2O3. In addition, the broadening of the diffraction peak gives insight into the size of the crystallites: sharp peaks indicate larger crystallites, while broader peaks indicate smaller crystallites. The Ni–silica sample has a very sharp XRD peak for NiO, suggesting a much larger particle size of Ni, compared to the Pd–silica or Rh–silica samples which show broader diffraction peaks. For the alumina supported samples, only Pd–alumina has some reflections originating form PdO, suggesting that this sample has larger particles compared to the other alumina supported samples investigated. For the ion-exchanged sample Ni/ZSM-5, no new reflections are observed compared to the pattern for ZSM-5 suggesting that Ni is highly dispersed in this sample.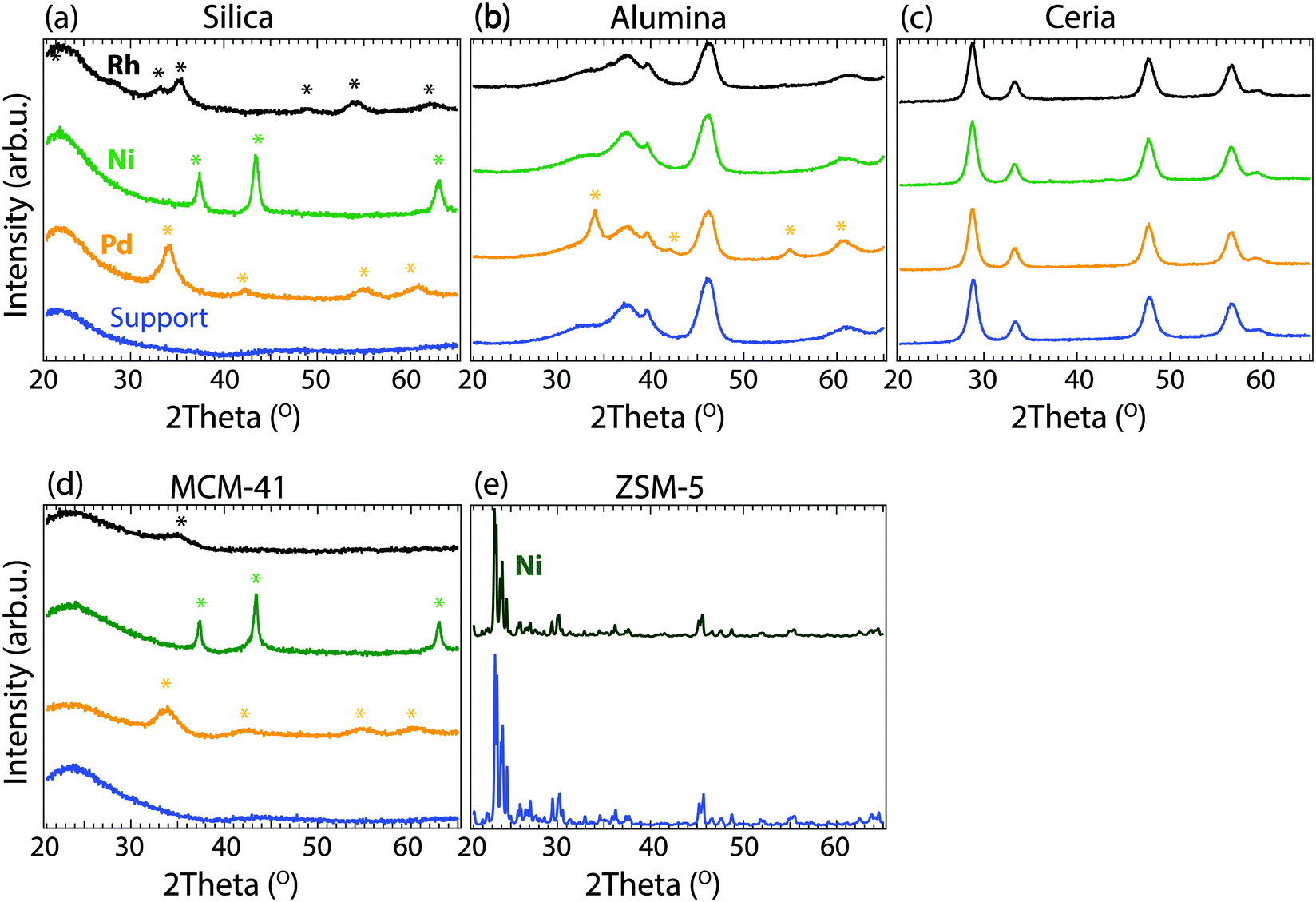 | ||
| Fig. 1 X-ray diffraction patterns of the support materials (blue) and as-prepared Pd (orange), Ni (green) and Rh (black) catalysts (3 wt%) supported on a) silica b) alumina c) ceria and d) MCM-41. e) Shows the XRD pattern for 1.5 wt% Ni (dark green) ion-exchanged into ZSM-5. | ||
3.2 Methanation reaction over Pd-, Ni-, and Rh-based catalysts
Fig. 2 shows the catalytic performance of the metal-promoted silica, alumina and ceria samples for CO2 hydrogenation. The inlet gas concentrations to the flow reactor are 1% CO2 and 5% H2 in Ar and each marker in the figure represents the measured average outlet concentration over a 10 min period. The main reaction products formed during CO2 hydrogenation are CH4, CO and H2O. No hydrocarbon compounds except methane are observed. Due to high background, the water signal is not included in Fig. 2.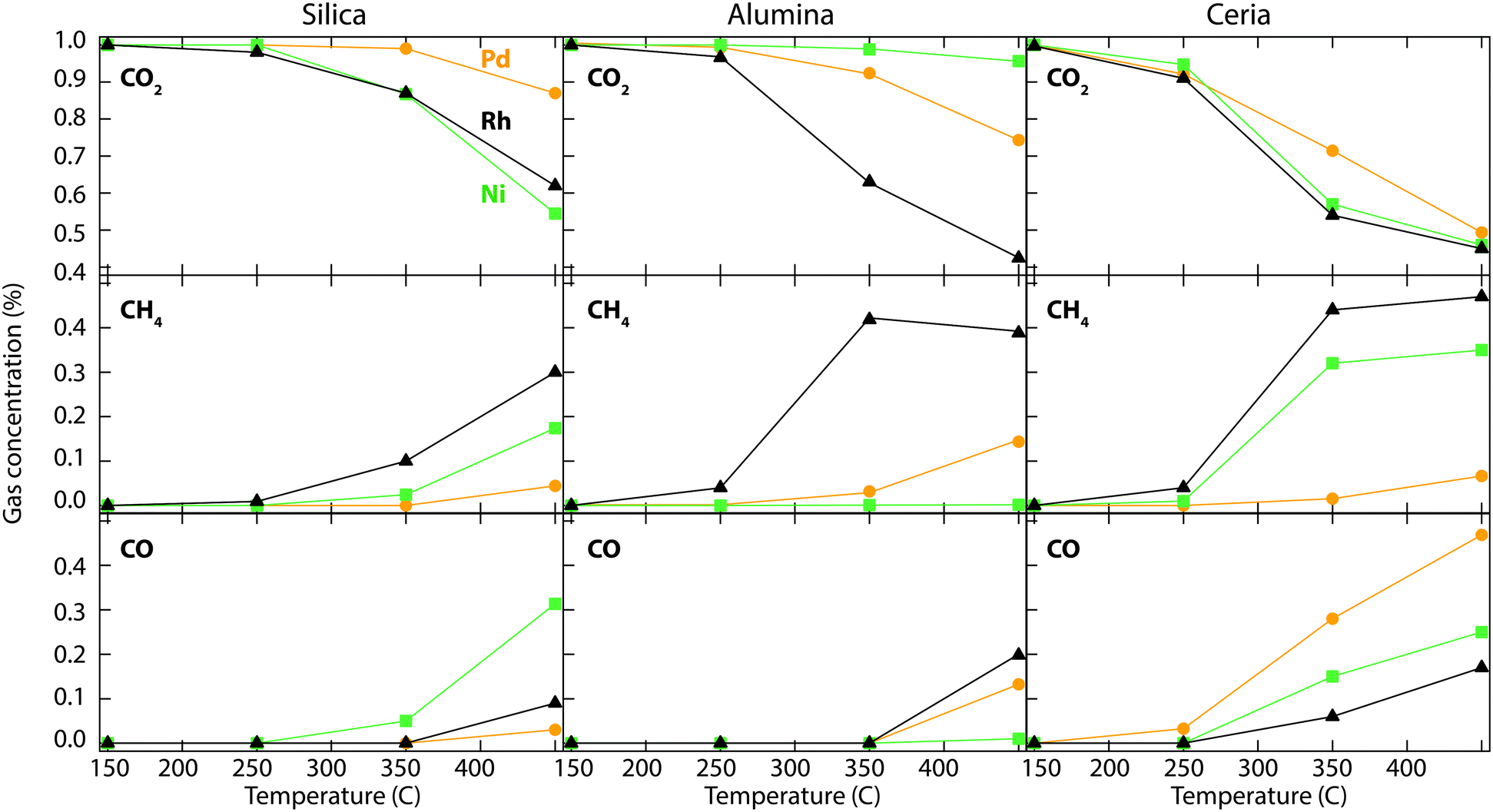 | ||
| Fig. 2 Steady-state outlet concentration of CO2 (top), CH4 (middle) and CO (bottom) as a function of temperature during the reaction between 1% CO2 and 5% H2 in Ar between 150 and 450 °C at a total flow rate of 2000 ml min−1 at atmospheric pressure over the silica, alumina and ceria supported Pd (orange), Ni (green) and Rh (black) catalysts. | ||
The temperature dependence of CO2 hydrogenation can clearly be seen for all samples. A clear trend is the increased formation of CO at high temperature, due to the reverse water gas shift reaction.16 The ceria supported catalysts show the highest CO2 conversion and CH4 production over the temperature range investigated, followed by the alumina supported catalysts. This can be attributed to metal particle size effects. As discussed above, the catalysts supported on ceria have no metal XRD peaks suggesting amorphous or very small metal particles, followed by the alumina supported catalysts. Among the studied catalysts, the Rh/Al2O3 catalyst shows the lowest CO production and highest methane selectivity, up to 350 °C. Over this temperature the CH4 production declines and the selectivity towards CO increases. The Ni/CeO2 sample shows the highest CO2 conversion compared to the other Ni catalysts investigated, in agreement with previous studies.13 The silica supported catalysts show the overall lowest methane formation also suggested by the larger metal particle sizes as obtained from the XRD measurements. Further, a significant difference in product selectivity between the active metals is observed.
Using MCM-41 as a support gives similar results as the silica supported catalysts described above, with a slightly higher selectivity towards CH4. However, for Ni/ZSM-5, almost no conversion of CO2 is observed. The results over the metal-promoted MCM-41 and ZSM-5 samples are shown in Fig. S1 (see ESI†). Due to the low catalytic activity for CO2 hydrogenation (at least below 400 °C), the results of silica, MCM-41 and ZSM-5 catalysts will not be further reported in this study.
The results of a more extended investigation of CO2 hydrogenation over the Rh/CeO2 catalyst are shown in Fig. 3. The experiment is carried out by measuring the steady-state outlet gas concentrations for inlet concentrations between 0.3 and 1.5% of CO2 (left panel), and between 1 and 5% H2 (right panel). Since no reaction was observed at 150 °C during the previous test, the investigation focused on measuring the activity at 250 (blue), 350 (orange) and 450 °C (grey). At 250 °C, the outlet concentration of CO2 is the same as the inlet concentration for both experiments, which means that no reaction occurs. At 350 and 450 °C the outlet concentrations of CO2 and the formation of methane are similar, whereas the formation of CO is considerably higher at the highest temperature. Above an inlet CO2 concentration of 1%, the increased CO formation gives rise to a slightly higher CO2 conversion. Initially, for low inlet concentrations of CO2, the left column shows a higher CH4 formation at 350 compared to 450 °C. The formation of CH4 then decreases slightly with increased inlet concentration of CO2 and becomes lower than at 450 °C for an inlet CO2 concentration of 1.5%. In the right column one can see a linear increase in CH4 formation with the same gradient at both 350 and 450 °C when the inlet H2 concentration is increased. At the same time, the formation of CO decreases linearly with a less steep slope. To obtain higher CO2 conversion and CH4 formation it is necessary to increase the H2:CO2 ratio.
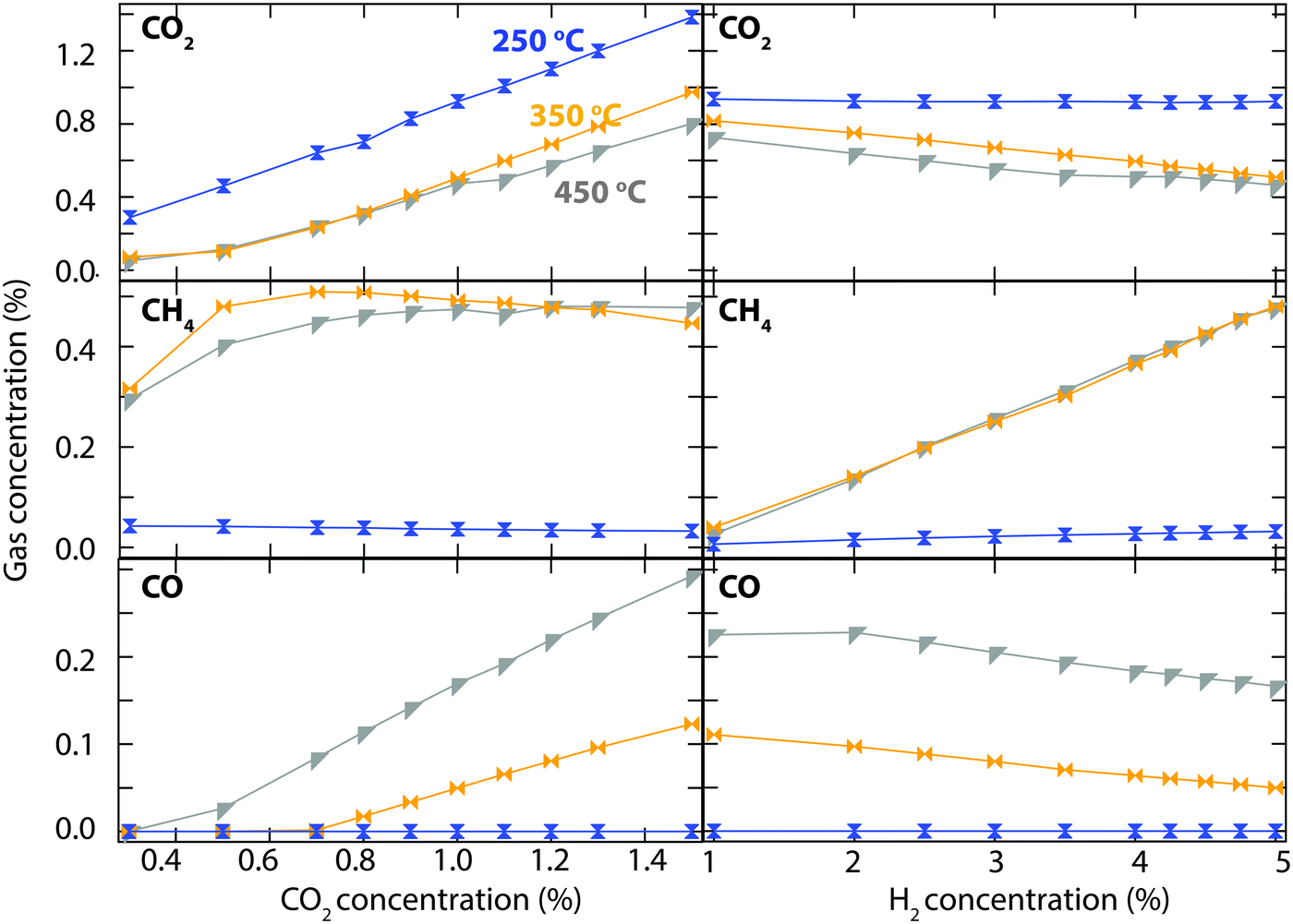 | ||
| Fig. 3 Steady-state outlet concentration of CO2 (top), CH4 (middle) and CO (bottom) over Rh/CeO2 as a function of inlet concentration of CO2 at constant H2 concentration of 5% (left column) and as a function of inlet concentration of H2 at constant CO2 concentration of 1% (right column) in Ar gas with a total flow rate of 2000 ml min−1 at atmospheric pressure. | ||
From all studied catalysts in this work, the ones supported on ceria are found to have the highest activity for CO2 hydrogenation, followed by the alumina supported catalysts. No considerable methanol production was observed over the studied catalysts under ambient pressure conditions and temperatures up to 450 °C. In addition, the results reveal that the catalysts preparation method and structure play an important role on the catalytic activity and/or selectivity. The catalysts prepared by impregnation show the highest CO2 conversion activity and the comparison between the different catalysts prepared by impregnation reveals that the catalysts with the smallest metal particles (i.e. the catalysts supported on ceria) show the highest activity and selectivity for CO2 methanation.
The most prominent result in this study is observed for the Rh/CeO2 catalyst that managed to convert 44% CO2 almost exclusively to CH4 at 350 °C. An even higher CO2 conversion is observed at 450 °C but, due to CO formation through the reverse water-gas shift reaction, the CH4 selectivity decreases. Further evaluation of the Rh/CeO2 catalyst revealed evidence of a maximum CH4 formation around 380 °C. A linear relationship between CH4 formation and H2:CO2 ratio was observed at both 350 and 450 °C (Fig. 3). The experiments for inlet CO2 concentrations below 0.5% confirm that a high H2:CO2 ratio leads to total conversion towards CH4 formation. Ceria-based materials have aroused increasing interest as supports or catalysts toward CO2 conversion due to the unique structural properties resulting from oxygen vacancies and reversible valence change (Ce4+ and Ce3+).17
3.3 Spectroscopic surface speciation during CO2 methanation
In situ FTIR spectroscopy in diffuse reflectance mode has been employed to study the surface interaction of CO2 and H2 for selected Rh- and Ni-based catalysts at temperatures between 250–400 °C, in order to provide insights to the CO2 methanation reaction mechanism. The DRIFTS results for the hydrogen pulse experiments under a continuous flow of CO2 for Rh/Al2O3 (left panels), Rh/CeO2 (middle panels) and Ni/CeO2 (right panels) at 350 °C are shown in Fig. 4. The top panels show the color coded intensities (blue corresponds to low intensity, red to high intensity) of the IR spectra in the interval 790–3800 cm−1versus time. The bottom panels show the time evolution of the integrated IR peak areas between 1200–1600 cm−1 corresponding to carbonates and formate species, and around 2020 cm−1 corresponding to carbonyls. By presenting the data in color maps the dynamics of the evolution of the IR adsorption bands during the different phases of the experiment is clearly displayed. A peak centred around 2350 cm−1, corresponding to gaseous carbon dioxide, is visible in all spectra.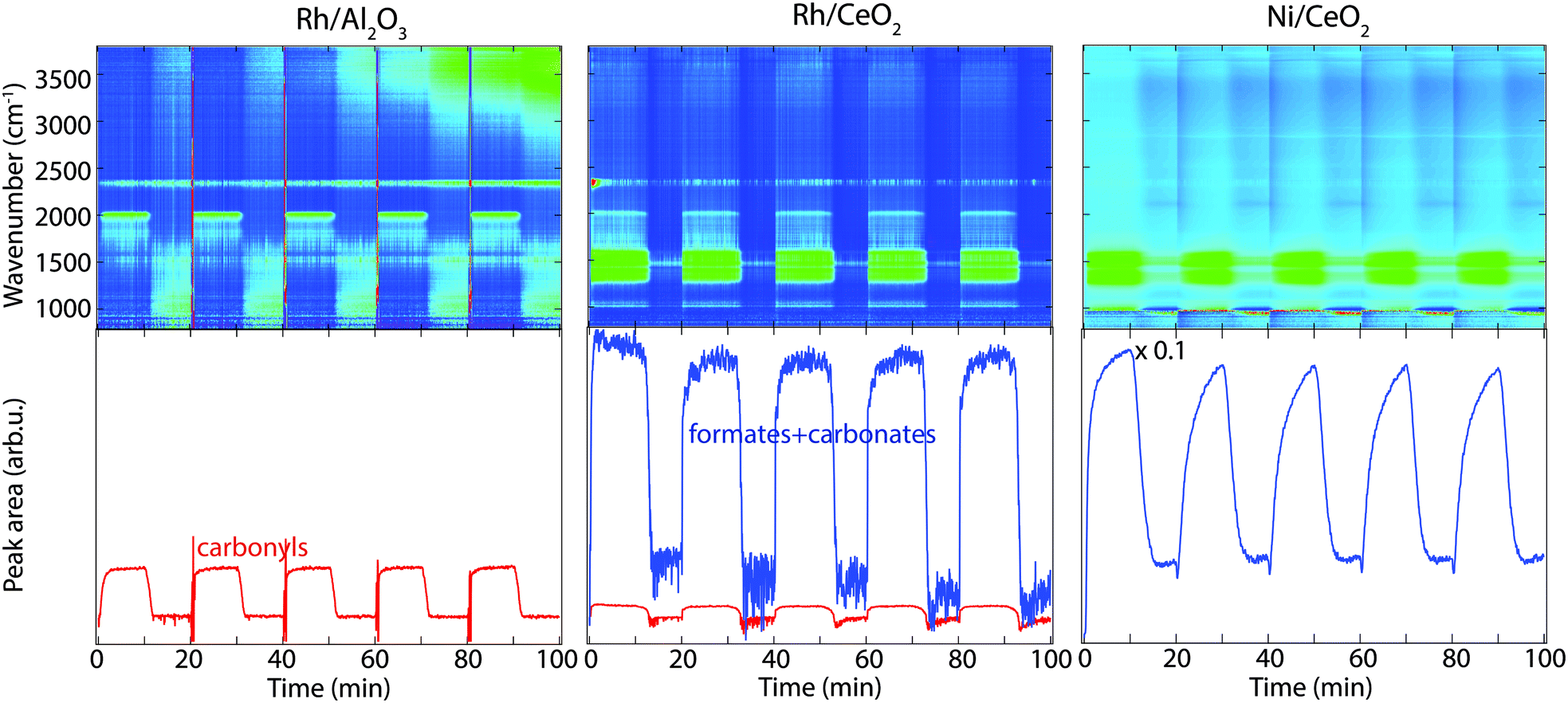 | ||
| Fig. 4 Transient hydrogenation of 0.2% CO2 over Rh/Al2O3 (left panels), Rh/CeO2 (middle panels) and Ni/CeO2 (right panels) during periodic variation of the feed gas composition between 0.8% H2 + 0.2% CO2 and 0.2% CO2 at 350 °C for 10 min. The top panels show the color coded intensities (blue corresponds to low intensity, red to high intensity) of the IR bands in the region 790–3800 cm−1versus time. The bottom panels show the IR peak areas between 1200 and 1600 cm−1 representing formate and carbonate species (blue lines) and around 2020 cm−1 representing carbonyls (red lines). | ||
The interaction of CO2 and H2 (in the feed) with the surface of the catalysts generates different surface species, which are assigned as illustrated in Table 2. For the Rh/Al2O3 sample, a strong peak around 2020 cm−1 corresponding to linearly adsorbed CO species on Rh (Rh0–CO, carbonyl),18,19 appears during CO2 hydrogenation, suggesting CO2 dissociation. The adsorbed CO species disappear about 2 min after the H2 flow has been turned off. In addition, some weak components at lower wavenumbers are observed and will be discussed below (see Fig. 5 description).
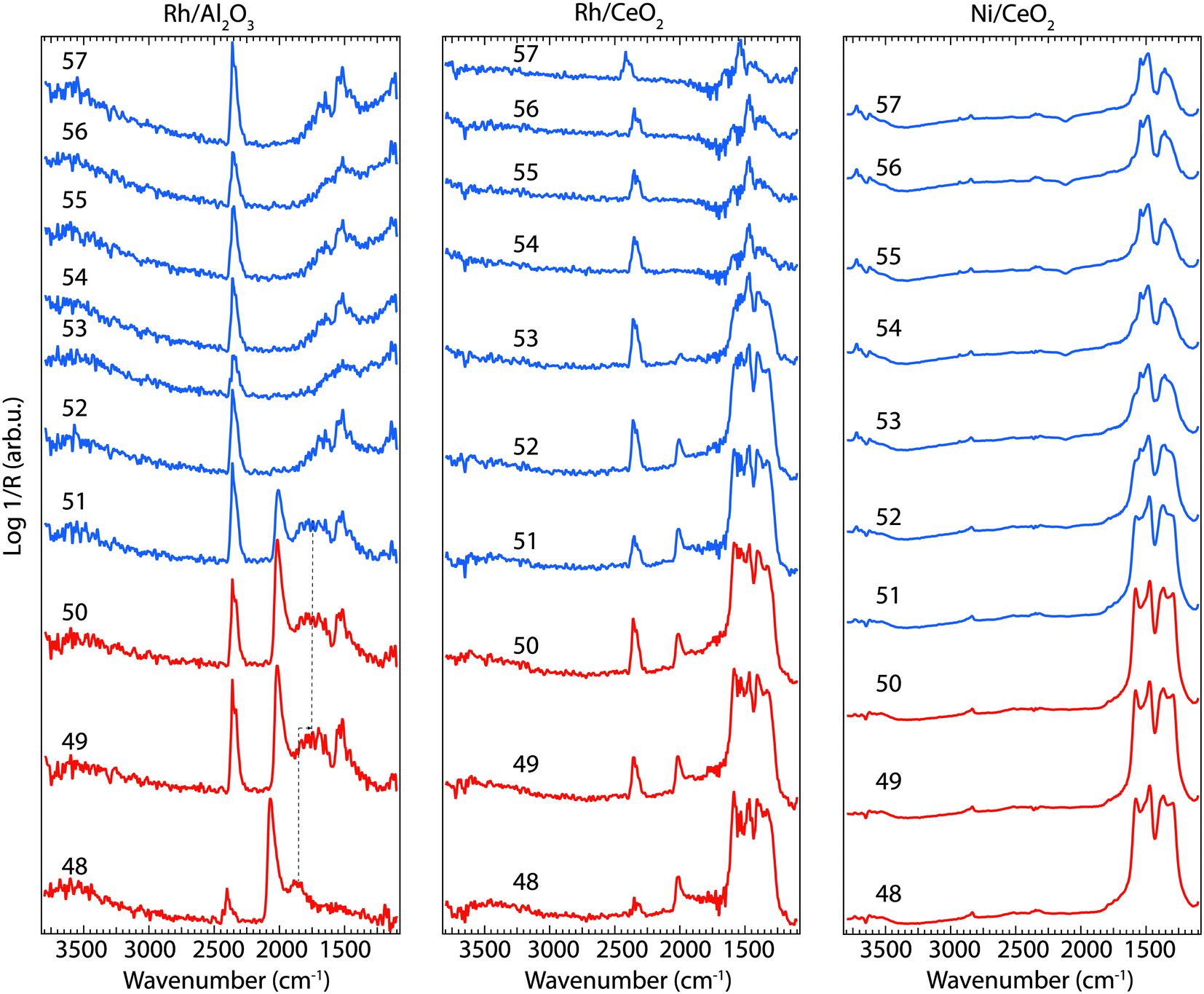 | ||
| Fig. 5 Evolution of IR absorption bands in the wavenumber region 1100–3800 cm−1 for the Rh/Al2O3 (left panel), Rh/CeO2 (middle panel) and Ni/CeO2 (right panel) catalysts exposed to CO2 and H2 while changing the feed gas composition from 0.8% H2 + 0.2% CO2 (48–50, red spectra) and 0.2% CO2 (51–57, blue spectra) at an inlet gas temperature of 350 °C. The insert numbers indicate the time (min) in the dynamic experiment where the spectra were recorded. | ||
The color map for the Rh/CeO2 sample in Fig. 4 (middle panels) shows a slightly different behaviour, formate and carbonate-like species18,23 form during the exposure to CO2 and H2 at 350 °C, which can be observed in the IR region 1200–1600 cm−1 (Fig. 4, middle panels). In addition, a smaller peak centred at 2020 cm−1 appears corresponding to CO species linearly adsorbed on Rh (carbonyl), forming at the same time with the formate and carbonate species. When the H2 flow is turned off the intensity of the formed surface species decreases. The bands plotted in the bottom panel (formates and carbonates/carbonyl) show a sharp increase at the switches from CO2 to CO2 + H2 flow followed by a sharp decrease about four minutes after the H2 flow is turned off.
The results for the Ni/CeO2 sample are similar as for the Rh/CeO2 sample with the strong peaks appearing between 1200–1600 cm−1 corresponding to formate and carbonate-like species (Fig. 4, right panels). No carbonyl species are observed for the Ni/CeO2 sample during CO2 hydrogenation. A slower formation rate of formates and carbonate species is observed for the Ni sample compared to the Rh sample, even though they are much stronger for the Ni/CeO2 sample. When the H2 flow is turned off, a continuous decline of the formate and carbonate band is observed for about five min. It is interesting to note the hysteresis behaviour for the formate and carbonate species observed for the CeO2 supported catalysts during the CO2 hydrogenation, which is likely due to the active ceria support.
In order to investigate possible effects of the support, the experiments were repeated for pure alumina and ceria samples at 350 °C and the results are shown in Fig. S2 (see ESI†). Some weak carbonate/formate species are observed to form on the ceria sample, while alumina shows no clear adsorption bands, suggesting that ceria can interact with CO2. Additionally, no clear changes are observed for the two samples during the CO2 + H2versus the CO2 periods.
To better understand the changes that the formed surface species undergo during the transient experiment, a number of IR spectra are shown in Fig. 5 when the flow of CO2 + H2 (48–50 min) is changed to only CO2 flow (51–57 min) for Rh/Al2O3 (left panel), Rh/CeO2 (middle panel), and Ni/CeO2 (right panel) at 350 °C. The spectra reveal noticeable changes of the position/intensity of the IR bands during the cycle. All significant changes observed in the spectra are contained in the region between 1200 and 2100 cm−1. The insert numbers indicate the time (min) in the dynamic experiment where the spectra were recorded. The major changes in the IR spectra take place after the switch from CO2 + H2 flow (48–50 min) to CO2 flow only (51–57 min).
For the Rh/Al2O3 sample (Fig. 5, left panel), a strong peak corresponding to linearly adsorbed CO species on Rh (IR band around 2020 cm−1), appears during CO2 hydrogenation in agreement to previous reports on CO2 methanation over Rh/Al2O3.11 A broad band around 1805 cm−1 is also detected, which previously has been attributed to bridge-bonded CO on Rh (Rh02–CO).19,20 This band broadens and seems to appear at lower wavenumbers by the end of the CO2 + H2 cycle (1720 cm−1, denoted by the dotted lines in Fig. 5) suggesting the formation of bridged bonded CO species positioned between Rh and Al atoms from the support, similar to previous reports on CO adsorption between Rh and Ce atoms.21 Some additional weaker peaks in the region 1700–1500 cm−1 appear towards the end of the CO2 + H2 cycle (49–50 min) corresponding to carbonate or formate-like species. Two minutes after the H2 flow is turned off, the band related to carbonyl species adsorbed on Rh disappears and the band around 1700 cm−1 shifts towards lower wavenumbers (1650 cm−1) indicating the formation of different surface carbonates. A band centred around 1520 cm−1 is clearly visible through the entire time the sample is exposed to CO2 (51–57 min).
For the Rh/CeO2 sample (Fig. 5, middle panel), strong peaks are observed in the IR region between 1590–1330 cm−1, together with a weaker absorption band around 2020 cm−1. The intensity of the formed surface species during CO2 hydrogenation (formate and carbonate, and carbonyl species) shows a very slow diminution directly after the H2 flow is turned off and only a broad peak centered around 1460 cm−1 remains when the sample is exposed to CO2 only, corresponding mostly to carbonate species.22
For the Ni/CeO2 sample (Fig. 5 right panel), during the transition between CO2 + H2 to only CO2 flow mostly the intensity of the formates and carbonates peaks in the region 1300–1600 cm−1 decreases. The spectra during the CO2 hydrogenation phase (48–50 min) resemble well the spectra recorded for the Rh/CeO2 sample without the formation of carbonyl species. There are differences between the two samples during the CO2 flow only (51–57 min). Stronger peaks are observed for the Ni/CeO2 sample during the entire experiment. In addition to the carbonate and formate species, new peaks appear around 2800 cm−1 and 3500–3700 cm−1, corresponding to C–H and O–H vibrations from CHx (ref. 23) and hydroxyl groups,24 respectively. The presence of CHx species on the Ni containing species suggest the formation of different hydrocarbonated compounds on the surface of the catalysts, in contrast to the Rh containing samples, where only methane in the gas phase is detected suggesting the direct desorption of CH4 as soon as it forms on the surface. The behaviour of the Ni/CeO2 sample during the CO2 flow is slightly different compared to the Rh/CeO2 and Rh/Al2O3 catalysts discussed above and is likely that more formate species form on this sample.
3.4 Reaction mechanism during CO2 methanation
Recently, it has been demonstrated that CO2 can react with H2 over Rh/γ-Al2O3 to produce methane at room temperature and atmospheric pressure.12 However, in the present study, we see that higher temperatures are needed for methane to be produced from CO2 and H2 over Rh/Al2O3. No significant methane production was observed below 250 °C (see Fig. 2).Different mechanisms have been proposed for CO2 methanation over a variety of noble metal-based catalysts. The first mechanism (eqn (4) below) involves the adsorption of CO2 on the support and its reaction with adsorbed H species formed on the metal which leads to a formate intermediate (COOH) at the metal–support interface. The formates can give rise to CO species adsorbed on the metal by the rWGS reaction, which subsequently are hydrogenated to methane.25 The second mechanism (eqn (5) below) involves the direct dissociation of CO2 to CO and O adsorbed on the metal surface, with adsorbed CO being subsequently hydrogenated to CH4.10,12,19,26
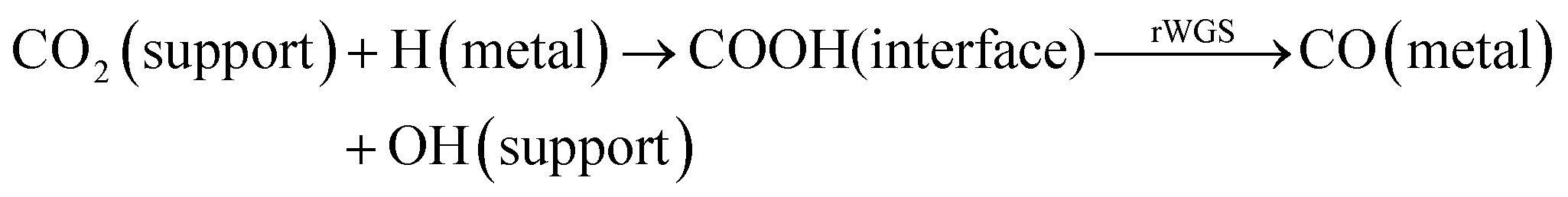 | (4) |
| CO2 → CO(metal) + O(metal) | (5) |
In contrast to several previous studies on the methanation mechanism, Upham et al.27 suggest another mechanism for CO2 methanation over Ru-doped ceria, where CH4 is not formed through intermediates of CO or a formate. IR spectroscopy measurements instead corresponds to a variety of surface carbonate spectra. This strengthened the idea of direct CO2 adsorption and formation of carbonates on ceria supported catalysts, without adsorption and dissociation on the metal phase. The formed carbonates react with hydrogen to produce methane that directly desorbs from the surface of the catalysts.
The DRIFTS results presented in this work give insights on the methanation reaction mechanism and differences are observed between the studied catalysts. In Fig. 4 and 5 the surface species formed when a mixture of CO2 + H2 followed by a flow of CO2 interacts with the surface of Rh/CeO2, Rh/Al2O3 and Ni/CeO2 are shown. As discussed above, for the Rh based catalysts, CO species adsorbed on metal are formed, suggesting that CO2 dissociates on these catalysts. Since no carbonyl species are formed under the flow of CO2 only, it is clear that CO2 dissociation is favored by the presence of hydrogen which is likely to be dissociated on the metallic Rh surface. In contrast, no adsorbed CO species are observed on the Ni catalyst, suggesting a different reaction mechanism over this sample. The ability of CO2 to dissociate over Rh/γ-Al2O3 has been evidenced by in situ DRIFTS experiments, which show the presence of bands that correspond to Rh–CO (carbonyls).12 Similar results have been observed over Rh/TiO2 catalysts.28
Due to the limited wavelength resolution we are unable to separate between the different formate and carbonate species in the IR region between 1200–1700 cm−1. However, the observed peak for C–H stretching at around 2800–2900 cm−1 for the Ni–ceria sample indicates that more formates are formed on this sample, while for the Rh containing samples the absence of this peak may suggest the predominance of carbonate species, which is also supported by the presence of a smaller peak around 840 cm−1 for these samples (IR region not shown), characteristic of carbonate species. We observe the formation of CO in the gas phase from the rWGS reaction over the Rh/CeO2 catalyst (Fig. 2). Additionally, the formation of formate and carbonate species is accompanied by the concomitant apparition of adsorbed CO species, suggesting the first reaction mechanism (through formate/carbonate species) over this sample. In contrast, no CO in the gas phase is observed for the Rh/Al2O3 catalyst up to 350 °C, suggesting the direct dissociation of CO2, in agreement with previous reports,10–12 where formates were proposed to be spectator species and to not contribute significantly to methane formation for the methanation reaction.11 No carbonyl bands are observed for the Ni/CeO2 catalyst, only formate and carbonate bands suggesting a formate/carbonate pathway reaction mechanism. The DRIFTS results suggest that the metal–support interface for the Rh/CeO2 and Ni/CeO2 catalysts may play an important role in the CO2 hydrogenation reaction, similar to the reports on Cu/CeOx/TiO2(111).29
4. Conclusions
Hydrogenation of CO2 at ambient pressure conditions has been studied over a series of supported Pd-, Rh- and Ni-based catalysts prepared by impregnation. High selectivity to methane and high CO2 conversion are obtained over the Rh-based catalysts. Our results show that it is possible to produce methane from CO2 and H2 at ambient pressure and relatively low temperature over Rh/CeO2, Rh/Al2O3 and NiCeO2 catalysts. Methane is the only hydrocarbon product observed in our analysis. The results of in situ DRIFTS experiments show that CO2 adsorption and dissociation occur over the Rh/Al2O3 catalyst in the presence of H2, resulting in the formation of linear Rh–CO species, which account for the majority of the adsorbed species formed during CO2 hydrogenation at 350 °C. In contrast the results show that the methanation reaction mechanism is different over the Rh/CeO2 catalyst where CO is formed through formate and carbonate intermediate species.The results, strengthened by previous studies indicate that a metal oxide like ceria can interact with CO2 and thus, be a part of and promote the hydrogenation reaction by direct formation of surface carbonates and formates.
Acknowledgements
This work was financially supported by the Swedish Research Council through the Röntgen–Ångström collaborations “Catalysis on the atomic scale” (No. 349-2011-6491) and “Time-resolved in situ methods for design of catalytic sites within sustainable chemistry” (No. 349-2013-567) and the Competence Centre for Catalysis, which is financially supported by Chalmers University of Technology, the Swedish Energy Agency and the member companies: AB Volvo, ECAPS AB, Haldor Topsøe A/S, Volvo Car Corporation, Scania CV AB, and Wärtsilä Finland Oy.References
- G. A. Olah, Catal. Lett., 2004, 93, 1–2 CrossRef CAS.
- I. Ganesh, Renewable Sustainable Energy Rev., 2014, 31, 221–257 CrossRef CAS.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- G. W. Busser, et al. , ACS Catal., 2015, 5(9), 5530–5539 CrossRef CAS.
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1(24), 3451–3458 CrossRef CAS.
- J. N. Park and E. W. McFarland, J. Catal., 2009, 266, 92–97 CrossRef CAS.
- G. C. Chinchen, P. J. Denny, J. R. Jennings, M. S. Spencer and K. C. Waugh, Appl. Catal., 1988, 36, 1–65 CrossRef CAS.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjær, J. S. Hummelshøj, S. Dahl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2014, 6, 320–324 CrossRef CAS PubMed.
- A. Beuls, C. Swalus, M. Jacquemin, G. Heyen, A. Karelovic and P. Ruiz, Appl. Catal., B, 2012, 113–114, 2–10 CrossRef CAS.
- A. Karelovic and P. Ruiz, Appl. Catal., B, 2012, 113–114, 237–249 CrossRef CAS.
- M. Jacquemin, A. Beuls and P. Ruiz, Catal. Today, 2010, 157, 462–466 CrossRef CAS.
- S. Tada, T. Shimizu, H. Kameyama, T. Haneda and R. Kikuchi, Int. J. Hydrogen Energy, 2012, 37, 5527–5531 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- P.-A. Carlsson, S. Mollner, K. Arnby and M. Skoglundh, Chem. Eng. Sci., 2004, 59, 4313–4323 CrossRef CAS.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- F. Wang, M. Wei, D. G. Evans and X. Duan, J. Mater. Chem. A, 2016, 4, 5773–5783 CAS.
- F. Solymosi, A. Erdohelyi and T. Bansagi, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 2645–2657 RSC.
- I. A. Fisher and A. T. Bell, J. Catal., 1996, 162, 54–65 CrossRef CAS.
- H. Y. Luo, H. W. Zhou, L. W. Lin, D. B. Liang, C. Li, D. Fu and Q. Xin, J. Catal., 1994, 145, 232–234 CrossRef CAS.
- A. Kiennemann, R. Breault, J.-P. Hindermann and M. Laurin, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 2119–2128 RSC.
- M. Marwood, R. Doepper and A. Renken, Appl. Catal., A, 1997, 151, 223–246 CrossRef CAS.
- R. J. Behm, S. Eckle and Y. Denkwitz, J. Catal., 2010, 269, 255–268 CrossRef.
- G. Busca, Phys. Chem. Chem. Phys., 1999, 1, 723–736 RSC.
- D. I. Kondarides, P. Panagiotopoulou and X. E. Verykios, J. Phys. Chem. C, 2011, 115, 1220–1230 Search PubMed.
- L. F. Liotta, G. A. Martin and G. Deganello, J. Catal., 1996, 164, 322–333 CrossRef CAS.
- D. C. Upham, A. R. Derk, S. Sharma, H. Metiu and E. W. McFarland, Catal. Sci. Technol., 2015, 5, 1783–1791 CAS.
- A. Karelovic and P. Ruiz, J. Catal., 2013, 301, 141–153 CrossRef CAS.
- J. Graciani, Science, 2014, 345, 546–550 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and S2. See DOI: 10.1039/c6cy02536f |
| This journal is © The Royal Society of Chemistry 2017 |