
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoprotection: extending lessons learned from studying natural sunscreens to the design of artificial sunscreen constituents
Lewis A.
Baker
a,
Barbara
Marchetti
b,
Tolga N. V.
Karsili
*c,
Vasilios G.
Stavros
*a and
Michael N. R.
Ashfold
 *d
*d
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: v.stavros@warwick.ac.uk
bDepartment of Chemistry, University of Pennsylvania, Philadelphia, USA
cDepartment of Chemistry, Temple University, Philadelphia, USA. E-mail: tug96217@temple.edu
dSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: mike.ashfold@bristol.ac.uk
First published on 5th June 2017
Abstract
Evolution has ensured that plants and animals have developed effective protection mechanisms against the potentially harmful effects of incident ultraviolet radiation (UVR). Tanning is one such mechanism in humans, but tanning only occurs post-exposure to UVR. Hence, there is ever growing use of commercial sunscreens to pre-empt overexposure to UVR. Key requirements for any chemical filter molecule used in such a photoprotective capacity include a large absorption cross-section in the UV-A and UV-B spectral regions and the availability of one or more mechanisms whereby the absorbed photon energy can be dissipated without loss of the molecular integrity of the chemical filter. Here we summarise recent experimental (mostly ultrafast pump–probe spectroscopy studies) and computational progress towards unravelling various excited state decay mechanisms that afford the necessary photostability in chemical filters found in nature and those used in commercial sunscreens. We also outline ways in which a better understanding of the photophysics and photochemistry of sunscreen molecules selected by nature could aid the design of new and improved commercial sunscreen formulations.
 Lewis A. Baker | Lewis Baker studied Physics at the University of Warwick, obtaining an MPhys in 2013. He went on to study Mathematical Biology and Biophysical Chemistry at the University of Warwick Molecular Organisation and Assembly in Cells Doctoral Training Centre, obtaining an MSc in 2014 and proceeding to a PhD with Drs Vasilios Stavros and Scott Habershon. His thesis focusses on understanding biological photoprotection through transient absorption spectroscopy and quantum dynamic simulations. |
 Barbara Marchetti | Barbara Marchetti obtained her undergraduate Bachelor and Masters degrees at the Università degli Studi di Perugia, Italy in 2010 and 2012, respectively. She completed her PhD degree at the University of Bristol in 2016 under the supervision of Prof. Michael Ashfold and will commence a Marie Skłodowska Curie international Post-Doctoral Research Fellowship in the group of Prof. Marsha Lester at the University of Pennsylvania, USA in the spring of 2017. Her research interests comprise the photochemistry and photophysics of biologically, catalytically and environmentally relevant chromophores. |
 Tolga N. V. Karsili | Tolga Karsili obtained his MSci degree from the University of Birmingham in 2010 and his PhD in 2014 from the University of Bristol, where he worked in the group of Prof. Michael Ashfold. He then undertook post-doctoral research in the Computational and Theoretical Chemistry group at the Technical University of Munich, Germany (supported in part by the award of a T. U. Munich Fellowship), and is now a research fellow at Temple University, USA. His research interests involve the photodynamics of organic and biological chromophores, supramolecular photochemistry and electron-induced reactions – in both the gas phase and in bulk solution. |
 Vasilios G. Stavros | Vasilios Stavros is a Reader at the University of Warwick. He completed his PhD in 1999 at King's College London, working in the group of Prof. Helen Fielding and remained at King's for a further three years as an EPSRC postdoctoral research fellow. In 2002, he undertook a postdoctoral position at the University of California Berkeley, working for Prof. Stephen Leone. He returned to the UK in 2005 as a Royal Society University Research Fellow. His research is centred on understanding photoprotection and photoactivation mechanisms in biologically related molecules using both gas- and solution-phase pump–probe spectroscopies. |
 Michael N. R. Ashfold | Mike Ashfold obtained his PhD degree in 1978 at the University of Birmingham in the group of Prof. John Simons. After postdoctoral research at the University of Oxford, he was appointed as a Lecturer at the University of Bristol, where he was promoted to a Chair in Physical Chemistry in 1992. He was elected to the Fellowship of the Royal Society in 2009. His research interests include molecular photophysics, spectroscopy, and plasma diagnosis (particularly in the context of diamond chemical vapour deposition). |
1. Introduction
The causal link between excessive sun exposure and the development of skin erythema and tanning has been known for millennia, even if the reason for this link has only begun to be understood much more recently. Ancient civilisations, such as the Egyptians and the Greeks, took steps to protect themselves with varying success; rice bran, jasmine and olive oil were applied to the skin, often for cosmetic reasons, but these formulations also provided some photoprotection and are thus amongst the earliest recorded sunscreens.1–3 Other landmark discoveries in this field include the identification of ultraviolet (UV) radiation (UVR) by Ritter (in 1801) and the experiments of Widmark in 1889 proving that UVR caused erythema solare (sunburn); suggestions to use chemical sunscreens in order to protect the skin were already gaining traction by the end of the 19th century.4–6One of the first successful commercial sunscreen products was developed by Schueller (in 1935), who went on to found the company now known as L'Oreal. The sunscreen was called ‘Ambre Solaire’, an oily substance containing the UV absorber benzylsalicylate. In subsequent years many other potential compounds were investigated spectroscopically, leading to the widespread adoption of para-aminobenzoic acid4,6–10 and, later, benzophenones.11 By now, many dozens of UV absorbing molecules find regular use in commercial products,12–14 which fuel a global industry, and international regulations control both the identities and the concentrations of specific UV absorbing molecules approved for use in such products. These regulations have developed, in part, as a result of concerns that some of the sunscreen components might have an adverse effect on the human skin or physiology. These concerns, often referred to as the ‘sunscreen controversy’,12,15 have caused a surge of research focussed on gaining a deeper understanding of how sunscreen constituents operate and provide photoprotection. Through understanding such photoprotective mechanisms, we might hope to learn lessons about the properties of what constitutes a good sunscreen molecule and, ultimately, apply this knowledge to the design of new or improved compounds.
The remainder of this Introduction overviews some of the ways photoprotection is achieved in nature and then outlines ways in which we can alleviate the deleterious effects of UVR. This is followed by a further four sections. The first is a short photochemistry primer, wherein we introduce photoexcited states and some of the many possible decay mechanisms for excited state molecules, then focus on mechanisms that enable (ultrafast) population transfer back to the starting ground state configuration – a key requirement for a chemical filter suitable for use in a commercial sunscreen. We then survey some of the contemporary experimental and theoretical methods by which the early time dynamics of photoexcited molecules can be probed and understood. This is followed by a description of several recent case studies of the ultrafast decay of natural and commercial molecular sunscreen components. Finally, we outline various experimental and theoretical prospects that can be expected to enhance this currently rather small but fast growing field of study.
Natural photoprotection
The UV wavelengths are the most energetic part of the solar spectrum reaching the surface of the Earth. These are usually sub-divided into three regions: UV-A (spanning the wavelength range 400–315 nm), UV-B (315–280 nm) and UV-C (280–100 nm). UV-C radiation is now almost completely absorbed and scattered by our ozone-rich atmosphere, but it has not always been so. In the early Archean period of the Earth's history (some 4 billion years ago), the atmosphere was dominated by carbon dioxide and nitrogen. The concentration of oxygen was very low.18 Thus there was nothing to attenuate the higher energy (UV-C) components within the solar spectrum prior to their reaching the Earth's surface. Early organisms (e.g. phototrophs) developing in these harsh conditions thus had two choices. One was to migrate away from the surface. The other was to develop protection mechanisms whereby damage from the short wavelength components within the incident solar radiation was minimised, whilst the longer wavelength components required for photosynthesis could still be harnessed. Similar selective pressures may well have led to many of the DNA-repair mechanisms observed in present-day organisms.19,20 This is a classic example of the so-called burden of disease,21 where solar radiation is required for photosynthesis, but can also lead to damage. Thus some equilibrium needs to be struck.A similar balance with regard to UVR exposure continues to exist across the Earth's biosphere – as illustrated by the burden of disease curve (for humans and plants) shown in Fig. 1. We start by considering the effects of UVR exposure and physiological responses in humans. More than 90% of the body's vitamin D requirements are satisfied by the UV-B mediated conversion of 7-dehydrocholesterol to previtamin D3, a precursor of vitamin D.22 Underexposure to UV-B can thus lead to a deficiency in vitamin D, which can lead to the development of skeletal disease and osteoporosis.22,23 Conversely, overexposure to UVR is the primary cause of skin cancers, e.g. basal and squamous cell carcinomas, and malignant melanomas. The latter are estimated to be responsible for some 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 fatalities world-wide, annually.21 Overexposure to UVR is also deemed responsible for some 3 million cases of cataracts per year, globally, and has even been linked to some mental illnesses.21
000 fatalities world-wide, annually.21 Overexposure to UVR is also deemed responsible for some 3 million cases of cataracts per year, globally, and has even been linked to some mental illnesses.21
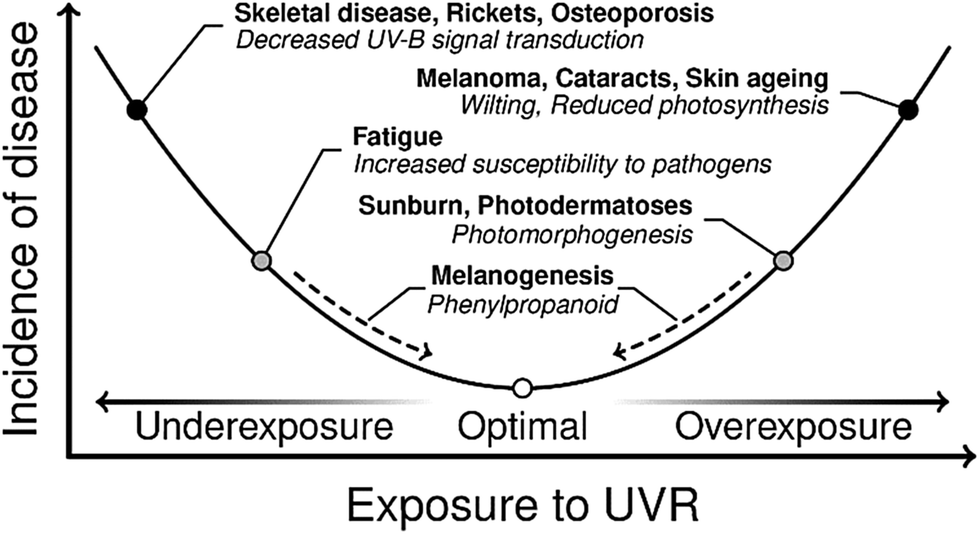 | ||
| Fig. 1 A burden of disease curve exhibited by humans and plants in response to exposure to UVR. There are a number of ailments due to acute and chronic under or overexposure to UVR. Gene-regulated pathways (dashed arrows) exist to respond to changing UVR exposure in order to perturb the incidence of disease to a minimum. This figure is adapted from ref. 24. | ||
The body has many mechanisms in place to reduce the effects of UVR exposure. For example, DNA checkpointing and repair processes can identify and rectify much of the damage caused by UVR.20,25 But these are post-UVR damage processes. The primary photoprotective mechanism is skin pigmentation which, among its many physiological roles,26,27 reduces UVR damage occurring in the first place. Human skin pigmentation consists of a class of UV absorbing molecules termed melanins. These sub-divide into eumelanin, a black-brown pigment, pheomelanin, a reddish-yellow pigment, and neuromelanin, a dark brown pigment found only in the brain.26,27 Eumelanin and pheomelanin (particularly the former) are mainly responsible for photoprotection against UVR, and we henceforth focus on these pigments.
Eumelanin and pheomelanin, the structures of which are shown in Fig. 2, are synthesised through a tyrosine-driven biochemical pathway. Tyrosine is first converted to dopaquinone enzymatically. If no cysteine is present, dopaquinone can be converted to dihydroxyindole or dihydroxyindole carboxylic acid. These polymerise to form eumelanin. In the presence of cysteine, however, dopaquinone is converted to isomers of cysteinyldopa before forming benzothiazine intermediates, which polymerise to form pheomelanin.28,29
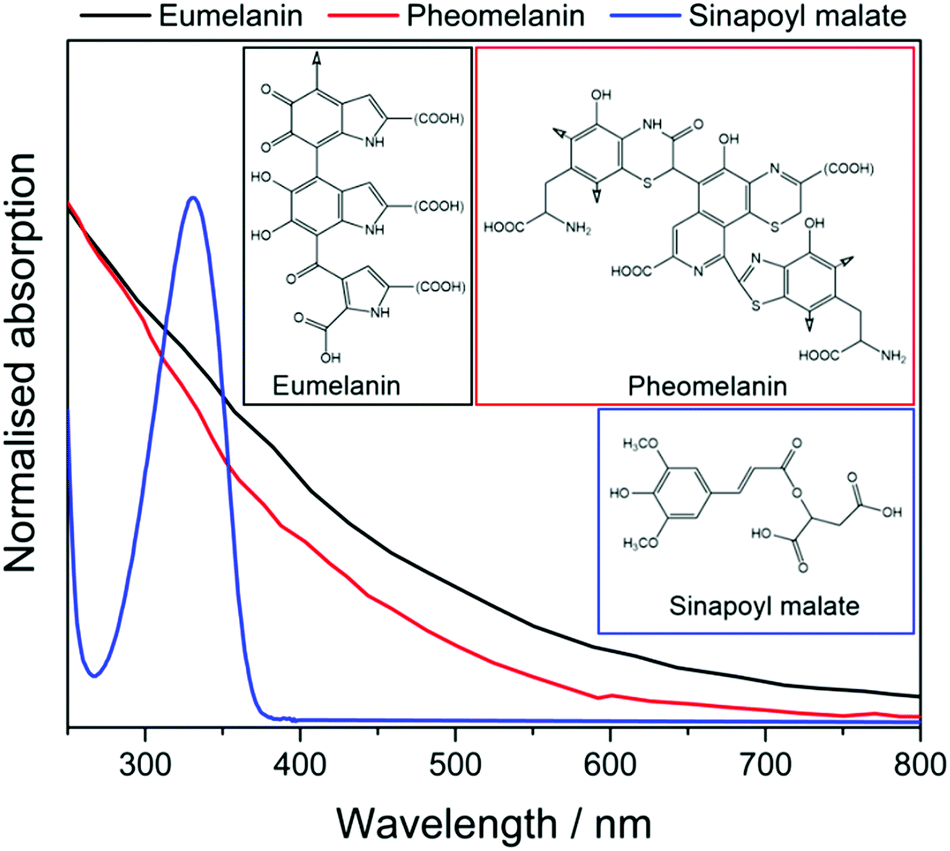 | ||
| Fig. 2 The UV-visible absorption spectra of eumelanin (black) and pheomelanin (red) in water.16 The high degrees of polymerisation of the chromophores give both pigments a broad absorption profile. Sinapoyl malate (blue) in dioxane,17 on the other hand exhibits a relatively narrow absorption band, mostly in the UV-B region. | ||
As Fig. 3 shows, specialised cells termed melanocytes are distributed throughout the stratum basale layer.30 These synthesise vesicles termed melanosomes which, in turn, synthesise melanin.30,31 Melanocytes form long projections (dendrites) out to surrounding skin keratinocytes (the dominant cell type in the epidermis) in the stratum basale and stratum spinosum layers; typically, one melanocyte reaches upwards of 40 keratinocytes.32 Melanin-containing melanosomes are transferred to the ends of the dendrites through cytoskeletal assisted processes, and thence to surrounding keratinocytes.32,33 Once the melanosomes are inside their host keratinocyte, they are positioned around the nucleus forming a supranuclear cap in the path of incident UVR. The overall photoprotection provided will depend on the concentration and distribution of melanocytes, and their size and morphology, as well as the concentration of melanin inside the melanosomes – properties which are regulated by a set of genes via processes collectively known as melanogenesis.25,30,34
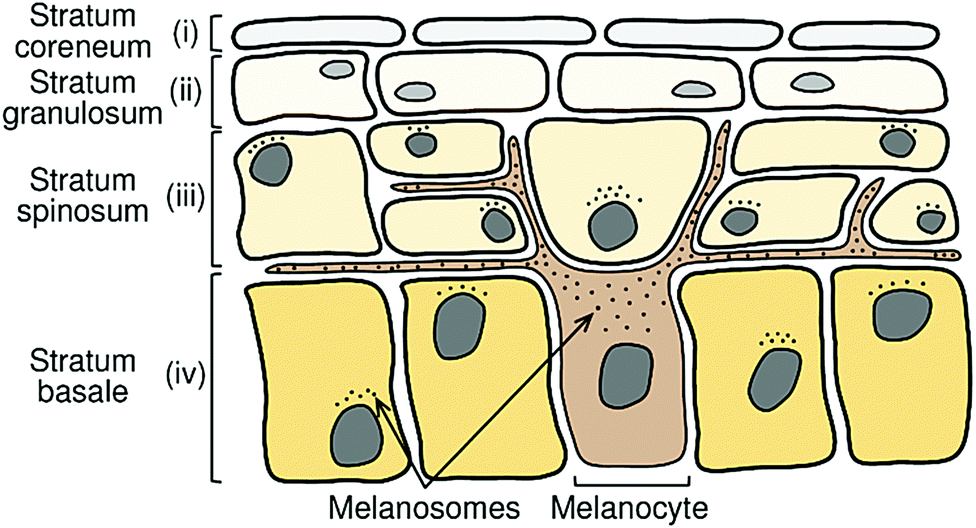 | ||
| Fig. 3 Schematic representation of the upper epidermal layers of the skin. Melanocytes in the stratum basale layer extend out to surrounding keratinocytes in the stratum basale and spinosum layers. Melanocytes synthesise melanin-containing melanosomes which are transferred to the surrounding keratinocytes. The melanosomes form a supranuclear cap above the nucleus of the host keratinocyte, in the path of incoming UVR. This figure is adapted from ref. 24. | ||
Melanogenesis turns a static picture of photoprotection into a dynamic, adaptive, photoprotective process. For example, when skin is subjected to high levels of UVR, against which the current level of skin pigmentation offers insufficient protection, signalling pathways up-regulate melanogenesis, thereby increasing the number of melanocytes and melanosome production, causing facultative skin colour (tanning).35 Melanogenesis thus enables the body to respond to changing levels of UVR as well as metabolic requirements, which helps to maintain an adventitious position within the burden of disease (Fig. 1). Whether natural sunscreens are fully optimised remains a moot point, however. For example, we note recent work highlighting the phototoxic properties of melanin.36 As such, the optimal position in the burden of disease may be a fine balance between photoprotection versus phototoxicity.
Plants have an analogous burden of disease relationship with UVR, as was also shown in Fig. 1. Exposure to too much UVR can, for example, damage vital photosynthetic machinery and thus reduce the efficacy of photosynthesis, whilst too little UVR (particularly UV-B) can render a plant more susceptible to invading pathogens and reduce the UV-B signal transduction pathways that underpin many physiological processes.38,39 Plants often employ sinapate ester derivatives as chemical filters to protect against overexposure to UVR. For example, sinapoyl malate (shown in Fig. 2) has been identified as such a sunscreen molecule in thale cress (Arabidopsis thaliana).40,41 In this case, the phenylpropanoid pathway is the gene regulated response to changing levels of UVR. When overexposed to UVR, the phenylpropanoid pathway can be up-regulated to synthesise more metabolites – e.g. sinapate esters like sinapoyl malate – and deposit them in the vacuoles of the upper epidermis, thereby increasing UVR protection for sensitive cells in lower layers of the plant leaf.
The foregoing serves as a brief overview of some of the natural photoprotective responses prevailing in both humans and plants to changing levels of UVR. There remains an obvious issue, however: the response is not immediate. Immediate tanning actually provides little additional photoprotection, since it involves oxidation of pre-existing melanin. Additional photoprotection develops over 3 days or more,42,43 by which time photodamage may well already have occurred. Further, given the recent growth in travel and tourism, as well as current cosmetic trends in tanning,43,44 people are increasingly becoming exposed to UVR environments against which their skin is not protected – necessitating additional photoprotection via other means.
Artificial photoprotection
Fortunately, there are many ways by which one can reduce the potentially damaging effects of UVR.45–47 Environmental factors – e.g. time of day, season, latitude, altitude and the terrestrial environment – all influence the local intensity of UVR. Clearly, avoiding high-UVR environments, or preparing for them appropriately by, for example, taking shade, wearing appropriate clothing, or using sun glasses, will substantially reduce the likelihood of UVR damage.Whilst such behaviours will obviously help mitigate against damage from UVR, there remains a demand for additional photoprotection – not least because we want to sunbathe. Sunscreen products are the almost universally accepted ‘solution’. These are applied to the upper epidermis of the skin and serve to complement the natural photoprotection provided by skin pigmentation. They contain components which are responsible for intercepting UVR and dissipating the energy through safe processes.
These components typically partition into two classes: chemical filters and inorganic particulate filters.13,48,49 Chemical filters (sometimes referred to as organic filters) are typically aromatic molecules with a high degree of charge conjugation. They display intense, broad absorptions across the UV-A and/or UV-B regions, typically associated with π* ← π electronic transitions. Following photoexcitation, an effective chemical filter molecule will dissipate the absorbed energy as heat to its immediate surroundings, via one or more non-destructive pathways24 of the types described later in this Review. The inorganic filters used in commercial sunscreens are typically nanosized particles that, again, display broad absorption across the UV-A/B wavelength range. These particulates also scatter incoming radiation away from the skin; indeed, they are sometimes termed inorganic scatterers. The relative efficiencies of absorption and scattering depends on the particle properties (e.g. diameter, surface coating, etc.).50
Appropriate combinations of chemical and inorganic particulate filters can obviously provide the broad spectral coverage that serves as one of the defining properties of any good sunscreen product. However, there are several other important considerations when designing a commercial sunscreen product: the texture, ease of application, degree of water-resistance, required frequency of application, suitability for a broad range of skin types and conditions, odour, and production costs all need to be factored in when designing the product. As such, a typical sunscreen product contains many ingredients for reasons other than simply providing protection from UVR, as shown in Fig. 4. Beyond these design criteria, there are further physiological requirements. It must be safe to apply the product to the skin. No harmful components must be absorbed into the blood stream. Such concerns are central to the aforementioned ‘sunscreen controversy’: incidences of skin cancer continue to rise,51 but there are also a growing number of studies that flag concerns regarding some of the commonly used filters.14,15,52–54
 | ||
| Fig. 4 The typical formulation of commercial sunscreen products.37 The European symbol of UV-A protection is shown. SPF defines the sun protection factor, a laboratory measure of the fraction of sun burning radiation that reaches the skin, e.g. an SPF of 25 means 1/25th of the radiation reaches the skin if a layer of 2 mg cm−2 sunscreen is applied, compared to skin with no sunscreen use. | ||
2. Requirements of a chemical filter
A basic photochemical perspective
The molecular constituents present within natural or commercial sunscreens are required to dissipate the absorbed photon energy efficiently and, as best as possible, without detriment to their integrity. Photoexcitation is viewed as the instantaneous promotion of an electron from one orbital to a higher energy orbital, resulting in an excited state (henceforth labelled S1) with an electronic configuration different from that of the ground state (S0). The excited state molecule will have a finite (short) lifetime before decaying. The mechanism of this decay is typically dependent on the molecule, the excitation energy, and the environment. Many of these possible decay mechanisms were first systematised by Jablonski, in the form of a diagram that still carries his name.55,56 This distinguishes radiative and non-radiative decay processes. Radiative decay between states of the same spin multiplicity is called fluorescence. The rate constant for the S1 → S0 fluorescence decay process is directly related to the strength of the corresponding S1 ← S0 absorption. Non-radiative decay processes recognised in a traditional Jablonski diagram include internal conversion (IC, i.e. an energy conserving transfer of population between states of the same spin multiplicity) and intersystem crossing (ISC, whereby population transfers to a state of different multiplicity, e.g. S1![[radiolysis arrow - arrow with voltage kink]](https://www.rsc.org/images/entities/char_e116.gif) T1, where T1 represents the lowest energy state of triplet spin multiplicity). Other possible non-radiative decay processes include ‘reaction’, e.g. isomerisation, bond fission, etc. (which will often be energetically feasible when exciting a molecule at near UV wavelengths) and, in solution, collisional quenching and even bimolecular reaction. The particular photophysical properties required of a chemical filter for potential use as a sunscreen are (i) a large absorption cross-section in the UV-A/B region, together with (ii) a high (ideally 100%) probability for the resulting excited state population to decay by IC to the S0 state, followed by (iii) relaxation to the original ground state geometry (i.e. minimal permanent conversion to another isomer).
T1, where T1 represents the lowest energy state of triplet spin multiplicity). Other possible non-radiative decay processes include ‘reaction’, e.g. isomerisation, bond fission, etc. (which will often be energetically feasible when exciting a molecule at near UV wavelengths) and, in solution, collisional quenching and even bimolecular reaction. The particular photophysical properties required of a chemical filter for potential use as a sunscreen are (i) a large absorption cross-section in the UV-A/B region, together with (ii) a high (ideally 100%) probability for the resulting excited state population to decay by IC to the S0 state, followed by (iii) relaxation to the original ground state geometry (i.e. minimal permanent conversion to another isomer).
Though informative, the traditional Jablonski diagram does not advise on the relative probabilities of the various decay mechanisms or on the eventual outcomes. Nowadays, a much clearer picture of the excited state dynamics can be obtained by monitoring the way in which the energy of a molecule changes as a function of variations in the nuclear geometry. Analogous calculations for ground state molecules can be traced back to the early days of quantum mechanics and the introduction of the Born–Oppenheimer approximation (the assumed separation of the electronic and nuclear contributions to the total energy of a molecule), and underpin traditional concepts like a reaction path from reactants to products proceeding through a transition state. Several of the figures later in this review show such potential energy curves (PECs), though these too are clearly over-simplifications. 3N-6 coordinates are required to describe fully the nuclear motions in a non-linear N-atom molecule, and a full description of the evolution from reactants to products thus requires a suitably multi-dimensional potential energy or free energy surface.
An excited state formed upon photoexcitation has a different electronic configuration to that of the ground state molecule, and the ways in which the energy of any particular excited state varies with changes in nuclear geometry will generally be different to that of other excited states and of the ground state. Given the different topographies of the ground and excited state potential energy surfaces (PESs), there will often be regions of configuration space where the energy difference between different electronic states becomes small or zero. Such points of degeneracy often constitute so-called conical intersections (CIs).
A full discussion of CIs is outside the scope of this review. Detailed descriptions can be found elsewhere.57–60 For current purposes, it suffices to recognise that CIs between PESs are ubiquitous in polyatomic molecules and that these are now recognised as pivotal in defining photochemically-driven phenomena, including (ultrafast) IC processes by which excited state population is channelled back to the S0 state in molecules defined as being photostable. Passage through a CI is termed non-adiabatic, since the instantaneous switch from one (adiabatic) PES to another involves intimate coupling between the electronic and nuclear degrees of freedom (which are otherwise viewed as separable within the Born–Oppenheimer approximation).
The probability of transition through a given CI depends on the strength of the coupling between the states involved in creating the CI. The reactive coordinate that drives a molecule towards a given CI contains components of one or more of the 3N-6 normal mode eigenvectors along which the potential gradient towards the CI is favourable. The dominant eigenvector of this nuclear coordinate is generally termed the gradient difference (g) vector. At the point of degeneracy, another normal mode is required to enhance the coupling between the electronic states in order to permit the radiationless transition. This derivative coupling vector (h) is orthogonal to the g vector. Put simply, small motions along the h dimension facilitate the change in electronic configuration and thus electronic state. The g and h vectors are the dimensions along which the double-cone topography of the CI exists. The remaining 3N-8 degrees of freedom belong to the seam space and do not promote electronic state changes at small nuclear displacements. Various geometries and types of CIs are possible. Their geometric positions are often dictated by symmetry but can also be altogether accidental in larger and more complex polyatomic molecules. We revisit some of these ideas when discussing specific examples in the next section.
Mechanisms of photostability
Here we describe several generic types of CI geometry that are known to facilitate the IC of excited state population to the ground state. In each case, we start with a simple ‘prototype’ molecule, and describe the ways in which a specific mechanism extrapolates and changes with increasing molecular complexity.Any energetically accessible CI between the excited and ground state PESs can facilitate non-radiative transfer of excited (e.g. S1) state population back to the S0 state and thus potentially reform the starting parent molecule. The probability of any such route to molecular photostability depends on several factors: (i) the topography of the excited state PES connecting the Franck–Condon region (i.e. the geometry with which the excited state molecule is ‘born’ upon photoexcitation from the S0 state) to the CI geometry, (ii) the topography of the ground state PES between this CI geometry and the S0 minimum energy geometry, and (iii) the vectors associated with the nuclear kinetic energy along a particular driving coordinate – the components of which largely depend on the potential gradients experienced prior to and following IC through the CI. Local environments that surround a given molecule (such as solvation) can influence the PES and thus the vectors associated with the nuclear kinetic energies. The overall dynamics in the presence of proximal solvent molecules will also be influenced by classical processes like thermal energy transfer and collisional deactivation.61,62
The first decay mechanism we consider is photodissociation, a primary process that runs counter to the requirements for photostability. Photodissociation is the fragmentation of a molecule following photon absorption, which we first illustrate by reference to water.63–67 The valence orbitals of water can be pictured as two O–H centred σ orbitals, the two corresponding O–H centred antibonding σ* orbitals and a 2px lone pair orbital perpendicular to the molecular plane. Considering just these orbitals and without invoking symmetry, the ground state electronic configuration of water is: (σ)2(σ)2(2px)2(σ*)0(σ*)0. Photoexcitation promotes an electron from the highest occupied 2px orbital to one of the σ* orbitals, yielding a 1nσ* excited state with configuration (σ)2(σ)2(2px)1(σ*)1(σ*)0 and 1Π term symbol (at linear geometry). Electronic excitation weakens one of the O–H bonds, to the extent that the excited state PES is dissociative with respect to extension of one O–H bond, yielding H + OH radicals. As Fig. 5(a) shows, the ground and excited state potentials intersect in this coordinate at linear geometries (i.e. H–O–H bond angle = 180°). Motion along the bending coordinate (i.e. along the h vector) lifts the degeneracy of the 1Π state, leads to the characteristic double cone topography of the symmetry allowed CI between the 1Π (A′) and S0(A′) states but has relatively little effect on the potential energy of the other 1Π (A′′) component (wherein the singly occupied px orbital is orthogonal to the nuclear plane). Other hydride molecules with similarly low lying, dissociative 1nσ* states include methanol (forming CH3O + H)68–72 and higher alcohols, hydrogen sulphide (forming SH + H),65,73 alkyl thiols, ammonia (forming NH2 + H)65,66,74 and alkyl amines.
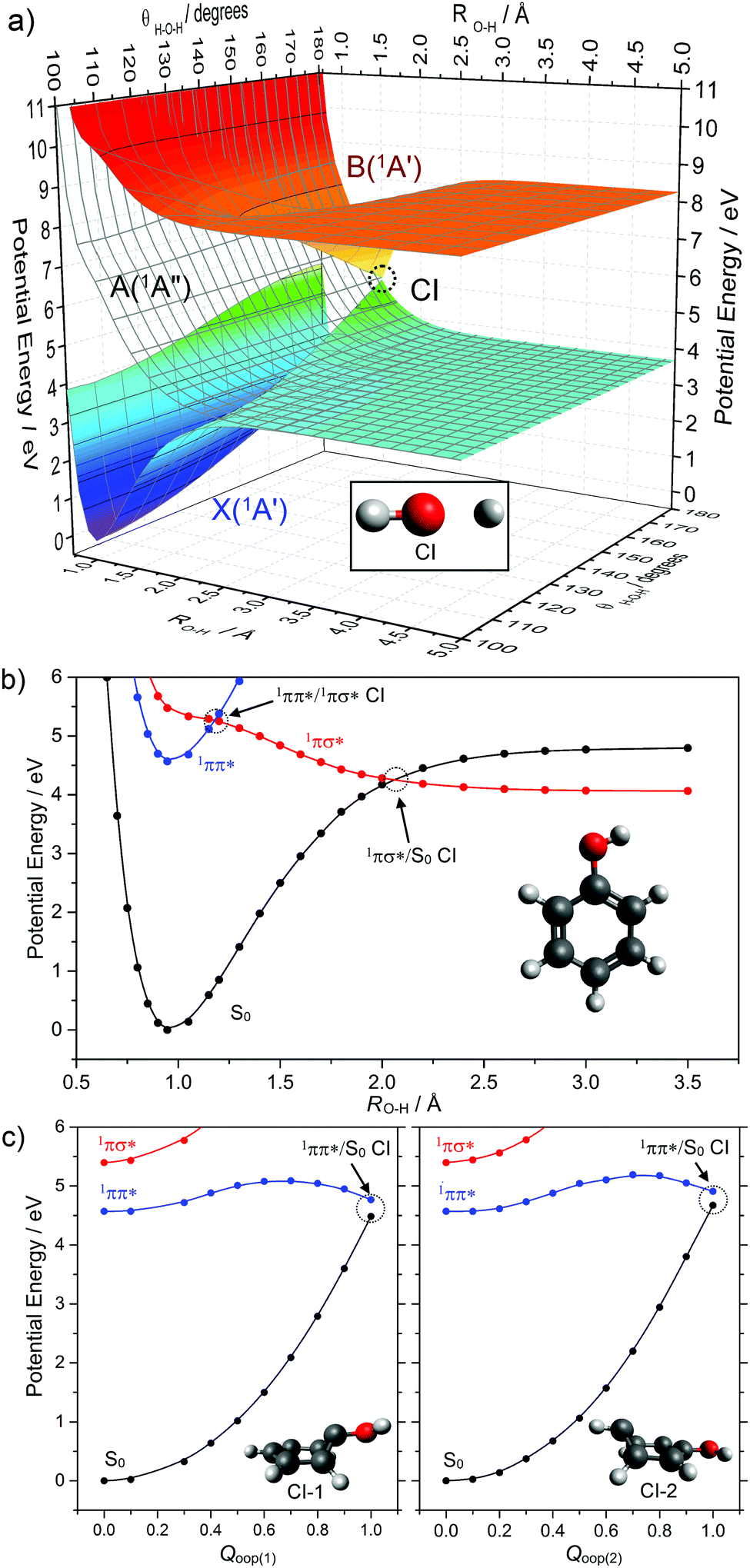 | ||
| Fig. 5 (a) Potential energy surfaces of the ground (X(1A′)) and lowest singlet excited states (A(1A′′) and B(1A′)) of water computed along the O–H stretch (RO–H) and H–O–H bending (θH–O–H) coordinates. The CI at a linear geometry is clearly identifiable at an extended O–H bond distance (RO–H ∼ 1.5 Å). (b) Potential energy curves for the ground and lowest singlet excited states (1ππ* and 1πσ*) for phenol along RO–H. (c) Potential energy curves for these same three singlet states of phenol along two out-of-plane (oop) ring deformation coordinates (Qoop(1) and Qoop(2)). The corresponding 1ππ*/S0 CIs are indicated by the grey circles with the relevant optimised geometries at each CI shown in each panel. | ||
Replacing one H atom in water with a phenyl group yields phenol, a molecule whose photodissociation dynamics have attracted much recent attention.75–79 The presence of the π system (cf. H2O) ensures that the first excited (i.e. S1) state has 1ππ* character. As Fig. 5(b) shows, this state is bound with respect to O–H bond elongation. The analogue of the 1nσ* state of water is the S2 state in phenol. This state is best viewed as having 1πσ* character, as the O(2px) lone pair is stabilised (and thus lowered in energy) via conjugation with the π system. As in water, the 1πσ* state is dissociative with respect to O–H bond extension and crosses the S0 potential at RO–H ∼ 2 Å, leading to a symmetry allowed 1πσ*/S0 CI – for which the h vector involves out-of-plane motion (notably O–H torsion). A second CI is also evident, between the 1ππ* and 1πσ* states at smaller RO–H. Experiments have shown that photoexcitation to both the S1 and S2 states of phenol results in O–H bond fission. The latter is unsurprising, and driven by direct dissociation. The former involves initial tunnelling from the 1ππ* state to the 1πσ* state, through the barrier created by the 1ππ*/1πσ* CI, and subsequent bond extension on the 1πσ* PES.80
Photodissociation acts counter to the requirements of photostability, and reversing the ballistic motion in the case of a light, fast, departing H atom is improbable. But Fig. 5 serves to illustrate a much wider family of molecules where a CI along a bond extension coordinate could facilitate IC between an excited state and the ground state. Firstly, the local topography in the region of the CI is important in determining the relative probabilities of bond fission (photodamage) and IC (and thus potential photostability). Second, non-adiabatic coupling in the vicinity of the CI may well be greater in the case of heavier, more slowly separating leaving groups. Third, the foregoing discussion has implicitly assumed collision-free conditions, but a surrounding solvent cage can encourage geminate recombination, re-sampling of the CI region and another opportunity to access the S0 potential.81
Even a molecule as small as phenol has sufficiently high dimensionality to ensure the existence of many other CIs between the S1 and S0 PESs (and between other excited states), any of which could facilitate IC if the molecule has sufficient internal energy to sample the relevant regions of configuration space. Many of these alternative CI geometries can be pictured more easily in the context of phenol's close brother: benzene.57Fig. 5(c) provides schematic depictions of two out-of-plane (oop) ring deformations in phenol that are representative CI geometries in many conjugated cyclic systems.
Motion towards such CIs in the specific case of phenol is hampered by a potential barrier between the Franck–Condon and CI geometries. But CIs with oop ring puckered geometries are known to facilitate the ultrafast IC of excited state population back to the S0 state (ultimately leading to photostability) in many larger and more complex molecules. Notable examples include the DNA and RNA nucleobases,82–84 for each of which the potential energy profile linking the Franck–Condon region and at least one CI with an oop ring puckered geometry is barrierless. This quite striking difference in the relative importance of oop CIs in enabling IC in phenol and in the nucleobases can be traced to a combination of effects arising from incorporating N atoms in the ring and substituent effects.85
These oop ring deformation CIs can be viewed as extensions of another well-known class of CI found in alkenes. Ethene,86 for example, shows a CI between its S1 and S0 PESs upon rotation about the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. Analogues of this CI enable the well-known E → Z photoisomerism intrinsic to all aliphatic alkenes, and such E → Z CIs are known to dominate the ultrafast excited state dynamics of many biologically and industrially relevant molecules.87 An equivalent twist about a CC bond in a cyclic aromatic system like benzene inevitably leads to the oop ring-deformation CI. Hence the frequent reference to oop ring deformation CIs in aromatic systems as ‘ethylenic’ CIs.88
C double bond. Analogues of this CI enable the well-known E → Z photoisomerism intrinsic to all aliphatic alkenes, and such E → Z CIs are known to dominate the ultrafast excited state dynamics of many biologically and industrially relevant molecules.87 An equivalent twist about a CC bond in a cyclic aromatic system like benzene inevitably leads to the oop ring-deformation CI. Hence the frequent reference to oop ring deformation CIs in aromatic systems as ‘ethylenic’ CIs.88
The final classes of CI geometry considered here are those arising via proton-transfer (PT) reactions. These can occur within or between molecules, yielding intra-89–91 and intermolecular PT,92–94 respectively. Regardless of this distinction, all PT reactions require an acidic proton donor (DH) and a basic acceptor (A), and are known to facilitate excited state/ground state curve-crossings in hydrogen-bonded complexes (i.e. DH–A) as depicted in Fig. 6. These are commonly termed proton coupled electron transfer (PCET) reactions. Photoinduced PCET in a hydrogen-bonded complex starts with promotion of an electron from an acidic donor (D) orbital to an unoccupied orbital localised on the basic acceptor (A). This creates a D+H–A− charge-separated or charge-transfer (CT) excited state, which is neutralised by the transfer of proton from D to A. A CT excited state almost always involves a favourable driving force for PT (i.e. a decrease in potential energy upon PT). The S0 state, in contrast, is stable in the closed-shell DH–A configuration, and its PES exhibits a large potential barrier to PT. Inevitably, therefore, the CT and S0 PESs cross along the PT coordinate and, as Fig. 6 shows, non-adiabatic coupling at the resulting CI could lead to formation of the D–HA adduct and/or reformation of the original DH–A pair. These limiting pathways can be viewed as photoreactivity and photostability, respectively.
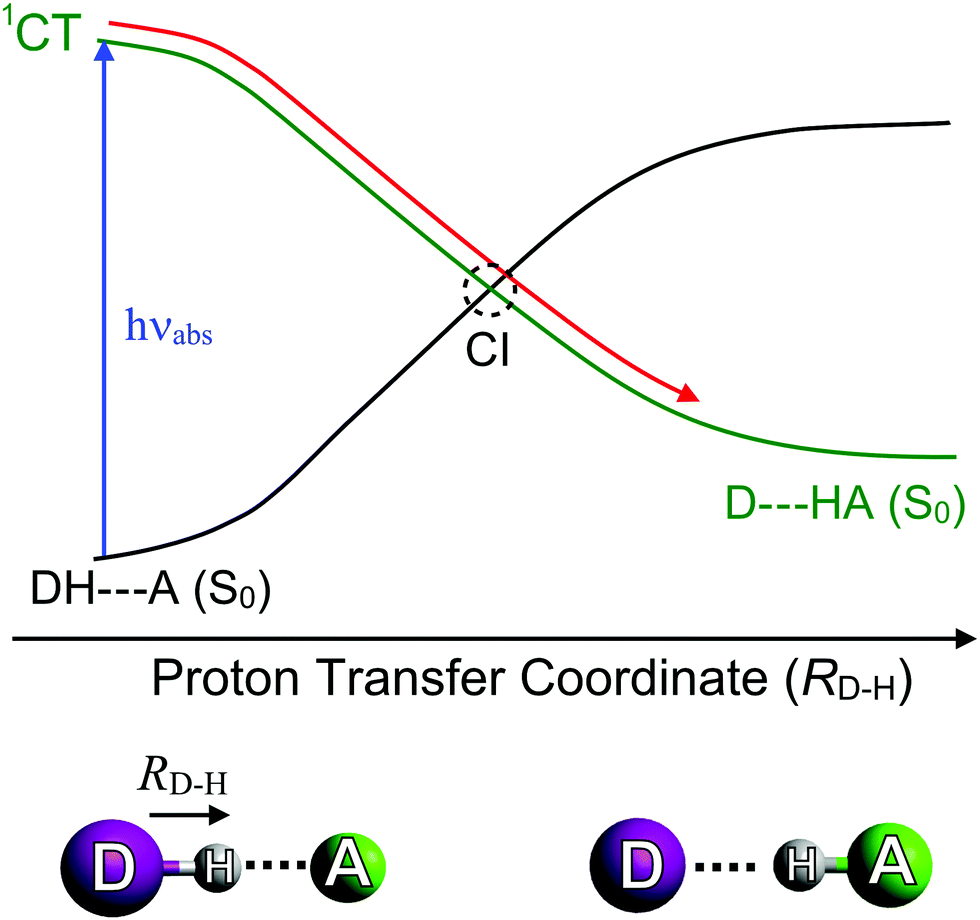 | ||
| Fig. 6 Potential energy curves illustrating the variation in energy of the ground (S0) state and a charge transfer (1CT) state as a function of the proton transfer coordinate (RD–H). | ||
3. Overview of experimental and theoretical methods
Experimental methods
Here we focus on contemporary methods designed to shed light on the early time dynamics that determine the efficacy (or otherwise) of a given chemical filter in a photoprotective role. A number of spectroscopic techniques have been used to probe the ultrafast excited state dynamics of such molecules in the gas-phase. These include both frequency- and time-domain methods. We choose not to discuss the former, but readily acknowledge the important role of such studies in revealing the energetics of relaxation processes and the vibrations that may facilitate these non-radiative decay pathways in related biomolecules; further discussion of such studies can be found in the literature.95–101 Studies in the solution-phase are somewhat closer to real life applications. Both absorption and fluorescence techniques can be applicable. Here we focus on absorption-based techniques, but recognise that fluorescence probe methods can offer advantage on occasion. Finally, we must emphasise that ‘more traditional’, steady-state methods (UV/Vis absorption, NMR spectroscopies, etc.) have provided, and continue to offer, many important and complementary insights into dynamical processes occurring on longer timescales, which may lead to photoproduct formation. | ||
| Fig. 7 Key features of a typical gas-phase experiment, including a molecular beam and a time of flight (TOF) mass spectrometer. The molecular beam is intercepted by the pump, hνpu, and probe, hνpr, laser pulses, which are represented, respectively, by the red and blue arrows. A suitably biased set of ion optics accelerates the ions formed by the probe laser towards a detector. The last section of such an apparatus consists of a field-free flight tube (see main text), so that ions reach the detector at different times depending on their m/z ratio. Figure adapted from ref. 95. | ||
The dynamical behaviour of the photoexcited molecule is revealed by monitoring how the yield of particular ion masses vary with the time delay between the pump and probe pulses (Δt).102,103 Such parent and/or fragment ion transients can then be modelled using appropriate functions (e.g. exponential rise and decay functions) to describe the observed kinetics. These kinetic analyses yield time-constants that can be related to the underlying photochemical and photophysical processes in operation.
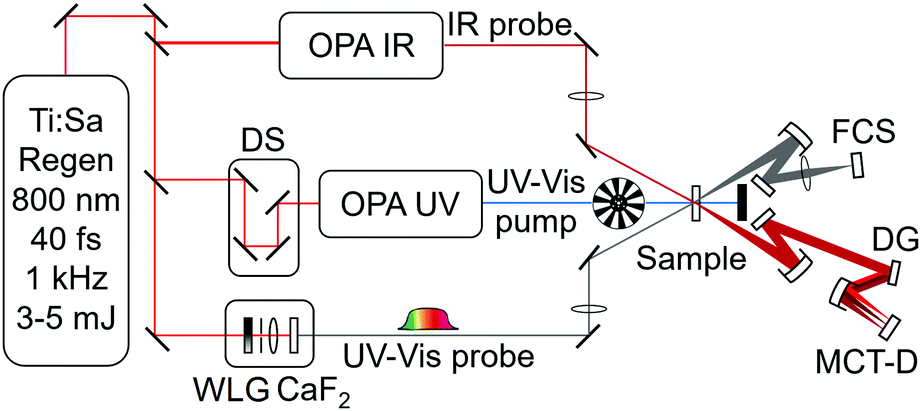 | ||
| Fig. 8 Schematic showing a typical ‘combined’ experimental set-up for transient electronic absorption and transient vibrational absorption spectroscopy (TEAS and TVAS, respectively). The experiment employs OPAs seeded by the output of a Ti:sapphire regenerative amplifier laser. Other components shown include: white light supercontinuum generation (WLG) in a CaF2 disk; delay stage (DS); dispersion grating (DG); mercury cadmium telluride detector (MCT-D); and fibre couple spectrometer (FCS). | ||
A good sunscreen filter should display a particularly simple transient spectrum. Oxybenzone (OB, a chemical filter featured as one of our case studies in Section 4) comes close to this ideal, as illustrated by Fig. 9, which shows transient UV/vis and IR absorption data obtained following 325 nm photoexcitation of OB in solution in cyclohexane.110 The former spectrum is dominated by an ESA feature that peaks ∼366 nm but spans almost all of the wavelength range accessible with the available WLC and exhibits an ultrafast decay (characterised by fs and ps time constants, the interpretation of which is discussed in Section 4). The transient IR spectrum is dominated by a GSB feature at ∼1620 cm−1, the bulk of which recovers with a ∼8 ps time constant. This latter time constant is largely determined by the rate at which photoexcited species relax by transferring vibrational energy to the surrounding solvent molecules and thereby repopulate the starting level of the S0 state. (No similar GSB signal is evident in Fig. 9(a) simply because the electronic absorption spectrum of OB in its S0 state barely extends to 340 nm, the short wavelength limit of the WLC probe.) Kinetic information is derived by modelling such transient absorption spectra recorded at many different pump–probe time delays using global fitting techniques.111–114
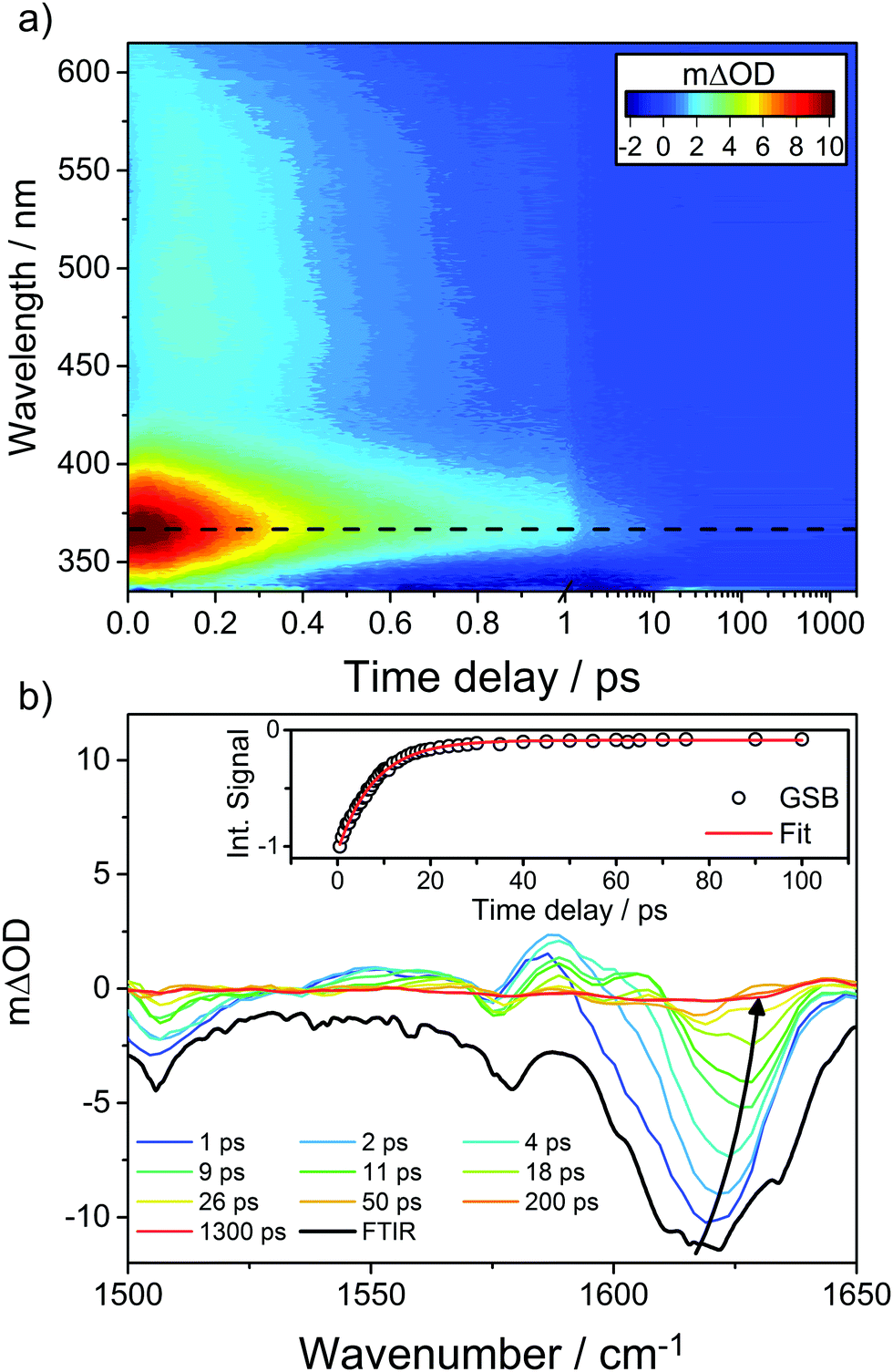 | ||
| Fig. 9 (a) False colour plot showing the transient electronic spectrum (ΔOD as a function of probe wavelength and pump–probe time delay) following 325 nm photoexcitation of OB in cyclohexane, with the decay of the transient ESA signal centred at ∼366 nm, represented by the black dashed line. (b) Transient IR spectra measured at different pump–probe delays (specified in the inset) in the range 1500–1650 cm−1 following 325 nm photoexcitation of OB in cyclohexane along with the steady-state Fourier transform IR spectrum of the precursor solution (shown by the bolder black line). The inset kinetic trace shows the best exponential fit to the integrated GSB signal (centred on ∼1620 cm−1, which blueshifts for larger pump–probe time delays as shown by the solid arrow) recovery. The original data are taken from ref. 110. | ||
We re-emphasise that several other techniques can provide dynamical insights complementary to those revealed by TR-MS and/or transient absorption spectroscopies. In the gas-phase, for example, time-resolved velocity map imaging methods allow one to track photofragment and/or photoelectron velocity distributions, the analysis of which offers a route to untangling at least some of the various relaxation pathways.115,116 In the solution-phase, time-resolved fluorescence utilising optical Kerr gating,117,118 and frequency up-conversion119,120 or stimulated Raman scattering121 methods could all provide insights complementary to those revealed by the transient absorption methods outlined above, but have yet to be applied decisively to studies of the kinds of chemical filters used in sunscreens.
We also emphasise that steady state measurements have long been used to explore photophysical properties of many sunscreen components, individually and in mixtures. Differences in the UV/vis absorption spectra of samples illuminated using, for example, an arc lamp and non-illuminated samples can provide information on photochemical stability, unwanted photoinduced production of singlet oxygen, and/or signatures of photodegradation products,17,122 with NMR spectroscopy offering another route to product identification.123 Such ‘classical’ photochemical studies have been crucial in the development of the many commercial sunscreens used today but are not discussed further since they do not report directly on the excited state photophysics that is the primary focus of this Review.
Computational and theoretical methods
Computational studies are equally necessary when it comes to any detailed description of likely IC mechanisms in the kinds of chemical filters used in sunscreens. Theoretical photochemists seek to determine (at least) approximate excitation energies, transition strengths and the likely dynamics along important nuclear coordinates, thereby offering an in-depth assessment of the feasibility of a given excited state reaction pathway. Existing computational methods are all challenged when it comes to calculating excited state energies, particularly in the regions of CI between electronic states that enable the ultrafast IC required for molecular photostability. The challenges stem from the multi-reference nature of CIs. The participating states (e.g. the S0 and S1 states) have comparable energies but different electronic configurations. The S0 state wavefunction represents the lowest energy occupied orbitals in a single Slater determinant. Most methods use this determinant to construct a new set of reference ground state orbitals, which are then used to compute excitation energies.However, the use of a single determinant reference configuration causes problems when states with different electronic configurations approach in energy. Multi-reference methods (such as CASSCF,124,125 CASPT2,126,127 MRCI128) circumvent this problem and have been used in many recent computational photochemical studies. Such calculations are expensive, however, and thus limited to relatively small molecular systems. Multi-reference calculations are not yet feasible for large systems, and we remain reliant on single-reference excited state methods (such as TD-DFT,129,130 EOM-CCSD,131 CC2132 and ADC(2)133) which, when used with suitable caution, can provide valuable qualitative pictures of excited state decay processes.134–137 These methods can be combined with classical theories in order to include environmental effects. Such calculations employ mixed quantum and classical (QM/MM) methods, where the chromophore is treated using high level electronic structure methods, while the surroundings (e.g. solvent molecules) are treated classically.138–142
The foregoing methods solve the time independent Schrödinger equation at selected nuclear geometries to determine the topography of the PES(s) for a given reaction path. They give no explicit information about the reaction dynamics. Timescales for an excited state reaction can be obtained by simulating the nuclear dynamics. Full quantum dynamics simulations, in which the nuclear degrees of freedom are tracked exactly by solving the time dependent Schrödinger equation (TDSE) represent the ‘gold standard’. Such quantum dynamics calculations have traditionally been limited to PESs of reduced dimensionality on account of the computational expense. Simulations on multi- and full-dimensional PES(s) are starting to appear, but are currently restricted to very short (∼100 fs) propagation times.
Much longer (>1 ps) timescales can be explored using semi-classical methods. Here, the electronic coordinates are computed by numerically integrating the TDSE (thereby retaining the possibility of non-adiabatic coupling between states), while the nuclear coordinates are driven classically by solving Newton's classical equations of motion. This is the premise behind Tully's fewest switches surface hopping (FSSH) algorithm,143 that is now included in several QM packages.144–146 FSSH can be used in combination with available PESs or can be programmed so that, at each time-step along the nuclear propagation, the energies and gradients of the various electronic states are computed ‘on-the-fly’ using a suitable QM or QM/MM electronic structure method(s).
Another important recent development has been the advent of the GPU-accelerated ab initio multiple spawning (AIMS) method for treating excited state dynamics. In AIMS, the nuclear dynamics and electronic structure problems are solved simultaneously, thereby allowing quantum behaviour (e.g. tunnelling) of the nuclei. GPU-accelerated AIMS has been successful in modelling the photoinduced ring-opening of an isolated molecule as large as provitamin D3 (a 51 atom system)147 and used to treat the excited state dynamics of a range of solvated chromophores.148 It is safe to predict that GPU-accelerated dynamics methods will become increasingly popular and important for simulating photochemical process in complex environments.
4. Case studies
Molecular constituents in natural sunscreens
Natural sunscreen molecules play a key role in protecting the vital genetic material contained within an organism. Thus, they tend to be present nearer the surface of most organisms. Natural sunscreen molecules will also exhibit the fundamental photophysical properties identified in Section 2 – namely (i) a high absorption cross-section for incident UVR (typically satisfied by a conjugated π system) and (ii) one or more mechanisms whereby the absorbed photon energy can be dissipated without loss of molecular integrity. Here we summarise results from recent studies that seek to unravel dynamical details of these dissipation mechanisms in both plant and mammalian sunscreens.Efforts to understand the photostability of lignin include gas-phase studies of the excited state dynamics of phenol, guaiacol and syringol following photoexcitation to their respective S1(1ππ*) states.152,153Fig. 10 compares these model chromophores (henceforth abbreviated as ArOH) with the relevant monolignols and a small section of lignin polymer. The measured quantities in this study were the respective S1 state lifetimes and the H atom yields (attributed to ArO–H bond fission) measured at the longest available pump–probe time delay (Δt = 1.2 ns). On the basis that ArO radical formation drives photodegradation,154 the relative photostabilities were found to be ordered: guaiacol > syringol > phenol. Intriguingly, this order matches that of the relative abundances of the monolignol building blocks in natural lignins,155 which are often dominated by coniferyl alcohol (which shares the same aromatic motif as guaiacol) with little (<10%) of the simplest phenol-based analogue, p-coumaryl alcohol.
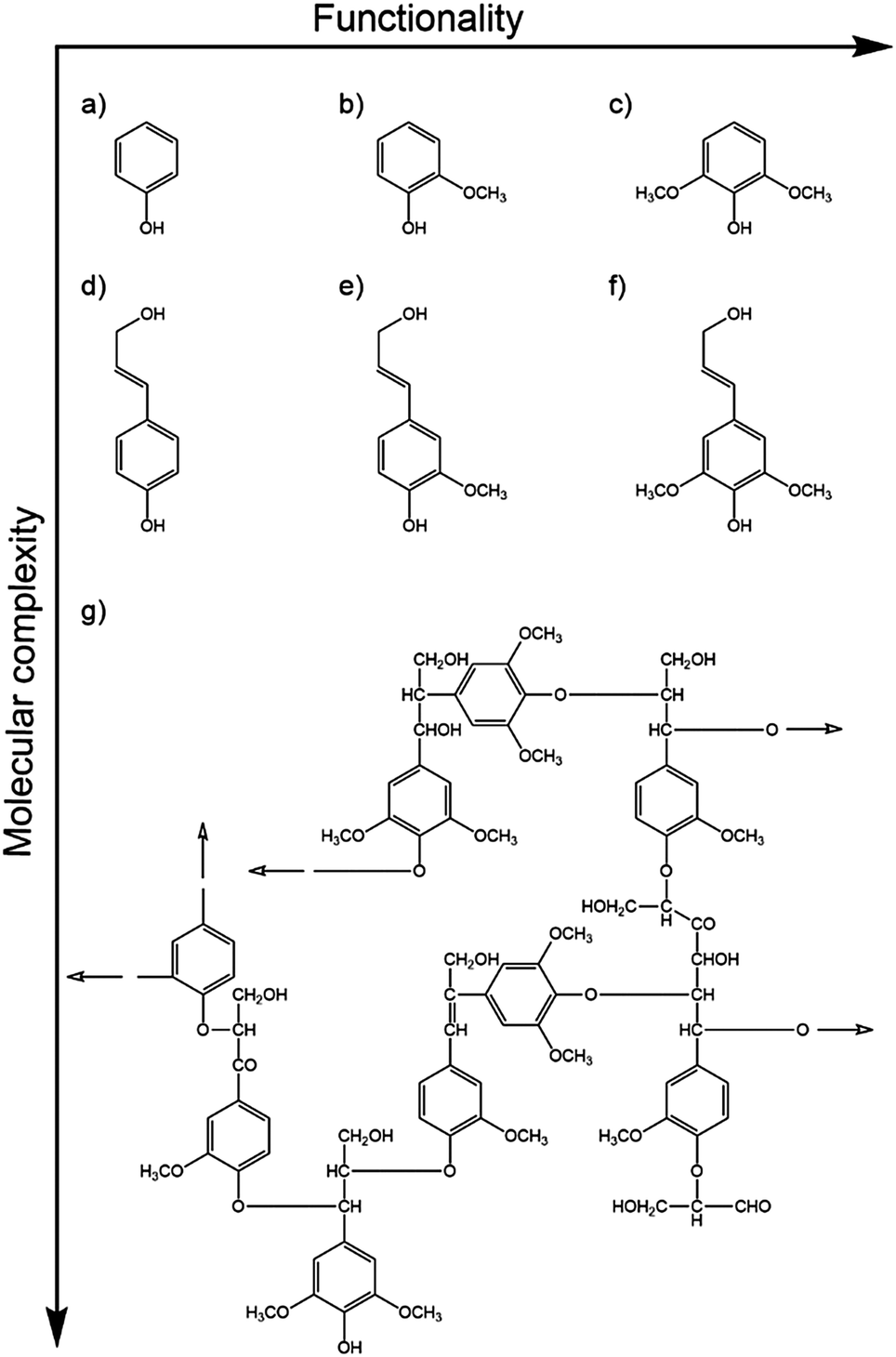 | ||
| Fig. 10 Model chromophores of the cross-linked phenolic polymer lignin, with increasing functionalisation and molecular complexity. Phenol-based chromophores: (a) phenol, (b) guaiacol, (c) syringol, and the more complex monolignols: (d) p-coumaryl, (e) coniferyl and (f) sinapyl alcohol. These form the building blocks of beech lignin,151 a subunit of which is shown in (g). Hollow arrows indicate the directions of further polymerisation. | ||
The relative photostabilities of these model chromophores are seen to correlate with the extent of intramolecular hydrogen bonding. O–H bond fission in each case involves tunnelling through the potential barrier associated with the CI between the 1ππ* and 1πσ* states (recall Fig. 5(b)), which offers greatest impediment at planar geometries. Excited state tunnelling probabilities are thus reduced if out-of-plane (torsional) motion of the O–H moiety is constrained by intramolecular H-bonding with the O atom in the one (in guaiacol) or two (in syringol) neighbouring –OCH3 groups. The extent to which these gas-phase findings translate into the condensed phase remains an open question. Transient absorption studies show that the relative behaviours persist in a weakly interacting solvent like cyclohexane: ArO radicals were clearly visible within 1 ns when exciting phenol in cyclohexane156 whereas, even at pump–probe delays Δt = 8 ns, it was hard to discern any ArO radical formation following UV photoexcitation of guaiacol in cyclohexane.61 Thus it is tempting to speculate that such behaviour might extrapolate to nature, given that solvation (by H2O) will be discouraged by the hydrophobic nature of the biopolymer (lignin).154
Sinapoyl malate was introduced earlier (Fig. 2) as a prominent example of a naturally occurring sunscreen molecule. It contains the syringol motif and is also a cinnamate derivative. Cinnamates are aromatic, unsaturated salts and esters derived from cinnamic acid – many of which have been studied both theoretically and experimentally.157 Gas-phase UV spectroscopy studies of sinapoyl malate, sinapic acid, and other sinapate esters show that they all absorb strongly in the UV-B region. Unlike the others, however, the absorption spectrum of sinapoyl malate is broad and featureless, even under jet-cooled conditions.158 In each case, the UV absorptions are attributed to π* ← π excitations, involving orbitals that are delocalised across the ring and the conjugated side-chain. IR-UV double resonance studies serve to exclude spectral congestion (e.g. from the presence of different conformers) as the cause of the apparently featureless absorption of sinapoyl malate. Rather, it is taken as evidence that the 1ππ* state is short-lived, and couples efficiently with a near resonant, optically ‘dark’ excited state (i.e. another excited state with a very small absorption cross-section from the S0 state). Evidence in support of this conjecture is provided by TD-DFT calculations, which suggest that the dark state is a 1nπ* state with substantial charge-transfer character.158
The steady-state fluorescence spectrum following excitation of an aqueous solution of sinapoyl malate at its UV absorption maximum shows a large Stokes shift and a low fluorescence quantum yield, ϕfl ∼ 3 × 10−3 – suggesting that population in this 1nπ* state also undergoes efficient non-radiative decay. Given the prior work on syringol, it is unlikely that the rate of O–H bond fission could be sufficient to account for this decay. Further insights into the decay mechanism have been provided by ultrafast UV pump – UV/vis absorption probe studies of sinapoyl malate in a range of solvents (dioxane, acetonitrile and methanol),17 which confirm that photoexcitation of the majority E-isomer at ∼330 nm results in ultrafast deactivation. Global fitting the measured transient absorption spectra revealed three decay processes with respective time constants τ1 (sub-ps), τ2 (∼1–5 ps) and τ3 (∼20–30 ps), the precise values of which are solvent dependent. Two possible relaxation mechanisms were proposed. Both assume initial photoexcitation to a strongly absorbing 11ππ* state. One assumes that the population evolves on the initially excited state, while the other assumes efficient non-adiabatic coupling to another excited state (e.g. the CT state suggested by Zwier and coworkers).158 In either scenario, the excited molecule is assumed to distort – most plausibly (by analogy with the documented photophysics of other cinnamates) – along the E–Z isomerisation coordinate towards a region of CI with the S0 PES. Vibrational energy transfer to the surrounding solvent cools the resulting S0 molecules. In no case did the parent absorption spectrum recover fully within the maximum time delay available (Δt = 2 ns), suggesting that some S0 molecules initially cool into the higher energy Z-isomer.
Though appealing, this description still leaves a lot of room for refinement. For example, recent excited state quantum dynamics calculations coupled with classical molecular dynamics (MD) simulations of the ultrafast non-radiative decay of sinapic acid – in isolation, and when microsolvated with methanol – offer additional insights into the early time excited state dynamics of this precursor to sinapoyl malate.159 Specifically, these simulations suggest an important role for proton transfer, enabled both by intramolecular (in the case of the bare molecule) and intermolecular (in the complex with methanol) hydrogen bonds in the photoexcited molecule.
The latter finding, in particular, may help account for the solvent dependent excited state decay kinetics observed following UV photoexcitation of sinapoyl malate. Whilst some mechanistic details remain to be resolved, there is no question that sinapoyl malate undergoes ultrafast decay to regenerate (predominantly) the original ground state molecule – justifying its use as a photoprotective sunscreen in plant leaves. As noted earlier, lignins are ubiquitous in nature. Could it be that evolution has favoured constituents that offer a multiplicity of ultrafast excited state decay pathways, capable of functioning in a range of different environments? Sinapoyl malate is not used in commercial sunscreen products, but further studies of this and related naturally occurring systems will surely provide further insights into the design of new, and/or the optimisation of existing, sunscreens.
We start with arguably the most famous class of photostable biomolecular systems: the DNA/RNA nucleobases and nucleosides. Adenine, thymine, guanine, cytosine and uracil (and their nucleosides) are DNA/RNA constituents and carry the genetic code in all biotic systems. Their primary role is to code for and subsequently generate proteins that maintain biotic life. However, the structural properties of the DNA/RNA nucleobases, nucleobase pairs and nucleosides offer secondary benefits, including acting as a sunscreen and thus protecting genetic information stored within the cell. DNA/RNA nucleobases are known to undergo intrinsic ring-centred oop deformations following UV photoexcitation that mediate ultrafast IC and efficient reformation of the ground state parent molecule.85,160–163 Such oop deformation-based IC routes are also intrinsic to the DNA/RNA base pairs and isolated nucleosides, but these can also undergo IC along PCET reaction paths – which are proposed to account for the much shorter excited state lifetimes of nucleosides compared to the isolated nucleobases.85,135,164 Within the cellular medium, strands of RNA, ADP or ATP, that contain analogous molecular moieties to that of DNA nucleobases and nucleosides, are also expected to show some degree of photostability – protecting their molecular integrity and the local environment by absorbing (and thus reducing) the incident UV flux. Nucleobases and nucleosides are generally contained well within the cellular environment, however, and are only exposed in abnormally harsh and penetrating UV-environments.
Other molecular sunscreens exist nearer the surface of the mammalian body. Consider the eye, for example. Its primary function is to direct light to photoreceptors located on the retina called cones and rods. The latter contain the protein rhodopsin, which houses the chromophore retinal. Retinal is extremely photostable and has been the focus of many experimental and theoretical studies. Retinal, and its protonated Schiff base, are both known to undergo ultrafast IC via low energy CIs located along Z/E photoisomerism coordinates.87,165–171
The lens, which focuses light onto the retina, contains many molecular constituents that help protect it (and the surrounding environment) from hard UV radiation. Kynurenines (see inset of Fig. 11) are one such class of molecular constituents, which are biosynthesised by the tryptophan dioxygenase enzyme. Experimentally, kynurenines have been shown to undergo fast excited state decay following photoexcitation.172 Theory suggests that the dominant mechanism in this case is IC via a CI located along a PCET coordinate as shown in Fig. 11.134 Kynurenines contain acidic amino donor and basic carbonyl acceptor groups which, in at least some of the low energy conformations, are linked by strong intramolecular hydrogen bonds that facilitate the proton transfer.
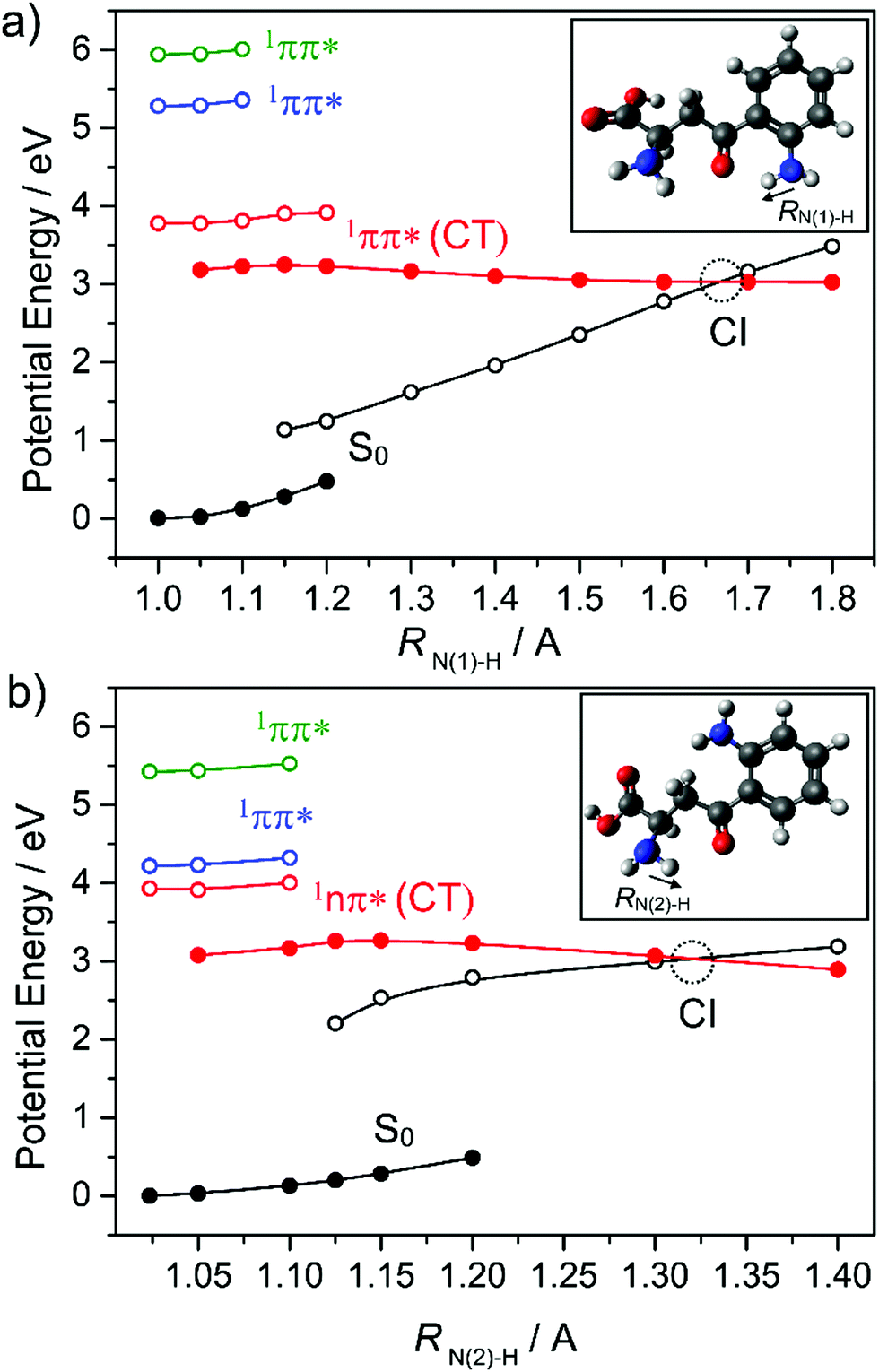 | ||
| Fig. 11 Potential energy curves of the ground and first three singlet excited states of (a) syn and (b) anti kynurenine obtained by a constrained relaxed scan along the N–H bond elongation coordinates (RN(1)–H and RN(2)–H) in the S0 state (full black circles) and in the 1nπ*(CT) state (full red circles) – representing intramolecular proton transfer. The vertical excitation energies of the lowest 1nπ* (empty red circles) and two 1ππ* states (empty blue and green circles) from the relaxed S0 geometries, and the vertical energies of the ground state (empty black circles) from the relaxed 1nπ*(CT) geometries are shown also. The data are taken from ref. 134. | ||
The part of a human or mammal that is most exposed to light is the skin. Molecular sunscreens thus play their most significant roles on and in the skin. Sunscreens present on the epidermis manifest either via synthesis from skin cells (see Section 1) or in sweat. An example of the latter is urocanic acid (UA) which, as Fig. 12 shows, has some structural similarities to sinapic acid – with an imidazole ring in place of the substituted benzene. UA absorbs strongly in the UV and displays an ultrashort excited state lifetime in both the gas-phase and in aqueous solution. Electronic structure calculations and FSSH MD simulations identify the dominant relaxation path as IC via CIs located along an E/Z isomerism coordinate.173,174 As Fig. 12(a) shows, population evolving through this CI can either reform UA in its global minimum ground state geometry (the E isomer) or branch into the minimum of the Z isomer. Both routes are thought to confer photostability, since the PE profiles suggest the reversible reformation of E or Z isomers by re-excitation.
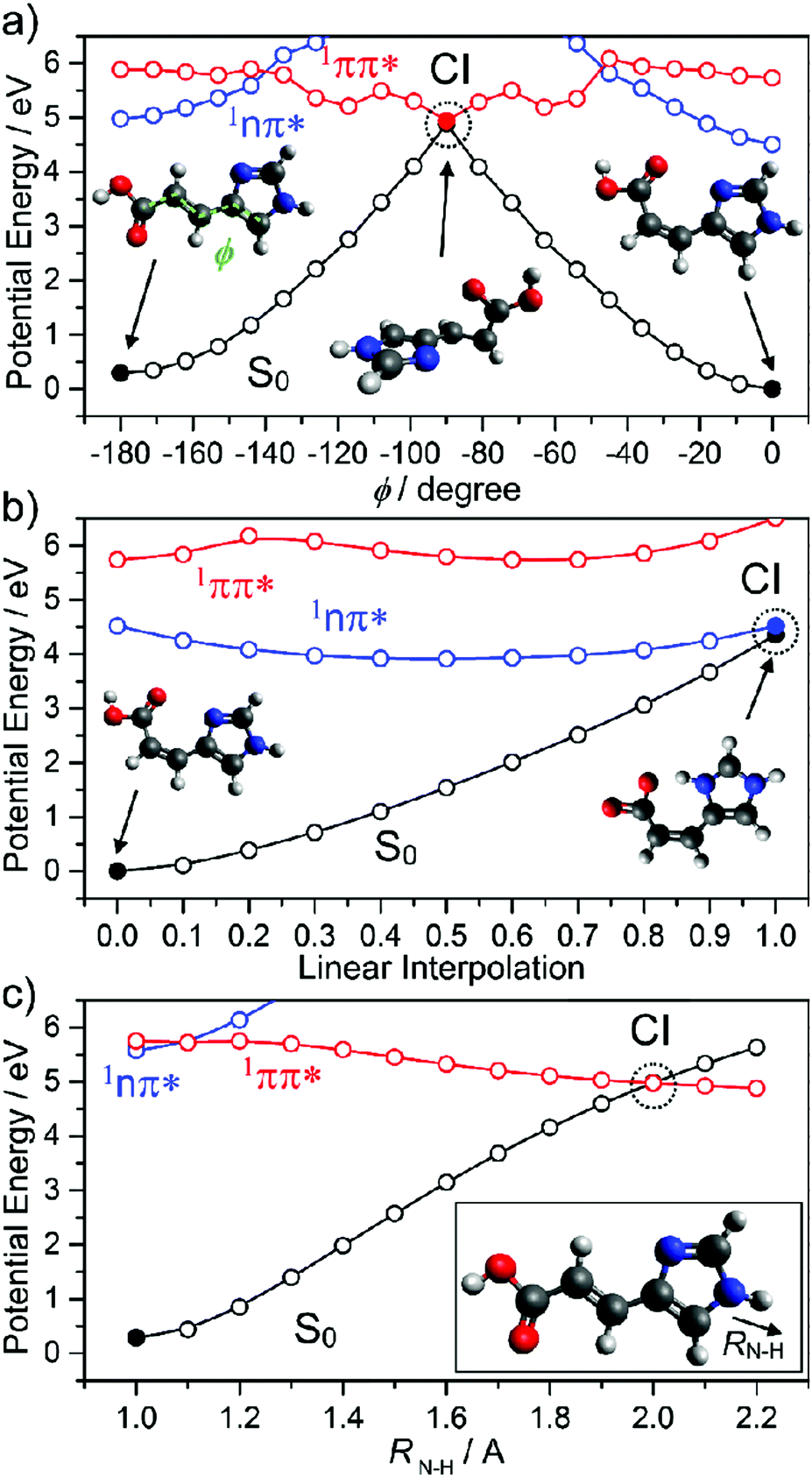 | ||
| Fig. 12 Potential energy curves of the lowest three singlet states of urocanic acid as functions of (a) E/Z isomerism (φ), (b) the intramolecular proton transfer that may occur after E/Z isomerisation and (c) the potentially deleterious N–H bond fission (RN–H). The data are taken from ref. 173. | ||
However, we note an additional caveat. The lowest energy Z isomer has an intramolecular hydrogen bond between the OH (of the carboxylic acid group) and an acceptor N atom in the imidazole ring. This introduces another possible excited state decay path, PCET, which could drive reformation of the ground state parent molecule (and thus photostability). But it could also lead to formation of an imino product, which would likely act to reduce the efficacy of UA as a sunscreen. Recalling Section 2, we recognise πσ* state-mediated N–H bond fission as another possible (and potentially harmful) excited state decay pathway if UA is exposed to shorter (i.e. UV-C) wavelengths.
The final but arguably the most important class of natural sunscreen constituents considered here are epidermal based melanins. As outlined in Section 1, eumelanin and pheomelanin are ubiquitous in the human skin and contain structural elements that appear to be specifically adapted to filtering UVR. Eumelanin is a dark pigment, with common polymeric chains of cross-linked 5,6-dihydroxyindole (DHI), indole-5,6-dione, hydroxyindolone and 5,6-dihydroxyindole-2-carboxylic acid (DHICA) motifs (recall Fig. 2). The structure(s) of eumelanin are still under debate,175 but oligomeric structures (shown in Fig. 13) are recognised with branched and π-stacked configurations. Many theoretical studies have sought to decompose and interpret the measured absorption profile in terms of weighted contributions from different plausible motifs.
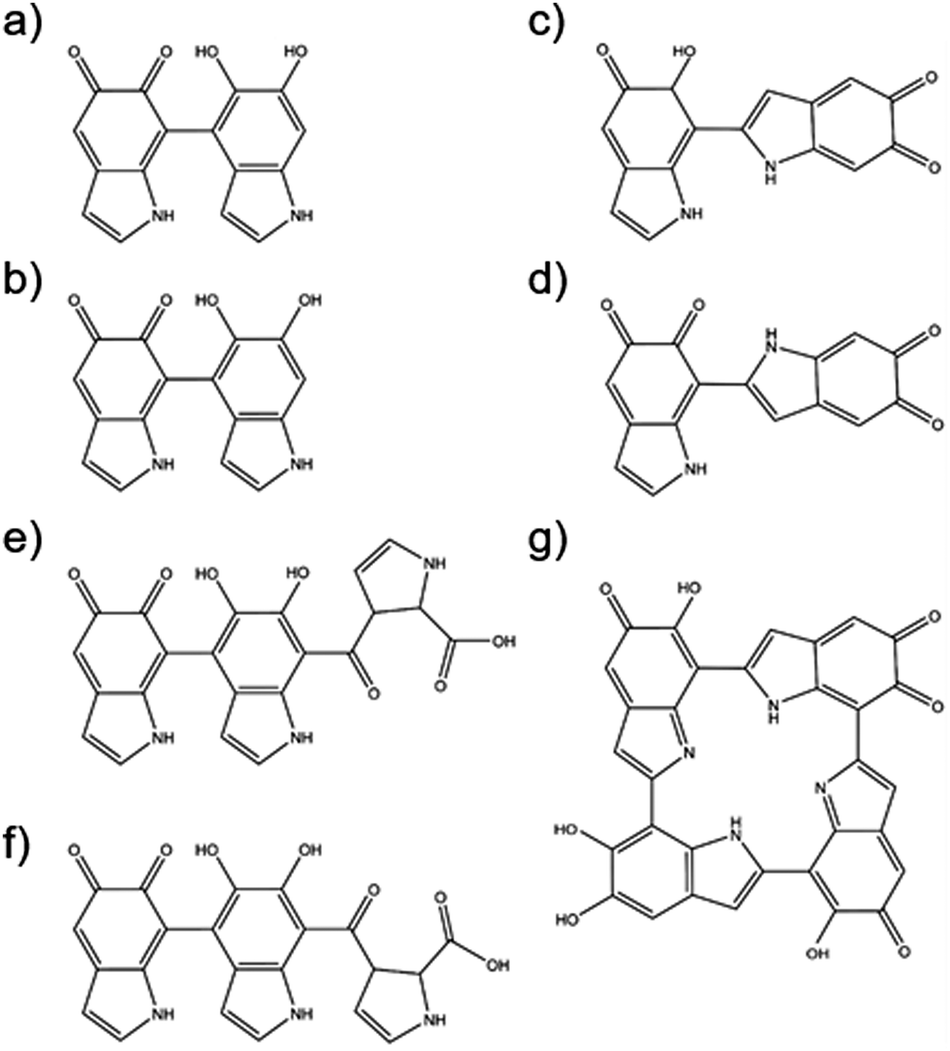 | ||
| Fig. 13 Structures associated with various polymeric configurations of eumelanin. | ||
Thus, for example, it is now concluded that absorption associated with oligomer chains containing fully hydrogenated DHIs lies in the near-UV, whereas that from partially (i.e. hydroxylindolone) or fully (i.e. indole-5,6-dione) dehydrogenated oligomers also contributes to the long wavelength tail of the measured absorption spectrum. Even the presence of just one non-fully hydrogenated form in the oligomer chain leads to a large bathochromic shift in the vertical excitation energy (cf. bare DHI).175
The excited state photophysics occurring following photoexcitation of small building blocks and/or oligomeric units present in melanins has been explored computationally and via solution-phase, ultrafast pump–probe studies.157,176–179 Electronic structure calculations on DHI180,181 suggest facile πσ*-state mediated O–H bond extension following π* ← π excitation (recall Section 2) which, in this case, has the net effect of migrating an H atom from the pendant OH to a neighbouring ring carbon atom. Photoprotection in this scenario is afforded primarily by the resulting isomeric species, 6-hydroxy-4-dihydro-indol-5-one, which is predicted to absorb strongly in the UV and visible spectral regions and to support excited states that undergo ultrafast IC to the S0 state via a PCET mechanism.
Recent ultrafast pump–probe studies of DHICA and DHICA-derived oligomers offer another insight into the potentially complex modus operandi of natural sunscreen components.182 These studies conclude that DHICA dimers are almost as efficient at dissipating UV-energy as the full polymeric pigment, and more than 1000-times more efficient in this role than the bare monomer – suggesting that the stellar photoprotective properties of black eumelanin pigments are a particular property of coupled DHICA units. Further, the authors identify two ultrafast excited state decay mechanisms (both intra- and intermolecular PT pathways) and find the dissipation rates to be relatively insensitive to the details of the sub-unit couplings.182 Again, this deduced multiplicity of efficient decay pathways accords with the view that nature will have selected a particularly robust photoprotection strategy.
However, we end this section by reiterating the point that natural sunscreens are unlikely to be completely free of deleterious side effects. As noted earlier in this Review, melanin – known for its photoprotective properties – has recently also been shown to be potentially carcinogenic.36 Similarly, the Z-isomer of urocanic acid has been observed to contribute to UV-induced skin cancer, likely through immunosuppression.183,184
Molecular constituents in commercial sunscreens
We now focus attention on chemical filters that find use in commercial sunscreen lotions designed to work alongside natural sunscreens and provide additional UV protection. Fig. 14 shows selected chemical filter molecules currently used in commercial sunscreens.185 These broadly partition into seven classes, i.e. derivatives of: para-aminobenzoates; cinnamates; salicylates; anthranilates; camphor; dibenzoyl methanes; and benzophenones. All bear at least some resemblance to components present in plant sunscreens.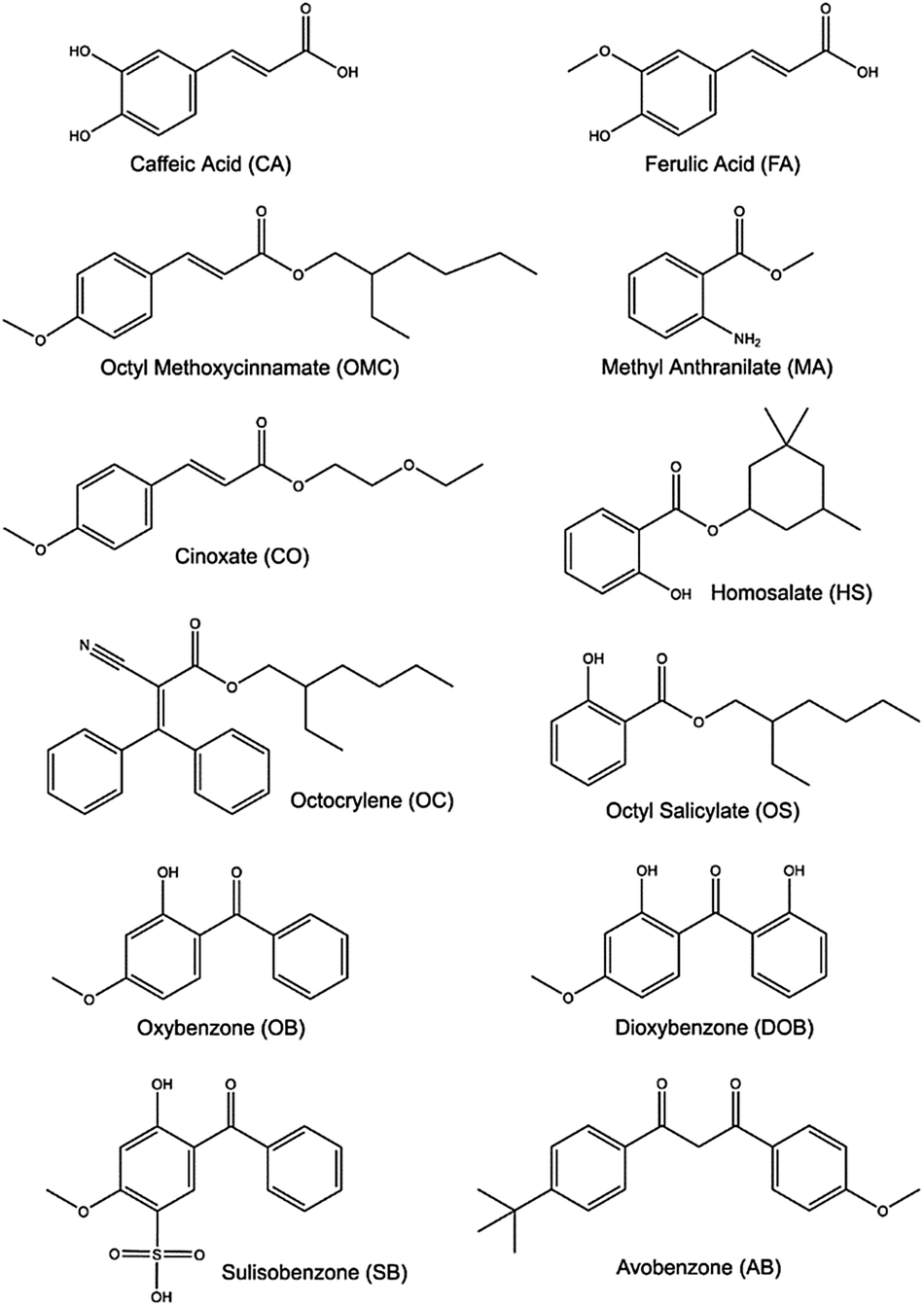 | ||
| Fig. 14 Structures of some of the molecular constituents present in commercially available sunscreen products. | ||
Aromatic acids are the basis for several of these chemical filters, including caffeic acid (CA) and ferulic acid (FA). The latter finds greater use in commercial sunscreens, on account of its superior photostability.186 Both can be viewed as substituted phenols and, as such, both could be prone to decay via πσ* state-mediated O–H bond fission following UV excitation. CA has two OH groups, only one of which can be ‘protected’ by an intramolecular H-bond, whereas the O–CH3 bond is the pendant bond in the lowest energy conformer of FA. Analogy with prior (gas-phase) photofragmentation studies of phenol, anisole and catechol suggests that the free O–H bond in CA will be most susceptible to photodissociation.187–189 Any such excited state bond fission will be in direct competition with non-radiative IC paths and thus reduce the photostability of CA. Several theoretical studies have addressed the strong near-UV absorption of FA,190–193 while others have used highly correlated multi-reference methods to explore its possible excited state decay mechanisms.191 These identify E → Z isomerism (Fig. 15), reminiscent of that prevailing in UA, as the dominant relaxation path following UV excitation of FA.
 | ||
| Fig. 15 Potential energy curves of the lowest four singlet states of ferulic acid computed along the E/Z isomerism coordinate (φ). The data are taken from ref. 191. | ||
CA and FA can be viewed as building blocks for the more complex aromatic esters included in many commercial sunscreen products. Examples of such esters include octyl methoxycinnamate (OMC), cinoxate (CO), methyl anthranilate (MA), octocrylene (OC), homosalate (HS), and octyl salicylate (OS). As Fig. 14 shows, the structures of CO, OMC and OC have some analogies with FA and CA, and all contain aliphatic side-chains with conjugated CC double bonds. Thus, it is reasonable to predict that similar E/Z isomerism pathways will contribute to the ultrafast decay of these species following absorption of UVR.
OMC is one of the most studied of these aromatic esters, and is chosen here as the first of three exemplars. Its excited state photophysics has been explored via both gas- and solution-phase experiments. Frequency-resolved gas-phase measurements reveal the existence of multiple rotamers.194 Linewidth analysis implies a sub-ps lifetime for the 11ππ* excited state. However, TR-MS measurements using a 193 nm probe to ionise excited state molecules formed following UV excitation to the 11ππ* origin level returned an excited state lifetime of ∼20 ns.194 These apparently contradictory findings were corroborated by later TR-MS studies with much higher pump–probe time resolution,123 and confirmed the previously proposed operation of two photophysical processes: ultrafast non-adiabatic coupling from the ‘bright’ 11ππ* state to an optically ‘dark’ (in terms of absorption from the S0 state) 1nπ* state, which then decays on a much slower timescale. Such a mechanism is reminiscent of the excited state behaviour summarised earlier in the case of sinapoyl malate, and consistent with the deduced energetic ordering of the 1ππ* and 1nπ* excited states in several other cinnamate derivatives.195–197 Neither TR-MS experiment used sufficiently energetic probe photons to confirm (or refute) the assumption that the slower decay provides a measure of the rate of E → Z isomerism required to couple population from the 1nπ* state to the S0 state. Such conclusions are also consistent with those advanced to explain TR-MS gas-phase results obtained for the simpler analogue, methyl-4-methoxycinnamate,198 though a recent combined experimental and computational (TD-DFT) study has also argued the need for a triplet state pathway to reconcile the observed dynamics.199
Efforts to extend such measurements to the solution-phase include gas-phase microsolvation studies involving MMC.194,197,198 TR-MS studies of MMC–H2O clusters show that microsolvation accelerates the excited state relaxation. This has been explained by assuming that the proximity of the polar H2O molecule destabilises the 1nπ* state, thereby reducing (or even switching off) the coupling between the 1ππ* and 1nπ* states and allowing the rival 1ππ* S0 IC process (again mediated by E → Z isomerism) to be kinetically competitive.196,198,200
Solution-phase studies have led to similar conclusions. The NMR data shown in Fig. 16(a) confirms that steady-state UV illumination of the lower energy (E-) isomer of OMC in cyclohexane and methanol results in some conversion to the Z-isomer.123Fig. 16(b) shows that the molar extinction coefficient of Z-OMC in the UV-A/B spectral region is significantly smaller than that of E-OMC. Thus the observed decline in the peak absorbance of samples comprising initially pure E-OMC in cyclohexane or methanol upon prolonged exposure to UVR provides further (indirect) evidence for E → Z isomerism.201 Time-resolved fluorescence measurements for both E-OMC and Z-OMC in cyclohexane solution returned excited state lifetimes <30 ps (the instrument response time).201,202 These studies served to encourage higher time-resolution pump–probe transient electronic absorption measurements that provide further insights into the early time dynamics following photoexcitation at the wavelength of peak absorption (λmax) of E-OMC, in both cyclohexane and in methanol.123 Specifically, the E-OMC (1ππ*) molecules prepared by photoexcitation are deduced to distort along the E → Z isomerisation coordinate and couple to a region of CI with the S0 state with a 1–2 ps time constant; a much longer (>ns) transient feature was assigned to the long-lived Z-OMC isomer. Again, we note the obvious similarities between the excited state decay mechanisms deduced for this synthetic chemical filter and for the natural sunscreen sinapoyl malate.
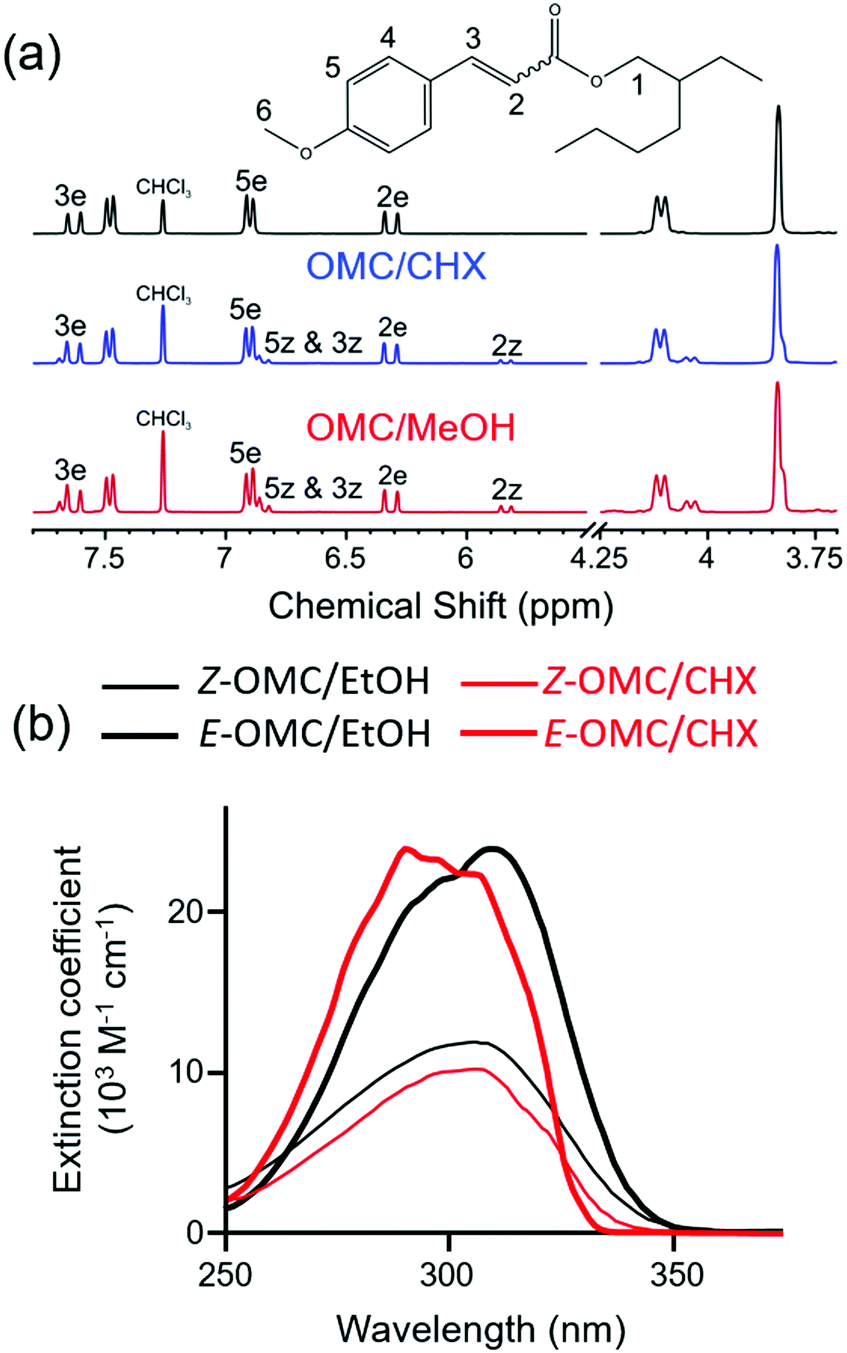 | ||
| Fig. 16 (a) 1H NMR spectra of E-OMC and peak assignments following irradiation at λmax = 290 nm (CHX) and 316 nm (methanol). Adapted from ref. 123. (b) Absorption spectra of E and Z OMC in cyclohexane (CHX) and ethanol (EtOH). Adapted from ref. 201. | ||
Benzophenones are generally the most abundant synthetic chemical filters in commercial sunscreens. Examples include oxybenzone (OB), dioxybenzone (DOB), sulisobenzone (SB) and avobenzone (AB) – see Fig. 14 – the simplest of which is OB, our second exemplar, for which we showed transient absorption data in Section 3. Recent theory191,203 and transient absorption experiments (exciting OB at 325 nm, in cyclohexane and in methanol solution)110,204 both conclude that PCET, wherein the OH centred proton migrates to the carbonyl O atom (i.e. enol–keto tautomerism), represents the dominant relaxation path. Photoexcitation populates the planar S2 (11ππ*) state, in which the π and π* orbitals are delocalised over the entire molecule. PCET is mediated by coupling and subsequent IC to a (optically dark) S1(11nπ*) state, which develops progressive charge transfer character upon migration of the proton from the OH to the CO, on a ∼100 fs timescale. The S1 state links to the S0 state via a 11nπ*/S0 CI, the minimum energy geometry of which requires loss of planarity (one ring twists relative to the other about the central aliphatic C–C bond). Following IC, the time constant for which is ∼400 fs, reverse H atom transfer on the S0 PES and vibrational energy transfer to the surrounding solvent results in reformation of the original enol isomer with a (solvent dependent) time constant in the range ∼5–8 ps. The ground state population does not fully recover within the maximum available experimental time delay (2 ns), leading to the suggestion that ∼10% of the excited state molecules form a photoproduct. Transient vibrational absorption studies identify the presence of the E-keto isomer (a logical product following full 180° rotation about the C–C bond).110,204 Other studies have suggested some phenoxyl radical formation.205 DOB and SB are structurally similar to OB, and we thus anticipate similarities in the decay mechanisms of their excited states formed upon exposure to UV-A/-B.205
Our final case study is avobenzone (AB). AB also has obvious structural similarities with OB but, as Fig. 14 shows, its lowest energy tautomer lacks an OH group. Nonetheless, AB tends to exist in a H-bonded enol form (characterised by an intramolecular H-bond),206 and the most detailed current picture regarding its excited state photophysics stems from transient absorption studies of AB in cyclohexane, acetonitrile and methanol by Crim and coworkers.207 The excited H-bonded enol form (termed a chelated enol in the original work)207 of AB formed by absorption of a 350 nm photon is deduced to decay on a sub-ps timescale, yielding several different non-H-bonded (or non-chelated) enols. One of the proposed non-H bonded structures simply involves 180° torsion of the O–H bond, but others involve E → Z isomerism about the C–C bond adjacent to one or other carbonyl group. The relative yields of these various non-H-bonded forms, and the rates with which they relax to the starting S0 (H-bonded) state are sensitive to the choice of solvent.206,207 As in so many of these examples, complete ground state recovery is not observed, implying some (low) probability for forming a long-lived photoproduct, possibly a stable keto-AB isomer.207,208
5. Discussion
Manipulating the photostability and efficacy of chemical filters
One key consideration in any sunscreen development is that a filter should absorb at the relevant UV-A/-B wavelengths, be photostable, and exhibit one or more efficient, non-radiative, photodeactivation pathways.48 The examples considered in this review display such pathways, but identifying and understanding what characteristics are key to ensuring the required photostability and photoprotective efficiency in one chemical filter could guide our manipulation of such properties in another. As shown in Section 4, details of the excited state photophysics can vary with choice of solvent and/or the solvent polarity,207,209 but this is unlikely to be a viable option for a commercial product.Another manipulation strategy could be to alter the electron density distribution in the filter molecule by, for example, chemical substitution. Simply introducing an electron donating or withdrawing group – at ring positions remote from the reaction coordinate – has been shown to have major impact on the rate of the πσ* state-mediated O–H bond fission process in a range of substituted phenols.210 It is not unrealistic to imagine that a similar aromatic substitution strategy might affect the UV photodynamics displayed by, for example, some of the cinnamate species used as chemical filters – boosting their deactivation efficiency by, for example, increasing the relaxation rate and/or the probability of parent ground state recovery.
Pursuing a synthetic line of thinking further, can we learn from nature and improve the photostability of a given chemical filter by coupling the monomer units – covalently, as in DHICA,182,211 or non-covalently,212 as in the case of the DNA-bases? Such questions apply not just to chemical filters currently used in sunscreen formulations but also, and perhaps more importantly, to potential filters that might have been investigated in the past and rejected on the basis that they displayed inadequate photostability when in monomer form.
Outlook of challenges and future prospects
This review began by summarising the natural photoprotective mechanisms shown by humans in response to changing levels of UVR, and highlighted situations when such mechanisms are inadequate and the burden-of-disease increases (Fig. 1). Sunscreens are now the almost universal solution to pre-empting overexposure to UVR. Given their prevalence, it is perhaps inevitable that the efficacy and the health consequences of sunscreen use continue to attract close scrutiny.12,14,15This review focuses on the photophysical properties of chemical filters, and how these are increasingly being revealed using combinations of theory and ultrafast laser chemistry. Gas-phase measurements together with ab initio electronic structure calculations can reveal the ultrafast excited state photophysics in isolated chemical filter molecules. Solution-phase studies take us a step closer to the actual environment in which such chemical filters are used as a sunscreen, and allow study of the full deactivation pathway back to the starting ground state molecule. Such studies provide a useful first cut at assessing the likely efficacy of a chemical filter, and to screen for the formation of rival photoproducts – which can be the root cause of some of the adverse effects associated with sunscreen use.54
Following such a ‘bottom-up’ approach has resulted in impressive advances in our understanding of the ultrafast decay following UV photoexcitation of many of these chemical filters, but many questions remain to be answered before we can start to claim to understand the full gamut of photoprotective mechanisms provided in nature. This review ends by outlining some of the outstanding questions.
(i) Given that sunscreen products contain many tens of individual components, how are the dynamics of one component affected by the presence of another? This question is not new. Many components are included in commercial sunscreens in particular combinations precisely because of the way they interact. Adding octocrylene, for example, is a recognised route to enhancing the photostability of avobenzone.48,213–216 The interactions between different ingredients need not be synergistic, however, so it remains important to examine the behaviour of one component in the presence of others. Extending the types of experimental study reviewed here to include multiple components is relatively simple if the signals from each are well separated – spectrally or temporally – as demonstrated, for example, by our own recent ultrafast UV pump–probe studies of oxybenzone in the presence of titanium dioxide (one of the commonly used inorganic filters).217
(ii) Are any photoproducts safe? Many studies deduce (via observation of an incomplete ground state bleach recovery) or actually observe photoproduct formation. Identifying that photoproduct can be challenging, however. NMR and GC-MS have shown success in identifying products after photoexcitation.123,218 Identification alone does not ensure adequate evaluation of the safety of the component in a sunscreen product, however. Any full assessment also requires that we investigate the fates of the (potentially reactive) excited states formed when these photoproducts themselves interact with UV light. This could be problematic if photoexcitation results in radical formation, for example. Armed with such knowledge, however, it may be possible to mitigate the problem by adding a suitable radical scavenger species to the blend. Further, we recognise that natural sunscreens such as melanins are continually replaced as they degrade. Conventional sunscreen products, in contrast, require frequent re-applications. A robust sunscreen that requires less frequent replacement would be particularly attractive to the end-user.
(iii) If, as has been demonstrated in several cases within this review, the excited state dynamics displayed by a chemical filter change upon switching from the gas- to solution-phase, what happens in an actual sunscreen product? Unarguably, studying the chemical filter in the solution-phase provides a better approximation (cf. the gas-phase) of the environment prevailing in a sunscreen, but it is still quite far from the case of the chemical filter in a formulation containing many other components in the form of an oil or paste. The challenge presented by multi-molecular systems has been touched on in (i) above, but the fact that the solution-phase remains a poor mimic of the native environment in a commercial product still needs to be addressed. Studies involving thin-film samples are a logical next step,157,219 while clinical trials involving skin samples will inevitably remain necessary for evaluating the safety and efficacy of any product in ways that are not assessable via any of the photophysical techniques considered in this review.
Acknowledgements
We are grateful to the Engineering and Physical Sciences Research Council for the following funding: EP/L005913 (Bristol), EP/J007153 (Warwick) and EP/F500378 (that provides a studentship for L. A. B.). V. G. S. is grateful to the Royal Society and the Leverhulme Trust for a Royal Society Leverhulme Trust Senior Research Fellowship, and B. M. and T. N. V. K. are both grateful to the Technical University of Munich for awards of T. U. Munich Fellowships.Notes and references
- A. S. Aldahan, V. V. Shah, S. Mlacker and K. Nouri, JAMA Dermatol., 2015, 151, 1316 CrossRef PubMed.
- C. D. Kaur and S. Saraf, Pharmacogn. Res., 2010, 2, 22–25 CrossRef CAS PubMed.
- A. Maciejewski, R. Naskrecki, M. Lorenc, M. Ziolek, J. Karolczak, J. Kubicki, M. Matysiak and M. Szymanski, J. Mol. Struct., 2000, 555, 1–13 CrossRef CAS.
- F. Urbach, J. Photochem. Photobiol., B, 2001, 64, 99–104 CrossRef CAS.
- P. E. Hockberger, Photochem. Photobiol., 2002, 76, 561–579 CrossRef CAS PubMed.
- M. Lorenzen, V. Racicot, D. Strack and C. Chapple, Plant Physiol., 1996, 112, 1625–1630 CrossRef CAS PubMed.
- A. Bachem and B. Fantus, Arch. Phys. Ther., 1939, 20, 69–76 CAS.
- U. Henschke, Strahlentherapie, 1940, 67, 659–667 Search PubMed.
- A. C. Giese, E. Christensen and J. Jeppard, J. Am. Pharm. Assoc., 1950, 39, 30–36 CrossRef CAS.
- N. J. Lowe, Dermatol. Clin., 2006, 24, 9–17 CrossRef CAS PubMed.
- J. M. Knox, A. C. Griffin and R. E. Hakim, J. Invest. Dermatol., 1960, 34, 51–58 CrossRef CAS PubMed.
- M. Lodèn, H. Beitner, H. Gonzalez, D. W. Edström, U. Åkerström, J. Austad, I. Buraczewska-Norin, M. Matsson and H. C. Wulf, Br. J. Dermatol., 2011, 165, 255–262 CrossRef PubMed.
- R. Jansen, U. Osterwalder, S. Q. Wang, M. Burnett and H. W. Lim, J. Am. Acad. Dermatol., 2013, 69, 867 CrossRef PubMed.
- S. E. Mancebo, J. Y. Hu and S. Q. Wang, Dermatol. Clin., 2014, 32, 427–438 CrossRef CAS PubMed.
- M. E. Burnett and S. Q. Wang, Photodermatol., Photoimmunol. Photomed., 2011, 27, 58–67 CrossRef CAS PubMed.
- M. Linh Tran, B. J. Powell and P. Meredith, Biophys. J., 2006, 90, 743–752 CrossRef PubMed.
- L. A. Baker, M. D. Horbury, S. E. Greenough, F. Allais, P. S. Walsh, S. Habershon and V. G. Stavros, J. Phys. Chem. Lett., 2016, 7, 56–61 CrossRef CAS PubMed.
- J. F. Kasting and J. L. Siefert, Science, 2002, 296, 1066–1068 CrossRef CAS PubMed.
- R. P. Sinha and D.-P. Häder, Photochem. Photobiol. Sci., 2002, 1, 225–236 CAS.
- A. Sancar, L. A. Lindsey-Boltz, K. Ünsal-Kaçmaz and S. Linn, Annu. Rev. Biochem., 2004, 73, 39–85 CrossRef CAS PubMed.
- R. Lucas, T. McMichael, W. Smith and B. Armstrong, Solar Ultraviolet Radiation. Global burden of disease from solar ultraviolet radiation, World Health Organization, Geneva, 2006 Search PubMed.
- M. F. Holick, Am. J. Clin. Nutr., 2004, 80, 1678S–1688S CAS.
- M. F. Holick, N. Engl. J. Med., 2007, 357, 266–281 CrossRef CAS PubMed.
- L. A. Baker and V. G. Stavros, Sci. Prog., 2016, 99, 282–311 CrossRef.
- R. S. Mason and J. Reichrath, Adv. Anticancer Agents Med. Chem., 2013, 13, 83–97 CrossRef CAS.
- A. Slominski, D. J. Tobin, S. Shibahara and J. Wortsman, Physiol. Rev., 2004, 84, 1155–1228 CrossRef CAS PubMed.
- H. Fedorow, F. Tribl, G. Halliday, M. Gerlach, P. Riederer and K. L. Double, Prog. Neurobiol., 2005, 75, 109–124 CrossRef CAS PubMed.
- S. Ito, Pigm. Cell Res., 2003, 16, 230–236 CrossRef CAS.
- K. Wakamatsu and S. Ito, Pigm. Cell Res., 2002, 15, 174–183 CrossRef CAS.
- M. Cichorek, M. Wachulska, A. Stasiewicz and A. Tymińska, Postępy. Dermatol. Alergol., 2013, 30, 30–41 CrossRef PubMed.
- M. Brenner and V. J. Hearing, Photochem. Photobiol., 2008, 84, 539–549 CrossRef CAS PubMed.
- X. Wu and J. A. Hammer, Curr. Opin. Cell Biol., 2014, 29, 1–7 CrossRef CAS PubMed.
- H. Ando, Y. Niki, M. Yoshida, M. Ito, K. Akiyama, J.-H. Kim, T.-J. Yoon, M. S. Matsui, D. B. Yarosh and M. Ichihashi, Cell. Logist., 2011, 1, 12–20 CrossRef PubMed.
- J. P. Ortonne, Br. J. Dermatol., 2002, 146, 7–10 CrossRef CAS PubMed.
- H. Y. Park, M. Kosmadaki, M. Yaar and B. A. Gilchrest, Cell. Mol. Life Sci., 2009, 66, 1493–1506 CrossRef CAS PubMed.
- S. Premi, S. Wallisch, C. M. Mano, A. B. Weiner, A. Bacchiocchi, K. Wakamatsu, E. J. H. Bechara, R. Halaban, T. Douki and D. E. Brash, Science, 2015, 347, 842–847 CrossRef CAS PubMed.
- U. Osterwalder, M. Sohn and B. Herzog, Photodermatol., Photoimmunol. Photomed., 2014, 30, 62–80 CrossRef.
- G. I. Jenkins, Annu. Rev. Plant Biol., 2009, 60, 407–431 CrossRef CAS PubMed.
- H. Frohnmeyer and D. Staiger, Plant Physiol., 2003, 133, 1420–1428 CrossRef CAS PubMed.
- K. Meyer, J. C. Cusumano, C. Somerville and C. C. S. Chapple, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 6869–6874 CrossRef CAS.
- C. M. Fraser and C. Chapple, Arabidopsis Book, 2011, vol. 9, p. e0152 Search PubMed.
- M. S. Eller and B. A. Gilchrest, Pigm. Cell Res., 2000, 13, 94–97 Search PubMed.
- J. A. Levine, M. Sorace, J. Spencer and D. M. Siegel, J. Am. Acad. Dermatol., 2005, 53, 1038–1044 CrossRef PubMed.
- N. A. Kasparian, J. K. McLoone and B. Meiser, J. Behav. Med., 2009, 32, 406–428 CrossRef PubMed.
- M. D. Palm and M. N. O'Donoghue, Dermatol. Ther., 2007, 20, 360–376 CrossRef PubMed.
- S. González, M. Fernández-Lorente and Y. Gilaberte-Calzada, Clin. Dermatol., 2008, 26, 614–626 CrossRef PubMed.
- S. Q. Wang, Y. Balagula and U. Osterwalder, Dermatol. Ther., 2010, 23, 31–47 CrossRef PubMed.
- S. Forestier, J. Am. Acad. Dermatol., 2008, 58, S133–S138 CrossRef PubMed.
- J. S. Adams and M. Hewison, J. Clin. Endocrinol. Metab., 2010, 95, 471–478 CrossRef PubMed.
- M. I. Cabrera, O. M. Alfano and A. E. Cassano, J. Phys. Chem., 1996, 100, 20043–20050 CrossRef CAS.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 7–30 CrossRef PubMed.
- T. G. Smijs and S. Pavel, Nanotechnol., Sci. Appl., 2011, 4, 95–112 CrossRef CAS PubMed.
- M. Krause, A. Klit, M. B. Jensen, T. Søeborg, H. Frederiksen, M. Schlumpf, W. Lichtensteiger, N. E. Skakkebaek and K. T. Drzewiecki, Int. J. Androl., 2012, 35, 424–436 CrossRef CAS PubMed.
- J. F. Nash and P. R. Tanner, Photoder, 2014, 30, 88–95 CrossRef CAS PubMed.
- A. Jabloński, Nature, 1933, 131, 839–840 CrossRef.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry: An Introduction, University Science Books, 2009 Search PubMed.
- F. Bernardi, M. Olivucci and M. A. Robb, Chem. Soc. Rev., 1996, 25, 321–328 RSC.
- G. A. Worth and L. S. Cederbaum, Annu. Rev. Phys. Chem., 2004, 55, 127–158 CrossRef CAS PubMed.
- S. Matsika and P. Krause, Annu. Rev. Phys. Chem., 2011, 62, 621–643 CrossRef CAS PubMed.
- D. R. Yarkony, Rev. Mod. Phys., 1996, 68, 985–1013 CrossRef CAS.
- S. E. Greenough, M. D. Horbury, J. O. F. Thompson, G. M. Roberts, T. N. V. Karsili, B. Marchetti, D. Townsend and V. G. Stavros, Phys. Chem. Chem. Phys., 2014, 16, 16187–16195 RSC.
- D. Murdock, S. J. Harris, T. N. V. Karsili, G. M. Greetham, I. P. Clark, M. Towrie, A. J. Orr-Ewing and M. N. R. Ashfold, J. Phys. Chem. Lett., 2012, 3, 3715–3720 CrossRef CAS PubMed.
- R. Schinke, Photodissociation Dynamics, Cambridge University Press, Cambridge, 1993 Search PubMed.
- V. Engel, V. Staemmler, R. L. Vanderwal, F. F. Crim, R. J. Sension, B. Hudson, P. Andresen, S. Hennig, K. Weide and R. Schinke, J. Phys. Chem., 1992, 96, 3201–3213 CrossRef CAS.
- M. N. R. Ashfold, D. H. Mordaunt and S. H. S. Wilson, Advances in Photochemistry, John Wiley and Sons, New York, 1996 Search PubMed.
- M. N. R. Ashfold and S. R. Langford, The Role of Rydberg States in Spectroscopy and Photochemistry, Kluwer Academic Press, Dordrecht, Netherlands, 1999 Search PubMed.
- S. A. Harich, D. W. H. Hwang, X. F. Yang, J. J. Lin, X. M. Yang and R. N. Dixon, J. Chem. Phys., 2000, 113, 10073–10090 CrossRef CAS.
- S. Satyapal, J. Park, R. Bersohn and B. Katz, J. Chem. Phys., 1989, 91, 6873–6879 CrossRef CAS.
- Y. Wen, J. Segall, M. Dulligan and C. Wittig, J. Chem. Phys., 1994, 101, 5665–5671 CrossRef CAS.
- S.-H. Lee, H.-I. Lee and Y. T. Lee, J. Chem. Phys., 2004, 121, 11053–11059 CrossRef CAS PubMed.
- S. Harich, J. J. Lin, Y. T. Lee and X. Yang, J. Phys. Chem. A, 1999, 103, 10324–10332 CrossRef CAS.
- K. J. Yuan, Y. Cheng, F. Y. Wang and X. M. Yang, Chin. J. Chem. Phys., 2008, 21, 301–307 CrossRef CAS.
- S. H. S. Wilson, J. D. Howe and M. N. R. Ashfold, Mol. Phys., 1996, 88, 841–848 CrossRef CAS.
- M. N. R. Ashfold, G. A. King, D. Murdock, M. G. D. Nix, T. A. A. Oliver and A. G. Sage, Phys. Chem. Chem. Phys., 2010, 12, 1218–1238 RSC.
- C. Xie, J. Ma, X. Zhu, D. R. Yarkony, D. Xie and H. Guo, J. Am. Chem. Soc., 2016, 138, 7828–7831 CrossRef CAS PubMed.
- X. Xu, J. Zheng, K. R. Yang and D. G. Truhlar, J. Am. Chem. Soc., 2014, 136, 16378–16386 CrossRef CAS PubMed.
- G. M. Roberts, A. S. Chatterley, J. D. Young and V. G. Stavros, J. Phys. Chem. Lett., 2012, 3, 348–352 CrossRef CAS PubMed.
- C. Ratzer, J. Kupper, D. Spangenberg and M. Schmitt, Chem. Phys., 2002, 283, 153–169 CrossRef CAS.
- M. G. D. Nix, A. L. Devine, B. Cronin, R. N. Dixon and M. N. R. Ashfold, J. Chem. Phys., 2006, 125, 133318 CrossRef PubMed.
- R. N. Dixon, T. A. A. Oliver and M. N. R. Ashfold, J. Chem. Phys., 2011, 134, 194303 CrossRef PubMed.
- Y. Zhang, T. A. A. Oliver, M. N. R. Ashfold and S. E. Bradforth, Faraday Discuss., 2012, 157, 141–163 RSC.
- S. Perun, A. L. Sobolewski and W. Domcke, J. Am. Chem. Soc., 2005, 127, 6257–6265 CrossRef CAS PubMed.
- S. Yamazaki, W. Domcke and A. L. Sobolewski, J. Phys. Chem. A, 2008, 112, 11965–11968 CrossRef CAS PubMed.
- V. B. Delchev, A. L. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 2010, 12, 5007–5015 RSC.
- B. Marchetti, T. N. V. Karsili, M. N. R. Ashfold and W. Domcke, Phys. Chem. Chem. Phys., 2016, 18, 20007–20027 RSC.
- R. J. Harrison, G. I. Fann, T. Yanai and Z. Gan, J. Chem. Phys., 2004, 121, 11587–11598 CrossRef CAS PubMed.
- S. Gozem, F. Melaccio, H. L. Luk, S. Rinaldi and M. Olivucci, Chem. Soc. Rev., 2014, 43, 4019–4036 RSC.
- A. L. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 2004, 6, 2763–2771 RSC.
- A. L. Sobolewski and W. Domcke, Chem. Phys. Lett., 1993, 211, 82–87 CrossRef CAS.
- A. L. Sobolewski, Chem. Phys. Lett., 1993, 211, 293–299 CrossRef CAS.
- A. L. Sobolewski and W. Domcke, Chem. Phys., 1994, 184, 115–124 CrossRef CAS.
- Z. Lan, L. M. Frutos, A. L. Sobolewski and W. Domcke, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12707–12712 CrossRef CAS PubMed.
- S. Perun and A. L. Sobolewski, J. Phys. Chem. A, 2006, 110, 9031–9038 CrossRef CAS PubMed.
- A. L. Sobolewski, W. Domcke and C. Hättig, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17903–17906 CrossRef CAS PubMed.
- N. das Neves Rodriguez, M. Staniforth and V. G. Stavros, Proc. R. Soc. A, 2016, 472, 0677 Search PubMed.
- E. G. Buchanan, W. H. James III, S. H. Choi, L. Guo, S. H. Gellman and C. W. Müller, J. Chem. Phys., 2012, 137, 094301 CrossRef PubMed.
- C.-M. Tseng, Y. T. Lee, M.-F. Lin, C.-K. Ni, S.-Y. Liu, Y.-P. Lee, Z. F. Xu and M. C. Lin, J. Phys. Chem. A, 2007, 111, 9463–9470 CrossRef CAS PubMed.
- T. S. Zwier, J. Phys. Chem. A, 2006, 110, 4133–4150 CrossRef CAS PubMed.
- X. M. Yang, Int. Rev. Phys. Chem., 2005, 24, 37–98 CrossRef CAS.
- J. Wei, A. Kuczmann, J. Riedel, F. Renth and F. Temps, Phys. Chem. Chem. Phys., 2003, 5, 315–320 RSC.
- D. A. Blank, S. W. North and Y. T. Lee, Chem. Phys., 1994, 187, 35–47 CrossRef CAS.
- H. Lippert, H.-H. Ritze, I. V. Hertel and W. Radloff, ChemPhysChem, 2004, 5, 1423–1427 CrossRef CAS PubMed.
- R. Montero, Á. P. Conde, V. Ovejas, M. Fernández-Fernández, F. Castaño, J. R. Vázquez de Aldana and A. Longarte, J. Chem. Phys., 2012, 137, 064317 CrossRef PubMed.
- A. Maciejewski, R. Naskrecki, M. Lorenc, M. Ziolek, J. Karolczak, J. Kubicki, M. Matysiak and M. Szymanski, J. Mol. Struct., 2000, 555, 1–13 CrossRef CAS.
- R. Berera, R. van Grondelle and J. T. M. Kennis, Photosynth. Res., 2009, 101, 105–118 CrossRef CAS PubMed.
- U. Megerle, I. Pugliesi, C. Schriever, C. Sailer and E. Riedle, Appl. Phys. B: Lasers Opt., 2003, 96, 215–231 CrossRef.
- M. Towrie, D. C. Grills, J. Dyer, J. A. Weinstein, P. Matousek, R. Barton, P. D. Bailey, N. Subramaniam, W. M. Kwok, C. Ma, D. Phillips, A. W. Parker and M. W. George, Appl. Spectrosc., 2003, 57, 367–380 CrossRef CAS PubMed.
- G. M. Roberts, H. J. B. Marroux, M. P. Grubb, M. N. R. Ashfold and A. J. Orr-Ewing, J. Phys. Chem. A, 2014, 118, 11211–11225 CrossRef CAS PubMed.
- Y. Zhang, K. de La Harpe, A. A. Beckstead, R. Improta and B. Kohler, J. Am. Chem. Soc., 2015, 137, 7059–7062 CrossRef CAS PubMed.
- L. A. Baker, M. D. Horbury, S. E. Greenough, P. M. Coulter, T. N. V. Karsili, G. M. Roberts, A. J. Orr-Ewing, M. N. R. Ashfold and V. G. Stavros, J. Phys. Chem. Lett., 2015, 6, 1363–1368 CrossRef CAS PubMed.
- A. S. Chatterley, C. W. West, V. G. Stavros and J. R. R. Verlet, Chem. Sci., 2014, 5, 3963–3975 RSC.
- S. C. Warren, A. Margineanu, D. Alibhai, D. J. Kelly, C. Talbot, Y. Alexandrov, I. Munro, M. Katan, C. Dunsby and P. M. W. French, PLoS One, 2013, 8, e70687 CAS.
- T. A. Roelofs, C.-H. Lee and A. R. Holzwarth, Biophys. J., 1992, 61, 1147–1163 CrossRef CAS PubMed.
- J. J. Snellenburg, S. Laptenok, R. Seger, K. M. Mullen and I. H. M. Van Stokkum, J. Stat. Softw., 2012, 49, 1–22 Search PubMed.
- G. M. Roberts and V. G. Stavros, Chem. Sci., 2014, 5, 1698–1722 RSC.
- V. G. Stavros and J. R. R. Verlet, Annu. Rev. Phys. Chem., 2016, 67, 211–232 CrossRef CAS PubMed.
- B. Schmidt, S. Laimgruber, W. Zinth and P. A. Gilch, Appl. Phys. B: Lasers Opt., 2003, 76, 809–814 CrossRef CAS.
- M. Sajadi, A. Dobryakov, E. Garbin, N. Ernsting and S. Kovalenko, Chem. Phys. Lett., 2010, 489, 44–47 CrossRef CAS.
- A. Cannizzo, O. Bräm, G. Zgrablic, A. Tortschanoff, A. A. Oskouei, F. van Mourik and M. Chergui, Opt. Lett., 2007, 32, 3555–3557 CrossRef CAS PubMed.
- M. Gerecke, G. Bierhance, M. Gutmann, N. P. Ernsting and A. Rosspeintner, Rev. Sci. Instrum., 2016, 87, 053115 CrossRef PubMed.
- P. Kukura, D. W. McCamant and R. A. Mathies, Annu. Rev. Phys. Chem., 2007, 58, 461–488 CrossRef CAS PubMed.
- K. M. Hanson, S. Narayanan, V. M. Nichols and C. J. Bardeen, Photochem. Photobiol. Sci., 2015, 14, 1607–1616 CAS.
- Y. Peperstraete, M. Staniforth, L. A. Baker, N. D. N. Rodrigues, N. C. Cole-Filipiak, W.-D. Quan and V. G. Stavros, Phys. Chem. Chem. Phys., 2016, 18, 28140–28149 RSC.
- B. O. Roos, P. R. Taylor and P. E. M. Siegbahn, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- L. M. Cheung, K. R. Sundberg and K. Ruedenberg, J. Am. Chem. Soc., 1978, 100, 8024–8025 CrossRef CAS.
- J. Finley, P.-Å. Malmqvist, B. O. Roos and L. Serrano-Andrés, Chem. Phys. Lett., 1998, 288, 299–306 CrossRef CAS.
- K. Andersson and B. O. Roos, Multiconfigurational second-order perturbation theory, Modern Electronic Structure Theory, World Scientific, 1995 Search PubMed.
- K. Ruedenberg, M. W. Schmidt, M. M. Gilbert and S. T. Elbert, Chem. Phys., 1982, 71, 41–49 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- J. F. Stanton and R. J. Bartlett, J. Chem. Phys., 1993, 98, 7029–7039 CrossRef CAS.
- P. Baudin, J. Sánchez Marín, I. García Cuesta and A. M. Sánchez de Merás, J. Chem. Phys., 2014, 140, 104111 CrossRef PubMed.
- A. Dreuw and M. Wormit, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 82–95 CrossRef CAS.
- D. Tuna, N. Došlić, M. Mališ, A. L. Sobolewski and W. Domcke, J. Phys. Chem. B, 2015, 119, 2112–2124 CrossRef CAS PubMed.
- D. Tuna, A. L. Sobolewski and W. Domcke, J. Phys. Chem. A, 2014, 118, 122–127 CrossRef CAS PubMed.
- S. Chaiwongwattana, M. Sapunar, A. Ponzi, P. Decleva and N. Došlić, J. Phys. Chem. A, 2015, 119, 10637–10644 CrossRef CAS PubMed.
- M. Barbatti, J. Am. Chem. Soc., 2014, 136, 10246–10249 CrossRef CAS PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- P. Ren and J. W. Ponder, J. Phys. Chem. B, 2003, 107, 5933–5947 CrossRef CAS.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel and L. Kalé, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- S. Metz, J. Kästner, A. A. Sokol, T. W. Keal and P. Sherwood, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 101–110 CrossRef CAS.
- J. N. Harvey, Faraday Discuss., 2004, 127, 165–177 RSC.
- J. C. Tully, J. Chem. Phys., 1990, 93, 1061–1071 CrossRef CAS.
- M. Barbatti, M. Ruckenbauer, F. Plasser, J. Pittner, G. Granucci, M. Persico and H. Lischka, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 26–33 CrossRef CAS.
- L. Du and Z. Lan, J. Chem. Theory Comput., 2015, 11, 1360–1374 CrossRef CAS PubMed.
- M. Richter, P. Marquetand, J. González-Vázquez, I. Sola and L. González, J. Chem. Theory Comput., 2011, 7, 1253–1258 CrossRef CAS PubMed.
- J. W. Snyder, B. F. E. Curchod and T. J. Martínez, J. Phys. Chem. Lett., 2016, 7, 2444–2449 CrossRef CAS PubMed.
- E. G. Hohenstein, J. Am. Chem. Soc., 2016, 138, 1868–1876 CrossRef CAS PubMed.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- D. V. Evtuguin and F. M. L. Amado, Macromol. Biosci., 2003, 7, 339–343 CrossRef.
- J. D. Gargulak, S. E. Lebo and T. J. McNally, Lignin, Kirk-Othmer Encyclopedia of Chemical Technology, 2015 Search PubMed.
- C. P. Rodrigo, W. H. James and T. S. Zwier, J. Am. Chem. Soc., 2011, 133, 2632–2641 CrossRef CAS PubMed.
- J. D. Young, M. Staniforth, J. C. Dean, G. M. Roberts, F. Mazzoni, T. N. V. Karsili, M. N. R. Ashfold, T. S. Zwier and V. G. Stavros, J. Phys. Chem. Lett., 2014, 5, 2138–2143 CrossRef CAS PubMed.
- B. George, E. Suttie, A. Merlin and X. Deglise, Polym. Degrad. Stab., 2005, 2, 268–274 CrossRef.
- M. Baucher, B. Monties, M. V. Montagu and W. Boerjan, Crit. Rev. Plant Sci., 1998, 2, 125–197 CrossRef.
- Y. Zhang, T. A. A. Oliver, M. N. R. Ashfold and S. E. Bradforth, Faraday Discuss., 2012, 157, 141–163 RSC.
- L. A. Baker, S. E. Greenough and V. G. Stavros, J. Phys. Chem. Lett., 2016, 7, 4655–4665 CrossRef CAS PubMed.
- J. C. Dean, R. Kusaka, P. S. Walsh, F. Allais and T. S. Zwier, J. Am. Chem. Soc., 2014, 136, 14780–14795 CrossRef CAS PubMed.
- F. Liu, L. Du, Z. Lan and J. Gao, Photochem. Photobiol. Sci., 2017, 16, 211–219 CAS.
- T. Gustavsson, R. Improta and D. Markovitsi, J. Phys. Chem. Lett., 2010, 1, 2025–2030 CrossRef CAS.
- R. Improta, F. Santoro and L. Blancafort, Chem. Rev., 2016, 116, 3540–3593 CrossRef CAS PubMed.
- B. Kohler, J. Phys. Chem. Lett., 2010, 1, 2047–2053 CrossRef CAS.
- C. E. Crespo-Hernández, B. Cohen, P. M. Hare and B. Kohler, Chem. Rev., 2004, 104, 1977–2020 CrossRef PubMed.
- K. Röttger, H. J. B. Marroux, M. P. Grubb, P. M. Coulter, H. Böhnke, A. S. Henderson, M. C. Galan, F. Temps, A. J. Orr-Ewing and G. M. Roberts, Angew. Chem., Int. Ed., 2015, 54, 14719–14722 CrossRef PubMed.
- R. W. Schoenlein, L. A. Peteanu, R. A. Mathies and C. V. Shank, Science, 1991, 254, 412–415 CAS.
- T. Kobayashi, T. Saito and H. Ohtani, Nature, 2001, 414, 531–534 CrossRef CAS PubMed.
- N. Ferré and M. Olivucci, J. Am. Chem. Soc., 2003, 125, 6868–6869 CrossRef PubMed.
- D. Polli, P. Altoè, O. Weingart, K. M. Spillane, C. Manzoni, D. Brida, G. Tomasello, G. Orlandi, P. Kukura, R. A. Mathies, M. Garavelli and G. Cerullo, Nature, 2010, 467, 440–443 CrossRef CAS PubMed.
- I. Schapiro, M. N. Ryazantsev, L. M. Frutos, N. Ferré, R. Lindh and M. Olivucc, J. Am. Chem. Soc., 2011, 133, 3354–3364 CrossRef CAS PubMed.
- S. Gozem, F. Melaccio, R. Lindh, A. I. Krylov, A. A. Granovsky, C. Angeli and M. Olivucci, J. Chem. Theory Comput., 2013, 9, 4495–4506 CrossRef CAS PubMed.
- G. Bassolino, T. Sovdat, A. S. Duarte, J. M. Lim, C. Schnedermann, M. Liebel, B. Odell, T. D. W. Claridge, S. P. Fletcher and P. Kukura, J. Am. Chem. Soc., 2015, 137, 12434–12437 CrossRef CAS PubMed.
- P. S. Sherin, J. Grilj, Y. P. Tsentalovich and E. Vauthey, J. Phys. Chem. B, 2009, 113, 4953–4962 CrossRef CAS PubMed.
- D. Tuna, A. L. Sobolewski and W. Domcke, J. Phys. Chem. B, 2014, 118, 976–985 CrossRef CAS PubMed.
- L. Zhao, P.-W. Zhou and G.-J. Zhao, J. Chem. Phys., 2016, 145, 044316 CrossRef PubMed.
- B. Marchetti and T. N. V. Karsili, Phys. Chem. Chem. Phys., 2016, 18, 3644–3658 RSC.
- J. Peon, G. C. Hess, J.-M. L. Pecourt, T. Yuzawa and B. Kohler, J. Phys. Chem. A, 1999, 103, 2460–2466 CrossRef CAS.
- M. Gauden, A. Pezzella, L. Panzella, A. Napolitano, M. d'Ischia and V. Sundström, J. Phys. Chem. B, 2009, 113, 12575–12580 CrossRef CAS PubMed.
- A. Huijser, A. Pezzella, J. K. Hannestad, L. Panzella, A. Napolitano, M. d'Ischia and V. Sundström, ChemPhysChem, 2010, 11, 2424–2431 CrossRef CAS PubMed.
- A. Corani, A. Pezzella, T. Pascher, T. Gustavsson, D. Markovitsi, A. Huijser, M. d'Ischia and V. Sundström, J. Phys. Chem. Lett., 2013, 4, 1383–1388 CrossRef CAS PubMed.
- A. L. Sobolewski and W. Domcke, ChemPhysChem, 2007, 8, 756–762 CrossRef CAS PubMed.
- A. Huijser, A. Pezzellab and V. Sundström, Phys. Chem. Chem. Phys., 2011, 13, 9119–9127 RSC.
- A. Corani, A. Huijser, T. Gustavsson, D. Markovitsi, P.-Å. Malmqvist, A. Pezzella, M. d'Ischia and V. Sundström, J. Am. Chem. Soc., 2014, 136, 11626–11635 CrossRef CAS PubMed.
- V. E. Reeve, G. E. Greenoak, P. J. Canfield, C. Boehm-Wilcox and C. H. Gallagher, Photochem. Photobiol., 1989, 49, 459–464 CrossRef CAS PubMed.
- S. Beissert and K. Loser, Photochem. Photobiol., 2008, 84, 29–34 CAS.
- D. Tuna, A. Udvarhelyi, A. L. Sobolewski, W. Domcke and T. Domratcheva, J. Phys. Chem. B, 2016, 120, 3493–3502 CrossRef CAS PubMed.
- C. Anselmi, M. Centini, M. Maggiore, N. Gaggelli, M. Andreassi, A. Buonocore, G. Beretta and R. M. Facino, J. Pharm. Biomed. Anal., 2008, 46, 645–652 CrossRef CAS PubMed.
- G. A. King, T. A. A. Oliver, R. N. Dixon and M. N. R. Ashfold, Phys. Chem. Chem. Phys., 2012, 14, 3338–3345 RSC.
- A. S. Chatterley, J. D. Young, D. Townsend, J. M. Żurek, M. J. Paterson, G. M. Roberts and V. G. Stavros, Phys. Chem. Chem. Phys., 2013, 15, 6879–6892 RSC.
- D. J. Hadden, C. A. Williams, G. M. Roberts and V. G. Stavros, Phys. Chem. Chem. Phys., 2011, 13, 4494–4499 RSC.
- G. Prampolini, I. Cacelli and A. Ferretti, RSC Adv., 2015, 5, 38513–38526 RSC.
- T. N. V. Karsili, B. Marchetti, M. N. R. Ashfold and W. Domcke, J. Phys. Chem. A, 2014, 118, 11999–12010 CrossRef CAS PubMed.
- G. Mazzone, N. Russo and M. Toscano, Comput. Theor. Chem., 2016, 1077, 39–47 CrossRef CAS.
- A. Urbaniak, M. Szeląg and M. Molski, Comput. Theor. Chem., 2013, 1012, 33–40 CrossRef CAS.
- E. M. M. Tan, M. Hilbers and W. J. Buma, J. Phys. Chem. Lett., 2014, 5, 2464–2468 CrossRef CAS PubMed.
- E. V. Gromov, I. Burghardt, J. T. Hynes, H. Köppel and L. S. Cederbaum, J. Photochem. Photobiol., A, 2007, 190, 241–257 CrossRef CAS.
- X.-P. Chang, C.-X. Li, B.-B. Xie and G. Cui, J. Phys. Chem. A, 2015, 119, 11488–11497 CrossRef CAS PubMed.
- E. M. M. Tan, S. Amirjalayer, B. H. Bakker and W. J. Buma, Faraday Discuss., 2013, 163, 321–340 RSC.
- Y. Miyazaki, K. Yamamoto, J. Aoki, T. Ikeda, Y. Inokuchi, M. Ehara and T. Ebata, J. Chem. Phys., 2014, 141, 244313 CrossRef PubMed.
- K. Yamazaki, Y. Miyazaki, Y. Harabuchi, T. Taketsugu, S. Maeda, Y. Inokuchi, S. Kinoshita, M. Sumida, Y. Onitsuka, H. Kohguchi, M. Ehara and T. Ebata, J. Phys. Chem. Lett., 2016, 7, 4001–4007 CrossRef CAS PubMed.
- D. H. William and I. Fleming, Spectroscopic Methods in Organic Chemistry, McGraw-Hill Education, London, 6th edn, 2008 Search PubMed.
- K. M. Hanson, S. Narayanan, V. M. Nichols and C. J. Bardeen, Photochem. Photobiol. Sci., 2015, 14, 1607–1616 CAS.
- T. M. Karpkird, S. Wanichweacharungruang and B. Albinsson, Photochem. Photobiol. Sci., 2009, 8, 1455–1460 CAS.
- C.-X. Li, W.-W. Guo, B.-B. Xie and G. Cui, J. Chem. Phys., 2016, 145, 074308 CrossRef PubMed.
- L. A. Baker, M. D. Horbury, S. E. Greenough, M. N. R. Ashfold and V. G. Stavros, Photochem. Photobiol. Sci., 2015, 14, 1814–1820 CAS.
- M. T. Ignasiak, C. Houée-Levin, G. Kciuk, B. Marciniak and T. Pedzinski, ChemPhysChem, 2015, 16, 628–633 CrossRef CAS PubMed.
- A. Cantrell and D. J. McGarvey, J. Photochem. Photobiol., B, 2001, 64, 117–122 CrossRef CAS.
- A. D. Dunkelberger, R. D. Kieda, B. M. Marsh and F. F. Crim, J. Phys. Chem. A, 2015, 119, 6155–6161 CrossRef CAS PubMed.
- M. Yamaji and M. Kida, J. Phys. Chem. A, 2013, 117, 1946–1951 CrossRef CAS PubMed.
- M. D. Horbury, L. A. Baker, W.-D. Quan, J. D. Young, M. Staniforth, S. E. Greenough and V. G. Stavros, J. Phys. Chem. A, 2015, 119, 11989–11996 CrossRef CAS PubMed.
- T. N. V. Karsili, A. M. Wenge, S. J. Harris, D. Murdock, J. N. Harvey, R. N. Dixon and M. N. R. Ashfold, Chem. Sci., 2013, 4, 2434–2446 RSC.
- M. Gauden, A. Pezzella, L. Panzella, M. T. Neves-Petersen, E. Skovsen, S. B. Petersen, K. M. Mullen, A. Napolitano, M. d'Ischia and V. Sundström, J. Am. Chem. Soc., 2008, 130, 17038–17043 CrossRef CAS PubMed.
- N. K. Schwalb and F. Temps, J. Am. Chem. Soc., 2007, 129, 9272–9273 CrossRef CAS PubMed.
- L. R. Gaspar and P. M. B. G. Maia-Campos, Int. J. Pharm., 2006, 307, 123–128 CrossRef CAS PubMed.
- B. Herzog, M. Wehrle and K. Quass, Photochemistry, 2009, 85, 869–878 CAS.
- S. Afonso, K. Horita, J. P. Sousa e Silva, I. F. Almeida, M. H. Amaral, P. A. Lobão, P. C. Costa, M. S. Miranda, J. C. G. Esteves da Silva and J. M. Sousa Lobo, J. Photochem. Photobiol., B, 2014, 140, 36–40 CrossRef CAS PubMed.
- V. Lhiaubet-Vallet, M. Marin, O. Jimenez, O. Gorchs, C. Trullas and M. A. Miranda, Photochem. Photobiol. Sci., 2010, 9, 552–558 CAS.
- L. A. Baker, L. C. Grosvenor, M. N. R. Ashfold and V. G. Stavros, Chem. Phys. Lett., 2016, 664, 39–43 CrossRef CAS.
- J. Kockler, L. Oelgemöller, S. Robertson and B. D. Glass, J. Photochem. Photobiol., C, 2012, 13, 91–110 CrossRef CAS.
- K. M. Hanson, E. Gratton and C. J. Bardeen, Free Radical Biol. Med., 2006, 41, 1205–1212 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |