
A mechanistic study of the Lewis acid–Brønsted base–Brønsted acid catalysed asymmetric Michael addition of diethyl malonate to cyclohexenone†
Yuri
Samoilichenko‡
a,
Veronica
Kondratenko§
a,
Mariam
Ezernitskaya
a,
Konstantin
Lyssenko
a,
Alexander
Peregudov
a,
Victor
Khrustalev
ab,
Victor
Maleev
a,
Margarita
Moskalenko
a,
Michael
North
c,
Alan
Tsaloev
d,
Zalina T.
Gugkaeva
a and
Yuri
Belokon
*a
aA. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, 28 Vavilov Street, Moscow 119991, Russian Federation. E-mail: yubel@ineos.ac.ru; Fax: +7 (495) 135 5085
bPeoples' Friendship University of Russia, 6 Miklukho-Maklay Street, Moscow 117198, Russian Federation
cGreen Chemistry Centre of Excellence, Department of Chemistry, The University of York, Heslington, York, YO10 5DD UK
dChemical Diversity Research Institute, 2a Rabochaya Street, Khimki, Moscow Region 141400, Russian Federation
First published on 14th November 2016
Abstract
The Michael addition of diethyl malonate (Michael Donor, MD) to cyclohexenone (Michael Acceptor, MA) catalysed by the mono-lithium salt of (S)- or (R)-BIMBOL in dichloromethane is shown to exhibit biomimetic behavior. A combination of kinetics, spectroscopic studies, synthesis of catalyst analogues, inhibition studies and DFT calculations are used to show that the catalyst activates both components of the reaction and uses a chain of proton transfers to facilitate the deprotonation of diethyl malonate. The initial reaction rate was first order relative to both MA and MD and 0.5 order relative to the catalyst, indicating that an equilibrium exists between monomeric and dimeric forms of the catalyst, with the dimer predominating, but only the monomeric form being catalytically active. This was supported by DOSY 1H NMR experiments. The importance of the Lewis acidic lithium cation in the catalytic step was established by complete inhibition of the reaction by lithium complexing agents. The importance of the number of OH-groups and their relative intramolecular orientation and acidities in the polyol catalyst was shown by studying the relative catalytic activities of catalyst analogues. DFT calculations allowed the relative energies and structures of the likely intermediates on the reaction coordinate to be calculated and indicated that the ionisation of MD was facilitated due to the Lewis acidity of the lithium cation and hydrogen bond formation between deprotonated MD (MD−1) and the OH groups of the BIMBOL moiety.
Introduction
Asymmetric catalysis using synthetic catalysts has advanced enormously over the last 25 years. Metal catalysed reactions are well established1 and asymmetric organocatalysis has blossomed over the last 15 years,2,3 with chiral Brønsted acid and hydrogen bond donor catalysis being amongst the most recent developments.3 Despite these impressive advances, synthetic catalysts still struggle to match the levels of activity and asymmetric induction achieved with enzymes. This is due to the presence of multiple, exquisitely orientated, functional groups within the active site of enzymes which facilitates both simultaneous activation of both reaction components; and intramolecular proton transfer to lower the energy of transition states between reaction intermediates.4 In this paper we show that a chiral synthetic catalyst containing both metal-based and non-metal based catalytic groups facilitates both intramolecular proton transfer and reactant activation in a biomimetic way.The introduction of basic groups into catalysts, alongside hydrogen bonding functions, results in highly efficient bifunctional chiral catalysts capable of simultaneous activation of both nucleophilic and electrophilic components of heterolytic reactions.3,5 The most popular catalytic motif of this type is represented by amine-thiourea organocatalysts.5 Recently, the combined use of metal-based and organocatalysts has been shown to catalyse tandem processes.6 As a result of these discoveries the established mechanisms of asymmetric catalysts have been re-evaluated and some well accepted cases of Lewis acid catalysis have been shown to originate from hidden Brønsted acid catalysis7 occurring via Lewis acid activated Brønsted acid catalysis.8
Another emerging multifunctional catalytic system for asymmetric C–C and C–H bond formation is exemplified by chiral alkali binaphtholate salts.9 These catalysts contain a Lewis acidic site, Brønsted basic and Brønsted acidic sites. The corresponding alkali metal salts of TADDOLs and bis-TADDOLs were also efficient in promoting asymmetric carbon–carbon bond forming reactions.10 The combination of both BINOL and TADDOL motifs in the same molecule to form BIMBOL lithium salt (Fig. 1) improved the catalytic performance of the system, compared to the corresponding salts of either BINOL or TADDOL in the benchmark reaction of the Michael addition of diethyl malonate (MD) to cyclohexanone (MA).11 The BIMBOL catalyst system has the potential to be trifunctional with the metal ion serving as a Lewis acid whilst the naphtholate ion and hydroxyls are Brønsted base and Brønsted acids respectively.
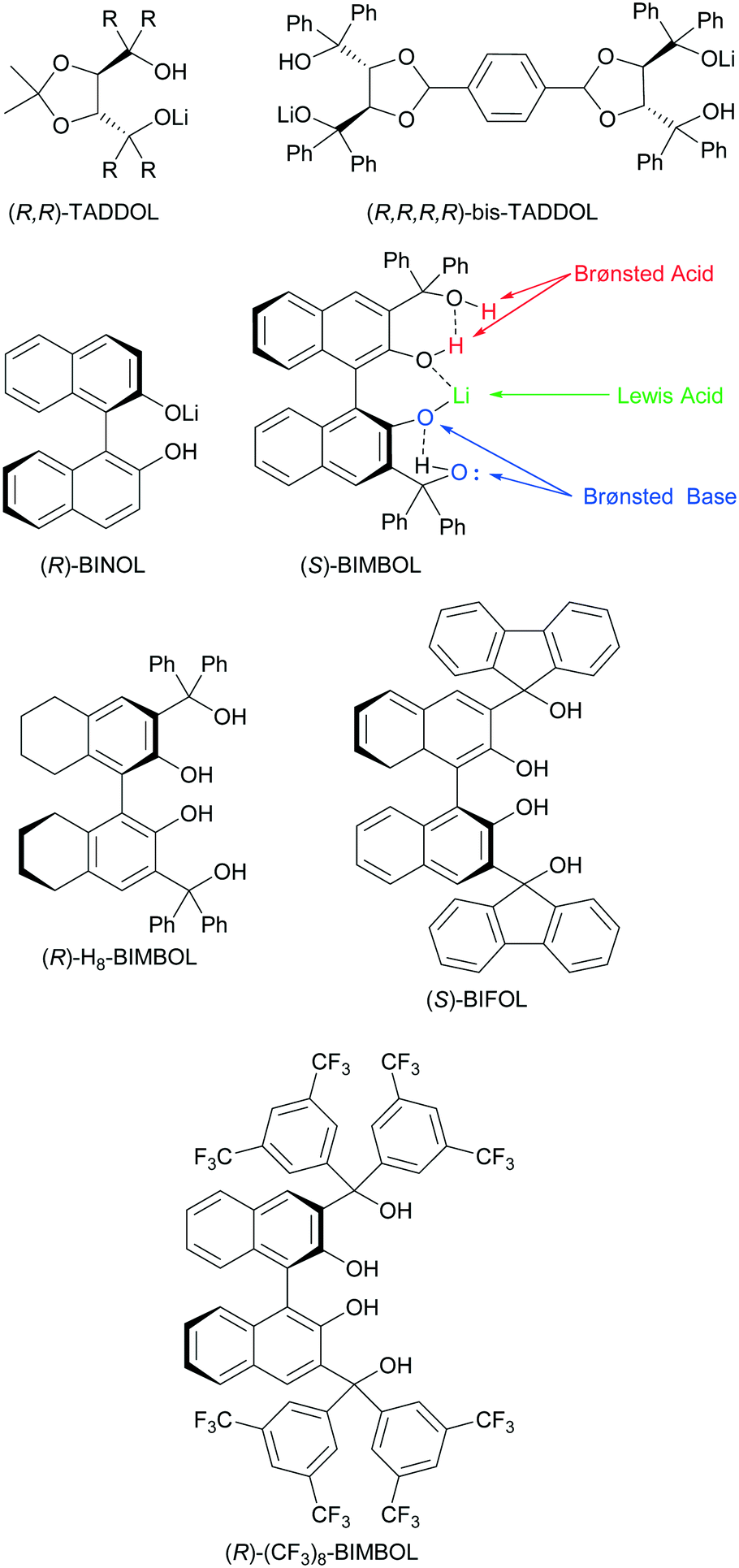 | ||
| Fig. 1 Mono-lithium-salts of chiral polyols: TADDOL, bis-TADDOL, BINOL and BIMBOL and the BINOL derivatives used in this work. | ||
The enantioselectivity and chemical yields of the BIMBOL catalysed reaction displayed concentration dependences, indicating that the catalyst self-associates to give species with different relative activities.11 The self-association of lithium derivatives is a well-known phenomenon12 as is the self-association of derivatives of thiourea,3i–k catalytically active silanediols13 and bis-TADDOLs.10c This self-association can be an impeding factor for the catalytic activity of amine-thioureas3i,j or a favourable one for electrostatically enhanced thioureas,3k silanediols13 and bis-TADDOLs.10c Therefore, we have carried out a combined experimental and computational study of the Michael addition of diethyl malonate to cyclohexanone catalysed by lithium salts of BIMBOL and related compounds and show that the reaction exhibits the positive catalytic features associated with enzymatic catalysis.
Results and discussion
Synthesis of (R)- and (S)-BIMBOL was achieved as reported earlier.14 The syntheses of (R)-H8-BIMBOL, (S)-BIFOL and (R)-(CF3)8-BIMBOL were achieved from (R)- or (S)-BINOL (S59†). H8-BIMBOL should have lower acidity (greater pKa value) than BIMBOL, whereas (CF3)8-BIMBOL should be more acidic (lower pKa value).The structure of (R)-BIMBOL (Fig. 2 top) has previously been determined by X-ray crystallography11 and shown to have two pairs of OH groups each involved in two pairs of intramolecular hydrogen bonds: naphthol–OH⋯OH–alcohol and vice versa and with both pairs of OH groups situated over the same face of the naphthyl moieties of BIMBOL (endo-type orientation).11 X-ray analysis of BIFOL¶ also showed that each pair of OH groups was involved in intramolecular hydrogen bond formation; but the triphenylmethanol groups were positioned on different faces of the naphthyl planes (exo-type orientation, Fig. 2 bottom). This raises the question as to whether rotation around the C3–C17 (or C11–C26) bond is hindered to such an extent as to make the exo-isomer observed in the solid state also the atropoisomer present in solution or whether the exo/endo transition is relatively facile in solution. Literature data on the rotation barriers for a series of closely related 2-methyl-1-naphthyl-fluorenes15 indicated that the energy of rotation around a naphthyl-fluorene bond is >26 kcal mol−1. Our calculation of the barrier to rotation in BIFOL from DFT data (S34†) gave a value of 16.6 kcal mol−1.
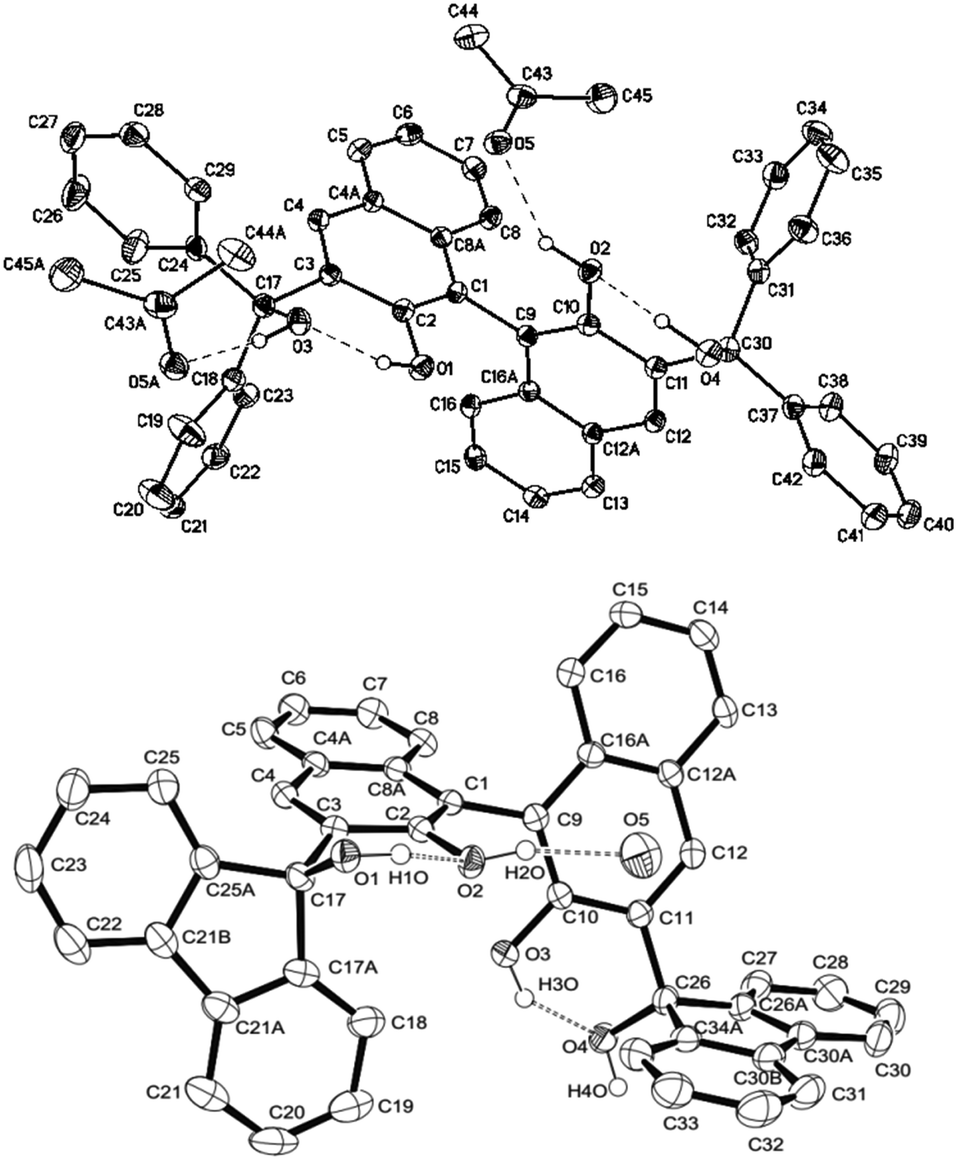 | ||
| Fig. 2 The structures of (R)-BIMBOL11 (top) and (S)-BIFOL (bottom) showing the different disposition (endo and exo) of the hydrogen bond coupled OH groups in the crystals of the compounds. | ||
The 1H NMR spectra of BIMBOL and BIFOL in CD2Cl2 between 30 °C and −60 °C differed drastically (S55 and S56†). While all the resonances of BIFOL (especially those of the OH groups) became narrower as the temperature was lowered, all the resonances of BIMBOL became broader and those of OH groups separated into several sets. Similarly, the 13C NMR spectra of BIFOL and BIMBOL recorded at 30 °C and −40 °C differed immensely (S57 and S58†). At 30 °C, the 13C NMR spectrum of BIFOL displayed broad signals for the ipso-carbon atoms bonded to the OH groups (157.7 and 91.7 ppm) and those connecting two binaphthyl moieties (122.7 ppm), whereas the analogous 13C resonances of BIMBOL (156.5, 88.3 and 119.7 ppm, respectively) at the same temperature were narrow. In contrast, at −40 °C, all the BIFOL resonances connected with C–OH moieties and those of the C1 and C9 atoms became narrow and no change in the number of other resonances took place. However, for BIMBOL at −40 °C, all the resonances became broader and most of them split into multiple signals (S55–S58†). Thus, it appears that BIFOL exists in solution at ambient temperature as an interconverting endo-exo-atropoisomer, probably with the isomer depicted in Fig. 2 as the major form. Decreasing the temperature slows the OH group proton exchange in BIFOL and as a result, the NMR resonances become progressively narrower and their resolution improves. In contrast, a decrease in temperature in case of BIMBOL caused, in addition to slowing the proton exchange, slow rotation around the sp3–sp2 bonds (C17–C3 or/and C11–C30) eventually producing a mixture of several atropoisomeric conformers, existing in a slowly established equilibrium at temperatures below −20 °C.
The benchmark Michael addition of MD to MA was conducted as shown in Scheme 1. The lithium salt of BIMBOL could be prepared by the reaction of lithium phenoxide with BIMBOL as the pKa of BINOL in DMSO is approximately 1316 whereas that of phenol is 18.17 In addition, it was shown previously that the catalyst generated in this manner was catalytically identical to one produced by the reaction of BIMBOL with BuLi.11
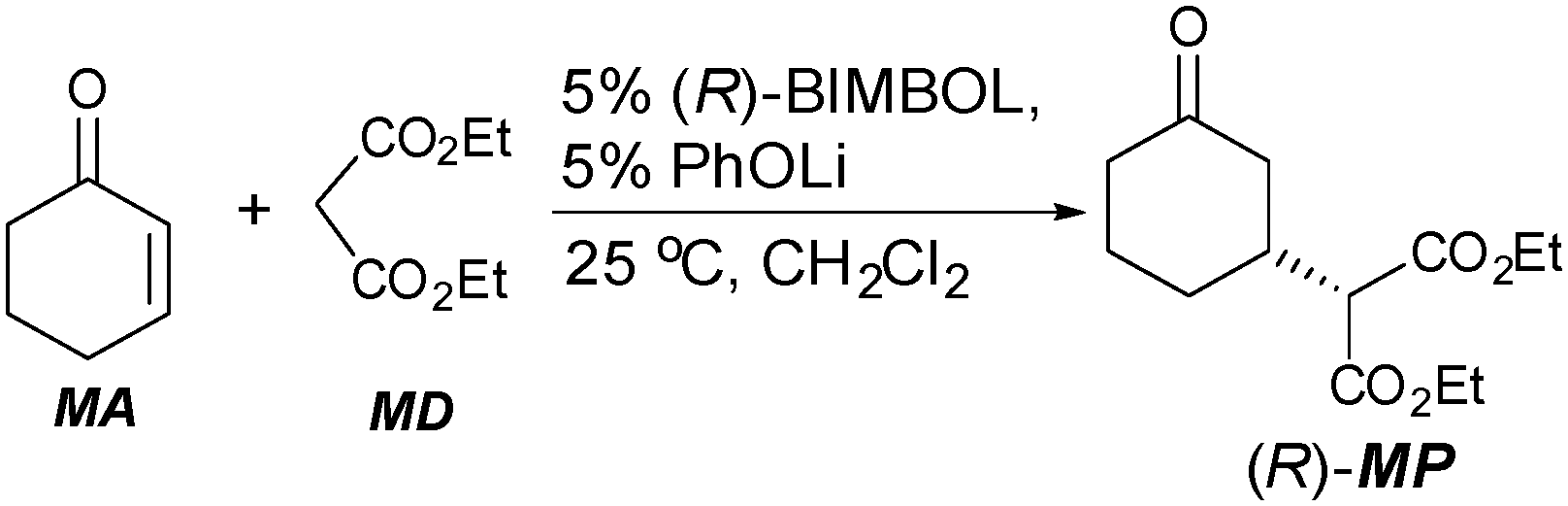 | ||
| Scheme 1 The addition of diethyl malonate (MD) to cyclohexanone (MA) using (R)-BIMBOL to give (R)-MP. | ||
The reaction could be easily monitored by IR spectroscopy, following the disappearance of the cyclohexenone absorptions at 1671 cm−1 (ε = 409) and 1685 cm−1 or the appearance of the carbonyl group of the adduct (MP) which absorbs at 1712 cm−1. Fig. 3 illustrates the progression of the changes of the IR spectra of the reaction mixture under the experimental conditions given in Scheme 1. An isobestic point at 1695 cm−1 was observed in the infrared spectra indicating that no side reactions occur during the Michael addition. To allow a quantitative analysis of the kinetic results, curve fitting of the observed spectra in the ν(CO) region was undertaken (see Experimental section for details) and the rate of formation of MP exactly matched the rate of disappearance of MA.
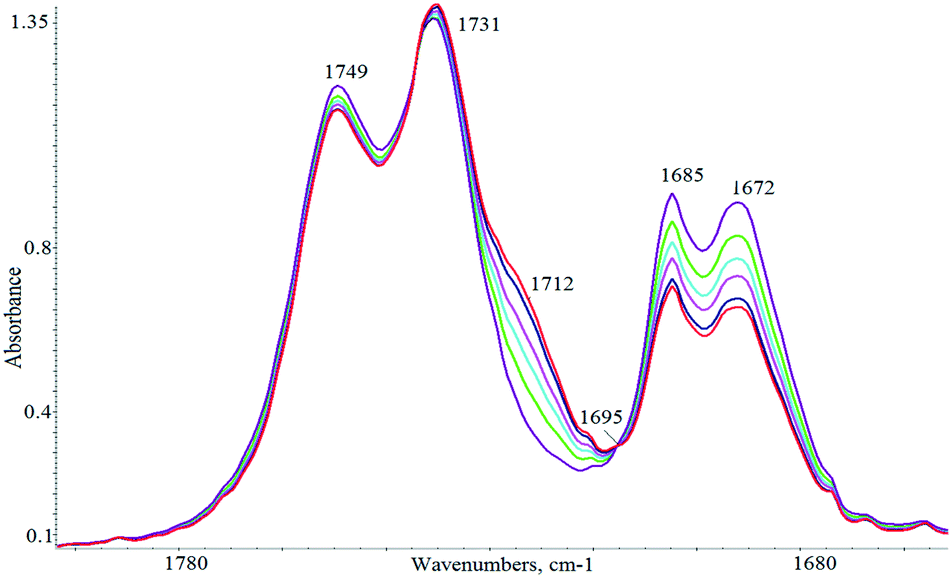 | ||
| Fig. 3 IR monitoring of the ν(CO) region under the reaction conditions: [BIMBOL] = [PhOLi] = 0.0057 M, [MA]o = [MD]o = 0.11 M, CH2Cl2, 25 °C. | ||
Previously we observed that the reaction took almost 48 hours to go to completion.11 However, the kinetic data obtained from the infrared spectra and displayed in Fig. 4 indicate that the reaction occurs in two kinetically distinct stages (Fig. 4, data points a). On the timescale shown in Fig. 4, the changes in reactant concentrations are sufficiently small that the data can be fitted to pseudo-zero order kinetics. The rapid, initial section of the reaction produced almost 20% of MP within 40 minutes. Then, the Michael addition was significantly slowed down. This effect was assumed to be due to catalyst inhibition by the reaction product and this was supported by the addition of 10–50% of MP relative to MA to the reaction mixture. A twofold excess of MP relative to the catalyst was sufficient to generate a new catalytic species, having lower catalytic efficiency (Fig. 4, data points b). A tenfold excess of MP in the reaction mixture had almost no additional negative effect on the reaction kinetics (Fig. 4, data points c).
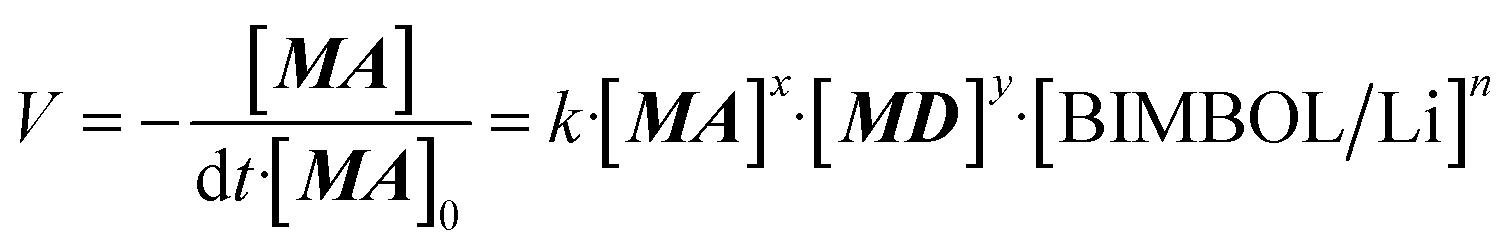 | (1) |
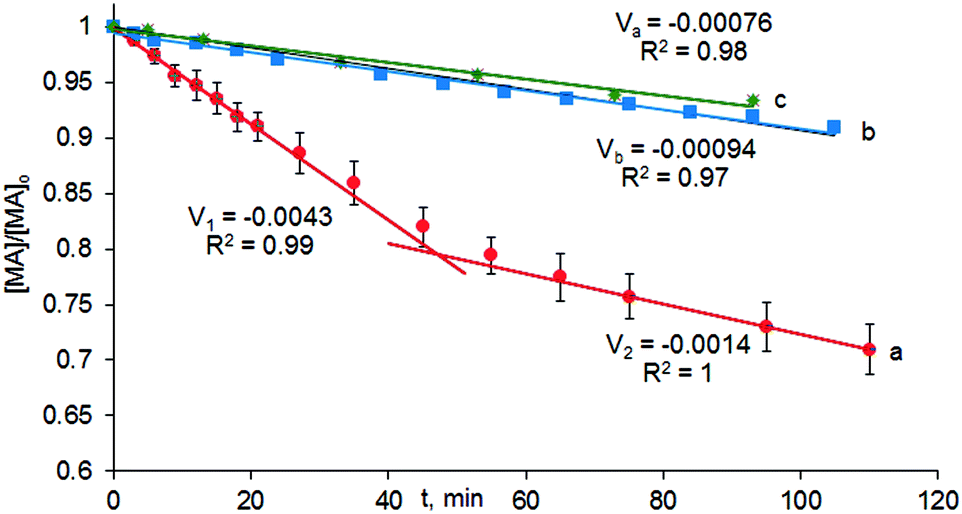 | ||
| Fig. 4 Reaction profile versus time plot for the conversion of cyclohexenone (MA) and malonic ester (MD) into MP catalysed by a catalyst prepared in situ from BIMBOL and PhOLi in CH2Cl2 under Ar at 25 °C: [BIMBOL] = [PhOLi] = 0.0057 M, [MA]0 = [MD]0 = 0.11 M, in CH2Cl2; reaction rates (V) are given in min−1. a) No additives b) 10% (relative to MA) of MP were added. c) Half an equivalent of MP was added. | ||
In order to test if some non-linear effects were present in the system, 10 mol% of MP with 90% enantiomeric purity was added to the reaction mixture at the beginning of the reaction and the enantiomeric purity of the resulting MP was checked at 10% and 98% of conversion. In each case the ee of MP was found to lie in the ee interval of 85–90%. In addition, 10 mol% of racemic MP was added to the reaction mixture and the final enantiomeric purity of the product was found to be 82% which corresponded to that of a mixture of 10% of a racemic additive and 90% of enantiomerically enriched MP with ee of 85–90%. Thus, no influence of the ee of MP on the asymmetric performance of the catalyst was detected.
To determine the order with respect to substrates MA and MD, reactions were carried out at three concentrations of each substrate whilst keeping the other component concentration constant. The initial zero order rates (up to 15% conversion) of the reactions were determined and the coefficients x and y in eqn (1) were found to be 0.87 and 0.76 respectively. Thus, there was some deviation from the expected second order behaviour, probably, reflecting competition of the substrate present in excess with the other substrate for the active sites of the catalyst.
To address the issue of the real composition of the catalytically active species, reactions were carried out at various ratios of (S)-BIMBOL to lithium phenoxide. The malonate carbanion formation should be the key stage of the addition sequence and the amount of basic groups is expected to be the most important feature of the catalyst. Since the number of basic groups in the case of di-Li salts is twice that of mono-Li-salts one could expect doubling the reaction rate. However, the catalytic performance of both was almost the same (Table 1, runs 1 and 2), this indicates that the presence of free hydroxyl groups in the mono-Li salt is an important boost of the catalytic performance of BIMBOL increasing its activity to approaching that of the di-Li salt. However, the ratio of (S)-BIMBOL to lithium phenoxide had almost no influence on the enantiomeric excess of the resulting MP (Table 1, runs 1–4) which supported the theory that the catalytically active species had the same composition in the reaction mixture whatever the ratio of the catalytic components.
| Run | (S)-BIMBOL, mol% | PhOLi, mol% | V,b min−1 | ee ofb (S)-MP |
|---|---|---|---|---|
| a The reaction was conducted under Ar at 25 °C, [MA]0 = [MD]0 = 0.11 M in CH2Cl2. b The initial rate and enantiomeric excess of MP (the estimated range of three experiments) at 90% conversion. c The enantiomeric excess of MP at 15% conversion (the estimated range of three experiments). | ||||
| 1 | 5 | 5 | 4.3 × 10−3 | 85–90 (86)c |
| 2 | 5 | 10 | 3.0 × 10−3 | 86 |
| 3 | 5 | 2.5 | 2.8 × 10−4 | 78–86 |
| 4 | 10 | 5 | 4.6 × 10−4 | 83–86 |
To determine the order with respect to the catalyst concentration, reactions were carried out in duplicate at four different catalyst concentrations. The average initial zero order rate constants (V, min−1) were used to construct a plot of ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) V against ln([BIMBOL/Li]). The resulting plot is shown in Fig. 5 and the best fit line through the data points has a slope of 0.51 ± 0.16, suggesting that the reaction had a 0.5 order dependence on the catalyst concentration (n in eqn (1)). This indicates that the catalytically active species is monomeric and exists in a rapid equilibrium with a catalytically inert dimeric species, and that the position of equilibrium is strongly shifted towards the dimer (Scheme 2). A very low concentration of the monomeric component in the reaction mixture was supported by the absence of any observable additional signals for either the malonic ester or cyclohexenone protons in their separate 1:1 mixtures with BIMBOL/Li in deuterated dichloromethane solutions (S37†). In contrast, the more acidic ethyl nitropropanoate had its spectrum significantly changed under the same conditions (S37†). The earlier observation of positive non-linear effects in the reaction, with racemic (S)- and (R)-BIMBOL forming an inert precipitate,11 provides further indirect evidence for the dimer being catalytically inactive.
V against ln([BIMBOL/Li]). The resulting plot is shown in Fig. 5 and the best fit line through the data points has a slope of 0.51 ± 0.16, suggesting that the reaction had a 0.5 order dependence on the catalyst concentration (n in eqn (1)). This indicates that the catalytically active species is monomeric and exists in a rapid equilibrium with a catalytically inert dimeric species, and that the position of equilibrium is strongly shifted towards the dimer (Scheme 2). A very low concentration of the monomeric component in the reaction mixture was supported by the absence of any observable additional signals for either the malonic ester or cyclohexenone protons in their separate 1:1 mixtures with BIMBOL/Li in deuterated dichloromethane solutions (S37†). In contrast, the more acidic ethyl nitropropanoate had its spectrum significantly changed under the same conditions (S37†). The earlier observation of positive non-linear effects in the reaction, with racemic (S)- and (R)-BIMBOL forming an inert precipitate,11 provides further indirect evidence for the dimer being catalytically inactive.
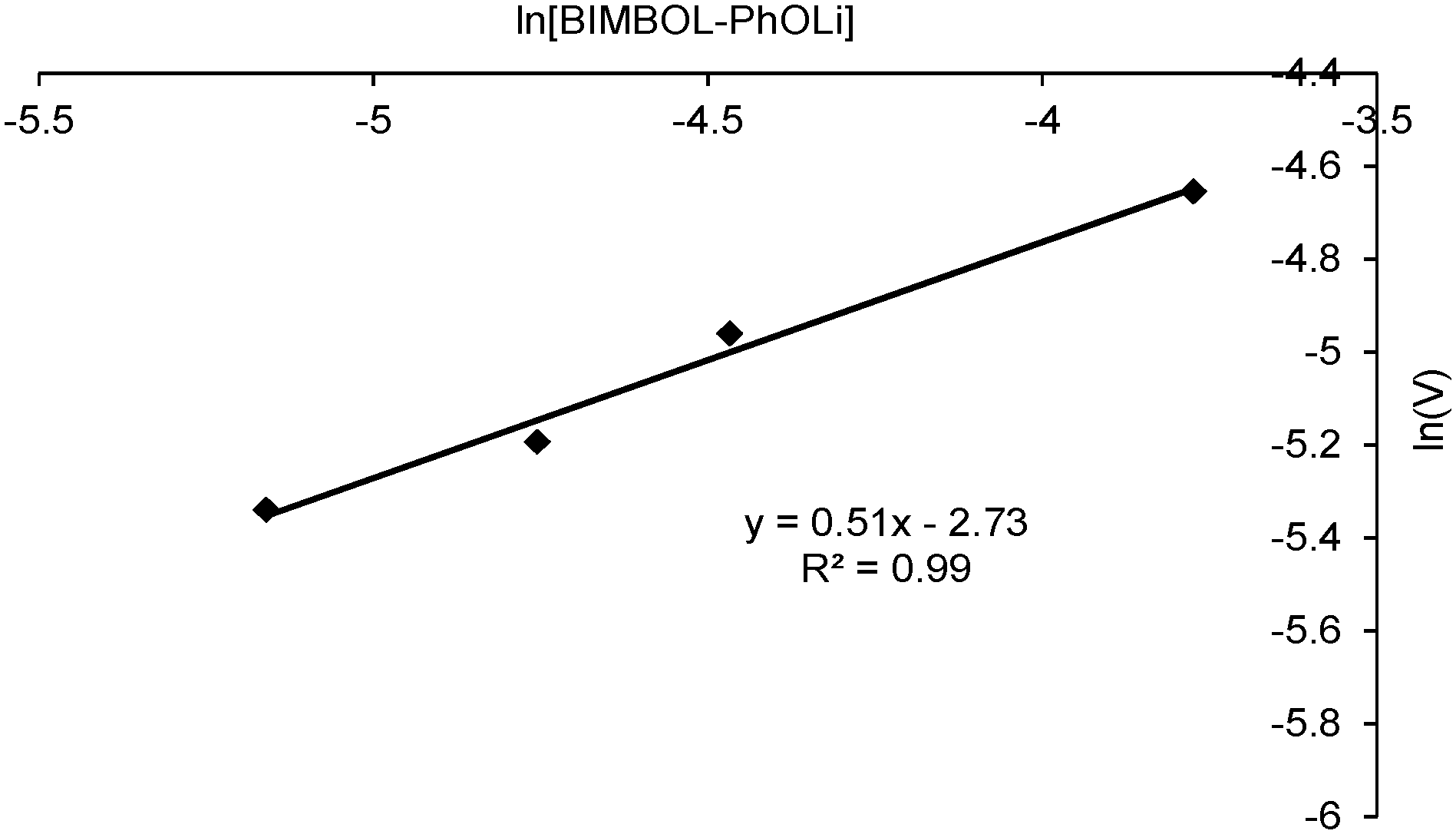 | ||
| Fig. 5 Plot of ln(V) versus ln[Cat] for the addition of diethyl malonate (MD) to cyclohexenone (MA): (S)-BIMBOL/PhOLi 1:1; [MA]0 = [MD]0 = 0.11 M, solvent CH2Cl2. | ||
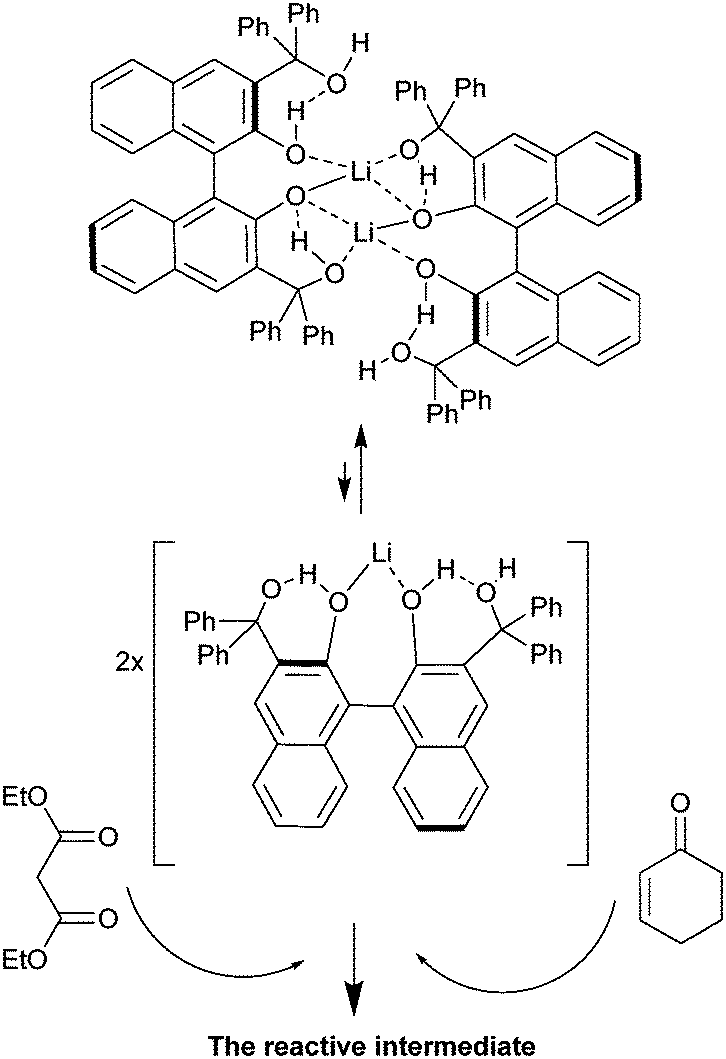 | ||
| Scheme 2 Illustration of a possible inactive dimer/active monomer equilibrium to rationalise the 0.5 order kinetics relative to the (S)-BIMBOL/Li concentration. | ||
In order to investigate the relative importance of Brønsted and Lewis acid centres within the catalyst, lithium complexing agents were added to the reaction mixture and the results are summarised in Table 2. The addition of two equivalents of TMEDA relative to the catalyst completely inhibited the reaction (run 2). The same effect was observed on the addition of just one equivalent of benzo-15-crown-5 (run 3). The addition of three equivalents of THF or morpholine also negatively influenced the yield of the product (runs 4 and 5) although to a lesser extent. On the other hand, three equivalents of 1,2-dimethoxyethane (DME) improved the yield of the product (run 6 and 7) whereas the addition of 20 equivalents of DME resulted in complete inhibition of the reaction.
| Run | Additive (mol% relative to MA) | Time, h | Yield of (S)-MP,a,b % (ee%) |
|---|---|---|---|
| The reaction was conducted under Ar at 25 °C in CH2Cl2, [(S)-BIMBOL] = [PhOLi] = 0.0124 M, [MA]0 = [MD]0 = 0.248 M. a The yield was estimated by 1H NMR. b The enantiomeric excess of the product was determined by chiral HPLC analysis of MP. | |||
| 1 | — | 48 | 55 (85–90) |
| 2 | TMEDA (10%) | 24 | 0 |
| 3 | Benzo-15-crown-5 (5%) | 24 | 0 |
| 4 | THF (15%) | 24 | 22 (n.d.) |
| 5 | Morpholine (15%) | 24 | 10 (n.d.) |
| 6 | DME (15%) | 24 | 60 (85–90) |
| 7 | DME (15%) | 48 | 98 (85–90) |
| 8 | DME (100%) | 24 | 0 |
According to DOSY data, summarised in Table 3, the formation of mono lithium salts of BIMBOL led to a reduction of its diffusion coefficient (D) relative to that of BIMBOL from 0.88 × 10−9 to 0.61 × 10−9 m2 s−1 (entries 1 and 2). The formation of a dimer of the Li-salt is a reasonable explanation of these results. The addition of benzo-15-crown-5 to BIMBOL/Li in a 1:1 ratio led to an increase of D in the case of the BIMBOL/Li salt (entry 3, D = 0.82 × 10−9) and decreased D for the crown ether (entries 3 and 4, D = 0.990 × 10−9 m2 s−1 and 1.32 × 10−9 m2 s−1 respectively). This can be explained by the establishment of a rapid equilibrium between species including significant amounts of monomeric BIMBOL-Li-benzo-15-crown-5. The effect of DME on the BIMBOL-Li was different. The addition of three equivalents of DME had almost no influence on the D of the lithium salt (runs 2 and 5), but the diffusion coefficient of DME itself somewhat decreased (run 5 and 6). This suggests that the interactions of DME were mostly with the lithium monomer present in small amounts in the solution.
| Run | Compound | logD |
D × 109 m2 s−1 |
|---|---|---|---|
| a DOSY experiments were carried out at 20 °C in CD2Cl2, [Polyol] = [PhOLi] = 0.03 M. The reaction conditions were the same as those given in the footnote to Table 2. | |||
| 1 | BIMBOL | −9.083 | 0.808 |
| 2 | BIMBOL + PhOLi | −9.21 | 0.61 |
| 3 | BIMBOL/Li + benzo-15-crown-5 (1:1 relative to Li) |
BIMBOL −9.0855 | 0.821 |
| Crown −9.0042 | 0.990 | ||
| 4 | Benzo-15-crown-5 | −8.88 | 1.32 |
| 5 | BIMBOL/Li/DME (3:1 relative to Li) |
−9.18 (BIMBOL) | 0.64 |
| −8.63 (DME) | 2.34 | ||
| 6 | DME | −8.52 | 3.07 |
These data show that the lithium cation constituted the core of the catalytic system and its coordination with strong external complexing agents led to the complete loss of the catalytic activity. However, some weakly coordinating agents such as DME could increase the catalytic activity in terms of both yield and enantioselectivity which might indicate a beneficial influence of the additive by thwarting the inhibiting effects of the product MP on the reaction kinetics and/or shifting the equilibrium of Scheme 2 towards the monomer components.
In order to investigate the relative importance of the BIMBOLate hydroxyl groups in the catalysis, the relative activities of the mono-lithium salts of BIMBOL, BINOL, H8-BIMBOL, BIFOL, and (CF3)8-BIMBOL were studied in the model reaction (Scheme 1) under identical conditions and the results are shown in Fig. 6 where the initial consumptions of MA are plotted versus time. As can be seen from Fig. 6, BINOL was much less active than BIMBOL. The superior activity of the later was not a consequence of the greater acidity of its phenolic hydroxyl groups, relative to those of BINOL, as the more acidic (CF3)8-BIMBOL also displayed a diminished activity relative to BIMBOL.
 | ||
| Fig. 6 Relative activities of the mono-lithium salts of (S)-BIMBOL, (R)-BINOL, (R)-H8BIMBOL, (S)-BIFOL, and (R)-(CF3)8BIMBOL in the model Michael addition reaction in CH2Cl2 at 25 °C: [tetraol] = [PhOLi] = 0.0057 M, [MA]0 = [MD]0 = 0.11 M. | ||
H8BIMBOL-Li had a different type of reactivity with an induction period observed at the beginning of the reaction, followed by a lower reaction rate than that of BIMBOL-Li. The induction period may reflect slow dissociation of the dimeric lithium salt, initiated by the substrates. Such processes were shown to become rate limiting in some reactions of lithium derivatives.12b (R)-H8-BIMBOL led to MP of (R)-configuration, as also observed for (R)-BIMBOL (see Table 1), though with a diminished enantiomeric excess of 25% after 1.5 h and 26% after 27 h. In addition, at longer time intervals, the rate of the H8BIMBOL-Li promoted reaction became faster than that of the BIMBOL-Li catalysed one. It took only 24 h for H8-BIMBOL-Li to bring the reaction to 80% completion, whereas BIMBOL-Li furnished only 55% of MP within that time interval. The difference in behaviour of BIMBOL and H8BIMBOL can be due to the variation in their acidity and in the torsion angles of their naphthyl moieties. The acidities of BINOL and H8BINOL in DMSO are 13.2 and 17.9 respectively17 and the minimum energy torsion angle is greater in case of H8-BINOL than for BINOL. The same trend could be expected to be retained in BIMBOL, and H8-BIMBOL with the torsional angle in the latter being greater by approximately six degrees, making the OH groups of the naphthyl moieties closer to each other (according to MM2 calculations). These two factors combined could produce the observed effects. One of the reasons for the greater activity of H8BIMBOL-Li at the later stages of the reaction could be the less likely formation of the non-productive complex with MP because of the combined acidity/steric properties of H8-BIMBOL-Li.
(S)-BIFOL was even less active than BINOL (Fig. 6) and even after 48 hours no MP formation was observed under the standard conditions with BIFOL-Li catalyst. Several explanations could be put forward to rationalise the lack of catalytic activity of BIFOL-Li:
1. The four OH group are unable to form hydrogen bond supported trimolecular MA and MD−1 (deprotonated MD) complexes with BIFOL in which the Michael acceptor and donor are situated in appropriate positions for carbon–carbon bond formation to take place, as hypothesised previously.11
2. The observation could be connected to the slow rotation of the hydroxyfluorenyl moieties around the sp2–sp3 bonds C(17)–C(3) or C(11)–C(26) (S34†). The release of MP from the catalytic site of the BIMBOLs and, thus, recovery of the catalyst should be coupled to the breaking of hydrogen bonds between the carbonyl groups of MP and the OH groups of BIMBOLs. This might necessarily involve the conformational rearrangement of the endo- to exo-orientation of the triphenylcarbinol moieties of BIMBOLs. In the case of BIFOL such conformational rearrangement is too slow (vide supra) and results in very slow catalyst release and slowing of the reaction.
3. Another explanation might be based on a slow Li-dimer-monomer dissociation in the case of BIFOL also linked to the slow conformational rearrangements.
Thus, the presence of four interconnected OH groups with endo-orientation inside the same chiral molecule is a prerequisite for the efficient performance of the catalyst family. The variation in their acidities, mutual orientation and the rate of the conformational rearrangements could be decisive factors in determining the catalyst activity of the system. Finally, the presence of a Lewis acidic lithium counter-cation is a prerequisite for efficient BIMBOL-Li catalysis.
In order to shed light on the details of the reaction mechanism, DFT calculations were performed on the system. For analysis of the intramolecular interactions QTAIM theory18 was used as that gives an opportunity to locate all the bonding interactions by a critical point search as well as to estimate the energy of the interactions by means of Espinosa correlation.19 Firstly, optimisation for two overall uncharged complexes Li1 and Li2 were carried out. The first complex Li1 contained the anion of BIMBOL, one lithium cation and a neutral malonic ester (Fig. 7, top) whereas Li2 contained a neutral BIMBOL, one lithium cation and a malonic ester anion (Fig. 7, bottom). In both complexes, the lithium cation is tetracoodinated by two oxygen atoms of BIMBOL and two oxygen atoms of the carbonyl groups of malonic ester. At the same time in Li1 the lithium cation is coordinated by oxygen atoms of the same naphthalene fragment (O(1) and O(2)) whereas in Li2 it forms bonds with both fragments (O(1) and O(3) atoms). As expected, in the Li1 complex, the Li–O distance with deprotonated O(2) atom is the shortest (1.852 Å), while the corresponding bonds with OH groups vary in the range of 1.892–1.917 Å. In contrast, the Li–O bonds with carboxy groups of malonic ester are shorter in the case of Li2. The additional stabilisation of the Li1 and Li2 salts is also due to O(3)–H⋯O(4), O(4)–H⋯O(7) and O(3)–H⋯O(4) hydrogen bonds respectively. Furthermore, the number of HO⋯OH and O(4)–H⋯C(2) interactions in Li2 have been established by a critical point search. The latter interactions can be interpreted as the O–H⋯OH hydrogen bonds and can serve as the channel of proton transfer from the BIMBOL molecule to the malonic ester anion and, according to the principle of microscopic reversibility, the chain of proton removal from the neutral malonic ester to the ionised BIMBOL. The energy of these C–H⋯O and O–H⋯O interactions are 2.0 and 3.2 kcal mol−1 respectively. Despite the significant difference in charge distribution and intramolecular interactions in Li1 and Li2 their total energy was almost equal, with Li2 being just 2.0 kcal mol−1 lower in energy.
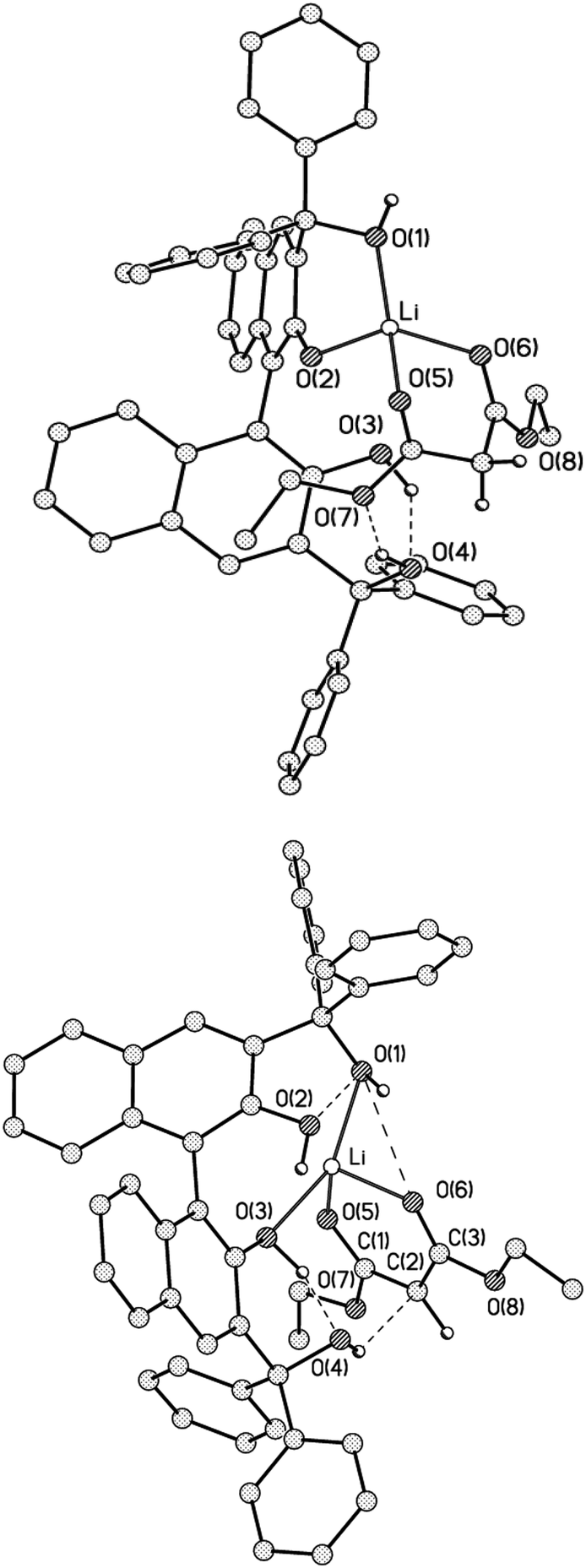 | ||
| Fig. 7 Views of Li1 (top) and Li2 (bottom) according to DFT calculations. Some hydrogen atoms are omitted for clarity. The intramolecular interactions are shown only for contacts for which the critical points (3, −1) of the electron density function were located. | ||
Generally, Li1 and Li2 differ mostly in the position of a proton on either BIMBOL or malonic ester moieties within their complexes. In other words, the two structures represent the limiting extremes of an acid-base equilibrium, involving BIMBOL-phenolate/BIMBOL conjugated acid and malonic-ester/malonate-anion. Taking into account that the acidities of BINOL and malonic esters in DMSO are 13.217 and 16.4,18 respectively; such a small difference in energy between Li1 and Li2 was unexpected. It could be an indication of a greatly increased acidity of the coordinated malonic ester by the stabilisation of malonate anion in Li2, as compared with the free CH-acid. Such stabilisation may originate from the lithium cation coordination of the malonate and also from the O(4)–H⋯C(2) hydrogen bond, linking the BIMBOL moiety and malonate.
As the next step, energy calculations on the MA adducts with both Li1 and Li2 (Li1MA, Li2MA and Li3MA) were conducted. In contrast to Li1 and Li2, the total energies of complexes Li1MA, Li2MA and Li3MA (Fig. 8) differed significantly, with the additional stabilisation of the latter two adducts by 13.6 and 13.5 kcal mol−1 respectively. The energies of interaction of MA with Li1 and Li2 were calculated to be 11.0 and 22.6 kcal mol−1 in Li1MA and Li2MA respectively. An alternative conformation of MA with L1 was also considered (S17†). In both complexes, the main force that binds MA is a O(1)–H⋯O(9)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond with an energy of 10.4 and 11.6 kcal mol−1 respectively. In addition to this hydrogen-bond, in Li2MA there are a number of additional interactions such as C⋯C (3.58 Å) and O⋯C (3.2–3.3 Å) between the malonate anion and MA with energy varying in the range of 0.7–1.2 kcal mol−1.
C bond with an energy of 10.4 and 11.6 kcal mol−1 respectively. In addition to this hydrogen-bond, in Li2MA there are a number of additional interactions such as C⋯C (3.58 Å) and O⋯C (3.2–3.3 Å) between the malonate anion and MA with energy varying in the range of 0.7–1.2 kcal mol−1.
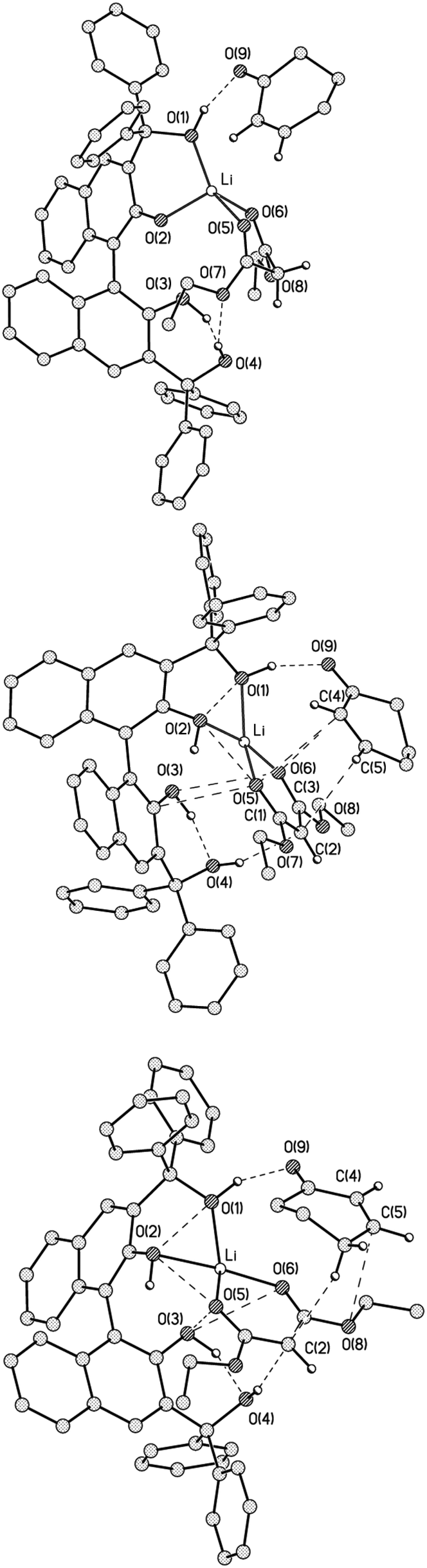 | ||
| Fig. 8 Views of Li1MA (top), Li2MA (middle) and Li3MA (bottom) according to the results of DFT calculations. Some hydrogen atoms are omitted for clarity. Intramolecular interactions are shown only for contacts for which critical points (3, −1) of electron density function were located. | ||
The Li3MA structure differed from Li2MA by rotation of the MA moiety by 180 degrees around the O(1)H⋯O(9)C bond (Fig. 8). Although the energy of this complex was almost equal to that of Li2MA (it is less stable by only 0.12 kcal mol−1), the type and number of interatomic interactions between the cyclohexenone and malonate were different. In contrast to the C(2)⋯(C5); O(5)⋯C(4) and O(6)⋯C(4) interactions in Li2MA, the atoms of the CC bond of MA in Li3MA interact only with the O(8) atom (O⋯C 3.68 Å) and with the C(2) atom of the malonate by one of the MA aliphatic protons. The distances between C(2) and C(5) are 3.58 Å in Li2MA and 4.2–4.28 Å in Li3MA. All other interactions in Li3MA are similar to those in Li2MA.
Thus, according to the DFT calculations, the ionisation of MD occurs by its α-proton being removed by the alcohol oxygen atom O(4) of BIMBOL-Li facilitated by proton distribution via the chain of hydrogen bonds inside Li1, leading to formation of Li2 existing in a facile equilibrium with Li1. However, the formation of the reactive intermediates BIMBOL0–Li+1–xMA–xMD−1 (Li2MA and Li3MA, Fig. 8) occurs by the addition of MA to Li2 and not Li1. Within adducts Li2MA and Li3MA the electrophilicity of the molecule of MA is greatly increased by the O(1)–H⋯O(9) hydrogen bond formation with the BIMBOL moiety. The mutual disposition of both MA and MD inside the intermediate complex Li2MA or Li3MA is ideal for the final carbon–carbon bond formation. The generation of MP can be easily portrayed as proceeding by the formation of a carbon–carbon bond between C(2) of MD−1 and C(5) of MA. The negative charge transfer is most likely being accompanied by a chain of proton transfers20 from O(1)–H to O(9) and from O(2)–H to O(1). Thus, there are no significant changes in the position of atoms and charges in the transition state of the carbon–carbon bond formation leading to MP.
It is the (R)-Li3MA intermediate that should lead to the expected R-configuration of MP (Tables 1 and 2) whereas (R)-Li2MA should give rise to MP with S-configuration. As the energy of both intermediates are almost equal, it is the energies of the relative transition states and not the initial intermediates that determine the stereochemical outcome of the carbon–carbon bond formation.
Conclusions
The family of tetraols, studied in this work, provide an interesting catalytic behaviour reminiscent of enzymatic catalysis. Features such as positioning of two substrates on the same catalytic centre, product inhibition of the catalysis and proton transfer through a chain of hydrogen bonds can all be found in typical enzymatic reactions. In addition, the trifunctional catalyst derived from BIMBOL-Li displays a sophisticated behaviour with its capabilities concealed by the intrinsic self-association phenomena. The BIMBOL scaffold should be a versatile unit for the generation of asymmetric catalysts and the mechanistic work reported herein not only allows a better understanding of this Michael addition, but will facilitate the design and development of other highly effective asymmetric catalysts based on BIMBOL and related polyol catalysts. Work in this direction is continuing in our group.Experimental
General experimental, crystallographic and computational details are given in the ESI.†(R)-BIMBOL
Synthesised by the previously reported method21 starting from (R)-BINOL. [α]25D +109.8 (c 1.00, CHCl3), lit.21 [α]20D +113.4 (c 1.00, CHCl3); Mp. 186–188 °C, (lit.22 mp. 176–179 °C); 1H NMR (300 MHz, CDCl3) δ 7.68–7.65 (m, 2H), 7.40–7.28 (m, 24H), 7.19 (s, 2H), 7.17–7.14 (m, 2H), 6.70 (s, 2H), 4.79 (s, 2H); found (%): C, 79.2; H, 5.8. C46H34O4·2H2O·(CH3)2CO. Calculated (%): C, 78.9; H, 5.9.(R)-H8-BINOL
(R)-BINOL (5 g, 17.5 mmol), Pd/C 10% wt (1.5 g), EtOH (39 ml) and H2O (1 ml) were charged into a 200 ml steel autoclave. The reaction was carried out at a pressure of 70 atm. of H2 and 120 °C for 5 hours. The product was purified by flash chromatography on SiO2 (n-hexane/EtOAc 5:1). Recrystallisation from n-hexane gave (R)-H8-BINOL (3.8 g, 76%) as a white powder. Measured: [α]25D +51.8 (c 2.00, CHCl3); mp. 149–151 °C; 1H NMR (400 MHz, CDCl3) δ 7.10 (d, J = 8.3 Hz, 2H), 6.85 (d, J = 8.3 Hz, 2H), 4.59 (s, 2H), 2.77 (t, J = 6.2 Hz, 4H), 2.40–2.08 (m, 4H), 1.87–1.64 (m, 8H). Literature data23 for (S)-enantiomer: [α]20D −49.3 (c 1.00, CHCl3), mp. 156–157 °C.
(R)-MOM-H8-BINOL
To a suspension of 55% NaH (1.2 g, 34.3 mmol) in THF (80 ml) under argon at 0 °C was added a solution of (R)-H8-BINOL (3.8 g, 13.3 mmol) in THF (18 ml). The reaction mixture was stirred at this temperature for 1 hour, then MOMCl (4.0 ml, 46 mmol) was added and the reaction left overnight. H2O (50 ml) was added to the reaction mixture which was then extracted with CH2Cl2 and washed with brine. The organic layer was dried over MgSO4, the solvent removed in vacuo and the residue was recrystallised from n-hexane to give (R)-MOM-H8-BINOL (4.66 g, 92%) as a white solid. Measured [α]25D +47.8 (c 2.00, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.07 (d, J = 8.5 Hz, 2H), 6.97 (d, 2H, J = 8.5 Hz), 5.03 (d, J = 6.7 Hz, 2H) 4.98 (dd, J = 6.7 Hz, 2H), 3.36 (s, 6H), 2.44–2.13 (m, 4H), 1.89–1.63 (m, 8H). Literature data for (S)-enantiomer24 [α]25D −46.6; 1H NMR corresponded to the lit. data.26(R)-MOM-H8-BIMBOL
To a solution of (R)-MOM-H8-BINOL (1.4 g, 3.6 mmol) in dry distilled THF (55 ml) under argon at −5 °C, a 2.5 M hexane solution of nBuLi (5.8 mL 14.6 mmol) was added dropwise with stirring. The reaction mixture was stirred at this temperature for 3 hours, then benzophenone (2.0 g, 11.0 mmol) was added and the reaction left stirring overnight at room temperature. The reaction was then neutralised by the addition of 0.1 M aqueous HCl. THF was removed in vacuo, the residue was extracted with CH2Cl2, the organic layer washed with water and then dried over MgSO4. The solvent was removed in vacuo. The product was recrystallised from MeOH to give white crystals of (R)-MOM-H8-BIMBOL (1.2 g, 45%). [α]25D +11.5 (c 1.00, CHCl3); mp. 222–224 °C; 1H NMR (400 MHz, CDCl3) δ 7.47–7.22 (m, 20H), 6.36 (s, 2H), 5.92 (s, 2H), 4.01 (d, J = 4.6 Hz, 2H), 3.96 (d, J = 4.6 Hz, 2H), 2.97 (s, 6H), 2.68–2.49 (m, 4H), 2.44–2.19 (m, 4H), 1.79–1.63 (m, 8H); 13C NMR (101 MHz, CDCl3) δ 150.51, 147.23, 146.95, 138.61, 136.97, 132.70, 131.54, 130.82, 127.94, 127.74, 127.72, 127.10, 97.55, 81.81, 56.88, 29.61, 27.72, 22.90 (2C).(R)-H8-BIMBOL
To a solution of (R)-MOM-H8-BIMBOL (1.2 g, 16.0 mmol) in THF (24 ml) was added 3 N aqueous HCl (4.7 ml) and the reaction heated under reflux for 5 hours. After cooling to room temperature, the reaction mixture was neutralized with 5% aqueous Na2CO3. Then it was extracted with CH2Cl2, dried over MgSO4, the solvent was removed in vacuo and the resulting solid purified by silica gel column chromatography (eluent: n-hexane/EtOAc 3:1) to give (R)-H8-BIMBOL (0.42 g, 40%) as a white solid. [α]25D +50 (c 0.5, CHCl3); mp. 116 °C (decomp.); 1H NMR (400 MHz, CDCl3) δ 7.42–7.16 (m, 20H), 6.26 (s, 2H), 5.99 (s, 2H), 4.60 (s, 2H), 2.58–2.50 (m, 4H), 2.32–2.12 (m, 4H), 1.79–1.61 (m, 8H); 13C NMR (101 MHz, CDCl3) δ 149.59, 146.21, 145.72, 136.98, 130.90, 129.74, 128.89, 127.93, 127.88, 127.79, 127.73, 127.31, 121.25, 82.64, 77.33, 29.38, 26.95, 22.98, 22.92; found (%): C, 83.3; H, 6.6. Calculated for C46H42O4 (%): C, 83.9; H, 6.4.
(S)-MOM-BINOL
The preparation of (S)-MOM-BINOL was carried out by the same method used for (R)-MOM-H8-BINOL and gave (S)-MOM-BINOL as a white solid, in 90% yield, m.p. 101–102 °C. 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 9.0 Hz, 2H), 7.90 (d, J = 8.0 Hz, 2H), 7.50–7.33 (m, 4H), 7.27–7.23 (m, 2H), 7.18 (d, J = 8.3 Hz, 2H), 5.17 (d, J = 6.24 Hz, 2H), 4.9 (d, J = 6.2 Hz, 2H), 3.19 (s, 6H). Literature data m.p. 102–103 °C.25(S)-BIFOL
To a solution of (S)-MOM-BINOL (2.0 g, 5.4 mmol) in dry distilled THF (50 ml) under argon at −5 °C, a 2.5 M hexane solution of nBuLi (7.5 mL, 18.7 mmol) was added dropwise with stirring. The reaction mixture was stirred at this temperature for 3 hours, then 9H-fluoren-9-one (2.4 g, 13.3 mmol) was added and the reaction left stirring overnight at room temperature. The reaction was quenched by the addition of saturated aqueous NH4Cl. THF was removed in vacuo, the residue extracted with CH2Cl2 and washed with water. The organic layer was dried over MgSO4, the solvent removed in vacuo to give (S)-MOM-BIFOL which was deprotected without further purification. The (S)-MOM-BIFOL was dissolved in dimethoxyethane (100 ml) and 6 N aqueous HCl (64 ml) added. The reaction was stirred at room temperature for 3 days, then neutralised with 5% aqueous Na2CO3, extracted with CH2Cl2 and dried over Na2SO4. The solvent was removed in vacuo and the product purified by silica gel column chromatography (eluting with petroleum ether/EtOAc 5:1, then 2:1). The resulting material was dried under vacuo over P2O5 and paraffin at the temperature of boiling toluene to give (S)-BIFOL (1.64 g, 47%) as a pale yellow crystalline compound. [α]25D +52.4 (c 0.21, CHCl3); mp. 200 °C (decomp.); 1H NMR (400 MHz, CDCl3) δ 9.37 (br s, 2H), 8.07 (d, J = 7.1 Hz, 2H), 7.96 (dd, J = 20.3, 9.1 Hz, 4H), 7.92–7.82 (m, 4H), 7.69 (dd, J = 14.5, 7.2 Hz, 6H), 7.62–7.41 (m, 10H), 4.62 (br s, 2H); 13C NMR (101 MHz, CDCl3) δ 152.26, 149.24, 148.44, 139.75, 139.32, 133.92, 129.66, 129.60, 129.42, 128.82, 128.72, 128.51, 128.42, 126.88, 126.82, 125.34, 124.94, 124.71, 123.70, 120.37, 120.32, 116.88, 86.38; Found (%): C, 85.3; H, 4.2. Calculated for C46H30O4 (%): C, 85.4; H, 4.7.
(R)-MOM-BINOL
The preparation of (R)-MOM-BINOL was carried out by the same method used for (R)-MOM-H8-BINOL and gave (R)-MOM-BINOL as a white solid in 91% yield.(R)-diethyl-MOM-BICBOL
To a solution of (R)-MOM-BINOL (2.8 g, 7.5 mmol) in dry distilled THF (110 ml) under argon at −5 °C, a 2.5 M hexane solution of nBuLi (12.7 ml, 31.8 mmol) was added dropwise with stirring. The reaction mixture was stirred at this temperature for 3 hours, then the resulting suspension was added dropwise to diethyl carbonate (70 ml) at 0 °C and left to stir overnight at room temperature. The reaction was then neutralised by addition of 0.1 M aqueous HCl. THF was removed in vacuo, the residue extracted with CH2Cl2 and washed with water. The organic layer was dried over MgSO4 and the solvent was removed in vacuo. The residue was purified by silica gel column chromatography (eluent: petroleum ether/EtOAc 4:1) to give (R)-diethyl-MOM-BICBOL (1.2 g, 31%) as a white solid. [α]25D +80.9 (c = 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.56 (s, 2H), 7.98 (d, J = 8.1 Hz, 2H), 7.48 (t, J = 7.5 Hz, 2H), 7.37 (m, 2H), 7.30 (d, J = 8.5 Hz, 2H), 4.86 (m, 4H), 4.47 (q, J = 7.1 Hz, 4H), 2.50 (s, 6H), 1.46 (t, J = 7.1 Hz, 6H). Literature data for (R)-enantiomer26 [α]20D +87.7 (c = 0.24, CHCl3). 1H NMR corresponded to the lit data.28
(R)-MOM-(CF3)8-BIMBOL
To a solution of (R)-diethyl-MOM-BICBOL (1.2 g, 2.3 mmol) in THF (22 ml) under an argon atmosphere at 0 °C, a 0.5 M THF solution of 3,5-di(trifluoro-methyl)phenylmagnesium bromide (25 ml, 12.5 mmol) was added, then the reaction was left overnight at room temperature. The reaction was quenched with saturated aqueous NH4Cl, extracted with CH2Cl2 and washed with water. The organic layer was dried over MgSO4 and solvent was removed in vacuo. The residue was purified by silica gel column chromatography (eluent: hexane, then hexane/EtOAc 5:1) to give (R)-MOM-(CF3)8-BIMBOL (2.6 g, 88%) as a mixture of conformers. Data for the major conformer: 1H NMR (300 MHz, CDCl3) δ 8.82–7.95 (m, 12H), 7.71 (d, J = 8.2 Hz, 2H), 7.50–7.38 (m, 4H), 7.18 (d, J = 8.3 Hz, 2H), 7.05 (s, 2H), 6.45 (s, 2H), 4.37 (d, J = 5.9 Hz, 2H), 4.01 (d, J = 5.9 Hz, 2H), 3.03 (s, 6H); 19F NMR (376 MHz, CDCl3) δ 14.94 (s), 14.70 (s); 13C NMR (101 MHz, CDCl3) δ 153.00 (s), 149.49 (s), 147.05 (s), 137.27 (s), 134.20 (s), 132.45–130.91 (m), 131.20 (s), 129.63 (s), 129.21 (s), 128.68 (s), 127.94 (s), 127.26 (d, J = 7.1 Hz), 126.43 (s), 125.64 (s), 125.17 (s), 124.54 (d, J = 7.2 Hz), 121.51–122.10 (m), 119.12 (d, J = 7.0 Hz), 99.73 (s), 80.01 (s), 57.53 (s); found (%): C, 54.1; H, 2.7; F, 35.4. Calculated for C58H34O6F24 (%): C, 54.3; H, 2.7; F, 35.5.
(R)-(CF3)8-BIMBOL
To a solution of (R)-MOM-(CF3)8-BIMBOL (2.6 g, 2.0 mmol) in dioxane (65 ml), 26% aqueous HCl (28 ml) was added and the reaction heated under reflux at 60 °C for 7 hours. The reaction mixture was quenched with aqueous Na2CO3 until it was just slightly acidic, extracted with CH2Cl2, washed with water, dried over MgSO4 and solvent was removed in vacuo. The residue was purified by silica gel column chromatography (hexane then hexane/acetone 1:1) to give (R)-(CF3)8-BIMBOL (1.5 g, 63%) as a beige crystalline solid. [α]25D −96.8 (c 1.00, CHCl3); mp. 175 °C; 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 20.3 Hz, 4H), 7.85 (d, J = 8.4 Hz, 8H), 7.76 (d, J = 7.2 Hz, 2H), 7.53–7.39 (m, 4H), 7.14–7.07 (m, 4H), 5.60 (s, 2H), 5.20 (s, 2H); 19F NMR (376 MHz, CDCl3) δ 14.93 (s), 14.85 (s). 13C NMR (101 MHz, CDCl3) δ 149.65 (s), 147.74 (s), 146.32 (s), 132.81 (s), 132.44 (s), 132.10 (s), 131.77 (s), 131.73 (s), 131.36 (s), 129.51 (s), 129.35 (s), 128.51 (s), 127.76 (br s), 127.21 (br s), 125.91 (s), 124.45 (s), 123.19 (s), 122.32 (br s), 121.74 (s), 112.76 (s), 80.89 (s); found (%): C, 53.9; H, 2.5; F, 37.8. Calculated for C54H26O4F24 (%): C, 54.3; H, 2.2; F, 38.2.
Diethyl (S)-2-(3-oxocyclohexyl)malonate (MP)
To a solution of (S)-BIMBOL (8.1 mg, 0.0124 mmol), PhOLi (1.3 mg, 0.013 mmol) and cyclohexenone (23.8 mg, 0.248 mmol) in anhydrous CH2Cl2 (1 ml) under argon at room temperature diethyl malonate (39.7 mg, 0.248 mmol) was added and the reaction was stirred for 48 hours. Then the reaction was quenched by adding acetic acid, the mixture was evaporated and diethyl (S)-2-(3-oxocyclohexyl)malonate isolated as a colourless oil (60.0 mg, 94%) by preparative thin layer chromatography eluting with petroleum/EtOAc 5:1. [α]25D −2.7 (c 0.5, CHCl3); 1H NM R (400 MHz, CDCl3) δ 4.22 (qd, J = 7.1, 4.1 Hz, 4H), 3.31 (d, J = 7.9 Hz, 1H), 2.55 (dtd, J = 15.2, 7.8, 3.7 Hz, 1H), 2.44 (t, J = 17.0 Hz, 2H), 2.36–2.20 (m, 2H), 2.14–2.04 (m, 1H), 1.98 (d, J = 12.7 Hz, 1H), 1.81–1.61 (m, 1H), 1.62–1.45 (m, 1H), 1.29–1,20 (m, 6H), found (%): C, 61.4; H, 7.8. Calculated for C13H20O5 (%): C, 60.9; H, 7.9. [α]25D −2.7 (c 1.00, CHCl3) Enantiomeric analysis: CHIRALPAK AS-H column (4.6 mm i.d. × 250 mm); eluent n-hexane/2-propanol = 90:10; flow rate 1.0 ml min−1; detection UV 210 nm; retention times: 18.3 min, 20.1 min, ee = 83%. Literature data for (R)-enantiomer27 [α]25D +3.3 (c 1, CHCl3) for 93% ee 1H NMR corresponded to the lit data.29
Kinetic measurements
Acknowledgements
Authors gratefully thank the financial support from RSF 15-13-00039 and RBFR 15-53-05014 grants.Notes and references
- For reviews see: Comprehensive Asymmetric catalysis, and Supplements 1 and 2, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, New York, 1999 CrossRef CAS; K. Ohmatsu and T. Ooi, Tetrahedron Lett., 2015, 56, 2043–2048 CrossRef CAS; Introduction to Frontiers in Transition Metal Catalyzed Reactions, Chem. Rev., ed. J. A. Gladysz, 2011, vol. 111, pp. 1167–2486 RSC; S. Matsunaga and M. Shibasaki, Chem. Commun., 2014, 50, 1044–1057 RSC.
- For reviews see: M. Waser, Progress in the Chemistry of Organic Natural Products. Asymmetric Organocatalysis in Natural Product Syntheses, Springer, New York, 2012 CrossRef CAS; R. Sutar and N. Joshi, Tetrahedron: Asymmetry, 2013, 24, 1345–1363 CrossRef CAS; A. Quintavalla, L. Cerisoli and E. Montroni, Curr. Organocatal., 2014, 1, 107–171 CrossRef; A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703–4832 CrossRef PubMed; S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312–4348 CrossRef PubMed; C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390–2431 CrossRef PubMed; Asymmetric Organocatalysis, Top. Curr. Chem, ed. B. List, Springer, New York, 2009, vol. 291 CrossRef PubMed; R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603–614 CrossRef PubMed.
- (a) S. Schenker, A. Zamfir, M. Freund and S. Tsogoeva, Eur. J. Org. Chem., 2011, 2209–2222 CrossRef CAS; (b) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed; (c) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS PubMed; (d) J. Yu, F. Shi and L.-Z. Gong, Acc. Chem. Res., 2011, 44, 1156–1171 CrossRef CAS PubMed; (e) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518–533 CrossRef CAS PubMed; (f) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed; (g) T. J. Auvil, A. G. Schafer and A. E. Mattson, Eur. J. Org. Chem., 2014, 2633–2646 CrossRef CAS; (h) C. Uyeda and E. N. Jacobsen, J. Am. Chem. Soc., 2011, 133, 5062–5075 CrossRef CAS PubMed; (i) H. B. Jang, H. S. Rho, J. S. Oh, E. H. Nam, S. E. Park, H. Y. Bae and C. E. Song, Org. Biomol. Chem., 2010, 8, 3918–3922 RSC; (j) G. Tarkanyi, P. Kiraly, T. Soos and S. Varga, Chem. – Eur. J., 2012, 18, 1918–1922 CrossRef CAS PubMed; (k) Y. Fan and S. R. Kass, Org. Lett., 2016, 18, 188–191 CrossRef CAS PubMed.
- Enzyme Catalysis in Organic Synthesis, ed. P. T. Anastas and R. H. Crabtree, Wiley-VCH, Stuttgart, 2009 Search PubMed.
- M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520–1543 CrossRef CAS PubMed; Y. Yamaoka, H. Miyabe and Y. Takemoto, J. Am. Chem. Soc., 2007, 129, 6686–6687 CrossRef PubMed; S. J. Zuend and E. N. Jacobsen, J. Am. Chem. Soc., 2007, 129, 15872–15883 CrossRef PubMed; J.-F. Bai, L.-L. Wang, L. Peng, Y.-L. Guo, L.-N. Jia, F. Tian, G.-Y. He, X.-Y. Xu and L.-X. Wang, J. Org. Chem., 2012, 77, 2947–2953 CrossRef PubMed; A. Quintavalla, L. Cerisoli and E. Montroni, Curr. Organocatalysis, 2014, 1, 107–171 CrossRef.
- G. Jindal, H. Kisan and R. Sunoj, ACS Catal., 2015, 5, 480–503 CrossRef CAS; J.-C. Wasilke, S. Obrey, R. Baker and G. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef PubMed.
- T. T. Dang, F. Boeck and L. Hintermann, J. Org. Chem., 2011, 76, 9353–9361 CrossRef CAS PubMed.
- H. Yamamoto and K. Futatsugi, Angew. Chem., Int. Ed., 2005, 44, 1924–1942 CrossRef CAS PubMed.
- H. Sasai, T. Arai, Y. Satow, K. N. Houk and M. Shibasaki, J. Am. Chem. Soc., 1995, 117, 6194–6198 CrossRef CAS; M. Shibasaki, M. Kanai, S. Matsunaga and N. Kumagai, Acc. Chem. Res., 2009, 42, 1117–1127 CrossRef PubMed; J. R. Robinson, J. Gu, P. J. Carroll, E. J. Schelter and P. J. Walsh, J. Am. Chem. Soc., 2015, 137, 7135–7144 CrossRef PubMed; R. Schiffers and H. Kagan, Synlett, 1997, 1175–1176 CrossRef; I. Holmes and H. Kagan, Tetrahedron Lett., 2000, 41, 7453–7456 CrossRef; M. Nakajima, Y. Orito, T. Ishizuka and S. Hashimoto, Org. Lett., 2004, 6, 3763–3765 CrossRef PubMed; T. Ichibakase, Y. Orito and M. Nakajima, Tetrahedron Lett., 2008, 49, 4427–4429 CrossRef; Y. Orito, S. Hashimoto, T. Ishizuka and M. Nakajima, Tetrahedron, 2006, 62, 390–400 CrossRef; M. Hatano, T. Ikeno, T. Miyamoto and K. Ishihara, J. Am. Chem. Soc., 2005, 127, 10776–10777 CrossRef PubMed; M. Hatano, T. Horibe and K. Ishihara, J. Am. Chem. Soc., 2010, 132, 56–57 CrossRef PubMed; H. Yan, J. S. Oh, J. W. Lee and C. E. Song, Nat. Commun., 2012, 3, 1212 CrossRef PubMed; T. Otani, A. Sugawara and Y. Tamai, Tetrahedron Lett., 2014, 55, 4923–4926 CrossRef; S. Kotani, M. Moritani and M. Nakajima, Asian J. Org. Chem., 2015, 4, 616–618 CrossRef.
- (a) Y. N. Belokon, K. A. Kochetkov, T. D. Churkina, N. S. Ikonnikov, A. A. Chesnokov, O. V. Larionov, I. Singh, V. S. Parmar, S. Vyskočil and H. B. Kagan, J. Org. Chem., 2000, 65, 7041–7048 CrossRef CAS PubMed; (b) Y. N. Belokon, Z. T. Gugkaeva, V. I. Maleev, M. A. Moskalenko, M. North and A. T. Tsaloev, Tetrahedron: Asymmetry, 2010, 21, 1793–1796 CrossRef CAS; (c) Y. N. Belokon, V. I. Maleev, Z. T. Gugkaeva, M. A. Moskalenko, A. T. Tsaloev, A. S. Peregudov, S. Ch. Gagieva, K. A. Lyssenko, V. N. Khrustalev and A. V. Grachev, Russ. Chem. Bull., 2007, 56, 1507–1514 CrossRef CAS; (d) Y. N. Belokon, S. Harutyunyan, E. V. Vorontsov, A. S. Peregudov, V. N. Chrustalev, K. A. Kochetkov, D. Pripadchev, A. S. Sagyan, A. K. Beck and D. Seebach, ARKIVOC, 2004, iii, 132–150 Search PubMed.
- Y. N. Belokon, Z. T. Gugkaeva, V. I. Maleev, M. A. Moskalenko, A. T. Tsaloev, V. N. Khrustalev and K. V. Hakobyan, Tetrahedron: Asymmetry, 2011, 22, 167–172 CrossRef CAS.
- (a) G. Kagan, W. Li, D. Li, R. Hopson and P. G. Williard, J. Am. Chem. Soc., 2011, 133, 6596–6602 CrossRef CAS PubMed; (b) J. Liang, A. C. Hoepker, A. M. Bruneau, Y. Ma, L. Gupta and D. B. Collum, J. Org. Chem., 2014, 79, 11885–11902 CrossRef CAS PubMed; (c) K. J. Jin and D. B. Collum, J. Am. Chem. Soc., 2015, 137, 14446–14455 CrossRef CAS PubMed.
- N. T. Tran, S. O. Wilson and A. K. Franz, Org. Lett., 2012, 14, 186–189 CrossRef CAS PubMed.
- Q. Wang, X. Chen, L. Tao, L. Wang, D. Xiao, X.-Q. Yu and L. Pu, J. Org. Chem., 2007, 72, 97–101 CrossRef CAS PubMed.
- T. H. Siddall and W. E. Stewart, J. Org. Chem., 1969, 34, 233–237 CrossRef CAS; W. T. Ford, T. B. Thompson, K. A. J. Snoble and J. M. Timko, J. Am. Chem. Soc., 1975, 97, 95–101 CrossRef.
- C. Yang, X.-S. Xue, X. Li and J.-P. Cheng, J. Org. Chem., 2014, 79, 4340–4351 CrossRef CAS PubMed.
- W. N. Olmstead and F. G. Bordwell, J. Org. Chem., 1980, 45, 3299–3305 CrossRef CAS.
- R. F. W. Bader, Atoms In molecules. A Quantum Theory, Clarendron Press, Oxford, 1990 Search PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS; E. Espinosa, I. Alkorta, I. Rozas, J. Elguero and E. Molins, Chem. Phys. Lett., 2001, 336, 457–461 CrossRef.
- J. M. Mayer, Acc. Chem. Res., 2011, 44, 36–46 CrossRef CAS PubMed; J. Bonin, C. Costentin, M. Robert, J.-M. Savéant and C. Tard, Acc. Chem. Res., 2012, 45, 372–381 CrossRef PubMed.
- Q. Wang, X. Chen, L. Tao, L. Wang, D. Xiao, X.-Q. Yu and L. Pu, J. Org. Chem., 2007, 72, 97–101 CrossRef CAS PubMed.
- Y.-L. Zhang and Q.-H. Fan, J. Chem. Res., 2005, 778–779 CrossRef.
- X. Li, Q. Ding, J. Ge, J. Xie and D. Xu, J. Org. Chem., 2009, 74, 1785–1787 CrossRef CAS PubMed.
- D.-W. Wang, S.-M. Lu and Y.-G. Zhou, Tetrahedron Lett., 2009, 50, 1282–1286 CrossRef CAS.
- Y. Yao, H. Shu, B. Tang, S. Chen, Z. Lu and W. Xue, Chin. J. Chem., 2015, 33, 601–609 CrossRef CAS.
- K.-X. Xu, L.-R. Yang, Y.-X. Wang, J. Zhao and C.-J. Wang, Supramol. Chem., 2010, 22, 563–570 CrossRef CAS.
- V. Wascholowski, R. K. Knudsen, E. T. C. Mitchell and V. S. Ley, Chem. – Eur. J., 2008, 14, 6155–6165 CrossRef CAS PubMed.
- F. Neese and F. Wennmohs, ORCA - An Ab Initio, DFT and Semiempirical SCF-MO Package, 2014 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, X-ray and computational details, NMR spectra and HPLC traces. CCDC 1409564. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cy01697a |
| ‡ Present address: Biotech Industry Ltd, 1 Leninskye Gory, Moscow 119992, Russian Federation. |
| § Present address: Drugs Technology, 2a Rabochaya Street, Khimki, Moscow Region, 141400, Russian Federation. |
| ¶ Crystallographic data for (S)-BIFOL·H2O has been submitted to CCDC and given code CCDC 1409564. |
| This journal is © The Royal Society of Chemistry 2017 |