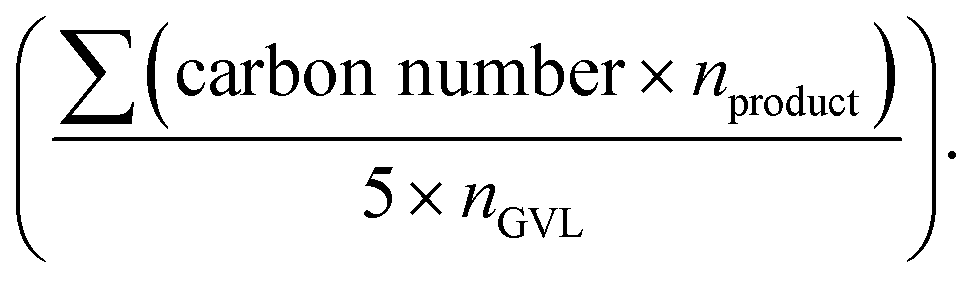Hydrodeoxygenation of gamma-valerolactone on transition metal phosphide catalysts†
Received
26th October 2016
, Accepted 30th November 2016
First published on 14th December 2016
Abstract
The catalytic hydrodeoxygenation (HDO) of the cyclic five-membered ester gamma-valerolactone (GVL-C5H8O2) on a series of supported metal phosphide catalysts and a commercial Pd/Al2O3 catalyst was studied at 0.5 MPa. Comparison of activities was based on turnover frequencies calculated from surface metal atoms determined from the chemisorption of CO at 50 °C. It was found that catalytic activity followed the order: Ni2P/MCM-41 ≫ CoP/MCM-41 ≫ Pd/Al2O3 ≈ MoP/MCM-41 > WP/MCM-41. On all catalysts, ring opening of the lactone to produce pentanoic acid was the main initial step with subsequent hydrogenation to form pentanal. On Ni2P/MCM-41, CoP/MCM-41 and Pd/Al2O3, this was followed mainly by decarbonylation to produce CO and saturated C4 hydrocarbons. On MoP/MCM-41 and WP/MCM-41, the principal subsequent step was deoxygenation to produce unsaturated C5 hydrocarbons. A possible reaction network is proposed based on product selectivity and selectivity versus conversion plots.
1. Introduction
Biomass has been recently studied as an alternative feedstock for chemicals and fuels to overcome environmental problems resulting from excessive use of fossil fuels.1,2 Several technologies such as hydrothermal liquefaction of woody biomass,3,4 pyrolysis upgrading of cedar chips and cellulose,5,6 and aqueous phase catalytic transformation of sugars and polyols7 have been developed for converting biomass into liquid fuels. Among them, fast pyrolysis gives high yields of liquids (pyrolysis bio-oils) of up to 75 wt% at moderate temperatures of around 500 °C and short hot vapor residence times of around 1 s.8 However, the bio-oil cannot be directly used as a fuel because it has a high oxygen content (35–40 wt%) with 15–30 wt% of water, which results in a low heating value, immiscibility with hydrocarbon fuels, high acidity, and chemical and thermal instability.9,10 Therefore, the oxygen in bio-oils needs to be removed to obtain a liquid fuel of required quality.
Catalytic hydrodeoxygenation (HDO) is one of the promising processes that aim at the production of usable liquid fuels by removal of oxygen in the presence of H2.11,12 Transition metal sulfide catalysts (CoMoS and NiMoS) have been widely tried as candidates due to their hydrogenation ability in commercial hydrodesulfurization (HDS) and hydrodenitrogenation (HDN) processes, which are closely related to HDO processes in the removal of a heteroatom with direct bonding to carbon. However, a major problem of the sulfides is that they are deactivated in the absence of sulfur, which is not normally present in bio-oils.13,14 Noble metals have also been tried as they are active in hydrogenation reactions under mild conditions.15,16 It has been found that Pd, Pt, Rh and Ru catalysts have high activity in the HDO of bio-oil model compounds such as dibenzofuran,17 phenol,18,19 guaiacol,16,20 and propanoic acid.21,22 However, their high cost and poor tolerance to sulfur and nitrogen are still problems.23,24
Recently, a number of studies of transition metal phosphide catalysts have reported that they are active for HDO processes25–27 as well as HDS and HDN.28,29 In a previous study, the HDO activity for 2-methyltetrahydrofuran (2-MTHF) was examined on a series of silica-supported transition metal phosphide catalysts and the activity order was found to be Ni2P > WP > MoP > CoP > FeP > Pd/Al2O3.30 Furthermore, the Ni2P phosphide catalyst showed higher activity for 2-MTHF HDO than Pd/Al2O3 at 300 °C and 1 atm. The catalytic performance of transition metal phosphide catalysts in the deoxygenation of methyl laurate was investigated, and the activity order followed the sequence: Ni2P > MoP > CoP–Co2P >WP > Fe2P–FeP; Ni2P > Ni12P5 > Ni3P. Various factors, including the surface metal site density, the electron density of metal sites and Brønsted acidity, give rise to the differences in catalytic activity.31
The compound GVL is found in bio-oils, and indeed, it is a so-called platform chemical because it is formed in large amounts in the degradation of cellulose and has potential use as a liquid fuel, an intermediate for fine chemicals, and a solvent.32,33 Thus, many approaches aiming to upgrade GVL to valuable chemicals have been reported. The production of C12 jet fuel from GVL was proposed in a dual reactor system involving the production of butene isomers via ring-opening and decarboxylation on SiO2/Al2O3 at 36 bar H2 with subsequent oligomerization of butene isomers on Amberlyst70. It was found that the yield of higher liquid alkenes reached 75%.34 Similarly, the conversion of GVL to 5-nonanone, a precursor to hydrocarbon fuels, was reported in a double-bed arrangement of Pd(5%)/Nb2O5 + ceria–zirconia in a single reactor. It was shown that Pd(5%)/Nb2O5 is responsible for ring opening and ceria–zirconia for further hydrogenation and subsequent ketonization.35 Also the formation of valerate esters from GVL was deemed to be promising since their energy density, polarity and volatility are compatible with either gasoline or diesel.36 The transformation required a bifunctional catalyst and it was found that 64% ethyl valerate would be formed on Co/HZSM-5.37
In this work, a series of transition metal phosphide catalysts (Ni2P, CoP, MoP, and WP) supported on MCM-41 is synthesized and characterized to investigate their activity in the HDO of GVL. This is the first time such a series has been prepared using the high surface area MCM-41 support, and the resulting materials are highly dispersed. The performance, including catalytic activity and product selectivity, is compared at various temperatures with that of a commercial Pd/Al2O3 catalyst, which has shown superior activity in HDO. Furthermore, based on the results of product selectivity, a possible reaction pathway for the HDO of GVL is proposed.
2. Experimental
2.1. Preparation of supported transition metal phosphide catalysts
A commercial MCM-41 support (Aldrich) was used as received. The supported catalysts were prepared by incipient wetness impregnation of aqueous metal phosphite precursors, followed by temperature-programmed reduction (TPR) under a hydrogen flow. A mixed solution was made by adding a certain amount of a desired metal salt into a phosphorus-containing solution.38,39 The amount of metal was 1.0 mmol gsupport−1 and the initial metal/P ratio was fixed at 1/2. The quantities of materials are summarized in Table 1. The mixed solution was used to impregnate the MCM-41 support by the incipient wetness technique. The obtained phosphite precursor was dried in air at 120 °C overnight, and then pelletized and sieved to a size of 650–1180 μm. The resulting precursor was reduced to the corresponding phosphides by TPR from room temperature to the reduction temperature at 3 °C min−1 in a quartz reactor under a hydrogen flow of 1000 cm3 min−1 gcatalyst−1. After reduction, the sample was cooled to room temperature under a helium flow and then was passivated under a 0.2% O2/He flow (100 cm3 min−1) for 4 h to prevent uncontrolled oxidation. For comparison, a 5 wt% Pd/Al2O3 commercial catalyst supplied by BASF Catalysts, Inc. was used.
Table 1 Quantities of materials used in catalyst preparation
| Sample |
Metal source/mmol |
Metal phosphide/wt% |
|
Metal loading level: 1.0 mmolmetal gsupport−1. MCM-41: 5 g. H3PO3: 0.828 g (10 mmol).
|
| POx/MCM-41 |
— |
— |
| Ni2P/MCM-41 |
Ni(OH)2 |
5.0 |
6.9 |
| CoP/MCM-41 |
Co(OH)2 |
5.0 |
8.2 |
| MoP/MCM-41 |
(NH4)6Mo7O24·4H2O |
0.7 |
11 |
| WP/MCM-41 |
(NH4)6W12O3·9H2O |
0.4 |
18 |
2.2. Characterization of prepared catalyst samples
The temperature-programmed reduction (TPR) method was used to determine the reduction characteristics of the material. First, 0.1 g of sample was loaded in a quartz reactor and then pretreated at 120 °C under a helium flow. After that, it was heated from 50 to 800 °C under a H2 flow of 100 cm3 min−1 while the effluent was monitored by a quadrupole mass spectrometer. Chemisorption uptakes of CO were measured on a sample re-reduced in a H2 flow at 550 °C for 3 h. Pulses of CO were passed at 50 °C over the sample to measure the total dynamic gas uptake. A BELSORP miniII micropore size analyzer was used to measure the specific surface area of the samples using N2 adsorption at 77 K from the linear portion of BET plots (P/P0 = 0.01–0.20). Before the measurements, the samples were degassed at 120 °C overnight to remove the adsorbed species from the sample. X-Ray diffraction (XRD) patterns of fresh and spent samples were measured on a diffractometer (Rigaku RINT 2400) operated at 40 kV and 100 mA, using a Cu-Kα monochromatic X-ray source. Data were collected over a Bragg angle (2Θ) range of 20–80° with a step size of 0.02°. X-ray absorption fine-structure (XAFS) spectra at the Ni K-edge (8.333 keV) were recorded at beam line 9C (BL9C) of the Photon Factory in the Institute of Materials Structure Science, High-Energy Accelerator Research Organization (KEK-IMSS-PF). The X-ray ring was operated at 2.5 GeV with a beam current of 450 mA. The XAFS spectra were taken in transmission mode using ionization chambers for the detection of the incident X-ray beam (I0, 100% N2) and transmitted beam (IT, 25% Ar in N2). The passivated disk sample with a mass of 40 mg was placed in the center of an in situ cell equipped with Kapton windows, and reduced at 550 °C for 2 h under a H2 flow in the same manner as for the activity tests. The EXAFS data were analyzed by Winxas3.1. Phase shift and amplitude functions of Ni2P were calculated by FEFF8. Curve fitting was carried out using the three dominant shells (2 Ni–P at 0.226 nm, 4 Ni–P at 0.2457 nm, 4 Ni–Ni at 0.2678 nm) and the reducing factor (So2) was fixed at 0.9, which was obtained from the Ni–Ni contribution of Ni foil.
2.3. Activity test for HDO of GVL
The catalytic tests of HDO for GVL were carried out in a continuous-flow reactor operated at 0.5 MPa and a temperature range of 250–350 °C. Quantities of catalyst corresponding to 10 μmol of sites as titrated by CO chemisorption were used. The catalyst was loaded in a section about 2 cm long in the middle of the reactor with quartz sand of the same particle size to disperse the catalyst. Before injecting the liquid feed, the phosphide catalyst was pretreated under a H2 flow of 100 cm3 min−1 gcatalyst−1 at 550 °C for 4 h. The reactant was introduced into the reactor with a liquid pump. A mixture of 98 wt% of GVL and 2 wt% of toluene as an internal standard was vaporized at 300 °C and mixed with a H2 gas stream to give a reactant stream of 2 mol% GVL in H2. The total pressure was fixed at 0.5 MPa using a back-pressure regulator. For stabilization, the catalysts were first maintained at 350 °C and 0.5 MPa for 12 h after introducing the reactant and then the temperature was varied downward and upward in the order: 350, 300, 250, 275, and 325 °C with each temperature maintained for 2 or 3 h. An on-line gas chromatograph (Shimadzu GC-14A, DB-624 UI, 60 m × 0.25 mm × 1.40 μm) equipped with a flame ionization detector (FID) and a thermal conductivity detector (TCD) was used for analyzing the products at 1 h intervals. In order to maintain the product in the gas phase, all of the lines were heated to 250 °C with ribbon heaters. Qualitative analysis of unknown products was carried out by gas chromatography–mass spectrometry (Shimadzu GCMS-QP2010 Ultra) by injecting the gas products which were collected from the outlet gas stream. A similar procedure was applied for the stability test of Ni2P/MCM-41. After the reaction, all the catalysts were treated under a N2 flow for 2 h and then collected for characterization by XRD, CO chemisorption and N2 physisorption.
The conversion of GVL, deoxygenation degree, product distribution and selectivity were calculated by using the following equations:
where
nGVL,f and
nGVL,e describe the moles of GVL in the feed and exit, respectively, and
ni is the moles of product i. The deoxygenation degree shows how much GVL is converted into hydrocarbons such as butane,
n-butenes, pentane,
n-pentenes, and
n-pentadienes.
3. Results and discussion
3.1. Characterization of the prepared catalysts
Table 2 shows the reduction temperatures, surface areas and CO-uptakes of the fresh and spent catalysts. The reduction temperature will be discussed in the following paragraph. The surface areas of the fresh catalysts ranged from 500 to 660 m2 g−1. After loading the metal, all the samples presented a decrease of surface area due to a reduction of pore volume, which is due to the presence of metal phosphide and excess phosphorus on the support. The spent catalysts exhibited a similar surface area to the fresh catalysts. The CO uptakes for fresh catalysts were between 30 and 74 μmol g−1 and for the spent catalysts, the CO uptakes did not change significantly. These results indicated that the structure of the catalysts was maintained during the reaction. The amounts of catalyst loaded in the reactor for catalytic testing corresponded to 10 μmol of sites, counted by CO adsorption (Table 2).
Table 2 Characterization of fresh and spent catalysts
| Sample |
Condition |
CO uptakeb/μmol g−1 |
Reduction temperature/°C |
BET surface area/m2 g−1 |
|
After activity test at 250–350 °C and 0.5 MPa for 24 h.
After in situ reduction at 550 °C for 2 h.
|
| MCM-41 |
As received |
— |
— |
997 |
| Ni2P/MCM-41 |
Fresh |
70 |
570 |
663 |
| Spenta |
67 |
— |
651 |
| CoP/MCM-41 |
Fresh |
59 |
705 |
582 |
| Spenta |
55 |
— |
578 |
| MoP/MCM-41 |
Fresh |
74 |
597 |
539 |
| Spenta |
71 |
— |
521 |
| WP/MCM-41 |
Fresh |
30 |
610 |
509 |
| Spenta |
27 |
— |
499 |
| Pd/Al2O3 |
As received |
80 |
— |
82 |
Fig. 1 shows the TPR profiles of the precursors of the Ni2P/MCM-41, CoP/MCM-41, MoP/MCM-41, and WP/MCM-41 catalysts. The H2O signal (m/z = 18) shown in Fig. 1(a) displays broad features between 150–210 °C due to desorption of strongly held water and dehydroxylation, and distinct features above 400 °C for reduction of the phosphite.30 The maximum peak temperature was chosen as the reduction temperature for large scale syntheses. The Ni2P catalyst precursor gave two main peaks at 400 and 570 °C to different stages in the reduction of Ni phosphite to Ni phosphide. In a previous study,40 it was suggested that the peak at lower temperatures (375–450 °C) is related to the formation of the Ni2P phase by the reaction between PH3 and metallic nickel, and the higher temperature peak (>400 °C) is related to the reduction of HPO3H− and nickel ions that interact strongly with the support. The CoP precursor showed peaks at 450 °C and 705 °C also corresponding to different steps in the reduction. It was reported that the formation of CoP occurred sequentially, where one of the intermediates was Co2P which reacts with PH3 to subsequently form CoP.39 The MoP catalyst presented a broad unresolved reduction peak at around 597 °C while the WP precursor showed a small peak at 530 °C and a major peak at 610 °C indicating that the stages for these group 6 metals occurred close to each other. The PH3 signal shown in Fig. 1(b) shows similar features to the H2O signal, indicating that both H2O and PH3 were formed simultaneously in the same reduction process. The PH3 signal was broader and occurred at a higher temperature for the Mo and W samples, indicating that metal reduction occurred before phosphite reduction. After reduction, phosphine kept being produced because of the use of excess phosphorus in the preparation method. All the trends are consistent with previous results, but in all cases the reduction temperatures are slightly higher than in the previous study with a silica support.30 This is because the present study uses a high surface area support, MCM-41, which enhances the dispersion of the metal precursors and leads to stronger interactions with the support so that the reduction temperatures are increased.41,42
 |
| | Fig. 1 TPR profiles for the precursors of phosphide catalysts: (a) mass signal (m/z) = 18 a.u. and (b) mass signal (m/z) = 34 a.u. | |
Fig. 2 shows the powder XRD patterns for the fresh and spent catalysts. The broad line centered at 2θ = 23° is typical for amorphous silica, which is observed in all catalysts. The Ni2P/MCM-41 shows broad peaks at 2θ = 40.7°, 44.6°, 47.4°, and 54.2° (PDF#74-1385) due to highly dispersed Ni2P particles on the MCM-41. The CoP/MCM-41 shows three main peaks at 2θ = 31.6°, 46.2°, and 48.1° (PDF#29-0497), corresponding to the characteristic XRD peaks of the CoP reference. The MoP presents a broad peak at 2θ = 43.1° (PDF#24-0771), indicating low crystallinity and small particle size. The WP/MCM-41 displays strong sharp peaks at 2θ = 31.1°, 43.2°, 44.6°, and 46.5° (PDF#29-1364), which are well matched with the WP reference. After the reaction, the XRD patterns of all the samples show a partial decrease of intensity, but the peaks of the phosphide phases are still visible indicating that the catalysts are stable under these reaction conditions. The crystallite size of Ni2P is below the detection limit for XRD (roughly 3 nm), so EXAFS measurements were adopted to confirm the presence of the Ni2P phase under reduction conditions (H2 flow and 550 °C). Fig. 3(a) and (b) show the Ni K-edge EXAFS spectra for the reduced Ni2P/MCM-41 sample. Although the experimental curve is not well resolved, there are two main peaks in the Fourier transforms located at 0.180 nm and 0.228 nm, corresponding to Ni–P and Ni–Ni, respectively.43 A three-shell curve-fitting analysis of the Fourier transform spectra was conducted as shown in Table 3.44 The distances for the Ni–P(I), Ni–P(II), and Ni–Ni bonds were 0.2207, 0.2344, and 0.2551 nm, respectively, and the corresponding coordination numbers were 1.8, 2.8, and 2.0, respectively. These values were similar to previous results which showed that smaller Ni2P crystallites have larger coordination numbers in Ni–P(II) and lower coordination numbers in Ni–Ni.27 This indicates that the Ni2P phase is present on the MCM-41 support.
 |
| | Fig. 2 XRD profiles for fresh and spent catalysts. | |
 |
| | Fig. 3 Ni K-edge EXAFS spectra (a) and Fourier transform graphs (b) of Ni2P/MCM-41. | |
Table 3 Curve-fitting results for Ni2P/MCM-41
|
|
Ni–P(I) |
Ni–P(II) |
Ni–Ni |
R-Factor/% |
| CN |
2 |
4 |
4 |
|
|
R/nm |
0.227 |
0.246 |
0.268 |
|
| Ni2P/MCM-41 |
|
|
|
1.4 |
| CN |
1.822 |
2.844 |
2.048 |
|
R/nm |
0.221 |
0.234 |
0.255 |
|
σ
2/10−5 nm2 |
0.975 |
2.334 |
1.351 |
| ΔE/eV |
−8.119 |
5.486 |
−1.669 |
3.2. Catalyst stability test
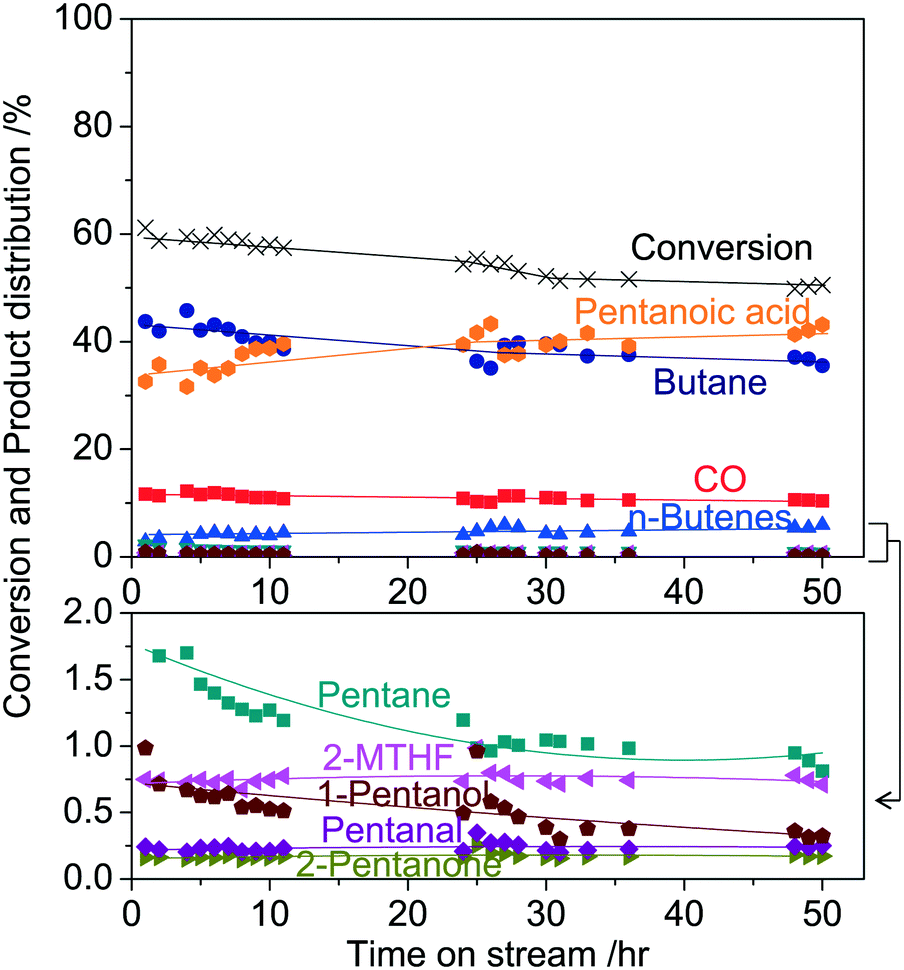
Fig. 4 shows the GVL conversion and product distributions over Ni2P/MCM-41 as a function of time on stream. A stability test was conducted at 300 °C and 0.5 MPa with 4% GVL in H2 for 50 h on stream. The main products were pentanoic acid (39%), 2-MTHF (0.7%), 1-pentanol (0.4%), pentanal (0.2%), 2-pentanone (0.2%), pentane (1%), butane (40%), n-butene (5%), and CO (10%). The conversion slightly decreased from 61% to 55% and then stabilized after 20 h, while the product distributions were not significantly changed. This result indicated that the Ni2P/MCM-41 catalyst was stable under the applied reaction conditions.
 |
| | Fig. 4 GVL conversion and product distributions over Ni2P/MCM-41 as a function of time on stream at 300 °C and 0.5 MPa with 4% GVL in H2. | |
3.3. Reactivity as a function of temperature over transition metal phosphide catalysts
Fig. 5 shows the GVL conversion, deoxygenation degree and turnover frequency (TOF) at 0.5 MPa as a function of temperature on Ni2P/MCM-41, CoP/MCM-41, MoP/MCM-41, WP/MCM-41, Pd/Al2O3, POx/MCM-41 and MCM-41. The TOF tracks the conversion because the amount of catalysts corresponding to equal quantities of sites was used in the measurements. The GVL conversion gives information about the amount of GVL that is converted into products, including both hydrocarbons and oxygen-containing compounds, while the deoxygenation degree (Deox) indicates how much GVL is transformed into hydrocarbons. Comparison of GVL conversion and deoxygenation degree clarifies the type of reaction that occurs. In all cases, the GVL conversion was higher than the deoxygenation degree because some products were oxygenates. In addition to the catalyst activity tests, blank tests were conducted with the support and phosphorus-loaded support. In the catalyst preparation, excess phosphorus was used so that some phosphorus likely was retained on the support, even though some was volatilized. Amounts of 1.0 g each of POx/MCM-41 and MCM-41 was evaluated at 0.5 MPa at various temperatures (250–350 °C). The MCM-41 exhibited a low activity for both GVL conversion (0.15%) and deoxygenation degree (0.1%) even at 350 °C. The POx/MCM-41 displayed a higher GVL conversion (6.9%) and deoxygenation degree (1.6%) at 350 °C, but these were still low. Therefore, the effects of support and phosphorus were negligible in the removal of oxygen under the conditions of study. 0.14 g of Ni2P/MCM-41, 0.17 g of CoP/MCM-41, 0.14 g of MoP/MCM-41, 0.33 g of WP/MCM-41 and 0.13 g of Pd/Al2O3 catalysts corresponding to 10 μmol of CO uptake sites were loaded and tested at a constant reactant flow rate of 1.6 μmol s−1. Thus the temperature variation results are considered to give the intrinsic catalytic activity of the catalysts. All samples had higher conversion and deoxygenation degree at higher temperatures as expected. At all temperatures, the GVL conversion and deoxygenation degree followed the same order: Ni2P/MCM-41 ≫ CoP/MCM-41 ≫ Pd/Al2O3 ≈ MoP/MCM-41 > WP/MCM-41, indicating that the catalytic activity of the phosphide catalysts was higher or close to that of Pd/Al2O3. In particular, Ni2P/MCM-41 presented remarkable activity attaining full conversion and deoxygenation degree at 350 °C, while MoP/MCM-41 and WP/MCM-41 were considerably less active, especially in the removal of oxygen. Interestingly, it was found that the iron group phosphides (Ni2P and CoP) were more active than the group 6 metal phosphides (MoP and WP). This might be related to the nature of the metal in phosphide catalysts, especially its electron density. In a previous work on deoxygenation of methyl laurate over transition metal phosphides, it was suggested that Ni, Co and Fe sites have higher electron density than Mo and W sites and so can favorably protonate O compounds, which leads to enhanced conversion.31 The right scale of Fig. 5 shows the turnover frequency (TOF) for the GVL reaction and the deoxygenation degree as a function of temperature on the transition metal phosphide catalysts and the commercial Pd/Al2O3 catalyst. The TOF value was calculated by the following equation:
 |
| | Fig. 5 GVL conversion (a), deoxygenation degree (b) and turnover frequency (right scale) as a function of temperature at 0.5 MPa. | |
With equal number of sites, the TOF is proportioned to conversion so the same curve applies with a different scale. The Ni2P/MCM-41 showed the highest activity with a TOF of 0.17 s−1 at 300 °C. In Ni2P/MCM-41, the conversion and TOF curves maintained in the temperature range of 325–350 °C. This was due to mass transfer limitations in the high conversion region (>70%). The Weisz–Prater criterion (CWP) is a method used to determine if internal diffusion is limiting the reaction. This value was calculated by the following equation:45,46
Internal mass transfer effects can be neglected if the value of CWP is less than 0.3.47 The parameters used for the criterion are summarized in Table 4. The observed rates  and the calculated CWP values are listed in Table 5. The values of CWP are less than 0.3 except for the Ni2P/MCM-41 sample at over 300 °C and the CoP/MCM-41 sample at 350 °C, which are exactly as expected from the shapes of the conversion curves, with mass transfer becoming important past the inflexion points in the curves. For the other samples, there are no diffusion limitations under all the reaction conditions. Also the Mears criterion for external diffusion was below 0.15 (see the ESI†)
and the calculated CWP values are listed in Table 5. The values of CWP are less than 0.3 except for the Ni2P/MCM-41 sample at over 300 °C and the CoP/MCM-41 sample at 350 °C, which are exactly as expected from the shapes of the conversion curves, with mass transfer becoming important past the inflexion points in the curves. For the other samples, there are no diffusion limitations under all the reaction conditions. Also the Mears criterion for external diffusion was below 0.15 (see the ESI†)  < 0.15, indicating that mass transfer from the bulk gas phase to the catalyst surface can be neglected.45,48
< 0.15, indicating that mass transfer from the bulk gas phase to the catalyst surface can be neglected.45,48
Table 4 Parameters in the Weisz–Prater criterion
|
R
|
Catalyst particle radius/cm |
0.09 |
|
ρ
c (Ni2P, CoP, Pd) |
Solid catalyst density/g cm−3 |
0.3 |
|
ρ
c (MoP and WP) |
Solid catalyst density/g cm−3 |
0.4 |
|
C
As (300 °C) |
Gas concentration at the catalyst surface/mol cm−3 |
4.11 × 10−7 |
|
D
e
|
Effective diffusivity/cm2 s−1 |
0.1 |
Table 5 Observed reaction rates and calculated Weisz–Prater criterion
|
|
Ni2P |
CoP |
MoP |
WP |
Pd |
| Temperature/°C |
Observed reaction rate/mol g−1 s−1 |
| 250 |
2.06 × 10−6 |
3.98 × 10−7 |
3.43 × 10−7 |
3.15 × 10−8 |
4.72 × 10−7 |
| 275 |
4.58 × 10−6 |
9.13 × 10−7 |
4.60 × 10−7 |
7.02 × 10−8 |
7.04 × 10−7 |
| 300 |
8.80 × 10−6 |
2.08 × 10−6 |
8.40 × 10−6 |
1.53 × 10−7 |
1.14 × 10−6 |
| 325 |
1.12 × 10−5 |
3.92 × 10−6 |
1.44 × 10−6 |
3.00 × 10−7 |
1.99 × 10−6 |
| 350 |
1.14 × 10−5 |
6.73 × 10−6 |
2.58 × 10−6 |
5.64 × 10−7 |
3.07 × 10−6 |
| Temperature/°C |
Weisz–Prater criterion |
| 250 |
0.12 |
0.02 |
0.03 |
0.00 |
0.03 |
| 275 |
0.27 |
0.05 |
0.04 |
0.01 |
0.04 |
| 300 |
0.52 |
0.12 |
0.07 |
0.01 |
0.07 |
| 325 |
0.66 |
0.23 |
0.11 |
0.02 |
0.12 |
| 350 |
0.68 |
0.40 |
0.20 |
0.04 |
0.18 |
The GVL conversion, deoxygenation degree, product distribution and carbon balance at 250 and 350 °C are summarized in Table 6. The products were classified into five groups: C4 (n-butenes and butane), C5 (n-pentadienes, n-pentenes, and pentane), C5–O (2-methyltetrahydrofuran, pentanal, 1-pentanol, and 2-pentanone), C5–O2 (pentanoic acid and n-pentenoic acids), and finally CO. At 250 °C, the GVL conversion was below 5% except for Ni2P/MCM-41 and followed the order Ni2P/MCM-41 (18%) > CoP/MCM-41 (4.1%) > Pd/Al2O3 (3.6%) > MoP/MCM-41 (1.2%) > WP/MCM-41 (0.6%). This order was the same as that of the deoxygenation degree, although deoxygenation was 0% for MoP/MCM-41 and WP/MCM-41 because no hydrocarbons were produced. At 250 °C, the main product was C5–O2 on all catalysts. At 350 °C, on the other hand, a different product distribution was observed. Among the hydrocarbons, the main product was C4 hydrocarbons on Ni2P/MCM-41, CoP/MCM-41 and Pd/Al2O3 and C5 hydrocarbons on MoP/MCM-41 and WP/MCM-41, indicating that cleavage of C–C bonds was favored on iron group phosphides and Pd/Al2O3. On Pd/Al2O3, a small amount of propane was produced by a cracking reaction, but its selectivity was below 0.5%, so it was excluded from the calculation of the product distribution. The carbon balance was within the error range (100 ± 5%) on all catalysts at 250 °C and 350 °C, indicating that carbon deposition on the applied catalysts or polymerization didn't occur significantly.
Table 6 GVL conversion, deoxygenation degree, product distribution and carbon balance at 250 °C and 350 °C at 0.5 MPa
| Temp./°C |
Sample |
Conv./% |
Deox/% |
TOF/s−1 |
Product distribution/% |
CBa/% |
| CO |
C4 |
C5 |
C5O |
C5–O2 |
Carbon balance (CB) is calculated by 
|
| 250 |
Ni2P/MCM-41 |
18.0 |
4.1 |
0.030 |
3.7 |
17.5 |
1.0 |
3.9 |
73.3 |
103 |
|
|
CoP/MCM-41 |
4.1 |
0.2 |
0.007 |
0.9 |
4.3 |
0.5 |
8.3 |
85.9 |
99 |
|
|
MoP/MCM-41 |
1.2 |
0.0 |
0.002 |
0.0 |
0.0 |
0.0 |
2.4 |
97.6 |
99 |
|
|
WP/MCM-41 |
0.6 |
0.0 |
0.001 |
0.0 |
0.0 |
0.0 |
0.0 |
100 |
101 |
|
|
Pd/Al2O3 |
3.6 |
0.04 |
0.006 |
0.2 |
0.9 |
0.0 |
19.9 |
79.1 |
97 |
| 350 |
Ni2P/MCM-41 |
100 |
99.5 |
0.166 |
16.8 |
81.3 |
1.4 |
0.3 |
0.1 |
101 |
|
|
CoP/MCM-41 |
69.9 |
35.8 |
0.115 |
8.2 |
40.7 |
0.9 |
2.0 |
47.4 |
98 |
|
|
MoP/MCM-41 |
21.3 |
7.9 |
0.036 |
3.0 |
14.1 |
19.5 |
17.2 |
46.1 |
97 |
|
|
WP/MCM-41 |
11.6 |
1.6 |
0.019 |
1.3 |
5.9 |
6.6 |
6.4 |
79.8 |
103 |
|
|
Pd/Al2O3 |
23.5 |
5.4 |
0.039 |
3.7 |
18.1 |
0.6 |
1.6 |
75.7 |
96 |
Fig. 6 shows the product selectivity at around 10% of GVL conversion (18% at 250 °C on Ni2P/MCM-41, 9% at 275 °C on CoP/MCM-41, 12% at 325 °C on MoP/MCM-41, 12% at 350 °C on WP/MCM-41, and 9% at 300 °C on Pd/Al2O3). The products were categorized into two groups: hydrocarbons (butane, n-butenes, pentane, n-pentenes and n-pentadienes) and oxygenates (2-MTHF, 2-pentanone, pentanal, 1-pentanol, pentanoic acid and pentenoic acid). All phosphides had a strong preference for either pentanoic acid or n-pentenoic acids among the oxygenates, which were produced via ring opening from GVL. The Ni2P/MCM-41, CoP/MCM-41 and Pd/Al2O3 catalysts followed a similar trend with a high affinity for the production of butane and pentanoic acid. The main products on Ni2P/MCM-41 were butane (21%) and pentanoic acid (73%), with very little n-butenes (1%), pentae (1%) and 2-MTHF (3%) while CoP produced more alkenes and less alkanes. Furthermore, Pd/Al2O3 produced mainly butane (4%) and very little pentane (0.1%) among the hydrocarbon products. Unlike the iron group phosphides, MoP/MCM-41 and WP/MCM-41 gave more C5 hydrocarbons in the hydrocarbon products, especially MoP/MCM-41. The main products on MoP/MCM-41 were n-butenes (8%), n-pentenes (6%), n-pentadienes (3%), pentanoic acid (47%) and pentenoic acid (19%), while WP/MCM-41 gave less C5 hydrocarbons and more pentenoic acid (43%). To summarize, the major differences in selectivity to hydrocarbons were that the iron-group catalysts (Ni2P/MCM-41 and CoP/MCM-41) and Pd/Al2O3 produced mainly C4 hydrocarbons, while group 6 phosphides catalysts (MoP/MCM-41 and WP/MCM-41) formed more unsaturated C5 hydrocarbons such as n-pentenes and n-pentadienes. This is also shown in Fig. 7 which shows the C5/C4 hydrocarbon ratio based on the product selectivity at various temperatures. The Mo and W phosphides may prefer to adsorb the GVL through the O atom of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group because of their larger electrophilicity than that of Ni and Co, and thus the CO group is more easily hydrogenated on Mo and W to produce the C5 hydrocarbons.31 Considering the energy content of the produced oil, the desired products are saturated hydrocarbons such as butane and pentane. Butane represents an undesired carbon loss, but this is offset by a decrease in required hydrogen amounts and the production of the reductant CO. This indicates that Ni2P/MCM-41 and CoP/MCM-41 catalysts present a noteworthy activity in terms of GVL conversion and deoxygenation degree to produce butane. Table 7 shows the mole ratio (C4H10 + C4H8)/CO at different temperatures for Ni2P/MCM-41, CoP/MCM-41 and Pd/Al2O3. As can be seen, the ratio is slightly above 1. This indicates that decarbonylation is a main side reaction that forms butane or n-butenes. The value of the ratio above 1 indicates that some decarboxylation could also be occurring, but this should be minor compared to decarbonylation, and very little CO2 was observed. A possible scheme is shown in Fig. 11.
O group because of their larger electrophilicity than that of Ni and Co, and thus the CO group is more easily hydrogenated on Mo and W to produce the C5 hydrocarbons.31 Considering the energy content of the produced oil, the desired products are saturated hydrocarbons such as butane and pentane. Butane represents an undesired carbon loss, but this is offset by a decrease in required hydrogen amounts and the production of the reductant CO. This indicates that Ni2P/MCM-41 and CoP/MCM-41 catalysts present a noteworthy activity in terms of GVL conversion and deoxygenation degree to produce butane. Table 7 shows the mole ratio (C4H10 + C4H8)/CO at different temperatures for Ni2P/MCM-41, CoP/MCM-41 and Pd/Al2O3. As can be seen, the ratio is slightly above 1. This indicates that decarbonylation is a main side reaction that forms butane or n-butenes. The value of the ratio above 1 indicates that some decarboxylation could also be occurring, but this should be minor compared to decarbonylation, and very little CO2 was observed. A possible scheme is shown in Fig. 11.
 |
| | Fig. 6 Product selectivity for HDO of GVL at around 10% of GVL conversion. | |
 |
| | Fig. 7 C5/C4 hydrocarbon ratio (selectivity ratio) as a function of temperature at 0.5 MPa. | |
Table 7 Mole ratio (C4H10 + C4H8)/CO at different temperatures for Ni2P/MCM-41, CoP/MCM-41 and Pd/Al2O3
| Temperature/°C |
Mole ratio (C4H10 + C4H8)/CO |
| Ni2P/MCM-41 |
CoP/MCM-41 |
Pd/Al2O3 |
| 250 |
1.18 |
— |
— |
| 275 |
1.12 |
— |
— |
| 300 |
1.09 |
1.09 |
— |
| 325 |
1.02 |
1.23 |
1.08 |
| 350 |
1.21 |
1.24 |
1.22 |
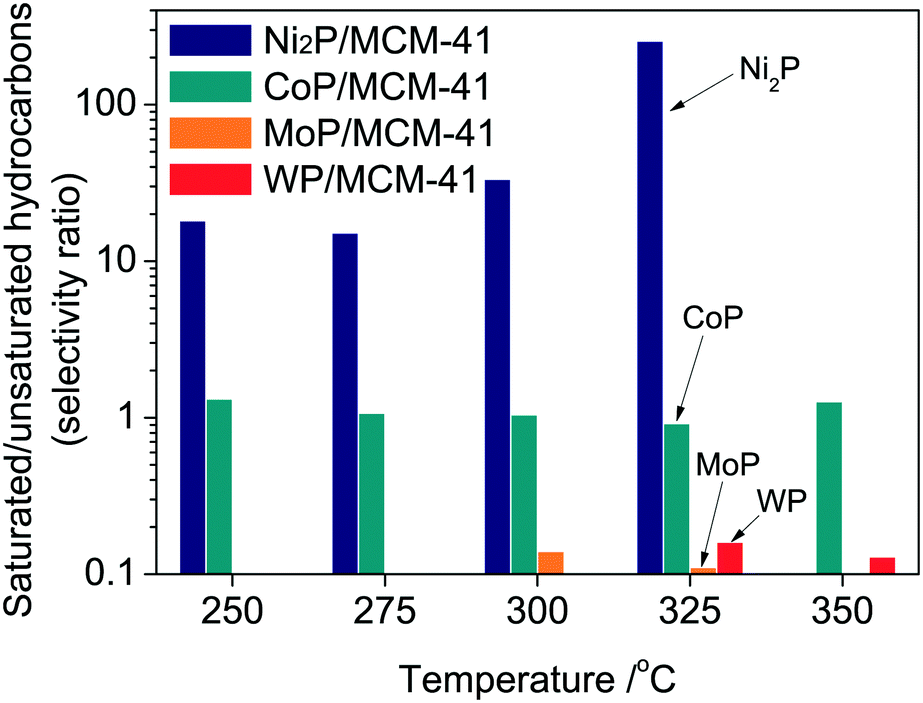
Fig. 8 presents the saturated hydrocarbon/unsaturated hydrocarbon selectivity ratio as a function of temperature on the prepared catalysts. The saturated hydrocarbons include the alkanes such as butane and pentane and the unsaturated hydrocarbons include n-alkenes such as 1-butene, cis-2-butene, trans-2-butene, 1-pentene, cis-2-pentene, trans-2-pentene and 1,3-pentadiene. Thus, this ratio indicates the hydrogenation ability of the catalysts. On Pd/Al2O3, only saturated hydrocarbons (butane and pentane) were observed at all temperatures, thus this ratio cannot be calculated. In the case of Ni2P/MCM-41, saturated hydrocarbons were only produced at 350 °C. The MoP/MCM-41 and WP/MCM-41 did not produce saturated hydrocarbons. Apart from the C5/C4 ratio in Fig. 6, the saturated/unsaturated hydrocarbon ratio was high for Ni2P/MCM-41 and CoP/MCM-41, and was low for MoP/MCM-41 and WP/MCM-41. In particular, Ni2P presented an extremely higher hydrogenation ability compared to CoP/MCM-41, MoP/MCM-41 and WP/MCM-41 with a saturated/unsaturated hydrocarbon ratio over 10.
 |
| | Fig. 8 Saturated/unsaturated hydrocarbon ratio (selectivity ratio) as a function of temperature at 0.5 MPa. | |
3.4. Reaction network for HDO of GVL on phosphide catalysts and Pd/Al2O3
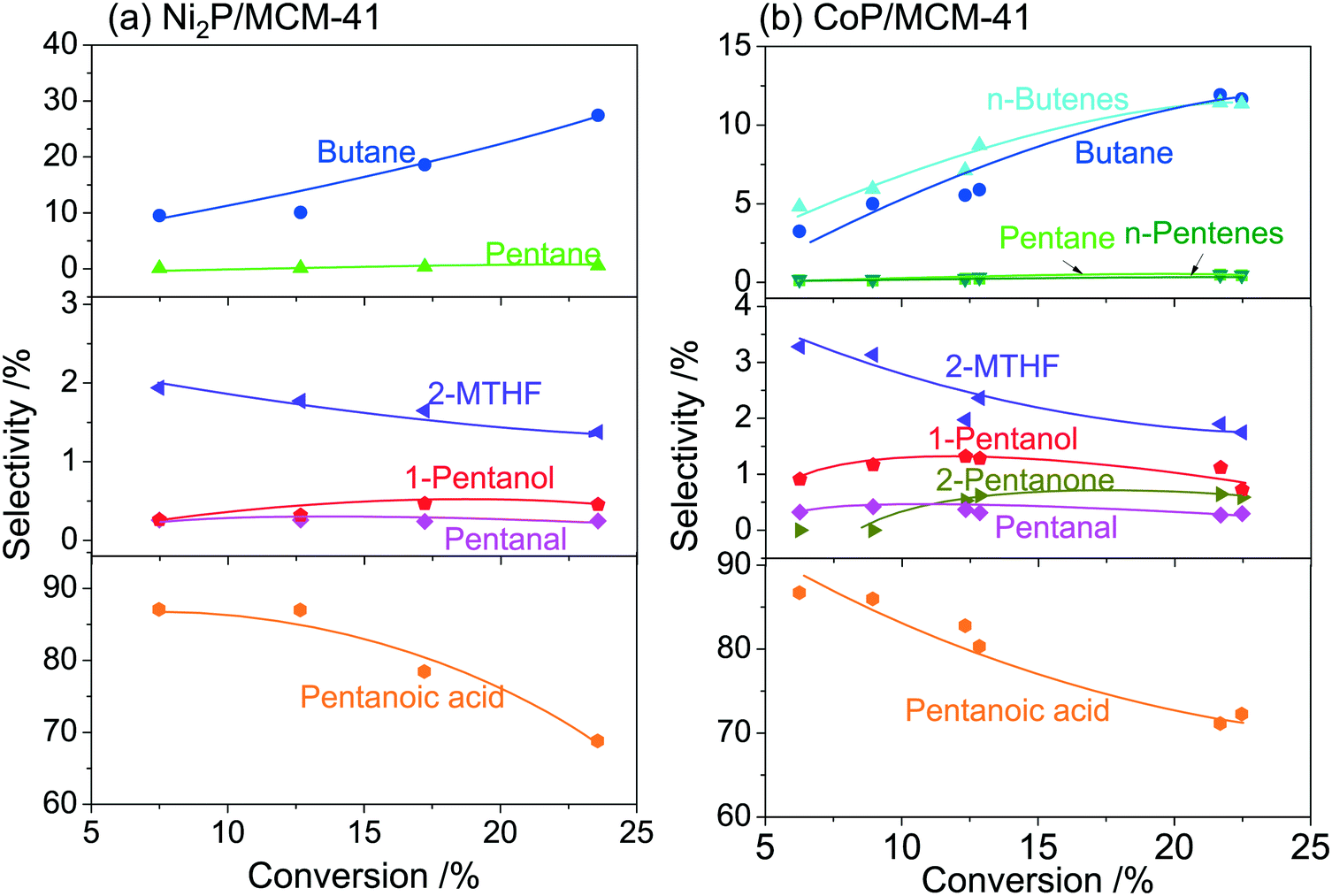
Fig. 9 shows the product selectivity as a function of GVL conversion at 300 °C and 0.5 MPa on (a) Ni2P/MCM-41 and (b) CoP/MCM-41. These catalysts were the most active in this reaction as shown in Fig. 5. The results were obtained by varying the feed flowrate of the 2 mol% GVL in H2. The selectivities to C4 hydrocarbons (butane and n-butenes) and C5 hydrocarbons (pentane and n-pentenes) increased with the conversion, while the selectivities to 2-MTHF and pentanoic acid decreased. These results indicate that the C4 and C5 hydrocarbons were the final products and that the 2-MTHF and pentanoic acid were the primary products. The selectivities to 1-pentanol, pentanal and 2-pentanone went through maxima and then decreased as the conversion increased, indicating that these products were intermediates.
 |
| | Fig. 9 Product selectivity as a function of GVL conversion at 300 °C and 0.5 MPa on (a) Ni2P/MCM-41 and (b) CoP/MCM-41. | |
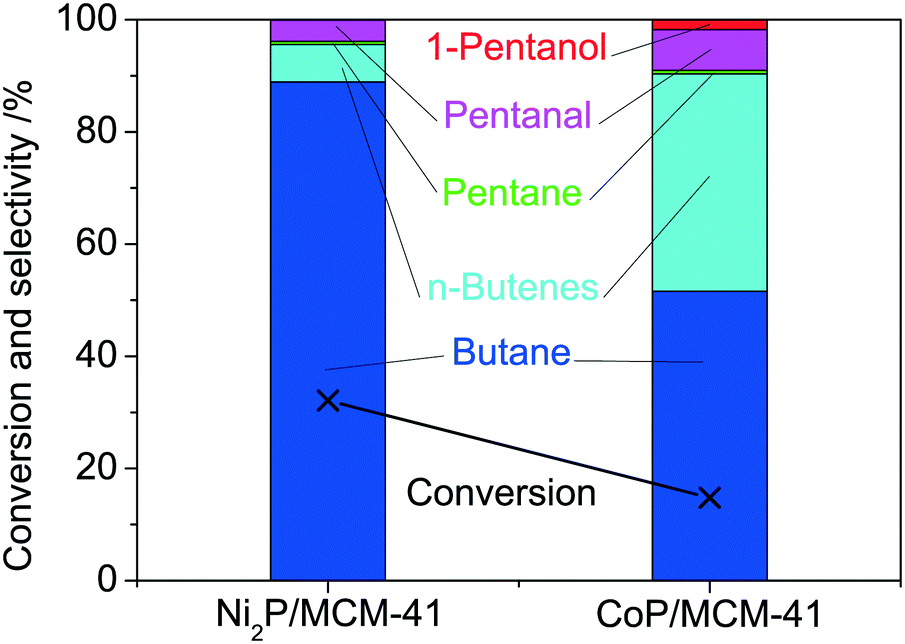
Furthermore, the major product, pentanoic acid, was used as a reactant in catalytic activity tests of Ni2P/MCM-41 and CoP/MCM-41 conducted at 0.5 MPa and 300 °C (Fig. 10). The main products on Ni2P/MCM-41 were butane (89%), n-butenes (7%), pentane (0.6%), pentanal (4%), and 1-pentanol (0.1%), while CoP/MCM-41 produced more n-butenes (39%). These results indicated that the main reaction of pentanoic acid on Ni2P and CoP was decarbonylation of pentanal to produce C4 hydrocarbons while a minor route proceeded by pentanal hydrogenation to 1-pentanol and the subsequent formation of pentane. Although corresponding studies of MoP and WP were not carried out, it can be surmised that they have less decarbonylation and more hydrogenation.
 |
| | Fig. 10 Pentanoic acid HDO on Ni2P/MCM-41 and CoP/MCM-41 at 300 °C and 0.5 MPa. | |
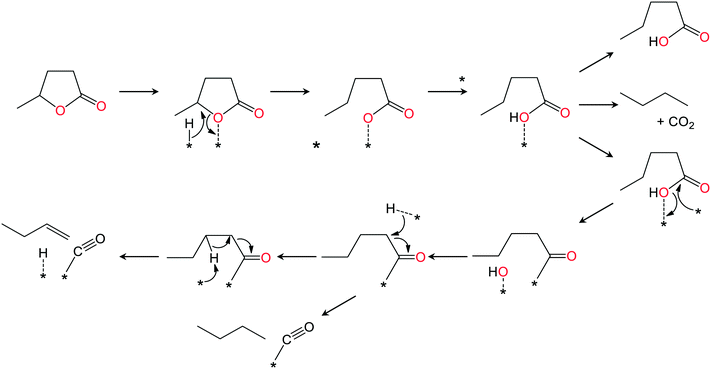 |
| | Fig. 11 Decarbonylation route to butane and butene. | |
A possible reaction network for the HDO of GVL is proposed (Fig. 12). Based on the results in Fig. 6, 9 and 10 and Table 6, it can be deduced that Ni2P/MCM-41, CoP/MCM-41 and Pd/Al2O3 catalysts follow the same reaction route, while MoP/MCM-41 and WP/MCM-41 involve a different reaction pathway. To begin, GVL produces either 2-MTHF by hydrodeoxygenation of the CO or pentanoic acid by ring-opening. In all catalysts, selectivity to pentanoic acid is always higher than that of 2-MTHF and the formation of 2-pentanone, which can be formed from 2-MTHF, is extremely low. Therefore, all catalysts follow a similar reaction route from GVL to pentanoic acid, involving a ring opening reaction and further hydrogenation to produce pentanal. On Ni2P/MCM-41, CoP/MCM-41, and Pd/Al2O3, the formation of pentanal is followed by decarbonylation to produce butane or n-butenes plus CO. In summary, all applied catalysts followed a similar initial reaction sequence from GVL to pentanal involving ring-opening and hydrogenation. After the formation of pentanal, the main reaction was decarbonylation on Ni2P/MCM-41, CoP/MCM-41 and Pd/Al2O3 and was hydrodeoxygenation on MoP/MCM-41 and WP/MCM-41.
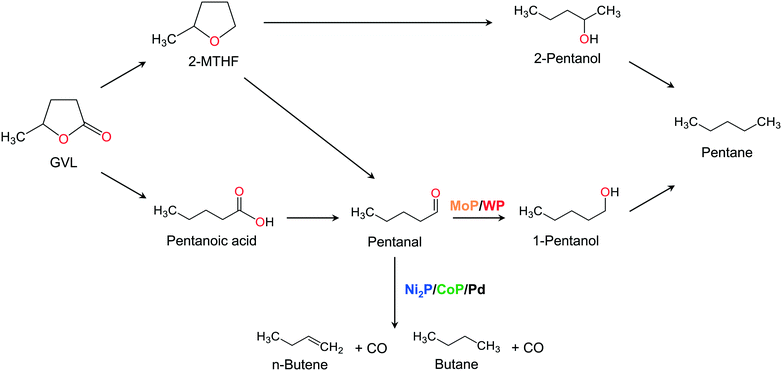 |
| | Fig. 12 A possible reaction network for HDO of GVL. | |
4. Conclusions
A series of transition metal phosphides supported on MCM-41 was prepared by temperature-programmed reduction and tested for the hydrodeoxygenation (HDO) of gamma-valerolactone (GVL). Highly dispersed phosphide catalysts were successfully synthesized, which were characterized by Brunauer–Emmett–Teller, X-ray diffraction, X-ray absorption fine-structure and CO-chemisorption measurements. The activity for GVL conversion and deoxygenation degree followed the order: Ni2P/MCM-41 ≫ CoP/MCM-41 ≫ Pd/Al2O3 ≈ MoP/MCM-41 > WP/MCM-41. The product distribution on the iron-group phosphides (Ni2P/MCM-41 and CoP/MCM-41) and Pd/Al2O3 differed from that on group 6 metal phosphides (MoP/MCM-41 and WP/MCM-41), especially in the selectivity to hydrocarbons. The main hydrocarbon product was butane on Ni2P/MCM-41, CoP/MCM-41, and Pd/Al2O3 and was n-pentenes on MoP/MCM-41 and WP/MCM-41. This indicates that iron group phosphide catalysts and Pd/Al2O3 follow a decarbonylation pathway whereas group 6 phosphides follow an HDO pathway in the removal of oxygen. The iron group phosphides and Pd/Al2O3 showed a high hydrogenation ability to form saturated hydrocarbons. Based on the product selectivity, there are two initial reaction routes, ring-opening and direct deoxygenation, both of which form pentanoic acid on all catalysts. Following that, pentanal is formed by hydrogenation of pentanoic acid and then as a final step decarbonylation occurs on Ni2P/MCM-41, CoP/MCM-41 and Pd/Al2O3.
Acknowledgements
This work was supported by the Development of Next-generation Technology for Strategic Utilization of Biomass Energy of the New Energy and Industrial Technology Development Organization (NEDO), Japan and by the National Science Foundation under Grant No. CHE-1361842. The XAFS experiments were conducted under approval of PF-PAC (Project No. 2014G619).
References
- H. Hernando, S. Jiménez-Sánchez, J. Fermoso, P. Pizarro, J. M. Coronado and D. P. Serrano, Catal. Sci. Technol., 2016, 6, 2829–2843 CAS.
- S. D. Stefanidis, K. G. Kalogiannis, P. A. Pilavachi, C. M. Fougret, E. Jordan and A. A. Lappas, Catal. Sci. Technol., 2016, 6, 2807–2819 CAS.
- C.-H. Zhou, X. Xia, C.-X. Lin, D.-S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- L. Nazari, Z. Yuan, S. Souzanchi, M. B. Ray and C. Xu, Fuel, 2015, 162, 74–83 CrossRef CAS.
- N. Koike, S. Hosokai, A. Takagaki, S. Nishimura, R. Kikuchi, K. Ebitani, Y. Suzuki and S. T. Oyama, J. Catal., 2016, 333, 115–126 CrossRef CAS.
- M. S. Mettler, A. D. Paulsen, D. G. Vlachos and P. J. Dauenhauer, Catal. Sci. Technol., 2014, 4, 3822–3825 CAS.
- T. Wang, S. Qiu, Y. Weng, L. Chen, Q. Liu, J. Long, J. Tan, Q. Zhang, Q. Zhang and L. Ma, Appl. Energy, 2015, 160, 329–335 CrossRef CAS.
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS.
- C. Perego and M. Ricci, Catal. Sci. Technol., 2012, 2, 1776–1786 CAS.
- I. D. V. Torri, V. Paasikallio, C. S. Faccini, R. Huff, E. B. Caramão, V. Sacon, A. Oasmaa and C. A. Zini, Bioresour. Technol., 2016, 200, 680–690 CrossRef CAS PubMed.
- J. Chen and Q. Xu, Catal. Sci. Technol., 2016, 6, 7239–7251 CAS.
- J. K. Satyarthi, T. Chiranjeevi, D. T. Gokak and P. S. Viswanathan, Catal. Sci. Technol., 2013, 3, 70–80 CAS.
- O. I. Senol, E.-M. Ryymin, T.-R. Viljava and A. O. I. Krause, J. Mol. Catal. A: Chem., 2007, 277, 107–112 CrossRef CAS.
- E.-M. Ryymin, M. L. Honkela, T.-R. Viljava and A. O. I. Krause, Appl. Catal., A, 2009, 358, 42–48 CrossRef CAS.
- L. Nie and D. E. Resasco, J. Catal., 2014, 317, 22–29 CrossRef CAS.
- H. Xu, K. Wang, H. Zhang, L. Hao, J. Xu and Z. Liu, Catal. Sci. Technol., 2014, 4, 2658–2663 CAS.
- L. Wang, H. Wan, S. Jin, X. Chen, C. Li and C. Liang, Catal. Sci. Technol., 2015, 5, 465–474 CAS.
- C. Zhao, J. He, A. A. Lemonidou, X. Li and J. A. Lercher, J. Catal., 2011, 280, 8–16 CrossRef CAS.
- C. Newman, X. Zhou, B. Goundie, I. T. Ghampson, R. A. Pollock, Z. Ross, M. C. Wheeler, R. W. Meulenberg, R. N. Austin and B. G. Frederick, Appl. Catal., A, 2014, 477, 64–74 CrossRef CAS.
- R. C. Runnebaum, T. Nimmanwudipong, D. E. Block and B. C. Gates, Catal. Sci. Technol., 2012, 2, 113–118 CAS.
- Y. K. Lugo-José, J. R. Monnier, A. Heyden and C. T. Williams, Catal. Sci. Technol., 2014, 4, 3909–3916 Search PubMed.
- Y. K. Lugo-José, J. R. Monnier and C. T. Williams, Appl. Catal., A, 2014, 469, 410–418 CrossRef.
- B. Dhandapani, T. St. Clair and S. T. Oyama, Appl. Catal., A, 1998, 168, 219–228 CrossRef CAS.
- Z. He and X. Wang, Catal. Sustainable Energy, 2012, 1, 28–52 Search PubMed.
- A. Iino, A. Cho, A. Takagaki, R. Kikuchi and S. T. Oyama, J. Catal., 2014, 311, 17–27 CrossRef CAS.
- A. Cho, H. Kim, A. Iino, A. Takagaki and S. T. Oyama, J. Catal., 2014, 318, 151–161 CrossRef CAS.
- A. Cho, A. Takagaki, R. Kikuchi and S. T. Oyama, Top. Catal., 2015, 58, 219–231 CrossRef CAS.
- L. Yang, X. Li, A. Wang, R. Prins, Y. Chen and X. Duan, J. Catal., 2015, 330, 330–343 CrossRef CAS.
- A. I. d'Aquino, S. J. Danforth, T. R. Clinkingbeard, B. Ilic, L. Pullan, M. A. Reynolds, B. D. Murray and M. E. Bussell, J. Catal., 2016, 335, 204–214 CrossRef.
- P. Bui, J. A. Cecilia, S. Ted. Oyama, A. Takagaki, A. Infantes-Molina, H. Zhao, D. Li, E. Rodríguez-Castellón and A. Jiménez López, J. Catal., 2012, 294, 184–198 CrossRef CAS.
- J. Chen, H. Shi, L. Li and K. Li, Appl. Catal., B, 2014, 144, 870–884 CrossRef CAS.
- F. Liguori, C. Moreno-Marrodan and P. Barbaro, ACS Catal., 2015, 5, 1882–1894 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS PubMed.
- J. C. Serrano-Ruiz, D. J. Braden, R. M. West and J. A. Dumesic, Appl. Catal., B, 2010, 100, 184–189 CrossRef CAS.
- J.-P. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., Int. Ed., 2010, 49, 4479–4483 CrossRef CAS PubMed.
- P. Sun, G. Gao, Z. Zhao, C. Xia and F. Li, ACS Catal., 2014, 4, 4136–4142 CrossRef CAS.
- J. A. Cecilia, A. Infantes-Molina, E. Rodríguez-Castellón and A. Jiménez-López, J. Catal., 2009, 263, 4–15 CrossRef CAS.
- J. A. Cecilia, A. Infantes-Molina, E. Rodríguez-Castellón and A. Jiménez-López, Appl. Catal., B, 2009, 92, 100–113 CrossRef CAS.
- J. A. Cecilia, A. Infantes-Molina, E. Rodríguez-Castellón and A. Jiménez-López, J. Phys. Chem. C, 2009, 113, 17032–17044 CAS.
- Y. Shu, Y.-K. Lee and S. T. Oyama, J. Catal., 2005, 236, 112–121 CrossRef CAS.
- Y.-K. Lee, Y. Shu and S. T. Oyama, Appl. Catal., A, 2007, 322, 191–204 CrossRef CAS.
- S. T. Oyama, X. Zhang, Y.-K. Lee and W.-J. Chun, J. Catal., 2004, 221, 263–273 CrossRef CAS.
- Y.-K. Lee and S. T. Oyama, J. Catal., 2006, 239, 376–389 CrossRef CAS.
- S. T. Oyama, X. Zhang, J. Lu, Y. Gu and T. Fujitani, J. Catal., 2008, 257, 1–4 CrossRef CAS.
- P. B. Weisz and C. D. Prater, Adv. Catal., 1954, 6, 143–196 CAS.
-
M. A. Vannice, Kinetics of catalytic reactions, Springer Science+Business Media, 2005, ch. 4, pp. 63–65 Search PubMed.
- D. E. Mears, Ind. Eng. Chem. Process Des. Dev., 1971, 10, 541–547 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cy02252a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 







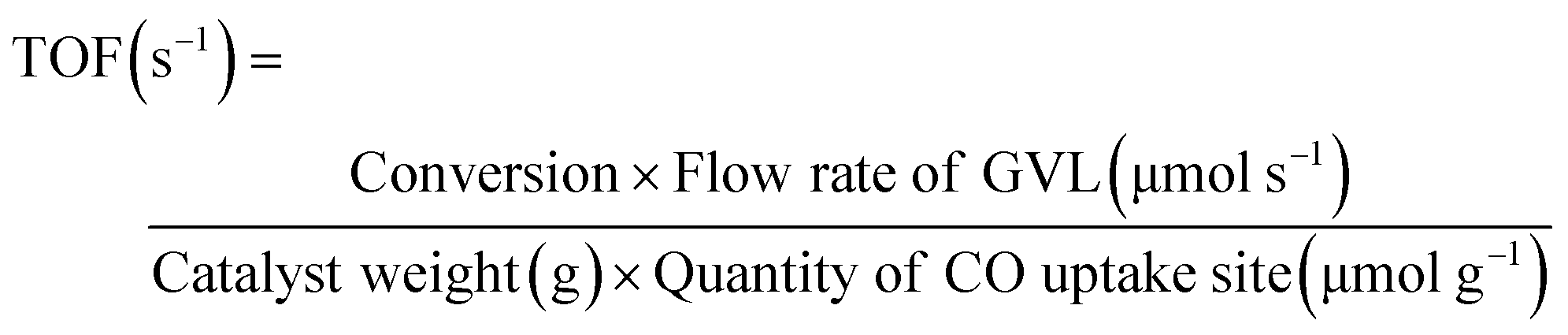


 and the calculated CWP values are listed in Table 5. The values of CWP are less than 0.3 except for the Ni2P/MCM-41 sample at over 300 °C and the CoP/MCM-41 sample at 350 °C, which are exactly as expected from the shapes of the conversion curves, with mass transfer becoming important past the inflexion points in the curves. For the other samples, there are no diffusion limitations under all the reaction conditions. Also the Mears criterion for external diffusion was below 0.15 (see the ESI†)
and the calculated CWP values are listed in Table 5. The values of CWP are less than 0.3 except for the Ni2P/MCM-41 sample at over 300 °C and the CoP/MCM-41 sample at 350 °C, which are exactly as expected from the shapes of the conversion curves, with mass transfer becoming important past the inflexion points in the curves. For the other samples, there are no diffusion limitations under all the reaction conditions. Also the Mears criterion for external diffusion was below 0.15 (see the ESI†)  < 0.15, indicating that mass transfer from the bulk gas phase to the catalyst surface can be neglected.45,48
< 0.15, indicating that mass transfer from the bulk gas phase to the catalyst surface can be neglected.45,48