
Titanium coordination compounds: from discrete metal complexes to metal–organic frameworks
Hala
Assi†
 a,
Georges
Mouchaham†
a,
Nathalie
Steunou
a,
Thomas
Devic
*ab and
Christian
Serre
*ac
a,
Georges
Mouchaham†
a,
Nathalie
Steunou
a,
Thomas
Devic
*ab and
Christian
Serre
*ac
aInstitut Lavoisier, UMR CNRS 8180, Université de Versailles St-Quentin en Yvelines, Université Paris Saclay, 45 Avenue des Etats-Unis, 78035 Versailles Cedex, France
bInstitut des Matériaux Jean Rouxel (IMN), UMR 6502, Université de Nantes, CNRS, 2 rue de la Houssinière, BP 32229, 44322 Nantes Cedex 3, France. E-mail: thomas.devic@cnrs-imn.fr
cInstitut des Matériaux Poreux de Paris, FRE 2000 CNRS Ecole Normale Supérieure, Ecole Supérieure de Physique et de Chimie Industrielles de Paris, PSL Research University, 75005 Paris, France. E-mail: christian.serre@ens.fr
First published on 24th May 2017
Abstract
Owing to their promise in photocatalysis and optoelectronics, titanium based metal–organic frameworks (MOFs) are one of the most appealing classes of MOFs reported to date. Nevertheless, Ti-MOFs are still very scarce because of their challenging synthesis associated with a poor degree of control of their chemistry and crystallization. This review aims at giving an overview of the recent progress in this field focusing on the most relevant existing titanium coordination compounds as well as their promising photoredox properties. Not only Ti-MOFs but also Ti-oxo-clusters will be discussed and particular interest will be dedicated to highlight the different successful synthetic strategies allowing to overcome the still “unpredictable” reactivity of titanium ions, particularly to afford crystalline porous coordination polymers.
 Hala Assi | Hala Assi, 26 years old, originates from Lebanon. She achieved her Master’s in Materials Chemistry in 2013 at Lebanese University of Beirut. She received her PhD in 2016 from the University of Versailles St Quentin-en-Yvelines/Université Paris Saclay, France, under the supervision of Dr Christian Serre and Dr Thomas Devic. In 2017, she joined the group of Dr Myrtil Kahn at Laboratoire de Chimie de Coordination CNRS, Toulouse, France, as postdoctoral researcher. Her current research focuses on the synthesis of nano-catalyst materials for water splitting. |
 Georges Mouchaham | Georges Mouchaham was born in Lebanon. He received his MSc (2009) and PhD (2012) degrees in Macro- and Supramolecular Chemistry from the University Paul Sabatier of Toulouse, France, under the supervision of Dr Jean-Pascal Sutter and Dr Nans Roques. In 2013, he joined the “Porous Solids” group at the Institut Lavoisier de Versailles, France, where he worked as a Postdoctoral Fellow under the supervision of Dr Christian Serre and Dr Thomas Devic. Since 2016, he has been a Postdoctoral Fellow at King Abdullah University of Science and Technology (KAUST), Saudi Arabia, working under the supervision of Prof. Mohamed Eddaoudi. His research interests mainly focus on the design, elaboration and study of robust MOFs. |
 Nathalie Steunou | Nathalie Steunou is professor at the Institut Lavoisier de Versailles (ILV) from the University of Versailles St Quentin-en-Yvelines/Université Paris Saclay. She completed her PhD on the synthesis of Ti-oxo clusters in 1997 in the Laboratory Chimie de la Matière Condensée de Paris at the University Pierre & Marie Curie (UPMC-Paris VI). After a postdoctoral position at the MPI for Colloid and Interfaces (Berlin, Germany), she rejoined the LCMCP as a lecturer for about 11 years working on the development of hybrid materials by assembling metal oxides and biomolecules (biopolymers, enzymes, etc.). In 2010, she moved to ILV reaching her present position. Her current research interests focus on the synthesis of nanomaterials and composites based on metal–organic frameworks for different applications (energy, biosensing, etc.). |
 Thomas Devic | Thomas Devic studied Chemistry at the Ecole Normale Superieure de Lyon and received his PhD in 2003 from the University of Angers, France, in the field of conducting molecular materials. After a postdoctoral stay at the Cornell University, USA, in the group of Prof. Stephen Lee (2003–2004), he was appointed as a CNRS researcher in the research group “Porous Solids” headed by Gérard Férey at the Institut Lavoisier de Versailles, France. In 2016, he moved to the Institut des Matériaux Jean Rouxel, Nantes, France. His research mainly deals with the synthesis, structural characterization and study of the properties of new hybrid solids, with a special emphasis on the use of non-conventional ligands, in situ studies and structure–property relationships, and more recently their applications in the field of electrochemical energy storage. |
 Christian Serre | Christian Serre obtained his PhD in 1999 from the University of Versailles St Quentin. After a post-doctoral fellowship in the USA, he became a CNRS researcher in the ‘Porous Solids’ group at Institut Lavoisier (Versailles), taking its lead in 2009 as research director. He moved recently to the Ecole Normale Supérieure and ESPCI (Paris). His research topics deal with the synthesis, structure and applications of porous solids. Christian has received the CNRS bronze medal, a ERC starting grant, the Solid State Chemistry award of the French chemical society and recently the Fondé de l’Etat award from the French academy of sciences. |
Introduction
Almost two decades after their popularization and uninterrupted exploration, metal–organic frameworks (MOFs) are still fascinating the research community. Due to their tunable crystalline structure, in terms of chemical nature, molecular arrangement as well as size and shape of cavities, MOFs have shown very promising potential applications in very diverse fields such as gas storage and separation, biomedicine, sensing, heterogeneous catalysis, etc.1,2 Among the numerous MOFs described in the literature, most systems are built up from divalent transition metal cations (Zn2+, Cu2+, Co2+, Ni2+, etc.), leading to thousands of open framework architectures with sometimes unprecedented porosity, but often unstable in the presence of water. Increasing the charge of the metal cation (together with its polarizing power: charge over ionic radius) is a powerful strategy to enhance the strength of the cation–ligand bond and thus the chemical stability of the coordination polymer.3,4 While the use of trivalent cations (Cr3+, Al3+, Fe3+, etc.) has been successful in obtaining porous water stable MOFs, tetravalent cation based MOFs are still scarce. This is mainly due to the high reactivity of such cations leading to the precipitation of their corresponding oxide or, in the best cases, to amorphous or poorly crystallized coordination polymers. One noticeable exception is the Zr(IV). The discovery of the first Zr-carboxylate MOF, UiO-66,5 built up from the very robust hexa-nuclear Zr-oxo-cluster, and the easy development of isoreticular or related solids, together with the possibility of using modulators6 (allowing tuning of the reactivity of the cations and, hence, the crystallization process), has paved the way to the extremely fast progress in the field of Zr(IV)-MOFs.7–10Developing renewable, sustainable and ecological resources has become one of the actual main concerns in order to overcome the problem of the controversial traditional energy sources. Mimicking the natural photosynthesis (in terms of photochemical conversion of light) seems to be a very appealing alternative. Titanium dioxide, TiO2, is considered nowadays as one of the most successful photocatalysts due to its high efficiency, high chemical stability, high abundance and rather low toxicity. Besides, MOFs, among other materials, have been proven to be very promising for heterogeneous catalysis11 because of their ability to ensure, concomitantly, high accessible surface area, high concentration of actives sites, confined spaces and very good diffusion towards the active sites.
In this context, titanium as a tetravalent Ti4+ cation appears to be a very attractive candidate to produce MOFs showing not only good chemical stability but also redox activity and photocatalytic properties (Ti4+ cation may be reduced to Ti3+ by photoreduction). While many research groups have focused their attention on the incorporation of this cation, for example in the form of titanium oxo clusters,12,13 into organic (block copolymers, polymers, dendrimers), inorganic and hybrid organic–inorganic matrices, porous or not,14–16 such materials do not generally exhibit any long-range order and are almost amorphous.
This review aims at giving an overview of the coordination chemistry of titanium cations and oxygenated ligands, with a specific focus on crystalline compounds. While extended coordination polymers, particularly porous solids (MOFs), will be exhaustively described, the most relevant molecular compounds will also be presented. Besides, attention will be paid to highlight synthetic routes allowing overriding the difficulty encountered with Ti-cations in order to obtain crystalline Ti-MOFs. Relevant photoredox properties of Ti-MOFs and selected Ti-oxo-clusters will also be discussed.
The chemistry of titanium cations
Even though Ti4+ is a highly attractive metal cation, its use to elaborate photoactive materials (other than TiO2) remains still rather challenging. The +4 oxidation state is the stable one in ambient (air, water) conditions (see Fig. 1), but due to the low electronegativity of Ti and the high polarizing power of Ti4+, the latter is prone to fast and spontaneous hydrolysis even in very acidic medium leading to uncontrolled precipitation of TiO2. This is demonstrated in the titanium Pourbaix diagram (Fig. 1) of Ti in water, where the area of titanium(IV) oxide is strongly dominating, the soluble molecular species occurring only in highly acidic media (pH ≤ 0).17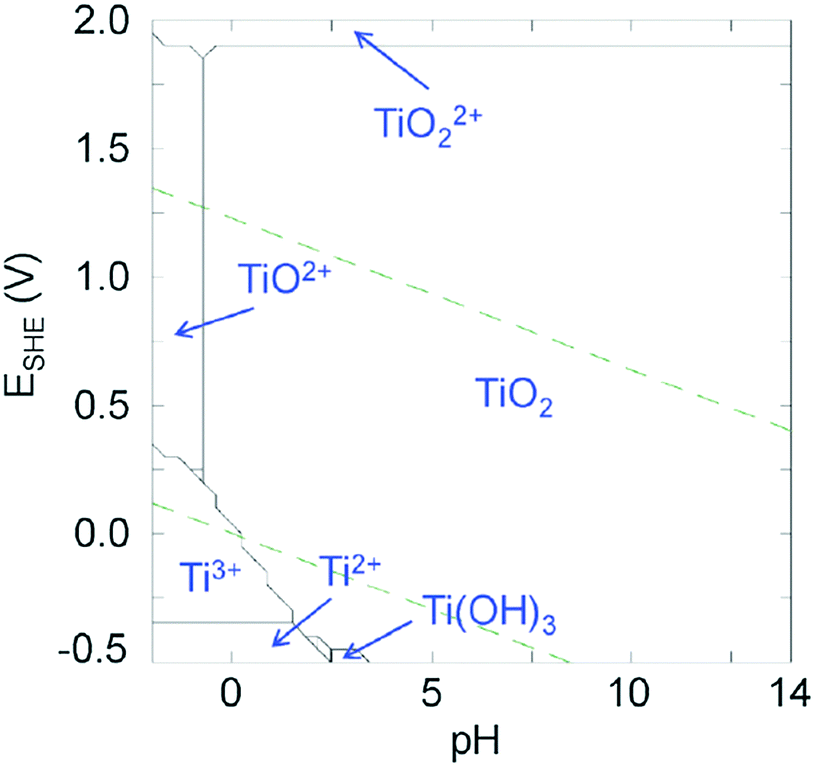 | ||
| Fig. 1 Pourbaix diagrams of titanium calculated for [Ti4+] = 10−3 mol L−1 at 25 °C using the Hydra and Medusa softwares.17 Green dashed lines: O2/H2O and H2O/H2 redox couples. | ||
Consequently, the choice of the titanium precursor and the power of the complexing ligand together with the pH of the media are key factors to stabilize this ion in solution. Titanium(IV) coordination chemistry is primarily based on the use of two types of precursors: titanium(IV) chloride and titanium(IV) alkoxides. It is noteworthy that for both precursors, the coordination number of Ti is lower than that of TiO2 (i.e. 6), which explains the high reactivity of these precursors towards water. Due to the violent hydrolysis and fast release of hydrochloric acid under ambient atmosphere, the use of TiCl4 is quite scarce, although it was used in the production of monodisperse and pure anatase, brookite or rutile nanoparticles.18
Because of the risky experimental manipulation of TiCl4, titanium alkoxides, Ti(OR)4, are often preferred since they are less readily hydrolyzed than TiCl4 due to the hydrophobic character of alkoxy groups and/or their steric hindrance. Moreover, Ti cations can also be complexed by a wide range of additional ligands (β-diketonate, carboxylates, phosphonates, catecholates, etc.) which, due to their bidentate characters, lead to an increase of the coordination number, thereby enhancing the stability of these species towards water.
Another way to control the hydrolysis-condensation reaction of Ti lies in the use of Ti-oxo-clusters of well-defined structure. These species of the general formula TinOm(OR/Cl)x(L)y are polynuclear titanium complexes that combine a metal oxo-core and organic complexing ligands (L, again alkoxide, β-diketonate, carboxylate and phenolate). They present a high diversity in terms of size, geometry, degree of O/Ti condensation, coordination number of titanium cations, and organic complexing ligands. These oxo-clusters are generally more stable than titanium alkoxides towards water since they present a higher nuclearity and condensation degree (O/Ti ratio) and possibly ligands (β-diketonates, carboxylates and/or phenolate) that are less hydrolysable than alkoxides. They are usually prepared either at the early stages of the corresponding titanium alkoxide polycondensation or from the controlled hydrolysis of the titanium alkoxide Ti(OR)x(L)y in solution.19 For numerous titanium-oxo clusters, the source of oxolation was also water generated in situ in the reaction medium through either esterification reactions20,21 or condensation–dehydration reactions of ketones22 while non-hydrolytic routes were also reported.23 As discussed later, such species can advantageously be used as precursors to build up extended coordination compounds.
Early structures: silicates, phosphates and phosphonates
Before analyzing in depth the case of hybrid Ti-based compounds, some of the first extended inorganic Ti-based structures need to be addressed. The reactivity of TiCl4 with inorganic anionic ligands such as fluorides, chlorides, sulfates and phosphates has indeed been thoroughly studied.24–28In the field of inorganic molecular sieves,29 the first reported microporous materials were zeolites, natural aluminosilicates, known since the 18th century. Since then the attention was turned to the development of new synthetic molecular sieve materials, composed from interconnected octahedral and tetrahedral oxide polyhedra, to obtain new topologies, novel compositions or larger pores.30 Particular attention has been devoted to the introduction of active heteroelements into the frameworks mainly for catalytic or separation purposes. Microporous titanium silicates have particularly attracted interest since the discovery of the first titanium silicate TS-1, an active catalyst in oxidation reactions.31 In 1989, a new family of microporous three dimensional titanosilicate frameworks was discovered and named ETS (ETS stands for Engelhard Corporation Titanosilicate). The structure of ETS-10 or A2TiSi5O13·4H2O (A = Na+ or K+) was reported in 1994 by Anderson et al.,32 and consists of a mixture of TiO6 octahedra and SiO4 tetrahedra connected together to form a microporous three dimensional structure. This material is an active base catalyst in the dehydration of tertbutyl alcohol,33–35 or in the dehydrogenation of alcohols.35,36 This is due to the presence of TiO6 bearing two negative charges balanced by K+ and Na+ cations. The effect of ionic-exchange has also been studied showing the fragility of this material upon treatments under acidic conditions.37 In addition, ETS-4, known also as a precursor for the synthesis of microporous titanium–niobium–silicate materials,38 has been investigated as a size-selective adsorbent.39 By adjusting the pore size of this material, it can be used for the separation of gas mixtures (N2/CH4, Ar/O2 and N2/O2).
Focusing on crystalline titanium phosphates, these compounds have been investigated for ion exchange, heterogeneous catalysis, proton conduction, etc.40 They are generally obtained by refluxing insoluble salts of amorphous titanium phosphates, already obtained from TiCl4 under acidic conditions in the presence of an excess of phosphoric acid. Alberti et al.40 reported almost 50 years ago the synthesis of monohydrated titanium phosphates of the formula Ti(HPO4)2·H2O while the di-hydrated form Ti(HPO4)2·2H2O was disclosed in 1977 by Alluli et al.41 The degree of hydration strongly affects the nature of the resulting structure and the arrangement of layers (αTiP and γTiP) – the interlayer distance is ca. 11.6 Å for the di-hydrated form and ca. 7.56 Å for the monohydrate – and the arrangement of the phosphate groups in the lattice. Similar observations have been reported later for zirconium phosphates by Clearfield et al.42 Noteworthily, these changes in the crystal structure lead to large differences in the resulting ionic exchange properties. The dihydrated titanium phosphates have shown high stability towards hydrolysis as well as a high ionic exchange capacity in a strongly acidic medium for sodium and strontium ions. At that time, the crystalline structure of these two arrangements of layers was not known. Though, in 1990, the structure of αTiP was solved from PXRD data,43 these two compounds were found to be very similar. Their structure consists of layers of TiO6 octahedra connected together by phosphate groups to form a two-dimensional structure. In the case of αTiP, HPO4 groups are distributed alternately above and below the Ti octahedral plane while in the case of γTiP these groups are either PO4 or H2PO4. Later, in 1997, Bortun et al.44 studied in detail the TiCl4/H3PO4/H2O system in order to discover new structures. Two new crystalline fibrous titanium(IV) oxophosphates were obtained, in addition to αTiP, denoted πTiP and ρTiP.
This work has been later extended through the in depth investigation of these systems in the presence of fluorine and/or amines or surfactants leading to a series of new structures denoted MIL-n (MIL stands for Materials of Institut Lavoisier). Hydrothermal conditions were selected to study the formation mechanisms of micro- or mesoporous titanium phosphates, oxyfluorinated or not. The TiO2/HF/H3PO4/amine/water system was the starting point of this work, and two layered oxyfluorinated titanium phosphates were isolated, Ti2(PO4)2F4·(N2C2H10) (MIL-62) and Ti2(PO4)2F4·(N2C3H12)·H2O (MIL-63) using either ethylenediamine or 1,3-diaminopropane, respectively.45 Their structures were solved from PXRD data and consist of TiO4F2 octahedra connected together by PO4 tetrahedra. The diammoniums are located in the interlayer space and strongly hydrogen bonded to the terminal fluorine atoms, further stabilizing these structures. A mixed valence metal solid, MIL-15 or TiIIITiIVF(PO4)2·2H2O, was also isolated; this three-dimensional framework is built up from TiO6 or TiO4(H2O)2 octahedra and PO4 groups delimiting small channels. Noteworthily, a microporous solid labeled MIL-18 or TiIV6O3(H2O)3–(PO4)7·(X)3·H2O (X = H3O or NH4) based on dimers of Ti octahedra was also reported however as an impurity preventing the study of its properties.46 Finally, MIL-28n or Ti3O2X2Hy(HPO4)x(PO4)y·(N2CnH2n+6)z·2H2O (n = 2, 3; x = 0, 2; y = 4; z = 3, 2) were obtained; these layered solids, built up from corner sharing chains of TiO5X octahedra and HPO4 groups, possess perforated layers where the protonated diamines interact with the terminal Ti–X groups (X = F and OH).47 However, in general, the use of charged templates raises several drawbacks, mainly the impossibility to access porosity due to the destruction of the structure upon removal of the organic moieties.
Thus, the use of diphosphonic acid instead of phosphoric acid, as initially proposed by Alberti, Clearfield et al.,40,42 has been shown to be an alternative route to develop open framework solids without the use of templates. This expansion to hybrid (coordination) compounds led to 3-D open framework structures such as MIL-22 or TiIV3O2(H2O)2(L)2·2H2O with L = (O3P-(CH2)-PO3)2 or methylenediphosphonate. Noteworthily, this solid built up from trimers of TiO6 or TiO5(H2O) octahedra and PO4 groups possesses a 2-D array of microporous channels filled with free and coordinated water molecules, although their removal does not lead to any accessible porosity towards nitrogen (Fig. 2a).48 Another series of 3-D pillared-titanium bisphosphonate structures, MIL-25n (n = 2, 3) formulated TiIV(L) with L = O3P–(CH2)n–PO3, although nonporous, has been reported by Serre et al. (MIL-252 in Fig. 2b and MIL-253 in Fig. 2c).46
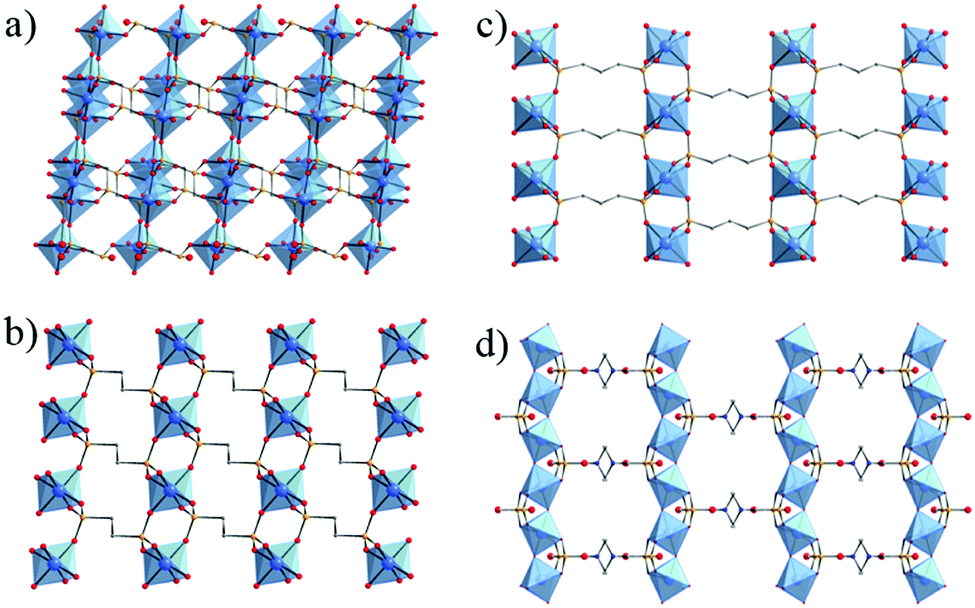 | ||
| Fig. 2 Crystalline structures based on diphosphonate linkers: (a) MIL-22,48 (b) MIL-252,46 (c) MIL-25346 and (d) MIL-91(Ti).49 Color code: Ti, blue; O, red; C, grey; P, orange; N, dark blue. H-atoms of C-atoms were omitted for the sake of clarity. | ||
In 2006, Serre et al.49 reported the first porous titanium-based metal–organic framework, denoted MIL-91 or TiIVO(H2-L)·nH2O (n ≈ 4.5) with L = O3P–CH2–NC4H8N–CH2–PO3 or N,N′-piperazinebismethylenephosphonate. Its structure is reminiscent of the tancoite architecture with corner-sharing chains of TiO6 octahedra surrounded by HPO3C groups (Fig. 2d). This delimits ≈4 Å diameter channels, associated with a noticeable BET surface area close to 500 m2 g−1. A recent study50 revealed that MIL-91 is a very promising candidate for the capture of CO2. Not only its hydrothermal stability was confirmed but a very high CO2/N2 selectivity was measured under post-combustion conditions due to the high adsorption enthalpy of CO2 (∼40 kJ mol−1) related to the strong confinement effect and the presence of polar P–OH groups. This first example has paved the way for the development of new crystalline porous titanium coordination polymers.
Ti-Based coordination compounds built up from C–O ligands
A considerable number of hybrid (organic–inorganic) compounds based on Ti-cation have been reported so far. As briefly mentioned in the introduction, a large family of Ti-oxo clusters including Ti-oxo-alkoxide, Ti-oxo-alkoxo-carboxylate and Ti-oxo-carboxylate was reported, mainly by the groups of Schubert,51 Klemperer52,53 and Sanchez.22,54 The reader interested in the field could refer to exhaustive recent reviews by Rozes et al.19,54 and Coppens et al.55 Unsurprisingly, Ti-oxo-clusters are, by far, more common compared to coordination polymers.Indeed, only a few examples of crystalline titanium polymeric coordination compounds, notably MOFs (based on multi-topic organic ligands), have been reported to date, although being desired since almost two decades. Their preparation being very challenging, different methodologies have been followed and, some of them appeared to be promising to overcome the high reactivity of Ti-cations.
The remainder of this review will encompass all Ti-MOFs reported to date classified according to the nature of organic ligands, namely, carboxylate, hydroxycarboxylate, phenolate and catecholate derivatives. As an introductory of each part, prominent examples of discrete Ti-oxo-clusters will be also described, especially those employed for the formation of Ti-MOFs.
Carboxylate ligands
Ti-carboxylate molecular clusters
The richness and the versatility of titanium cation chemistry (highly oxophilic) is clearly reflected by the large number Ti-oxo-clusters of diverse nuclearity reported to date. Indeed, their nuclearity spans from two56,57 to fifty-two.58Generally, Ti-oxo-carboxylate clusters (general formula TinOm(OR)x(OOCR′)y) are synthesized in solution from a metal cation precursor (very often Ti(OR)4) in the presence of the carboxylate ligand and/or a controlled amount of water. The main parameters to be controlled are the nature and the amount of the metal cation precursor and/or the ligand (R′COOH), the solvent (nature and amount), as well as the reaction conditions (time, pressure, presence of water or not, inert conditions or not, etc.).51,54
The titanium oxo core of these TinOm(OR)x(OOCR′)y complexes is formed from water either purposely introduced or generated in situ through esterification. Indeed, the substitution of alkoxide by carboxylate on the Ti ion competes with an esterification reaction that takes place either between the free carboxylic acid in excess and the released alcohol for a carboxylic acid/titanium ratio R′COOH/Ti (R) ≥ 1, or in an intramolecular fashion for R = 1. Moreover, the esterification–polycondensation reactions are kinetically and thermodynamically favoured by increasing the temperature; higher temperature consequently leads to a higher hydrolysis ratio, hence to Ti-oxo-clusters of higher degree of condensation.59 The R value plays a key role in the formation of titanium-oxo-carboxylate clusters since it determines the nuclearity of the cluster, its composition and thus its stability in solution. While 1 < R < 3 often results in oxo-carboxylate clusters with condensation rate (O/Ti) between 0.33 and 1, a higher degree of condensation (O/Ti > 1) has also been reported for clusters of higher nuclearity (greater than 12).55 Crystal structures of selected examples of oxo-carboxylate clusters are shown in Fig. 4.
Ti4+ adopts, generally, a six-fold octahedral environment where TiO6 units are connected through μ2-oxo and μ3-oxo bridges. Nonetheless, Ti-oxo-clusters showing under- (tetra- and penta-) and over- (hepta-) coordination of Ti4+,60 or even μ4-oxo61–63 have also been reported. TinOm(Cl)x(OOCR′)y are much less common than TinOm(OR)x(OOCR′)y and present only low nuclearities (i.e. 2,66–71 357,64 and 465). In some cases, using TiCl4 allowed the number of carboxylate ligands (OOCR′/Ti) in the oxo-cluster to be increased. This is the case of the tri-nuclear oxo-cluster [Ti3(μ2-O)(μ3-O)Cl3(OOCR′)5] (Fig. 4a; where R′ = ethyl57 and p-methyl, p-ethyl, p-fluoro- or p-iodo-phenyl64 groups), prepared by reaction of TiCl4 and carboxylic acid under inert conditions. These clusters present five bridging carboxylate ligands whereas Ti3-oxo-alkoxo clusters show only two or three.
‘Pure’ Ti-oxo-carboxylate clusters (without alkoxide or chloride groups) are however still very scarce with only two structural types based on mono-topic carboxylate ligands reported to date (Fig. 4g and h). Both are synthesized in the presence of a large excess of carboxylic acids (ca. 10 equivalents) and built up of hexa-coordinated Ti-cations. In the first, Ti8O8(OOCR′)16,66 Ti-cations are connected through μ2-oxo bridges and arranged in a wheel-shape (Fig. 4g). The second, Ti8O10(OOCR′)12,67 shows a different packing where μ2-oxo and μ3-oxo are bridging Ti-cations (Fig. 4h). As may be expected, due to their quite high condensation degree and the absence of alkoxide or chloride groups, both Ti8O8(OOCR′)16 and Ti8O10(OOCR′)12 present relatively interesting stability towards moisture and nucleophilic species.67
These two oxo-clusters also clearly illustrate the lability of the Ti-O iono-covalent bond. For example, Ti8O10(OOCR′)12 can be prepared from Ti8O8(OOCR′)16 under mild conditions, indicating the ability to reconstruct the TinOm core. More interestingly, different derivatives of Ti8O8(OOCR′)16 and Ti8O10(OOCR′)12 can be prepared by simple ligand exchange without altering the Ti8-core,66–68 as already exemplified with other Ti oxo clusters of high nuclearity and condensation degree.14
Comparable work of carboxylate ligand exchange has been reported recently on the Ti-oxo-alkoxo-carboxylate-phosphonate-cluster.69,70 Liu et al.71 have also succeeded in preparing [Ti6O6(OiPr)10(OOCR′)2(O3P-Ph)2] with 14 different carboxylate functional ligands by exploiting labile coordination sites occupied originally by acetate groups (Fig. 3a). This has allowed tuning rationally the optical bandgap (Fig. 3b) to drive the absorption of light into the visible range with the final aim of optimizing the photocatalytic activity.
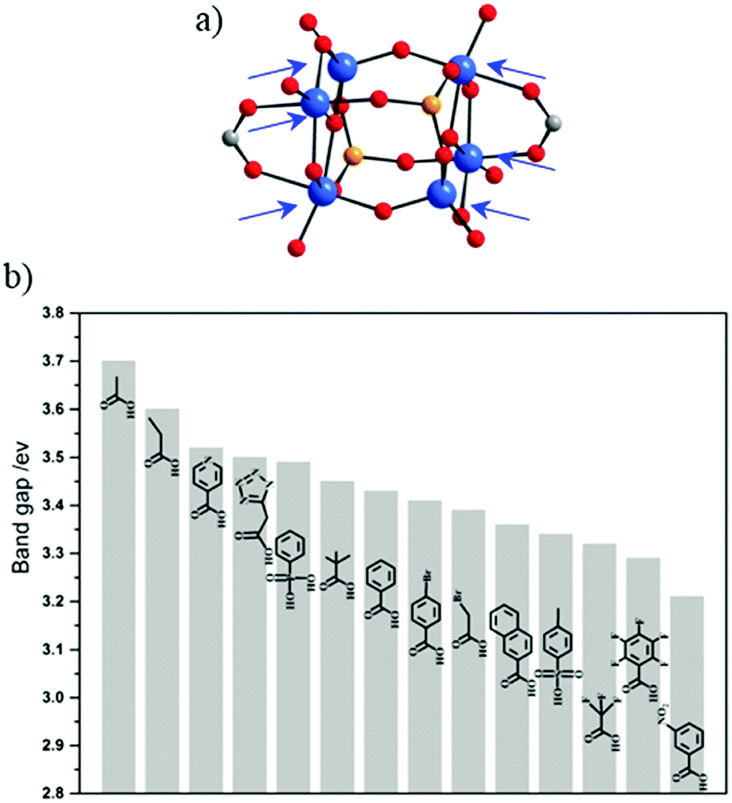 | ||
| Fig. 3 (a) Molecular structure of the [Ti6O6(OiPr)10(OAc)2(O3PR)2] cluster. Arrows point labile sites. (b) Bandgap summary of the different Ti-oxo-alkoxo-carboxylate-phosphonate-clusters obtained after exchanging acetate ligands by the corresponding electron donor ligands. Color code: Ti, blue; O, red; C, grey; P, orange.71 Adapted with permission from ref. 71. Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Overall, carboxylate based Ti-oxo-clusters show a large panel of nuclearities and geometries. Fig. 4 illustrates the molecular structures of selected carboxylate based Ti-oxo-clusters (including Cl-based one) of various nuclearity (3 to 10) and with the carboxylate/alkoxide and carboxylate/Cl ratio ≥ 1. Among them, Ti6O6(OR)6(OOCR′)6 (Fig. 4d) has been used in the synthesis of MOFs (see below). It can be described as two staggered Ti-trimers connected through μ3-oxo bridges and μ3-OOCR′.
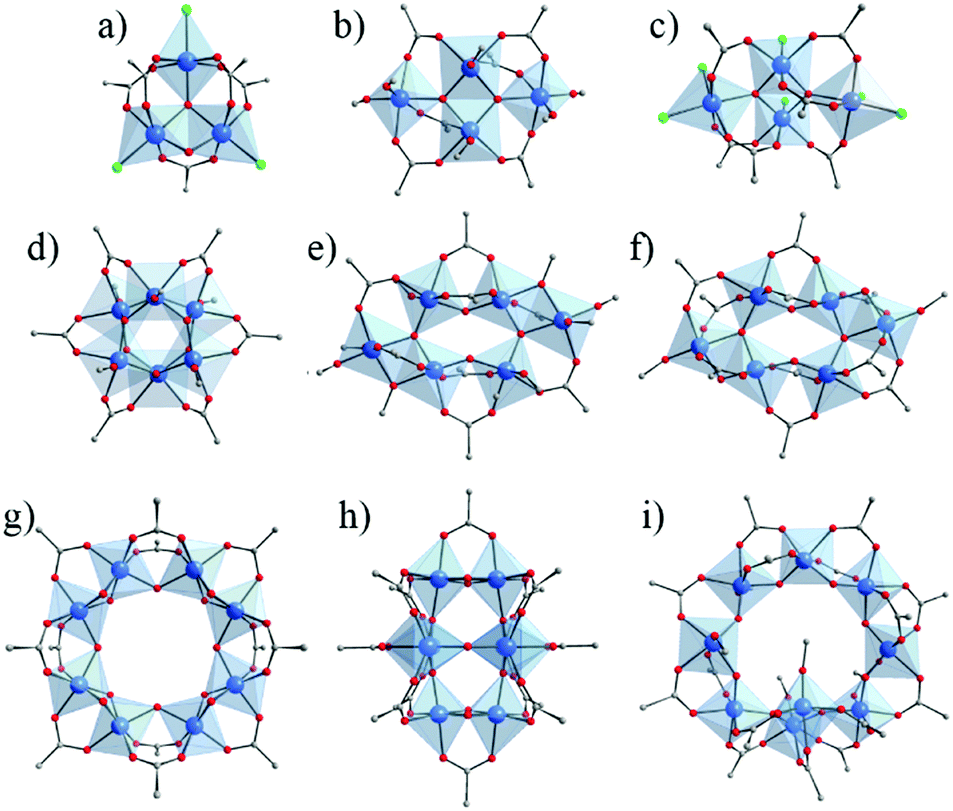 | ||
| Fig. 4 Crystalline structures of Ti-oxo-carboxylate clusters (including Cl-based ones) of nuclearity ranging between 3 and 10, showing ratio carboxylate/alkoxide (or Cl) ≥ 1. (a) Ti3O2Cl3(OOCR)5;56 (b) Ti4O2(OR)6(OOCR)6;21 (c) Ti4O2Cl6(OOCR)6;65 (d) Ti6O6(OR)6(OOCR)6;56,62,72–78 (e) Ti6O4(OR)8(OOCR)8;21,73,79–84 (f) Ti6O6(OR)2(OOCR)10;85 (g) Ti8O8(OOCR)16;66 (h) Ti8O10(OOCR)12;67 (i) Ti9O8(OR)4(OOCR)16.86 Color code: Ti, blue; O, red; C, grey; Cl: green. H-atoms of C-atoms are omitted for the sake of clarity. | ||
Recently, Chun and collaborators87,88 have shown that supramolecular frameworks built up from the molecular entities Ti6O6(OR)6(OOCR′)6 (R′ = p-tert-butyl-phenyl or p-amino-phenyl) selectively adsorb CO2 towards CH4 and N2. Similar results have also been obtained for Ti6O6(OR)2(OOCR′)10 (R = p-tert-butyl-phenyl) and Ti8O10(OOCR′)12 (R′ = p-amino-phenyl).
The high diversity (nuclearity and geometry) and the possible tunable character of Ti-oxo-carboxylate clusters (ligand exchange) make them very promising candidates for pre-designed and controlled synthesis of porous coordination polymers. The following part summarizes the state of the art of existing Ti-carboxylate MOFs. Light will be particularly shed on the difficulties encountered in the elaboration of Ti-MOFs and how they can be overcome.
Ti-carboxylate MOFs
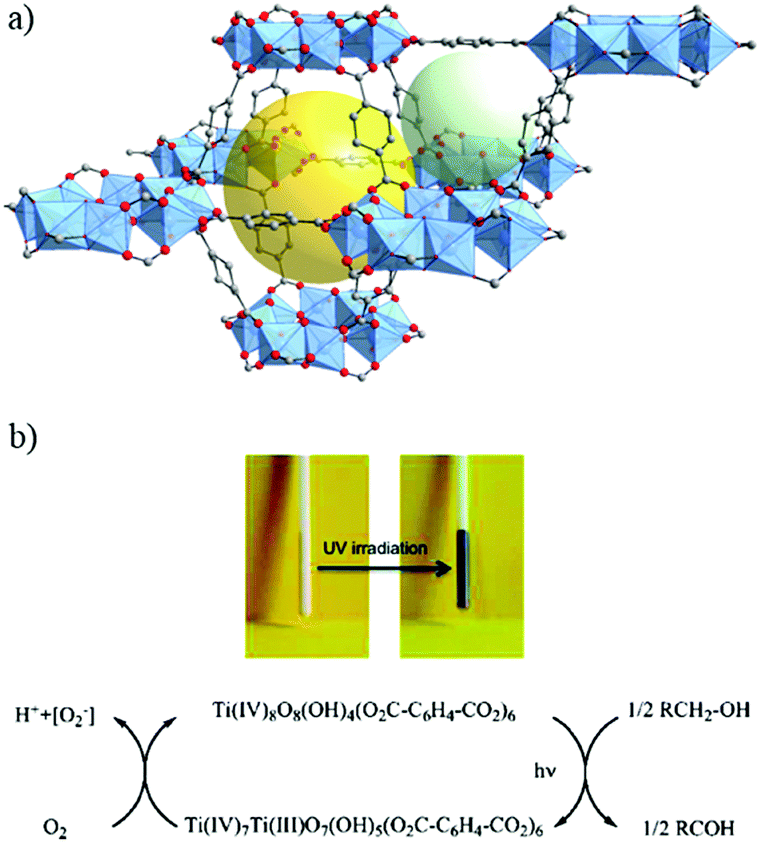 | ||
| Fig. 5 Crystalline structure of MIL-125:91 (a) SBU, Ti8-oxo-cluster, and (b) overall view showing one octahedral cage (yellow sphere) and one tetrahedral cage (light green sphere). (c) Powder of MIL-125 in the presence of benzyl alcohol before and after UV-light irradiation and the corresponding mechanism proposed for the reduction of MIL-125 under irradiation. Color code: Ti, blue; O, red; C, grey. | ||
Indeed, the discovery of MIL-125 was the trigger for various studies not only to obtain new porous Ti-MOFs but also to develop new applications based on their photoactivity.93 Since it was not obvious to synthesize new crystalline Ti-MOFs, efforts have been first focused on tailoring the photocatalytic properties of MIL-125(Ti) by organic functionalization. MIL-125, reported initially with the benzene dicarboxylate ligand, possesses photocatalytic activity only under UV irradiation with an optical bandgap of ca. 3.6 eV (λ = 345 nm).94 In order to drive the photoactivity to the visible domain, lowering the highest occupied molecular orbital–lowest unoccupied molecular orbital (HOMO–LUMO) gap of the carboxylated linker was a first approach. Fu et al.95 prepared the monoaminated analogue of MIL-125 reported initially by Zlotea et al.,96 NH2-MIL-125, which is a light yellow colored solid with an extra absorption band in the visible-light region, extending the absorption band to ca. 550 nm. As expected, this colored analogue has shown photocatalytic activity under visible-light irradiation for the photoreduction of CO2 into formate ions in the presence of acetonitrile with triethanolamine (TEOA) as the sacrificial agent.
Similarly, NH2-MIL-125 showed reversible photochromism during the photocatalytic process, highlighting the reduction of Ti4+ to Ti3+ induced by charge transfer from the organic linker to the inorganic cluster TiO5(OH) (Ligand-to-Metal Charge Transfer (LMCT)) at the origin of the photocatalytic activity (Fig. 6). As already observed in the literature for NH2-UiO-66(Zr),97 this behavior in the visible range is driven by the NH2 group of the organic linker and is associated with a reduction of the optical bandgap, experimentally deduced, down to 2.6 eV.
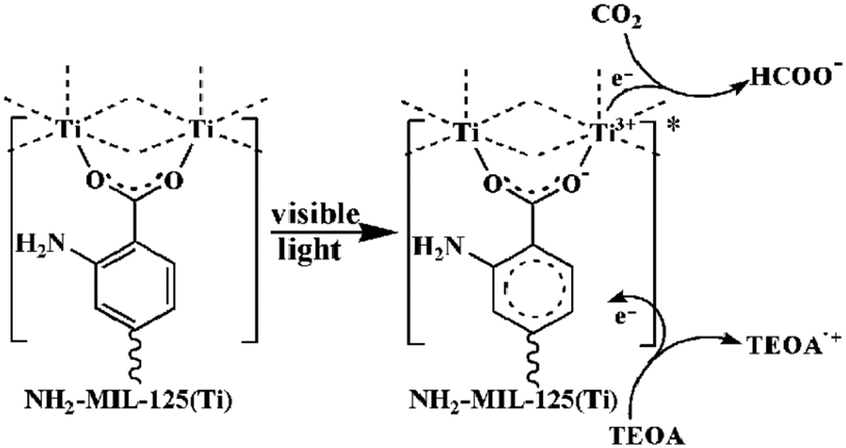 | ||
| Fig. 6 Proposed mechanism for the photocatalytic reduction of CO2 with NH2-MIL-125 under visible light irradiation.95 Reprinted with permission from ref. 95. Copyright 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
This lowering of the gap induced by –NH2 groups was further exploited through a combined experimental and computational study by Hendon et al.94 The authors confirmed that grafting a –NH2 group on the phenyl ring resulted in lowering of the optical gap, while the hypothetical diaminated (NH2)2-MIL-125 would exhibit an even lower gap, theoretically estimated, of ca. 1.3 eV.
Besides, in order to improve the photocatalytic activity under visible-light irradiation, Nasalevich et al.98 have succeeded in post-modifying NH2-MIL-125 by converting up to 30% of the linkers to dye-like molecular fragments. The obtained MR-MIL-125(Ti) [MR stands for the dye methyl red or N2-C6H4-N(CH3)2] shows a significant improvement of its absorption in the visible domain (reaching almost 700 nm) due to the extended conjugation of the aromatic system (Fig. 7).
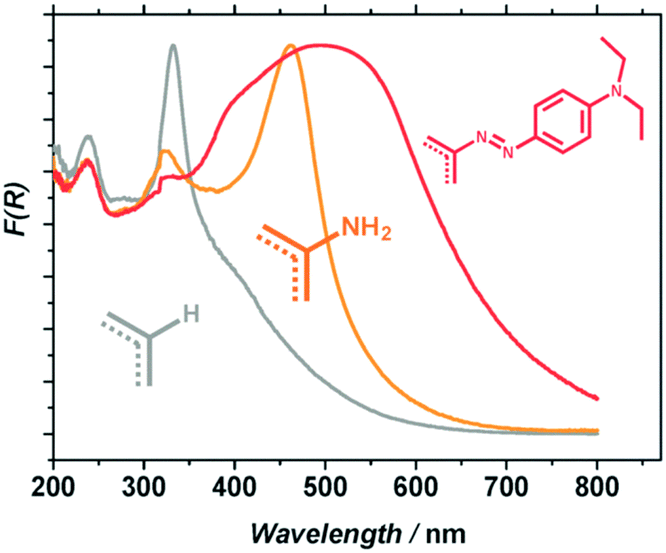 | ||
| Fig. 7 Diffuse reflectance spectra of MIL-125 (grey), NH2-MIL-125 (orange) and MR-MIL-125 (red).98 Reprinted with permission from ref. 98. Copyright 2013 The Royal Society of Chemistry. | ||
This solid was later evaluated in the oxidation reaction of benzyl alcohol. An improvement in the photocatalytic performance of 40% in the visible range was observed in comparison to the pristine NH2-MIL-125. However, this is still lower than the 100% improvement observed in photon absorption for MR-MIL-125. This difference in performance is attributed to other factors affecting the photocatalytic activity such as the molecular adsorption/diffusion properties of the MOF or the absorption of photons of energy too low to drive the photocatalytic transformation.
Horiuchi and collaborators followed another approach to improve photocatalytic performance by encapsulating active species, i.e. platinum nanoparticles, on the surface of NH2-MIL-125 to obtain Pt/NH2-MIL-125 (Fig. 8). The composite material showed significant photocatalytic activity in the visible (ca. 500 nm) towards H2 production.99
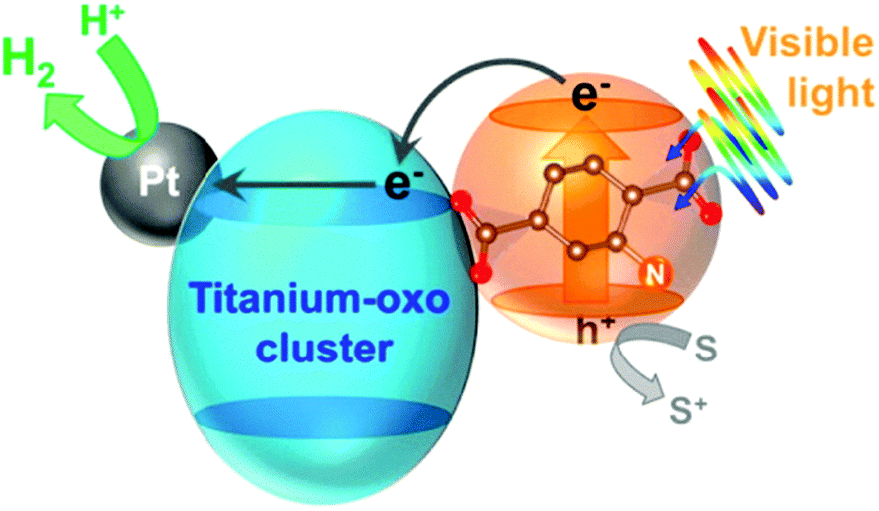 | ||
| Fig. 8 Diagram of the photocatalytic H2 production in the presence of Pt/NH2-MIL-125.99 Reprinted with permission from ref. 99. Copyright 2012 American Chemical Society. | ||
Owing to the very promising photocatalytic properties of MIL-125 and its derivatives, discovering new titanium MOFs became even more attractive. However, new porous Ti-MOFs started to appear only a few years after the discovery of MIL-125s. This is certainly due to the rather high reactivity and fast polycondensation of titanium cations in solution, controlling this step being a key prerequisite for obtaining highly crystalline Ti-MOFs. This poor control of the polymerization of titanium is illustrated by the very different synthetic procedures followed for the few Ti-MOF reported to date. Two main strategies can however be highlighted:
(1) The control of the kinetics of the coordination reaction and/or polycondensation of Ti, through the use of either very complexing solvents129 or less reactive titanium precursors such as organometallic complexes100 and preformed polycondensed titanium oxo-clusters.19,101,102
(2) The post-synthetic metathesis (PSM) of metal ions and post-synthetic metathesis and oxidation (PSMO).103–108
Very recently, Bueken et al.100 reported a new Ti-MOF based on a trinuclear titanium-oxocarboxylate [TiIV3(μ3-O)(O)2(COO)6] cluster. This MOF, denoted COK-69 and formulated TiIV3O3(L)3·DMF with L = OOC–C6H10–COO or 1,4-cyclohexanedicarboxylate (CBC), was prepared by reacting dicyclopentadienyl titanium(IV) dichloride (Cp2TiIVCl2) and 1,4-cyclohexanedicarboxylic acid (H2-CBC) in DMF and acetic acid under an inert atmosphere and solvothermal conditions. The reduced form was first obtained [COK-69(TiIII)] and later oxidized under air exposure [COK-69(TiIV)]. The crystalline structure of COK-69 showed that it resembles closely the iron or chromium dicarboxylates MIL-88 and presents also a flexible 3-D framework, although such flexibility arises from a different mechanism (Fig. 9). Note that because of the close pore phase obtained after activation, COK-69 shows a very low porosity towards N2 (SBET = 29 m2 g−1). The authors postulate that the synthesis of this MOF is induced first by the preformation of the cyclopentadienyl-capped Ti3(μ3-O) oxo-cluster preventing the spontaneous hydrolysis of titanium and favouring the direct formation of the [TiIV3(μ3-O)(μ1-O)2(COO)6] units.
 | ||
| Fig. 9 Crystalline structure of COK-69s:100 (a) SBU, Ti3-oxo-cluster (corresponding to closed-pore phase); (b, c) view along the channels of COK-69: (b) closed-pore and (c) open-pore. Color code: Ti, blue; O, red; C, grey. | ||
COK-69 (theoretically estimated optical gap 3.77 eV) showed, under UV irradiation and without any loss of crystallinity, a reversible photoreduction and reoxidation activity in the presence of ethanol and upon air exposure, respectively. It was demonstrated that this process involves simultaneous transfer of both electrons and protons (proton-coupled electron transfer (PCET)).
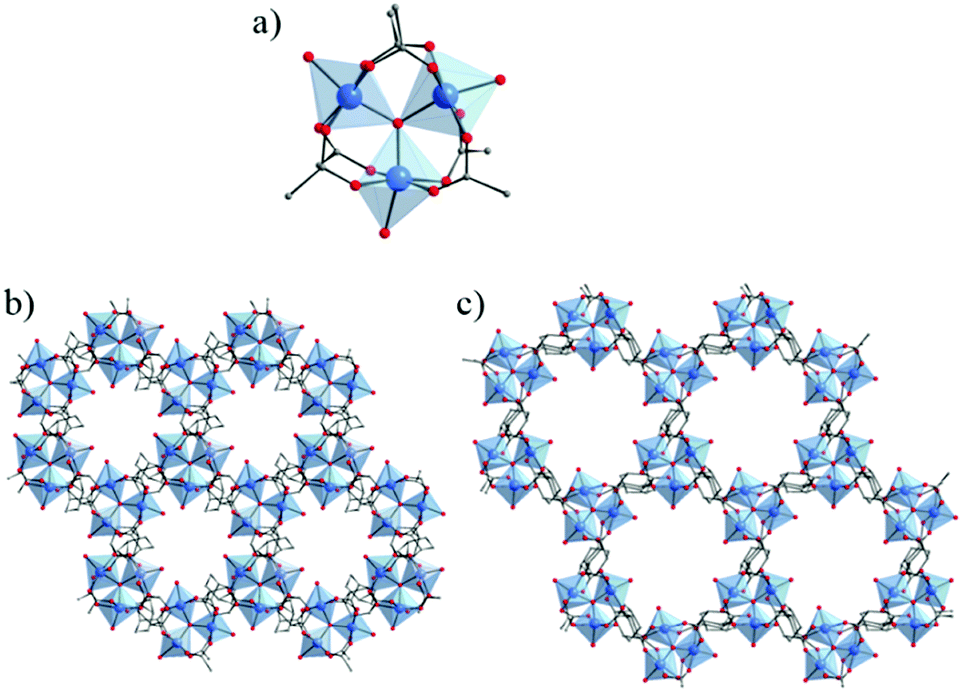
Using a more reactive titanium precursors, Long et al.109 have succeeded in preparing the Ti(III) analogue of MIL-101, a MOF based on the trinuclear oxo-carboxylate M(III) oxo-cluster. As for COK-69, Ti(III)-MIL-101 or TiIII3O(OCH2CH3)(L)3·2DMF with L = OOC–C6H4–COO or terephthalate (BDC) was synthesized under an inert atmosphere in DMF under solvothermal conditions, but by reacting that time H2-BDC with TiCl3. Illustration of the proposed composition of the Ti3O clusters is shown in Fig. 10. Each titanium is coordinated to five oxygen atoms in addition to one labile ligand (solvent molecule). The unsaturated five-coordinated TiIII (obtained after activation) has shown its ability to react with O2 molecules generating TiIV superoxo and peroxo species. Note that this allows irreversible steep adsorption of O2.
 | ||
| Fig. 10 Illustration of the proposed composition of the Ti3O clusters in MIL-101 after (a) heating at 150 °C in vacuum, (b) reacting with O2 to form titanium(IV) superoxide and (c) reacting with O2 to form titanium(IV) peroxide. Note that the cluster likely distorts.109 Reprinted with permission from ref. 109. Copyright 2015 American Chemical Society. | ||
If the direct synthesis (one pot reaction) using titanium salts or organometallic complexes (generally, isolated Ti-cation) as precursors under inert conditions has led to two additional new Ti-carboxylated MOFs initially with Ti ions under a trivalent state, attempts using preformed titanium(IV) oxo-clusters have also been a fruitful alternative. As for metallocene complexes, using Ti-oxo-clusters as a source of titanium should allow a slower reaction, as both ligands (the monotopic one initially on the oxo-cluster and the polytopic one used to build the extended network) show similar coordination ability towards titanium. Two possibilities have been considered. In the best case, a slow exchange between the monotopic and polytopic ligands might take place without altering the nuclearity and the geometry of the preformed oxo-cluster.19,102,110–113 Contrarily, the oxo-cluster could act as a reservoir of matter and undergo complete or partial reorganization but still permitting to slow down the reaction between cations and ligands and lead to the crystallization of a coordination polymer.101
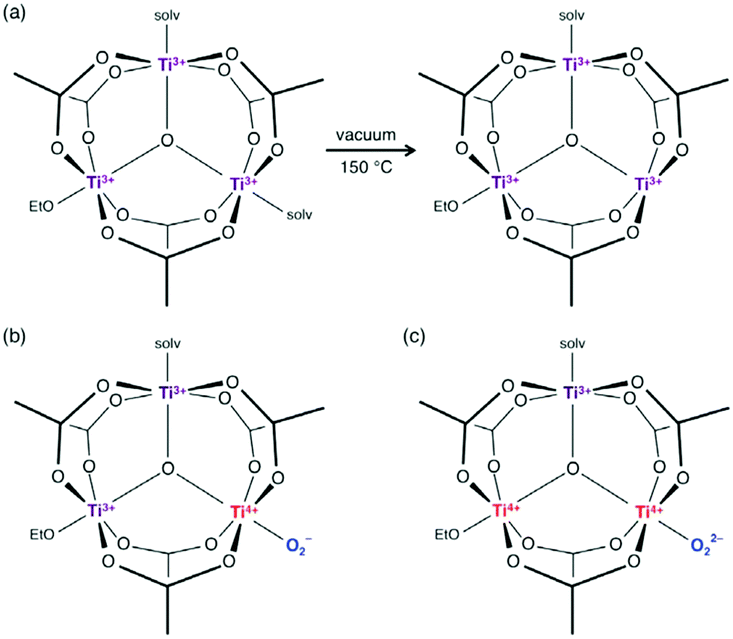
This last strategy, initially proposed for MIL-125 (see above), was used by Yuan et al.101 to prepare a new porphyrinic carboxylated Ti-MOF. PCN-22 (PCN stands for porous coordination network), formulated TiIV7O6(L)12·2DEF with L = tetrakis(4-carboxyphenyl)porphyrin, was obtained by mixing, under solvothermal conditions, a preformed titanium oxo-carboxylate cluster Ti6O6(OiPr)6(abz)6 (with abz = H2N-C6H4-COO or 4-aminobenzoate) (see Fig. 4d) with a porphyrin-based linker H4-TCPP (tetrakis(4-carboxyphenyl)porphyrin) in the presence of benzoic acid. The structure of PCN-22 revealed that the Ti6 oxo-cluster was completely transformed during the reaction and the obtained 3-D framework is based on unprecedented Ti7O6 oxo-clusters connected to each other by TCPP linkers (Fig. 11). Ti7O6 could be described as a pair of Ti3O3 units bridged by a Ti-cation. It is a 12-connected cluster composed of seven Ti4+ ions, two μ3-O2− ions, two terminal O2− ions, two terminal OH− ions and two bridging solvent molecules (DEF).
 | ||
| Fig. 11 (a) Crystalline structure of PCN-22:101 (a) SBU, Ti7-oxo-cluster (only O-atoms of DEF molecules coordinated to the central Ti4+ are represented); (b) view of the framework along channels. Color code: Ti, blue; O, red; C, grey; N, blue; H, white. H-atoms of C-atoms are omitted for the sake of clarity. | ||
PCN-22 presents a high permanent porosity towards N2 (SBET = 1284 m2 g−1). Besides, thanks to the TCPP moieties, this Ti-MOF shows a broad range of light absorption from 200 nm to 640 nm with an experimentally deduced optical gap of 1.93 eV, the smallest reported to date for pure carboxylate based Ti-MOFs. In addition, the photocurrent measurements indicated that PCN-22 is photoactive under visible-light illumination (>450 nm). Finally, studies revealed that PCN-22 acts as a heterogeneous photocatalyst capable of promoting, under visible-light, alcohol oxidation reaction converting benzyl alcohol to the corresponding aldehyde (conversion rate of 28% after two hours and TON over 100). The proposed mechanism is depicted in Fig. 12.
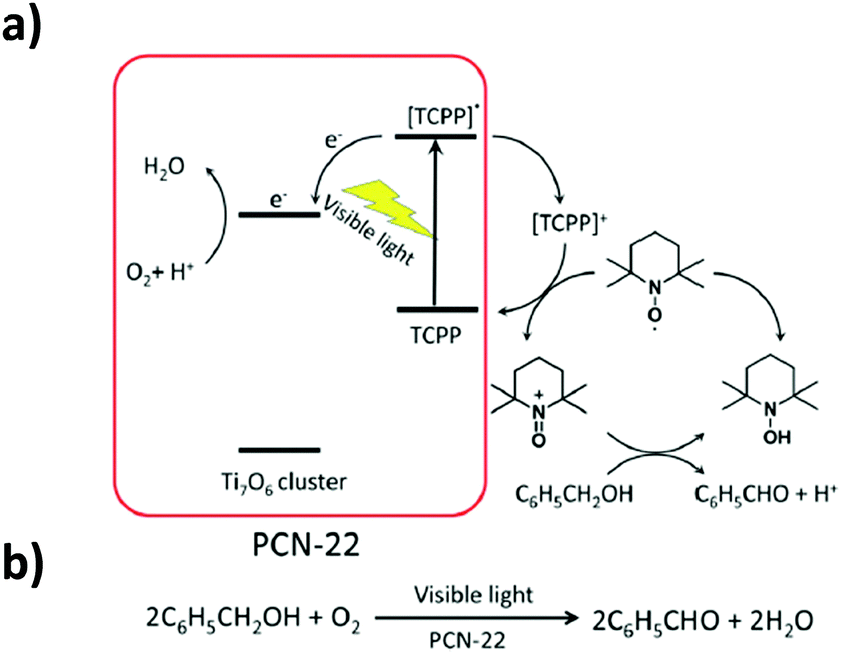 | ||
| Fig. 12 Proposed mechanism for the photocatalytic oxidation reaction of benzyl alcohol for the PCN-22/TEMPO system (TEMPO: 2,2,6,6-tetramethylpiperidinyloxyl).101 Reprinted with permission from ref. 101. Copyright 2015 The Royal Society of Chemistry. | ||
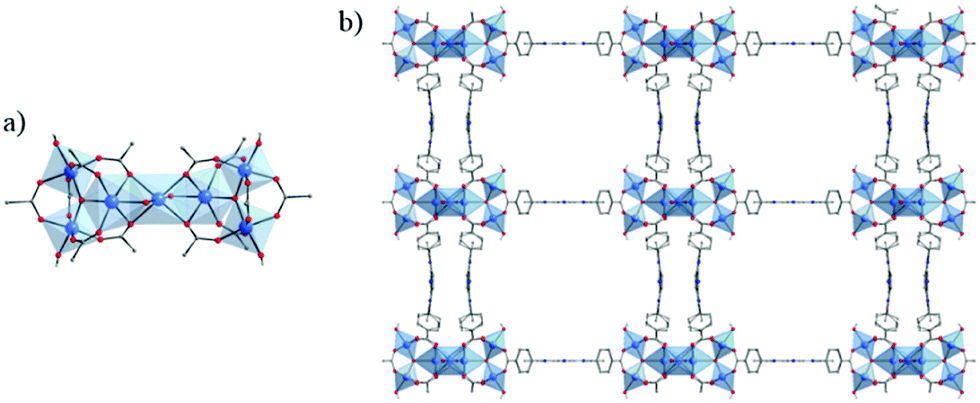
The same hexameric Ti-oxo-cluster, Ti6O6(OiPr)6(abz)6, was also used very recently to elaborate an original Ti-MOF. Reported by Nguyen et al., MOF-901, formulated TiIV6O6(OCH3)6(L)3 with L = OOC-N2C20H14-COO or 1,4-phenylenebis(methanylylidene)bis(azanylylidene)dibenzoate, is the first MOF obtained by combining the preparation strategies of metal- and covalent-organic frameworks.102 Its synthesis consists of connecting preformed Ti6 oxo-carboxylate clusters through imine functions resulting from condensation reactions between terminal –NH2 groups decorating the aforementioned clusters and carbonyl groups of benzene-1,4-dialdehyde spacer. The overall structure of MOF-901 can be described as perforated 2-D layers, showing triangular apertures, stacked in an AB fashion (Fig. 13). MOF-901 exhibits a permanent porosity towards N2 with an experimentally deduced SBET of 550 m2 g−1. It is noteworthy that the best crystallinity was nevertheless achieved by mixing all reactants in one-pot (see Fig. 13a) while using the preformed hexameric cluster led to a less crystalline solid.
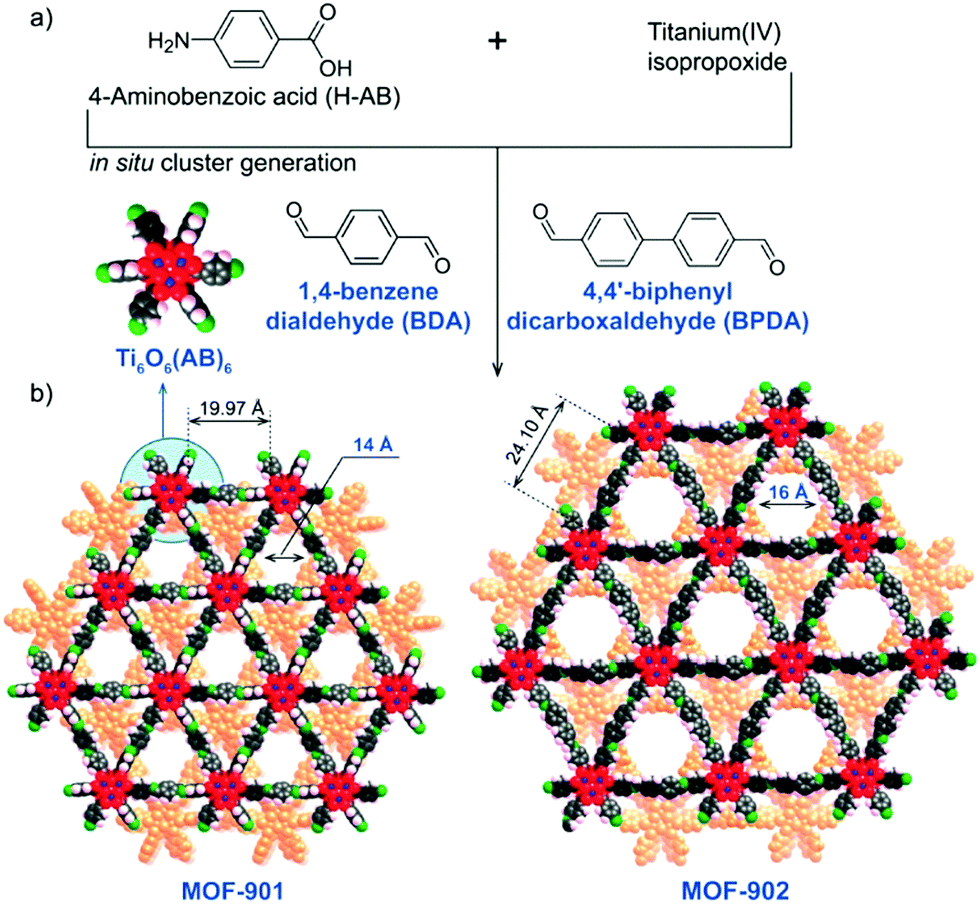 | ||
| Fig. 13 MOF-901102 and MOF-902.114 Scheme summarizing the synthetic procedure. (b) Space filling views of crystalline structures of isoreticular MOFs. The discrete hexameric cluster is shown in the inset of (b). Color code: Ti, blue; C, black; O, red; N, green; H, pink; and second layer, orange. Capping methoxide moieties have been removed for clarity. Reprinted with permission from ref. 114. Copyright 2016 American Chemical Society. | ||
The yellow colored MOF-901 possesses a large absorption spectrum ranging from 340 to 550 nm with a maximum located at 360 nm (experimentally deduced optical gap = 2.65 eV), slightly higher than that measured for PCN-22. Moreover, MOF-901 was successfully used, without loss of crystallinity, as a heterogeneous photocatalyst under visible-light irradiation for the radical polymerization of methyl methacrylate (MMA) with ethyl α-bromophenylacetate as co-initiator. It was found to outperform P-25 TiO2 and Ti6O6(OiPr)6(abz)6, VNU-1 (VNU stands for Vietnam National University), NH2-MIL-125 and NH2-UiO-66.
More recently, MOF-902, isoreticular to MOF-901 with a longer organic linker (bearing two benzene rings instead of one), was reported.114 It was synthesized using the same strategy as that employed for MOF-901 with a slightly modified procedure. It has a similar layered structure with larger triangular apertures of ca. 16 Å but a lower experimental surface area (SBET of 400 m2 g−1). No explanation was provided concerning the latter point.
MOF-902 showed a red-shifted optical absorption band, ranging from 340 nm to 640 nm with a maximum located at 390 nm, probably induced by the higher conjugation system of the organic backbone. Its experimentally deduced band-gap energy was found to be ca. 2.50 eV.
The photoactivity under visible light of MOF-902 was first demonstrated using the same catalytic reaction of MMA polymerization tested for MOF-901. It resulted in the production of polyMMA with a higher molecular weight (31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 465 g mol−1vs. 26850 g mol−1) and a lower polydispersity index (1.11 vs. 1.6) compared to MOF-901. Additionally, MOF-902 showed a significantly better performance than MOF-901 in the photocatalytic polymerization of benzyl methacrylate and styrene monomers without significant loss of performance over consecutive cycles.
465 g mol−1vs. 26850 g mol−1) and a lower polydispersity index (1.11 vs. 1.6) compared to MOF-901. Additionally, MOF-902 showed a significantly better performance than MOF-901 in the photocatalytic polymerization of benzyl methacrylate and styrene monomers without significant loss of performance over consecutive cycles.
Following the PSM strategy, Cohen and collaborators first succeeded in partially transforming existing MOFs into their titanium analogues, among others. They prepared Zr/Ti-UiO-66103 (Zr/Ti < 0.1) and two aminated derivatives: NH2-Zr/Ti-UiO-66104 (Zr/Ti = 3.03) and (NH2)2-NH2-Zr/Ti-UiO-66 (Zr/Ti = 2.52). These Ti-analogues were obtained by reacting crystalline Zr-MOFs with a solution of TiCl4(THF)2 (THF = tetrahydrofuran) at 85 °C for 5 days (Fig. 14). Various techniques (PXRD, ICP, N2 sorption measurements) were combined to prove the insertion of Ti, although the exact nature of the coordination sphere of the Ti ions within this solid was not discussed (e.g. substitution of Zr by Ti would lead to unexpected 8-fold coordinated Ti, except if such transformation is accompanied by the formation of defects of the Zr/Ti oxo-cluster).
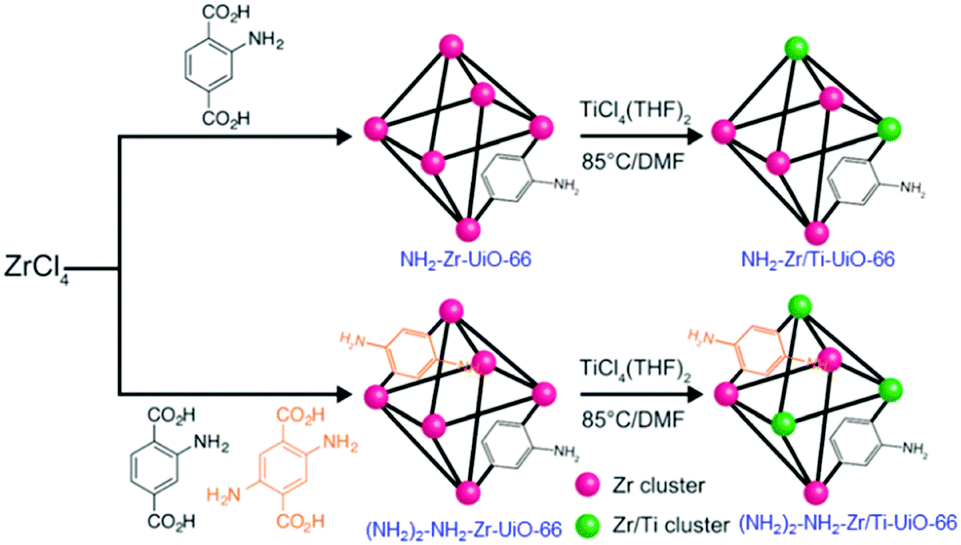 | ||
| Fig. 14 Synthesis of aminated Zr- and Zr/Ti-MOF 1(Zr): NH2-Zr/Ti-UiO-66 (Zr4.5Ti1.5O4(OH)4(C8H7O4N)6), and (NH2)2-NH2-Zr/Ti-UiO-66 (Zr4.3Ti1.7O4(OH)4 (C8H7O4N)5.17(C8H8O4N2)0.83).104 Reprinted with permission from ref. 104. Copyright 2015 The Royal Society of Chemistry. | ||
If no photocatalytic study was reported for Ti-UiO-66, both aminated Ti-analogues showed photocatalytic CO2 reduction under visible-light irradiation in the presence of triethanolamine (TEOA) and 1-benzyl-1,4-dihydronicotinamide (BNAH) as sacrificial donor reagents. (NH2)2-NH2-Zr/Ti-UiO-66 showed a higher activity than NH2-Zr/Ti-UiO-66 (turnover numbers after 6 hours of 6.27 and 4.66, respectively). Such enhancement was attributed to the existence of two absorption paths suitable for the reduction of CO2 in (NH2)2-NH2-Zr/Ti-UiO-66, while only one in NH2-Zr/Ti-UiO-66.
A few other studies on post-synthetic exchange (PSE) with the Ti-ion have also been reported.101,106,120,121 Among them, Zhou et al.105 performed a systematic study where two metal(III) carboxylate MOFs (based on MIII3O3 trimers: MIL-100(Sc), PCN-333(Sc)) and two metal(II) hydroxycarboxylate solids (based on MIIO chains: MOF-74(Zn) and MOF-74(Mg)) were converted partially into their corresponding Ti-analogues, and finally tested as photocatalysts. The synthetic strategy, termed high valence metathesis and oxidation (HVMO), consists of post-exchanging cations with Ti3+ to obtain an intermediate Ti(III)-MOF analogue followed by mild oxidation under air to yield Ti(IV)-MOF (Fig. 15).
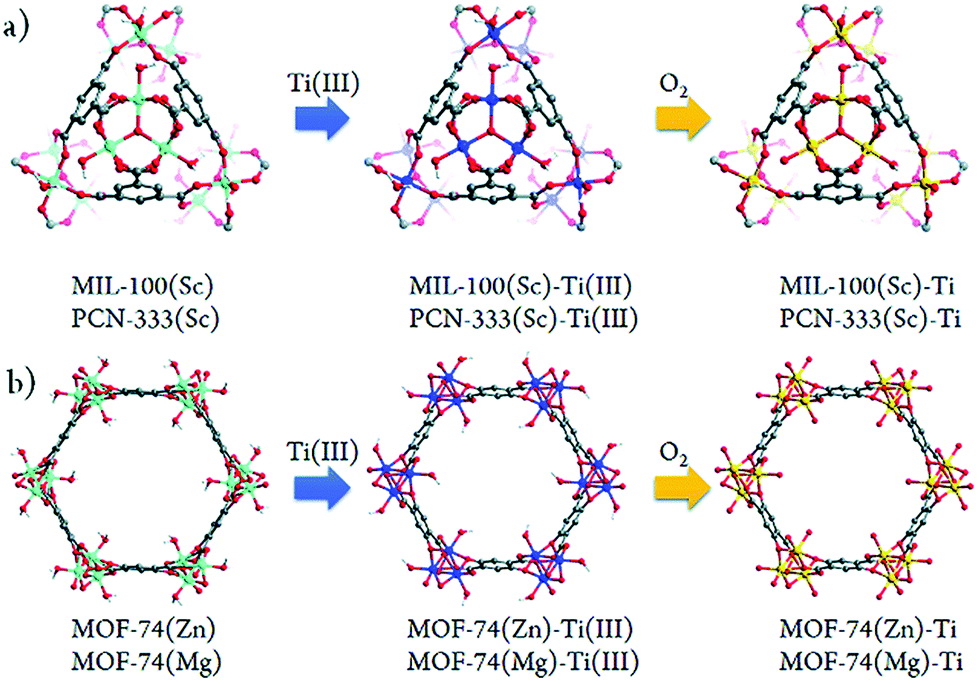 | ||
| Fig. 15 Schematic illustration of the stepwise HVMO procedure for the design of Ti-MOFs from the template MOFs: (a) MIL-100(Sc) and PCN-333(Sc) metal metathesis with Ti3+ followed by metal node oxidation in the air (PCN-333(Sc) and MIL-100(Sc) have the same topology but different structures) and (b) similar process for MOF-74(Zn) and MOF-74(Mg).105 Reprinted with permission from ref. 105. Copyright 2015 The Royal Society of Chemistry. | ||
The crystalline structure integrity of the obtained Ti-MOFs was confirmed by PXRD and N2 sorption measurements. The metal exchange ratio indicates variable exchange rate, from 37.9% to 94.7%. The resulting solids exhibit a slightly lower N2 sorption capacity than the pristine materials, attributed to the presence of extra anions balancing the charge. Diffuse reflectance UV-Vis absorption spectroscopy showed absorption edge at 380 nm for PCN-333(Sc)-Ti and MIL-100(Sc)-Ti and extended absorption up to 660 nm for MOF-74(Ti)s in agreement with the LMCT occurring between 2,5-dihydroxyterephthalic acid (H2-DOBDC) and Ti centers (see the next section). Therefore, the photocatalytic potential of the Ti-MOFs was examined based on the photo-degradation, under visible-light irradiation, of methylene blue (MB) as a probe reaction. While PCN-333(Sc)-Ti and MIL-100(Sc)-Ti showed only moderate photo-degradation activity after 9 minutes (35% and 64%, respectively), both MOF(74)-Ti revealed excellent conversions up to 98% only after 3 minutes of irradiation (Fig. 16a). The excellent photocatalytic performance of Ti-MOF-74 was attributed to its capability of absorbing more broadly the visible light and a longer excitation lifetime, most probably due to the chain-like connectivity of the Ti-ion. The proposed mechanism is depicted in Fig. 16b.
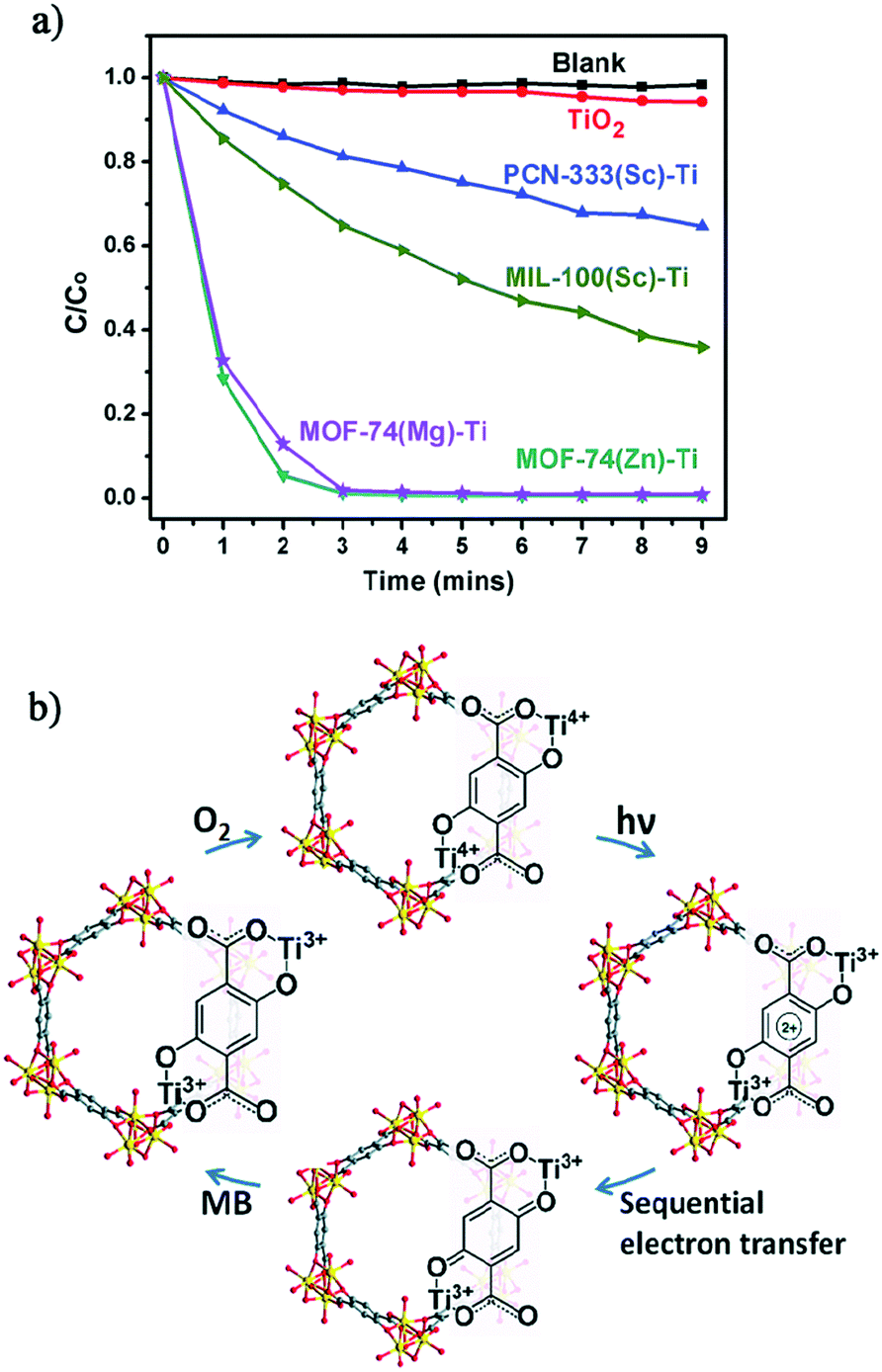 | ||
| Fig. 16 (a) Photodegradation of MB using no catalyst (blank), TiO2, PCN-333(Sc)-Ti, MIL-100(Sc)-Ti, MOF-74(Zn)-Ti and MOF-74(Mg)-Ti with 300 W xenon light irradiation and (b) proposed mechanism of MB degradation of Ti-MOF-74 in the presence of air.105 Reprinted with permission from ref. 105. Copyright 2015 The Royal Society of Chemistry. | ||
Phenolate based ligands
Although molecular Ti oxo-alkoxo are well studied (see above), crystalline extended networks are far more limited. To the best of our knowledge, a single 3-D Ti alkoxide (i.e. built up from alkyl-derived alcoholate) built up from 1,4-butandiol was reported, albeit soluble (and hence unstable) in most common organic solvents.122 As detailed below, aryloxide (phenolate) ligands have been more studied.As demonstrated by the very good photoactivity of Ti-MOF-74 under visible-light illumination, such ligands succeeded in driving the photoactivity of Ti complexes to the visible-light range. This can be accounted for by the TiIV–Ophenolate bonds, known to lead to ligand-to-metal charge transfer (LMCT; between the lone pairs of oxygen atoms and the vacant 3d-orbitals of Ti atoms), giving rise to strong absorption bands in the visible range.123–127 In addition to providing red-shift in the light absorption spectrum, the high basicity of phenolates (much higher than that of carboxylates) combined with the high oxophilic character of Ti-cations might lead to a strengthening of the interaction between Ti-centers and linkers yielding a better chemical stability. Studies have indeed already proven that, generally, combining a metal cation of high charge density (Zr, Ti) and a ligand showing high pKa values is an efficient strategy towards obtaining highly chemically stable MOFs.7,128,129
It is worth reminding that the combination between highly reactive species (highly basic phenolates and highly oxophilic Ti-cations) might turn the crystal growth of extended frameworks, i.e. MOFs, into a more complicated task, particularly when considering one-pot synthesis. However, a few crystalline coordination polymers and MOFs based on either carboxyphenolate or pure phenolate derivatives have been reported so far, and will be described below.
Hydroxycarboxylate derivatives
Crystalline compounds based on titanium and hydroxycarboxylate ligands remain very scarce. To our knowledge, only one type of complex prepared with purely α-hydroxybenzoic acid (salicylic acid, H2-Sal or (OH)-C6H4-COOH) has been reported so far. In any case (neutral [Ti(Sal)(H-Sal)2](BunOH)2123 or salts130–132), tris(salicylato)titanate(IV) is constructed from the isolated Ti-center with an octahedral geometry. The Ti-cation is hexacoordinated by three chelating Sal ligands through one Ophenolate and one Ocarboxylate atoms (Fig. 17a).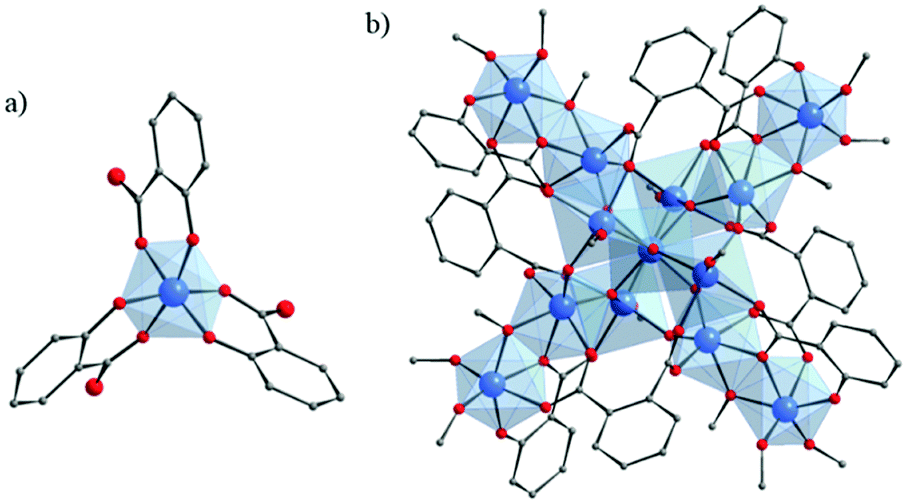 | ||
| Fig. 17 (a) Crystalline structure of tris(salicylato)titanate(IV);123 (b) crystalline structure of Ti13O10(o-BDC)4(Sal)4(OiPr)16.133 Color code: Ti, blue; O, red; C, grey. H-atoms are omitted for the sake of clarity. | ||
Very recently Hou et al.133 reported two isostructural Ti-oxo-clusters with a higher nuclearity, Ti13O10(o-O2C-C6H4-CO2)4(Sal)4(OiPr)16 and Ti13O10(o-O2C-C6H4-CO2)4(Sal-Cl)4(OiPr)16, prepared with a mixture of two ligands (Sal and o-BDC) under solvothermal conditions. Their crystal structures revealed that all the Ti4+ ions are octahedrally coordinated and assembled together through μ2-O and μ3-O bridges, oxygen atoms of chelating o-BDC, and Sal ligands with OiPr acting as terminal ligands (Fig. 17b). Despite the fact that these Ti13-clusters are not bearing only Sal ligands, they showed under a full-wavelength light source, similar photocurrent densities of ca. 0.30 μA cm−2.
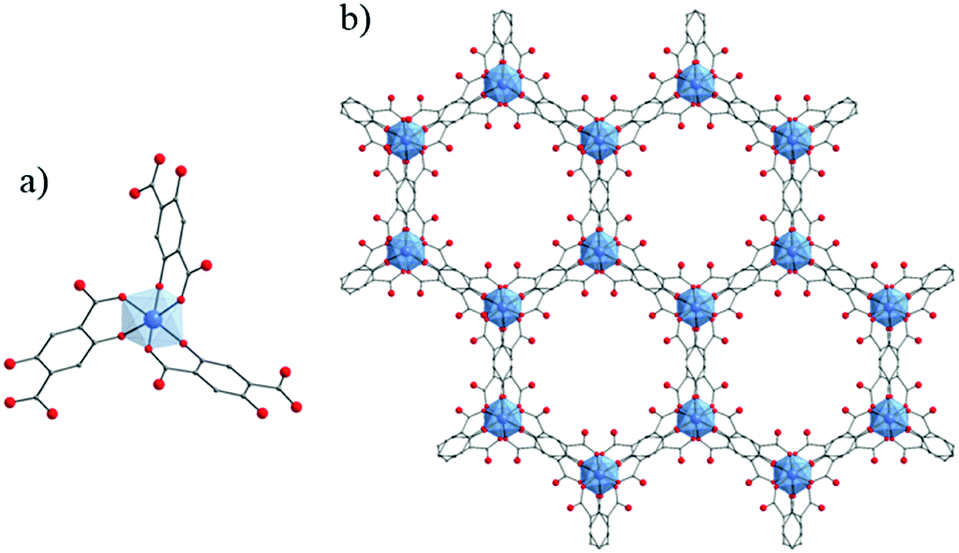
Due to their potential benefits for photocatalysis in the visible-light range, the use of carboxyphenolate ligands has expanded to extended coordination frameworks. The first cases are heterometallic Ti/Zn MOFs, labeled ZTOF-1134 and ZTOF-2,135 obtained using asymmetrical linkers, 2-hydroxyterephthalic acid and 3-hydroxy-2,7-naphthalenedicarboxylic acid, respectively. Though their crystalline structures totally differ, the connectivity of their Zn6Ti2 oxo-clusters to the linker remains similar where Ti-cations are coordinated by one Ophenolate and one Ocarboxylate atoms (Fig. 18). Both MOFs showed air-stable frameworks and permanent porosity towards different gases (N2, Ar, CO2 and H2) where calculated surface areas are SBET ZTOF-1 = 1045 m2 g−1 and SBET ZTOF-2 = 1878 m2 g−1.
 | ||
| Fig. 18 Crystalline structures of ZTOF-1134 (left) and ZTOF-2135 (right): (a and c) coordination mode between Zn- and Ti-cations and organic linkers (only one complete linker is represented); (b and d) general views of ZTOF-1 and ZTOF-2, respectively. Color code: Ti, blue; Zn, purple; O, red; C, grey. H-atoms are omitted for the sake of clarity. | ||
NTU-9129 (NTU stands for Nanyang Technological University) is one of the scarce MOFs constructed from purely Ti4+ cations and hydroxycarboxylate derivatives (2,5-dihydroxyterephthalic acid). It is obtained as deep-red single crystals after reacting, under solvothermal conditions, Ti(OiPr)4 with H4-DOBDC or HOOC-C6H2(OH)2-COOH in acetic acid. The structure of NTU-9 or TiIV2(H-L)2(H2-L)n with L = OOC-C6H2(O)2-COO or 2,5-dihydroxyterephthalate (DOBDC) is built up from Ti-cations octahedra, the six oxygen atoms being issued from phenolate and carboxylate groups, connected together via the organic ligands to form two dimensional perforated honeycomb-like layers. The coordination geometry of titanium ions is very similar to that of tris(salicylato)titanate described above. The stacking of these layers generates 1-D channels showing 11 Å × 11 Å hexagonal apertures (Fig. 19). Despite its stability under air, in water and common organic solvents and after guest removal at 120 °C, the permanent porosity of NTU-9 has not yet been assessed. Nevertheless, owing to the LMCT (confirmed by the density of states (DOS) calculation) at the origin of its deep-color, NTU-9 shows a broad range of light absorption in the visible region with a maximum at ca. 520 nm associated with an experimentally deduced optical gap of 1.72 eV which correlated with the theoretical calculations. Photoelectrochemical studies revealed that the aforesaid MOF shows a rather very weak cathodic photocurrent of ca. 60 nA cm−2 (measured on the NTU-9/FTO electrode). Finally, NTU-9 showed, under visible-light, a good photocatalytic activity towards the degradation of organic dyes (rhodamine B and methylene blue; degradation completed after 80 min and 20 min, respectively).
 | ||
| Fig. 19 Crystalline structure of NTU-9:129 (a) coordination mode between Ti-cation and organic linker; (b) view along the channels of NTU-9. Color code: Ti, blue; O, red; C, grey. H-atoms are omitted for the sake of clarity. | ||
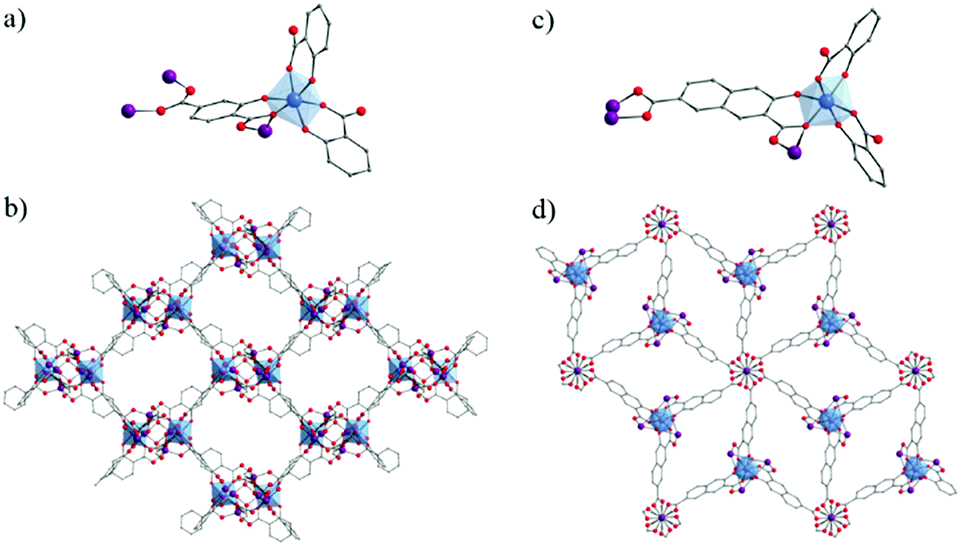
Very recently, the reactivity of 2,5-dihydroxyterephthalic acid (DOBDC or L in the following for MIL-167, MIL-168 and MIL-169) with titanium has been thoroughly investigated by some of us.127 Under solvo/hydro-thermal conditions and upon systematic variation of different parameters (solvent, titanium precursors, stoichiometry, modulator, etc.), four crystalline phases have been identified (Fig. 20).
 | ||
| Fig. 20 Crystalline structures of MIL-167, MIL-168 and MIL-169:127 (a, c and e) coordination mode between Ti-cation and organic linkers, respectively. Global views showing (b) two interpenetrated 3-D nets in MIL-167, (d) packed 1-D chains in MIL-168 and (f) layers connected through piperazinium cations via H-bonds (dashed blue lines) in MIL-169. Color code: Ti, blue; O, red; C, grey; N, dark blue; H, white. H-atoms of C-atoms are omitted for the sake of clarity. | ||
First, when using Ti(OiPr)4 as a precursor and pure N,N-diethylformamide (DEF) as a solvent, two phases were isolated: a first one very similar to NTU-9129 and a second one labelled MIL-168, formulated as TiIV(HxL)1.5(DEAH)2−1.5x·nsolv and TiIV(L)(cat)·2DEAH, respectively.
The addition of methanol to the reaction medium ultimately leads to the formation of MIL-167, formulated as TiIV(L)1.5(Et2MeNH)2·nH2O. This structure consists of isolated TiO6 octahedra, with the oxygen atoms arising solely from DOBDC ligands, connected to each other by the ditopic linkers through chelating six-membered rings involving both phenolic and carboxylic oxygen atoms to afford a 3-D chiral network exhibiting 10,3-a topology. Two enantiomeric networks are interpenetrated to afford the final solid, which holds cavities of ca. 6–7 Å free diameter. Interestingly, MIL-167 and NTU-9 are built up from the same coordination motif and hence, without taking into account the counter-ions, are simply polymorphs. The difference between both solids arises from a different alternation of the absolute configuration of the neighboring octahedral metal centers.
Adding catechol (H2-cat) in the reaction mixture allowed the crystallization of MIL-168. Its structure is built up from isolated TiO6 octahedra, with the coordinating oxygen atoms arising this time from two DOBDC ligands and one catechol moiety. This defines 1-D zigzag chains running parallel to each other and separated by N–H⋯O hydrogen-bonded diethylammonium (DEAH) cations, arising from the degradation of DEF.
In order to favor the formation of more condensed inorganic units, a tetrameric preformed titanium oxalate cluster, Ti4O4(OOC-COO)7(N2C4H10)3,136 was used as the precursor. Under hydrothermal conditions, MIL-169 was obtained. In this structure, each titanium ion is chelated by two DOBDC ligands through the six-membered ring involving phenolate and carboxylate groups already observed in MIL-167, MIL-168 and NTU-9. It is worth noting that the titanium cations are associated in dimers (Ti2O11) of octahedra bridged by a μ2-oxo group and bearing terminal water molecules. Dimers are connected through four DOBDC ligands to afford anionic layers separated by hydrogen bonded piperazinium cations and free water molecules.
For all these solids, no noticeable adsorption of N2 was however detected. However, MIL-167 shows a modest microporous character after adsorption of CO2 under near-ambient conditions (15 °C) with a maximal adsorption capacity of ca. 2.1 mmol·g−1 at 20 bar. This is due to both the interpenetration and the presence of entrapped bulky Et2MeNH cations inside the pores, arising again from the degradation of DEF into DEA and then the reaction of DEA with methanol.
All solids present strong absorption in the visible range, much broader than that of the titanium carboxylate systems,94,95,98,137 associated again with LMCT.123–127 Their optical spectra have shown a band centered at 250 nm, together with a broad band covering the 300–600 nm window. Noteworthily, under UV irradiation (λ = 280 nm), a noticeable production of hydrogen was detected for MIL-167 with a reaction rate initially ca. 2.6 higher than for MIL-125-NH2.99,138 Nevertheless, no activity was detected under visible light irradiation.
Pure phenolate derivatives
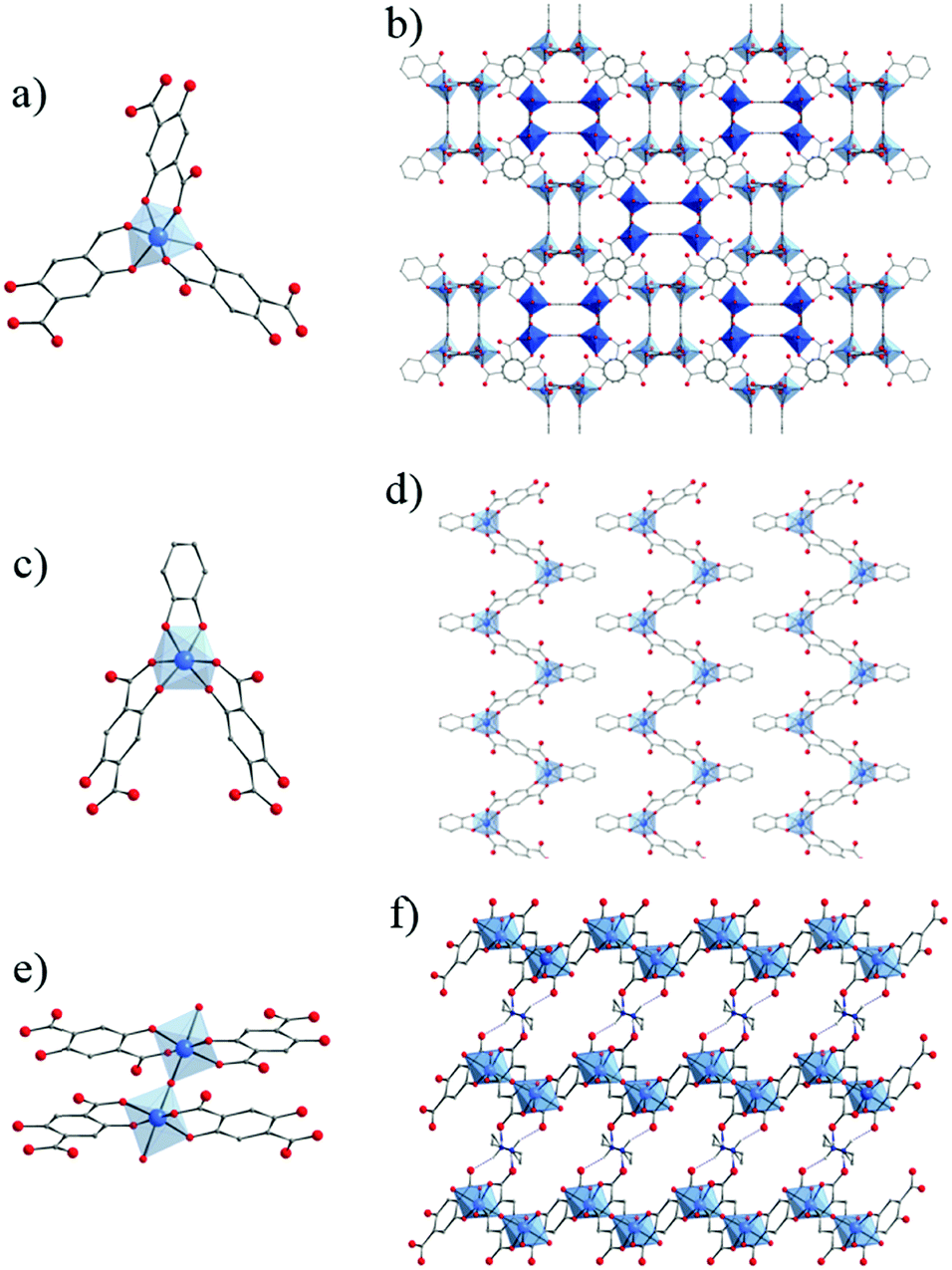
Several crystalline coordination complexes based on titanium and monotopic phenolate derivatives139 have been reported. They range from mono-,140–142 di-, tri-nuclear140 oxo-clusters up to polymeric arrangement (showing μ2-Cl and μ2-O bridges)143 (Fig. 21). The most common is the di-nuclear one of which different analogues have been reported: hexa-coordinated to only phenolates (Fig. 21a)144 or phenolate and terminal ligands,140,145,146 and penta-coordinated147 (phenolates and chlorine; Fig. 21b).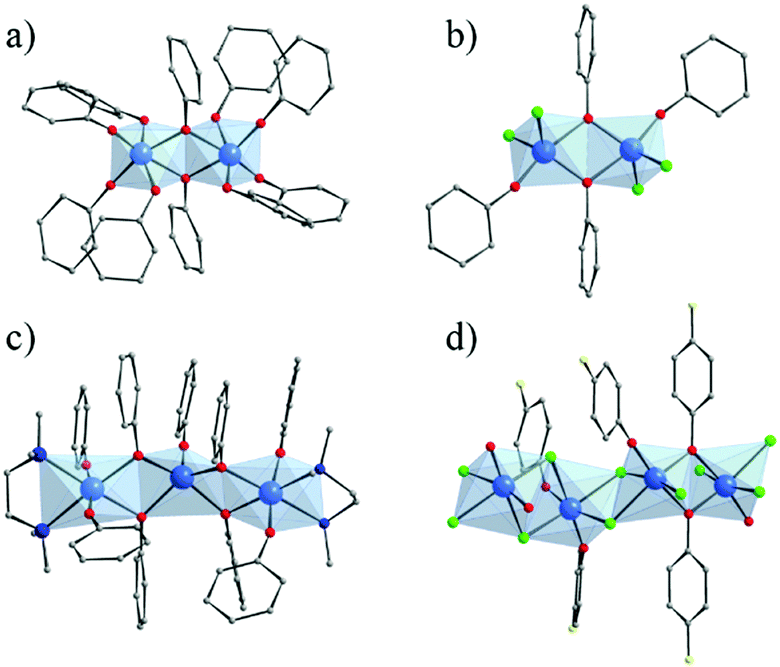 | ||
| Fig. 21 Crystalline structures of di-nuclear (a) 6-coordinated144 and (b) 5-coordinated147; (c) tri-nuclear 5-coordinated140 phenolate oxo-clusters; and (d) polymeric143 Ti-phenolate chain. Color code: Ti, blue; O, red; C, grey; N, dark blue; Cl, green; F, yellow. H-atoms are not represented for the sake of clarity. | ||
Ditopic phenolate ligands were first used to build up amorphous Ti based hybrid materials as early as in 1990.148,149 A few years later, Wolczanski and collaborators reported a series of crystalline Ti-oxo-phenolate coordination polymers. Prepared under inert conditions and quoted ‘covalent networks’, they present two- or three-dimensional networks based on isolated Ti-centers or dinuclear oxo-clusters (with or without terminal ligands) connected through various phenolate derivatives (see details in Table 1).150–154 The nature of the organic solvent seemed to play a role in the coordination mode and, thus, in the resulting coordination network. Among these coordination polymers, the one obtained by reacting Ti(OiPr)4 and 4,4′-dihydroxybiphenyl in THF under an inert atmosphere showed a 3-D open framework based on Ti-dimers (Fig. 22).153 If a large pore opening of the 3-D networks is of interest, the potential porosity is once again strongly reduced due to the presence of an interpenetrated network.
| Name | Molecular formula | Organic linker (L) | Inorganic unit nuclearity | Network dimensionality | Porosity | Photoredox property | Ref. |
|---|---|---|---|---|---|---|---|
| MIL-22 | TiIV3O2(H2O)2(L)2·2H2O | Methylenediphosphonate | 3 | 3-D | — | — | 48 |
| MIL-252 | TiIV(L) | Ethylenediphosphonate | 1 | 3-D | — | — | 46 |
| MIL-253 | TiIV(L) | Propylenediphosphonate | 1 | 3-D | — | — | 46 |
| MIL-91 | TiIVO(H2-L)·nH2O (n ∼ 4.5) | N,N′-piperazinebismethylenephosphonate | Infinite chain | 3-D | S BET = 500 m2 g−1 (N2 77 K) | — | 49 |
| MIL-125 | TiIV8O8(OH)4(L)6 | Terephthalate | 8 | 3-D | S BET = 1550 m2 g−1 (N2 77 K) | Alcohol oxidation (UV light) | 91 |
| COK-69op&c | TiIV3O3(L)3·DMF | 1,4-Cyclohexanedicarboxylate | 3 | 3-D | S BET = 29 m2 g−1 (N2 77 K) | Alcohol oxidation (UV light), Eg = 3.77 eV | 100 |
| MIL-101(Ti) | TiIII3O(OCH2CH3)(L)3·2DMF | 1,4-Benzenedicarboxylate | 3 | 3-D | S BET = 2970 m2 g−1 (N2 77 K) | — | 109 |
| PCN-22 | TiIV7O6(L)12·2DEF | Tetrakis(4-carboxyphenyl)porphyrin | 7 | 3-D | S BET = 1284 m2 g−1 (N2 77 K) | Alcohol oxidation (visible light), Eg = 1.93 eV | 101 |
| MOF-901 | TiIV6O6(OCH3)6(L)3 | 1,4-Phenylenebis(methanylylidene) bis(azanylylidene)dibenzoate | 6 | 2-D | S BET = 550 m2 g−1 (N2 77 K) | Mediated radical polymerization of methyl methacrylate (visible light), Eg = 2.65 eV | 102 |
| MOF-902 | TiIV6O6(OCH3)6(L)3 | Biphenyl-4,4-diylbis-(methanylylidene) bis(azanylylidene)dibenzoate | 6 | 2-D | S BET = 400 m2 g−1 (N2 77 K) | Mediated radical polymerization of methyl methacrylate (visible light), Eg = 2.50 eV | 114 |
| NTU-9 | TiIV2(H-L)2(H2-L)n | 2,5-Dihydroxyterephthalate | 1 | 2-D | — | Degradation of organic dyes (visible light), Eg = 1.72 eV | 129 |
| MIL-167 | TiIV(L)1.5(Et2MeNH)2·nH2O | 2,5-Dihydroxyterephthalate | 1 | 3-D | Maximal adsorption capacity ∼ 2.1 mmol g−1 (CO2 20 bar) | Water splitting (UV light) | 127 |
| MIL-168 | TiIV(L)(cat)·2DEAH | 2,5-Dihydroxyterephthalate | 1 | 1-D | — | — | 127 |
| MIL-169 | TiIVO0.5(L)(H2O)(H2-pip)0.5·nH2O | 2,5-Dihydroxyterephthalate | 2 | 2-D | — | — | 127 |
| Ti-phenol | [TiIV(L)(H-L)]2(μ-H-L)2 | Hydroquinone | 2 | 3-D | — | — | 150 |
| Ti-phenol | TiIV(L)2·py2 | Hydroquinone | 1 | 2-D | — | — | 150 |
| Ti-phenol | TiIV2(μ1,4-L)2(μ1,4-H-L)2(μ-H-L)2 | Hydroquinone | 2 | 3-D | — | — | 151 |
| Ti-phenol | TiIV2(μ1,4-L)2(μ1,4:η2,η1-L)2(OH2)2(H2O)2(H2-L)·x(CH3CN) (x = 1 or 2) | Hydroquinone | 2 | 3-D | — | — | 151 |
| Ti-phenol | TiIV2(μ1,7-L)2(μ1,7:η2,η1-H-L)2(OiPr)2 | 2,7-Dihydroxy-naphthalene | 2 | 3-D | — | — | 151 |
| Ti-phenol | TiIV(μ1,4-L)2py2 | Hydroquinone | 1 | 2-D | — | — | 152 |
| Ti-phenol | TiIV(μ1,4-L)2·py2·py | Hydroquinone | 1 | 2-D | — | — | 152 |
| Ti-phenol | TiIV(μ1,4-L)2·(4-Ph-py)2 | Hydroquinone | 1 | 2-D | — | — | 152 |
| Ti-phenol | TiIV(μ1,3-L)2·py2 | Resorcinol | 1 | 2-D | — | — | 152 |
| Ti-phenol | TiIV(μ1,3-L)2·(4-Ph-py)2 | Resorcinol | 1 | 2-D | — | — | 152 |
| Ti-phenol | TiIV(μ1,6:η2,η1-4,4′-L)0.5(μ1,6:η2,η1-4,4′-L)(OiPr)(HOiPr)2·THF | 4,4′-Dihydroxy-biphenyl | 2 | 3-D | — | — | 153 |
| Ti-phenol | TiIV(μ1,3-1,4-L)(μ1,3-1,3-L)(1,3-H-L)(HOiPr)2 | Resorcinol | 2 | 2-D | — | — | 153 |
| Ti-phenol | TiIV(μ2,7-L)2·py2 | 2,7-Dihydroxy-naphthalene | 1 | 1-D | — | — | 154 |
| Ti-phenol | TiIV(μ2,7-L)2(4-picoline)2.5 | 2,7-Dihydroxy-naphthalene | 1 | 2-D | — | — | 154 |
| Ti-CAT-5 | TiIV(L)·2DMA | 2,3,6,7,9,11-Hexahydroxytriphenylene | 1 | 3-D | S BET = 450 m2 g−1 (N2 77 K) | — | 176 |
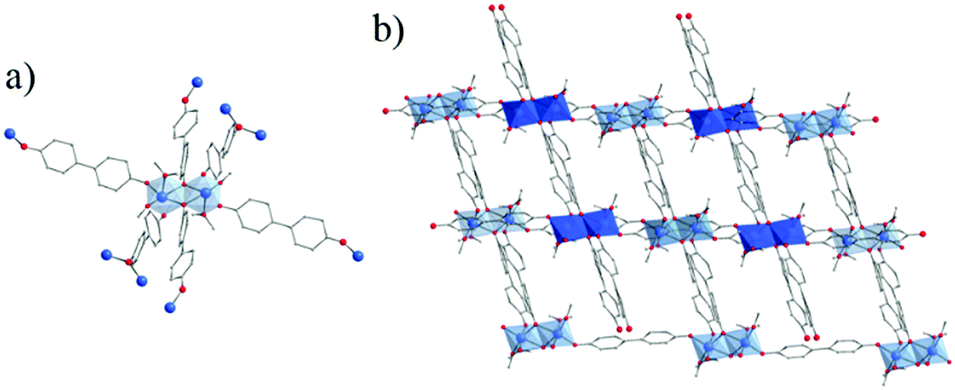 | ||
| Fig. 22 Crystal structure of Ti-oxo-biphenolate:153 (a) view of the dinuclear oxo-cluster and (b) two interpenetrated nets. Color code: Ti, blue; O, red; C, grey; H, white. H-atoms of C-atoms and pore filling molecules are omitted for the sake of clarity. | ||
Catecholate derivatives
Among the different possible phenolate derivatives, catechol (H2-cat or C6H4(OH)2) is an interesting candidate due to its interest in biology, electrochemistry and catalysis. In addition, being rather cheap and easy to handle, H2-cat is ideal for industrial applications. In biology,155 one can cite siderophores, a class of functionalized catechols. In catalysis, titanium aryloxides are used as homogeneous catalysts for instance for olefine polymerization,156 oxidation,157 epoxidation158 or the enantioselective formation of carbon–carbon bonds.159 In molecular coordination chemistry, catechol ligands can strongly stabilize various cations in solution,123 and this allows tuning their electrochemical properties. Catechols have also been used to graft Ru(II) photosensitizers on the surface of TiO2 photoelectrodes in photovoltaic cells.160 The use of polycatechol derivatives to build up new Ti-MOFs and benefit from the optical/electrochemical properties already demonstrated at the molecular scale is of strong interest. In this part, we will highlight some of the most relevant reported titanium–catecholate complexes that could be considered as secondary building units for the preparation of new hybrid porous solids (MOFs) based on titanium. Metallo-supramolecular self-assembled Ti-polycatecholate complexes161,162–165,166,167 (i.e. cages, helicates) are not discussed in this review.According to the literature, catechol can coordinate metal ions in different fashions (chelation or μ2-coordination mode), which are summarized in Fig. 23.123
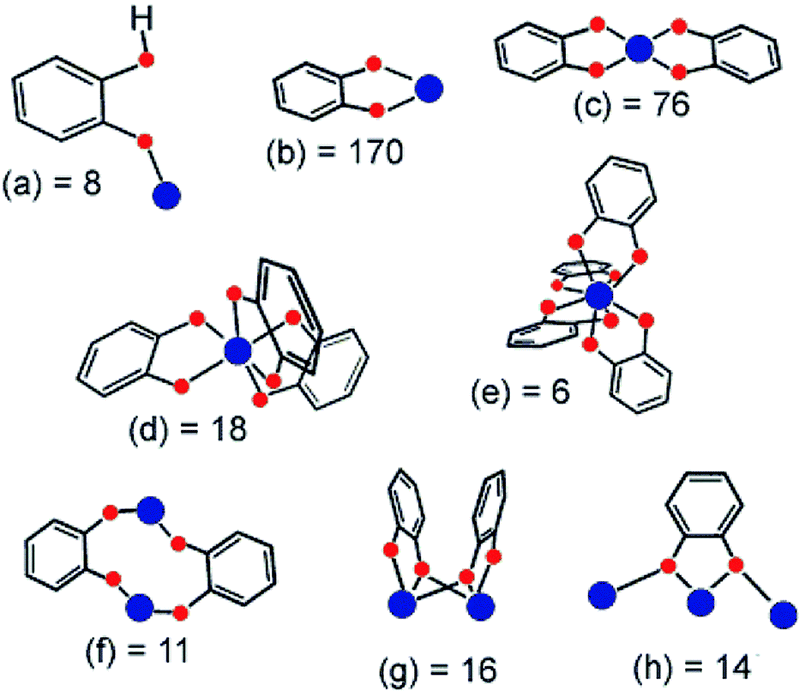 | ||
| Fig. 23 Coordination modes of the catecholato ligand characterized through single-crystal X-ray diffraction. The number after the letter is the occurrence found in the 5.21 release (January 2001) of the Cambridge Crystallographic Database. Metal and oxygen atoms are colored blue and red, respectively.123 Reprinted with permission from ref. 123. Copyright 2001 American Chemical Society. | ||
In 1920, Rosenheim et al.168 observed that catechol has a very high affinity for Ti4+, considered as a strong Lewis acid. They have reported the synthesis of [NH4]2[Ti(cat)3]·H2O (Fig. 24a), prepared from TiCl4 in HCl 1 M solution of catechol in a deoxygenated solution of NH4OH. They pointed out that a catechol solution is able to dissolve hydrated titanium oxides (relatively insoluble) illustrating the high affinity of catechol to strong Lewis acids. According to the authors, this system appeared to be limited to acidic medium, probably because of subsequent hydrolysis and polymerization of titanium(IV).
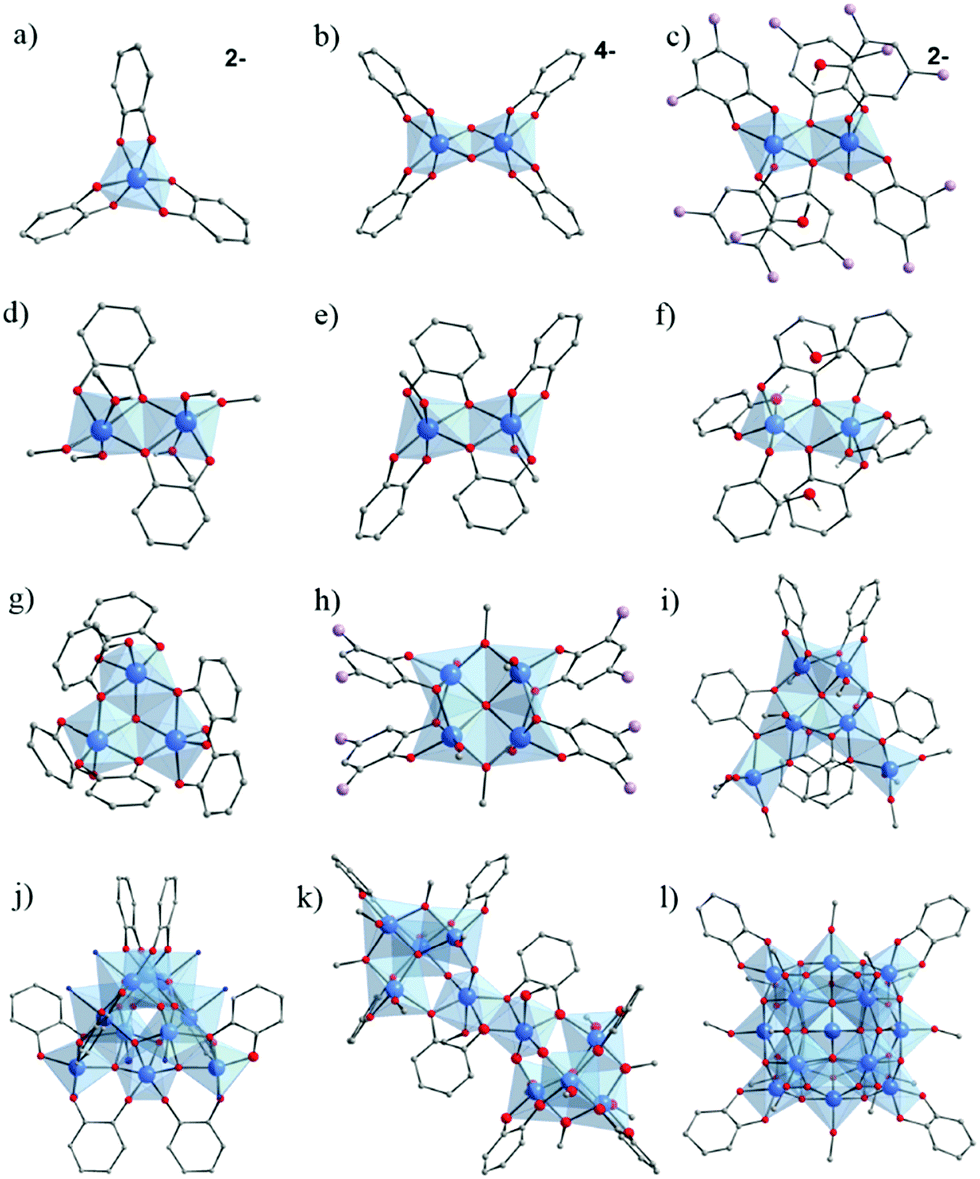 | ||
| Fig. 24 Molecular structures of the core of Ti–catechol/ate complexes and clusters cited in this review. (a) [Ti(cat)3];2–168,169 (b) [Ti2O2(cat)4];4–169 (c) [Ti2(DTBC)4(HDTBC)2];2–169 (d) Ti2(OiPr)4(HOiPr)2(cat)2;60,170 (e) Ti2(HOiPr)2(cat)4;170 (f) Ti2(cat)2(cat-H)4;60 (g) Ti3O(cat)4(cat-H)2;172 (h) Ti4O(OiPr)6(DTBC)4:173 (i) Ti6O(cat)6(OiPr)10;60 (j) Ti10O12(cat)8(py)8;174 (k) Ti10O6(OBun)12(HOBun)2(cat)8123 and (l) Ti17O24(OiPr)16(cat)4.60 Color code: Ti, blue; O, red; C, grey; N, dark blue; C(CH3)3, lavender. Counter-ions (ammonium in (a) and (c), and potassium in (b)), C-atoms in alcohol/-ate groups, and all H-atoms of C-atoms are not represented for the sake of clarity. N-atoms represent pyridine groups in (j). | ||
In 1984, Borgias et al.169 reported two new Ti–catecholate complexes K4[Ti2(μ2-O)(cat)4]·9H2O (Fig. 24b) and [NH(CH3CH2)3]2[Ti2(DTBC)4(HDTBC)2]·2CHCl3 (DTBC = 3,5-Di-tert-butylcatecholate or [(CH3)3C]2C6H2(OH)2) (Fig. 24c), in addition to the complex [NH(CH3CH2)3]2[Ti(cat)3], similar to the one reported earlier by Rosenheim et al.168 All compounds were here prepared in basic conditions, illustrating the effect of the complexing group on the synthetic conditions. If both dimeric complexes are anionic, only in the first Ti4+ are connected through μ2-oxo bridges while in the second two O-atoms of two catechol are bridging.
UV-vis spectroscopic studies have shown that Ti4+ ions are able to stabilize the cat2− ions more strongly than the Fe3+ under acidic conditions.169 Moreover, the absorption spectrum of [NH4]2[Ti(cat)3] does not change from pH 6 to pH 12 even in the absence of an excess of catechol, suggesting that the complex is preserved. Only strong basic conditions lead to the [TiO(cat)2]24− dimer.
Dinuclear Ti–catecholate clusters have also been reported by Davidson et al.170 Two different molecular structures, Ti2(μ-OiPr)4(μ-HOiPr)2(cat)2 (Fig. 24d) and Ti2(μ-HOiPr)2(cat)4 (Fig. 24e), have been isolated by reacting under inert conditions Ti(OiPr)4 with one and two equivalents of catechol, respectively. A series of Ti2(μ-OiPr)4(μ-HOiPr)2(R)2 has also been obtained by performing similar reactions using substituted catechol derivatives (H2R) where R = 4-methyl-, 4-tertbutyl-, 3,5-tertbutyl-, 3-methoxy-, 4-nitro-, or 3-methyl-catechol. In all these Ti2–catecholate dimers, catechol bridges between two titanium centers. In contrast to the anionic dimers reported by Borgias et al.,169 these complexes are neutral. Due to the potential active sites after removal of coordinated alcohol groups, the catalytic activity of both series (activated in vacuum) for the ring-opening-polymerization of caprolactone to afford polycaprolactone (PCL) was evaluated.171 Ti2(μ-OiPr)4(μ-HOiPr)2(cat)2 has shown the highest activity.
In 2005, Boyle et al.172 reported a trinuclear Ti-oxo-catecholate cluster. Single crystals of Ti3(μ3-O)(μ-cat)3(cat)(cat-H)2 have been isolated by recrystallizing, from pyridine, the red powder of titanium catecholate obtained after the reaction of catechol with [Ti(μ-ONep)(ONep)3]2 (ONep = OCH2CMe3) in anhydrous THF. Its structure (Fig. 24g) consists of a three hexa-coordinated Ti4+ connected through a centered μ3-O bridge, one chelating catechol and two bridging-chelating catechols, and is, to a certain extent, reminiscent of the trimeric unit found in carboxylate MOFs.
Very recently, a tetra-nuclear Ti-oxo-catecholate based cluster has been reported. Ti4(μ4-O)(OiPr)6(DTBC)4 (DTBC = 3,5-di-tert-butylcatecholate) was obtained under solvothermal conditions by reacting Ti(OiPr) and H2-DTBC in isopropanol.173 The four Ti-centers adopt a square-shaped arrangement connected through a central μ4-oxo bridge and bridging bidentate chelate DTBC (Fig. 24h). XPS measurements revealed that it is a mixed valence oxo-cluster with a Ti4+/Ti3+ ratio of ca. 1:1.8. Thanks to the bidentate chelate coordination mode of catecholate derivatives, the oxo-cluster shows strong visible absorption promoted by LMCT, and resulting in an optical gap of ca. 2.46 eV (lower than that calculated from DFT, 2.82 eV).
In the quest of elaborating new Ti–catechol based clusters, Benedict and collaborators60 succeeded in isolating new titanium oxo-clusters of different nuclearities. Two di-nuclear, Ti2(OiPr)4(HOiPr)2(cat)2 and Ti2(cat)2(cat-H)4 (Fig. 24f), and one hexa-nuclear Ti6(μ4-O)(cat)6(OiPr)10 (Fig. 24i) were obtained by direct reaction between Ti(OiPr)4 and catechol under inert conditions. If the structure of Ti2(cat)2(OiPr)4(HOiPr)2 is a novel polymorph of a previously reported Ti–catechol dimer,170 Ti2(cat)2(cat-H)4 (Fig. 24f) is the first titanium–catecholate polynuclear complex bearing purely catechol/ate ligands (showing three coordination modes: monodentate, bidentate and μ2-(O, O′, O′)). In the Ti6(μ4-O)(cat)6(OiPr)10 oxo-cluster, the six catechol ligands are connecting Ti4+ through two coordination modes, μ2-(O, O′, O′) for two ligands and μ2-(O, O′, O, O′) for the others, as well as one μ4-oxo-bridge.
Besides, an oxo-cluster, Ti17(μ4-O)4(μ3-O)16(μ2-O)4(OiPr)16(cat)460 (Fig. 24l), of high nuclearity has also been isolated by reacting the pre-formed [Ti17O24(OiPr)20] cluster.59 The latter consists of one central tetra-coordinated Ti-center surrounded by sixteen Ti centers, among which a few are five-coordinated and bear isopropoxide groups. Upon reaction with catechol, isopropoxide groups are partially exchanged by bidentate catechols, leading to hexa-coordinated Ti.
Theoretical optical gap calculation limited to the aforementioned Ti-oxo-clusters showed no correlation between the size of the cluster and the gap values (Ti2cat2: 3.78 eV, Ti2cat6: 3.51 eV, Ti6cat6: 3.41 eV and Ti17cat6: 4.71 eV).
Another example of Ti-oxo-catecholate cluster of high nuclearity has been reported by Gigant et al.123 In agreement with previous studies,175 it was not possible to obtain single crystals from a molar ratio R of catechol to Ti of greater than 1. Depending on the amount of water added (h = H2O/Ti) to the resulting clear solution, a good yield of microcrystalline precipitate was observed for h = 0.3 and R = 0.75 with Ti(OBun)4 or h = 0.4 and R = 0.8 for Ti(OiPr)4. The crystalline structure of the complex obtained with Ti(OBun)4 is [Ti10O6(OBun)12(HOBun)2(cat)8].123 It consists of two tetrahedral (μ4-O)Ti4 building blocks sharing corners with one central edge-sharing dioctahedral unit and leading to a bowtie-shaped molecule. TiO6 octahedra are linked together through three catechol and two butoxy ligands.
Recently, Chaumont et al.125,174 prepared several complexes incorporating Ti–O bonds (phenolate) which absorb again between 300 nm and 500 nm (red-orange). They first studied the reactivity of catechol with a titanium oxo-carboxylate complex of the formula Ti8O8(OOCCH2C(CH3)3)16 (see Fig. 4g) having at its periphery carboxylato ligands substituted with tert-butyl groups, such groups being used to significantly increase the solubility of the complex in solution.125 At a low catechol ratio (1 to 7 equivalent of catechol per Ti8O8 cluster), it was possible to form titanium complexes incorporating both carboxylate and catecholate ligands. With an excess of catechol (32 equivalents), the complex was completely converted in the presence of DMAc (dimethylacetamide) and DMF to produce other complexes exclusively containing catechol as ligand, and formulated Ti2(cat)4(DMAc)2 or Ti2(cat)4(DMF)2. Noteworthily, from the recrystallization of these complexes, they were also able to isolate a new [Ti10O12(cat)8(py)8] oxo-cluster.174 NMR DOSY studies further revealed the lability of the pyridine ligands present at the periphery of the cluster, which could be exchanged by other nitrogen ligands (picoline, 4-phenylpyridine or 2-methoxy-5-(pyridin-4-yl)-benzaldehyde). This exchange is however associated with a reorganization of the original cluster.
These results have highlighted the richness of the titanium–catecholate molecular chemistry. At the sight of these works, the use of phenolate linkers allows obtaining various Ti-oxo-clusters with geometries different from those obtained with carboxylate ligands that can be potentially used as SBUs for the design of MOFs.
However, only one example of titanium-based polycatecholate MOF has been reported to date. Published very recently by Nguyen et al., MOF Ti-CAT-5 was obtained as a crystalline powder after reacting, under solvothermal conditions, the tritopic catechol derivative ligand (2,3,6,7,9,11-hexahydroxytriphenylene, H6-HHTP or C18H6(OH)6) and Ti(OiPr)4 under basic conditions in DMF.176 The structure of this MOF (Fig. 25) consists of an anionic 3-D framework based on isolated Ti-centers octahedrally coordinated to 3 chelating HHTP ligands. The large aperture of this network is occupied by an identical interpenetrated network and organic cations (DMAH) leading to a strong reduction of the accessible surface area (SBET = 450 m2 g−1). Noteworthily, iron and vanadium analogues of Ti-CAT-5 could be isolated. Proton conductivity measurements revealed lower conductivity values for Ti-CAT-5 in comparison with the iron-based structure (σ = 8.2.10−4 and 5.0.10−2 S cm−1, respectively, at 25 °C and 98% RH). The authors attributed this to the differences of pore content between the two forms (DMAH vs. sulfates in addition to water).
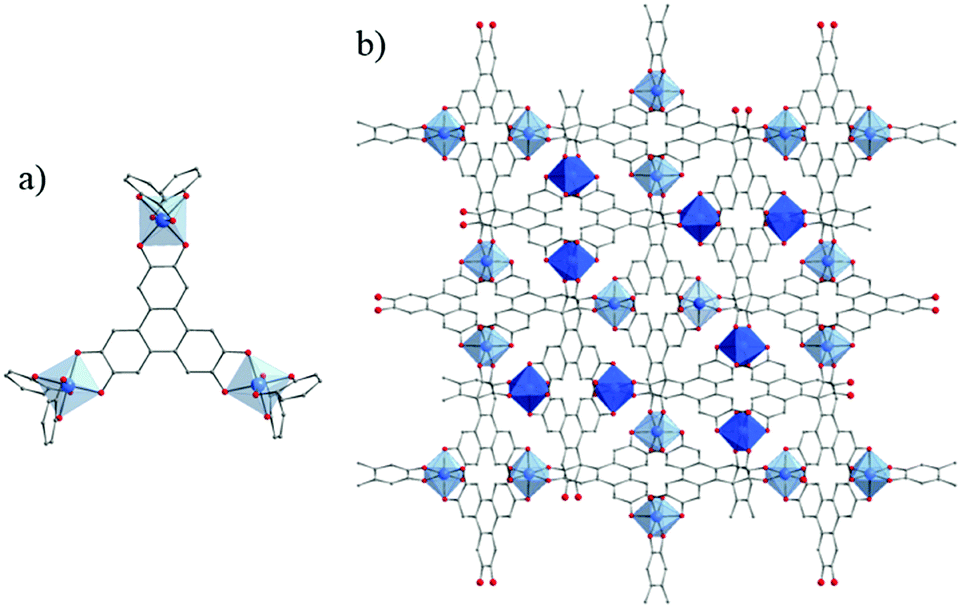 | ||
| Fig. 25 Crystalline structure of Ti-CAT-5:176 (a) coordination mode between Ti-cation and organic linker; (b) view of the two interpenetrated networks. Ti, blue; O, red; C, grey. H-atoms of C-atoms and pore filling molecules are omitted for the sake of clarity. | ||
Conclusion
If the chemistry of titanium based coordination compounds (molecular and polymers) could appear as not “new” in a sense that it has been initiated more than a few decades ago, there has been since then a continuous effort to develop new architectures with a high degree of structural and chemical diversity.This review proposes a summary of the solid state coordination chemistry of Ti with oxygenated ligands. On the opposite, nitrogen based ligands are far less explored. To our knowledge only one example of coordination polymer based on isolated Ti4+ ions connected through N-atoms has been reported to date.177 Its structure consists of 2-D networks based on TiIVCl4 units linked by tetrahedral ligands bearing four nitrile groups. The scarceness of such compounds could be mainly attributed to the high oxophilic character of Ti-cations rendering their crystallization under ambient conditions and/or in the presence of any oxygenated source (alkoxide, water, etc.) very challenging.
The structural and chemical diversity of Ti-based compounds has been significantly expanded so far through a careful control of the synthetic parameters (precursor, hydrolysis rate, etc.); the incorporation of many types of organic oxygenated linkers (i.e. phosphonate, carboxylate, hydroxycarboxylate, phenolate and catecholate) in polymeric hybrid crystalline materials (coordination polymers, MOFs, etc.) appears now feasible. One could raise several intermediate conclusions from this study: (i) on a qualitative basis, the design of Ti coordination compounds is still hardly achievable and/or predictable due to a lower level of control compared to the other systems based on di-, tri-valent and Zr cation based MOFs, (ii) from the structural point of view, depending mainly on the nature of the ligand and the hydrolysis ratio, the nuclearity and condensation rates strongly vary. More specifically, while increasing the complexing ability of the ligand allows preventing the competitive formation of TiO2 during the synthesis, it also disfavors the formation of large oxo-clusters. For instance, when Ti alkoxides are considered, the nuclearity of the oxo-cluster ranges from Ti256 to Ti3455 while Ti oxo-carboxylate clusters span from tri-, hexa-, hepta-, and nona-nuclear clusters (ratio carboxylate/alkoxide ≥ 1); when turning to phenolate based systems, most structures exhibit a lower degree of nuclearity, typically from single Ti octahedra to dimers or trimers of octahedra, and only very specific strategies can lead to larger clusters.
In terms of optical properties, which are one of the key targets here, the consequences of their chemical or structural characteristics, particularly in the visible-light region, have been discussed. If one shall mainly follow post-synthetic functionalisation strategies (ligand, inclusion of metal complexes or nanoparticles) to introduce visible light absorption in most cases (phosphonates, carboxylates), the LMCT occurring in Ti–Ophenolate bonds is strongly beneficial to the Ti phenolate based systems with a significant enhancement in light absorption in the visible-light range. Although these solids have still been less explored and suffer from a lower thermal stability, this makes such systems very promising in photocatalysis or opto-electronics.
Finally, if one considers more specifically Ti-based MOFs, the recent progress made within the past decade with a series of new microporous solids, from phosphonates, carboxylates to phenolates, through specific direct synthesis or post-synthetic strategies, confirmed the potential benefits of these systems. Although still one of the most challenging domains of MOF chemistry, there is certainly still strong interest to pursue efforts to rationalize and further expand the scope of chemical and structural diversity of Ti based MOFs or molecular solids in the view of new potential applications.
As highlighted in this review, controlling the kinetics of crystallization remains one of the most prominent strategies allowing crystalline and porous Ti-MOFs to be obtained. Nevertheless, the highly unpredictable polycondensation of titanium cations still renders the design of MOFs a quasi-impossible task so far. In this context, in order to exert more control on the elaboration of new MOFs in the future, two possible strategies can be envisioned. The first one would be to better take advantage of the tools of organic chemistry in combination with the plethora of existing Ti-oxo-clusters to form new porous Ti-MOFs;102,114 in such a case one should however consider that most Ti-oxo-clusters are not very chemically stable (e.g. alkoxide or carboxylate based complexes) and thus one challenge would be to rely on the most stable ones (e.g. catecholates). Another “dream” of the MOF chemists would be, as it was done during the past decade with trivalent based MOFs (e.g. V, Fe or Al) or more recently with zirconium(IV), to find new chemical routes enabling a certain degree of control over the inorganic polycondensation in the presence of the complexing ligands. It was indeed proposed previously with bis-phosphonate ligands leading however to a very limited number of open framework materials, probably due to the intrinsic character of metal–phosphonate connectivity. Very recently a few interesting results were also reported with phenolate based ligands including the synthesis of new Ti-MOFs as single crystals; although once again these solids exhibit a very limited porous character, mainly due to interpenetration issues, the latter approach shall be pursued in order to reach a higher degree of rationale in the synthesis of porous Ti-MOFs.
Acknowledgements
The authors acknowledge past and present coworkers involved in the synthesis and characterization of new MOFs, especially those involved in the chemistry of Ti-MOFs (H. Dan-Hardi, V. Guillerm, N. Guillou, P. Horcajada, T. L. A. Nguyen, F. Nouar, L. C. Pardo-Perez, F. Ragon and G. Férey).Notes and references
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed
.
- H.-C. “Joe” Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC
- J. J. Low, A. I. Benin, P. Jakubczak, J. F. Abrahamian, S. A. Faheem and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 15834–15842 CrossRef CAS PubMed
- T. Devic and C. Serre, Chem. Soc. Rev., 2014, 43, 6097–6115 RSC
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem. – Eur. J., 2011, 17, 6643–6651 CrossRef CAS PubMed
- G. Mouchaham, L. Cooper, N. Guillou, C. Martineau, E. Elkaïm, S. Bourrelly, P. L. Llewellyn, C. Allain, G. Clavier, C. Serre and T. Devic, Angew. Chem., Int. Ed., 2015, 54, 13297–13301 CrossRef CAS PubMed
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC
- V. Guillerm, F. Ragon, M. Dan-Hardi, T. Devic, M. Vishnuvarthan, B. Campo, A. Vimont, G. Clet, Q. Yang, G. Maurin, G. Férey, A. Vittadini, S. Gross and C. Serre, Angew. Chem., Int. Ed., 2012, 51, 9267–9271 CrossRef CAS PubMed
- M. Kim and S. M. Cohen, CrystEngComm, 2012, 14, 4096–4104 RSC
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC
- N. Steunou, S. Förster, P. Florian, C. Sanchez and M. Antonietti, J. Mater. Chem., 2002, 12, 3426–3430 RSC
- A. Strachota, K. Rodzeń, V. Raus, F. Ribot, M. Janata and E. Pavlova, Eur. Polym. J., 2015, 68, 366–378 CrossRef CAS
- G. Fornasieri, L. Rozes, S. Le Calvé, B. Alonso, D. Massiot, M. N. Rager, M. Evain, K. Boubekeur and C. Sanchez, J. Am. Chem. Soc., 2005, 127, 4869–4878 CrossRef CAS PubMed
- L. Nicole, C. Boissière, D. Grosso, A. Quach and C. Sanchez, J. Mater. Chem., 2005, 15, 3598 RSC
- C. Sanchez, B. Julián, P. Belleville and M. Popall, J. Mater. Chem., 2005, 15, 3559 RSC
- Hydra/Medusa Chemical Equilibrium, Database and Plotting Software, KTH Royal Institute of Technology, 6th edn, 2004
- S. Cassaignon, M. Koelsch and J.-P. Jolivet, J. Mater. Sci., 2007, 42, 6689–6695 CrossRef CAS
- L. Rozes and C. Sanchez, Chem. Soc. Rev., 2011, 40, 1006–1030 RSC
- N. Steunou, F. Robert, K. Boubekeur, F. Ribot and C. Sanchez, Inorg. Chim. Acta, 1998, 279, 144–151 CrossRef CAS
- B. Moraru, N. Hüsing, G. Kickelbick, U. Schubert, P. Fratzl and H. Peterlik, Chem. Mater., 2002, 14, 2732–2740 CrossRef CAS
- N. Steunou, F. Ribot, K. Boubekeur, J. Maquet and C. Sanchez, New J. Chem., 1999, 23, 1079–1086 RSC
- R. Schmid, A. Mosset and J. Galy, J. Chem. Soc., Dalton Trans., 1991, 1999–2005 RSC
-
N. N. Greenwood and A. Earnshaw, Chemistry of the elements, Butterworth-Heinemann, Oxford, 2nd edn, 1997 Search PubMed
-
P. Pascal, Nouveau traité de chimie minérale, Masson, Paris, 2nd edn, 1962 Search PubMed
-
F. A. Cotton and G. Wilkinson, Advanced inorganic chemistry, Wiley, New York, 5th edn, 1988 Search PubMed
-
J.-P. Jolivet, De la solution à l’oxide, InterEditions, CNRS, Paris, 1994 Search PubMed
- H. Auterhoff, Arch. Pharm., 1965, 298, 400 CrossRef
- D. W. Breck, J. Chromatogr. Sci., 1975, 13, 18A CrossRef
- D. M. Chapman and A. L. Roe, Zeolites, 1990, 10, 730–737 CrossRef CAS
-
M. Taramasso, G. Perego and B. Notari, US Pat., US 4410501A, 1983 Search PubMed
- M. W. Anderson, O. Terasaki, T. Ohsuna, A. Philippou, S. P. MacKay, A. Ferreira, J. Rocha and S. Lidin, Nature, 1994, 367, 347–351 CrossRef CAS
- A. Philippou, M. Naderi, J. Rocha and M. W. Anderson, Catal. Lett., 1998, 53, 221–224 CrossRef CAS
- J. Rocha, P. Brandão, J. D. Pedrosa de Jesus, A. Philippou and M. W. Anderson, Chem. Commun., 1999, 471–472 RSC
- P. Brandão, A. Philippou, A. Valente, J. Rocha and M. Anderson, Phys. Chem. Chem. Phys., 2001, 3, 1773–1777 RSC
- S. Lima, A. S. Dias, Z. Lin, P. Brandão, P. Ferreira, M. Pillinger, J. Rocha, V. Calvino-Casilda and A. A. Valente, Appl. Catal., A, 2008, 339, 21–27 CrossRef CAS
- Y. K. Krisnandi, E. E. Lachowski and R. F. Howe, Chem. Mater., 2006, 18, 928–933 CrossRef CAS
- J. Rocha, P. Brandão, Z. Lin, A. P. Esculcas, A. Ferreira and M. W. Anderson, J. Phys. Chem., 1996, 100, 14978–14983 CrossRef CAS
- S. M. Kuznicki, V. A. Bell, S. Nair, H. W. Hillhouse, R. M. Jacubinas, C. M. Braunbarth, B. H. Toby and M. Tsapatsis, Nature, 2001, 412, 720–724 CrossRef CAS PubMed
- G. Alberti, P. Cardini-Galli, U. Costantino and E. Torracca, J. Inorg. Nucl. Chem., 1967, 29, 571–578 CrossRef CAS
- S. Allulli, C. Ferragina, A. La Ginestra, M. A. Massucci and N. Tomassini, J. Inorg. Nucl. Chem., 1977, 39, 1043–1048 CrossRef CAS
- A. Clearfield and J. A. Stynes, J. Inorg. Nucl. Chem., 1964, 26, 117–129 CrossRef CAS
- A. N. Christensen, E. K. Andersen, I. G. K. Andersen, G. Alberti, M. Nielsen, M. S. Lehmann and T. Tokii, Acta
Chem. Scand., 1990, 44, 865–872 CrossRef CAS
- A. I. Bortun, S. A. Khainakov, L. N. Bortun, D. M. Poojary, J. Rodriguez, J. R. Garcia and A. Clearfield, Chem. Mater., 1997, 9, 1805–1811 CrossRef CAS
- C. Serre and G. Férey, J. Mater. Chem., 1999, 9, 579–584 RSC
- C. Serre and G. Férey, Inorg. Chem., 2001, 40, 5350–5353 CrossRef CAS PubMed
- C. Serre, F. Taulelle and G. Férey, Chem. Mater., 2002, 14, 998–1003 CrossRef CAS
- C. Serre and G. Férey, Inorg. Chem., 1999, 38, 5370–5373 CrossRef CAS
- C. Serre, J. A. Groves, P. Lightfoot, A. M. Z. Slawin, P. A. Wright, N. Stock, T. Bein, M. Haouas, F. Taulelle and G. Férey, Chem. Mater., 2006, 18, 1451–1457 CrossRef CAS
- V. Benoit, R. S. Pillai, A. Orsi, P. Normand, H. Jobic, F. Nouar, P. Billemont, E. Bloch, S. Bourrelly, T. Devic, P. A. Wright, G. de Weireld, C. Serre, G. Maurin and P. L. Llewellyn, J. Mater. Chem. A, 2016, 4, 1383–1389 CAS
- U. Schubert, J. Mater. Chem., 2005, 15, 3701–3715 RSC
- V. W. Day, T. A. Eberspacher, W. G. Klemperer and C. W. Park, J. Am. Chem. Soc., 1993, 115, 8469–8470 CrossRef CAS
- V. W. Day, T. A. Eberspacher, W. G. Klemperer, C. W. Park and F. S. Rosenberg, J. Am. Chem. Soc., 1991, 113, 8190–8192 CrossRef CAS
- L. Rozes, N. Steunou, G. Fornasieri and C. Sanchez, Monatsh. Chem., 2006, 137, 501–528 CrossRef CAS
- P. Coppens, Y. Chen and E. Trzop, Chem. Rev., 2014, 114, 9645–9661 CrossRef CAS PubMed
- A. Pandey, V. D. Gupta and H. Nöth, Eur. J. Inorg. Chem., 2000, 1351–1357 CAS
- H. Barrow, D. A. Brown, N. W. Alcock, W. Errington and M. G. Wallbridge, J. Chem. Soc., Dalton Trans., 1994, 3533–3538 RSC
- W.-H. Fang, L. Zhang and J. Zhang, J. Am. Chem. Soc., 2016, 138, 7480–7483 CrossRef CAS PubMed
- N. Steunou, G. Kickelbick, K. Boubekeur and C. Sanchez, J. Chem. Soc., Dalton Trans., 1999, 3653–3655 RSC
- J. B. Benedict and P. Coppens, J. Am. Chem. Soc., 2010, 132, 2938–2944 CrossRef CAS PubMed
- Z. Liu, J. Lei, M. Frasconi, X. Li, D. Cao, Z. Zhu, S. T. Schneebeli, G. C. Schatz and J. F. Stoddart, Angew. Chem., Int. Ed., 2014, 53, 9193–9197 CrossRef CAS PubMed
- T. J. Boyle, T. M. Alam, C. J. Tafoya and B. L. Scott, Inorg. Chem., 1998, 37, 5588–5594 CrossRef CAS PubMed
- R. Ghosh, M. Nethaji and A. G. Samuelson, Chem. Commun., 2003, 2556–2557 RSC
- A. Bashall, D. A. Brown, M. McPartlin and M. G. H. Wallbridge, J. Chem. Soc., Dalton Trans., 1992, 2529–2530 RSC
- N. W. Alcock, D. A. Brown, S. M. Roe and M. G. H. Wallbridge, J. Chem. Soc., Chem. Commun., 1992, 846–848 RSC
- T. Frot, S. Cochet, G. Laurent, C. Sassoye, M. Popall, C. Sanchez and L. Rozes, Eur. J. Inorg. Chem., 2010, 5650–5659 CrossRef CAS
- T. Frot, J. Marrot, C. Sanchez, L. Rozes and C. Sassoye, Z. Anorg. Allg. Chem., 2013, 639, 2181–2185 CrossRef CAS
-
T. Frot, PhD thesis, University of Pierre et Marie Curie, 2009
- M. Czakler, C. Artner and U. Schubert, Eur. J. Inorg. Chem., 2014, 2038–2045 CrossRef CAS PubMed
- W.-H. Fang, L. Zhang and J. Zhang, Dalton Trans., 2017, 46, 803–807 RSC
- J.-X. Liu, M.-Y. Gao, W.-H. Fang, L. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2016, 55, 5160–5165 CrossRef CAS PubMed
- K. Hong, W. Bak and H. Chun, Inorg. Chem., 2014, 53, 7288–7293 CrossRef CAS PubMed
- K. Hong and H. Chun, Inorg. Chem., 2013, 52, 9705–9707 CrossRef CAS PubMed
- T. J. Boyle, R. P. Tyner, T. M. Alam, B. L. Scott, J. W. Ziller and B. G. Potter, J. Am. Chem. Soc., 1999, 121, 12104–12112 CrossRef CAS
- P. S. Ammala, S. R. Batten, C. M. Kepert, L. Spiccia, A. M. van den Bergen and B. O. West, Inorg. Chim. Acta, 2003, 353, 75–81 CrossRef CAS
- P. Piszczek, M. Richert and A. Wojtczak, Polyhedron, 2008, 27, 602–608 CrossRef CAS
- P. Piszczek, A. Grodzicki, M. Richert and A. Wojtczak, Inorg. Chim. Acta, 2004, 357, 2769–2775 CrossRef CAS
- R. Papiernik, L. G. Hubert-Pfalzgraf, J. Vaissermann and M. C. H. B. Goncalves, J. Chem. Soc., Dalton Trans., 1998, 2285–2288 RSC
- G. A. Seisenbaeva, E. Ilina, S. Håkansson and V. G. Kessler, J. Sol–Gel Sci. Technol., 2010, 55, 1–8 CrossRef CAS
- A. Rammal, F. Brisach and M. Henry, C. R. Chim., 2002, 5, 59–66 CrossRef CAS
- I. Gautier-Luneau, A. Mosset and J. Galy, Z. Kristallogr., 1987, 180, 83–95 CrossRef CAS
- S. Cochet, L. Rozes, M. Popall and C. Sanchez, Mater. Sci. Eng., C, 2007, 27, 1401–1405 CrossRef CAS
- P. Heinz, M. Puchberger, M. Bendova, S. O. Baumann and U. Schubert, Dalton Trans., 2010, 39, 7640–7644 RSC
- Y. Gao, F. R. Kogler, H. Peterlik and U. Schubert, J. Mater. Chem., 2006, 16, 3268–3276 RSC
- T. Frot, C. Sassoye, C. Sanchez and L. Rozes, private communication, 2010 Search PubMed.
- G. Kickelbick and U. Schubert, Eur. J. Inorg. Chem., 1998, 159–161 CrossRef CAS
- K. Hong, W. Bak and H. Chun, Inorg. Chem., 2014, 53, 7288–7293 CrossRef CAS PubMed
- K. Hong and H. Chun, Inorg. Chem., 2013, 52, 9705–9707 CrossRef CAS PubMed
- M. Sabo, W. Böhlmann and S. Kaskel, J. Mater. Chem., 2006, 16, 2354–2357 RSC
- C. Maurer, B. Baumgartner, S. Pabisch, J. Akbarzadeh, H. Peterlik and U. Schubert, Dalton Trans., 2014, 43, 950–957 RSC
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Férey, J. Am. Chem. Soc., 2009, 131, 10857–10859 CrossRef CAS PubMed
- A. Walsh and C. R. A. Catlow, ChemPhysChem, 2010, 11, 2341–2344 CrossRef CAS PubMed
- Note that the terminology of the field of semiconductors (gap, valence band, conduction band) is often used to describe the optoelectronic properties of MOFs. Nevertheless, most MOFs are indeed not semiconductors, and can be better described from an electronic standpoint as an array of electronically isolated, molecular entities whose HOMO and LUMO levels define absorption bands rather than from the band theory.
- C. H. Hendon, D. Tiana, M. Fontecave, C. Sanchez, L. D’arras, C. Sassoye, L. Rozes, C. Mellot-Draznieks and A. Walsh, J. Am. Chem. Soc., 2013, 135, 10942–10945 CrossRef CAS PubMed
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed
- C. Zlotea, D. Phanon, M. Mazaj, D. Heurtaux, V. Guillerm, C. Serre, P. Horcajada, T. Devic, E. Magnier, F. Cuevas, G. Férey, P. L. Llewellyn and M. Latroche, Dalton Trans., 2011, 40, 4879–4881 RSC
- C. Gomes Silva, I. Luz, F. X. Llabrés i Xamena, A. Corma and H. García, Chem. – Eur. J., 2010, 16, 11133–11138 CrossRef PubMed
- M. A. Nasalevich, M. G. Goesten, T. J. Savenije, F. Kapteijn and J. Gascon, Chem. Commun., 2013, 49, 10575–10577 RSC
- Y. Horiuchi, T. Toyao, M. Saito, K. Mochizuki, M. Iwata, H. Higashimura, M. Anpo and M. Matsuoka, J. Phys. Chem. C, 2012, 116, 20848–20853 CAS
- B. Bueken, F. Vermoortele, D. E. P. Vanpoucke, H. Reinsch, C.-C. Tsou, P. Valvekens, T. De Baerdemaeker, R. Ameloot, C. E. A. Kirschhock, V. Van Speybroeck, J. M. Mayer and D. De Vos, Angew. Chem., Int. Ed., 2015, 54, 13912–13917 CrossRef CAS PubMed
- S. Yuan, T.-F. Liu, D. Feng, J. Tian, K. Wang, J. Qin, Q. Zhang, Y.-P. Chen, M. Bosch, L. Zou, S. J. Teat, S. J. Dalgarno and H.-C. Zhou, Chem. Sci., 2015, 6, 3926–3930 RSC
- H. L. Nguyen, F. Gándara, H. Furukawa, T. L. H. Doan, K. E. Cordova and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 4330–4333 CrossRef CAS PubMed
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed
- Y. Lee, S. Kim, J. K. Kang and S. M. Cohen, Chem. Commun., 2015, 51, 5735–5738 RSC
- L. Zou, D. Feng, T.-F. Liu, Y.-P. Chen, S. Yuan, K. Wang, X. Wang, S. Fordham and H.-C. Zhou, Chem. Sci., 2016, 7, 1063–1069 RSC
- C. K. Brozek and M. Dincă, J. Am. Chem. Soc., 2013, 135, 12886–12891 CrossRef CAS PubMed
- D. H. Son, Science, 2004, 306, 1009–1012 CrossRef CAS PubMed
- A. S. Yasin, J. Li, N. Wu and T. Musho, Phys. Chem. Chem. Phys., 2016, 18, 12748–12754 RSC
- J. A. Mason, L. E. Darago, W. W. Lukens and J. R. Long, Inorg. Chem., 2015, 54, 10096–10104 CrossRef CAS PubMed
- V. Guillerm, S. Gross, C. Serre, T. Devic, M. Bauer and G. Férey, Chem. Commun., 2010, 46, 767–769 RSC
- S. Surblé, C. Serre, C. Mellot-Draznieks, F. Millange and G. Férey, Chem. Commun., 2006, 284–286 RSC
- S. Hausdorf, F. Baitalow, T. Böhle, D. Rafaja and F. O. R. L. Mertens, J. Am. Chem. Soc., 2010, 132, 10978–10981 CrossRef CAS PubMed
- C. Serre, F. Millange, S. Surblé and G. Férey, Angew. Chem., Int. Ed., 2004, 43, 6285–6289 CrossRef PubMed
- H. L. Nguyen, T. T. Vu, D. Le, T. L. H. Doan, V. Q. Nguyen and N. T. S. Phan, ACS Catal., 2017, 7, 338–342 CrossRef CAS
- J. Zhao, L. Mi, J. Hu, H. Hou and Y. Fan, J. Am. Chem. Soc., 2008, 130, 15222–15223 CrossRef CAS PubMed
- H. Fei, M. R. Bresler and S. R. J. Oliver, J. Am. Chem. Soc., 2011, 133, 11110–11113 CrossRef CAS PubMed
- H. Fei, C. H. Pham and S. R. J. Oliver, J. Am. Chem. Soc., 2012, 134, 10729–10732 CrossRef CAS PubMed
- S. Das, H. Kim and K. Kim, J. Am. Chem. Soc., 2009, 131, 3814–3815 CrossRef CAS PubMed
- T.-F. Liu, L. Zou, D. Feng, Y.-P. Chen, S. Fordham, X. Wang, Y. Liu and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 7813–7816 CrossRef CAS PubMed
- R. J. Comito, K. J. Fritzsching, B. J. Sundell, K. Schmidt-Rohr and M. Dinca, J. Am. Chem. Soc., 2016, 138, 10232–10237 CrossRef CAS PubMed
- A. Wang, Y. Zhou, Z. Wang, M. Chen, L. Sun and X. Liu, RSC Adv., 2016, 6, 3671–3679 RSC
- C. J. Chuck, M. G. Davidson, M. D. Jones, G. Kociok-Köhn, M. D. Lunn and S. Wu, Inorg. Chem., 2006, 45, 6595–6597 CrossRef CAS PubMed
- K. Gigant, A. Rammal and M. Henry, J. Am. Chem. Soc., 2001, 123, 11632–11637 CrossRef CAS PubMed
- P. Persson, R. Bergström and S. Lunell, J. Phys. Chem. B, 2000, 104, 10348–10351 CrossRef CAS
- C. Chaumont, E. Huen, C. Huguenard, P. Mobian and M. Henry, Polyhedron, 2013, 57, 70–76 CrossRef CAS
- D. M. Weekes, C. Diebold, P. Mobian, C. Huguenard, L. Allouche and M. Henry, Chem. – Eur. J., 2014, 20, 5092–5101 CrossRef CAS PubMed
- H. Assi, L. C. Pardo Pérez, G. Mouchaham, F. Ragon, M. Nasalevich, N. Guillou, C. Martineau, H. Chevreau, F. Kapteijn, J. Gascon, P. Fertey, E. Elkaim, C. Serre and T. Devic, Inorg. Chem., 2016, 55, 7192–7199 CrossRef CAS PubMed
- L. Cooper, N. Guillou, C. Martineau, E. Elkaim, F. Taulelle, C. Serre and T. Devic, Eur. J. Inorg. Chem., 2014, 6281–6289 CrossRef CAS
- J. Gao, J. Miao, P.-Z. Li, W. Y. Teng, L. Yang, Y. Zhao, B. Liu and Q. Zhang, Chem. Commun., 2014, 50, 3786–3788 RSC
- Y.-L. Fu, Z.-W. Xu, J.-L. Ren and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m1733–m1734 CAS
- Y.-L. Fu, Z.-W. Xu, J.-L. Ren and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m1730–m1732 CAS
- A. K. Babko, O. I. Volkova and T. E. Get’man, Zh. Neorg. Khim., 1961, 6, 354–359 CAS
- J.-L. Hou, W. Luo, Y.-Y. Wu, H.-C. Su, G.-L. Zhang, Q.-Y. Zhu and J. Dai, Dalton Trans., 2015, 44, 19829–19835 RSC
- K. Hong, W. Bak and H. Chun, Inorg. Chem., 2013, 52, 5645–5647 CrossRef CAS PubMed
- K. Hong and H. Chun, Chem. Commun., 2013, 49, 10953–10955 RSC
- Y. Fu, Y. Liu, Z. Shi, B. Li and W. Pang, J. Solid State Chem., 2002, 163, 427–435 CrossRef CAS
- M. A. Nasalevich, C. H. Hendon, J. G. Santaclara, K. Svane, B. van der Linden, S. L. Veber, M. V. Fedin, A. J. Houtepen, M. A. van der Veen, F. Kapteijn, A. Walsh and J. Gascon, Sci. Rep., 2016, 6, 23676 CrossRef CAS PubMed
- M. A. Nasalevich, R. Becker, E. V. Ramos-Fernandez, S. Castellanos, S. L. Veber, M. V. Fedin, F. Kapteijn, J. N. H. Reek, J. I. van der Vlugt and J. Gascon, Energy Environ. Sci., 2015, 8, 364–375 CAS
- Note that our research on the CSD data base (version 5.37) was limited to phenol/-ate derivatives where o-positions were strictly considered as H-atoms.
- R. Minhas, R. Duchateau, S. Gambarotta and C. Bensimon, Inorg. Chem., 1992, 31, 4933–4938 CrossRef CAS
- A. J. Nielson, J. A. Harrison, C. Shen and J. M. Waters, Polyhedron, 2006, 25, 1729–1736 CrossRef CAS
- A. N. Desnoyer, B. Fartel, K. C. MacLeod, B. O. Patrick and K. M. Smith, Organometallics, 2012, 31, 7625–7628 CrossRef CAS
- J. A. Suttil, D. S. McGuinness, M. Pichler, M. G. Gardiner, D. H. Morgan and S. J. Evans, Dalton Trans., 2012, 41, 6625–6633 RSC
- G. W. Svetich and A. A. Voge, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 1760–1767 CrossRef CAS
- R. R. Gowda, D. Chakraborty and V. Ramkumar, Eur. J. Inorg. Chem., 2009, 2981–2993 CrossRef CAS
- P. J. Fischer, P. Yuen, V. G. Young and J. E. Ellis, J. Am. Chem. Soc., 1997, 119, 5980–5981 CrossRef CAS
- K. Watenpaugh and C. N. Caughlan, Inorg. Chem., 1966, 5, 1782–1786 CrossRef CAS
- R. R. Burch, Chem. Mater., 1990, 2, 633–635 CrossRef CAS
- Y. Zhao, S.-Y. Lee, N. Becknell, O. M. Yaghi and C. A. Angell, J. Am. Chem. Soc., 2016, 138, 10818–10821 CrossRef CAS PubMed
- T. P. Vaid, E. B. Lobkovsky and P. T. Wolczanski, J. Am. Chem. Soc., 1997, 119, 8742–8743 CrossRef CAS
- T. P. Vaid, J. M. Tanski, J. M. Pette, E. B. Lobkovsky and P. T. Wolczanski, Inorg. Chem., 1999, 38, 3394–3405 CrossRef CAS PubMed
- J. M. Tanski, T. P. Vaid, E. B. Lobkovsky and P. T. Wolczanski, Inorg. Chem., 2000, 39, 4756–4765 CrossRef CAS PubMed
- J. M. Tanski, E. B. Lobkovsky and P. Wolczanski, J. Solid State Chem., 2000, 152, 130–140 CrossRef CAS
- J. M. Tanski and P. T. Wolczanski, Inorg. Chem., 2001, 40, 2026–2033 CrossRef CAS PubMed
- K. N. Raymond and C. J. Carrano, Acc. Chem. Res., 1979, 12, 183–190 CrossRef CAS
- D. C. H. Oakes, B. S. Kimberley, V. C. Gibson, D. J. Jones, A. J. P. White and D. J. Williams, Chem. Commun., 2004, 2174–2175 RSC
- M. Fujiwara, Q. Xu, Y. Souma and T. Kobayashi, J. Mol. Catal. A: Chem., 1999, 142, 77–84 CrossRef CAS
- Y. Pérez, I. del Hierro, M. Fajardo and A. Otero, J. Organomet. Chem., 2003, 679, 220–228 CrossRef
- Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155–3212 CrossRef CAS PubMed
- W. R. Duncan and O. V. Prezhdo, J. Phys. Chem. B, 2005, 109, 365–373 CrossRef CAS PubMed
- C. Diebold, P. Mobian, C. Huguenard, L. Allouche and M. Henry, Inorg. Chem., 2010, 49, 6369–6371 CrossRef CAS PubMed
- M. Albrecht, H. Röttele and P. Burger, Chem. – Eur. J., 1996, 2, 1264–1268 CrossRef CAS
- M. Albrecht, M. Schneider and R. Fröhlich, New J. Chem., 1998, 22, 753–754 RSC
- M. Albrecht, M. Schneider and H. Röttele, Angew. Chem., Int. Ed., 1999, 38, 557–559 CrossRef CAS
- M. Albrecht, I. Janser, J. Runsink, G. Raabe, P. Weis and R. Fröhlich, Angew. Chem., Int. Ed., 2004, 43, 6662–6666 CrossRef CAS PubMed
- H. Senouci, B. Millet, C. Volkringer, C. Huguenard, F. Taulelle and M. Henry, C. R. Chim., 2010, 13, 69–96 CrossRef CAS
- D. Van Craen, M. Albrecht, G. Raabe, F. Pan and K. Rissanen, Chem. – Eur. J., 2016, 22, 3255–3258 CrossRef CAS PubMed
- A. Rosenheim and O. Sorge, Ber. Dtsch. Chem. Ges. B, 1920, 53, 932–939 CrossRef
- B. A. Borgias, S. R. Cooper, Y. B. Koh and K. N. Raymond, Inorg. Chem., 1984, 23, 1009–1016 CrossRef CAS
- M. G. Davidson, M. D. Jones, M. D. Lunn and M. F. Mahon, Inorg. Chem., 2006, 45, 2282–2287 CrossRef CAS PubMed
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS PubMed
- T. J. Boyle, L. J. Tribby, T. M. Alam, S. D. Bunge and G. P. Holland, Polyhedron, 2005, 24, 1143–1152 CrossRef CAS
- J. Hou, Q. Zhang, Y. Wu, Y. Liu, L. Du, C.-H. Tung and Y. Wang, Inorg. Chim. Acta, 2016, 443, 279–283 CrossRef CAS
- C. Chaumont, P. Mobian and M. Henry, Dalton Trans., 2014, 43, 3416–3419 RSC
- X. Lei, M. Shang and T. P. Fehlner, Organometallics, 1996, 15, 3779–3781 CrossRef CAS
- N. T. T. Nguyen, H. Furukawa, F. Gándara, C. A. Trickett, H. M. Jeong, K. E. Cordova and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 15394–15397 CrossRef CAS PubMed
- F.-Q. Liu, F.-Q. Liu and T. Don Tilley, Chem. Commun., 1998, 103–104 RSC
Footnote |
| † Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |