
Advancing porphyrin's biomedical utility via supramolecular chemistry
M. A.
Rajora
ab,
J. W. H.
Lou
ac and
G.
Zheng
 *abc
*abc
aPrincess Margaret Cancer Centre, University Health Network, 101 College Street, Toronto, Ontario M5G 1L7, Canada. E-mail: gang.zheng@uhnresearch.ca
bInstitute of Biomaterials and Biomedical Engineering, University of Toronto, 164 College Street, Toronto, Ontario M5S 3G9, Canada
cDepartment of Medical Biophysics, University of Toronto, 101 College Street, Toronto, Ontario M5G 1L7, Canada
First published on 19th October 2017
Abstract
Porphyrins are organic heterocyclic macrocycles with photophysical properties well-suited for clinical phototherapy and cancer imaging. However, their wider application in the clinical management of disease is barred by poor aqueous solubility, bioavailability, tumour accumulation and skin phototoxicity. These limitations instigated the development of supramolecular platforms that improved porphyrin pharmacokinetics and tumour-homing. The supramolecular formulation of porphyrins also facilitates single agent-mediated deeper tissue photoactivation, extended imaging and theranostic multimodality, and synergistic application of multiple therapies. Supramolecular porphyrin structures can overcome additional limitations of porphyrin-mediated photodynamic therapy (PDT), including low depths of tissue penetration that restrict PDT to superficial lesions, inability to treat hypoxic tumours, and incomplete tumour damage. In this review, we discuss the photophysical properties of porphyrins, and overview the clinically-relevant advantages and challenges arising from their incorporation within supramolecular platforms. Specifically, fundamentals underlying the ability of these platforms to ameliorate passive and active porphyrin delivery to tumours, achieve deeper tissue PDT via red-shifted porphyrin Q-bands, energy transfer and sonodynamic effects, and enable new porphyrin-mediated theranostics and synergistic therapeutic capabilities will be explained and exemplified with seminal and cutting-edge in vivo studies.
 M. A. Rajora | Maneesha Rajora is an MD/PhD candidate and Vanier Scholar at the University of Toronto. She completed her MASc in Biomedical Engineering in 2013, prior to which she obtained her BSc(H) in Chemistry from Dalhousie University. She is currently completing her doctoral studies under the supervision of Dr Gang Zheng within the Institute of Biomaterials and Biomedical Engineering. Her research focuses on the development of blood–brain barrier permeating biomaterials for cancer theranostics. |
 J. W. H. Lou | Jenny Lou obtained her BSc in Neuroscience at the University of Alberta in 2015. Presently, she is a Vanier Scholar and a PhD candidate in the Department of Medical Biophysics at the University of Toronto. Under the supervision of Dr Gang Zheng, she is investigating the use of porphyrin nanoparticles for tracking the migration of T-cells and enhancing their cytotoxicity for cancer immunotherapy. |
 G. Zheng | Dr Gang Zheng is a Professor at the University of Toronto and a Senior Scientist at the Princess Margaret Cancer Center. He received his PhD in 1999 from SUNY Buffalo in Medicinal Chemistry. He joined the University of Pennsylvania in 2001 as an Assistant Professor of Radiology and moved to Canada in 2006. His research focuses on developing clinically translatable technologies to combat cancer. His lab discovered ‘porphysome’, a nontoxic nanoparticle applicable for cancer imaging and therapy. Dr Zheng is an Associate Editor for Bioconjugate Chemistry and a Fellow of the American Institute of Medical and Biological Engineering. |
Key learning points• Biomedically-explored classes of porphyrin supramolecular structures, including their advantages and disadvantages.• Passive, active and controlled approaches for porphyrin delivery. • Development of platforms that allow for porphyrin activation with 700–900 nm near infrared light, X-ray or ultrasound for deeper tissue access relative to porphyrin monomers. • Biomedical utility of extending porphyrin theranostic capabilities. • Supramolecular structures that enable the synergistic combination of photodynamic therapy with photothermal, chemo, gene or immune therapy. |
1. Introduction: porphyrin monomers, their properties and biomedical applications
Porphyrins comprise a class of heterocyclic organic molecules that are pivotal in sustaining plant and mammalian life. These pigmented macrocycles derive their name from “porphura”, a Greek term referring to the colour purple, which aptly alludes to the photophysical properties that drive the biological functions of porphyrins and their derivatives. Of these, oxygen transport via hemoglobin and photosynthesis in chloroplasts are ubiquitously familiar and long-standing roles, wherein the corresponding heme and chlorophyll moieties represent a naturally-occurring porphyrin and porphyrin derivative respectively. In the last century, the applications of porphyrins have expanded into the realms of disease treatment and imaging.The photophysical properties that give rise to these biomedical utilities are rooted in the aromatic macrocyclic structure of porphyrins (Fig. 1). Free base porphyrins contain four bridged pyrrole groups consisting of 22 π-electrons, of which 18 are thought to be conjugated. This gives rise to facile π → π* transitions, yielding two optical signatures within the visible spectrum of light: (1) a strong Soret, or B band, at ∼400 nm, resulting from a ground state to second excited singlet state electronic transition (S0 → S2), and (2) four lower energy and less intense Q-bands between ∼450–650 nm resulting from ground state to first excited singlet state transitions (S0 → S1). Reduction of one or two opposing pyrrole double bonds yields chlorin and bacteriochlorin porphyrin derivatives respectively, with associated red-shifted higher absorptivity Q-bands between 650–800 nm as a result of disrupted tetrapyrrole symmetry. Upon absorbing light, porphyrin electrons are excited to a short-lived (nanosecond-length) electronic singlet state (Sn). Following internal conversion to S1, porphyrins can return to ground state via non-radiative vibrational relaxation, fluorescence emission, or may undergo a non-radiative spin-forbidden electronic transition to a triplet state (T1). The relatively longer lifetime of the triplet state (∼micro to milli-seconds) allows porphyrins to undergo radiative decay via phosphorescence, or interact with their surroundings to generate reactive oxygen species (ROS) through two routes. The first, termed a Type I reaction, involves the transfer of an electron or proton to neighbouring cellular substrates and molecules to form radical species, which then interact with molecular oxygen to form cytotoxic ROS, including hydroxyl radicals and hydrogen peroxide. The second, and more dominant Type II reaction, involves a direct transfer of energy from the porphyrin triplet excited state to the triplet ground state of molecular oxygen (3O2) to form highly reactive singlet oxygen (1O2).
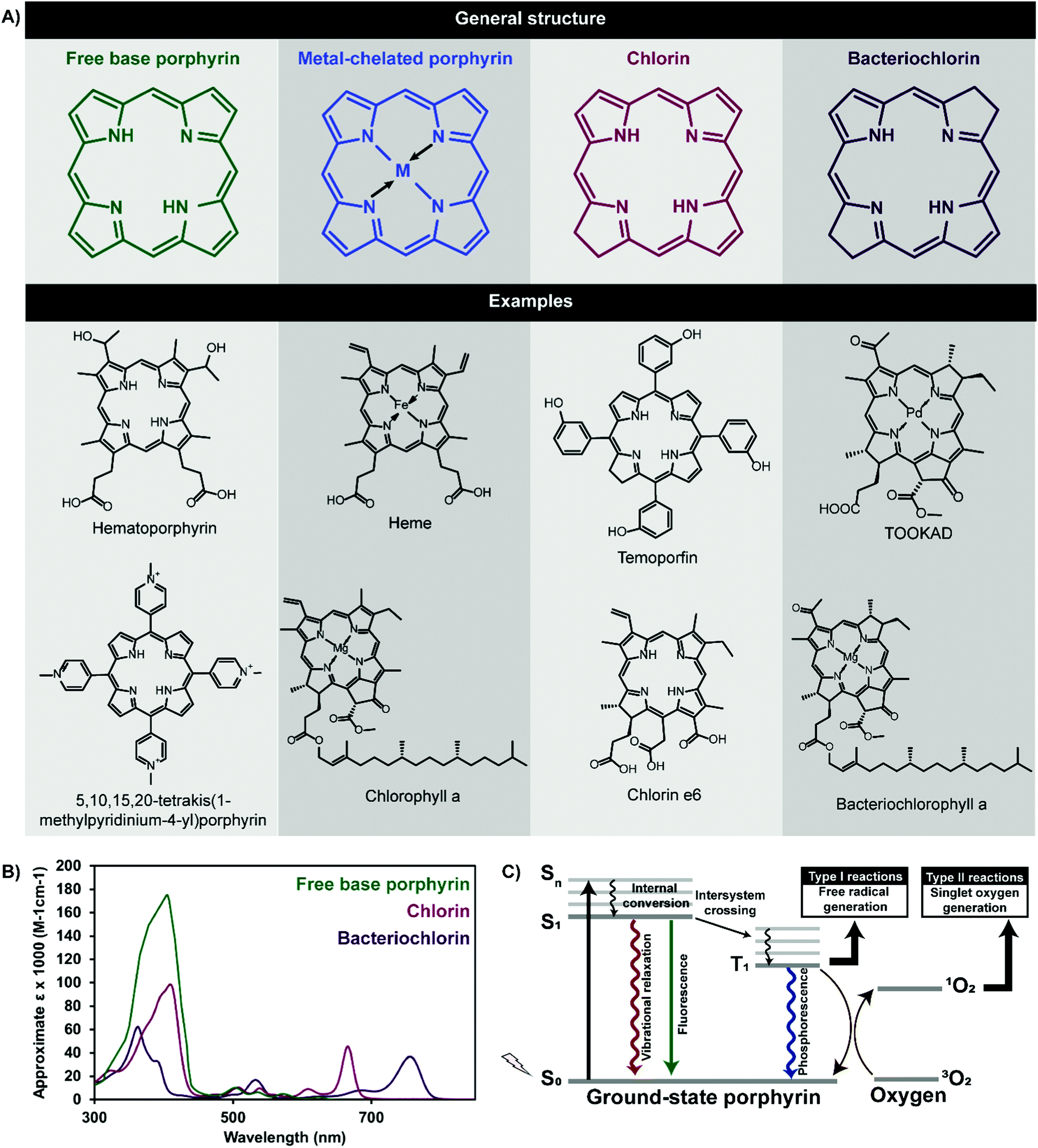 | ||
| Fig. 1 General chemical structures and examples of porphyrins and their derivatives (A), and associated absorbance spectra (B). Porphyrin excitation by visible or near infrared light yields a number of decay pathways (C) that dictate their biomedical application. | ||
This latter electronic transition underlies photodynamic therapy (PDT), a treatment modality that triggers spatially-confined cell death through the photoactivation of photosensitizers (PS) in the presence of oxygen. Porphyrins and their derivatives are the most ubiquitously-explored PS. Photoinduced 1O2 generation leads to cell membrane, mitochondrial, protein and deoxyribonucleic acid (DNA) damage, depending on the region of porphyrin cellular localization. The resulting cellular apoptosis and/or necrosis has led to the application of porphyrins as PDT agents for diverse indications, including cancer, infectious diseases, cardiovascular disease, and acne. The therapeutic benefits of porphyrin PS lies in their: (1) minimally-invasive photoactivation with red or near infrared (NIR) light within the 700–900 optically-clear window in tissue, via their low energy Q-bands, (2) propensity to accumulate in malignant versus healthy cells, and (3) relatively high 1O2 quantum yields, enabling the 10–55 nm diffusion distance of 1O2 in tissue to be exploited for localized therapy. Collectively, these traits enable porphyrins to potentially mediate minimally-invasive, localized, healthy tissue-sparing, non-ionizing, controlled and target-specific cancer therapy; a combination unachievable by surgery, radiation therapy or chemotherapy. As such, porphyrins are currently clinically-approved for the treatment of lung, skin and esophageal cancers as summarized in Table 1.
| Photosensitizer | Porphyrin type | Approval | Application | Excitation wavelength and extinction coefficient |
|---|---|---|---|---|
| Hematoporphyrin derivative | Porphyrin | World-wide | Lung, esophageal, bile duct, bladder, brain, ovarian, and cervical cancer PDT, myopic maculopathy |
630 nm
3000 M−1 cm−1 |
| 5-Aminolevulinic acid | Porphyrin pro-drug | US, EU | Actinic keratosis, basal-cell carcinoma, head and neck and gynaecological cancer PDT. Brain, head and neck, bladder cancer imaging. |
635 nm
<10 |
| 5-Aminolevulinic acid esters | Porphyrin pro-drug | World-wide | Actinic keratosis, Bowen's disease, basal cell carcinoma PDT. Bladder cancer diagnosis. |
635 nm
<10 |
| Verteporfin | Benzoporphyrin | US, EU, Canada | Age-related macular degeneration, pathologic myopia, histoplasmosis |
690 nm
35 |
| Talaporfin | Chlorin | Japan | Lung cancer PDT |
664 nm
45 |
| Temoporfin | Chlorin | EU | Head and neck, prostate, pancreatic cancer PDT |
652 nm
30 |
Porphyrin accumulation within malignant cells can be leveraged alongside their red/NIR fluorescence emissions for image-guided tumour resection. The central chelation of porphyrins by transition metals can expand this imaging repertoire to include positron emission tomography (PET), single-photon emission computed tomography (SPECT), and magnetic resonance (MR) imaging, as reviewed by Bryden et al. and summarized in Table 2.1 Transition metal chelation increases the symmetry of porphyrins, and can raise their π* energy level through charge transfer, leading to two hypsochromically-shifted Q-bands relative to free base porphyrin. However, chelation of a number of metals, including Zn, Cu, Mn and Fe, quenches porphyrin fluorescence and 1O2 generation efficiency. Thus, multimodal imaging cannot be effectively nor simultaneously derived from a single monomeric porphyrin contrast agent.
| Central atom | Application |
|---|---|
| Abbreviations: PAI (photoacoustic imaging), SPECT (single photon emission computed tomography), PET (positron emission tomography), MRI (magnetic resonance imaging). | |
| H2 (free base) | Fluorescence, PAI |
| Cu | 64Cu SPECT, PET |
| Fe | 52Fe SPECT |
| Ga | 67Ga SPECT, 68Ga PET |
| Gd | Gd MRI |
| In | 111In gamma imaging, SPECT |
| Mn | 54Mn SPECT, 51Mn PET, 55Mn MRI |
| Pd | Phosphorescence |
| Zn | 52Zn PET, PAI |
| Tc | 99mTc SPECT |
Metal chelation can further influence the photophysical properties of porphyrins by promoting their ordered aggregation. For example, Mg2+ central chelation mediates bacteriochlorin J-aggregation, which consists of face-to-face porphyrin assembly, leading to bathochromically-shifted higher absorptivity Q-bands. Contrarily, side-to-side H-aggregation of porphyrins shifts Q-bands hypsochromically. As will be further discussed, J-aggregation-induced red-shifts in porphyrin Q-bands can be beneficial in facilitating the photoactivation of porphyrins via deeper tissue penetrating NIR light. However, the practical utility of this porphyrin monomer self-assembly is limited by aqueous insolubility of porphyrin aggregates.
High hydrophobicity and the resulting aggregation of porphyrins in aqueous solutions in either an ordered or disordered manner reduces their bioavailability and accumulation at target lesions. Systemically administered porphyrins induce skin phototoxicity, which restricts dose escalation. Furthermore, NIR light used to excite porphyrins only permeates through 1–2 cm deep tissue, thereby confining porphyrin-PDT treatment to superficial lesions. As an oxygen-dependent phenomenon, porphyrin-PDT is further constrained to the treatment of hypoxic tissue. Thus, despite bearing therapeutic advantages over conventional cancer therapies, the widespread clinical use of porphyrins is hindered.
2. Addressing molecular porphyrin limitations through supramolecular chemistry
As summarized in Fig. 2, the above clinical challenges associated with monomeric porphyrin delivery can be overcome by supramolecular porphyrin structures. Assembly of porphyrins via intermolecular forces can transform their biological and physiochemical properties. This is exemplified naturally, wherein bacteriochlorophyll self-assembly within chlorosomes via J-aggregation increases the Q-band absorption coefficient relative to the monomer, and facilitates highly efficient bacterial photosynthesis in light-deficient environments. Heme exists naturally as a four protein sub-unit supramolecular assembly that enables oxygen binding and transport. Accordingly, synthetic supramolecular assembly can alter and even enhance the clinical utility of monomeric porphyrins. To this end, diverse porphyrin supramolecular structures, illustrated in Fig. 2, have been explored to both advance existing therapeutic and imaging properties of monomeric porphyrin, and to introduce new and synergistic therapeutic functionalities. Here, we describe how supramolecular porphyrin structures can increase porphyrin delivery to tumours, facilitate deeper tissue access, broaden multimodality, and deliver new therapeutic paradigms in vitro and in vivo relative to molecular porphyrin.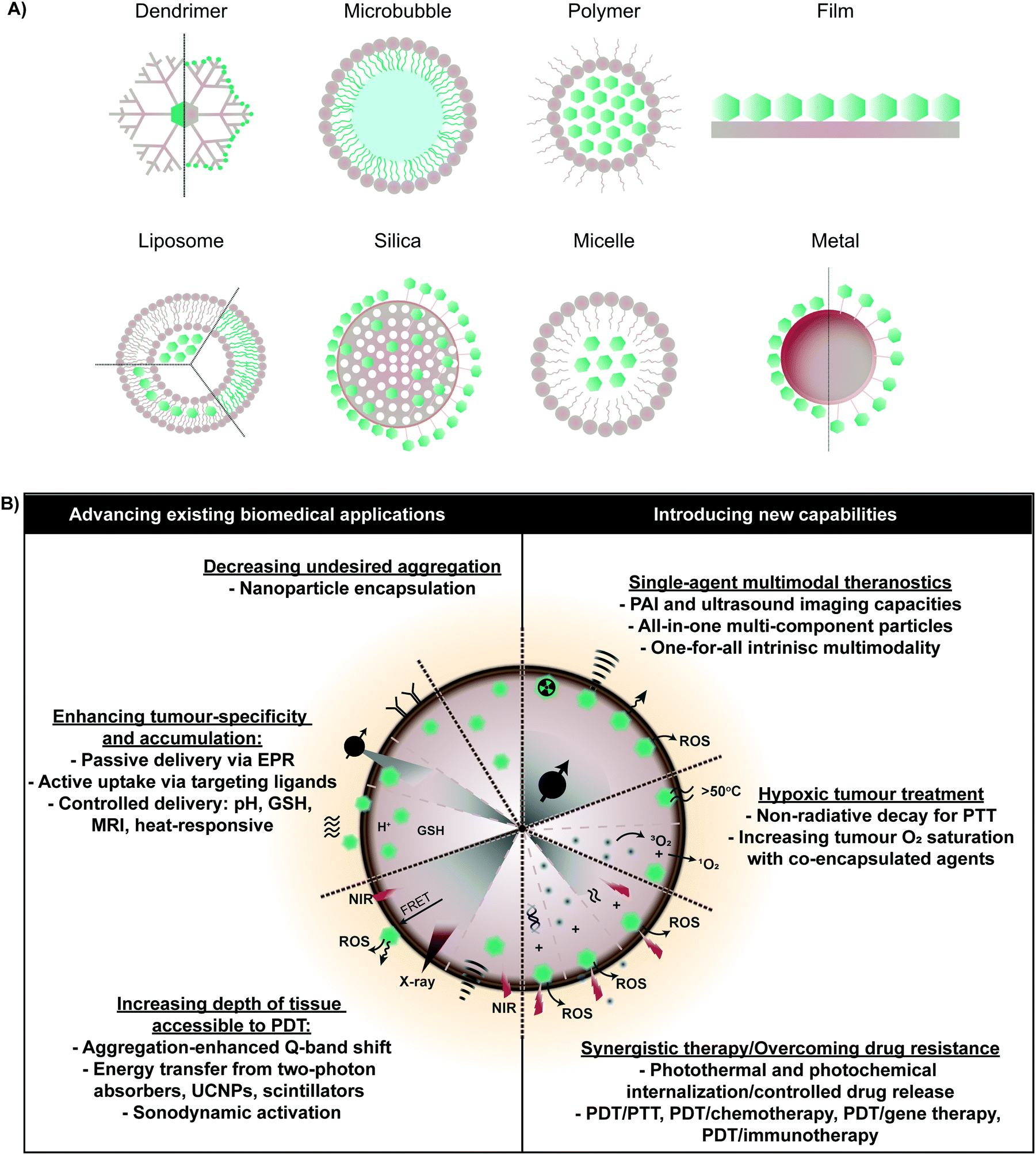 | ||
| Fig. 2 Overview of porphyrin supramolecular structures (A) explored for therapeutic and medical imaging purposes pre-clinically. Incorporation within supramolecular platforms allows for more effective implementation of existing functionalities of monomeric porphyrin (shown in green), but also introduces new therapeutic and imaging capabilities within a single porphyrin agent, which, as shown (B) can address current clinical barriers in PDT. | ||
3. Enhancing porphyrin delivery via supramolecular chemistry
Nanoparticles that augment PS delivery to tumours encompass the largest class of biomedically-relevant porphyrin supramolecular structures explored thus far. Nanoparticles can increase tumour accumulation of porphyrins by: (1) increasing porphyrin solubility and thereby bioavailability, (2) extending porphyrin circulation half-lives, (3) protecting against premature photodegradation, and (4) facilitating site-specific delivery. The latter two features in particular hold the potential of decreasing off-target PDT toxicity, and are rooted in the exploitation of the tumour microenvironment and activatable PDT; a strategy in which porphyrin fluorescence and singlet oxygen generation are quenched upon particle administration, and restored upon application of a stimulus.3.1 Tumour microenvironment
The multifaceted tumour microenvironment (TME) plays a major role in the corruption of healthy, differentiated cells into malignant cells. The TME comprises: (1) tumour cells, (2) the extracellular matrix, composed of proteoglycans, hyaluronic acid, and fibrous proteins, (3) stromal cells such as fibroblasts, mesenchymal and immune cells, (4) peptides such as chemokines and cytokines, and (5) metabolites from tumour and stromal cells (Fig. 3A). A growing tumour has an overwhelming demand for oxygen and nutrients that outstrips supply. Consequently, neovascularization is induced, where the resulting vasculature is characterized by disorganized branching with a greater vascular density at the tumour's periphery. Other characteristics of tumour neovasculature include discontinuous, fenestrated capillaries with unevenly distributed pericytes, elevated interstitial fluid pressure, slow venous return, and poor lymphatic drainage. Thus, oxygen perfusion within tumours varies, such that hypoxic and necrotic regions are found at the tumour core. Due to the paucity of oxygen, tumour cells rely on glycolysis for ATP generation. The subsequent accumulation of lactate results in a TME pH of 6.7–7.1. While the complexity of the TME poses major challenges for effective therapy, it also provides exploitable avenues to facilitate drug delivery.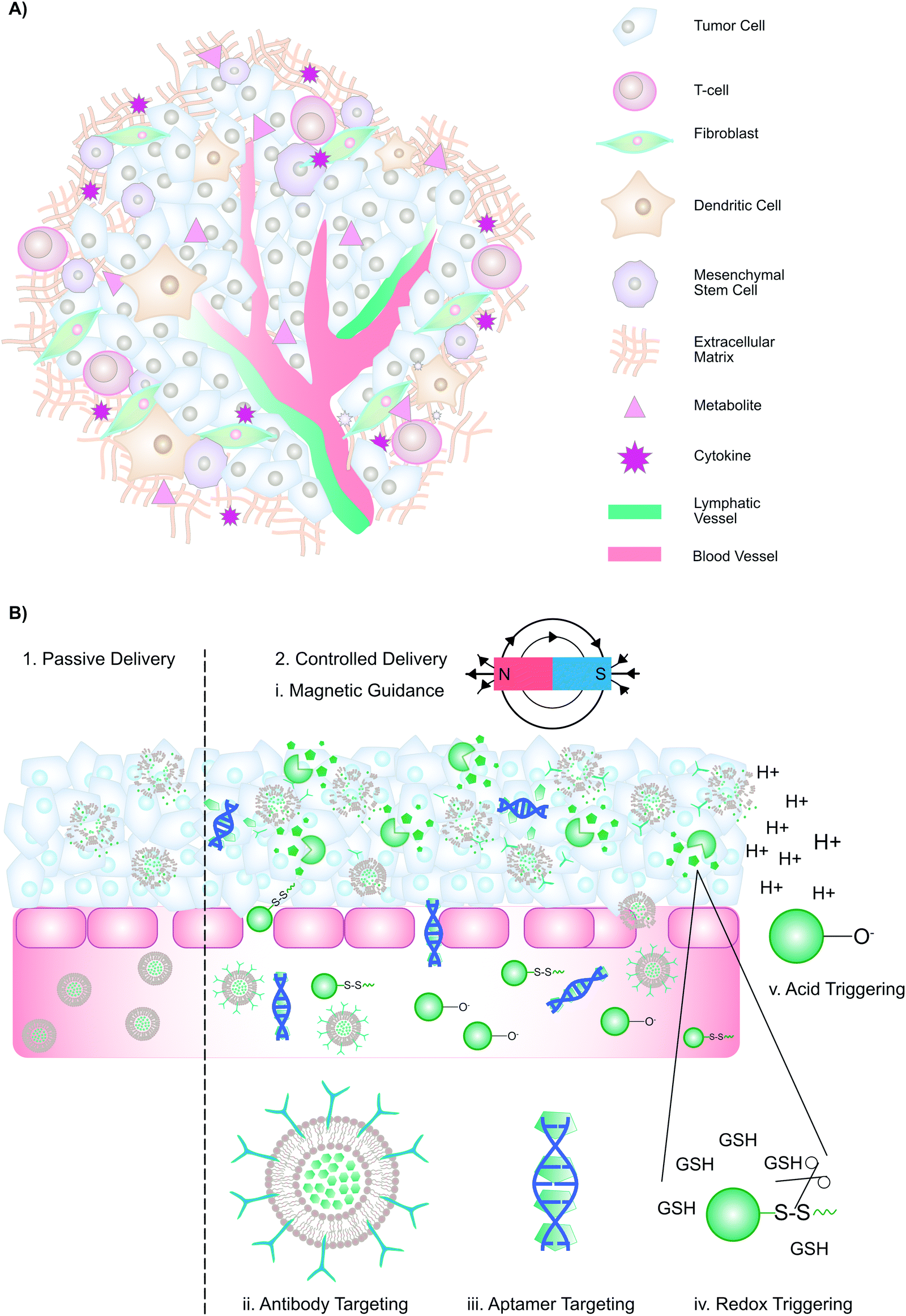 | ||
| Fig. 3 Passive and controlled delivery strategies that promote porphyrin accumulation in the tumour interstitium. (A) Schematic representation of the tumour microenvironment. (B) Passive and controlled delivery methods rely upon the enhanced permeability and retention effect, with the exception of external magnetic guidance. Upon arrival at the tumour, strategies such as antibody-mediated receptor, aptamer targeting, redox- and pH- triggered release promote cell-specific porphyrin uptake. | ||
3.2 Passive and active drug delivery
Conventionally, drug delivery methods are categorized as either “passive” or “active” (Fig. 3B). Passive drug delivery to tumours relies on the enhanced permeability and retention (EPR) effect: Because of the discontinuous, fenestrated capillaries in the TME, macromolecules (>30 kDa) and nano-scaled drug delivery vehicles can extravasate into the interstitium, where they are entrapped due to elevated interstitial fluid pressure, slow venous return, and poor lymphatic drainage, ultimately leading to preferential accumulation in the tumour interstitium. To maximize drug accumulation, passive delivery methods aim to extend the circulation time of macromolecules and nanoparticles. To improve cell-selective uptake upon passive delivery, drug delivery systems can be functionalized with targeting moieties such as antibodies, sugars, aptamers, or peptides, which enable selective particle binding to cells expressing a biomarker of interest (“active targeting”). Alternatively, controlled delivery can be achieved through magnetic guidance, cell-mediated delivery, or stimuli-responsive nanomaterials. These methods exploit inherent TME features such as acidic pH and higher levels of glutathione, or external triggers such as temperature, to trigger drug release. Collectively, drug delivery vehicles employing these strategies may improve drug accumulation at tumour sites. As outlined in Tables 3–5, supramolecular porphyrin structures that facilitate passive, active and controlled PS delivery have been investigated widely for their potential to enhance tumour accumulation of porphyrins, and augment PDT efficacy.| Supramolecular structure | No. of publications | In vitro applications | In vivo applications |
|---|---|---|---|
| Silica | 13 |
Cancer (skin, breast, liver, bile duct, cervix, ovary) PDT
Cancer fluorescence imaging (ovary, cervix, macrophages) |
Cancer (breast) and chick embryo choroidal neovasculature PDT
Macrophage NIR fluorescence tracking |
| Metal oxides | 9 | Rheumatoid arthritis (synovial fibroblasts), cancer (prostate, breast, ovary) and anti-bacterial (E. coli) PDT | Rheumatoid arthritis PDT |
| Gold | 10 | Cancer (cervix, breast, colon, ovaries, rectum, head, neck, brain, leukemic T-cells) and anti-bacterial (E. coli) PDT | Cancer (head, neck, leukemia) PDT |
| Metal organic framework | 2 | Cancer (liver, cervix, head, neck) PDT | Cancer (head, neck) PDT |
| Other metals | 5 |
Cancer (cervix, macrophages, rectum, synovium) and antibacterial (S. aureus, P. aeruginosa, E. coli, S. epidermis, M. fortuitum) PDT
Fluorescence imaging (fibroblasts) |
|
| Liposomes | 30 |
Cancer (cervix, ovaries, mast cells, colon, liver, larynx, blood vessels, lung), anti-parasitic (leishmaniasis), anti-bacterial (methicillin-resistant bacterium, S. aureus) and anti-angiogenic PDT
Fluorescence imaging of macrophages, liver cells |
Cancer (mast cells, skin, connective tissue, bladder), rabbit ciliary body, choroidal neovascularization (chick embryos, rabbits, monkeys, humans), and ocular (monkey, rabbit, mice chick embryo) PDT
Fluorescence imaging of lymph nodes |
| Polymer | 28 |
Cancer (lung, prostate, colon, skin, bile duct, breast, ovary, brain, liver, rectum), and anti-bacterial (S. aureus, S. epidermidis) PDT
Cancer fluorescence imaging (breast) |
Cancer (cervix, lung, skin, colon, ovary, skeletal muscle, brain, skinpig), anti-bacterial (skinpig), chick embryo choroidal neovasculature PDT, and fluorescence imaging of liver cancer |
| Micelles | 13 |
Cancer (colon, rectum, breast, lymphoma, lung, mast cells, head, neck, ovary) PDT
Cancer fluorescence imaging (lung, liver) |
Cancer (mast cells) and choroidal neovascularization PDT
Cancer (liver cancer, pancreatic cancer) fluorescence imaging |
| Polysaccharides | 13 | Cancer (liver, cervix, skin, leukemic T-cells, oral, blood) and antimicrobial (methicillin-resistant bacterium, S. aureus, E. coli) PDT | Chick embryo PDT |
| Carbon | 4 | Cancer (lung, T-cells, larynx), anti-bacterial (S. aureus) and anti-viral (influenza A) PDT | |
| Dendrimers | 3 | Cancer (cervix, lung) PDT | |
| Albumin | 2 | Cancer (T-cells) PDT | |
| Films | 3 | Oxygen sensor (cervical cancer, skin) | |
| Lipid | 2 | Cancer (brain, ovary) PDT | Cancer (brain) PDT |
| Marine atellocollagen | 2 | Cancer (cervix) and anti-malarial PDT | |
| Miscellaneous | 13 |
Cancer (skin, cervix, brain, bladder, leukemia), antibacterial (E. coli, S. aureus, methicillin-resistant S. aureus, P. aeruginosa) and anti-fungal (Candida albicans) PDT
Drug delivery (breast cancer, human umbilical vein endothelial cells, stem cells) |
Cancer (skin, bladder, brain) PDT
Cancer (brain) fluorescence imaging Antibacterial (methicillin-resistant S. aureus, E. coli, P. aeruginosa) PDT |
| Supramolecular host/structure | Targeting ligand/target | Photosensitizer | Application |
|---|---|---|---|
| Abbreviations: CNV: choroidal neovascularization; PPh-a: pyropheophorbide-a; HPD: hematoporphyrin derivative; ATX-70: gallium porphyrin analogue; THPP: 5,10,15,20-tetrakis(4-hydroxyphenyl)-21H,23H-porphine; PdTPTBP: Pd(II) meso-tetraphenyl-tetrabenzoporphyrin; TMP: meso-tetra(N-methyl-4-pyridyl) porphine tetra tosylate; mTHPP: 5,10,15,20-tetrakis(3-hydroxyphenyl)porphyrin; PR-SH: 5-[4-(11-mercaptoundecyloxy)phenyl]-10,15,20-triphenylporphyrin; TMPyP4: 5,10,15,20-tetrakis(1-methylpyridinium-4-yl)porphyrin; NMM: N-methylmesoporphyrin IX; PdTPP: Pd-porphyrin; BchlBOA: bacteriochlorin e6 bisoleate; TPPS: tetraphenylporphyrin tetrasulfonic acid hydrate; mTHPC: meta-tetra(hydroxyphenyl)chlorine; mitoTPP: (5-(p-(4-trimethylammonium)butoxyphenyl)-10,15,20-triphenylporphyrin bromide); TMPyP: tetrakis(1-methylpyridinium-4-yl)-porphyrin; T4P: tetra(4-aminophenyl)porphyrin; TP-Zn-P: 5,10,15,20-tetrakis(4′-propargyloxyphenyl)-Zn(II)-porphyrin 22; mTPPS: meso-tetrakis (4-sulfonatophenyl) porphyrin. | |||
| Antibody-mediated targeting | |||
| mAb-PS conjugate |
UCD/AB 6.01/keratin 8
F11-39/carcinoembryonic antigen Cetuximab/EGFR Rituximab/CD20 2C5 antinuclear antibody/nucleosome Endoglobulin Trastuzumab/HER2 |
HPD, ATX-70, Verteporfin, PPh-a, THPP
Porphyrin azide |
In vitro PDT: vulvar squamous cell carcinoma cells (A-431), epidermoid carcinoma (A-341), B lymphoma (Ramos), acute T-cell leukemia (Jurkat), breast adenocarcinoma (MCF-7, MDA-MB-468, SK-BR-3), small cell lung cancer (H69), ovarian adenocarcinoma (OVCAR-5), ovarian cystadenocarcinoma (SK-OV-3), ductal carcinoma (BT-474), CNV (MS1)
in vivo PDT: vulvar squamous cell carcinoma (A-431), colon carcinoma (CACO-2), colorectal carcinoma (COLO-205), prostate carcinoma (PC-3), colon adenocarcinoma (LS-174T), small cell carcinoma (H69), ovarian cystadenocarcinoma (SK-OV-3), breast adenocarcinoma (MDA-MB-321), ductal carcinoma (BT-474) |
| Cyclodextran–porphyrin conjugate | Endoglobulin | Zinc porphyrin | In vivo PDT: amelanotic melanoma (C32) |
| Polystyrene | Herceptin/HER2 | PdTPTBP |
In vitro hypoxia imaging: murine alveolar macrophages (MH-S), breast adenocarcinoma (SK-BR-3, MDA-MB-231), pancreatic adenocarcinoma (AsPC1)
In vivo hypoxia imaging: pancreatic adenocarcinoma (AsPC1) |
| Polyclonal Ab-PS conjugate | VEGF antibody/VEGF | Verteporfin | In vitro PDT: CNV (MS1 murine endothelial cells) |
| Hydrogel | Anti-DR5 antibody/Death receptor 5 | TMP | In vitro PDT: colon carcinoma (HCT116) |
| Bispecific antibody | HEA125 × OKT3 antibody/Epcam | mTHPP |
In vitro combination chemotherapy and adoptive cell therapy: lung adenocarcinoma (A549), ovarian cystadenocarcinoma (SK-OV-3)
In vitro PDT: lung adenocarcinoma (A549), ovarian cystadenocarcinoma (SK-OV-3) |
| Gold | Anti-erbB2 antibody/erbB2 receptor | PR-SH | In vitro PDT: breast adenocarcinoma (SK-BR-3) |
| Aptamer | |||
| G-quadruplex | AS1411/nucleolin | TMPyP4 | In vitro PDT: breast adenocarcinoma (MCF-7) |
| Virus capsid | Phenylene diamine modified DNA aptamers/tyrosine kinase 7 receptors | Porphyrin maleimide | In vitro PDT: acute T cell leukemia (Jurkat) |
| Gold | AS1411/nucleolin | NMM | In vitro PDT and fluorescence imaging: cervical adenocarcinoma (HeLa) |
| Amino acid | |||
| Polymer micelles | L-Phenylalanine | THPP | In vitro PDT: breast adenocarcinoma (MCF-7) |
| Peptide | |||
| Silica | cRGDyK peptides/αvβ3 integrins | PdTPP | In vitro PDT: glioblastoma (U87-MG), breast adenocarcinoma (MCF-7) |
| Micelle |
[(C18)2K]2KR8 GRGS/integrin
Biotin |
Porphyrin
Verteporfin |
In vitro PDT: cervical adenocarcinoma (HeLa)
In vitro fluorescence imaging: prostate adenocarcinoma (PC3), breast adenocarcinoma (MCF-7) In vitro PDT: cervical adenocarcinoma (HeLa) |
| Upconversion nanoparticle | RGD peptide c(RGDyK)/integrin | PPh-a | In vitro and in vivo PDT: glioblastoma (U87-MG) |
| HDL-mimetic | ApoA-1 mimetic R4F/SR-B1 | PPh-a-lipid |
In vivo PET imaging: orthotopic prostate adenocarcinoma (PC-3), orthotopic ovarian cystadenocarcinoma (SK-OV-3), ovarian metastases
In vitro PDT: glioblastoma (U87-MG) In vivo PDT: orthotopic ovarian cystadenocarcinoma (SK-OV-3) orthotopic glioma (9L-luc+), orthotopic glioblastoma (U87-MG), orthotopic prostate adenocarcinoma (PC-3), cervical adenocarcinoma (KB) In vivo fluorescence imaging: cervical adenocarcinoma (KB), glioma (9Lluc), glioblastoma (U87GFP) In vivo fluorescence guided surgery: glioblastoma (U87GFP) |
| Protein | |||
| Lipoprotein-mimetics |
LDL/LDL receptor
ApoE3/LDL receptor ApoA-1/SR-BI |
Verteporfin
PPh-a-lipid BChlBOA |
In vivo PDT: CNV (cynomolgus monkeys), orthotopic Greene amelanotic melanoma in albino rabbits, orthotopic glioblastoma (U87-GFP), cervical adenocarcinoma (KB)
In vitro PDT: glioblastoma (U87-MG), Chinese hamster ovary ldlA7 cells, cervical adenocarcinoma (KB) |
| Core/shell nanoparticle | Pullulan/asialoglycoprotein receptor | TPPS | In vitro PDT: cervical adenocarcinoma (HeLa), hepatocellular carcinoma (HepG2) |
| Enzyme | |||
| Micelles | Lipase | mTHPC | In vitro PDT: 14C carcinoma |
| Folate and hyaluronic acid | |||
| Liposome | Folate/folate receptor | PPh-a-lipid |
In vitro PDT: cervical adenocarcinoma (KB), fibrosarcoma (HT1080)
In vivo PDT: cervical adenocarcinoma (KB) In vitro fluorescence imaging: macrophages (RAW264.7) In vivo fluorescence imaging: myocardial infarcted mice In vivo PET/CT imaging: myocardial infarcted mice |
| Silica | Folate/folate receptor | TCPP | In vivo PTT and PDT: orthotopic myeloma (RPMI 8226) |
| Micelles | Folate/folate receptor | MitoTPP | In vitro PDT: cervical adenocarcinoma (HeLa) |
| Graphene oxide | Folate/folate receptor | MitoTPP | In vitro PDT: cervical adenocarcinoma (HeLa) |
| Metal organic framework | Folate/folate receptor | TMPyP | In vitro PDT: cervical adenocarcinoma (HeLa) |
| Iron oxide | Folate/folate receptor | Ph-a | In vitro PDT and MRI: breast adenocarcinoma (MDA-MB-231, MCF-7) |
| Gold |
Hyaluronic acid/CD44 receptor
Folate/folate receptor |
T4P
Verteporfin |
In vitro fluorescence imaging: glioblastoma (U-87), cervical adenocarcinoma (HeLa)
In vitro PDT: glioblasoma (U-87), cervical adenocarcinoma (HeLa), lung adenocarcinoma (A549) |
| Albumin | Folate/folate receptor | Ph-a |
In vitro PDT: mouse melanoma (B16F10), cervical adenocarcinoma (HeLa), breast adenocarcinoma (MCF-7)
In vivo fluorescence imaging: breast adenocarcinoma (MCF-7) In vivo PDT and fluorescence imaging: breast adenocarcinoma (MCF-7), mouse melanoma (B16F10) |
| Core–shell nanoparticles | Hyaluronic acid/CD44 receptor | TPPS | In vitro PDT: breast adenocarcinoma (MDA-MB-231), MCF-7 |
| Saccharides | |||
| Porphyrin–bile acid conjugates | Bile acid/saccharides (glucose, sialic acid, hyaluronic acid, heparan sulfate) |
Porphyrin
TP-Zn-P |
In vitro PDT: vulvar squamous cell carcinoma (A431NS), cervical adenocarcinoma (HeLa), mammary carcinoma (4T1), pancreatic adenocarcinoma (MIA PaCa-2)
In vitro fluorescence microscopy: vulvar squamous cell carcinoma (A431NS) cervical adenocarcinoma (HeLa), mammary carcinoma (4T1) In vivo PDT: mammary carcinoma (4T1) |
| Virus-like particle | Sialoside ligand/CD22 receptor | Zinc tetraaryl porphyrin | In vitro PDT: Chinese hamster ovary cells |
| Albumin | Galactosyl human serum albumin/lectin receptor | NMP1 | In vitro and in vivo fluorescencine imaging: ovarian cystadenocarcinoma (SHIN3) |
| Sugar-PS conjugate |
Glucosamine
Mannose |
PPh-a |
In vitro bimodal PDT and gene delivery: breast adenocarcinoma (MCF-7), Chinese hamster ovary
In vitro combination chemotherapy + PDT: breast adenocarcinoma (MCF-7) |
| Cell-mediated | |||
| Liposome | Hematopoietic stem cells | Gadophrin-2 | In vivo optical and MRI imaging of healthy nude mice |
| Core shell nanoparticles | Mesenchymal stem cells | mTPPS | In vitro PDT: osteosarcoma (U2OSTubRFP, U2OS) |
| Supramolecular host/structure | Photosensitizer | Application |
|---|---|---|
| H2TPPS4(HCl)2: sulfonated tetraphenylporphine dihydrochloride; FPP: 5-(pentafluorophenyl)-10,15,20-tris(4-pyridyl)porphyrin; FPPI: 5-(pentafluorophenyl)-10,15,20-tris(1-methylpyridinium-4-yl)porphyrin tri-iodide; HP: 5-(pentafluorophenyl)-10,15,20-triphenylporphyrin hematoporphyrin; PHPP: 2,7,12,18-tetramethyl-3,8-di-(1-propoxyethyl)-13,17-bis-(3-hydroxypropyl) porphyrin; TPP: tetraphenylporphine; CAAP: 5,10,15,20-tetrakis(4-N-carbonylacrylic aminophenyl)porphyrin; ATPPn: 9-acetoxy-tetra-n-propylporphycene; TAPP: 5,10,15,20-tetrakis-(4-aminophenyl)-21H,23H-porphyrine; ZnTPP: zinc tetra(progaryloxy-phenyl) porphyrin; MitoTPP: (5-(p-(4-trimethylammonium)butoxyphenyl)-10,15,20-triphenylporphyrin bromide; PMC16: buckminsterfullerene(C60)-2-(butadiene-1-yl)-tetra(o-γ-aminobutyryl-o-phtalyl)porphyrin(Porphylleren-MC16); TMPyP: meso-tetra(N-methyl-4-pyridyl)porphine; TCPP: 5,10,15,20-tetrakis(carboxyphenyl) porphyrin; M1P1MPP: 5,10,15-tri(1-methylpyridinium-4-yl)-20-(4-methoxyphenyl)porphyrin; TMPyP4: 5,10,15,20-tetrakis(1-methylpyridinium-4-yl)porphyrin; M1P2MPP: 5,15-Di(1-methylpyridinium-4-yl)-10,20-di(4-methoxyphenyl)porphyrin; 4M3MPP: 5-(4-Methoxyphenyl)-10,15,20-tri(1-methylpyridinium-4-yl)porphyrin; Sn-mTCPP: SnCl2-meso-tetra(4-carboxyphenyl)porphine; TCAPP: 5,10,15,20-tetrakis(4-N-carbonylacrylic aminophenyl)porphyrin; PTPP-OH: 6-(5′-(4′-phenoxyl)-10′,15′,20′-triphenylporphyrin)-1-hexanol; TPP-OH: 5-(4-hydroxylphenyl)-10,15,20-triphenylporphyrin; TPPC6-SS-Py: disulfide-modified pyridinium terminal porphyrin; TPPC6SA: 6-(5′-(4′-phenoxyl)-10′,15′,20′-triphenylporphyrin) succinate; TPPS: tetraphenylporphyrin tetrasulfonic acid; mHPP: meso-tetra(p-hydroxyphenyl) porphine; mTHPP: 5,10,15,20-tetrakis(3-hydroxyphenyl)porphyrin. | ||
| Magnetism | ||
| Magnetite–porphyrin complex | H2TPPS4(HCl)2, photodithazine, FPP, FPPI, HP | In vitro PDT: hepatocellular carcinoma (HepG2), cervical adenocarcinoma (HeLa), breast adenocarcinoma (MCF-7), prostate adenocarcinoma (PC-3), Escherichia coli, Enterococcus faecalis, T4-like phage |
| Iron oxide | Hematoporphyrin | In vitro PDT: cervical adenocarcinoma (HeLa), breast adenocarcinoma (MCF-7), prostate adenocarcinoma (PC-3) |
| Graphene oxide | Hematoporphyrin | In vitro PDT: cervical adenocarcinoma (HeLa) |
| Chitosan nanoparticles | PHPP | In vitro and in vivo PDT: colorectal adenocarcinoma (SW480) |
| Temperature | ||
| Polymer | TPP | In vitro PDT: cervical adenocarcinoma (HeLa) |
| Hydrogel | CAAP | In vitro PDT: mammary carcinoma (A453) |
| pH | ||
| Liposome | ATPPn | In vitro PDT: breast adenocarcinoma (MCF-7), bladder carcinoma (WAF) |
| Micelle | Dendrimer Zn-porphyrin, TAPP, ZnTPP, MitoTPP | In vitro PDT: cervical adenocarcinoma (HeLa) |
| Fullerene | PMC16 | In vivo hypoxia imaging of rats with medicinal hypoxia syndromes A & B |
| Silica | TPP, TMPyP, PPh-a, zinc-TCPP, TCPP | In vitro PDT: cervical adenocarcinoma (HeLa), breast adenocarcinoma (SK-BR-3, MCF-7), squamous cell carcinoma (Nt-8e) |
| Superparamagnetic iron oxide | TMPyP4, M1P1MPP, M1P2MPP, 4M3MPP | In vitro PDT: cervical adenocarcinoma (HeLa) |
| Polysaccharide–porphyrin complex | Chlorin e6 | In vitro and in vivo PDT: cervical adenocarcinoma (HeLa) |
| Copolymer | Tetrahydroxyethyl-terminated porphyrin | In vitro PDT: breast adenocarcinoma (MCF-7) |
| Dendrimer | Ph-A | In vitro fluorescence imaging: breast adenocarcinoma (MCF-7, MDA-MB-231, 293T) |
| Hydrogel | TPP, Sn-mTCPP, TCAPP |
In vitro PDT: cervical adenocarcinoma (HeLa), mammary carcinoma (A453)
In vivo fluorescence imaging of healthy mice |
| Conjugate | Gold porphyrin |
In vitro and in vivo combination chemotherapy and PDT: colon carcinoma (HCT116)
In vitro PDT: bladder carcinoma (T24), cervical adenocarcinoma (HeLa) |
| Metal organic framework | TCPP |
In vivo fluorescence imaging and combination chemotherapy and PDT: hepatocellular carcinoma (HepG2)
In vitro PDT: hepatocellular carcinoma (HepG2) |
| Gold | Verteporfin | In vitro fluorescence imaging and PDT: cervical adenocarcinoma (HeLa) |
| Redox | ||
| Gold | Ph-A | In vitro and in vivo PDT: lung adenocarcinoma (A549) |
| Chitosan | Ph-A |
In vitro PDT: cervical adenocarcinoma (KB)
In vivo PDT: rectosigmoidal adenocarcinoma (HT-29) |
| Conjugate | Ph-A, PTPP-OH | In vitro PDT: cervical adenocarcinoma (HeLa), breast adenocarcinoma (MCF-7), hepatocellular carcinoma (HepG2) |
| Micelles |
Dendrimer porphyrin, TPP-OH
adamantane-terminated porphyrin, porphyrin disulfide, TPPC6-SS-Py, TPPC6SA, TPPS, |
In vitro PDT: breast adenocarcinoma (MCF-7), lung adenocarcinoma (A549) |
| Dendrimer | mHPP, TPP-OH | In vitro PDT: breast adenocarcinoma (MDA-MB-231, MCF-7) |
| Dimer | TPP–OH | In vitro PDT: hepatocellular carcinoma (HepG2) |
| Polymer | mTHPP | In vitro PDT: breast adenocarcinoma (MDA-MB-231) |
3.3 Passive delivery
Successful passive delivery of porphyrins to tumours is contingent upon particle water solubility, extension of circulation time, and extravasation into the tumour interstitium. Given the challenges associated with porphyrin aggregation in solution, various organic (liposomes, micelles, albumin, and dendrimers, Fig. 4) and inorganic (gold, metal organic frameworks, and silica, Fig. 5) nanoparticles have been simultaneously employed to better disperse porphyrins in aqueous media, and increase porphyrin delivery to tumours (Table 3). These supramolecular porphyrin structures exhibit improved pharmacokinetic profiles and biomedically-relevant photophysical properties relative to monomeric porphyrin, and exploit the EPR effect to home porphyrin delivery for enhanced PDT. Here, we overview the beneficial properties imparted to molecular porphyrins by these nano-scaled supramolecular structures, and outline their associated disadvantages.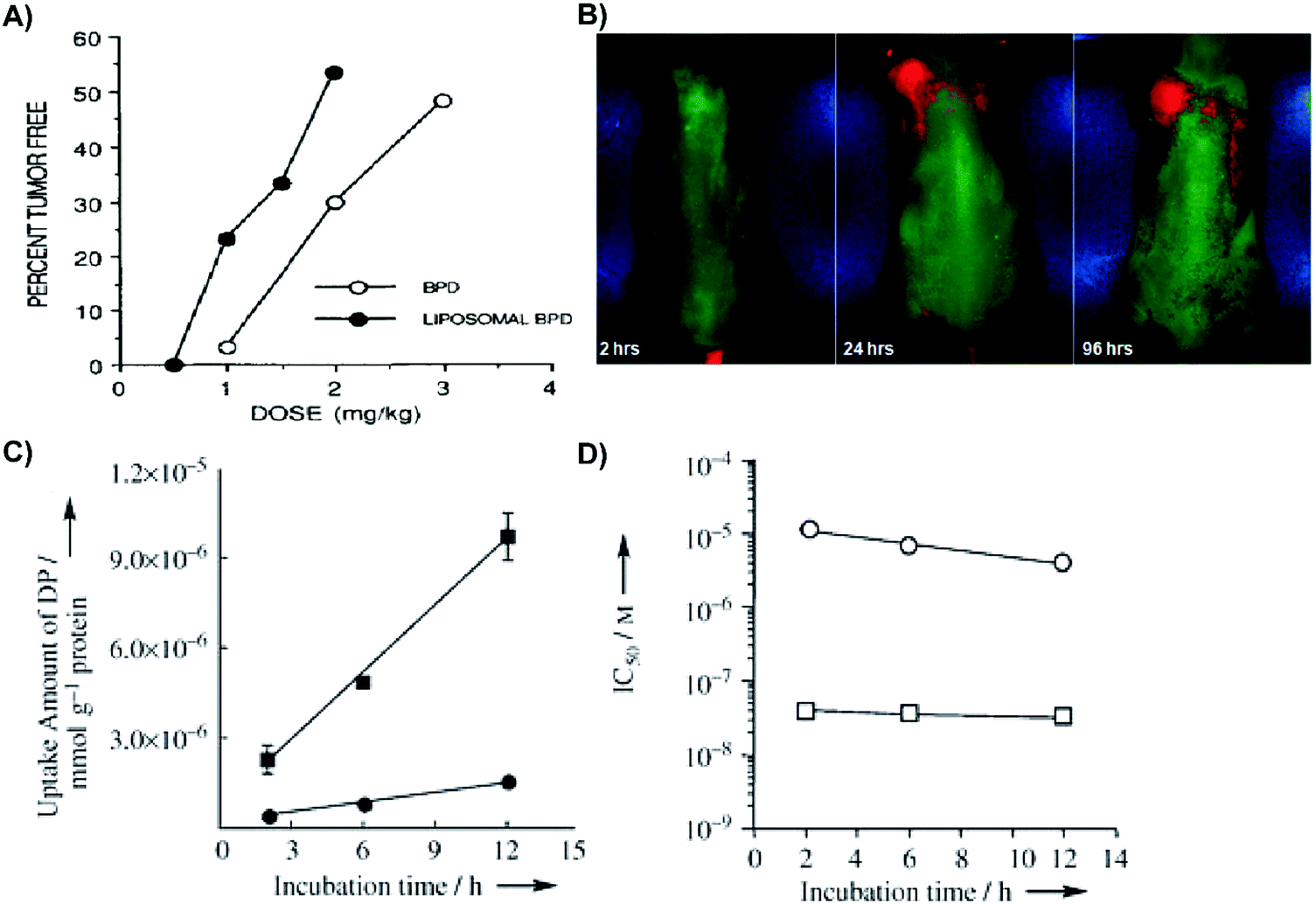 | ||
| Fig. 4 Merits of organic supramolecular porphyrin carriers for passive delivery. (A) Rhabdosarcoma M1-tumour bearing mice were administered liposomal or aqueous verteporfin, and treated with 690 nm laser irradiation at 150 J cm−2 or 210 J cm−2, respectively. Twenty days post PDT, the percentage of tumour-free mice was assessed. Copyright (1993) Wiley. Used with permission from Richter et al.2 (B) Phosphorescence luminesce images of mice bearing subcutaneous PANC-1 pancreatic tumours 2, 24, and 96 hours post intravenous injection of Pt(II)-tetraphenyltetranaphthoporphyrin/DSPE-PEG/phosphatidyl choline micelles. Reprinted with permission from Kumar et al.3 Copyright (2009) American Chemical Society. (C) Incubation time dependent Lewis lung carcinoma cell uptake of 12 μM-equivalent concentrations of dendrimer porphyrins (filled circles) and dendrimer porphyrins encapsulated in micelles (filled squares) over time. Copyright (2011) Wiley. Used with permission from Park et al.6 (D) Photoirradiation of Lewis lung carcinoma cells (10 min; 150 W; 180 kJ cm−2) with subsequent cell viability assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. 50% growth inhibitory concentration (IC50) of dendrimer porphyrins (open circles) and dendrimer porphyrins in micelles (open square) was dependent on time. Copyright (2011) Wiley. Used with permission from Park et al.6 | ||
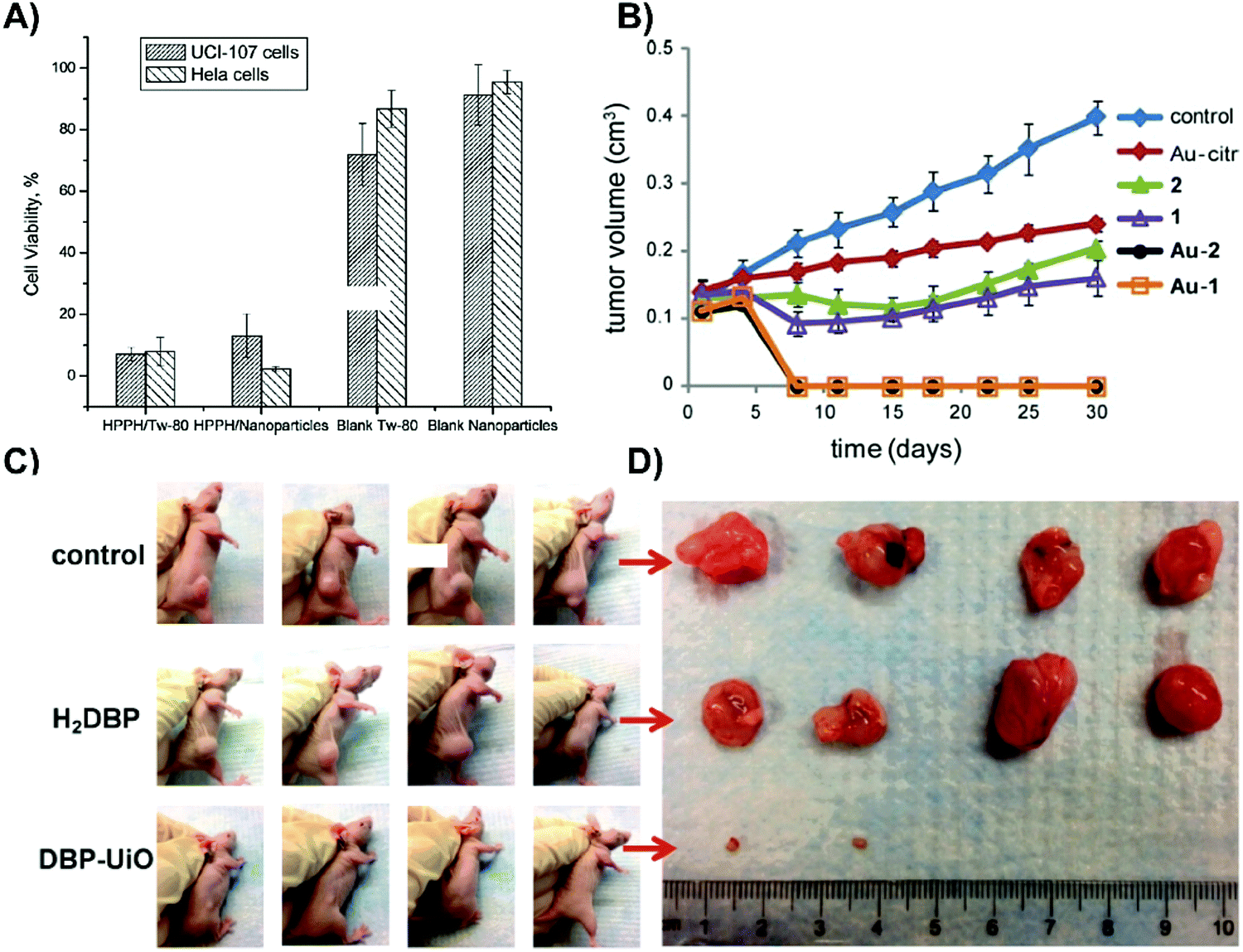 | ||
| Fig. 5 Merits of inorganic supramolecular porphyrin carriers for passive delivery. (A) Photocytotoxicity of micellar HPPH (HPPH/Tw-80), HPPH-doped mesoporous silica nanoparticles (HPPH/Nanoparticles), control micelles (Blank Tw-80) and mesoporous silica nanoparticles lacking HPPH (blank nanoparticles) on UCI-107 ovarian and HeLa cervical tumour cells. Cells were treated with 0.25% Tween micellar HPPH, HPPH-doped mesoporous silica nanoparticle, 0.25% Tween-80/water micelles, or mesoporous silica nanoparticles, and irradiated (650 nm; 10 minutes; 1.4 mW cm−2). Cell viability was assessed with an MTT assay after overnight incubation. Reprinted with permission from Roy et al.7 Copyright (2003) American Chemical Society. (B) Photodynamic efficacy of porphyrin–brucine conjugates immobilized on gold nanoparticles. Subcutaneous PE/CA-PJ34 basaloid squamous cell carcinoma-bearing mice (n = 7) were injected with either free porphyrin–brucine conjugates (1, 2), porphyrin/AuNP conjugates (Au-1, Au-2) or no porphyrin (control). Six hours post-injection, tumours were irradiated (500–700 nm; 100 J cm−2; 200 mW cm−2). Reproduced from Zaruba et al.8 with permission from the Royal Society of Chemistry. (C) Photographs of SQ20B tumours of mice (n = 4) 8 days post PDT. Subcutaneous SQ20B laryngeal squamous cell carcinoma bearing mice were administered either PBS control, free porphyrin control (H2DBP), or DBP-UiO. Twelve hours post injection, the tumour site was irradiated (640 nm; 30 minutes; 100 mW cm−2). Reprinted with permission from Lu et al.9 Copyright (2014) American Chemical Society. (D) Ex vivo SQ20B tumours 8 days post PDT, in which DBP-UiO treatment eradicated tumours in two mice. Reprinted with permission from Lu et al.9http://pubs.acs.org/doi/abs/10.1021%2Fja508679h. Further permissions related to these figures should be directed to the American Chemical Society. | ||
Micelles are spherical self-assembling complexes comprised of amphiphilic lipids and surfactants that encapsulate PS within a hydrophobic core. Micelles produced with amphiphilic co-polymers are particularly promising for porphyrin delivery because of their biocompatibility, capacity to solubilize hydrophobic compounds, and potential for functionalization with targeting moieties. These properties were exemplified by Kumar et al. via the encapsulation of Pt(II)-tetraphenyltetranaphthoporphyrin into DSPE-PEG/phosphatidyl choline micelles.3 Administration of these ∼130–170 nm micelles to PANC1 pancreatic xenograft-bearing mice yielded strong phosphorescence signal at the tumour site for up to 96 hours post injection (Fig. 4B). These observations were indicative of in vivo stability, long circulation time, and appreciable tumour accumulation. Furthermore, the micelles enabled identification of visually-undetectable subcutaneous tumours in mice 24 hours post injection, presumably due to preferential porphyrin accumulation. However, common shortcomings of micelles include rapid payload release, poor cell uptake, and immunogenicity imparted by the incorporation of emulsifying agents such as Tween-80 or Cremophor-EL. Thus, micelles are a suboptimal choice as supramolecular porphyrin drug delivery vehicles.
Albumin is the most prevalent serum protein in humans, and demonstrates immense potential as a supramolecular porphyrin carrier due to its long blood circulation half-life of 19–22 days, and endogenous binding of hydrophobic PS within its hydrophobic binding pockets. Upon arrival at the target site, albumin can engage cellular receptors, like glycoprotein 60, to initiate cell uptake and facilitate non-lysosomal delivery. The advantages of albumin (considerable structural stability, high aqueous solubility, and exceptional biocompatibility) were demonstrated by Ramachandran et al., who loaded a porphyrin derivative, 5,10,15,20-tetrakis(meso-hydroxyphenyl)porphyrin (mTHPP), into a poly(lactic-co-glycolic acid) polymer core encapsulated by a human serum albumin shell, that was functionalized with the chemotherapeutic tyrosine kinase inhibitor, dasatinib.4 The albumin shell imparted in vivo biocompatibility; nanocarrier administration at 25 mg kg−1 affected neither the eating and drinking behavior, nor the weight of Wistar rats. Follow-up histology revealed no significant pathological changes to the liver, kidneys, spleen, lungs, and brain 96 hours post injection as compared to saline-injected control rats. Furthermore, serum differential leukocyte and erythrocyte counts were within normal ranges, suggestive of immunocompatibility. However, the biocompatibility and safety of albumin nanoparticles is contingent upon the source of albumin: ovalbumin from egg whites is highly immunogenic, while bovine serum albumin can induce mild immune reactions.5 Thus, protein supplies must be considered for the successful translation of albumin-porphyrin supramolecular structures.
Dendrimers are highly branched polymers that can accommodate PS loading within their core or peripheral dendrons. Dendrimers are advantageous as PS delivery agents due to their tunable hydrophobicity, facile functionalization with targeting moieties, and ability to evade the mononuclear phagocyte system (MPS). The latter is an important feature, as cells of the MPS uptake, degrade and subsequently clear nanoparticles. Jang et al. assembled a dendrimer porphyrin (DP) consisting of a zinc porphyrin core within a third generation poly(benzyl ether) dendrimer.6 The DP was then encapsulated by a poly(ethylene glycol)-poly(L-lysine) block copolymer to form complex polyionic micelles. Compared to free DP, micellar DP demonstrated stronger fluorescence emission at 610 nm, indicative of reduced porphyrin self-quenching. More impressively, when compared to free DP, the micellar DP formulation increased porphyrin uptake by Lewis lung carcinoma cells by 8-fold (Fig. 4C), and photocytotoxicity by 280-fold upon laser irradiation (Fig. 4D). The separation of the core porphyrin by dendrimer branches prevents intracellular aggregation, thereby increasing porphyrin potential for 1O2 generation. Despite these promising results, dendrimer clinical translation is challenged by difficulties in achieving synthesis scale-up, efficient removal of reaction side products, and favourable product yields.
Gold nanoparticles (AuNPs) serve as ubiquitous foundations for multimodal theranostic agents, and have functioned as carriers for passive delivery of porphyrin PS incorporated therein. Their optical properties are facilely tunable through modulation of shape, size, and composition of other layered or alloyed materials, such as silica and silver, into the gold nanostructure. Localized surface plasmon resonance, a coherent oscillation of conduction band electrons about the ionic cores of the nanoparticle, is responsible for the high absorbance and scattering cross-sections of plasmonic nanoparticles, which can be shifted into the NIR range, with resulting implications in delivering dual PDT/photothermal therapy, as will be discussed later. Additionally, drugs and small molecule PS can be physisorbed or chemisorbed directly onto the gold surface, or incorporated into the passivating layer, which allows the tunable AuNP properties to be applied for enhancing porphyrin distribution and photosensitization. This was exemplified by Zaruba et al. who used 14.7 nm AuNPs as PS carriers by immobilizing porphyrin–brucine quaternary ammonium salts directly to the nanoparticle surfaces.8 Despite facilitating poorer porphyrin photoactivation in vitro, the AuNP/porphyrin conjugates enabled the complete eradication of basaloid squamous cell carcinoma PE/CA-PJ34 murine tumours without relapse for up to 30 days, whereas control mice treated with free porphyrin–brucine experienced ∼2-fold increase in tumour volumes (Fig. 5B). These results underscore the unmet need for in vivo characterization of supramolecular structures employed in porphyrin delivery. In particular, biodegradability of all nanoparticle delivery systems need to be scrupulously investigated. This is especially true for AuNPs, which when larger than ∼5 nm in diameter, cannot be renally excreted and thus accumulate in the liver and spleen.
Nanoscale metal organic frameworks (NMOFs) are supramolecular structures constructed from single or clustered metal ions chelated by linking organic bridging ligands. NMOFs are characterized by high porosity, biodegradability, and facile tunability of chemical composition, structure, and PS loading capacity. For example, Lu et al. synthesized a highly porous, nanoscale UiO MOF with a remarkable 77 weight% loading achieved via a porphyrin-derived bridging ligand, 5,15-di(p-benzoato)porphyrin (H2DBP).9 The NMOF structure isolated the H2DBP units thereby preventing aggregation-induced fluorescence quenching. The 1O2 generation was enhanced through the coordinated Hf4+ ions, as compared to the free-base H2DBP. Moreover, 1O2 generation was observed to be at least twice as efficient for the NMOF compared to free H2DBP. ROS readily diffused out of the highly porous NMOF nanostructure, which was hypothesized to improve cytotoxicity. Mice bearing subcutaneous SQ20B xenografts that were treated with DBP-UiO and irradiated with DBP-resonant laser exhibited either complete tumour eradiation or experienced 50-fold decreases in tumour volume, while free H2DBP-injected controls experienced continual tumour growth (Fig. 5C and D). Given these encouraging results, NMOF-porphyrin supramolecular structures warrant further development.
3.4 Controlled porphyrin delivery
Some of the most intriguing supramolecular porphyrin delivery strategies investigated in vivo involve active targeting and stimuli-responsive release. Active targeting via antibodies, and aptamers can yield more specific cell uptake of PS relative to molecular porphyrin. Meanwhile, stimuli-responsive nanoparticles can both enhance porphyrin delivery specificity and protect porphyrins from premature release through external magnetic triggers, or pH and redox reaction-triggers endogenous to the hypoxic tumour environment for activatable PDT. Supramolecular structures that exploit these strategies for enhancing porphyrin delivery to tumours are summarized in Tables 4 and 5, and are discussed in greater detail by Karimi et al.5 and Lucky et al.10Antibody-mediated targeting. Antibodies are large ∼150 kDa proteins comprised of a constant region that mediates immune responses, and a variable region that can bind targets with high specificity and affinity. Napp et al. exploited these properties to apply the clinically-approved anti-HER2 antibody, Herceptin, for active tumour cell targeting of their ratiometric dual wavelength fluorescent oxygen sensor.11 Their tumour hypoxia sensor consisted of polystyrene nanoparticles doped with oxygen-sensitive palladium meso-tetraphenylporphyrin (PdTPTBP) and an inert, oxygen-insensitive reference cyanine dye, DY-635, both of which are excitable at 635 nm. Compared to free PdTPTBP in hypoxic conditions, incorporation of PdTPTBP into the nanoparticles led to a ∼3-fold increase in absolute fluorescence quantum yield, giving rise to stronger contrast for hypoxia detection. Cell specificity of the sensor was observable via high porphyrin fluorescence intensity emanating from treated HER2/neu-over expressing breast caner SK-BR-3 cells within 1.5 hours in conjunction with the absence of observable porphyrin fluorescence from HER2 receptor(−) MDA-MB-231 after 4 hours of incubation. For a proof-of-concept demonstration of sensor efficacy in vivo, subcutaneous pancreatic AsPC-1 adenocarcinoma xenograft-bearing mice were intravenously injected with the nanoprobe. This gave rise to strong fluorescence signal with an intensity ∼2-fold higher than the reference cyanine dye (Fig. 6A). Ex vivo analysis showed that the fluorescence intensity ratio of the PdTPTBP was ∼3.6 fold greater than that of the reference dye in tumours (Fig. 6B). Thus, antibody-mediated targeting can increase site-specific porphyrin delivery for imaging purposes. Disadvantages of antibody functionalization include their immunogenicity, sensitivity to denaturation and structural dependence on environmental pH, salt concentration, and temperature. Accordingly, surface functionalization of porphyrin nanoparticles by antibodies is restricted to the use of mild synthesis conditions, all-the-while generating particles that can still have reduced in vivo stability.
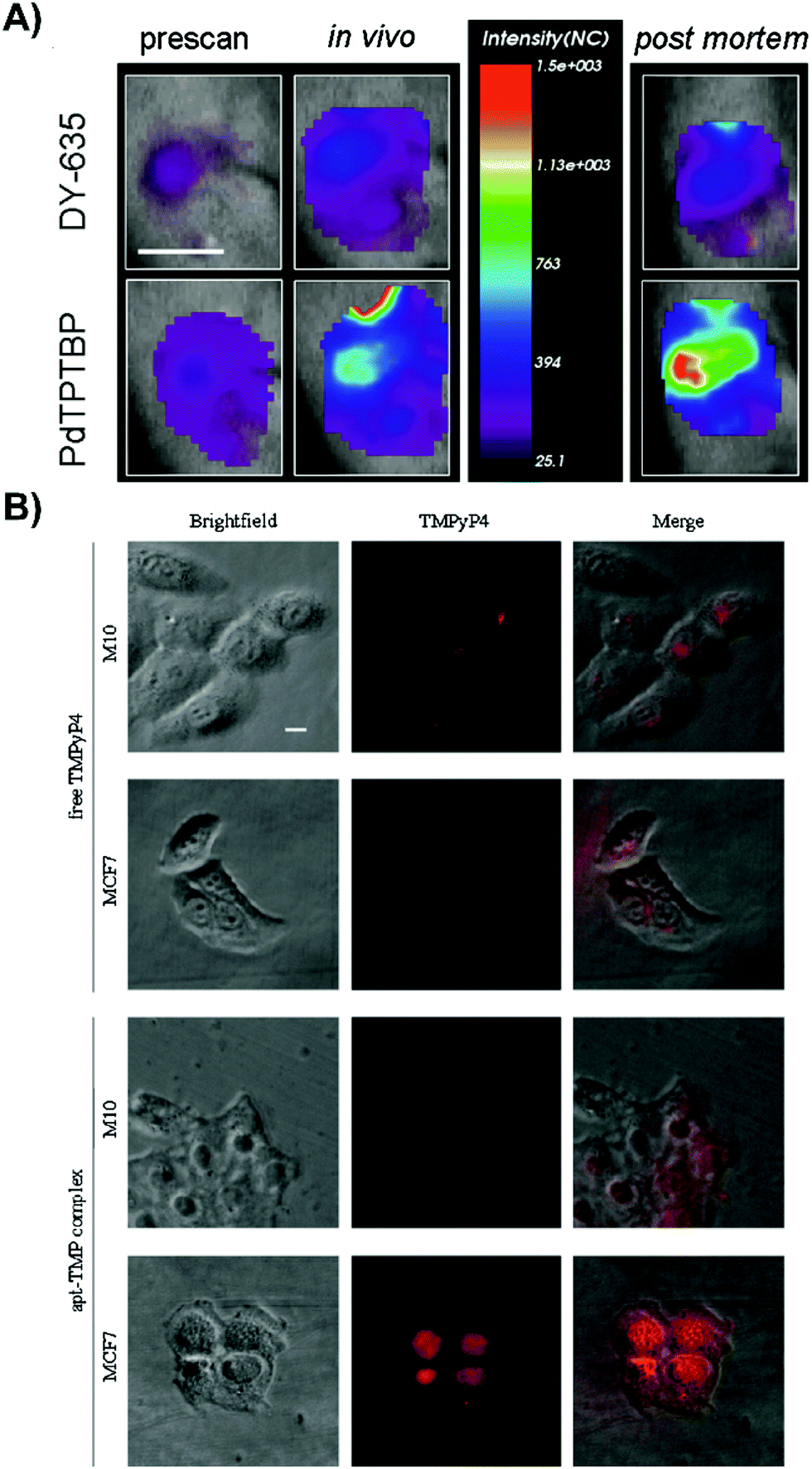 | ||
| Fig. 6 Active targeting strategies can be employed to enhance uptake of porphyrin nanoprobes and supramolecular porphyrin structures. (A) Tumor oxygenation, as demonstrated by tumor uptake of the herceptin-targeted oxygen polystyrene nanoparticle probe that is co-encapsulated with oxygen-sensitive palladium meso-tetraphenylporphyrin (PdTPTBP) and an inert, oxygen-insensitive, reference cyanine dye, DY-635. Near infrared fluorescence imaging of a subcutaneous AsPC-1 pancreatic tumour bearing mice intravenously injected with Ox-PS-NPs was performed 5 hours post intravenous injection, and ∼10 minutes post-mortem. Scale bar indicates one cm. Reprinted with permission from Napp et al.11 Copyright (2011) American Chemical Society. (B) Fluorescence imaging of intracellular localization of free TMPyP4, and TMPyP4 equivalent of the apt-TMP complex, upon a 2 h incubation of MCF-7 breast tumour and normal M10 breast epithelial cells. Reprinted with permission from Shieh et al.12 Copyright (2010) American Chemical Society. | ||
Aptamers are single-stranded oligonucleotides that can bind their targets with high affinity and specificity. Compared to antibodies, aptamers are associated with reduced immunogenicity, and lower fabrication costs due to easy bulk synthesis. Previously, Shieh et al. complexed a G-quadruplex AS1411 aptamer to six cationic 5,10,15,20-tetrakis(1-methylpyridinium-4-yl)porphyrin (TMPyP4) molecules via electrostatic interactions (apt-TMP).12 The authors aimed to use AS1411 to target nucleolin, a shuttle molecule involved in RNA transcription, and DNA replication. Nucleolin is overexpressed in various tumour types, including breast cancer. As such, the in vitro uptake of apt-TMP by MCF7 breast cancer cells was 3.8-fold higher than that in non-malignant M10 epithelial cells. This cell uptake specificity was re-affirmed by fluorescence imaging of MCF7 cells treated with apt-TMP; intracellular red fluorescence signal emanating from cells treated with apt-TMP exceeded that of molecular TMPyP4 (Fig. 6B). The nuclei-specific uptake of Apt-TMP in MCF7 cells was thought to account for the ∼1.5× greater phototoxicity compared to free TMPyP4, which was retained within the cytoplasm of MCF7 cells. This observation shed light on the utility of porphyrin/aptamer supramolecular complexes in facilitating cell and organelle-specific porphyrin delivery. Despite the promise of aptamers, they remain limited by their facile degradation by nucleases in physiological environments.13
pH-Triggered release. The higher acidity (and lower pH) exhibited by tumours versus healthy tissue, and by lysosomes/endosomes (pH of 4.5–5 and 5.5–6 respectively) versus the cytoplasm can be exploited by supramolecular porphyrin carriers to selectively trigger drug release via: (1) polymers that change conformation or solubility upon ionization, leading to exposure of encapsulated drug or targeting ligand, or (2) through the cleavage of acid-sensitive bonds resulting in the release of otherwise protected molecules. Accordingly, premature porphyrin release is reduced and uptake specificity is increased.
This potential was demonstrated by Park et al., who designed a polysaccharide/drug conjugate composed of a glycol chitosan backbone, a 3-diethylaminopropyl isothiocyanate (DEAP) block, a photosensitizing chlorin e6 (Ce6) block, and a poly(ethylene glycol) block (GCS-g-DEAP-g-Ce6-g-PEG).14 The pKb value of the resulting GCS-g-DEAP-g-Ce6-g-PEG particle was ∼6.8, resulting in the protonation of the conjugate upon arrival at the tumour and subsequent disentanglement of the originally self-quenched porphyrin assembly. This conformational change restored 1O2 generation to levels comparable to free Ce6, and DEAP-free conjugate. These observations were translated in vitro, wherein similar PDT effects were observed with GCS-g-DEAP-g-Ce6-g-PEG or dose-equivalent free Ce6 treatment, demonstrating successful activation of 1O2 generation. Fluorescence imaging of subcutaneous HeLa xenograft-bearing mice demonstrated more intense focal fluorescence contrast at the tumour site with GCS-g-DEAP-g-Ce6-g-PEG treatment versus treatment with free Ce6 or DEAP-free conjugate (Fig. 7A). These results demonstrated successful pH-triggered release of porphyrins, resulting in selective, higher tumour uptake relative to molecular PS. Efforts are ongoing to enhance tumour and organelle-specific delivery through dual targeting systems, in which nanoparticles are sensitive to tumour interstitium and endosomal pH.
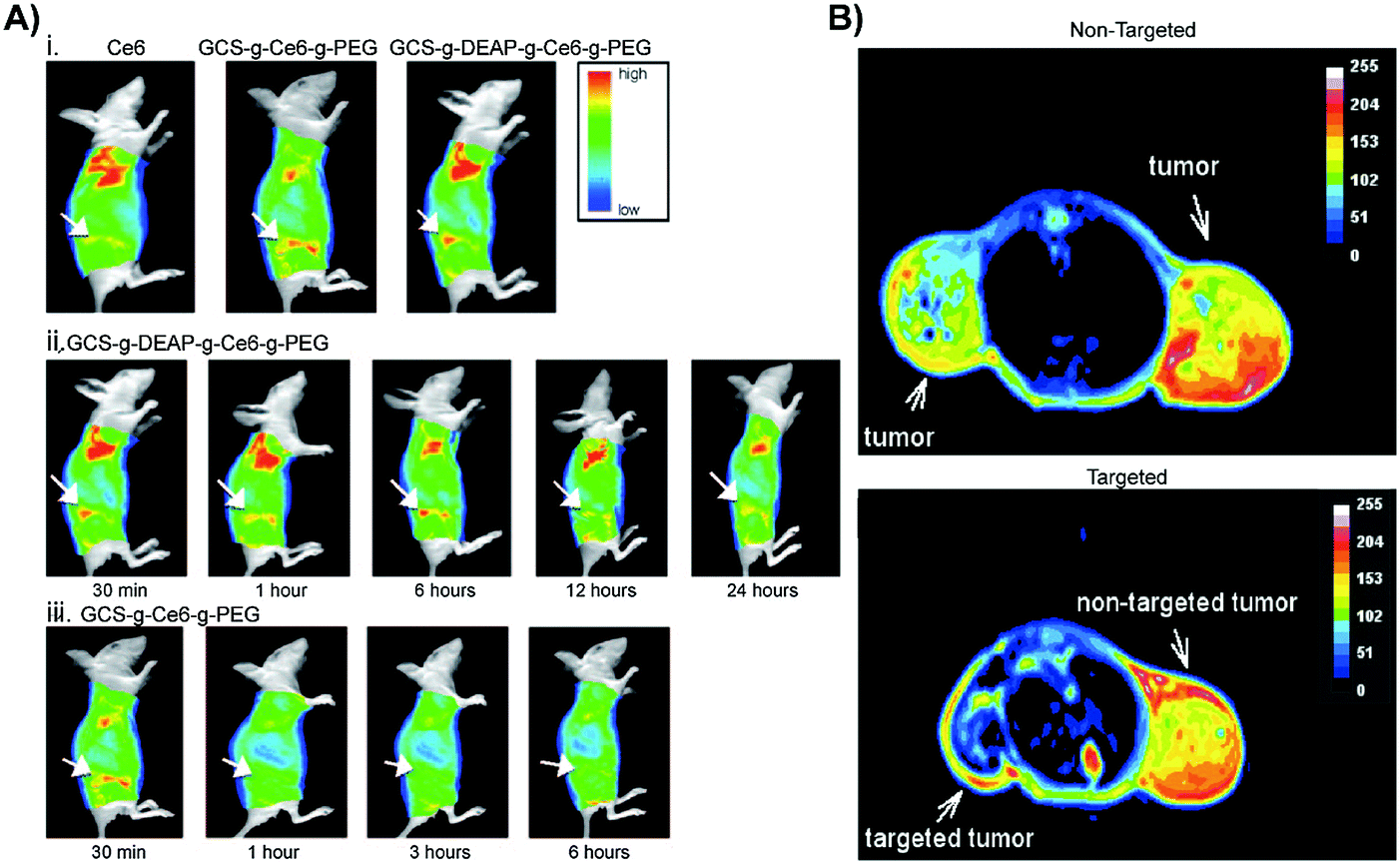 | ||
| Fig. 7 In vivo applications of controlled targeting strategies. (A) (i) Fluorescence images acquired 30 minutes after HeLa cervical tumour-bearing mice were intravenously administered 2.5 mg kg−1 free Ce6, control non-pH responsive conjugate GCS-g-Ce6-g-PEG (0.1 mg kg−1 equivalent of Ce6), and GCS-g-DEAP-g-Ce6-g-PEG (0.1 mg kg−1 equivalent of Ce6) (from left to right). In vivo fluorescence images of a HeLa cervical tumour bearing mice 30 minutes, 1, 6, 12, and 24 hours post injection with GCS-g-DEAP-g-Ce6-g-PEG (ii) and GCS-g-Ce6-g-PEG (iii). Copyright (2011) Wiley. Used with permission from Park et al.14 (B) MRI of SW480 colon tumours. Top image: T2-Weighted MRI of an intradermal, double-grafted tumour bearing mice injected with PHPP functionalized magnetic iron oxide nanoparticles (PHPP-MTCNP) in the absence of external magnetic field guidance. Bottom image: T2-Weighted MRI of subcutaneous, double grafted tumour-bearing mouse, with a 1 T external magnetic field applied to the left flank for 8 hours, after injection with PHPP-MTCNP. The control right flank did not receive magnetization. Copyright (2009) IOP Publishing. Used with permission from Sun et al.15 | ||
Magnetic guidance. Since magnetic stimuli do not interfere with any physical interactions within the body, magnetism is considered a safe candidate force to direct nanocarriers and trigger payload release. A magnetic field can either be applied via a permanent magnet positioned on the biological target or via an alternating magnetic field to concentrate magnetic nanoparticles at target sites, subsequent to which payloads can be released passively, or actively through the application of a magnetic pulse. Magnetic nanoparticles can generate heat upon exposure to an external high frequency alternating magnetic field, which can trigger drug release through nanoparticle structural disruption or a pumping effect.
Given this intriguing dual-targeting capacity of magnetic guidance, Sun et al. developed a core–shell nanoparticle (PHPP-MTCNPs), in which an iron oxide core was encapsulated by the biocompatible and non-immunogenic polymer, chitosan, which in turn was functionalized with 2,7,12,18-tetramethyl-3,8-di-(1-propoxyethyl)-13,17-bis-(3-hydroxypropyl) porphyrin (PHPP).15 PHPP-MTCNPs were administered to dual-subcutaneous SW480 colon tumour-bearing mice, subsequent to which a guiding 1 T magnetic pulse sequence was applied to one of the inoculated tumours. T2-Weighted MRI revealed lower T2-values in the targeted tumour versus the contralateral tumour, demonstrating successful guidance of particles to the desired tissue site (Fig. 7B). This targeting elicited significantly reduced tumour burden in mice treated with magnetic localization of PHPP-MTCNPs, compared to those treated without magnetic localization. Their ability to guide nanoparticles up to a distance of a few centimeters, and subsequently trigger PS release make magnetic-responsive nanocarriers promising drug delivery vehicles. However, practical parameters such as depth of the target, magnetic field strength and geometry, duration of magnetic field application, and vascular supply need to be considered prior to use.
Redox-triggered release. Redox, the process by which electrons are transferred between molecules, atoms or ions, can be leveraged by supramolecular porphyrin delivery systems to trigger site-specific PS release. In reducing tissue and intracellular environments such as the tumour interstitium, cytoplasm, nucleus, and mitochondria, high concentrations of glutathione (GSH) enables the ready reduction of disulfide bonds incorporated in nanoparticle drug delivery systems, resulting in drug release. To this end, Li et al. linked gold nanoparticles to a conjugate of heparin/pheophorbide-a (PhA-H/AuNP) via a thiol bond, that was cleaved in the presence of GSH to restore fluorescence and 1O2 generation.14 The 1O2 quantum yield increased from 0.03 to 0.47 following the exposure of PhA-H/AuNP to GSH. This GSH-dependent unquenching of porphyrin yielded robust cytoplasm and nuclear membrane-specific fluorescence contrast in PhA-H/AuNP-treated A549 lung adenocarcinoma cells. In turn, this site-specific delivery of PS contributed to a ∼1.5-fold reduced tumour volume in A549 tumour-bearing mice, compared to mice treated with free PhA. However, the ubiquitous, constitutive expression of GSH lends to the potential of premature drug loss prior to arrival at the target site. Furthermore, studies investigating redox-triggered release have largely employed micelles, the relative instability of which can promote further premature drug loss.
3.5 Key learning points, challenges and future directions
• Supramolecular drug delivery systems can enhance porphyrin delivery to tumours relative to molecular porphyrin administration via passive and active delivery methods. Diverse supramolecular structures can solubilize porphyrins, improve their photophysical properties, and enhance their phototoxicity.• Successful passive delivery techniques extend the circulation time of supramolecular porphyrin structures, allowing preferential accumulation in the tumour presumably via the EPR effect.
• Supramolecular porphyrin structures can also enable active and stimuli-responsive PS delivery to tumours when functionalized with various targeting ligands that improve cell-specific porphyrin uptake, including antibodies, aptamers, and peptides. Alternatively, magnetic guidance can steer nanoparticles to the tumour. Stimuli-responsive nanoparticles can facilitate triggered release of porphyrin payloads through magnetic pulsing, or through alterations in pH and redox. To improve porphyrin delivery, there is great interest in combining targeting strategies within one nano-carrier.
• The field of supramolecular porphyrin delivery must confront many challenges, including the abnormal distribution of neovasculature within tumours that restricts core-penetration of porphyrins, in addition to a lack of in vitro studies being translated in vivo. These may be addressed by: (1) exploring combination therapy with tumour normalizing agents, (2) overcoming the ubiquitous use of the EPR effect as a homing strategy by facilitating delivery across intact tumour vasculature, and (3) employing more clinically-relevant tumour models for better assessment of agent translational potential.
4. Alternative photoactivation strategies
In addition to enhancing PS delivery to target lesions, supramolecular porphyrin structures also address another crux in PDT: the restricted tissue penetration of light. Successful light transmission and subsequent activation of PS in vivo are contingent upon the wavelength of the delivered light, sensitizer extinction coefficient at the irradiated wavelength and tissue makeup, as shown in Fig. 8.16 Though porphyrins feature high absorptivity Soret bands (ε > 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 in saline), the near ultraviolet wavelength of these bands restricts light delivery to depths of <1 mm. As such, the lower energy and less intense Q-band is exploited for PDT. For example, Photofrin is excited for PDT at 630 nm at its highest intensity Q-band (Qmax) band with an associated absorptivity of only 3000 M−1 cm−1 and light tissue penetration depth of up to 4 mm.17 Consequently, to-date, the success of porphyrin-PDT has been limited to the treatment of superficial lesions and those accessible endoscopically, including esophageal and endobronchial cancers. Recently, supramolecular porphyrin chemistry was leveraged to deepen tissue photoactivation via Q-band modulation, non-radiative energy transfer, and sonodynamic effects, as summarized in Table 6.
000 M−1 cm−1 in saline), the near ultraviolet wavelength of these bands restricts light delivery to depths of <1 mm. As such, the lower energy and less intense Q-band is exploited for PDT. For example, Photofrin is excited for PDT at 630 nm at its highest intensity Q-band (Qmax) band with an associated absorptivity of only 3000 M−1 cm−1 and light tissue penetration depth of up to 4 mm.17 Consequently, to-date, the success of porphyrin-PDT has been limited to the treatment of superficial lesions and those accessible endoscopically, including esophageal and endobronchial cancers. Recently, supramolecular porphyrin chemistry was leveraged to deepen tissue photoactivation via Q-band modulation, non-radiative energy transfer, and sonodynamic effects, as summarized in Table 6.
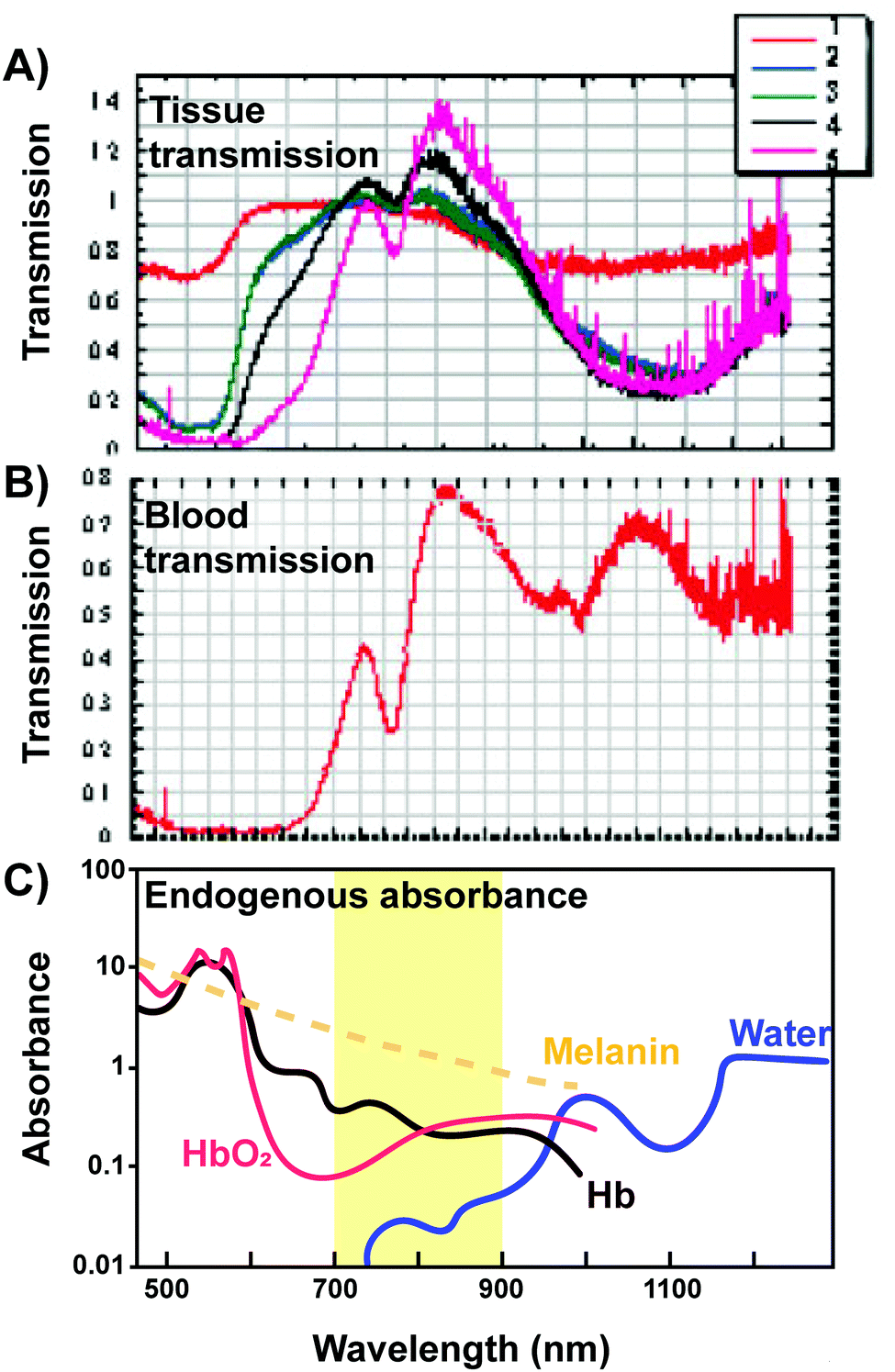 | ||
| Fig. 8 Transmission of light through various tissues. (A) Transmission of light in relation to its wavelength through (1) skin, (2) loose connective tissue, (3) dense connective tissue, (4) muscle, (5) vertebral column and spinal cord tissue and (B) blood. Copyright (2005) Wiley. Adapted with permission from Byrnes et al.16 (C) The “optical window” amenable for phototherapy with minimal absorbance and scattering of light from endogenous absorbers is highlighted. | ||
| Supramolecular host/structure | Porphyrin | Treatment or imaging parameters | Disease model |
|---|---|---|---|
| Abbreviations: BPh-a: bacteriopheophorbide-a, PPh-a: pyropheophorbide-a; TCPP: tetrakis(4-carboxyphenyl)porphyrin, Ce6: chlorin e6, TPPS4: tetrasodiummeso-tetra(sulfonatophenyl)porphine, TMPP: 5-(4-acetamidophenyl)-10,15,20-tris(4-methoxylphenyl)porphyrin, TSPP: tetrakis(4-sulphonatophenyl) porphyrin, MTAP: meso-tetra-(o-amino phenyl) porphyrin, TPP: tetraphenylporphyrin, MTCP: meso-tetra(4-carboxyphenyl) porphyrin, ITMP: 5-(4-iso-thiocyanatophenyl)-10,15,20-tris(4-N-methylpyridiniumyl), HPPH: 2-devinyl-2-(1-hexyloxyethyl)pyropheophorbide, TPTBP: meso-tetraphenyltetrabenzoporphyrinatozine, HP: hematoporphyrin, PPIX: protoporphyrin IX; H2TPACPP: tetra(N-propynyl-4-aminocarbonylphenyl)porphyrin; H2DBC: 5,15-di(p-benzoato)-chlorin. | |||
| Q-band modulation 18–20 | |||
| Liposomes |
BPh-a-lipid
Zn-Ph-a-lipid |
725–825 nm photoacoustic imaging | In vivo: cervical adenocarcinoma (KB), chemically-induced hamster cheek pouch cancer |
| Polymersomes | Zn–porphyrin linear arrays | 705–765 excitation/805–880 nm emission fluorescence imaging | In vivo: rat glioma (9L) |
| Low density lipoprotein | Zn–porphyrin linear arrays | 488 excitation/700 nm long pass fluorescence imaging | In vitro: melanoma (B16) |
| Silica nanoparticles | TCPP | 675 nm excitation/740 nm emission fluorescence imaging | In vivo: lymphoma (RPMI 8226) |
| Iron oxide nanoparticles | Ce6 | 704 nm PDT(9 J cm−2) | In vitro/in vivo: mammary carcinoma (4T1) |
| Polymeric nanoparticles | Zn–porphyrin linear arrays |
808 nm PTT (150–300 J cm−2)
UV PTT |
In vitro: cervical adenocarcinoma (HeLa), breast adenocarcinoma (MDA-MB-231)
In vivo: zebrafish liver hyperplasia |
| Porphyrin nanotubes | TPPS4 | Not provided | In vitro: cervical adenocarcinoma (HeLa) |
| Nanoscale metal organic framework | H2DBC | 650 nm PDT (in vitro: 90 J cm−2, in vivo: 90 or 180 J cm−2) |
In vitro: colon carcinoma (CT26, HT-29)
In vivo: colon carcinoma (CT26, HT-29) |
| One-photon FRET | |||
| Quantum dots* | Ce6 | 405 nm PDT (24 J cm−2) | In vitro: Ehrlich-Lettre ascites carcinoma cells |
| Fullerenes* | TMPP | 350–800 nm lamp PDT (54 J cm−2) | In vitro: Larynx carcinoma (Hep-2) |
| ZnO* | TSPP, MTAP | 365 nm or UV-Vis PDT | In vitro: Staph. aureus, E. coli, ovarian carcinoma (NIH:OVAR-3) |
| Two-photon FRET 21,23 | |||
| Gold nanorods* | Pd-TPP, MTCP | 800 or 900 nm (250 J cm−2) PDT |
In vivo: mammary adenocarcinoma (MDA-MB-231)
In vitro: hepatocellular carcinoma (HepG2) |
| Carbon nanodots* | TMPyP | 700 nm PDT (432 J mm−2) | In vitro: cervical adenocarcinoma (HeLa) |
| Polymeric nanoparticles* | Pt-MTCP, TPP |
740 nm or 800 nm PDT (640–900 J cm−2)
740 nm excitation/420–480 emission fluorescence imaging |
In vitro: hepatocellular carcinoma (HepG2) |
| Silica nanoparticles* | ITMP, HPPH | 800 nm or 850 nm PDT | In vitro: mammary adenocarcinoma (MCF-7), cervical adenocarcimona (HeLa) |
| UCNP FRET 22,24,25 | |||
|
NaYF4 nanocrystals*
(dopants: Yb3+, Er3+, Mn2+, Tm3+) |
Ce6, PPh-a, Zn-TPTBP, endogenous porphyrin, pyropheophorbide-a methyl ester |
980 nm PDT (in vitro 150–1459, 3000 J cm−2
in vivo: 180, 900–1200 J cm−2) 980 nm excitation photoluminescence imaging/641.5-708.5 nm emission fluorescence imaging |
In vitro: lung carcinoma (NCI-H460), mammary adenocarcinoma (4T1, MCF-7), carcinoma (KB), P acnes, melanoma (A-375), glioblastoma (U87 MG), atherosclerosis (THP-1 macrophage), hepatocellular carcinoma (QGY-7703)
In vivo: lung carcinoma (NCI-H460), breast adenocarcinoma (4T1), glioblastoma (U87MG), colon carcinoma (CT 26) |
| NaGdF4 nanocrystals* (dopants: Yb3+, Er3+) | Ce6, HP | 980 nm PDT (in vitro: 960–1500 J cm−2, in vivo: 7200 J cm−2) | In vitro and in vivo: cervical adenocarcinoma (HeLa) |
| NaYF4/NaGdF4 nanocrystals* | Ce6 |
980 nm PDT (180 J cm−2)
980 nm excitation/850 nm emission fluorescence imaging |
In vivo: glioblastoma (U87MG) |
| LiYF6 nanocrystals* | Temoporfin | 980 nm PDT | In vitro: cervical adenocarcinoma (HeLa) |
| NaYbF4/NaGdF4 nanocrystals* | Ce6 | 808 nm PDT (300–1800 J cm−2) | In vitro: cervical adenocarcinoma (KB) and lung adenocarcinoma (A549) |
| X-ray energy transfer 26,27 | |||
| CeF3 nanoparticles* | Verteporfin | 6 MeV, 1–6 Gy X-ray | In vitro: pancreatic carcinoma (Panc 1) |
|
LaF3 nanoparticles*
(dopants: Ce, Tb3+) |
Ce6, MTCP | 80–250 keV, 2–10 Gy X-ray | In vitro: melanoma (B16), rat glioma (9L) |
| LaF3* polymer microspheres | PPIX | 90 keV, 3 Gy X-ray | In vitro: prostate adenocarcinoma (PC3) |
| SiC/SiOx nanowires* | H2TPACPP | 6 MeV, 2 Gy X-ray | In vitro: lung adenocarcinoma (A549) |
| ZnO nanoparticles* | MTCP | 70 keV, 0.94 Gy | In vitro: prostate carcinoma (DU147), mammary carcinoma (T-47D) |
| SDT 28,29 | |||
| Silica nanoparticles | PPIX | 1 MHz, 1.5–2.3 W cm−2 ultrasound | In vitro and in vivo: mammary carcinoma (4T1) |
| Antibody conjugates | ATX-70 | 1 MHz, 1 W cm−2 ultrasound |
In vitro: gastric carcinoma (KATO-III)
In vivo: gastric carcinoma (MKN-45 sc) |
| Polymeric nanoparticles | TPPS | 4 Hz, 0.43–0.88 mJ mm−2 (90 MPa) ultrasound |
In vitro: neuroblastoma (SH-SY5Y)
In vivo: orthotopic rat Mat B III mammary cancer |
4.1 Bathochromically-shifted Q-bands
The prime window for light delivery through tissue exists between 700–900 nm, in which light absorbance and scattering from endogenous absorbers such as oxy- and deoxy-hemoglobin, water and lipids are minimized (Fig. 8). Indeed, Stolik et al. demonstrated that 780 and 835 nm laser light permeated upwards of 2× the distance in malignant and healthy tissue compared to 633 nm light.17 This optical window therefore demands the use of NIR and infrared (IR) light for optimal porphyrin photoactivation. As such, linear porphyrin arrays have come under investigation for NIR optical applications. These arrays are composed of porphyrin monomers linked covalently via ethyne or butadiyene bridges, yielding a bathochromic shift of porphyrin Qmax bands into the NIR and IR region through the formation of an extensively π-conjugated oligomer backbone. However, the hydrophobicity of these porphyrin arrays hinders their biomedical use. To address this limitation while preserving the biomedically-relevant NIR optical properties of porphyrin oligomers, Ghoroghchian et al. introduced water-soluble NIR polymersomes.18 Polymersomes consist of amphiphilic di-block co-polymers that self-assemble into nanoparticles with an aqueous core and an amphiphilic bilayer shell (Fig. 9), much like liposomes. The use of polymeric components bestows the added advantage of generating a nanoparticle shell twice the thickness of that of a liposome, allowing for the stable incorporation of nanometer-sized zinc-chelated porphyrin oligomers (PZn). Ultimately, this facilitated the aqueous dispersion of these porphyrin arrays and rendered their NIR Q-bands amenable for imaging and therapeutic purposes. By increasing the number of porphyrin units within each PZn, changing the porphyrin meso substituents, varying the di-block co-polymers, and tuning PZn loading, the polymersome Q-band and corresponding emission maxima could impressively be bathochromically-shifted and modulated from 650 to 900 nm. The extension of the PZn chain from two to five porphyrin units also increased the NIR (794 nm) extinction coefficient of the polymersomes from 70000 to 200000 M−1 cm−1. Collectively, these properties enabled the application of polymersomes for high contrast in vivo NIR fluorescence imaging of heterotopic subcutaneous murine tumours. To further substantiate their utility as contrast agents, evaluation of polymersome biodistribution and imaging capacity in more clinically-relevant, orthotopic tumour models is needed.
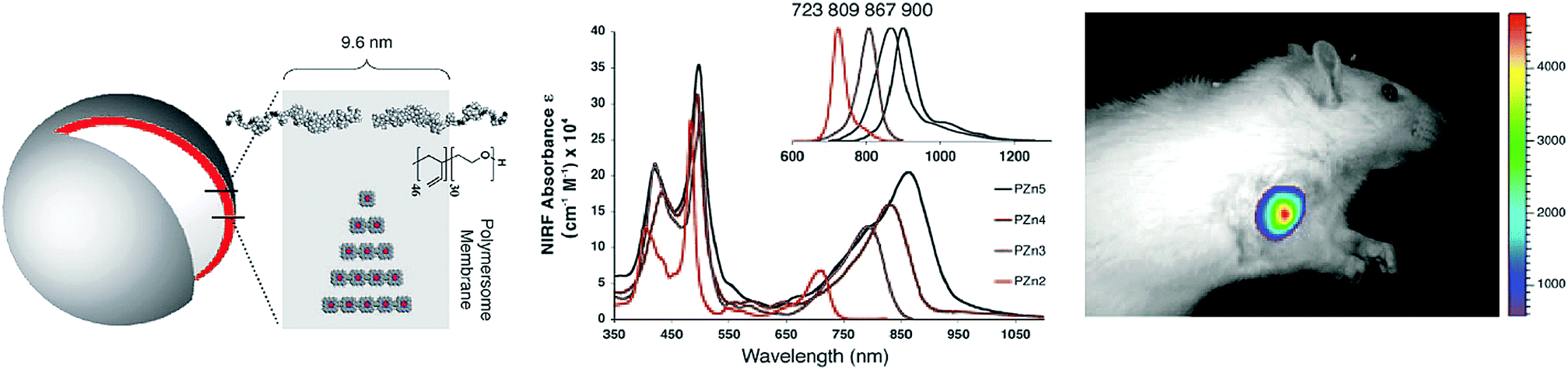 | ||
| Fig. 9 Polymerosome mediated imaging. Illustration of polymersome structure, wherein porphyrin linear arrays, PZn, are loaded within the particle polymer membrane to impart aqueous solubility.18 Increasing linear chain length of porphyrins within the polymersomes red-shifted the particle Q-bands, and associated fluorescence emissions (see inset spectrum), allowing for NIR fluorescence imaging of subcutaneous 9L rat xenografts 10 minutes post intratumoural injection of porphysomes (705–765 nm excitation, 805–880 nm emission). Copyright (2005) National Academy of Sciences. | ||
As previously introduced, bathochromic shifting of molecular porphyrin Q-bands can be instigated via the reduction of one or two tetrapyrrole double bonds, forming chlorins and bacteriochlorins respectively. Chlorins and bacteriochlorins feature one order of magnitude higher Q-band extinction coefficients relative to their porphyrin counterparts, wherein chlorins display deep-red (650–700 nm) Qmax bands, while bacteriochlorins display NIR/IR (700–800) Qmax bands. Consequently, as summarized in Table 1, a number of chlorins have been essayed clinically for PDT. The directed supramolecular assembly of chlorins and bacteriochlorins can further enhance the light-absorbing properties of these PS, as evidenced from nature. Chlorins and bacteriochlorins respectively constitute the chromophores in chloroplasts and chlorosomes, the light-harvesting complexes of plants and bacteria responsible for photosynthesis. Chlorosomes are exceptionally efficient reactions centres, allowing for photosynthesis to occur in green sulfur bacteria in light-deficient environments underwater at depths of 50–100 m. This light harvesting efficiency is rooted in the self-assembled, high density packing of bacteriochlorophyll monomers into lipid-enclosed tubular nanostructures. Specifically, it is thought that bacteriochlorophylls form J-aggregates through the coordination of their centrally-chelated Mg2+ to the 3′ hydroxy group, that in turn can undergo hydrogen-bonding with the 13′ keto group. Combined with π–π stacking, this ultimately yields strong exciton coupling and a high absorptivity Qy band red-shifted >800 nm. Recently, we mimicked and exploited this supramolecular assembly for in vivo photoacoustic imaging (PAI); a modality suited to imaging tissue at 5–8 cm in depth, wherein imaging contrast arises from acoustic waves generated from the thermoelastic expansion of tissue following the vibrational relaxation of optically-excited chromophores. By conjugating 3′-modified pyropheophorbide-a to a phospholipid backbone, we induced stable J-aggregation of the chlorin monomers within self-assembled water-soluble nanovesicles. Accordingly, we exploited the NIR optical properties of porphyrin J-aggregates while simultaneously overcoming their limited aqueous solubility (Fig. 10A).19 This aggregation relied on the combined presence of centrally-chelated zinc and a 3′ methoxy group within the chlorin-lipid conjugate, giving rise to a relatively narrow Qy band at 725 nm, red-shifted by 72 nm relative to the corresponding monomeric chlorin-lipid. Furthermore, the self-assembly of the chlorin-lipid monomers yielded fluorescently-quenched nanovesicles, presumably alluding to non-radiative thermal decay of excited state chlorins. The supramolecular assembly-enabled high (98%) quenching efficiency and red-shifting of the chlorin Q-band into the tissue-transparent optical window facilitated the preliminary exploration of these nanovesicles in a hamster cheek pouch tumour model as PAI contrast agents that could be spectrally-unmixed from endogenous absorbers. Additionally, we generated J-aggregates through the self-assembly of bacteriochlorophyll-lipid monomers into nanovesicles, which similarly featured a 75 nm bathochromic shift of the Qy band (825 nm), but largely unquenched fluorescence, allowing for both PAI and NIR fluorescence imaging of VX-2 rabbit metastatic lymph nodes.20 Collectively, these results demonstrate the promising, yet largely unexplored, potential of using supramolecular assemblies for bathochromic-shifting of chlorin and bacteriochlorin Q-bands for optically-driven deep lesion imaging and therapy.
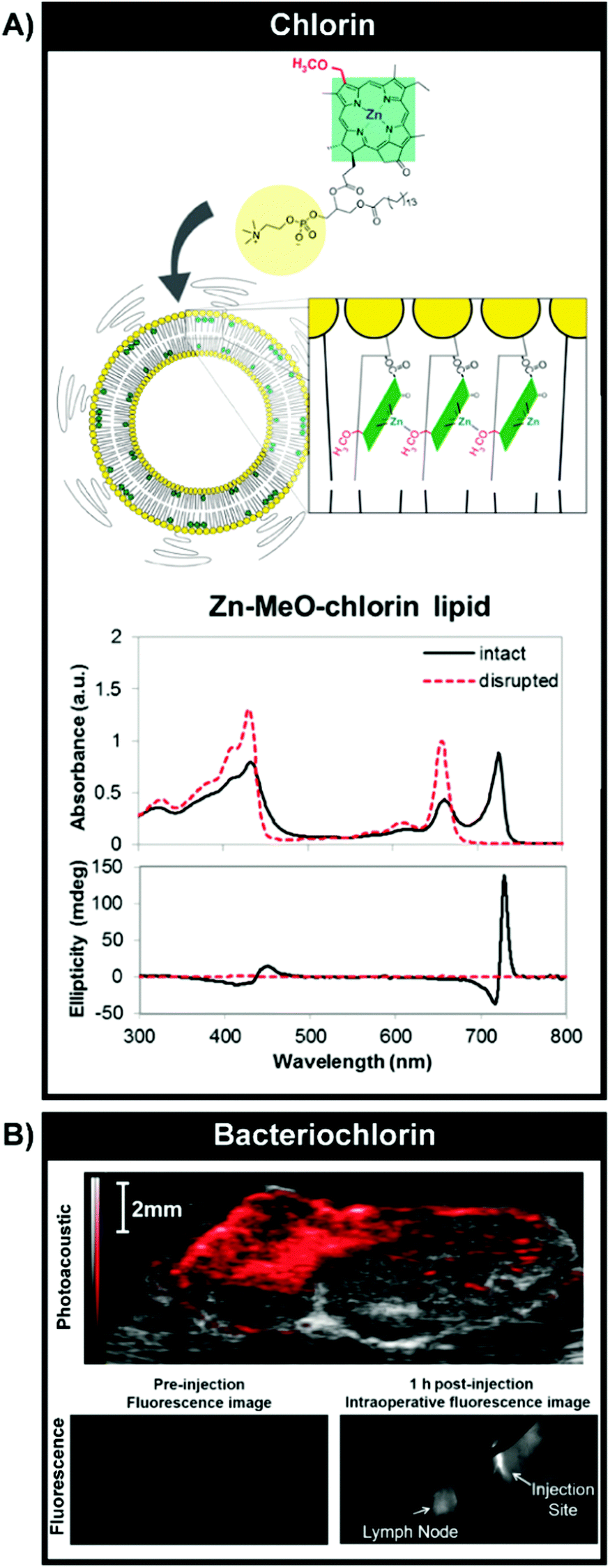 | ||
| Fig. 10 Optical properties and imaging mediated by J-aggregation. (A) Bathochromic shifting of chlorin Q-bands by inducing J-aggregation through self-assembly of Zn–chlorin–lipid into chlorosome-mimetic vesicles. A 72 nm shift was observed relative to monomeric Zn–chlorin–lipid. Reprinted with permission from Ng et al.19 Copyright (2016) American Chemical Society. Vesicular self-assembly of bacteriochlorin–lipid (B) could also yield J-aggregation but without complete fluorescence quenching, enabling intraoperative fluorescence imaging of VX-2 rabbit buccal tumours (1 h post subcutaneous injection of particles, 806 nm excitation) and ex vivo tumour PAI (image shown overlaid with B-mode ultrasound image, 680–900 nm PA signal collection). Reproduced from Shakiba et al.20 with permission from the Royal Society of Chemistry. | ||
4.2 Photoactivation via non-radiative energy transfer
The use of non-radiative energy transfer represents the most extensively explored strategy to enable NIR and IR light-mediated porphyrin activation via supramolecular structures. Here, an energy donor molecule or nanostructure able to efficiently absorb NIR light is excited by >700 nm laser light (Fig. 11). The resulting excited donor undergoes decay by transferring energy non-radiatively to an acceptor porphyrin molecule, which can then return to ground state through luminescence or via energy transfer to oxygen to generate ROS. This process, known as Förster resonance energy transfer (FRET), requires strong overlap between the donor fluorescence and porphyrin absorbance spectra, as well as sub-10 nm spacing between the donor and porphyrin acceptor. To this end, nanoscaled supramolecular structures that spatially-confine donors and porphyrins are of interest. These nanoparticles capitalize upon the optical properties of NIR light-absorbing donors while conserving the physiochemical features and 1O2 generating capabilities of porphyrins by evading porphyrin chemical modification. In doing so, they facilitate deeper tissue porphyrin photoactivation. Although the use of single-photon excitation of donors has been studied, most of the literature surrounding NIR activation of porphyrins by FRET involves multiphoton sensitization via two-photon absorbers and upconverting nanoparticles (Table 6). Here, we provide a concise overview of the fundamentals, limitations and seminal proceedings driving this field, as several recently-published review articles, including those by Shen et al.21 and Idris et al.,22 already provide a comprehensive summary of porphyrin supramolecular structures for multiphoton PDT.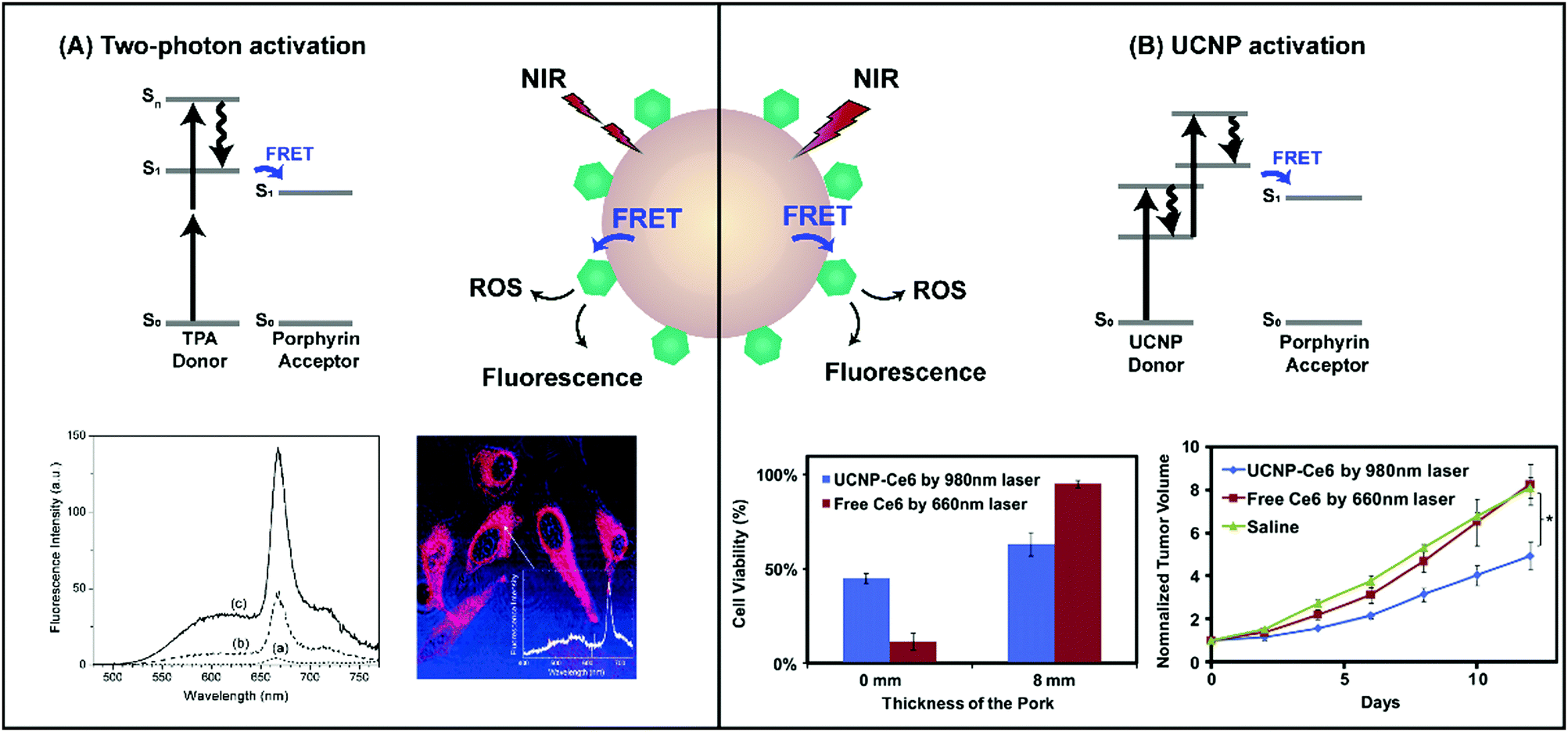 | ||
| Fig. 11 Schematic representation of two-photon and upconversion nanoparticle excitation, and subsequent energy transfer to co-assembled porphyrins. (A) Co-encapsulation of HPPH with the two-photon absorber, BDSA, increased two-photon emission at the HPPH Q-band (spectrum c) relative to BDSA-free particles (spectrum a) when excited at 850 nm. This allowed for two-photon confocal microscopy of HeLa cells following incubation with particles. Reprinted with permission from Kim et al.23 Copyright (2007) American Chemical Society. (B) NIR photoactivation of Ce6 loaded onto NaYF4-UCNPs with a 980 nm laser (0.5 W cm−2) yielded higher 4T1 mammary carcinoma cell cytotoxicity across 8 mm of tissue, and more significantly delayed tumour growth (n = 10, *p < 0.05) relative to Ce6 PDT with a 660 nm laser (0.5 W cm−2). Reprinted with permission from Wang et al.24 Copyright (2011) Elsevier B.V. | ||
Two-photon excitation, as the name suggests, involves the concurrent absorption of two equivalent low-energy photons to reach an excited singlet state, Sn (Fig. 11A). This rapid, near-simultaneous multiphoton absorption requires the use of high energy femto-second laser pulses. The non-linearity of the two-photon excitation process (namely, increasing the intensity of the delivered light results in a quadratic increase in fluorescence) endows it with high spatial confinement of light delivery to deeper tissue, which combined with femto-second laser pulsing, minimizes non-target photoexcitation or photobleaching. However, monomeric porphyrins exhibit low (<50 GM at NIR wavelengths of light) two-photon absorption cross-sections. The non-covalent dimerization of conjugated porphyrin linear arrays yields two orders of magnitude enhanced cross sections, but this increased two-photon absorption comes at the expense of porphyrin solubility and synthetic yield. Consequently, a variety of organic and inorganic nanoparticles consisting of efficient NIR two-photon absorbers (TPAs) have been explored over the last decade for the indirect activation of porphyrin photodynamic effects via: (1) co-encapsulation of molecular TPAs and porphyrin within nanospheres, or (2) loading porphyrins onto inherently two-photon absorbing nanoparticles such as gold, silica, and conjugated polymer nanoparticles, and quantum dots.
These nanostructures have been predominantly evaluated for 1O2 generation and FRET efficiency in solution, with one of the first in vitro applications introduced by Kim et al. in 2007.23 The authors developed a unique TPA, 9,10-bis[4′-(4′′-aminostyryl)styryl]anthracene (BDSA), which displayed an NIR two-photon absorption cross-section of 217 GM, and strong two-photon fluorescence even when aggregated; a beneficial trait in facilitating high TPA loading into nanoparticles while preventing competing non-radiative decay through vibrational relaxation. BDSA was co-loaded alongside HPPH into 25 nm silica nanoparticles, yielding a 73% FRET efficiency. This elicited a 30-fold enhancement in two-photon (850 nm excitation) fluorescence emission, and accelerated the in solution generation of 1O2 relative to silica nanoparticles loaded solely with HPPH, which exhibited negligible two-photon stimulated 1O2 generation and fluorescence. These promising in solution results translated into the rapid TPA-dependent death of cervical cancer HeLa cells. However, despite continued in vitro assessments of various two-photon responsive porphyrin nanoparticles (Table 6), there lacks significant in vivo evaluation. The translation of two-photon PDT is largely hindered by its dependence on femtosecond lasers, which require lengthy irradiation procedures to account for the small laser spot size, and thereby the administration of 10-fold higher total light doses compared to conventional PDT.
This limitation can be partially overcome by upconversion nanoparticles (UCNPs). Upconversion excitation is a multiphoton process, which unlike two-photon excitation, exploits metastable excited states to facilitate sequential and not simultaneous absorption of two or more low energy photons, allowing for the use of continuous wave lasers and shorter irradiation times (Fig. 11B). The absorption of NIR light by UCNPs is associated with large anti-Stokes shifts, resulting in photon emission in the visible region of light with minimal tissue autofluorescence, making UCNPs interesting agents to explore for in vivo applications. These properties of UCNPs arise from the use of metal-doped lanthanide crystals, the composition of which can be modulated to tailor excitation and emission wavelengths. For example, the use of NaYF4 nanocrystals doped with Yb3+ and Er3+ gives rise to efficient upconversion following 980 nm light irradiation, and sharp emission peaks in the blue, green and red visible regions of light, of which the latter overlaps with porphyrin and chlorin Q-bands. As such, the supramolecular assembly of porphyrins on the surface of UCNPs provides the opportunity for NIR-triggered PDT.
This potential was exemplified in vivo by Wang et al. in 2011 through the application of chlorin e6 (Ce6)-NaYF4:Yb,Er UCNPs as an NIR PS in a heterotopic model of murine breast cancer.24 Upon treatment with 980 nm light following the intratumoural injection of Ce6-UCNPs, tumour growth was largely delayed over a 2-week period, resulting in a ∼70% survival rate over 60 days following treatment (all control treatment groups succumbed within 30 days). Importantly, the authors demonstrated that the use of NIR light and Ce6-UCNPs enhanced photosensitization across 8 mm tissue penetration depths in vitro and in vivo relative to the administration of 660 nm light and monomeric Ce6. These results validated the continued exploration of porphyrin-UCNPs for deeper tissue PDT, which has subsequently been conducted predominantly for the treatment of heterotopic murine tumours via 980 nm light activation of NaY4:Yb,Er UCNPs surface-modified with porphyrin.
Despite these initially-promising results, the use of porphyrin-UCNP supramolecular structures for deep tissue PDT has major limitations. Firstly, like other metallic nanoparticles, lanthanide UCNPs are not biodegradable, wherein associated yttrium is detectable within MPS organs even 60 days following administration,24 posing a major roadblock towards clinical translation. Secondly, though the use of UCNPs reduces the irradiation time relative to two-photon excitation, the explored fluences exceed those used in the clinic, giving rise to the possibility of undesired tissue heating. Thirdly, the 980 nm excitation band of the explored UCNPs coincides with the strong absorbance band of water in biological tissue. Consequently, the application of 980 nm light promotes undesired tissue heating and cell death,25 while restricting light transmission in tissue by approximately 50% relative to the delivery of 700–900 nm light (Fig. 8). Accordingly, attention has been directed towards generating porphyrin/lanthanide UCNPs with compositions amenable to excitation by 800 nm light.25 Even so, photosensitization at this “optically transparent” NIR wavelength of light is restricted to a 1–2 cm tissue depth using the photoactivated porphyrin supramolecular structures discussed thus far.
4.3 Deep tissue (>1 cm) sensitization of porphyrins
In order to extend the utility of porphyrin-PDT for the management of deep-seated (>1 cm) lesions, activation of porphyrins by alternative energy sources is of interest. To this end, supramolecular structures have opened avenues of using chemiluminescence self-lighting PDT, X-ray scintillation and ultrasound to activate porphyrin-mediated ROS generation for deep PDT. Of these, X-ray and ultrasound (i.e. sonodynamic) excitation have been most prominently explored as alternatives to porphyrin photoactivation.X-rays are ionizing forms of radiation that can generate scintillation (short bursts of light) when absorbed by heavy atoms. X-ray-activated PDT makes use of these scintillators to absorb ionizing radiation, creating electron–hole pairs and excitons that can transfer energy non-radiatively to neighbouring porphyrin molecules to incite ROS generation (Fig. 12A). When using high energy (>4 MeV) X-ray beams, this energy transfer allows for photosensitization to occur at depths of >2 cm. Like FRET, this energy transfer requires spectral overlap between the scintillator luminescence and porphyrin Soret or Q-bands, as well as spatial confinement of the scintillator and porphyrin. As such, the co-encapsulation of porphyrins and heavy atom scintillators within a single nanoparticle is beneficial for enabling deep-tissue PDT. The conception of this supramolecular porphyrin utility is credited to Chen and Zheng, who in 2006 published a seminal proceeding launching the in vitro exploration of porphyrin-inorganic nanoparticle conjugates for X-ray-mediated PDT [ref. 26 and ref. 29 therein]. Most of these studies apply lanthanide crystals as the scintillators due to their high densities and radiation absorption coefficients. The most effective in vitro cytotoxicity has thus far been demonstrated recently by Chen et al., who achieved a ∼70% decrease in 9L glioma cell viability following the administration of low energy (80 kV) X-rays and LaF3:Tb nanoparticles with surface-adsorbed mesotetra(4-carboxyphenyl)porphyrin.27 However, despite the use of relatively low energy irradiation, X-ray treatment in the absence of nanoparticles decreased cell viability by ∼30% (Fig. 12A). These observations are consistent with outcomes obtained from studies across the field, wherein the X-ray or nanoparticle doses required to incite appreciable cytotoxicity are toxic in and of themselves. Combined with the known ability of porphyrins to be directly activated by X-ray and a lack of adequate photophysical particle characterization in the field, this uncontrolled toxicity fuels the existing debate of whether energy transfer from scintillators to porphyrins is truly the source of ROS generation following X-ray irradiation, and brings into question the utility of safely using X-rays to incite PDT; a therapeutic modality which distinguishes itself based on the localization of tissue damage to target sites.
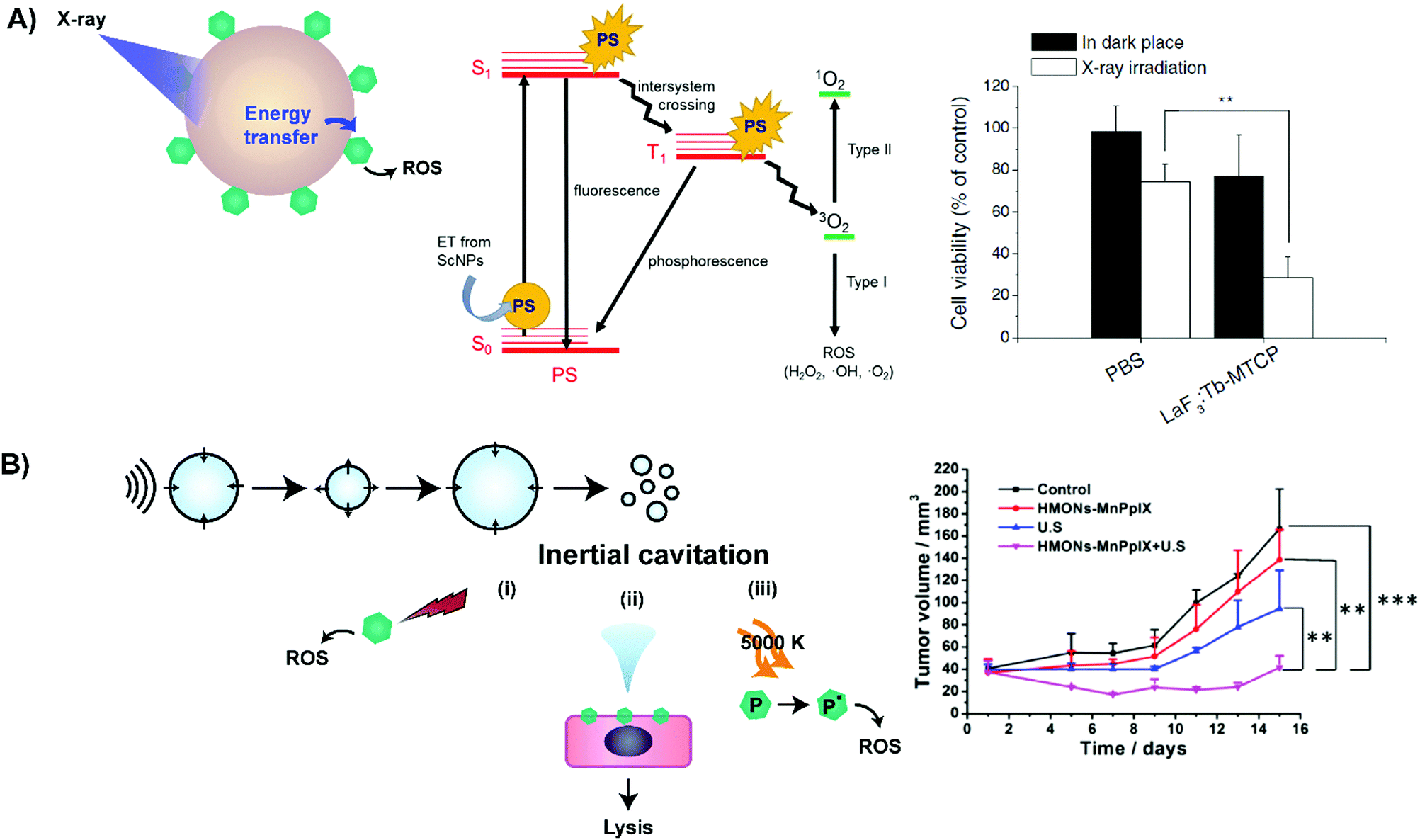 | ||
| Fig. 12 Deep-tissue photoactivation of porphyrin via X-ray (A) and ultrasound (B). Excitation of scintillation nanoparticles with X-rays leads to energy transfer to adjacent adsorbed porphyrins, resulting in ROS generation. Chen et al. used this strategy to treat 9L glioma cells following 80 kVp irradiation of cells treated with LaF3Tb scintillation nanoparticles loaded with MTCP (n = 5, p < 0.01). Adapted with permission from Kamkaew et al.26 (Copyright (2016) American Chemical Society) and Chen et al.27 (Creative commons license CC BY 4.0 http://creativecommons.org/licenses/by/4.0/). (B) Sonodynamic porphyrin sensitization begins with inertial cavitation of microbubbles, initiated by low frequency ultrasound. This cavitation can generate (i) sonoluminescence for photoactivation of porphyrins through a PDT mechanism, (ii) microjets that cause cell lysis following membrane destabilization by porphyrins, or (iii) focal heat leading to porphyrin pyrolysis. Sonodynamic activation (1.0 MHz, 2.3 W cm−2, 50% duty cycle, 5 minutes) of subcutaneous 4T1 mammary carcinoma mouse xenografts following intravenous injection of animals with mesoporous silica nanoparticles (HMONs) loaded with metal-cleated PpIX led to significant tumour growth delay versus controls (n = 5; **p < 0.01, ***p < 0.001). Reprinted with permission from Huang et al.29 Copyright (2017) American Chemical Society. | ||
To this end ultrasound may be a more clinically-viable energy source for deep tissue activation of porphyrins. Therapeutic ultrasound is rooted in the generation and oscillation of microbubbles in vasculature, a process known as cavitation. In the presence of porphyrins, this cavitation can lead to synergistic localized cell death, a phenomenon described as sonodynamic therapy (SDT): ultrasonic cavitation leading to microbubble collapse releases energy that can activate the cytotoxic effects of neighbouring sonosensitive drugs, including porphyrins. This cytotoxicity is proposed to be a consequence of porphyrin-derived ROS generation either from a PDT-type pathway activated by sonoluminescent light, or by sonosensitizer pyrolysis following heat generation from microbubble inertial cavitation (Fig. 12B). Alternatively, porphyrin sonosensitizers may also destabilize cell membranes to enhance the susceptibility of cells to damage by microbubble cavitation-induced shear stress. Thus, when considering the ability of focussed ultrasound to access deep-seated tissue at depths beyond 10 cm, SDT may localize porphyrin-mediated tumouricidal effects to deep-seated tumours.
The clinical translation of SDT is challenged by underwhelming sonosensitization achieved by monomeric porphyrin agents. To this end, porphyrin nanoparticles have shown enhanced SDT effects relative to molecular porphyrin administration, presumably by enhancing porphyrin delivery to cancer cells [ref. 28, and ref. 46 and 206 therein]. This has recently translated into achieving decreases in cell viability as high as 80% in vitro against 4T1 mammary carcinoma cells, and reduction in tumour burden by 75% in subcutaneous 4T1 tumour-bearing mice administered low-intensity (1 MHz, 2.3 W cm−2, 50% duty cycle) ultrasound and Mn-chelated protoporphyrin silica nanoparticles (Fig. 12B).29 Though the use of porphyrin nanoparticles has improved SDT effects, only delays rather than complete suppression of tumour growth have been achieved thus far. As such, exogenously-delivered microbubbles, which lower the energy of cavitation, may allow this relatively new field of study to advance.
4.4 Key learning points, challenges and future directions
• Low tissue penetration of light remains a challenge in extending the biomedical utility of porphyrin-mediated imaging and PDT.• Supramolecular assembly of porphyrins enables NIR/IR light or X-ray photoactivation through Q-band modulation or energy transfer, facilitating deeper tissue PDT versus visible light-mediated activation of existing molecular porphyrins. Incorporation of porphyrins within nanostructures can also open the doorway to enhanced ultrasound activation of porphyrin sensitization for deep tissue SDT.
• These photoactivation strategies have only recently been introduced, and thus require further investigation to establish clinical utility. Such exploration should include the use of orthotopic tumour models, intravenous administration of agents, and more in-depth and longitudinal characterization of particle distribution and bio-effects in vivo; characterization which is largely lacking in studies conducted thus-far.
• Clinical translation of these supramolecular structures is currently challenged by the reliance of energy transfer strategies on the use of non-biodegradable inorganic materials and passive tumour accumulation of the explored nanoparticles.
• Therapeutic effects and porphyrin activation efficiency may be enhanced via exogenous microbubble delivery for SDT or through the development of energy donors with spectral overlap with the Soret versus the lower absorptivity Q-band for energy transfer to porphyrins. Ongoing efforts to enhance the imaging and therapeutic capacity of NIR-absorbing supramolecular porphyrin particles include the development of agents able to deliver multimodal imaging contrast and synergistic therapy as discussed in the next section.
5. Extending the imaging and therapeutic multimodality of porphyrins via supramolecular chemistry
In addition to improving the existing biomedical utilities of porphyrins, supramolecular structures can also bestow novel therapeutic properties upon porphyrins, and increase their intrinsic multimodality. To this end, supramolecular porphyrin structures have been studied as multimodal imaging agents, theranostic platforms, and mediators of synergistic therapeutic paradigms.5.1 New theranostic paradigms
While monomeric porphyrins can be used for separate imaging applications (Table 2), their intrinsic fluorescence imaging capacities cannot be effectively combined with PET, SPECT and MR imaging due to fluorescence quenching from the very metal-chelation that endows these additional contrast capabilities. By co-encapsulating free base porphyrins with MRI and PET contrast agents, these metal-dependent porphyrin imaging capabilities can be achieved without compromising fluorescent properties. For example, super paramagnetic iron oxide nanoparticle (SPION) conjugation to porphyrins can facilitate SPION-mediated T2-weighted MRI and porphyrin-mediated fluorescence imaging from a single agent; a feat unattainable with MR-susceptible Gd3+-porphyrin monomers. This multimodal imaging allows for complimentary information to be obtained from a single agent while overcoming the limitations of each individual imaging technique (summarized in Table 7).| Imaging modality | Advantages | Limitations |
|---|---|---|
| Fluorescence |
• Can perform molecular imaging
• High sensitivity (nM, μM concentrations) • Can be performed internally via endoscopy • Inexpensive • No ionizing radiation |
• Limited tissue depth penetration
• Qualitative analyses • Autofluorescence can result in high background |
| Photoacoustic |
• Can assess blood flow, perfusion
• Overcomes optical diffusion limit • Inherent contrast between different organs • High resolution • No ionizing radiation • Often in combination with ultrasound to attain complementary information |
• Emerging modality; probes require more development |
| Ultrasound |
• Can assess blood flow, perfusion
• High spatial, temporal resolution • Non-invasive • Economical • Easily accessible • Quick • No ionizing radiation |
• Low contrast
• Imaging and interpretation variability between ultrasonographers |
| Positron emission tomography/single photon emission computed tomography |
• Images biological processes and metabolic activity
• High sensitivity • Quantitative analyses • Whole body imaging • Can be combined with CT, MRI for complementary information |
• Radiation exposure
• Expensive • Low spatial resolution |
| Magnetic resonance imaging |
• High spatial resolution
• High soft tissue contrast • Unlimited tissue depth penetration • No ionizing radiation |
• Expensive
• Long acquisition time (minutes) • Limited molecular sensitivity |
| Computed tomography |
• Unlimited tissue depth penetration
• 3D, whole body imaging • High spatial resolution • Rapid scanning time (seconds) |
• Lacks soft tissue contrast
• Limited molecular sensitivity • Radiation exposure |
In addition to restoring fluorescence emission and 1O2 generation, supramolecular structures can endow porphyrin platforms with unprecedented imaging modalities, including ultrasound, and PAI (Table 8). The former cannot be accomplished with monomeric porphyrins, while the latter cannot readily differentiate exogenous monomeric porphyrin contrast from endogenous oxy/deoxy-hemoglobin. Through the self-assembly and aggregation of bacteriochlorophyll-a-lipid within a gas-enclosed microbubble lipid shell (Fig. 13A), Huynh et al. delivered ultrasound, PA and fluorescence imaging contrast from a single porphyrin supramolecular platform (Fig. 13C). The authors discovered that the application of low frequency ultrasound pulses mediated an in situ microbubble-to-nanoparticle conversion and subsequent porphyrin extravasation to tumour tissue, likely due to the transient, non-lethal permeabilization of the tumour vasculature (Fig. 13B).30 These microbubbles illustrate both the imaging and drug-delivery facilities of supramolecular porphyrin structures that extend beyond that of passive EPR effect-dependent drug carriers.
| Modality | Number of reports | Supramolecular structure | Porphyrin imaging agent |
|---|---|---|---|
| Dual modal | |||
| Ultrasound + PAI | 2 |
Microbubble
Nanodroplet |
Pyropheophorbide-lipid |
| Fluorescence + PAI | 1 | Nanovesicle | Bacteriopheophorbide-lipid |
| Fluorescence + PET | 3 |
Porphysomes
Albumin Polymer |
Pyropheophorbide-lipid
Pheophorbide-a meso-Tetra(4-carboxyphenyl) porphyrin |
| Fluorescence + MRI | 2 | Polymer | Phenyl porphyrin |
| Liposome | Gadophrin-2 | ||
| Trimodal | |||
| Fluorescence + PAI + ultrasound | 1 | Microbubble | Pyropheophorbide-lipid and bacteriochlorophyll-lipid |
| Hexamodal | |||
| CT + fluorescence + PAI + PET + Cerenkov luminescence + NIR-to-NIR UC luminescence | 1 | Upconversion nanoparticle | Porphyrin–phospholipid |
 | ||
| Fig. 13 Utility of porphyrin microbubbles for drug delivery, and as a contrast agent for photoacoustic and ultrasound imaging.30 (A) Assembly of bacteriochlorophyll–lipid into porphyrin microbubbles that encapsulate perfluorocarbon gas. (B) Application of ultrasound waves at the conversion frequency disrupts porphyrin microbubbles, resulting in the formation of porphyrin nanoparticles. (C) Ultrasound and photoacoustic imaging of a tumour cross-section from a KB-cervical tumour bearing mouse, pre- and post-injection with porphyrin microbubbles, with and without the application of ultrasound at the conversion frequency. Conversion of porphyrin microbubbles to nanoparticles resulted in the delivery and persistence of nanoparticles in the tumour. | ||
With their multimodal imaging capabilities and photosensitive nature, porphyrin supramolecular structures demonstrate great potential for theranostics: imaging and diagnostics from a single agent. This was illustrated by Fan et al., who developed a triple layer nanoparticle (tNP) for fluorescence/MRI-guided PDT.31 The Gd-chelated inner polymeric core encapsulated Ce6, which was protected by 2 outer layers. The inner layer contained anionic and cationic polymers that were dismantled by the acidic TME, resulting in deshielding of Gd3+. This facilitated site-specific enhancement of fluorescence and T1-weighted MRI contrast in BxPC-3 mice pancreatic tumours compared to free Ce6, pH-unresponsive control nanoparticles and the clinically-approved gold-standard MRI agent, Gd-DTPA. This strong contrast was indicative of tNP tumour accumulation, which reduced tumour volume by 30% upon photoactivation, whereas pH-non-responsive nanoparticles were unable to suppress tumour growth. Such a platform could thus enable image-guided PDT, allowing clinicians to direct laser irradiation to targets that may be otherwise visually indiscernible.
This combination of MRI with porphyrin phototherapy represents the most widely explored strategy for porphyrin-derived multimodal theranostics, though a plethora of supramolecular porphyrin platforms have also combined PET, PAI, CT, luminescence and phototherapy as summarized in Table 9 and reviewed by Zhou et al.32 Porphyrin theranostic systems can be categorized into “all-in-one” or “one-for-all” systems. The “all-in-one” approach combines multiple constituents, each with a unique function, to attain multifunctionality. Presently, this design underlies most investigated theranostic systems, and is exemplified by Gong et al. in their multifunctional assembly (Fig. 14A).33 PEG-polymer conjugates were self-assembled into micelles that encapsulated IR825, a NIR photothermal sensitizer, and Ce6. The presence of Ce6 and Gd3+ chelation granted the micelle trimodal fluorescence, PA and MR imaging capabilities (Fig. 14C–E). Notably, the chelation of Gd3+ to Ce6 resulted in a 7-fold higher T1 relaxation time (r1) of 29.91 mM−1 s−1 relative to the clinically-used Magnevist Gd contrast agent. In 4T1 tumour-bearing mice, this impressive r1 translated to vivid contrast of the tumour 1–24 hours post injection. Meanwhile, IR825 and Ce6 enabled synergistic PDT and photothermal therapy that halted tumour growth (Fig. 14B).
| Modality | Number of reports | Nanoparticle | Photosensitizers |
|---|---|---|---|
| Abbreviations: HPPH: 3-devinyl-3-(1′-hexyloxyethyl)pyropheophorbide, PPh-a: pyropheophorbide-a, Ce6: chlorin e6, Yb-HP: Yb-2,4-dimethoxyhematoporphyrin, TSPP: meso-tetra(4-sulfonatophenyl) porphyrin dihydrochloride, TCPP: 5,10,15,20-tetrakis (4-carboxyphenyl) porphyrin. | |||
| PDT | |||
| MRI | 7 | Iron oxide, silica, dyad, micelle | Ce6, TSPP, Mn-porphyrin, free porphyrin, PPh-a |
| PET | 1 | Albumin | Hematoporphyrin |
| PAI | 1 | Calcium carbonate | Ce6 |
| Infrared luminescence | 1 | Metal nanocage | Yb-HP |
| Fluorescence | 1 | Iron oxide | PPh-a |
| Upconversion luminescence + MRI | 1 | Upconversion nanoparticle | Ce6 |
| Near infrared luminescence + MRI | 2 | Polymeric, dextran | Ce6 |
| Fluorescence + MRI | 6 | Iron oxide, upconversion nanoparticles, polymeric | Ce6, gadophrin-2 |
| Fluorescence + PET | 5 | Graphene oxide, lipoprotein-like, polymeric, micelles; HPPH, PPh-a-lipid, chlorophyll-1 | Ce6 |
| Fluorescence + PAI | 3 | Ceramide gold, micelles | Ce6, HPPH |
| Fluorescence + PAI + MRI | 1 | Albumin | Ce6 |
| PTT | |||
| PAI | 1 | Nanofiber | Purpurin |
| MRI | 3 | Porphysome, polymeric | PPh-a-lipid, Mn-porphyrin |
| Fluorescence | 2 | Porphysome | PPh-a-lipid |
| Fluorescence + PAI | 2 | Porphysome | PPh-a-lipid |
| Chemotherapy | |||
| Fluorescence | 1 | Polymeric micelle | Tetrakis-(4-aminophenyl)-terminated porphyrin |
| MRI | 3 | Polymeric, metal oxide, upconversion nanoparticle | Mn-porphyrin, Ce6 |
| PDT + PTT | |||
| Fluorescence + PAI | 1 | Carbon | Ce6 |
| Fluorescence + MRI + PET | 1 | Nanoporphyrin | Pyropheophorbide cholic acid telodendrimer |
| Fluorescence + MRI + PAI | 2 | Polymeric micelle | TCPP, IR825, Ce6 |
| Upconversion luminescence + CT + PAI + MRI | 1 | Upconversion | Ce6 |
| PDT + Chemo | |||
| MRI | 1 | Albumin | Ce6 |
 | ||
| Fig. 14 Example of an “all-in-one” nanoparticle with dual modal therapy and triple modal imaging due to the incorporation of many functional building blocks. (A) Schematic of IR825@C18PMH-PEG-Ce6-Gd. Gd3+ chelated chlorin e6 is conjugated to a PEG polymer that assembles into a nanomicelle, encapsulating the photothermal agent, IR825. (B) In vivo dual modal PDT and PTT efficacy. 4T1 mammary carcinoma-bearing mice were intravenously injected with either saline or IR825@C18PMH-PEG-Ce6-Gd. Twelve hours post injection, mice were administered PTT (808 nm laser; 0.3 W cm−2) and/or PDT (660 nm; 2 mW cm−2). The combination of PDT and PTT significantly reduced relative tumour volume, regardless of order (n = 5; n.s = not significant, * p < 0.05, ** p < 0.01). In vivo fluorescence (C), T1-weighted MR (D), and photoacoustic tomography (E) of 4T1 mammary carcinoma bearing mice at various times post intravenous injection of IR825@C18PMH-PEG-Ce6-Gd. Dashed circles and arrows indicate the tumour, and heart respectively. Copyright (2014) Wiley. Used with permission from Gong et al.33 | ||
This study exemplified one of the key advantages of an “all-in-one” approach: the ability to facilitate strong imaging contrast and synergistic therapy through readily-available constituents. Consequently, advances in material development associated with each individual field can be exploited. Additionally, all-in-one systems benefit from the use of established targeting ligands, and magnetic, acid or GSH-susceptible moieties for targeting particle delivery. However, the multi-constituent construction of these systems poses translational challenges. These “all-in-one” systems often require multistep syntheses, which can reduce yield and hinder scale-up. Furthermore, to enter the clinic, each new constituent requires pharmacokinetic, biodistribution, and toxicity evaluation.
This clinical translation challenge may be circumvented through “one-for-all” nanoparticles, which are assembled from one inherently multifunctional constituent. To create one such multifunctional agent, Lovell et al. acylated the glycerol backbone of lysophosphatidylcholine and pyropheophorbide-a in a 1:1 ratio to generate pyropheophorbide-a-lipid, which self-assembled into nanovesicles termed “porphysomes” (Fig. 15A).34 The high-density packing of porphyrin (∼80000 units per particle) within the porphysome bilayer induced fluorescence self-quenching that could be exploited for activatable fluorescence imaging upon passive tumour accumulation, as shown in Fig. 15B. Fluorescence quenching and the high particle extinction coefficient (2.9 × 109 M−1 cm−1) enabled non-radiative decay of porphyrin excitons for PAI of rat lymph nodes and photothermal therapy (PTT). Mice bearing KB subcutaneous xenografts exhibited complete tumour remission and 100% survival 28-days post porphysome and laser administration, while control mice reached humane endpoints within 21 days (Fig. 15C and D). This photothermal capability could be supplemented therapeutically via 90% loading efficiency of the chemotherapeutic agent doxorubicin into the porphysomes (Fig. 15E). Since this seminal proceeding, porphysomes have been radiolabelled with 64Cu at an efficiency of 99%, allowing for PET imaging of micrometastases <1–2 mm in size, and lymph node metastases in orthotopic rat prostate and rabbit head and neck cancers. Additionally, porphysomes can be functionalized with targeting ligands, as exemplified by folic acid-functionalized porphysomes for activatable PDT.35
 | ||
| Fig. 15 Example of a “one-for-all” nanoparticle with dual modal imaging and therapy capabilities.34 (A) Schematic representation of the porphysome, a nanovesicle composed of one inherently multifunctional building block, pyropheophorbide a-lipid. (B) Lymphatic mapping of rats via photoacoustic imaging (top), prior to and 15 minutes after intradermal injection of porphysomes into the left forepaw. Secondary lymph vessels, lymph nodes, and inflowing vessels are denoted in cyan, red, and yellow respectively. Scale bar represents 5 mm. In vivo activatable fluorescence imaging (bottom) of subcutaneous KB cervical tumour bearing mice administered 7.5 pmol of porphysomes, whereupon porphysomes are quenched 15 minutes post-intravenous injection. At 48 hours post intravenous injection, porphysomes are disrupted and fluorescence is restored. (C) In vivo efficacy of PTT mediated by porphysomes. Subcutaneous KB cervical tumour bearing mice were intravenously injected with porphysomes or PBS. 24 hours post injection, mice were irradiated (660 nm; 1.9 W cm−2; 60 s). (D) Survival of mice after PTT (n = 5). When tumours grew to 10 mm in size, mice were euthanised. (E) Active loading of porphysomes with the chemotherapeutic drug, doxorubicin, as confirmed by measuring fluorescence intensities of gel filtration fractions of 50 molar% cholesterol porphysomes in detergent. Porphysomes were incubated with 0.45 mg mL−1 of doxorubicin hydrochloride and 155 mM ammonium sulphate at pH 5.5 for 2 hours at 37 °C, and free doxorubicin was removed with gel filtration. Pyropheophorbide-a-lipid, and doxorubicin fluoresce blue, and green respectively. | ||
As a prime display of its multimodality in a supramolecular form, the coating of NaYBF4:Tm-NaYF4 UCNPs with porphyrin-phospholipid, and subsequent porphyrin 64Cu radiolabeling yielded an agent with hexamodal imaging capabilities: fluorescence, PAI, computed tomography, NIR upconversion luminescence, PET and Cerenkov luminescence.36 Particle accumulation within the first draining lymph node was clearly detectable across all modalities one hour post-injection of the PoP-UCNPs into the rear left footpad of mice.
Inspired by the porphysome, Li et al. developed another “one-for-all” porphyrin nanoparticle.37 They synthesized a multifunctional telodendrimer constituent, which comprised linear PEG conjugated to dendritic oligomers of pyropheophorbide-a and cholic acid (PEG-Por4-CA-4). Assembly of PEG-Por4-CA-4 into the nanoporphyrin platform resulted in trimodal NIR fluorescence, MR, and PET imaging, and PDT, PTT and doxorubicin triple therapy. To date, there are few “one-for-all” systems, due to the challenge of synthesizing an inherently multifunctional nanomaterial constituent.
Despite its infancy, the field of supramolecular porphyrin theranostics has demonstrated immense potential. To achieve this promise, modality selection within novel platforms should go beyond proof-of-concept demonstration of multifunctionality to reflect clinical needs. In general, the field of porphyrin nanotheranostics begs of answers to the following questions: (1) Which imaging modalities and therapies are optimal for diagnostics and to plan or guide treatment? (2) At what point does complementary imaging reach a point of diminishing returns? (3) Is combination therapy and imaging in one supramolecular system more efficacious than individual administrations of separate contrast and therapeutic agents? Additionally, theranostic building blocks that favour imaging modalities with existing infrastructure, low costs, and ease of access is desirable. With the pre-existing knowledge of the multimodality achievable from a single porphyrin theranostic system, efforts can now focus on the rational selection of functionalities to develop systems that become clinical mainstays.
5.2 Expanding the scope of porphyrin phototherapy
The ability of porphysomes to deliver PTT via the photoactivation of porphyrins represents a paradigm shift that expands the biomedical utility of porphyrin supramolecular structures; PTT does not require molecular oxygen to incite cytotoxicity, and thus, unlike PDT, can treat hypoxic tumours. This was exemplified by Jin et al.,38 who demonstrated the ability of porphysome-mediated PTT to successfully eradicate hypoxic subcutaneous KB mouse cervical cancer xenografts, which were otherwise unamenable to effective treatment by molecular porphyrin-PDT or porphysome-PDT. Porphyrin supramolecular structures can additionally facilitate porphyrin-mediated phototherapy of hypoxic tumours via the co-delivery of oxygen bubbles, or agents which stimulate tumour oxygenation. For example, metformin is known to decrease tumour oxygen consumption, thereby augmenting oxygenation status. As such, by co-delivering metformin and Ce6 within a single liposome, Song et al. improved oxygen saturation of subcutaneous mouse 4T1, CT26 and SCC-7 tumours relative to free metformin delivery, yielding stronger photodynamic inhibition of 4T1 tumour growth relative to metformin-free Ce6 liposomes-PDT or free metformin/Ce6-PDT combination therapy (Fig. 16).39 The above studies demonstrate how incorporation of porphyrin within supramolecular structures expands the scope of porphyrin phototherapy beyond its limited use in oxygenated tumour treatment, thereby addressing a major crux in PDT delivery. | ||
| Fig. 16 Synergistic drug delivery for PDT of oxygen-deficient tumours, as demonstrated by Song et al.39 Liposomes loaded with metformin and Ce6 respectively increase tumour oxygen saturation relative to free metformin in subcutaneous CT26 colon carcinoma and SCC-7 head and neck carcinoma mouse xenografts, and mediate more potent PDT (660 nm, 63 J cm−2) in subcutaneous 4T1 mammary carcinoma-bearing animals relative to controls. All treatment groups (n = 5) received laser irradiation and intravenous particle or drug administration. Copyright (2017) Springer International Publishing. Used with permission from Song et al.39 | ||
5.3 Synergistic therapies and porphyrin-mediated drug delivery
In addition to enabling multimodal theranostics and hypoxic tumour treatment, supramolecular porphyrin chemistry also facilitates synergistic cancer therapy via the co-loading of porphyrin and other therapeutic agents within a single platform. This relatively new arena of research may hold great promise in augmenting the photosensitization response of tumours, overcoming tumour drug resistance, and introducing new paradigm-changing drug-delivery strategies.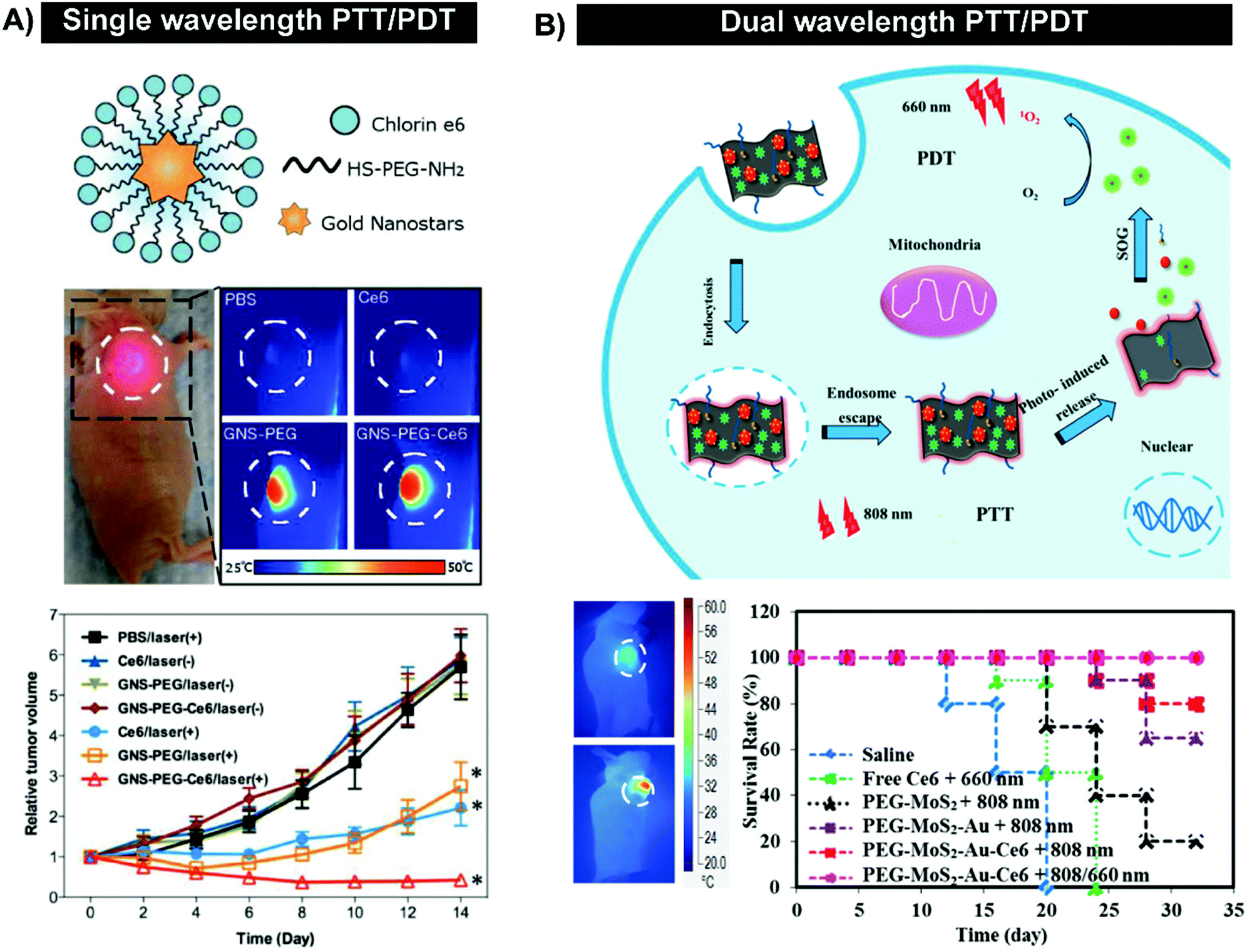 | ||
| Fig. 17 Synergistic PTT/PDT through single (A) or dual wavelength irradiation (B). Single wavelength PTT/PDT was performed by Wang et al.41 using gold nanostars decorated with Ce6. Irradiation of subcutaneous MDA-MB-435 melanomas generated temperatures of 50 °C at the tumour site (671 nm, 1 W cm−2) upon intravenous administration of Ce6-gold nanostars. This led to regression of tumours over 14 days (n = 4–6, p < 0.05). Copyright (2013) Wiley. Modified with permission from Wang et al.41 (B) Dual wavelength PTT/PDT operates with the principle that PTT facilitates particle uptake and endolysosomal release for greater concentration of photoactive porphyrins in tumours. Irradiation of subcutaneous 4T1 mammary carcinoma -bearing mice with 808 nm (1.5 W cm−2, 5 minutes) led to >50 °C tumour temperatures, unlike saline controls.42 When combined with subsequent 660 nm (1 W cm−2, 3 minutes) irradiation, a synergistic effect was observed on animal survival (n = 6). Reproduced from Liu et al.42 with permission from the Royal Society of Chemistry. | ||
Tian et al. were the first to demonstrate the utility of single-agent synergistic phototherapy in 2011.40 The authors loaded Ce6 onto graphene oxide nanoparticles, which could be irradiated at 808 nm (360 J cm−2) to generate mild heating (5–6 °C temperature increase) prior to chlorin photoactivation. This hyperthermia itself did not cause cellular toxicity, but when combined with subsequent PDT (660 nm, 15 J cm−2), led to near complete KB cell death and enhanced cytotoxicity relative to PDT-only cell treatment. Contrarily, hyperthermia/PDT treatment with dose-equivalent Ce6 or graphene oxide did not cause significant cell death, demonstrating the therapeutic advantage of supramolecular structures amenable to both hyperthermia and PDT. The observed synergistic phototherapy effects were credited to heat-induced enhancement of cellular permeability and thereby intracellular Ce6 particle delivery.
The utility of supramolecular structure-facilitated, heat-amplified PDT was further explored in vivo. Wang et al. treated MDA-MB-435 breast cancer cells with Ce6-decorated gold nanostars and applied PTT/PDT using a single wavelength to excite both the chlorin and the gold.41 This treatment paradigm resulted in higher cellular toxicity compared to free Ce6, undecorated gold nanostars or a mixture of free Ce6 and gold nanostars, thereby demonstrating supramolecular structure-dependent PDT/PTT. These results translated in vivo to gold-dependent generation of MD-MB-435 xenograft temperatures above 50 °C leading to photoablative effects, which combined with simultaneous Ce6 photosensitization, resulted in synergistic tumour regression (Fig. 17A).
This demonstration of synergistic PDT and PTT from a single porphyrin agent was followed by studies using single or dual wavelength irradiation. While the use of a single irradiation step ensures spatial co-localization of PTT and PDT, it also restricts the type of photothermal agents that can be co-irradiated with porphyrin: gold nanoparticles are predominantly applied, as their localized surface plasmon can be tuned to match the Q-band of porphyrin. Accordingly, most porphyrin PDT/PTT platforms explored thus far are activated by a two-step process where the photothermal agent is irradiated at a different wavelength of light than porphyrin. Dual-wavelength PDT/PTT facilitates the exploration of a more diverse arsenal of supramolecular structures (Table 10) and provides the feasibility of conducting PDT before or after PTT, giving rise to the following differential observations: (1) conducting PDT prior to PTT can elicit stronger synergistic effects via pre-sensitization of tissue to heat, and thermal protection of particles and PS, (2) heat generated by PTT can enable tissue and cell membrane permeabilization to enhance particle delivery and enable stronger subsequent PDT in deeper tissue, and (3) synergy is unaffected by the treatment order (ref. 33 and ref. 26–28 therein). The second school of thought is more widely accepted and has been advanced with the use of activatable phototherapy systems. For example, Liu et al. recently developed gold/molybdenum nanosheets that quenched the fluorescence and ROS-generating abilities of the adsorbed Ce6 (Fig. 17B).42 Photothermal activation of the nanosheets with an 808 nm laser released and unquenched the Ce6, which was subsequently activated by a 660 nm laser to generate ROS in 4T1 cells. When applied in vivo to subcutaneous 4T1 tumour-bearing mice, the PTT + PDT platform led to tumour regression over two weeks, while Ce6-PDT or gold/molybdenum nanosheet-PTT did not suppress tumour growth. This exemplifies the therapeutic potential of supramolecular porphyrin structures that deliver combination phototherapy. However, further investigations exploring the clearance of these predominantly metallic platforms are needed.
| Synergistic agent | Number of reports | Porphyrin | Nanoparticle | Cancer model |
|---|---|---|---|---|
| Abbreviations: PPh-a: pyropheophorbide-a, H2TPyP: 5,10,15,20-tetra(4-pyridyl)porphyrin, TAPP: 5,10,15,20-tetrakis-(4-aminophenyl)-porphyrin, Ce6: chlorin e6, HP: hematoporphyrin, TCPP: 5,10,15,20-tetrakis(carboxyphenyl) porphyrin, H4TBC: 5,10,15,20-tetra(benzoato) chlorin. | ||||
| Chemotherapy + PDT (co-encapsulation, general) | ||||
| Cisplatin | 1 | PPh-a | Cisplatin-Zn coordinate | Head and neck |
| Curcumin | 1 | H2TPyP | Curcumin/perylene matrix | Lung |
| Docetaxel | 1 | TAPP | Polymer | Cervical |
| Dox | 11 | Ce6, HP, TCPP, PPh-a | Polymer micelle, metal organic framework, CaCO3, graphene oxide, dendrimer, red blood cells, silica | Fibrocarcinoma, lung, breast, liver, ovarian |
| Oxaliplatin | 1 | Ce6 | Polymer micelle | Orthotopic breast |
| Paclitaxel | 3 | Ce6, PPh-a | Polymer, cyclodextran | Breast, colon |
| Chemotherapy + PDT (ROS-responsive nanoparticles/pro-drugs) | ||||
| Dox/lipid oxidation | 4 | Hexyloxyethy-PPh-a, PPh-a, Ce6 | Liposomes | Pancreatic, cervical, colon |
| Dox/disulfide bond cleavage | 2 | Ce6 | Mesoporous silica, polysaccharide | Pancreatic |
| Irinotecan/lipid oxidation | 1 | PPh-a | Liposomes | Pancreatic |
| Camptothecin pro-drug cleavage | 1 | Ce6 | Upconversion nanoparticle | Lung |
| PDT + Immunotherapy | ||||
| TBC-Hf | 1 | H4TBC | Metal–organic framework | Colon (primary and distant) |
| Imiquimod | 1 | Ce6 | Upconversion nanoparticle | |
| Oxaliplatin | 1 | Pph-a | Polymer | |
| PTT + PDT (dual wavelength) | ||||
| Gold | 4 | Ce6 | Gold (rod, sphere, nanogel, Pd/silica-coated, UCNP-conjugate, MoS2 hybrid) | Head and neck, cervical, breast, sarcoma |
| IR825 | 2 | Ce6, TCPP | Polymer, polymer micelle | Breast |
| MoS2 | 1 | Ce6 | MoS2 nanosheet | Breast |
| Conjugated polymer | 2 | Ce6 | Polymer | Breast, liver |
| Graphene oxide | 2 | Ce6, sinoporphyrin sodium | Graphene oxide | Breast, lung |
| CuS | 1 | Ce6 | Liposome | Cervical |
| Iron oxide | 1 | TCPP | Iron oxide (carbon shell) | Breast |
| Carbon | 1 | Ce6 | Single-walled carbon nanotube | Head and neck |
| PTT + PDT (single wavelength) | ||||
| Gold | 4 | Ce6 | Gold (hollow spheres, nanostars, graphene-coated) | Breast, cervical, skin |
| Pd | 1 | Ce6 | Pd nanosheets | Sarcoma |
| PTT + chemotherapy + PDT | ||||
| Doxorubicin/carbon | 1 | Ce6 | Carbon dots | Uterine |
| Doxorubicin/gold | 1 | Ce6 | Gold nanospheres | Liver |
| Radiotherapy + PDT | ||||
| Hafnium | 1 | TCPP | Metal–organic framework | Breast |
Overcoming drug resistance. Down-regulation of cellular pro-apoptotic pathways and upregulation of survival pathways and drug efflux pumps are mechanisms by which tumour cells develop drug resistance. To this end, co-administration of PDT and chemotherapy via porphyrin/drug supramolecular structures is favoured for drug resistance reversal through (1) the delivery of tumouricidal effects through two mechanisms (for example ROS generation and DNA intercalation), (2) PDT-enabled efflux pump damage, (3) concealment of drugs from efflux pumps via nanoparticle encapsulation, (4) application of nanoparticle passive and active targeting strategies for site-specific enhanced chemotherapy delivery, and (5) PDT-mediated cellular lipid peroxidation causing endolysosomal breakdown and drug escape, allowing drugs to more effectively reach their target sites in the nucleus.43–45 These features promise strong synergistic treatment effects between PDT and chemotherapy, as shown by Park et al. The authors observed drastic reductions in tumour burden in UV-2237 subcutaneous fibrosarcoma-bearing mice, which were otherwise resistant to doxorubicin or PDT treatment (Fig. 18A).44 Such synergy has since been established using diverse porphyrin nanoparticles (Table 10), and amplified with activatable PDT and nanoparticle targeting strategies.43,45
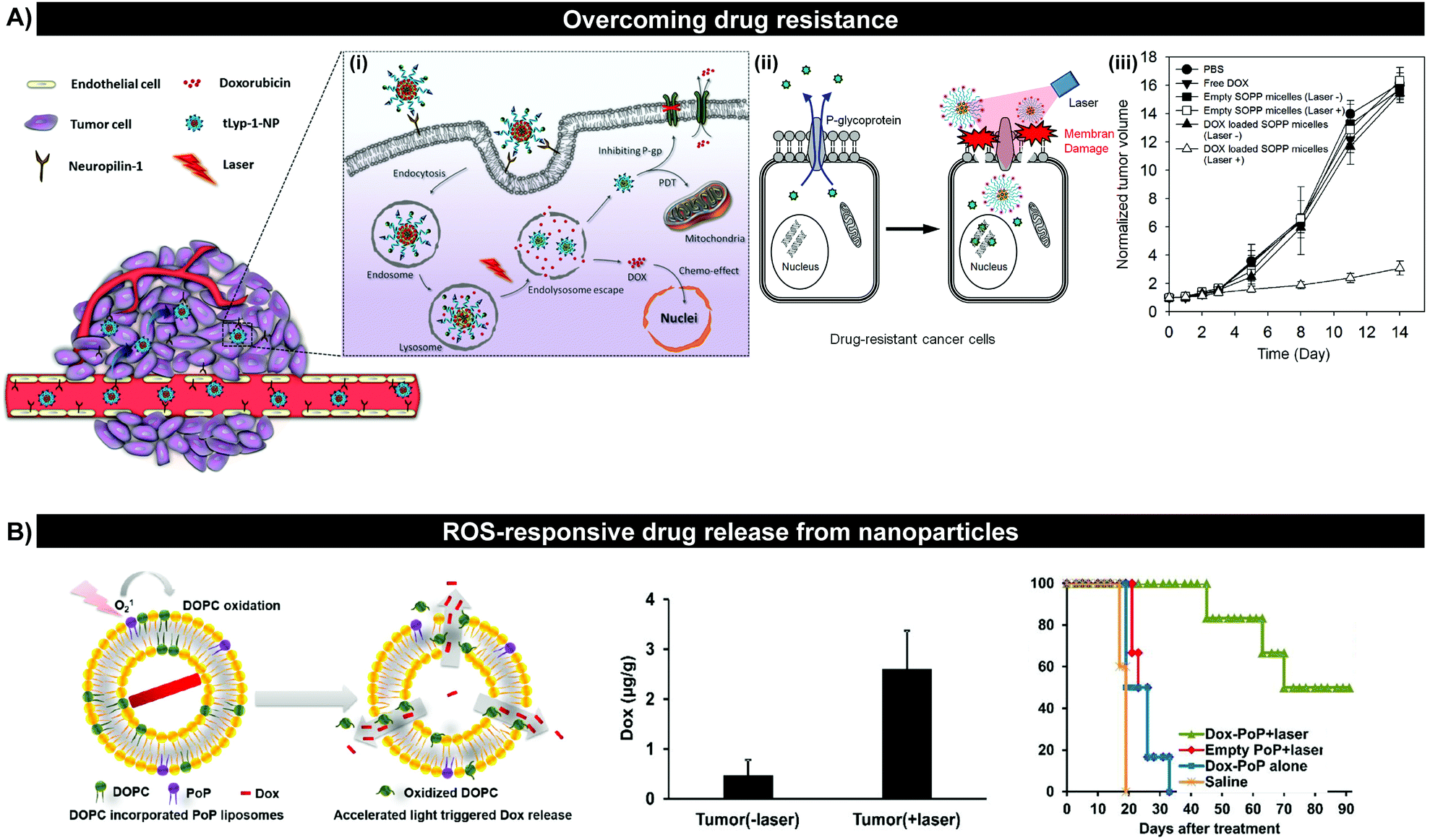 | ||
| Fig. 18 PDT/chemotherapy combination therapy can overcome drug resistance via photochemical internalization of drugs (A), and facilitate controlled release of drugs from ROS-responsive nanoparticles (B). (A) Park et al.44 demonstrated PDT/chemotherapy drastically delayed drug-resistance in colorectal adenocarcinoma HCT-8 mouse xenografts following the intravenous administration of micelles loaded with doxorubicin and Ce6 and PDT (670 nm, 100 J cm−2, n = 4). Reproduced from Jiang et al.45 with permission from the Royal Society of Chemistry and with permission from Park et al.24 Copyright (2014) Elsevier B.V. (B) Liposomal formulation of porphyrin–lipid and doxorubicin facilitated light-triggered doxorubicin release and enhanced tumour accumulation following laser irradiation (250 mW cm−2) for 40 minutes in subcutaneous MIA PaCa-2 pancreatic carcinoma xenograft-bearing mice (n = 4).46 This translated into ∼3–4 fold extended survival of mice compared to controls. Copyright (2013) Wiley. Modified with permission from Luo et al.46 | ||
ROS-mediated controlled chemotherapy delivery. The slow and uncontrolled release of encapsulated drugs limits the efficacy of many nano drug delivery systems. PDT-triggered release of drugs from porphyrin nanoparticles can address these limitations: PDT generates 1O2 which reacts with ROS-sensitive nanoparticles or pro-drugs, facilitated by the spatial confinement of porphyrin, drug and labile nanoparticle moieties. The resulting de-stabilized nanoparticles release encapsulated drug at tumour sites, allowing for synergistic chemotherapy/PDT. Examples of ROS-labile groups explored in this regard include lipids and disulfide bonds (Table 10). Particularly noteworthy are porphyrin-phospholipid (PoP) liposomes recently developed by Luo et al. (Fig. 18B, ref. 46 and ref. 26, 27, and 33 therein). Laser irradiation of PoPs caused oxidation of associated lipid and cholesterol constituents, leading to transient permeabilization of the liposome bilayer and complete release of the otherwise stably encapsulated doxorubicin into solution. This translated in vivo to a 3-7-fold increase in drug accumulation at tumour sites, and ∼3-4 fold longer median survival (80.5 days) compared to controls following PDT and PoP administration to Mca P1 xenograft -bearing mice. This efficacy, combined with higher and faster drug delivery may improve the therapeutic index of chemotherapeutics. Light as an external trigger for drug release may facilitate more spatio-temporal control over drug release than systems triggered by ubiquitous endogenous stimuli, and may be favoured over heat-stimulated drug release systems for the treatment of tumours within heat-sensitive tissue. However, this strategy is also restricted to the treatment of superficial and readily accessible tumours due to the limited 1–2 cm penetration of NIR light through tissue.
These concepts were illustrated by Xu et al., who assessed the efficacy of PDT administered via supramolecular PS (UCNP-Ce-6-R837) in combination with the clinically-approved immune checkpoint blockade inhibitor αCTLA-4, a cytotoxic T-lymphocyte-associated protein 4 antibody (Fig. 19).48 This immunostimulant was delivered in conjunction with UCNPs loaded with Ce6, and imiquimod (R837), an adjuvant that stimulates dendritic cells. The incorporation of R837 into the nanoparticle facilitated localization of R837 to the tumour via the EPR effect, and protected against systemic immune reactions, and off-target toxicities. Impressively, PDT + UCNP-Ce-6-R837 + αCTLA-4 eliminated primary murine CD26 colorectal subcutaneous tumours, resulting in tumour remission for 50 days, significantly longer than all controls. This synergistic immunotherapy/PDT was mirrored in distant tumours that did not receive laser irradiation, indicative of a robust abscopal effect. Upon immune re-challenging of the mice with a second inoculation of tumour cells, the PDT + UCNP-Ce6-R837 + αCTLA-4 treatment group remained tumour-free and had an impressive 100% survival over 93 days. This finding suggests that memory T cells were activated and induced tumour cell death. Taken together, immunotherapy in combination with PDT can induce the abscopal effect and immune memory. As such, this emerging combination therapy demonstrates potential for distant metastases to be targeted and tumour recurrences to be prevented, thereby warranting further investigation.
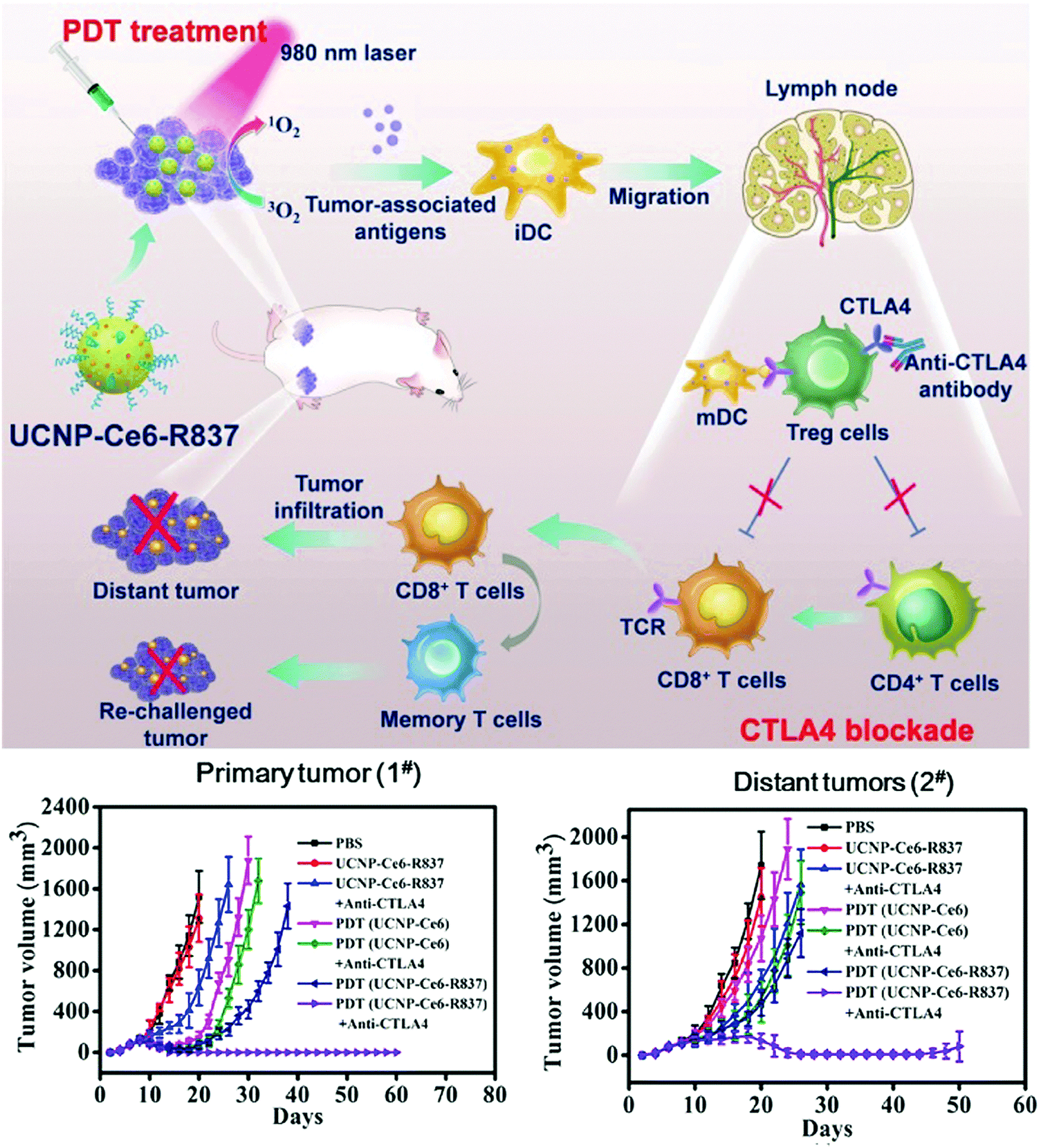 | ||
| Fig. 19 Demonstration of PDT/immunotherapy synergy by Xu et al.48 UCNPs loaded with Ce6, Toll-like-receptor-7 agonist, R837, and checkpoint blockade inhibitor, ∝CTLA-4, were administered to dual, subcutaneous CT26 colorectal tumour-bearing mice, which was followed by irradiation of only one tumour with light (980 nm, 0.5 W cm−2, 30 minutes). Tumour regression was observed at the primary irradiated tumour, and the non-irradiated tumour. Reprinted with permission from Xu et al.48 Copyright (2017) American Chemical Society. | ||
5.4 Key learning points, challenges and future directions
• Incorporation of monomeric porphyrins into supramolecular structures expands their theranostic scope; it allows their intrinsic multimodal imaging capabilities to be simultaneously used, while also expanding the repertoire of imaging and therapeutic modalities porphyrin agents can engage, including ultrasound imaging and PTT. This theranostic value particularly lies in the delivery of image-guided phototherapy.• Most theranostic porphyrin systems are designed to be “all-in-one”, where individual constituents with different functions are combined into one system. While successful in preclinical animal models, advancing these “all-in-one” systems into the clinic may be challenging. On the otherhand, the “one-for-all” design involves one building block that is inherently multifunctional. As such, regulatory approval may be easier to obtain.
• Supramolecular structures allow for co-assembly of porphyrins with drug, photothermal agents, oligonucleotides and immune modulators, enabling synergistic PDT/chemotherapy, PDT/PTT, PDT/gene and PDT/immunotherapy respectively. This synergy stems in part from enhanced, nanoparticle-enabled controlled drug delivery through PDT-mediated photochemical internalization.
• Supramolecular porphyrin-mediated synergistic PDT represents a novel field of study with great potential in overcoming limitations of individual therapies. However, given the depth limitations of PDT, evaluation of synergistic therapies should be directed towards lesions amenable to light irradiation in clinically-relevant, orthotopic tumour models.
6. Summary, challenges and future promise
Supramolecular porphyrin chemistry is an emerging field poised to address limitations that impede the wider applications of porphyrins for biomedical imaging and therapy. Namely, supramolecular formulations of porphyrin can change PS photophysical and biological properties, yielding increased PS solubility, circulation half-life, tumour deposition, fluorescence quantum yields and phototherapeutic potential relative to their molecular counterparts. Initially pursued in medicine as a means to alter PS pharmacokinetics, supramolecular porphyrin chemistry has flourished and now encompasses new theranostic and phototherapy paradigms. The translational potential of theranostic phototherapy will depend on addressing existing limitations of supramolecular porphyrin platforms, while expanding upon exciting advances.6.1 Overarching limitations of nanoparticles
The ubiquitous inclusion of targeting ligands and endogenous stimuli (for example GSH and pH)-responsive moieties within all classes of porphyrin supramolecular structures facilitates activatable PDT, which may protect against non-specific porphyrin release and cell uptake, and reduce off-target phototoxicity. However, these targeting strategies still rely on passive particle extravasation into tumour interstitium via the EPR effect, which as discussed, is an inefficient and often clinically-irrelevant strategy. Paired with the propensity of nanoparticles to be uptaken primarily by the MPS, this limits the value of using supramolecular porphyrin structures to selectively increase PS tumour accumulation and subsequent photochemical tumour killing; one of the key rationales of exploring supramolecular porphyrin. The sequestration of nanoparticles by MPS organs is of further concern when using non-biodegradable building blocks, such as gold, lanthanide crystals and metal–organic frameworks, which otherwise have demonstrated immense potential in facilitating potent photothermal synergy and NIR activation of porphyrin-PDT. Though an array of UCNP and other NIR-activatable structures have been designed to increase tissue depths accessible by PDT, few studies have clearly demonstrated this capacity.6.2 Future directions
These limitations can be addressed by ongoing developments within the fields of supramolecular porphyrin chemistry and nanomedicine at large. Strategies that employ external stimuli to guide nanoparticles to tumour sites and subsequently trigger their extravasation are particularly interesting future avenues for EPR-independent tumour homing. For example, magnetic guidance, the use of vascular targets, and stimulation of hyperthermia can facilitate particle localization and permeation through tumour extracellular matrices. To this end, an ultrasound-mediated micro-to-nano conversion of porphyrin microbubbles can capitalize upon the sonosensitivity of porphyrins, and the recent impressive advances made in microbubble-enhanced, MRI-guided focused ultrasound in transiently, focally and safely permeabilizing vasculature to facilitate EPR-independent porphyrin delivery and deep tissue activation. Synergistic PDT also represents an exciting new field in supramolecular porphyrin chemistry that can broaden the scope of porphyrin-treatable tumours to include hypoxic tumours, deeper lesions and metastases. Often, this utility resides in the use of metal particles to incite hyperthermia or photoablation. Thus, new strategies that prevent uptake of nanoparticles by MPS cells may also advance the utility of supramolecular porphyrin phototherapy. Alternatively, potent and efficient NIR activation of photothermal effects may be induced with J-aggregation or porphyrins within organic supramolecular platforms, including chlorosome mimetics. In general, supramolecular porphyrin theranostics have been largely unexplored for applications beyond cancer phototherapy, such as image-guided resection, and particularly anti-microbial therapy, which may benefit from synergistic therapy to overcome multidrug resistance for the treatment of localized infections.6.3 Translational potential
Extensive work remains to be conducted prior to realizing the clinical utility of supramolecular porphyrin platforms. Firstly, the use of more clinically-relevant orthotopic and genetic preclinical animal models must become mainstay once proof-of-principle evaluations of particle efficacy have been established. Due to ease of access, there exists a heavy reliance on subcutaneous tumour models. However, these models often do not recapitulate physiologically- and clinically-relevant tumours, and may engender a false sense of optimism regarding treatment efficacy and imaging utility of the supramolecular platform under investigation. Therefore, clinical applications and particle designs within each study should be better rationalized to ensure that unmet clinical needs are being realistically addressed. As skin phototoxicity remains a fundamental barrier in clinical PDT, the field of supramolecular porphyrin chemistry would benefit from more thorough toxicity evaluations. Lastly, as with the field of nanomedicine at large, the exploration of novel porphyrin supramolecular platforms requires more robust experimental design, better controls for clear evaluation against conventional molecular porphyrin use, and standardization against clinical gold standards.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Christina MacLaughlin for her technical expertise. They also thank their funding sources: Vanier Canada Graduate Scholarship, Mr Robert and Ms Francine Ruggles Innovation Award, Government of Alberta Sir James Lougheed Award, Canadian Institutes of Health Research, Natural Sciences and Engineering Research Council of Canada, Terry Fox Research Institute, Canadian Cancer Society Research Institute, Canada Foundation for Innovation, Princess Margaret Cancer Foundation, Prostate Cancer Canada, and the Joey and Toby Tanenbaum/Brazillian Ball Chair in Prostate Cancer Research.References
- F. Bryden and R. W. Boyle, Adv. Inorg. Chem., 2016, 68, 141–221 CrossRef CAS.
- A. M. Richter, E. Waterfield, A. K. Jain, A. J. Canaan, B. A. Allison and J. G. Levy, Photochem. Photobiol., 1993, 57, 1000–1006 CrossRef CAS PubMed.
- R. Kumar, T. Y. Ohulchanskyy, I. Roy, S. K. Gupta, C. Borek, M. E. Thompson and P. N. Prasad, ACS Appl. Mater. Interfaces, 2009, 1, 1474–1481 CAS.
- R. Ramachandran, G. L. Malarvizhi, P. Chandran, N. Gupta, D. Menon, D. Panikar, S. Nair and M. Koyakutty, J. Biomed. Nanotechnol., 2014, 10, 1401–1415 CrossRef CAS PubMed.
- M. Karimi, A. Ghasemi, P. Sahandi Zangabad, R. Rahighi, S. M. Moosavi Basri, H. Mirshekari, M. Amiri, Z. Shafaei Pishabad, A. Aslani, M. Bozorgomid, D. Ghosh, A. Beyzavi, A. Vaseghi, A. R. Aref, L. Haghani, S. Bahrami and M. R. Hamblin, Chem. Soc. Rev., 2016, 45, 1457–1501 RSC.
- W. D. Jang, N. Nishiyama, G. D. Zhang, A. Harada, D. L. Jiang, S. Kawauchi, Y. Morimoto, M. Kikuchi, H. Koyama, T. Aida and K. Kataoka, Angew. Chem., Int. Ed., 2005, 44, 419–423 CrossRef CAS PubMed.
- I. Roy, T. Y. Ohulchanskyy, H. E. Pudavar, E. J. Bergey, A. R. Oseroff, J. Morgan, T. J. Dougherty and P. N. Prasad, J. Am. Chem. Soc., 2003, 125, 7860–7865 CrossRef CAS PubMed.
- K. Záruba, J. Králová, P. Øezanka, P. Pouèková, L. Veverková and V. Král, Org. Biomol. Chem., 2010, 8, 3202–3206 Search PubMed.
- K. Lu, C. He and W. Lin, J. Am. Chem. Soc., 2014, 136, 16712–16715 CrossRef CAS PubMed.
- S. S. Lucky, K. C. Soo and Y. Zhang, Chem. Rev., 2015, 115, 1990–2042 CrossRef CAS PubMed.
- J. Napp, T. Behnke, L. Fischer, C. Würth, M. Wottawa, D. M. Katschinski, F. Alves, U. Resch-Genger and M. Schäferling, Anal. Chem., 2011, 83, 9039–9046 CrossRef CAS PubMed.
- Y. A. Shieh, S. J. Yang, M. F. Wei and M. J. Shieh, ACS Nano, 2010, 4, 1433–1442 CrossRef CAS PubMed.
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS PubMed.
- S. Y. Park, H. J. Baik, Y. T. Oh, K. T. Oh, Y. S. Youn and E. S. Lee, Angew. Chem., Int. Ed., 2011, 50, 1644–1647 CrossRef CAS PubMed.
- Y. Sun, Z. L. Chen, X. X. Yang, P. Huang, X. P. Zhou and X. X. Du, Nanotechnology, 2009, 20, 135102 CrossRef PubMed.
- K. R. Byrnes, R. W. Waynant, I. K. Ilev, X. Wu, L. Barna, K. Smith, R. Heckert, H. Gerst and J. J. Anders, Lasers Surg. Med., 2005, 36, 171–185 CrossRef PubMed.
- S. Stolik, J. A. Delgado, A. Pérez and L. Anasagasti, J. Photochem. Photobiol., B, 2000, 57, 90–93 CrossRef CAS.
- P. P. Ghoroghchian, F. P. R. K. Susumu, D. Blessington, A. K. Brannan, F. S. Bates, B. Chance, D. A. Hammer and M. J. Therien, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2922–2927 CrossRef CAS PubMed.
- K. K. Ng, M. Takada, K. Harmatys, J. Chen and G. Zheng, ACS Nano, 2016, 10, 4092–4101 CrossRef CAS PubMed.
- M. Shakiba, K. K. Ng, E. Huynh, H. Chan, D. M. Charron, J. Chen, N. Muhanna, F. S. Foster, B. C. Wilson and G. Zheng, Nanoscale, 2016, 8, 12618–12625 RSC.
- Y. Shen, A. J. Shuhendler, D. Ye, J. J. Xu and H. Y. Chen, Chem. Soc. Rev., 2016, 45, 6725–6741 RSC.
- N. M. Idris, M. K. Jayakumar, A. Bansal and Y. Zhang, Chem. Soc. Rev., 2015, 44, 1449–1478 RSC.
- S. Kim, T. Y. Ohulchanskyy, H. E. Pudavar, R. K. Pandey and P. N. Prasad, J. Am. Chem. Soc., 2007, 129, 2669–2675 CrossRef CAS PubMed.
- C. Wang, H. Tao, L. Cheng and Z. Liu, Biomaterials, 2011, 32, 6145–6154 CrossRef CAS PubMed.
- F. Ai, Q. Ju, X. Zhang, X. Chen, F. Wang and G. Zhu, Sci. Rep., 2015, 5, 10785 CrossRef CAS PubMed.
- A. Kamkaew, F. Chen, Y. Zhan, R. L. Majewski and W. Cai, ACS Nano, 2016, 10, 3918–3935 CrossRef CAS PubMed.
- M. H. Chen, Y. J. Jenh, S. K. Wu, Y. S. Chen, N. Hanagata and F. H. Lin, Nanoscale Res. Lett., 2017, 12, 62 CrossRef PubMed.
- X. Qian, Y. Zheng and Y. Chen, Adv. Mater., 2016, 28, 8097–8129 CrossRef CAS PubMed.
- P. Huang, X. Qian, Y. Chen, L. Yu, H. Lin, L. Wang, Y. Zhu and J. Shi, J. Am. Chem. Soc., 2017, 139, 1275–1284 CrossRef CAS PubMed.
- E. Huynh, B. Y. Leung, B. L. Helfield, M. Shakiba, J. A. Gandier, C. S. Jin, E. R. Master, B. C. Wilson, D. E. Goertz and G. Zheng, Nat. Nanotechnol., 2015, 10, 325–332 CrossRef CAS PubMed.
- F. Fan, Y. Yu, F. Zhong, M. Gao, T. Sun, J. Liu, H. Zhang, H. Qian, W. Tao and X. Yang, Theranostics, 2017, 7, 1290–1302 CrossRef PubMed.
- Y. Zhou, X. Liang and Z. Dai, Nanoscale, 2016, 8, 12394–12405 RSC.
- H. Gong, Z. Dong, Y. Liu, S. Yin, L. Cheng, W. Xi, J. Xiang, K. Liu, Y. Li and Z. Liu, Adv. Funct. Mater., 2014, 24, 6492–6502 CrossRef CAS.
- J. F. Lovell, C. S. Jin, E. Huynh, H. Jin, C. Kim, J. L. Rubinstein, W. C. Chan, W. Cao, L. V. Wang and G. Zheng, Nat. Mater., 2011, 10, 324–332 CrossRef CAS PubMed.
- M. S. Valic and G. Zheng, Curr. Opin. Chem. Biol., 2016, 33, 126–134 CrossRef CAS PubMed.
- J. Rieffel, F. Chen, J. Kim, G. Chen, W. Shao, S. Shao, U. Chitgupi, R. Hernandez, S. A. Graves, R. J. Nickles, P. N. Prasad, C. Kim, W. Cai and J. F. Lovell, Adv. Mater., 2015, 27, 1785–1790 CrossRef CAS PubMed.
- Y. Li, T. Y. Lin, Y. Luo, Q. Liu, W. Xiao, W. Guo, D. Lac, H. Zhang, C. Feng, S. Wachsmann-Hogiu, J. H. Walton, S. R. Cherry, D. J. Rowland, D. Kukis, C. Pan and K. S. Lam, Nat. Commun., 2014, 5, 4712 CrossRef CAS PubMed.
- C. S. Jin, J. F. Lovell, J. Chen and G. Zheng, ACS Nano, 2013, 7, 2541–2550 CrossRef CAS PubMed.
- X. Song, L. Feng, C. Liang, M. Gao, G. Song and Z. Liu, Nano Res., 2017, 10, 1200–1212 CrossRef CAS.
- B. Tian, C. Wang, S. Zhang, L. Feng and Z. Liu, ACS Nano, 2011, 5, 7000–7009 CrossRef CAS PubMed.
- S. Wang, P. Huang, L. Nie, R. Xing, D. Liu, Z. Wang, J. Lin, S. Chen, G. Niu, G. Lu and X. Chen, Adv. Mater., 2013, 25, 3055–3061 CrossRef CAS PubMed.
- L. Liu, J. Wang, X. Tan, X. Pang, Q. You, Q. Sun, F. Tan and N. Li, J. Mater. Chem. B, 2017, 5, 2286–2296 RSC.
- C. He, D. Liu and W. Lin, ACS Nano, 2015, 9, 991–1003 CrossRef CAS PubMed.
- H. Park, W. Park and K. Na, Biomaterials, 2014, 35, 7963–7969 CrossRef CAS PubMed.
- D. Jiang, X. Gao, T. Kang, X. Feng, J. Yao, M. Yang, Y. Jing, Q. Zhu, J. Feng and J. Chen, Nanoscale, 2016, 8, 3100–3118 RSC.
- D. Luo, L. Nasi, K. A. Carter, C. Lin, J. Geng, S. Shao, W. C. Huang, Y. Qin, G. E. Atilla-Gokcumen and J. F. Lovell, Small, 2016, 12, 3039–3047 CrossRef CAS PubMed.
- X. Wang, K. Liu, G. Yang, L. Cheng, L. He, Y. Liu, Y. Li, L. Guo and Z. Liu, Nanoscale, 2014, 6, 9198–9205 RSC.
- J. Xu, L. Xu, C. Wang, R. Yang, Q. Zhuang, X. Han, Z. Dong, W. Zhu, R. Peng and Z. Liu, ACS Nano, 2017, 11, 4463–4474 CrossRef CAS PubMed.
- A. Srivatsan, J. R. Missert, S. K. Upadhyay and R. K. Pandey, J. Porphyrins phthalocyanines, 2015, 19, 109–134 CrossRef CAS.
- D. Luo, K. A. Carter, D. Miranda and J. F. Lovell, Adv. Sci., 2017, 4, 1600106 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2017 |