
Organic-free synthesis of {001} facet dominated BiOBr nanosheets for selective photoreduction of CO2 to CO†
Dan
Wu
a,
Liqun
Ye
*ab,
Ho Yin
Yip
a and
Po Keung
Wong
*a
aSchool of Life Sciences, The Chinese University of Hong Kong, Shatin, N.T, Hong Kong SAR, China. E-mail: pkwong@cuhk.edu.hk
bCollege of Chemistry and Pharmaceutical Engineering, Nanyang Normal University, Nanyang 473061, China. E-mail: yeliquny@163.com
First published on 1st December 2016
Abstract
{001} facet dominated BiOBr nanosheets are fabricated via a facile hydrothermal method in the presence of nitric acid without any organic additive and applied for CO2 photoreduction. The concentration of nitric acid easily regulates the thickness of the obtained BiOBr nanosheets. When employing concentrations of 0, 0.1, 0.5, 1 and 4 M nitric acid, the corresponding surface area and percentage of exposed {001} facets of BiOBr-0, BiOBr-0.1, BiOBr-0.5, BiOBr-1 and BiOBr-4 nanosheets significantly increase in sequence. BiOBr-0 nanosheets are incapable of converting CO2 into CO. However, BiOBr-0.1, BiOBr-0.5, BiOBr-1 and BiOBr-4 nanosheets show successively enhanced CO2 photoreduction performance. Surprisingly, they exhibit high selectivity for converting CO2 into CO with negligible generation of CH4. In particular, BiOBr-4 shows the highest CO production rate of 4.45 μmol g−1 h−1 under simulated sunlight irradiation. The electronic structure analysis demonstrates that the conduction band minimum is significantly raised to endow BiOBr-4 with reduction power for CO2/CO conversion, in comparison with the incapability of BiOBr-0. The breakthrough in CO2 reduction of BiOBr-4 nanosheets is ascribed to the larger active surface area, higher electron transfer, more effective charge carrier separation and significantly raised reduction ability.
Introduction
The capture and efficient conversion of CO2 into value-added chemical fuels using renewable solar energy is receiving particular attention as a long-term promising solution to address CO2-mediated global warming and the depletion of fossil fuels.1,2 In principle, CO2 can be reduced to form reusable CO via a 2e−/2H+ reduction process. With further water-gas shift reaction, syngas (CO/H2 mixture) can be achieved and utilized to yield synthetic liquid fuels for direct energy storage and distribution.3 Therefore, it is of significance to discover efficient approaches to selectively convert CO2 into CO at normal atmospheric pressure. Among various currently identified methods, photocatalytic reduction of CO2 powered by renewable solar energy is considered as a potentially desirable “green” strategy.To succeed in photocatalytic CO2 reduction and conversion, a great deal of effort has been devoted to developing efficient semiconductors as photocatalysts. Among various studied photocatalysts, numerous efforts have been attempted on a variety of layered nanomaterials due to their electrical polarization effect, which can facilitate the separation and migration of photoexcited charge carriers.4,5 Bismuth oxyhalogen BiOX (X = Cl, Br and I) nanostructures have attracted considerable attention owing to their unique structure-dependent photocatalytic activities under UV or visible light (VL) irradiation. In particular, because the [Bi2O2] unit interleaved with a double halogen layer tends to stack along the [001] axis, BiOX easily forms two-dimensional (2D) nanosheets with exposed {001} facets. Pioneering studies have revealed that the {001} facets are highly active for BiOX 2D materials in photocatalytic applications, such as dye degradation,6 microbial disinfection,7,8 organics decomposition9 and nitrogen fixation.10 However, it is difficult to realize CO2 conversion on {001}-facet exposed BiOX 2D nanostructures due to their relatively low conduction band minimum energy. Given this predicament, two strategies, including halogen deletion to form a new phase11 and oxygen vacancy creation,12–14 were usually employed to construct BiOX to achieve photocatalytic CO2 reduction activity. However, few reports have been attempted on applying the defect-free BiOX nanosheets themselves for CO2 photoreduction.15 In particular, the CO2 photoreduction activity of {001}-facet exposed BiOBr nanosheets without oxygen vacancies has not been addressed.
Since highly reactive crystal facets functionalize compounds with fantastic chemical activities, considerable efforts have been devoted to obtaining a high percentage of exposed active facets of semiconductors.16–18 As for the BiOX photocatalysts, the typically used hydrothermal routes can easily endow them with highly active {001} facets, but usually with large thicknesses. Therefore, during the synthetic procedure, various organics are employed to act as capping agents, such as cetyltrimethylammonium bromide,11 poly(vinyl pyrrolidone)19,20 and polyethylene glycol21 to control the growth rate of the {001} facets. Besides, due to the hydrolysis characteristics of Bi3+ ions in aqueous solution, various kinds of organics, such as ethanol,11,15 ethylene glycol,9,12,14 oleic acid13 and octadecylene,22 are also applied as solvents instead of water to retard the hydrolysis. In fact, both capping agents and organic solvents are included in the aforementioned cases. Nevertheless, polar organics may have specific molecular interactions with respect to the preferential growth orientations of crystals,23 further affecting the final exposed facets. Moreover, organics prefer to physically or chemically attach onto the surface of photocatalysts to strongly cover the corresponding active sites, which is detrimental to the photocatalytic performance. Specially, CO2 is slightly acidic, and thus the surface properties of photocatalysts play a significant role in achieving efficient CO2 capture and its subsequent conversion.24 Hence, simple synthesis of a high percentage of {001}-facet exposed BiOBr nanosheets with reduced lateral size in the absence of any organics is still challenging.
In this work, BiOBr nanosheets with exposed {001} facets were synthesized via a hydrothermal method without any organic additive and the thickness of the obtained nanosheets can be simply modulated by different concentrations of nitric acid. The CO2 photoreduction activities of a series of BiOBr nanosheets were investigated under simulated sunlight irradiation. The electronic structure and photogenerated charge carrier transfer and separation of photocatalysts were systematically studied to elucidate their superior CO2 conversion ability.
Experimental
Synthesis of BiOBr nanosheets
BiOBr nanosheets were prepared via a hydrothermal method. Typically, equivalent molar amounts of Bi(NO3)3·5H2O and KBr were dissolved in 30 mL diluted HNO3 (x M) and 30 mL distilled water, respectively. The two solutions were mixed together under stirring for 30 min at room temperature (ca. 25 °C). Subsequently, the mixture was transferred into a Teflon-lined steel autoclave and maintained at 160 °C for 12 h. Finally, the precipitate was centrifuged, washed with distilled water and dried at 60 °C for 24 h. The obtained product was denoted as BiOBr-x, where x refers to the concentration of diluted HNO3 solution (x = 0, 0.1, 0.5, 1 and 4).Characterization
The crystal information was provided by a SmartLab X-ray diffraction (XRD) diffractometer (Rigaku, Japan) with Cu Kα radiation. The morphology information was obtained on a Quanta 400F field-emission scanning electron microscope (SEM, FEI, USA) and a Tecnai F20 high resolution transmission electron microscope (HRTEM, FEI, USA). Nitrogen adsorption–desorption experiments were conducted at 77 °K on an Autosorb-1 sorption analyzer (Quantachrome, USA). The surface area was calculated based on the Brunauer–Emmett–Teller (BET) method. X-ray photoelectron spectroscopy (XPS) was performed on an AXIS-ULTRA DLD-600 W spectrometer (Shimadzu-Kratos, Japan). The electrochemical measurements were conducted using a CHI760C electrochemical working station (CH Instruments, China) in a three-electrode quartz cell. UV-vis diffuse reflectance spectra were measured by a Lambda 35 UV-vis spectrophotometer (PerkinElmer, USA) using BaSO4 as the background. Room-temperature photoluminescence (PL) spectra were obtained using a FP-6500 fluorescence spectrometer (Jasco, Japan).Photocatalytic CO2 reduction
The photocatalytic CO2 reduction activity of the as-prepared samples was measured in a 500 mL PLS-SXE300 Labsolar-IIIAG closed gas system (Perfectlight, China). Fifty mg of the photocatalyst powder was evenly dispersed on a watch-glass with an area of ca. 28 cm2. A xenon lamp (300C, Perfectlight, China) with a focus intensity of 0.21 W cm−2 was used to simulate sunlight. To obtain CO2 gas with 1 atm pressure, 1.7 g NaHCO3 was put into the reaction cell followed by thorough vacuum treatment, and then 4 M H2SO4 (5 mL) was injected into the reaction cell. The reaction temperature was kept at 20 °C by a DC-0506 low-temperature thermostat bath system (Sunny Hengping Scientific Instrument Co., Ltd., China). At given time intervals, 1 mL of the generated gas was collected by a syringe injector and then measured by a GC9790II gas chromatograph (Zhejiang Fuli Analytical Instrument Co., Ltd., China) equipped with a GDX-502 flame ionization detector and a TDX-01 thermal conductivity detector. The outlet gases were determined to be CH4, CO and CO2, respectively, and their corresponding yields were estimated according to a standard curve.Results and discussion
The XRD patterns of the obtained samples are shown in Fig. S1.† All the diffraction peaks can be indexed to tetragonal BiOBr (PDF 01-078-0348), indicating that BiOBr-0 and BiOBr-4 samples were successfully synthesized with pure phases. The morphology of the two samples was characterized by SEM. As shown in Fig. 1a, BiOBr-0 exhibits a two-dimensional nanosheet-like shape with a diameter mainly ranging from 1.9 to 4.8 μm. The average thickness of these nanosheets is estimated to be 319 nm (Fig. 1b). As can be seen, the nanosheet shape is also found for BiOBr-4 (Fig. 1c). However, the BiOBr-4 nanosheets are much larger with diameters from 6.8 to 26.4 μm in comparison with the BiOBr-0 nanosheets. Fig. 1d reveals that the thick sheets are piled up by several layers of thin nanosheets. This is because no capping agent was used during the synthetic process so that the thin layers tend to stack together. It is also found that the BiOBr-4 nanosheets are easily wrinkled, further confirming the high ratio of diameter-to-thickness induced by the thin layers. | ||
| Fig. 1 SEM images of (a, b) BiOBr-0 and (c, d) BiOBr-4 nanosheets. | ||
The HRTEM image in Fig. 2a further indicates the pilling characteristics of BiOBr-4 nanosheets. Notably, due to the high ratio of diameter over thickness, BiOBr-4 nanosheets are prone to warping. As it can be seen in Fig. 2b, the clear perpendicular lattice fringes with an interplanar spacing of about 0.28 nm correspond to the theoretical value of the (110) atomic plane of tetragonal BiOBr. The corresponding selected-area electron diffraction (SAED) pattern reveals a 45° angle between the (200) and (110) planes of BiOBr, suggesting that the preferred growth direction is along the [001] orientation. The side view in Fig. 2c confirms the nanosheet stacked feature of the BiOBr-4 sample. The thickness of a single nanosheet is 20 nm at a rough estimate. The corresponding lattice spacing in Fig. 2d is about 0.82 nm, which agrees well with the spacing between the (001) planes of BiOBr crystals. Therefore, the predominantly exposed top and bottom surfaces are {001} facets for BiOBr-4 nanosheets. Similarly, BiOBr-0 nanosheets are also well evidenced to exhibit exposed {001} facets on the top and bottom surfaces.25
 | ||
| Fig. 2 HRTEM images of (a, b) top view (the inset is the corresponding SAED pattern) and (c, d) side view (the inset is an enlarged image) of BiOBr-4 nanosheets. | ||
Obviously, nitric acid significantly affects the thickness and subsequent {001} facet areas of BiOBr nanosheets. In the synthetic process, once Bi3+ is dispersed in a large quantity of water, it immediately hydrolyses to form a white precipitate (Fig. S2a†). The XRD pattern in Fig. S3a† indicates that this white precursor is monoclinic bismuth oxide hydroxide nitrate hydrate Bi6O5(OH)3(NO3)5(H2O)3 with a pure phase (PDF 01-070-1226, a = 17.152 Å, b = 9.181 Å and c = 17.752 Å). The crystal structure is composed of polycations, in which the [Bi6O5(OH)3] groups are joined across the symmetry centres through two bridging O atoms, along with nitrate anions and loosely bounded water molecules.26 As shown in the SEM images (Fig. S4a and S4b†), Bi6O5(OH)3(NO3)5(H2O)3 is shaped as nanoplates. At the beginning of Br− addition, the colour of the suspension quickly changes into yellow (Fig. S2b†). After further stirring for 30 min, the yellow precipitates are collected (Fig. S2c†) and identified as BiOBr (PDF 01-078-0348) by the XRD pattern (Fig. S3b†). Comparatively, the addition of nitric acid can greatly suppress the hydrolysis of Bi3+ in water. The reaction mixture remains transparent even at the initial stage of Br− addition (Fig. S2a and S2b†). With further stirring, the yellow precipitates gradually form (Fig. S2c†) and are also confirmed to be BiOBr (PDF 01-078-0348) as indicated by the XRD pattern (Fig. S3c†). Obviously, the diameter and thickness of these BiOBr nanosheet precursors are different between BiOBr-0 (Fig. S4c and S4d†) and BiOBr-4 (Fig. S4e and S4f†). Moreover, the strongest diffraction peak of the precursors in the XRD pattern (Fig. S3†) significantly differs from those of BiOBr-0 and BiOBr-4 (Fig. S1†), suggesting that the following hydrothermal treatment alters the preferential growth direction of the two BiOBr precursors. In addition, when the concentration of nitric acid changes, the thickness of the final BiOBr nanosheets also changes. As shown in Fig. S5,† the average thickness of the BiOBr-0.1, BiOBr-0.5 and BiOBr-1 nanosheets decreases in sequence as 236, 205 and 186 nm, respectively. Therefore, the presence of nitric acid affects not only the hydrolysis process of Bi3+ but also the subsequent ion exchange and further growth during hydrothermal treatment, resulting in various sizes of BiOBr nanosheets.
XPS analysis was applied to determine the elemental compositions and valence states of the as-prepared materials. The carbon C 1s peak at the binding energy of 285 eV was used as a reference (Fig. S6a†). High-resolution XPS Bi 4f spectra of BiOBr-0 and BiOBr-4 nanosheets are presented in Fig. 3a. The two peaks at 164.80 and 159.48 eV associated with Bi 4f5/2 and Bi 4f7/2 indicate the Bi3+ valence state in BiOBr-0 nanosheets. Comparatively, an asymmetry profile of the Bi 4f spectra is observed for BiOBr-4, indicating different chemical states of Bi on the BiOBr-4 surface. The spectral profile can be deconvoluted into two bimodal peaks. In addition to the bimodal peaks for Bi3+, the shoulder peaks appearing at higher binding energies (165.46 and 160.16 eV) infer a more oxidative valence state of Bi than Bi3+.27,28 Generally, it is stable Bi5+,25 which probably resulted from the nitric acid induced surface oxidation during the synthetic process. The high-resolution XPS O 1s spectra are displayed in Fig. S6b.† There is a slight shift to higher binding energy for the peak of lattice O for BiOBr-4 (530.38 eV) in comparison with BiOBr-0 (530.24 eV), which is due to the chemical environment changes of Bi–O bonds on the surface. Similarly, the binding energy shift of the Br 3d spectral profile between BiOBr-0 and BiOBr-4 (Fig. S6c†) can also be an indication of the influence of surface Bi atoms. Since nitric acid may also introduce impurities into the BiOBr crystals, the XPS N 1s spectra of the BiOBr-0 and BiOBr-4 samples are examined. As shown in Fig. 3b, no peak is observed for the two samples, which excludes the influence of N atoms introduced by nitric acid on the BiOBr crystals. It is worth noting that a gas mixture is easily formed from nitric acid under high temperature and high pressure conditions, so no concentration higher than 4 M nitric acid was used to ensure the safety of the experiments.
 | ||
| Fig. 3 XPS spectra of (a) Bi 4f and (b) N 1s for BiOBr-0 and BiOBr-4 nanosheets. | ||
Fig. 4 displays the UV-vis diffuse reflectance spectra of the two samples. BiOBr-0 and BiOBr-4 nanosheets possess optical absorption from UV to visible light wavelengths, with absorption edges of 431 and 438 nm, respectively. On the one hand, BiOBr-4 nanosheets possess a larger portion of uncoordinated surface atoms to facilitate light absorption. On the other hand, their stacked structure feature would promote light reflection and refraction among single thin layers. Therefore, BiOBr-4 nanosheets can harvest more visible light than BiOBr-0. Their corresponding bandgap energies (Eg) were estimated from the plots of (αhυ)1/2versus the energy of absorbed light. Accordingly, the Eg value of BiOBr-4 is 2.83 eV, slightly lower than that of BiOBr-0 (2.88 eV). The decreased Eg value mainly arises from the size difference between the two nanosheets. Compared to the BiOBr-0 sample, BiOBr-4 nanosheets with a much larger diameter-to-thickness ratio would increase the amplitude of atomic vibrations, resulting in decreased band gap energies.29
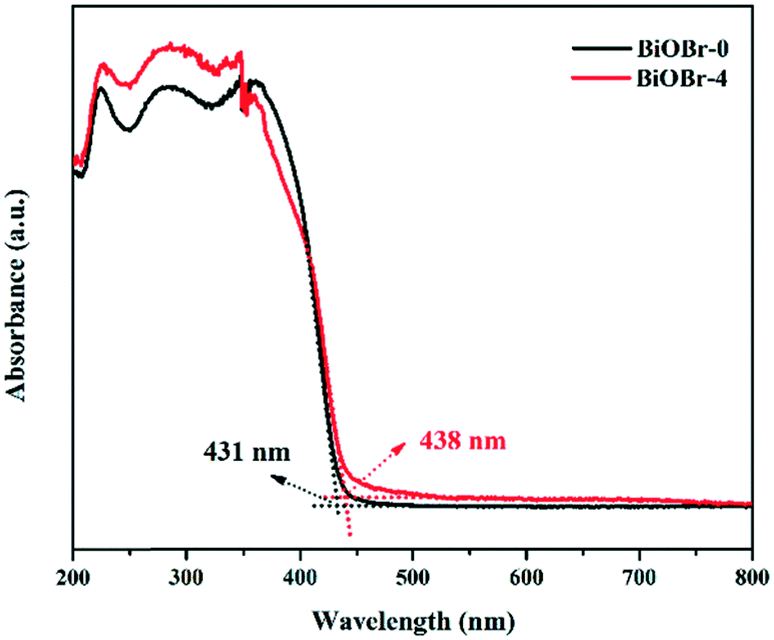 | ||
| Fig. 4 UV-vis diffuse reflectance spectra of BiOBr-0 and BiOBr-4 nanosheets. | ||
The as-prepared BiOBr nanosheets were applied to convert CO2 into renewable energy gas under simulated sunlight (xenon light) irradiation. A series of control experiments demonstrate that CO2 could not be photoreduced into energy gas (CH4 or CO) without photocatalysts or light irradiation. As shown in Fig. 5a, only negligible amounts of CO and CH4 gas are generated by the BiOBr-0 nanosheets. When the BiOBr-4 nanosheets were employed as photocatalysts to convert CO2, no detectable CH4 gas can be detected. Surprisingly, BiOBr-4 shows a significantly increased production of CO gas with prolonged irradiation time, achieving a yield of 8.56 μmol g−1 at 2 h. The corresponding production rate is about 4.45 μmol g−1 h−1. Particularly, the CO gas generated by BiOBr-4 accumulates linearly as a function of time. And no change of Eg is observed before and after the reaction (Fig. S7†), revealing that BiOBr-4 is quite stable during the reaction. Moreover, the CO converted from CO2 by BiOBr-0.1, BiOBr-0.5, BiOBr-1 and BiOBr-4 gradually increases in sequence, with production rates of 1.55, 2.64, 3.33, 4.45 μmol g−1 h−1 (Fig. 5b). More importantly, the generation of CO gas is only observed for all the samples synthesized in the presence of nitric acid, revealing a high selectivity for converting CO2 to CO for the as-prepared photocatalysts. As aforementioned, the concentration of nitric acid can regulate the thickness of the BiOBr samples, which further affects the surface area of the final products. Expectedly, the Brunauer–Emmett–Teller (BET) surface area of BiOBr-0, BiOBr-0.1, BiOBr-0.5, BiOBr-1 and BiOBr-4 follows an increasing order of 0.73, 1.55, 2.65, 3.36 and 4.45 m2 g−1. Notably, the CO production rate is linearly dependent on the BET surface area (Fig. S8†). Since the photocatalytic conversion of CO2 was conducted at a solid–gas interface, a large specific surface area provides more surface adsorption sites for CO2 and more active sites for photoreduction, leading to an increased rate of CO generation.
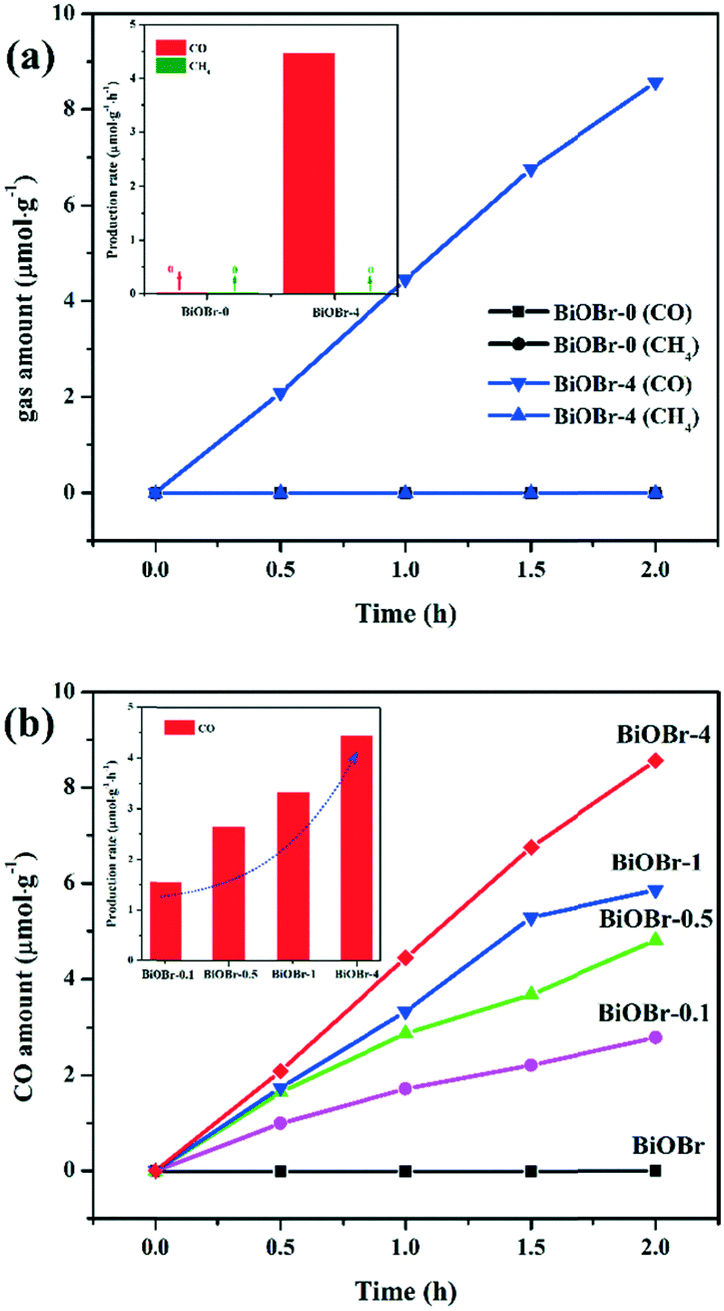 | ||
| Fig. 5 (a) Production rate of CH4 and CO gas and (b) selective photoreduction of CO2 into CO under xenon light irradiation for BiOBr photocatalysts. | ||
Since no oxygen vacancy is introduced into the two samples (Fig. S9†), the breakthrough of CO2 conversion from BiOBr-0 to BiOBr-4 nanosheets strongly depends on their reduction abilities. In order to understand the difference in the CO2 conversion ability between these two photocatalysts, it is crucial to locate the band edges of BiOBr-0 and BiOBr-4 nanosheets. Mott–Schottky experiments of film electrodes on fluorinated tin oxide glass were conducted at frequencies of 1 and 2 kHz. The flat-band potentials can be obtained from the intercept intersections of the extrapolated lines. As shown in Fig. 6a and b, positive slopes of Mott–Schottky plots suggest that both samples are n-type semiconductors. The flat-band potential for the BiOBr-0 and BiOBr-4 is 0.15 and −0.33 V, respectively, relative to the Ag/AgCl electrodes. For n-type semiconductors, the Fermi level (Ef) is equal to the flat-band potential.30–32 Accordingly, the Ef of BiOBr-0 and BiOBr-4 nanosheets is estimated to be 0.36 and −0.12 V relative to the normal hydrogen electrode (NHE).33 The valence band XPS spectra can estimate the energy gaps from Ef to the valence band maximum (VBM) of semiconductors.34 The linear intersection in Fig. 6c is situated at 2.23 and 2.34 eV for the two samples. As a result, the position of VBM for BiOBr-0 and BiOBr-4 is located at about 2.59 and 2.22 eV, respectively. Based on their optical properties, the conduction band maximum (CBM) for BiOBr-0 and BiOBr-4 occurs at −0.29 and −0.62 eV, respectively. In the presence of Bi3+, hybridization between Bi 6s2 and O 2p orbitals mainly contributes to VB formation.35 As evidenced by the XPS spectra, Bi5+ exists on the surface of BiOBr-4. The empty 6s orbital of Bi5+ can affect both the CBM and VBM of semiconductors.36 Herein, the upshift VBM and CBM of BiOBr-4 may be due to the band transition and electronic configuration in the presence of Bi5+. The corresponding schematic of the band energy alignment is illustrated in Fig. 6d. In principle, the CBM position of photocatalysts plays a predominant role in the reduction power for CO2 conversion. The redox potential of CO2/CO is about −0.52 eV (vs. NHE).37 From the viewpoint of redox potential, BiOBr-4 obviously can convert CO2 into CO whereas BiOBr-0 cannot. Regarding the reduction pathway for CH4 generation, it is proposed that CH4 is converted from the adsorbed carbon-containing intermediates (such as CO) through a direct eight-electron transfer process37 or multiple proton-coupled electron transfer processes.38,39 The difficulty to continuously deliver available electrons and/or protons probably leads to an extremely low yield of CH4. Therefore, the BiOBr-4 nanosheets exhibit high selectivity for conversion of CO2 into CO.
 | ||
| Fig. 6 Mott–Schottky plot of (a) BiOBr-0 and (b) BiOBr-4 film electrodes at frequencies of 1 and 2 kHz, (c) valence band XPS spectra and (d) schematic of the band energy alignment for BiOBr-0 and BiOBr-4 nanosheets. | ||
As photogenerated electrons have a crucial influence on the CO2 reduction process, electrochemical measurements were performed in a three-electrode electrochemical setup to evaluate the charge carrier separation and transfer efficiency between the two samples. The transient photocurrent responses of the BiOBr-0 and BiOBr-4 electrodes were recorded through multiple on/off light irradiation cycles. As exhibited in Fig. 7a, the photocurrent of the BiOBr-4 electrode is significantly higher than that of the BiOBr-0 electrode. Because the photocurrent response infers the separation efficiency of photogenerated electron–hole pairs within electrodes,40 the higher photocurrent suggests a low electron–hole pair recombination and a higher photogenerated electron transfer efficiency for BiOBr-4 nanosheets. Moreover, the electrochemical impedance spectra are compared between the BiOB-0 and BiOBr-4 electrodes (Fig. 7b). Compared to BiOBr-0, the smaller arc radius in the Nyquist plot of BiOBr-4 upon light on and off means lower electron transfer resistance, indicating a faster interfacial electron transfer process. The lower recombination and superior separation of the photogenerated charge carriers of BiOBr-4 nanosheets are also confirmed by room-temperature PL spectra (Fig. 7c), with a much lower intensity compared to BiOBr-0. A thin layer can facilitate the charge transfer, leading to the suppression of the recombination of charge carriers. Hence, the more efficient charge carrier separation and electron transfer of BiOBr-4 are eventually beneficial for the corresponding CO2 reduction reaction.
 | ||
| Fig. 7 (a) Transient photocurrent responses, (b) electrochemical impedance Nyquist plots of BiOBr-0 and BiOBr-4 nanosheets under xenon light irradiation (NaSO4: 0.1 M) and (c) room-temperature PL spectra (λex = 315 nm) of the two samples. | ||
Conclusions
In summary, BiOBr nanosheets with exposed {001} facets are successfully synthesized in the presence of nitric acid without any organic surface capping agent. The thickness of the as-prepared BiOBr nanosheets is modulated by different concentrations of nitric acid. The BiOBr nanosheets prepared in the absence of nitric acid (BiOBr-0) are incapable of converting CO2 under simulated sunlight irradiation. However, the BiOBr nanosheets prepared with nitric acid addition show high selectivity with effective photocatalytic activities for reducing CO2 into CO. In particular, the CO production rate of ultrathin BiOBr nanosheets (BiOBr-4) reaches 4.45 μmol g−1 h−1. In comparison with BiOBr-0, BiOBr-4 nanosheets with a high specific surface area provide not only a high percentage of active {001} facets, but also more surface adsorption sites for CO2 reduction. In addition, BiOBr-4 nanosheets can generate electrons with efficient charge carrier separation and transfer efficiency. In particular, the significantly raised CBM position of BiOBr-4 endows it with reduction power for converting CO2 into CO. This work not only demonstrates a controllable pathway to fabricate ultrathin BiOX (X = Cl, Br and I) nanosheets, but also provides the possibility of utilizing BiOX materials for noble-metal-free reduction of CO2 into energy gas.Acknowledgements
This project was supported by research grants from the Research Grant Council, Hong Kong SAR Government (GRF141000115) given to P. K. Wong, and from the National Natural Science Foundation of China (No. 51502146, U1404506) to L. Q Ye. P. K. Wong was also supported by the CAS/SAFEA International Partnership Program for Creative Research Teams of Chinese Academy of Sciences.References
- Y. H. Fu, D. R. Sun, Y. J. Chen, R. K. Huang, Z. X. Ding, X. Z. Fu and Z. H. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- J. C. Matsubu, V. N. Yang and P. Christopher, J. Am. Chem. Soc., 2015, 137, 3076–3084 CrossRef CAS PubMed.
- J. L. DiMeglio and J. Rosenthal, J. Am. Chem. Soc., 2013, 135, 8798–8801 CrossRef CAS PubMed.
- Z. F. Huang, L. Pan, J. J. Zou, X. W. Zhang and L. Wang, Nanoscale, 2014, 6, 14044–14063 RSC.
- K. Teramura, S. Iguchi, Y. Mizuno, T. Shishido and T. Tanaka, Angew. Chem., Int. Ed., 2012, 51, 8008–8011 CrossRef CAS PubMed.
- J. Jiang, K. Zhao, X. Y. Xiao and L. Z. Zhang, J. Am. Chem. Soc., 2012, 134, 4473–4476 CrossRef CAS PubMed.
- D. Wu, B. Wang, W. Wang, T. An, G. Li, T. W. Ng, H. Y. Yip, C. Xiong, H. K. Lee and P. K. Wong, J. Mater. Chem. A, 2015, 3, 15148–15155 CAS.
- D. Wu, S. T. Yue, W. Wang, T. C. An, G. Y. Li, H. Y. Yip, H. J. Zhao and P. K. Wong, Appl. Catal., B, 2016, 192, 35–45 CrossRef CAS.
- J. L. Han, G. Q. Zhu, M. Hojamberdiev, J. H. Peng, X. Zhang, Y. Liu, B. Ge and P. Liu, New J. Chem., 2015, 39, 1874–1882 RSC.
- H. Li, J. Shang, Z. H. Ai and L. Z. Zhang, J. Am. Chem. Soc., 2015, 137, 6393–6399 CrossRef CAS PubMed.
- L. Ye, X. Jin, C. Liu, C. Ding, H. Xie, K. H. Chu and P. K. Wong, Appl. Catal., B, 2016, 187, 281–290 CrossRef CAS.
- L. Zhang, W. Z. Wang, D. Jiang, E. P. Gao and S. M. Sun, Nano Res., 2015, 8, 821–831 CrossRef CAS.
- J. Jin, Y. Wang and T. He, RSC Adv., 2015, 5, 100244–100250 RSC.
- L. Ye, H. Wang, X. Jin, Y. Su, D. Wang, H. Xie, X. Liu and X. Liu, Sol. Energy Mater. Sol. Cells, 2016, 144, 732–739 CrossRef CAS.
- L. Ye, X. Jin, X. Ji, C. Liu, Y. Su, H. Xie and C. Liu, Chem. Eng. J., 2016, 291, 39–46 CrossRef CAS.
- J. X. Zhu, Z. Y. Yin, D. Yang, T. Sun, H. Yu, H. E. Hoster, H. H. Hng, H. Zhang and Q. Y. Yan, Energy Environ. Sci., 2013, 6, 987–993 CAS.
- J. F. Xie, J. J. Zhang, S. Li, F. Grote, X. D. Zhang, H. Zhang, R. X. Wang, Y. Lei, B. C. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 17881–17888 CrossRef CAS PubMed.
- C. X. Bi, C. Feng, T. T. Miao, Y. H. Song, D. Y. Wang and H. B. Xia, Nanoscale, 2015, 7, 20105–20116 RSC.
- M. Guan, C. Xiao, J. Zhang, S. Fan, R. An, Q. Cheng, J. Xie, M. Zhou, B. Ye and Y. Xie, J. Am. Chem. Soc., 2013, 135, 10411–10417 CrossRef CAS PubMed.
- G. J. Zhang, A. T. Su, J. W. Qu and Y. Xu, Mater. Res. Bull., 2014, 55, 43–47 CrossRef CAS.
- M. Guerrero, S. Pané, B. J. Nelson, M. D. Baró, M. Roldán, J. Sort and E. Pellicer, Nanoscale, 2013, 5, 12542–12550 RSC.
- Y. Wu, B. Yuan, M. Li, W.-H. Zhang, Y. Liu and C. Li, Chem. Sci., 2015, 6, 1873–1878 RSC.
- C. C. Weber, A. F. Masters and T. Maschmeyer, Green Chem., 2013, 15, 2655–2679 RSC.
- S. UkáYu, S. HoáKim, Y. SeobáYoon, D. YoungáKim and A. HyunáKwon, Chem. Commun., 2012, 48, 735–737 RSC.
- D. Wu, L. Ye, S. Yue, B. Wang, W. Wang, H. Y. Yip and P. K. Wong, J. Phys. Chem. C, 2016, 120, 7715–7727 CAS.
- F. Lazarini, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 3169–3173 CrossRef.
- F. M. Toma, J. K. Cooper, V. Kunzelmann, M. T. McDowell, J. Yu, D. M. Larson, N. J. Borys, C. Abelyan, J. W. Beeman, K. M. Yu, J. H. Yang, L. Chen, M. R. Shaner, J. Spurgeon, F. A. Houle, K. A. Persson and I. D. Sharp, Nat. Commun., 2016, 7, 12012 CrossRef PubMed.
- Y. G. Zhou, Y. F. Zhang, M. S. Lin, J. L. Long, Z. Z. Zhang, H. X. Lin, J. C. S. Wu and X. X. Wang, Nat. Commun., 2015, 6, 8340 CrossRef PubMed.
- S. J. Peng, L. L. Li, P. N. Zhu, Y. Z. Wu, M. Srinivasan, S. G. Mhaisalkar, S. Ramakrishna and Q. Y. Yan, Chem. – Asian J., 2013, 8, 258–268 CrossRef CAS PubMed.
- D. E. Scaife, Sol. Energy, 1980, 25, 41–54 CrossRef CAS.
- X. M. Tu, S. L. Luo, G. X. Chen and J. H. Li, Chem. – Eur. J., 2012, 18, 14359–14366 CrossRef CAS PubMed.
- H. F. Li, H. T. Yu, X. Quan, S. Chen and Y. B. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 2111–2119 CAS.
- Q. L. Xu, J. G. Yu, J. Zhang, J. F. Zhang and G. Liu, Chem. Commun., 2015, 51, 7950–7953 RSC.
- J. Wang, H. C. Yao, Z. Y. Fan, L. Zhang, J. Wang, S. Q. Zang and Z. Li, ACS Appl. Mater. Interfaces, 2016, 8, 3765–3775 CAS.
- A. Kudo, K. Omori and H. Kato, J. Am. Chem. Soc., 1999, 121, 11459–11467 CrossRef CAS.
- R. A. He, S. W. Cao, P. Zhou and J. G. Yu, Cuihua Xuebao, 2014, 35, 989–1007 CAS.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- L. J. Liu, H. L. Zhao, J. M. Andino and Y. Li, ACS Catal., 2012, 2, 1817–1828 CrossRef CAS.
- J. J. Wu, R. M. Yadav, M. J. Liu, P. P. Sharma, C. S. Tiwary, L. L. Ma, X. L. Zou, X. D. Zhou, B. I. Yakobson, J. Lou and P. M. Ajayan, ACS Nano, 2015, 9, 5364–5371 CrossRef CAS PubMed.
- Y. B. Xie, Adv. Funct. Mater., 2006, 16, 1823–1831 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cy02040b |
| This journal is © The Royal Society of Chemistry 2017 |