Tetronics/cyclodextrin-based hydrogels as catalyst-containing media for the hydroformylation of higher olefins†
Received
29th September 2016
, Accepted 7th November 2016
First published on 10th November 2016
Abstract
The rhodium-catalyzed hydroformylation of alkenes has been investigated under biphasic conditions using combinations of α-cyclodextrin (α-CD) and poloxamines (Tetronics®). Thermo-responsive hydrogels containing the Rh-catalyst are formed under well-defined conditions of concentration. Hydrogels consisting of the reverse-sequential Tetronic® 90R4 prove to be more effective than those containing the conventional sequential Tetronic® 701. The presence of α-CD is crucial to provoke the decantation of the multiphasic system once the reaction is complete. Optimized conditions (CO/H2 pressure, Rh-precursors, phosphanes, etc.) show that the catalytic system is especially applicable to the hydroformylation of terminal alkenes. The catalytic performance remains unchanged upon recycling as the hydrogel matrix prevents the oxidation of the phosphane.
Introduction
Following the seminal work of O. Roelen,1 a range of improvements have been made in transition-metal-catalyzed hydroformylation of alkenes (Scheme 1). Various experimental conditions have been studied,2 including single-phase homogeneous systems,3 biphasic systems with aqueous4–7 fluorous,8,9 or ionic liquid phases,10–13 thermomorphic solvent mixtures and microemulsions,14–16 catalysts immobilized on a solid support,17–20 supercritical fluid–ionic liquid biphasic systems,21–28 amphiphilic nanogels,29 or continuous liquid–vapor reactors.30,31
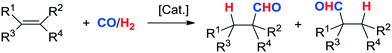 |
| | Scheme 1 Rh-catalyzed hydroformylation of alkenes. | |
Concurrently, we have been developing the use of alternative reaction media capable of ensuring high catalytic performance and reusability of the organometallic catalyst.32,33 We especially focused our attention on cyclodextrin (CD)-based hydrogels which are 3D networks containing large amounts of water.34 CDs are cyclic oligosaccharides consisting of glucopyranose units (Fig. 1). The most common ones are α-CD, β-CD and γ-CD. They consist of 6, 7 and 8 glucose units, respectively, linked by α-D-1,4-glycosidic bonds. Although they are water soluble, their cavity is hydrophobic and can host guest molecules to form inclusion complexes. We recently showed that hydrogels consisting of CDs and polyethylene glycol (PEG) were effective in the hydroformylation of very hydrophobic alkenes (>C12).35,36 In such catalytic systems, CDs are multifunctional entities. They are constitutive building blocks of the hydrogels and participate in the alkene conversion as molecular receptors, supramolecular hosts and fluidifiers of the aqueous/organic interface.
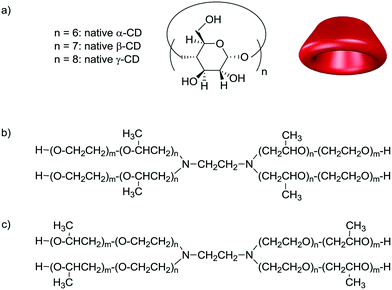 |
| | Fig. 1 Structure of a) cyclodextrins (CDs), b) conventional sequential poloxamines and c) reverse-sequential poloxamines. | |
Given the very good results obtained with the CD/hydrogel combination, we have been exploring the catalytic properties of other CD-based hydrogels under biphasic conditions. We especially focused our interest on poloxamines (also known as Tetronic® macromolecules). They consist of block copolymers based on hydrophilic poly(ethylene oxide) (PEO) and hydrophobic poly(propylene oxide) (PPO) attached on an ethylene diamine spacer. They differ from their linear counterparts (poloxamers, also known as Pluronic® macromolecules) by their X-shaped structure. Conventional sequential Tetronics® consist of PEO blocks at the outer sphere and PPO blocks attached to the diaminoethylene core, while reverse-sequential poloxamines show PPO blocks at the periphery and PEO attached to the central diamino moiety (Fig. 1). Their amphiphilic characteristic can be modulated by the number of PEO and PPO units. Depending on the PEO/PPO ratio and the length of the PEO–PPO arms, various phases with specific molecular architecture can be obtained.37,38 Combined with CDs, Tetronics® form hydrogels through non-covalent interactions.39,40 CDs slide along the PEO/PPO polymer chains, and selectively accommodate the PEO blocks. Further intermolecular interactions between threaded α-CDs form stacked nanocylinders that aggregate into nanosized columnar α-CD domains (Fig. 2).
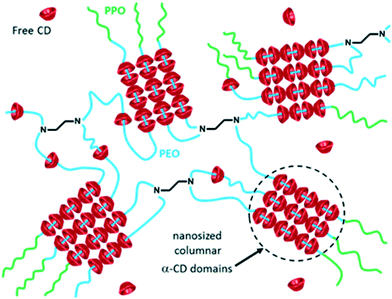 |
| | Fig. 2 Polypseudorotaxane aggregates of α-CD/reverse-sequential poloxamines (Tetronic® 90R4). | |
The rich phase behavior of the resulting hydrogels prompted researchers to investigate their applications, including enzyme, protein and gene delivery,41–44 sustained release of drugs,45,46 tissue engineering,47 and as P-glycoprotein inhibitors,48 stabilizers of metallic nanoparticles49,50 and templates in the preparation of mesoporous materials.51–53 However, nothing has been described so far on the utilization of Tetronics® in biphasic catalysis in the presence of organometallic catalytic species. Herein we describe how the conventional sequential Tetronic® 701 (Mw 3600, 2.1 EO and 14 PO units per arm) and the reverse-sequential Tetronic® 90R4 (Mw 7200, 16 EO and 18 PO units per arm) (ESI†) can retain Rh-catalysts within their 3D networks for the hydroformylation of very hydrophobic alkenes under biphasic conditions. We especially highlight the benefit of using Tetronics® in combination with CDs to form thermo-responsive hydrogels. We show that the latter clearly ensure the reusability of the catalytic system. Other parameters such as the nature of the rhodium precursor, the reaction temperature or the CO/H2 pressure are also discussed.
Results and discussion
Conditions for obtaining hydrogels
The first set of experiments consisted of defining the range of Tetronic® concentrations in water to ensure the existence of a mixture of liquid phases (sol phase + organic phase) at the reaction temperature (80 °C) and a two-phase system (gel phase + organic phase) at room temperature. The goals are twofold: i) favor contacts between the organic substrate and the catalyst at high temperature and ii) recover both the product and the Rh-catalyst in two different phases when cooling the system at room temperature once the reaction is complete. To elaborate such thermo-responsive hydrogels, Tetronic® 90R4 was first mixed in water in various proportions at 80 °C (temperature of the studied hydroformylation reaction). As expected, no gel could be obtained whatever the concentration. A turbid solution was observed for low concentration Tetronic® 90R4 solutions while demixing took place for high concentration Tetronic® 90R4 solutions (ESI,† Table S2). On the other hand, addition of α-CD to Tetronic® 90R4/water mixtures gave hydrogels under well-defined concentration and temperature ranges. Tetronic® 90R4 was mixed in various proportions in aqueous saturated α-CD solutions (870 mg α-CD (0.9 mmol) dissolved in 6 mL water). Upon heating at 80 °C for 30 min, α-CD threaded onto the PEO–PPO chains of Tetronic® 90R4 leading to Tetronic® 90R4/α-CD polypseudorotaxanes in water (Fig. 2). The mixtures were then cooled at 4 °C overnight and the vials were inverted. Below 10 wt% Tetronic® 90R4 (600 mg Tetronic® 90R4 (0.8 mmol) in 6 mL water; tube 10, Table 1), particles flocculated in the aqueous solutions. On the contrary, hydrogels were observed for mixtures containing at least 600 mg Tetronic® 90R4. As expected, high proportions of Tetronic® 90R4 were required for the formation of hydrogels.54 The latter was driven by the micro-crystallization of inclusion complexes of α-CD and PEO blocks and the micellization of the PPO blocks.55 Moreover, above 10 wt% Tetronic® 90R4 in saturated solutions of α-CDs, the hydrogels showed thermo-responsivity. Below 25 °C, they behaved as gels. Above 25 °C, they were liquids and formed a biphasic system (ESI,† Table S4). The gel phases were recovered at room temperature for solutions containing more than 10 wt% Tetronic® 90R4 (ESI,† Table S5). The gel-to-sol transition of Tetronic® 90R4/α-CD/water mixtures was observed by inverting the vials and was more accurately monitored using a rheometer. Increasing the temperature led to a sharp decrease in the viscosity. Fig. 3 is illustrative of the drop in viscosity around 25 °C for a mixture containing 1250 mg Tetronic® 90R4 (0.17 mmol, 25 wt%), 725 mg α-CD (0.75 mmol) and 5 mL water. To better define the limits of the system, the amount of α-CD was also varied from an aqueous solution containing 1250 mg Tetronic® 90R4 (0.17 mmol) dissolved in 5 mL water. Below 600 mg α-CD (0.61 mmol), a clear solution was observed whatever the temperature. Above 600 mg α-CD, a gel phase was formed at room temperature and evolved to a sol phase above 25 °C. Note that the formation of hydrogels was specific to the native α-CD.
Table 1 Optical photos of Tetronic® 90R4/α-CD/water solutions (870 mg α-CD, 6 mL water) as a function of the Tetronic® 90R4 weight percentage at 20 °C. P: precipitate; G: gel; D: demixing
| Tube |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
19 |
20 |
21 |
22 |
23 |
| Weight% |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
15 |
20 |
25 |
30 |
35 |
40 |
45 |
50 |
55 |
60 |
65 |
70 |
75 |

|
| Observation |
P |
P |
P |
P |
P |
P |
P |
P |
P |
P |
G |
G |
G |
G |
G |
G |
G |
G |
G |
G |
G |
G |
G |
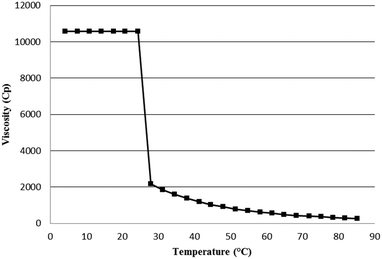 |
| | Fig. 3 Variation of the viscosity with the temperature for a mixture containing 1250 mg Tetronic® 90R4 (0.17 mmol, 25 wt%), 725 mg α-CD (0.75 mmol) and 5 mL water. | |
For example, no gel could be obtained in the presence of the native β-CD whatever the concentration (up to 3 g (2.6 mmol) in 5 mL water), even if β-CD is well-known to selectively accommodate PPO blocks.56
The formation of hydrogels from α-CD and Tetronic® 701 followed the same trend. However, higher CD concentrations were required to trigger the formation of hydrogels as exemplified in Fig. 4. As CDs can be easily removed from the external PEO blocks, the Tetronic® 701/α-CD/water hydrogel is less stable than the Tetronic® 90R4-based hydrogel. More CDs (35 vs. 10 wt%) are then necessary to form hydrogel textures (ESI,† Table S7).
 |
| | Fig. 4 a) 50 mg Tetronic® 701/725 mg α-CD/5 mL water; b) 1250 mg T701/725 mg α-CD/5 mL water. | |
Hydroformylation of 1-dodecene using Tetronic® 90R4
The performance of the Tetronic® 90R4-based hydrogels was assessed in the rhodium-catalyzed hydroformylation of 1-dodecene (model substrate). To this effect, Tetronic® 90R4 (20 wt% in water) was dissolved in water with or without α-CD upon stirring for 20 min at 60 °C under an inert atmosphere. The solution was allowed to stand for 30 min at room temperature before Rh(CO)2(acac) and the sodium salt of the trisulfonated triphenylphosphane (TPPTS) were added under nitrogen. The resulting mixture was stirred at 60 °C for another 30 min. 1-Dodecene was then added, and the mixture was stirred at 80 °C under 50 bar of CO/H2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) pressure for 1 h. Without any phosphane, a moderate 25%-conversion was obtained (Table 2, entry 1). However, such an experiment is irrelevant as the reaction takes place in the organic phase as suggested by the colored upper phase of the recovered biphasic system. This is indirect proof that the Tetronic® 90R4 nitrogens are unable to stabilize the catalytic species within the gel. In the presence of TPPTS, Table 2 clearly shows that the conversions are lower without α-CD (entries 2–4), indicative of the contribution of the Tetronic® 90R4/α-CD assembly at the interfacial layer. These results are in line with our previous results on α-CD/POE hydrogels.35,36 Moreover, without α-CD, the multiphasic system remains emulsified once the reaction is complete and no decantation is observed even after 12 h. Recovering the reaction products then requires the use of an organic solvent (diethyl ether).
:1) pressure for 1 h. Without any phosphane, a moderate 25%-conversion was obtained (Table 2, entry 1). However, such an experiment is irrelevant as the reaction takes place in the organic phase as suggested by the colored upper phase of the recovered biphasic system. This is indirect proof that the Tetronic® 90R4 nitrogens are unable to stabilize the catalytic species within the gel. In the presence of TPPTS, Table 2 clearly shows that the conversions are lower without α-CD (entries 2–4), indicative of the contribution of the Tetronic® 90R4/α-CD assembly at the interfacial layer. These results are in line with our previous results on α-CD/POE hydrogels.35,36 Moreover, without α-CD, the multiphasic system remains emulsified once the reaction is complete and no decantation is observed even after 12 h. Recovering the reaction products then requires the use of an organic solvent (diethyl ether).
Table 2 Hydroformylation of 1-dodecenea
| Entry |
Tetronic® |
Tetronic®/water (w/w) |
α-CD |
Conv.b (%) |
Aldehyde sel.b (%) |
l/bb |
Alkene isomers (%) |
|
Conditions: 1-dodecene (1.63 mmol), Rh(CO)2(acac) (3 mg, 0.012 mmol), TPPTS (33 mg, 0.058 mmol), α-CD (870 mg, 0.90 mmol), 6 mL H2O, 80 °C, 50 bar CO/H2, 1 h.
Conversions and selectivities were determined by 1H NMR spectroscopy of the crude reaction mixture.
Without TPPTS.
|
| 1c |
90R4 |
10 |
No |
25 |
85 |
2 |
15 |
| 2 |
90R4 |
10 |
No |
13 |
78 |
3.5 |
22 |
| 3 |
90R4 |
25 |
No |
26 |
74 |
2.8 |
26 |
| 4 |
90R4 |
40 |
No |
64 |
73 |
3.4 |
27 |
| 5 |
90R4 |
10 |
Yes |
36 |
92 |
2.5 |
8 |
| 6 |
90R4 |
25 |
Yes |
67 |
92 |
2.4 |
8 |
| 7 |
90R4 |
40 |
Yes |
84 |
96 |
2.4 |
4 |
| 8 |
90R4 |
75 |
Yes |
92 |
99 |
2.5 |
<1 |
| 9 |
701 |
40 |
No |
31 |
48 |
2.5 |
52 |
| 10 |
701 |
40 |
Yes |
51 |
68 |
2.6 |
32 |
The conversion regularly increased with the proportion of Tetronic® 90R4, indicative of the effect of the structure of Tetronic® 90R4 at the interface between the catalyst-containing aqueous phase and the organic substrate. Low conversions were measured for low quantities of Tetronic® 90R4 (<20%), meaning that the flocculation of polypseudorotaxanes in the aqueous solutions was not appropriate to favor contacts between the Rh-catalyst and the substrates. Conversely, high conversion values were found for high proportions of Tetronic® 90R4 (>40%) for which a liquid–liquid two-phase system was identified at 80 °C (Table 1). As shown in Fig. 5, the α-CD/Tetronic® 90R4 polypseudorotaxane aggregates partially cover the droplets of the organic substrate resulting in a Pickering-like emulsion that favors the contacts between 1-dodecene and the Rh-catalyst at the aqueous/water interface.36 The studied Tetronic® 90R4-based catalytic system gave better conversions than those obtained using our previous hydrogel-based system consisting of α-CD and PEO under the same catalytic conditions.36
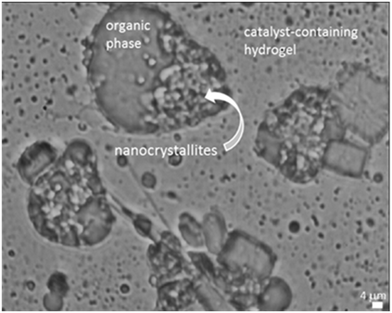 |
| | Fig. 5 Pickering-like emulsion sampled at 80 °C and observed at 20 °C. Tetronic® 90R4 (1500 mg, 0.21 mmol), α-CD (870 mg, 0.90 mmol), 6 mL H2O. | |
Indeed, the reaction appeared to approach 100% completion using very concentrated Tetronic® 90R4/α-CD/water solutions within 1 h while only 37% conversion was obtained with the α-CD/PEO-based hydrogel within the same reaction time. The catalytic activity obtained with the Tetronic® 90R4/α-CD/water combination compared more favorably with the α-CD/PEO/water system doped with a fluidifier (RAME-β-CD).36 Yet, the main advantage of Tetronic® 90R4 over the fluidifier-based system lies in the molecular structure of Tetronic® 90R4 that already contains the interfacial properties required to overcome the mass transfer limitation at the aqueous/organic interface. Indeed, hydrophilic moieties (such as PEO or α-CD/PEO assemblies) and PPO blocks are well-known to self-assemble into micelles at the interface.57,58 Consequently, no fluidifier is required in that case.
The conversion vs. time diagram (Fig. 6) illustrates the dependence of the conversion on the reaction time. The high initial conversion of 1-dodecene regularly decreased with time. Note that the catalyst is not deactivated as this would terminate the conversion and not just cause a decrease in the conversion rate. Moreover, no significant change in the kinetic regime was noted, indicative of the stability of the Rh-catalysts over the course of the reaction. No induction period was observed, suggesting that the formation of the crystallites is fast under catalytic conditions. The regular decrease in the slope of the curve reflects the dependence of the catalytic rate toward the substrate concentration. Note that, throughout the reaction, the aldehyde selectivity and the linear to branched aldehyde ratio remained constant, also indicative of the stability of the Rh-catalyst.
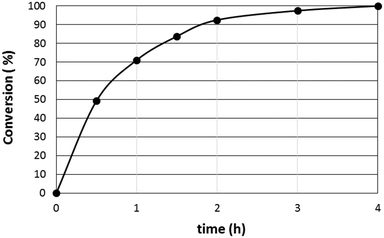 |
| | Fig. 6 Conversion profile of the Rh-catalyzed hydroformylation of 1-dodecene in time. Conditions: 1-dodecene (1.63 mmol), Rh(CO)2(acac) (3 mg, 0.012 mmol), TPPTS (33 mg, 0.058 mmol), Tetronic® 90R4 (25 wt%, 1500 mg, 0.21 mmol), α-CD (870 mg, 0.90 mmol), 6 mL H2O, 80 °C, 50 bar CO/H2. | |
The chemoselectivity of the hydroformylation reaction was also influenced by the Tetronic® 90R4 concentration. While only 73% aldehydes were produced using 25 wt% Tetronic® 90R4, the percentage of aldehydes reached 99% using 35 wt% Tetronic® 90R4 (ESI,† Table S3). This result is in contrast with what was previously obtained using α-CD/PEO-based hydrogels for which a poor 77% aldehyde selectivity was measured.36 This suggests a more selective catalytic process at the interface between the Tetronic® 90R4-containing aqueous compartment and the organic phase. This is in line with previous results showing that the confined environment of a micelle-like structure in which the Rh-catalyst operates is crucial to finely control the catalytic coordination sphere and orient the catalytic reaction in a given direction.59–62 The regioselectivity, however, did not vary upon changing the amount of Tetronic® 90R4. The linear to branched aldehyde ratio (l/b) remained constant (2.3 < l/b < 2.5), suggesting that the equilibria between the Rh-species were not perturbed under such catalytic conditions whatever the Tetronic® 90R4 concentration.
Not only does the proportion of Tetronic® 90R4 influences the catalytic performances, but it also influences the formation of gel or sol phases. As expected, the presence of α-CD is crucial in the studied catalytic system as it ensures the formation of a gel upon cooling once the reaction is complete. The separation of the liquid products and the catalyst could then be easily performed.
Comparison between Tetronic® 90R4 and Tetronic® 701
The reverse-sequential Tetronic® 90R4 was compared to the conventional sequential Tetronic® 701 in saturated aqueous solutions of α-CD. Using 35 wt% Tetronic® in water, both Tetronic®/α-CD/water combinations formed hydrogels at room temperature and were in the sol phase at 80 °C. As shown in Table 2 (entries 8 and 9), the nature of the poloxamine significantly affects the catalytic activity and the selectivity. Tetronic® 701 is far less effective than Tetronic® 90R4 in terms of conversion and aldehyde selectivity. The explanation lies in their respective structures. Tetronic® 90R4 is a reversed poloxamine, meaning that PEO blocks are directly attached to the central diamino core while the PPO blocks are at the periphery and form micelles.63 Once threaded on the PEO blocks of Tetronic® 90R4, α-CDs cannot be easily “de-threatened” from the PEO blocks upon heating because PPO blocks act as stoppers. In that case, α-CD/Tetronic® 90R4 crystallites favorably interact with linear substrate molecules at the interfacial layer to convert alkenes into aldehydes as already described in our previous papers.64,65
Conversely, the conventional sequential Tetronic® 701 consists of PPO blocks attached to the diamino core with PEO blocks at the periphery of the poloxamine structure. Accordingly, α-CDs can be easily removed upon heating from the PEO chains to be released in the aqueous compartment. The polypseudorotaxane aggregates were then less effective at the aqueous/organic interface, resulting in lower conversion (51% vs. 81% for Tetronic® 90R4).
Optimization of the catalytic conditions
Effect of the CO/H2 pressure.
Once the variation of the conversion with the Tetronic® 90R4 concentration was established, we focused our attention on other reaction parameters. To ensure the presence of a liquid–liquid biphasic system at 80 °C (temperature of the hydroformylation reaction) and the formation of a hydrogel phase at room temperature (to recover the Rh-catalyst), a 25 wt% Tetronic® 90R4 concentration was chosen for further investigations. First, the CO/H2 pressure was varied from 10 to 90 bar to assess its influence on the catalytic performances. In a saturated aqueous solution of α-CD (870 mg in 6 mL), no significant change in the conversion nor in the chemoselectivity was noticed, indicating that the diffusion of CO and H2 in the biphasic system was not the limiting factor of the reaction rate (Table 3). However, a slight decrease in the l/b ratio was observed. This logically correlates with the formation of CO-coordinated Rh-species (such as HRh(CO)2(TPPTS)) which are more prone to yield branched aldehydes;66 however, the extent of the effect was relatively small. Varying the CO and H2 partial pressure slightly affects the catalytic activity and selectivity. A higher percentage of H2 logically led to a decrease in aldehyde selectivity (87%) as more hydrogenated products are formed during the course of the reaction (entry 8). Conversely, a higher proportion of CO yields a lower proportion of linear aldehydes (l/b = 2.1) because of the presence of increased amounts of phosphane low-coordinated Rh-species in the medium (entry 9).
Table 3 Conversions and selectivities in the Rh-catalyzed hydroformylation of 1-dodecene at various CO/H2 pressuresa
| Entry |
CO/H2 pressure (bar) |
Conv.b (%) |
Aldehyde sel.b (%) |
l/bb |
|
Conditions: 1-dodecene (1.63 mmol), Rh(CO)2(acac) (3 mg, 0.012 mmol), TPPTS (33 mg, 0.058 mmol), Tetronic® 90R4 (25 wt%, 1500 mg, 0.21 mmol), α-CD (870 mg, 0.90 mmol), 6 mL H2O, 80 °C, 1 h.
Conversions and selectivities were determined by 1H NMR.
|
| 1 |
5/5 |
81 |
90 |
2.5 |
| 2 |
10/10 |
73 |
91 |
2.4 |
| 3 |
15/15 |
69 |
93 |
2.4 |
| 4 |
20/20 |
76 |
94 |
2.3 |
| 5 |
25/25 |
71 |
93 |
2.3 |
| 6 |
35/35 |
76 |
93 |
2.3 |
| 7 |
45/45 |
86 |
95 |
2.2 |
| 8 |
17/33 |
69 |
87 |
2.3 |
| 9 |
33/17 |
74 |
93 |
2.1 |
Effect of the reaction temperature.
While the effect of the CO/H2 pressure did not change substantially the catalytic performances, the influence of the temperature on the reaction rate was much more pronounced (Table 4). With an increase in the temperature from 30 to 150 °C, the conversion was found to drastically increase from 1.4 to 87%, in line with both the higher mobility of the polymer chain at the aqueous/organic interface and the enhanced rhodium catalytic activity at high temperature. However, exceedingly increasing the reaction temperature up to 150 °C had a detrimental effect on both the aldehyde selectivity and the linear to branched aldehyde ratio (entry 6) suggesting a partial decomposition of the Rh-catalyst. Indeed, the temperature influences both the distribution of products due to enhanced isomerization of the carbon–carbon double bond of 1-dodecene and the dissociation of ligands from the catalytic complexes.67
Table 4 Conversions and selectivities in the Rh-catalyzed hydroformylation of 1-dodecene for different reaction temperatures using Tetronic® 90R4 for 1 ha
| Entry |
T (°C) |
Conv.b (%) |
Aldehyde sel.b (%) |
l/bb |
TOF (h−1) |
|
Conditions: 1-dodecene (1.63 mmol), Rh(CO)2(acac) (3 mg, 0.012 mmol), TPPTS (33 mg, 0.058 mmol), Tetronic® 90R4 (25 wt%, 1500 mg, 0.21 mmol), α-CD (870 mg, 0.90 mmol), 50 bar of CO/H2, 6 mL H2O, 1 h.
Conversions and selectivities were determined by 1H NMR.
|
| 1 |
30 |
1 |
73 |
2.4 |
1 |
| 2 |
50 |
14 |
99 |
2.4 |
19 |
| 3 |
70 |
54 |
95 |
2.4 |
73 |
| 4 |
90 |
75 |
90 |
2.5 |
95 |
| 5 |
110 |
81 |
90 |
2.4 |
110 |
| 6 |
150 |
87 |
47 |
2.0 |
118 |
With the proportion of phosphane low-coordinated Rh-species being more important, the linear to branched aldehyde ratio logically dropped at high reaction temperatures. From the initial catalytic activity values (TOF) obtained over the 30–150 °C temperature range (Table 4), the activation energy Ea was calculated from a classical Arrhenius plot. The resulting graph consists of two straight-line segments that intersect at about 60 °C (Fig. 7). The calculated Ea is significantly higher at low temperature (86.6 kJ mol−1) than at high temperature (4.3 kJ mol−1). Ea at low temperatures (below 60 °C) is relatively close to that reported for hydroformylation of 1-dodecene in the presence of homogeneous catalytic systems (57.3 kJ mol−1).68–70 The apparent Ea obtained below 60 °C is far too high to represent a mass transfer process, and likely corresponds to a chemical rate determining step (e.g. H2 oxidative addition). Indeed, similar values have been reported in the literature for rhodium-catalyzed hydroformylation of olefins in homogeneous media (40–75 kJ mol−1).71 Conversely, the low Ea value obtained above 60 °C is typical of a process in which the mass transfer is the limiting step.72
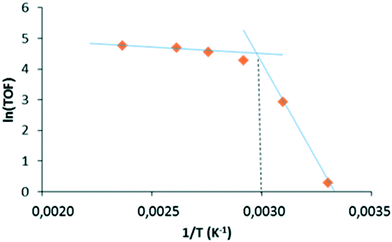 |
| | Fig. 7 Initial rate dependence on the reaction temperature. | |
Rhodium precursors
[Rh(CO)2(acac)] was compared with other Rh-precursors (Table 5) in the catalyzed hydroformylation of 1-dodecene. [Rh(CO)2(acac)] was the best rhodium precursor in terms of catalytic activity and aldehyde selectivity (entry 1). Astonishingly, comparable catalytic results were obtained using RhCl3 as the rhodium precursor (entry 2), indicating that the Rh-catalyst was not affected by the HCl released from RhCl3 under CO/H2 pressure. In addition to the positive effect of the Tetronic® 90R4/α-CD/water combination on the interface, this catalytic result underscores another advantage in the use of Tetronics® in Rh-catalyzed hydroformylation of alkenes. Indeed, contrary to standard observations showing that halides perturb (or even inhibit) the catalytic activity under hydroformylation conditions,2,73 Tetronic® nitrogen atoms probably act as halide scavengers and help neutralize the released HCl in the form of ammonium chloride. The cationic Rh(COD)2BF4 precursor, for its part, led to a lower 62% conversion (entry 3). Note that, whatever the rhodium precursor, the chemo-/regioselectivity ratio remained rather constant over time.
Table 5 Effect of the catalytic rhodium precursor in the metal-catalyzed hydroformylation/hydrogenation of 1-dodecenea
| Entry |
Rhodium precursor |
Conv.b (%) |
Aldehyde sel.b (%) |
l/bb |
|
Conditions: 1-dodecene (1.63 mmol), rhodium precursor (0.012 mmol), TPPTS (33 mg, 0.058 mmol), Tetronic® 90R4 (25 wt%, 1500 mg, 0.21 mmol), α-CD (870 mg, 0.90 mmol), 50 bar of CO/H2, 6 mL H2O, 80 °C, 1 h.
Conversions and selectivities were determined by 1H NMR.
|
| 1 |
Rh(CO)2(acac) |
71 |
93 |
2.3 |
| 2 |
RhCl3 |
71 |
91 |
2.4 |
| 3 |
Rh(COD)2BF4 |
62 |
89 |
2.5 |
Phosphanes
Having established favorable conditions for 1-dodecene hydroformylation using TPPTS as a water soluble ligand, hydroformylation reactions were investigated using other phosphanes. A survey of phosphanes depicted in Fig. 8 revealed that the nature of the phosphane significantly influenced the catalytic performance (Table 6). Tris(oOMe)TPPTS led to a meager conversion (<10%) and low chemoselectivity because of its poor ability to coordinate the rhodium complex (entry 3). Conversely, DBPPTS and TrisBiph provided high conversions and aldehyde selectivities (entries 5 and 7). Compared to our previously published results on the utilization of Pickering-like emulsions in the presence of RAME-β-CD,36 the chemo- and regioselectivities were improved under the same experimental conditions suggesting a more discriminating process during the catalytic cycle. For example, while the aldehyde selectivity and l/b ratio were 94% and 1.8, respectively, in the hydroformylation of 1-dodecene using Pickering emulsions, the α-CD/Tetronic® 90R4 combination led to an aldehyde selectivity of 98% and a l/b ratio of 2.3 in the presence of DBPPTS.
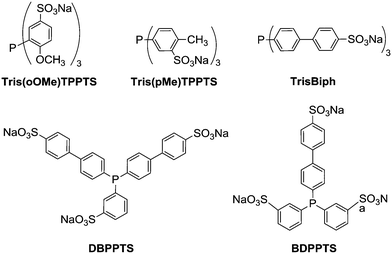 |
| | Fig. 8 Studied phosphanes in hydroformylation of 1-dodecene. | |
Table 6 Rh-catalyzed hydroformylation of 1-dodecene using various phosphanesa
| Entry |
Phosphanes |
Conv.b (%) |
Aldehyde sel.b (%) |
l/bb |
|
Conditions: substrate (1.63 mmol), Rh(CO)2(acac) (3 mg, 0.012 mmol), phosphane (33 mg, 0.058 mmol), Tetronic® 90R4 (1500 mg, 0.21 mmol), α-CD (870 mg, 0.90 mmol), 6 mL H2O, 80 °C, 50 bar CO/H2, 1 h.
Conversions and selectivities were determined by 1H NMR.
|
| 1 |
— |
5 |
61 |
1.5 |
| 2 |
TPPTS |
65 |
83 |
2.3 |
| 3 |
Tris(oOMe)TPPTS |
4 |
66 |
2.9 |
| 4 |
Tris(pMe)TPPTS |
92 |
91 |
2.3 |
| 5 |
DBPPTS |
99 |
98 |
2.3 |
| 6 |
BDPPTS |
87 |
92 |
2.4 |
| 7 |
TrisBiph |
100 |
96 |
2.3 |
By using Tris(pMe)TPPTS and BDPPTS, Rh-catalyzed hydroformylation of 1-dodecene delivered the corresponding products in 92% and 87% conversion, respectively, and similar aldehyde selectivity (entries 4 and 6). Note that the l/b ratio remained rather constant (2.3–2.4) with phosphane-stabilized rhodium catalytic species with high catalytic activity and was significantly improved to 2.9 by using phosphane-stabilized rhodium species with lower catalytic activity.
Hydroformylation of other substrates
To broaden the scope of the utilization of Tetronic® 90R4 in the biphasic Rh-catalyzed hydroformylation of alkenes, other substrates were considered (Table 7). The catalytic activity of the hydroformylation reaction decreases with increasing chain length of the substrate because the higher hydrophobic character of 1-octadecene disfavored contacts with the water-soluble catalyst (entry 3). However, the 80% conversion obtained within the 4 h reaction time favorably compared with our previous results.36 More surprisingly, the chemo- and regioselectivity were also affected, indicative of a less discriminating catalytic process for longer alkenyl chains. Styrene and 2-vinylnaphtalene were fully converted within 4 h with a high proportion of branched aldehydes attributed to the formation of an η3-benzyl-Rh complex (entries 4 and 5).74 Note that the catalytic system proves to be ineffective to hydroformylate internal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds (entry 6).
C double bonds (entry 6).
Table 7 Hydroformylation of hydrophobic substratesa
| Entry |
Substrate |
Conv.b (%) |
Aldehyde sel.b (%) |
l/bb |
|
Conditions: substrate (1.63 mmol), Rh(CO)2(acac) (3 mg, 0.012 mmol), TPPTS (33 mg, 0.058 mmol), Tetronic® 90R4 (1500 mg, 0.21 mmol), α-CD (870 mg, 0.90 mmol), 6 mL H2O, 80 °C, 50 bar CO/H2, 4 h.
Conversions and selectivities were determined by 1H NMR.
|
| 1 |
1-Dodecene |
100 |
95 |
2.2 |
| 2 |
1-Tetradecene |
100 |
88 |
2.2 |
| 3 |
1-Octadecene |
80 |
80 |
1.9 |
| 4 |
Styrene |
100 |
99 |
0.1 |
| 5 |
2-Vinylnaphthalene |
100 |
99 |
0.1 |
| 6 |
Methyl oleate |
0 |
— |
— |
Reusability of the Rh-catalyst
The reusability of the catalytic system was assessed over three consecutive runs. After each run, the organic phase was recovered and the catalyst-containing hydrogel phase was recycled. The main advantage of the process lies in the easy separation of the liquid organic phase and the gel phase once the reaction mixture was cooled down. Very good reusability of the Rh-catalyst was obtained after the second and the third run. Conversions and selectivities remained unchanged after each run (Table 8). The stability of the Rh-complex embedded in the hydrogel matrix was confirmed by 31P NMR measurements. Samples of HRh(CO)(TPPTS)3 (synthesized from Rh(CO)2(acac) and excess TPPTS, see the ESI†) in water or in Tetronic® 90R4/α-CD/water mixtures were left in air. In water, a regular decrease in the intensity of the doublet at 43 ppm related to the coordination of TPPTS onto Rh (1JRh–P = 155 Hz) was rapidly observed over a period of 120 h (Fig. 9 and 10a). Conversely, good stability of the Rh-complex was observed at 40 °C in the sol phase of a Tetronic® 90R4/α-CD/water mixture (Fig. 9 and 10b). Gratifyingly, excellent stability of HRh(CO)(TPPTS)3 was observed at 25 °C in the gel phase (Fig. 9 and 10c).
Table 8 Reusability of the catalyst-containing Tetronic® 90R4/α-CD/water gel phasea
| Entry |
Conv.b (%) |
Aldehyde sel.b (%) |
l/bb |
|
Conditions: 1-dodecene (1.63 mmol), Rh(CO)2(acac) (3 mg, 0.012 mmol), TPPTS (33 mg, 0.058 mmol), Tetronic® 90R4 (1500 mg, 0.21 mmol), α-CD (870 mg, 0.90 mmol), 6 mL H2O, 80 °C, 50 bar CO/H2, 1 h.
Conversions and selectivities were determined by 1H NMR.
|
| 1 |
65 |
83 |
2.3 |
| 2 |
64 |
84 |
2.2 |
| 3 |
64 |
82 |
2.3 |
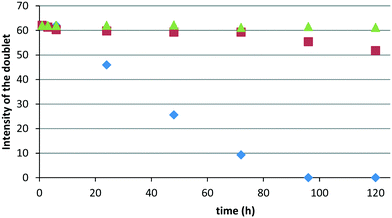 |
| | Fig. 9 Change in the intensity of the doublet (Rh–P coupling) over time a) in water at 25 °C ( ), in the sol phase at 40 °C ( ), in the sol phase at 40 °C ( ), and in the gel phase at 25 °C ( ), and in the gel phase at 25 °C ( ). Conditions for ). Conditions for  : HRh(CO)(TPPTS)3 (3 mg, 0.012 mmol), 6 mL H2O. Conditions for : HRh(CO)(TPPTS)3 (3 mg, 0.012 mmol), 6 mL H2O. Conditions for  and and  : HRh(CO)(TPPTS)3 (3 mg, 0.012 mmol), Tetronic® 90R4 (1500 mg, 0.21 mmol), α-CD (870 mg, 0.90 mmol), 6 mL H2O. : HRh(CO)(TPPTS)3 (3 mg, 0.012 mmol), Tetronic® 90R4 (1500 mg, 0.21 mmol), α-CD (870 mg, 0.90 mmol), 6 mL H2O. | |
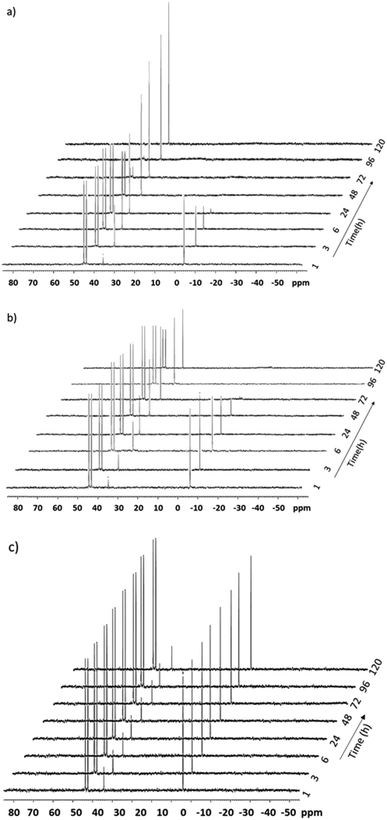 |
| | Fig. 10 Stacked 31P NMR spectra of HRh(CO)(TPPTS)3 in the presence of excess TPPTS over time a) in neat water at 25 °C, b) in the sol phase at 40 °C, and c) in the gel phase at 25 °C. | |
Concurrently, while excess TPPTS was rapidly oxidized in water, the oxidation process was moderate in the sol phase at 40 °C and remarkably low in the gel phase at room temperature (<5%), thus revealing the protecting role of the hydrogel matrix toward the phosphane against oxidation and thus toward the Rh-catalyst. Obviously, the stability of the HRh(CO)(TPPTS)3 complex is far higher under hydrogen, even in the sol phase (ESI,† Fig. S1). Also note that no displacement of the phosphane ligands by the nitrogens of Tetronic® 90R4 could be detected by 31P NMR. Accordingly, once embedded in the Tetronic® 90R4/α-CD/water gel phase, the Rh-catalyst does not undergo significant alteration over time resulting in unchanged catalytic performances upon recycling. Moreover, only traces of Tetronic® 90R4 were detected in the organic phase (0.3 mol%) after each run (ESI,† Fig. S2).
Conclusions
In our constant effort to develop green chemical processes, the studied Tetronic®-based catalytic systems represent an alternative to classical aqueous biphasic hydroformylation systems. The best result was obtained using the reverse-sequential Tetronic® 90R4 and DBPPTS under 50 bar CO/H2 pressure at 80 °C. Under such conditions, 1-dodecene was fully hydroformylated within 1 h with an aldehyde selectivity of 98% and a linear to branched ratio of 2.3. When associated with α-CD, Tetronics® contain within their structure the amphiphilic properties allowing the mass transfer limitation to be overcome at the aqueous/organic interface. The presence of α-CD is essential to provoke the decantation of the multiphasic system once the reaction is complete. The catalytic performance was significantly influenced by the reaction temperature while attempts to enhance the performance of the Rh-catalyst through variation of CO/H2 pressure did not result in additional improvement. The main advantage of Tetronic®/α-CD assemblies in biphasic catalysis lies in the easy recovery of the organic product in one phase and the Rh-catalyst in another phase. While the Rh-catalyst embedded into the Tetronic®/α-CD/water mixture remains in the flask (in the gel sate at room temperature), the liquid aldehydes are recovered by simply inclining the flask containing the reaction mixture. Additionally, the stability of the Rh-catalyst is remarkable as the Tetronic®/α-CD/water gel phase prevents the oxidation of the Rh-coordinated phosphane at room temperature.
Acknowledgements
Chevreul Institute (FR 2638), Ministère de l'Enseignement Supérieur et de la Recherche, Région Nord – Pas de Calais and FEDER are acknowledged for supporting and partially funding this work. We also thank Dr. Nicolas Kania and D. Prevost for technical assistance.
Notes and references
-
O. Roelen, Ruhrchemie AG, Patent DE 849548, 1938.
- L. Obrecht, P. C. J. Kamer and W. Laan, Catal. Sci. Technol., 2013, 3, 541–551 CAS.
-
P. C. J. Kamer, J. N. H. Reek and P. W. N. M. van Leeuwen in Rhodium Catalyzed Hydroformylation, Kluwer Academic Publishers, 2000 Search PubMed.
- C. W. Kohlpaintner, R. W. Fischer and B. Cornils, Appl. Catal., A, 2001, 221, 219–225 CrossRef CAS.
-
E. Wiebus, K. Schmid and B. Cornils, In Science of Synthesis: Water in Organic Synthesis, ed. S. Kobayashi, Georg Thieme Verlag, Stuttgart, Germany, chap 7.1, 2012 Search PubMed.
- C. Li, H. Fu, R. Zhang, W. Liu, C. Yang, H. Chen and X. Li, Chin. J. Catal., 2008, 29, 895–900 CAS.
- H. Bricout, F. Hapiot, A. Ponchel, S. Tilloy and E. Monflier, Curr. Org. Chem., 2010, 14, 1296–1307 CrossRef CAS.
- I. T. Horváth and J. Rábai, Science, 1994, 266, 72–75 Search PubMed.
- E. Perperi, Y. Huang, P. Angeli, G. Manos, C. R. Mathison, D. J. Cole-Hamilton, D. J. Adams and E. G. Hope, Dalton Trans., 2004, 2062–2064 RSC.
- S. L. Desset, S. W. Reader and D. J. Cole-Hamilton, Green Chem., 2009, 11, 630–637 RSC.
- A. Riisager, R. Fehrmann, S. Flicker, R. van Hal, M. Haumann and P. Wasserscheid, Angew. Chem., Int. Ed., 2005, 44, 815–819 CrossRef CAS PubMed.
- M. Haumann, K. Dentler, J. Joni, A. Riisager and P. Wasserscheid, Adv. Synth. Catal., 2007, 349, 425–431 CrossRef CAS.
- S. L. Desset, D. J. Cole-Hamilton and D. F. Foster, Chem. Commun., 2007, 1933–1935 RSC.
- H. Nowothnick, A. Rost, T. Hamerla, R. Schomäcker, C. Müller and D. Vogt, Catal. Sci. Technol., 2013, 3, 600–605 CAS.
- A. Rost, Y. Brunsch, A. Behr and R. Schomäcker, Chem. Eng. Technol., 2014, 37, 1055–1064 CrossRef CAS.
- M. Schwarze, T. Pogrzeba, K. Seifert, T. Hamerla and R. Schomäcker, Catal. Today, 2015, 247, 55–63 CrossRef CAS.
- S. C. Stouten, T. Noël, Q. Wang and V. Hessel, Chem. Eng. Process., 2014, 83, 26–32 CrossRef CAS.
- M. Janssen, J. Wilting, C. Müller and D. Vogt, Angew. Chem., Int. Ed., 2010, 49, 7738–7741 CrossRef CAS PubMed.
- J. Fang, R. Jana, J. A. Tunge and B. Subramaniam, Appl. Catal., A, 2011, 393, 294–301 CrossRef CAS.
- A. Schönweiz, J. Debuschewitz, S. Walter, R. Wölfel, H. Hahn, K. M. Dyballa, R. Franke, M. Haumann and P. Wasserscheid, ChemCatChem, 2013, 5, 2955–2963 CrossRef.
- D. Koch and W. Leitner, J. Am. Chem. Soc., 1998, 120, 13398–13404 CrossRef CAS.
- D. R. Palo and C. Erkey, Organometallics, 2000, 19, 81–86 CrossRef CAS.
- P. B. Webb, M. F. Sellin, T. E. Kunene, S. Williamson, A. M. Z. Slawin and D. J. Cole-Hamilton, J. Am. Chem. Soc., 2003, 125, 15577–15588 CrossRef CAS PubMed.
- T. E. Kunene, P. B. Webb and D. J. Cole-Hamilton, Green Chem., 2011, 13, 1476–1481 RSC.
- A. C. Frisch, P. B. Webb, G.-Y. Zhao, M. J. Muldoon, P. J. Pogorzelec and D. J. Cole-Hamilton, Dalton Trans., 2007, 5531–5538 RSC.
- U. Hintermair, Z.-X. Gong, A. Serbanovic, M. J. Muldoon, C. C. Santini and D. J. Cole-Hamilton, Dalton Trans., 2010, 39, 8501–8510 RSC.
- U. Hintermair, G.-Y. Zhao, C. C. Santini, M. J. Muldoon and D. J. Cole-Hamilton, Chem. Commun., 2007, 1462–1464 RSC.
- T. Pogrzeba, D. Müller, M. Illner, M. Schmidt, Y. Kasaka, A. Weber, G. Wozny, R. Schomäcker and M. Schwarze, Chem. Eng. Process., 2016, 99, 155–166 CrossRef CAS.
- E. Lobry, A. F. Cardozo, L. Barthe, J.-F. Blanco, H. Delmas, S. Chen, F. Gayet, X. Zhang, M. Lansalot, F. D'Agosto, R. Poli, E. Manoury and C. Julcour, J. Catal., 2016, 342, 164–172 CrossRef CAS.
- S. L. Bourne, M. O'Brien, S. Kasinathan, P. Koos, P. Tolstoy, D. X. Hu, R. W. Bates, B. Martin, B. Schenkel and S. V. Ley, ChemCatChem, 2013, 5, 159–172 CrossRef CAS.
- M. L. Abrams, J. Y. Buser, J. R. Calvin, M. D. Johnson, B. R. Jones, G. Lambertus, C. R. Landis, J. R. Martinelli, S. A. May, A. D. McFarland and J. R. Stout, Org. Process Res. Dev., 2016, 20, 901–910 CrossRef CAS.
- M. Ferreira, F. Jérôme, H. Bricout, S. Menuel, D. Landy, S. Fourmentin, S. Tilloy and E. Monflier, Catal. Commun., 2015, 63, 62–65 CrossRef CAS.
- F. Jérôme, M. Ferreira, H. Bricout, S. Menuel, E. Monflier and S. Tilloy, Green Chem., 2014, 16, 3876–3880 RSC.
- L. Voorhaar and R. Hoogenboom, Chem. Soc. Rev., 2016, 45, 4013–4031 RSC.
- J. Potier, S. Menuel, E. Monflier and F. Hapiot, ACS Catal., 2014, 4, 2342–2346 CrossRef CAS.
- J. Potier, S. Menuel, M.-H. Chambrier, L. Burylo, J.-F. Blach, P. Woisel, E. Monflier and F. Hapiot, ACS Catal., 2013, 3, 1618–1621 CrossRef CAS.
- J. González-López, C. Alvarez-Lorenzo, P. Taboada, A. Sósnik, I. Sández-Macho and A. Concheiro, Langmuir, 2008, 24, 10688–10697 CrossRef PubMed.
- E. Larrañeta and J. R. Isasi, Langmuir, 2013, 29, 1045–1053 CrossRef PubMed.
- S. Tan, K. Ladewig, Q. Fu, A. Blencowe and G. G. Qiao, Macromol. Rapid Commun., 2014, 35, 1166–1184 CrossRef CAS PubMed.
- E. Larrañeta and J. R. Isasi, Langmuir, 2012, 28, 12457–12462 CrossRef PubMed.
-
C. Álvarez-Lorenzo, A. Concheiro and A. Sosnik, in Handbook of Hydrogels: Properties, Preparation & Applications, Nova Science Publishers Inc., Hauppauge, NY, 2011, pp. 449–484 Search PubMed.
- J. Zhang, A. Sen, E. Cho, J. S. Lee and K. Webb, J. Tissue Eng. Regener. Med., 2014 DOI:10.1002/term.1906.
- C. A. Estévez, J. R. Isasi, E. Larrañeta and I. Vélaz, Beilstein J. Org. Chem., 2014, 10, 3127–3135 CrossRef PubMed.
- E. Larrañeta and J. R. Isasi, Carbohydr. Polym., 2014, 102, 674–681 CrossRef PubMed.
- S. M. Moghimi and A. C. Hunter, Trends Biotechnol., 2000, 18, 412–420 CrossRef CAS PubMed.
- J. Shen, G. Xu, X. Xin, L. Wang, Z. Song, H. Zhang, L. Tonga and Z. Yang, RSC Adv., 2015, 5, 40173–40182 RSC.
- A. Sosnik, B. M. Leung and M. V. Sefton, J. Biomed. Mater. Res., Part A, 2007, 86, 339–353 Search PubMed.
- C. Alvarez-Lorenzo, A. Rey-Rico, J. Brea, M. I. Loza, A. Concheiro and A. Sosnik, Nanomedicine, 2010, 5, 1371–1383 CrossRef CAS PubMed.
- Y. Y. Yu, W. W. Zhu, H. Li, H. M. Wang and Z. S. Hou, J. Colloid Interface Sci., 2014, 415, 117–126 CrossRef CAS PubMed.
- B. Jing, X. Chen, X. Wang, Y. Zhao and H. Qiu, ChemPhysChem, 2008, 9, 249–252 CrossRef CAS PubMed.
- B. C. Ledesma, V. A. Vallés, L. P. Rivoira, M. L. Martínez, O. A. Anunziata and A. R. Beltramone, Catal. Lett., 2014, 144, 783–795 CrossRef CAS.
- J. Ma, L. S. Qiang, J. F. Wang, D. Y. Tang and X. B. Tang, Catal. Lett., 2011, 141, 356–363 CrossRef CAS.
- J. Yang, L. Lukashuk, H. Li, K. Föttinger, G. Rupprechter and U. Schubert, Catal. Lett., 2014, 144, 403–412 CrossRef CAS PubMed.
- S. M. N. Simões, F. Veiga, J. J. Torres-Labandeira, A. C. F. Ribeiro, M. I. Sandez-Macho, A. Concheiro and C. Alvarez-Lorenzo, Eur. J. Pharm. Biopharm., 2012, 80, 103–112 CrossRef PubMed.
- J. Li, X. Li, Z. Zhou, X. Ni and K. W. Leong, Macromolecules, 2001, 34, 7236–7237 CrossRef CAS.
- A. Harada and M. Kamachi, Macromolecules, 1990, 23, 2821–2823 CrossRef CAS.
- P. Alexandridis and T. Hatton, Colloids Surf., A, 1995, 96, 1–46 CrossRef CAS.
- L. Trong, M. Djabourov and A. Ponton, J. Colloid Interface Sci., 2008, 328, 278–287 CrossRef CAS PubMed.
- C. A. G. Carter, R. T. Baker, S. P. Nolan and W. Tumas, Chem. Commun., 2000, 347–348 RSC.
-
Molecular Encapsulation: Organic Reactions in Constrained Systems, ed. U. H. Brinker and J.-L. Mieusset, Wiley, New York, 2010 Search PubMed.
- G. I. Vagapova, F. G. Valeeva, G. A. Gainanova, V. V. Syakaev, I. V. Galkina, L. Y. Zakharova, S. K. Latypov and A. I. Konovalov, Colloids Surf., A, 2013, 419, 186–193 CrossRef CAS.
- T. Dwars, E. Paetzold and G. Oehme, Angew. Chem., Int. Ed., 2005, 44, 7174–7199 CrossRef CAS PubMed.
- J. Gonzalez-Lopez, C. Alvarez-Lorenzo, P. Taboada, A. Sosnik, I. Sandez-Macho and A. Concheiro, Langmuir, 2008, 24, 10688–10697 CrossRef CAS PubMed.
- M. Ferreira, H. Bricout, N. Azaroual, D. Landy, S. Tilloy, F. Hapiot and E. Monflier, Adv. Synth. Catal., 2012, 354, 1337–1346 CrossRef CAS.
- N. Six, A. Guerriero, D. Landy, M. Peruzzini, L. Gonsalvi, F. Hapiot and E. Monflier, Catal. Sci. Technol., 2011, 1, 1347–1353 CAS.
- L. Gonsalvi, A. Guerriero, E. Monflier, F. Hapiot and M. Peruzzini, Top. Curr. Chem., 2013, 342, 1–48 CrossRef CAS PubMed.
-
P. W. N. M. van Leeuwen, in Homogeneous Catalysis: Understanding the Art, Kluwer Academic Publishers, ch. 7, 2004 Search PubMed.
- B.-M. Bhanage, S.-S. Divekar, R.-M. Deshpande and R.-V. Chaudhari, J. Mol. Catal., 1997, 115, 247–257 CrossRef CAS.
- S.-S. Divekar, R.-M. Deshpande and R.-V. Chaudhari, Catal. Lett., 1993, 21, 191–200 CrossRef CAS.
- In Aqueous-Phase Organometallic Catalysis-Concepts and Applications, ed. R. V. Chaudhari, B. Bhanage, B. Cornils and W.A. Herrmann, VCH, Weinheim, 1998, pp. 283–294 Search PubMed.
- G. Fremy, E. Monflier, J. F. Carpentier, Y. Castanet and A. Mortreux, J. Catal., 1996, 162, 339–348 CrossRef CAS.
- T. Mathivet, C. Méliet, Y. Castanet, A. Mortreux, L. Caron, S. Tilloy and E. Monflier, J. Mol. Catal. A: Chem., 2001, 176, 105–116 CrossRef CAS.
- J. C. Bayón, J. Real, C. Claver, A. Polo and A. Ruiz, J. Chem. Soc., Chem. Commun., 1989, 1056–1057 RSC.
-
P. W. N. M. van Leeuwen and C. Claver, Rhodium Catalyzed Hydroformylation, Kluwer Academic Publishers, Dordrecht, 2000, pp. 29–31 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods – catalytic experiments, physico-chemical properties of poloxamines, optical photos. See DOI: 10.1039/c6cy02070d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
F.
Hapiot
*
and
F.
Hapiot
*











 ), in the sol phase at 40 °C (
), in the sol phase at 40 °C ( ), and in the gel phase at 25 °C (
), and in the gel phase at 25 °C ( ). Conditions for
). Conditions for  : HRh(CO)(TPPTS)3 (3 mg, 0.012 mmol), 6 mL H2O. Conditions for
: HRh(CO)(TPPTS)3 (3 mg, 0.012 mmol), 6 mL H2O. Conditions for  and
and  : HRh(CO)(TPPTS)3 (3 mg, 0.012 mmol), Tetronic® 90R4 (1500 mg, 0.21 mmol), α-CD (870 mg, 0.90 mmol), 6 mL H2O.
: HRh(CO)(TPPTS)3 (3 mg, 0.012 mmol), Tetronic® 90R4 (1500 mg, 0.21 mmol), α-CD (870 mg, 0.90 mmol), 6 mL H2O.