
Fluorescent chemical probes for accurate tumor diagnosis and targeting therapy
Min
Gao
ab,
Fabiao
Yu
*ac,
Changjun
Lv
c,
Jaebum
Choo
*d and
Lingxin
Chen
 *ac
*ac
aKey Laboratory of Coastal Environmental Processes and Ecological Remediation, Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences, Yantai 264003, China. E-mail: fbyu@yic.ac.cn; lxchen@yic.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cDepartment of Respiratory Medicine, Affiliated Hospital of Binzhou Medical University, Binzhou 256603, China
dDepartment of Bionano Engineering, Hanyang University, Ansan 426-791, South Korea. E-mail: jbchoo@hanyang.ac.kr
First published on 20th March 2017
Abstract
Surgical resection of solid tumors is currently the gold standard and preferred therapeutic strategy for cancer. Chemotherapy drugs also make a significant contribution by inhibiting the rapid growth of tumor cells and these two approaches are often combined to enhance treatment efficacy. However, surgery and chemotherapy inevitably lead to severe side effects and high systemic toxicity, which in turn results in poor prognosis. Precision medicine has promoted the development of treatment modalities that are developed to specifically target and kill tumor cells. Advances in in vivo medical imaging for visualizing tumor lesions can aid diagnosis, facilitate surgical resection, investigate therapeutic efficacy, and improve prognosis. In particular, the modality of fluorescence imaging has high specificity and sensitivity and has been utilized for medical imaging. Therefore, there are great opportunities for chemists and physicians to conceive, synthesize, and exploit new chemical probes that can image tumors and release chemotherapy drugs in vivo. This review focuses on small molecular ligand-targeted fluorescent imaging probes and fluorescent theranostics, including their design strategies and applications in clinical tumor treatment. The progress in chemical probes described here suggests that fluorescence imaging is a vital and rapidly developing field for interventional surgical imaging, as well as tumor diagnosis and therapy.
 Min Gao | Min Gao is currently a doctoral candidate, under the guidance of Prof. Lingxin Chen, at Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences, and has been since 2015. She received her MS degree in analytical chemistry at Department of Chemistry, Qufu Normal University, in 2015. Her current research interests focus on the development of functional fluorescent probes for bioimaging, cancer specific imaging and targeted therapy. |
 Fabiao Yu | Fabiao Yu is currently an associate professor at Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences. He received his PhD, joint-educated, at Dalian University of Technology, and Dalian Institute of Chemical Physics, Chinese Academy of Sciences, in 2013. His research interests focus on functional probe molecules (fluorescence and phosphorescence analysis), theranostics, and organic functional materials. |
 Changjun Lv | Changjun Lv is now a vice president in Binzhou Medical University. He is a professor and serves in Affiliated Hospital of Binzhou Medical University as a chief physician. He received his MD from China Medical University in 1995. He is skilled in the diagnosis and treatment of respiratory diseases, especially in the diagnosis of interstitial lung diseases. He is an expert with China's State Council special allowance due to his excellent performance in clinical work. His research interests include diagnosis and therapy of idiopathic pulmonary fibrosis, drug developments, and theranostics. |
 Jaebum Choo | Jaebum Choo has been in the faculty of the Bionano Engineering Department at Hanyang University, since 1995. He obtained his PhD in laser-induced spectroscopy at Texas A&M University, in 1994. In 2015, He became a Baik Nam Distinguished Professor due to his excellent academic achievements. He served as a President of Korea Biochip Society, in 2015. His current research programs are centered on the development of ultra-sensitive optical detection systems for rapid and accurate in vitro diagnostics. He has given 120 invited lectures in the USA, Europe and Asia, and has published over 220 research papers in refereed journals and 4 book chapters. |
 Lingxin Chen | Lingxin Chen has been a professor at Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences, since 2009. He obtained his PhD in analytical chemistry at Dalian Institute of Chemical Physics, Chinese Academy of Sciences, in 2003. During 2004–2009, he worked at Department of Chemistry, Tsinghua University, and Department of Applied Chemistry, Hanyang University, respectively. His research interests include the studies of novel properties of materials such as functionalized nanoparticles & functional probe molecules for developing nanoscale biochemical analysis methods and molecular imprinting-based sample pretreatment technology. He has published 3 academic books, 200 research papers and topical reviews. |
1. Introduction
Cancer is an extremely aggressive disease that involves uncontrolled cell growth and cell division. The incidence and death rate associated with cancer remain high,1 and the mechanisms of the development and recurrence of tumors in humans are still not yet fully understood.2 Therefore, cancer therapy faces huge challenges in modern medical technology. Precise detection of tumors should be the first consideration. The primary challenge for the diagnosis and treatment of tumors is finding effective ways to discriminate the boundaries between the tumor and healthy surrounding tissue at the cellular level. Early diagnosis and treatment of a tumor can increase the survival rate to 90%. However, a precancerous lesion has a small volume and its morphology is atypical. Therefore, the lesion is difficult to identify with the naked eye. Technologies that can indicate the molecular boundary between tumor and healthy tissue are necessary to precisely identify diseased tissue and exploit the unique characteristics of tumors. Traditional clinical approaches of surgery, radiotherapy, chemotherapy, and immunotherapy need to be further improved because untargeted therapies can damage both normal cells and cancerous cells, thereby causing serious side effects and poor quality of life and prognosis. Therefore, accurate diagnosis of the tumor is required before starting therapy.It seems likely that bioimaging technologies will meet this challenge. To date, in vivo medical imaging has made great advances in locating and discriminating tumor lesions as a result of developments in the engineering of imaging devices and the chemistry of imaging probes.3 Several imaging diagnostic technologies have been applied in clinical medicine to reveal underlying disease and to assess prognosis and treatment, including magnetic resonance imaging (MRI), X-ray radiography, computed tomography (CT), positron emission tomography (PET), ultrasonography (US), and optical imaging. However, novel imaging tools with high sensitivity are urgently required because of the relatively low concentrations of target analytes in vivo. Traditional imaging modalities like CT, MRI, and US lack sufficient specificity and sensitivity, which can be attributed to background tissue noise, tissue metabolism, and the limited resolution and depth of signal penetration. PET is highly sensitive, but widespread application of this imaging modality is limited by its poor spatial and temporal resolution and the stringent safety regulations for radioactive compounds.4 Compared with the conventional strategies of CT, MRI, and radioisotope imaging, an optical-based imaging approach can increase the target-to-background ratio by employing optical probes with unique features of (1) simultaneous multicolor imaging and (2) signal activation in the tumor. Therefore, optical imaging, such as fluorescence and bioluminescence imaging, offers the promise of accurate tumor diagnosis through non-invasive, real-time, and high-resolution imaging. Fluorescence bioimaging as a technique to visualize specific organelles in live cells5–10 and whole animals11,12 has become a powerful supporting tool for biological research,13–16 and even for clinical utilization such as in the emerging field of fluorescence-guided surgery.17–21 Additionally, fluorescence bioimaging can capture specific molecular information on tumor structure and tumor metabolism. Fortunately, these probes are also low cost, non-radioactive, simple and quick to use. In particular, near-infrared (NIR) fluorescence bioimaging probes with NIR absorption and emission profiles can maximize tissue penetration while minimizing the absorbance of heme in hemoglobin and myoglobin, water, and lipids.22–29 Recently developed NIR probes for fluorescence-guided surgery have shown great progress in determining the tumor margin and executing lesion resection.
The other challenge for tumor therapy is how to improve the drug therapeutic efficacy with minimum side effects due to the non-specific distribution of small molecular drugs in vivo. Undoubtedly, the rapidly growing class of chemotherapeutic drugs has achieved clinical success. However, the severe side effects of many drugs, including poor bioavailability, rapid blood/renal clearance, non-selective accumulation, uncontrollable drug release, bone marrow depression, severe multidrug resistance, and gastrointestinal disorders, have decreased the drug efficacy and caused tremendous pain to patients. The new nano-carriers that have rapidly emerged as drug delivery systems may overcome these limitations.30–38 The drug delivery mechanism of nano-carriers depends on enhanced permeability and retention (EPR) effects. The anticancer drugs are either conjugated to the nano-carriers or packaged as nano-capsules. These combinations demonstrate better tumor penetration and controllable drug release at the target site compared with free drugs.39–41 However, these conjugates have several disadvantages that cannot be ignored, such as the potential cytotoxicity of the heavy metal component or surface-coated materials, high cost, and difficulties in reproducibility and quantification.42,43 Compared with nanoparticle-based therapeutic agents, small molecular fluorescent probe-based therapeutic agents are becoming preferred chemotherapeutic candidates.3,44–50 These small fluorescent theranostic agents exhibit improved photophysical properties and can be easily modified by chemical synthesis.51,52 The structural architecture of these systems is often relatively simple and compact: a desirable fluorophore, a tumor targeting ligand, and a masked antitumor drug. The intact theranostic agent is delivered through the circulation to accumulate in the tumor through the action of the targeting ligand. After being triggered to release drug in situ, the fluorophore emits fluorescence for monitoring of pharmacokinetics, tumor therapy, and tumor prognosis (Fig. 1).
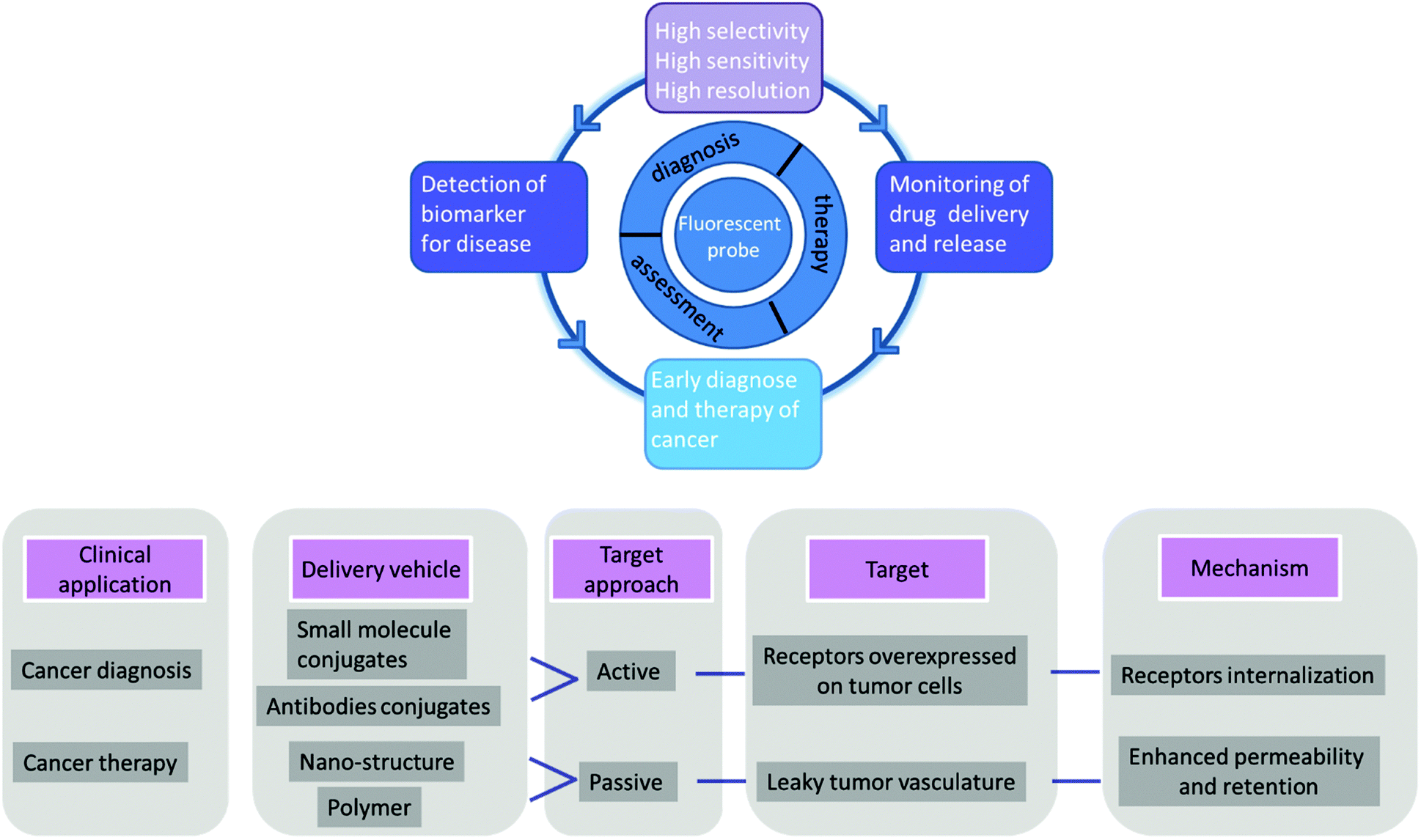 | ||
| Fig. 1 Summary of applications of fluorescent probes in diagnosis and therapy. | ||
In this review, we describe the entities that have been elegantly established to date, and aim to provide an up-to-date and concise overview of the design, application, and development of small molecular-weight fluorescent imaging agents and fluorescent theranostics for tumor diagnosis and therapy. We summarize the criteria governing the choice of the targeted receptor, modification of the targeting ligand, employment of the conjugating linker, utilization of the fluorophore, and optimization of the therapeutic drug. We discuss the strategies for integrating a targeting ligand with a suitable fluorophore and the desired linker-cleavage chemical reaction for selective release of the masked cytotoxic drug after endocytosis into tumor cells. We also address the issue that the development of fluorescent imaging agents is currently limited by the availability of NIR fluorophores.
2. Fluorescent probes for tumor diagnosis
The early diagnosis of preinvasive and metastatic tumor cells in patients is critical for the success of tumor therapy and improvement of survival rates. The fluorescence bioimaging approach, which is emerging as a promising non-invasive, real-time, and high-resolution modality, can be employed for the early diagnosis of tumors. Fluorescent signals can provide molecular information on tumor tissues that is related to the tumor anatomical structure and metabolism. There is an urgent need to establish stable, efficient, and safe multifunction fluorescence systems to accelerate the achievement of accurate and personalized medicine. A successful fluorescent probe must meet several requirements for medical imaging, including wavelength, brightness, biostability, photostability, specific tissue accumulation, and pharmacokinetics. Conventionally, one approach is to simply and directly conjugate the fluorophore with a ligand that can bind to a certain cellular surface receptor: a so-called active imaging probe (Fig. 2). The imaging mechanism relies on accumulation and retention of the probe at the target site as a contrast agent. This approach is helpful for tumor imaging, but the non-bound probe must be quickly cleared from the circulation via excretion. An activatable imaging probe can be employed to achieve highly specific delivery; this type of probe is only activated within the targeted tumor cells, for example by a tumor-specific enzyme. Such probes are non-fluorescent or weakly fluorescent in the unactivated state, but become strongly fluorescent after activation by the relevant molecular trigger. Thus, the target-to-background ratio is higher than that of active probes. The activation mechanisms of activatable probes mainly depend on enzyme digestion and quenching effects. After removal of the quencher moiety, the released fluorophores will immediately emit fluorescence (Fig. 2). However, the fluorescent probes that are currently employed in intraoperative imaging systems for clinical fluorescence imaging are untargeted fluorophores including indocyanine green,53 methylene blue,54 5-aminolevulinic acid,55 and fluorescein.56 In this section, we classify these probes according to their conceived structures and functions.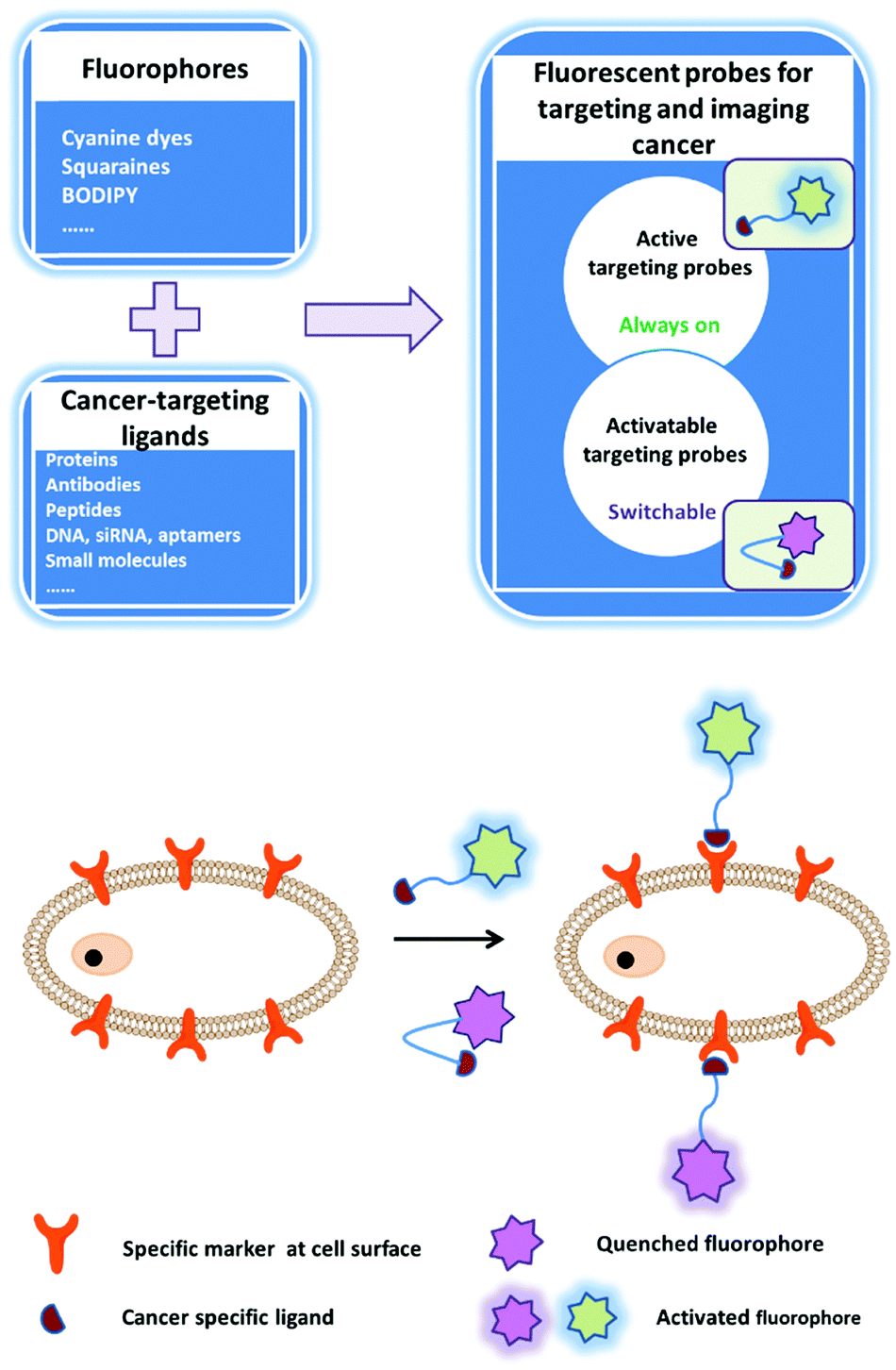 | ||
| Fig. 2 Strategies of fluorescent probes for tumor diagnosis. | ||
2.1 NIR imaging fluorophores
Near-infrared (NIR) light can penetrate tissue more deeply and minimize the interference from background autofluorescence, which greatly facilitates in vivo imaging of the molecular processes.22–24,57–59 However, suitable fluorophores for tumor imaging must possess good hydrophilicity, stable photostability, high quantum yield, and excellent sensitivity in biological systems. Recent developments in cyanine dyes, squaraine, phthalocyanines, porphyrin derivatives, and BODIPY dyes have made them good candidates for tumor imaging (Fig. 3).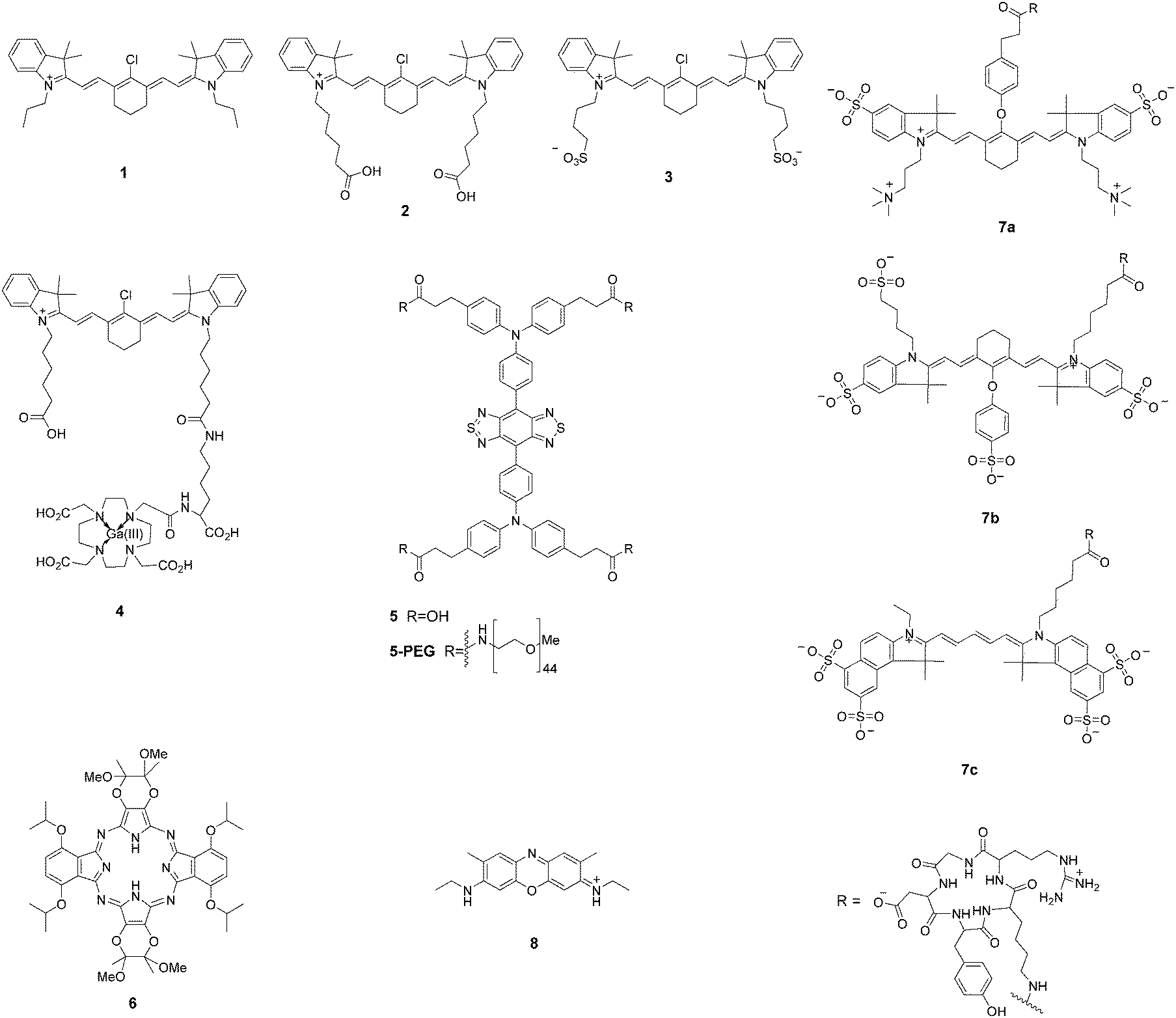 | ||
| Fig. 3 NIR imaging fluorophores. | ||
Shi and Chung et al. identified a class of NIR fluorescence heptamethine cyanines (1–3) for tumor imaging.60,61 These small molecular dyes could be taken up and accumulated in tumor cells without requiring chemical conjugation with tumor-specific targeting ligands. The recognition mechanism was mediated by hypoxia-induced HIF-1α and specific organic anion transporting polypeptides (OATPs). This simple and direct strategy had no limitation to specific tumor cell types. The unique properties of 1 for selective imaging of the sentinel lymph node (SLN) in animals are very important because the SLN is the first barrier that prevents tumor cell metastasis from the lymph node. Real-time images from the injection site toward the SLN were achieved within 5 min, and the retention time could be up to 2 weeks. After conjugation of 2 with a positron-emitting radionuclide, the NIR dye 4 showed improved deep-tissue tumor detection with fluorescence imaging and PET/CT scans.62 The above NIR dyes can be actively taken up and accumulated in cancer cells within 30 min. In vivo NIR imaging was achieved over a period of 24 h.
The fluorescence emission that lies in the first near-infrared window (NIR-I; 650–900 nm) is far superior to visible wavelengths, but a fluorophore that emits within the second near-infrared window (NIR-II; 1000–1700 nm) exhibits a greater improvement in imaging quality, such as decreased tissue autofluorescence, reduced photon scattering, and low levels of photon absorption.63–66 However, NIR-II fluorophores are often constrained by slow metabolism and long retention in the reticuloendothelial system. Hong et al. synthesized a soluble NIR-II emitting probe (5) for imaging the mouse lymphatic vasculature and sentinel lymphatic mapping near the tumor.66 The fluorescent signal in 4T1 xenograft tumors was observed within 10 min after intradermal injection of 5-PEG. The probe 5-PEG was excreted through the kidneys within 24 h. Dye 5 also allowed targeted imaging of tumors in vivo when conjugated with an anti-epidermal growth factor receptor (EGFR) antibody.
Hoffman et al. reported a chiral NIR porphyrazine (6) for accumulation in tumor cells.67 The porphyrin derivative was composed of four bridged pyrrole subunits linked through nitrogen atoms. Compound 6 was taken up by cells into the lysosomes through low-density lipoprotein (LDL) receptor-mediated endocytosis. LDL is preferentially associated with highly proliferative tumor cells over normal cells, therefore this NIR fluorescent probe enabled excellent contrast imaging between tumor and surrounding tissue. In vivo studies revealed that 6 was preferentially taken up and retained in MDA-MB-231 human breast cell xenografts in mice. The retention of 6 in the tumors continued to increase up to 48 h after injection.
A chemical compound with well-balanced charge would result in a more hydrophilic molecule with low non-specific targeting properties. Frangioni et al. introduced zwitterionic properties into NIR fluorophores to improve the signal-to-background ratio for diagnostic and therapeutic applications.68 Conjugates of the zwitterionic heptamethine indocyanine with cyclic peptide cyclo(RGD-yK) (7a–c) showed good behaviors in vitro for cell binding assays, histology, and immunoblotting and in vivo for xenograft tumor targeting and image-guided surgery. Real-time intraoperative melanoma detection was achieved 4 h after intravenous injection into mice. A real-time intraoperative thrombus could be detected at 30 min.
Accidental transection or injury of nerves leads to significant morbidity of numbness, pain, or paralysis. Choi et al. evaluated an oxazine derivative (8) to navigate peripheral nerve structures in rats and swine.69 The pharmacokinetic data suggested that the targeted fluorophore rapidly penetrated and was retained in nerves. The fluorescent labeled nerve was clear enough for discrimination for 3–4 h during complicated nerve surgery. The dye provided a nerve-targeted signal in the brachial plexus, sciatic nerve, and recurrent laryngeal nerve for up to 12 h after a single intravenous injection.
2.2 Probes integrating tumor-targeting ligands
Tumor targeting capability of fluorescent imaging probe is crucial for the accurate diagnosis of tumors. Common strategies for targeting ligand-based imaging probes involve exploitation of certain cell surface receptors that are overexpressed in cancerous cells (Fig. 2). There are a number of ligands (e.g., small molecules, peptides, proteins, antibodies, and aptamers) with a high intrinsic affinity for tumor targets that can be considered in the design of imaging probes. The probe is targeted to tumor cells through conjugation of a certain ligand, which accordingly reduces the delivery to normal cells and the associated collateral toxicity. Such strategies involve direct conjugation between the desirable fluorescent dyes and the ligands to produce a suitable imaging probe. This type of fluorescent probe can be targeted and bind to the tumor, while surplus probe will be excreted from the blood circulation. In this regard, an activatable imaging probe has the potential to further improve the signal-noise ratio.A. Prostate-specific membrane antigen. Prostate-specific membrane antigen (PSMA) is a type II membrane glycoprotein that is highly expressed in the neovasculature of tumors.70,71 PSMA-specific antibodies,72 aptamers,73 peptides,74 peptide derivatives,75 and other small molecules76,77 have been developed as targeting ligands. Slusher et al.78 described a high-affinity, single nucleophile-containing, small-molecule imaging probe (9) targeting the active site of the PSMA enzyme. The probe was obtained by conjugation of a tetra-sulfonated NIR heptamethine indocyanine fluorescent derivative with (2-[((3-amino-3-carboxypropyl)(hydroxy)phosphinyl)-methyl]pentane-1,5-dioic acid) (GPI). GPI itself was a potent inhibitor with a Ki of 9.0 nM, but the probe showed greater than 20-fold improvement in affinity (0.4 nM). The specificity of binding of 9 to cell-surface PSMA was tested not only in human prostate cancer cell lines LNCaP and PC-3, but also in human prostate cancer xenograft tumors in athymic mice.
Berkman et al.79,80 reported fluorescent probes 10 and 11 for imaging PSMA-expressing cells. The fluorophore of compound 10 was fluorescein. Compound 10 could effectively label cell membranes of LNCaP cells due to interactions with PSMA. Co-localization studies of 10 with transferrin-Texas Red showed that 10 was located in the perinuclear region. Moreover, 10 could be retained within endosomes for up to 150 min without loss of the signal. The IC50 values of GPI and fluorescent conjugate 10 were 14 nM and 0.35 nM, respectively. Obviously, the dye-conjugate showed greater inhibitory potency against PSMA. The fluorescent probe 11 was developed by conjugation of NIR Cy5.5 with GPI. The probe exhibited high potency against PSMA with IC50 of 0.55 nM.
In addition, Pomper et al.81 described a PSMA-binding NIR probe 12 for targeting PSMA in mice. The fluorophore of compound 12 was a heptamethine indocyanine derivative. Probe 12 demonstrated a PSMA inhibitory activity of 0.37 nM. The pharmacokinetic behavior of 12in vivo was acquired at an array of post-injection time points. The probe had clearly accumulated in the tumor at 18.5 h, and the fluorescence could be imaged repeatedly over 70.5 h.
Kozikowski et al.82 outlined a strategy of directly conjugating a PSMA inhibitor and doxorubicin (13) for locating prostate cancer cells by targeting PSMA. The antiproliferative action of 13 was poor without compromising the binding affinity.
B. Cyclooxygenase-2. Cyclooxygenase-2 (COX-2) is a crucial biological mediator in the etiology of inflammation and cancer. This enzyme is absent or present at low levels in normal cells but has high expression levels in inflamed tissues as well as many premalignant and malignant tumors.83 COX-2 has been used as an ideal imaging biomarker for cancer cells. Many fluorescent probes have been engineered to target COX-2.
Marnett et al. reported a series of fluorescent probes (14, 15) that efficiently targeted COX-2 in cells in vitro and in vivo.84–87 The design strategy for these optimized candidates was based on a rhodamine-derived fluorophore with selective COX inhibitors (Fig. 4). In vitro experiments in 1483 head and neck squamous cell carcinoma (HNSCC) cells showed that compound 14 localized at the perinuclear regions of membraneous structures that appeared to be endoplasmic reticulum or Golgi. Lipopolysaccharide-activated RAW264.7 macrophages showed stronger fluorescence labeling due to immunological stress. These probes showed preferential accumulation in inflammatory lesions and tumors. Inflammation was induced with carrageenan in C57BL/6 mice and images were acquired 3 h after injection of the probes. The probes were then employed to image human tumor xenografts. Levels of fluorescence in the tumor required 3 h to reach near maximal levels and remained relatively high for at least 24 h. The authors also summarized the structure–activity relationship (SAR) effects between fluorophores and linkers.85 The SAR study revealed that indomethacin conjugates were superior COX-2-targeted agents compared with other carboxylic acid-containing nonsteroidal anti-inflammatory drugs (NSAIDs) or COX-2-selective inhibitors. The 2′-trifluoromethyl analog of indomethacin is a potent and selective COX-2 inhibitor. The same group synthesized and evaluated the fluorescence imaging probe 15 with improved ability to inhibit COX-2 in inflammatory tissues and human tumor xenografts.86 Kinetic analysis revealed that 15 was a slow and tightly binding inhibitor of COX-2. Application of the NIR COX-2-targeted probe 15 could improve imaging signal-to-noise in cancer detection. Probe 15 exhibited selective and potent COX-2 inhibition of purified protein and in human cancer cell lines. Time course imaging studies conducted from 3 h to 7 days post-injection showed a gradual decrease of the fluorescent signal in 1483 HNSCC tumors.
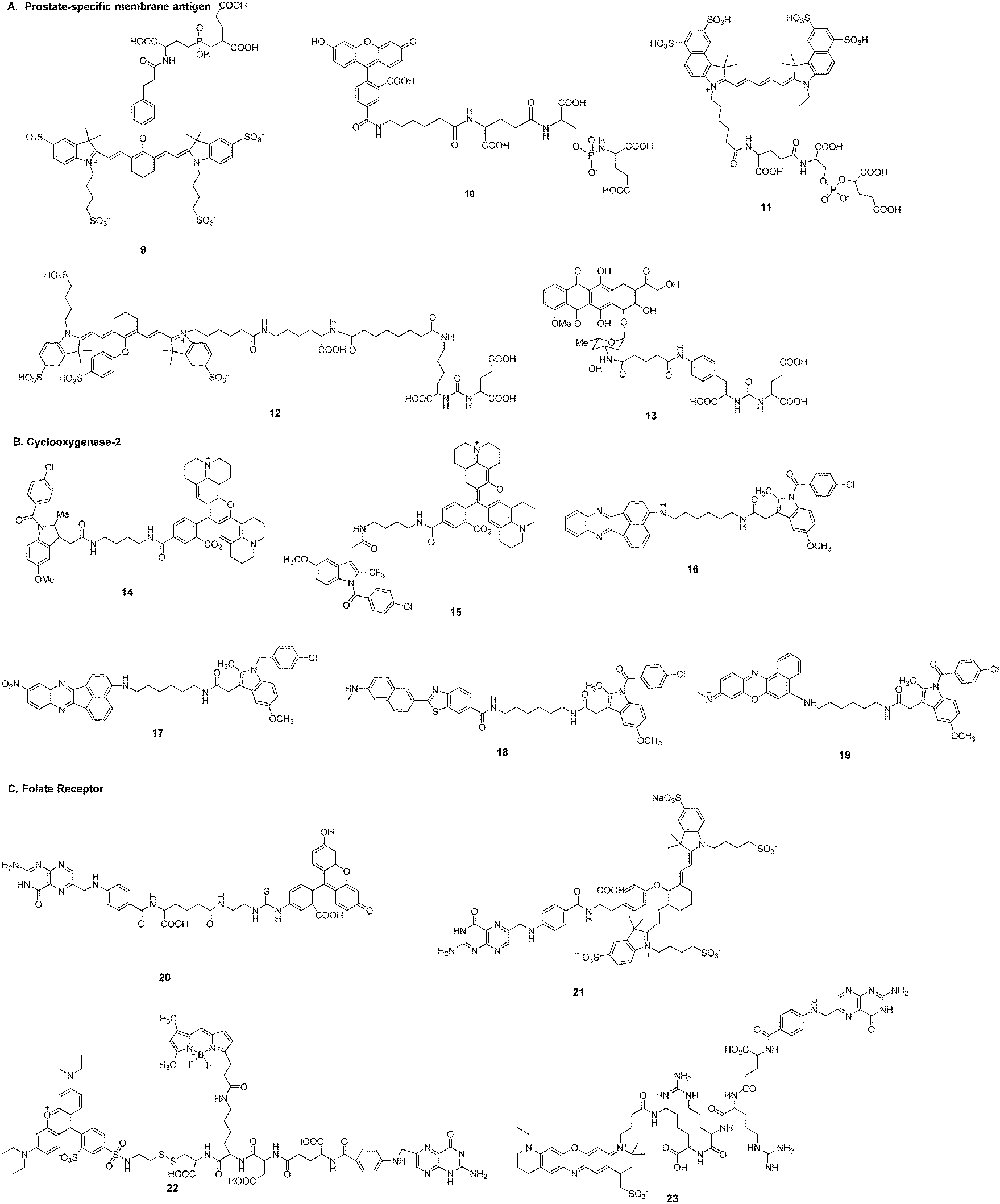 | ||
| Fig. 4 Small molecules as tumor targeting ligands: A–C. | ||
Peng et al. developed two-photon activatable fluorescent probes 16–19 for imaging COX-2 in cells and in vivo.88–91 All of these free probes existed in the folded state. This molecular configuration led to quenching of fluorescence due to photoinduced electron transfer (PET) between the fluorophores and the inhibitor of COX-2. Strong fluorescence would be emitted after binding of the probe to COX-2. The imaging probe 16 could target the Golgi apparatus of tumor cells.88 It stained cancer cells within 0.5 h and fluorescence intensity remained almost unchanged in cells for at least 6 h. This probe allowed imaging of sarcoma 180 tissue slices at a depth from 0 to 650 μm. The enzyme COX-2 is expressed at low levels (<0.085 μg mL−1) as a monomer in inflammatory lesions. Each COX-2 monomeric unit is composed of three distinct structural domains. However, in tumor cells and tumor tissues COX-2 is expressed at high levels (>0.085 μg mL−1) and exists as a homodimer. The binding site of enzyme inhibitors is located at the end of a hydrophobic cavity. Peng et al. improved their probe (17) for a sensitive fluorescence response to environmental changes.89 The fluorescence of 17 was “turned on” in both inflammation and cancer but the fluorescent emission was quite different. Moreover, cancerous tissues, inflamed tissues, and normal tissues could be discriminated in vivo by the naked eye. Probe 18 afforded high sensitivity and selectivity for COX-2 with a detection limit of 1.0 nM.90 Probe 19 was a COX-2-specific NIR fluorescent probe91 that could be directly applied to native polyacrylamide gel electrophoresis analysis and in-gel fluorescence analysis. This utilization facilitated rapid and sensitive screening of cancer cells without the need for a time-consuming enzyme-linked immunosorbent assay (ELISA).
C. Folate receptor. Folate is essential for the proliferation and maintenance of all cells. Folate has a high affinity for its cell surface folate receptor (FR, Kd: 10−10).92 The FR is primarily expressed on the healthy apical surface of polarized epithelial cells where it does not readily encounter folate from the bloodstream.93 Once malignant transformation occurs, these apically restricted receptors will become accessible to folate because intercellular junctions are lost during tumorigenesis and the FR is overexpressed over the entire cell surface. This significant upregulation of the FR on tumor cells has already become the main design strategy for folate-based tumor imaging probes.44,49,92,93
Low et al. described a tumor-targeting folate–fluorescein conjugate (20) for imaging of folate receptor-expressing tumors.93 This probe could be visualized in peritoneal, subcutaneous, and metastatic murine tumor models with 2 h intravenous administration. Folate receptor-mediated transcytosis could not be exploited to deliver this folate conjugate into the brain.94 Tumor masses exhibited significantly more fluorescence than adjacent normal tissues regardless of autofluorescence of non-injected controls. Excretion of the fluorescein conjugate from non-targeted tissues was achieved mainly through the kidneys within 2 h. Tumor nodules less than 1 cm in size could not be accurately detected with either computer tomography or ultrasound, but the fluorescent probe could locate malignant lesions smaller than 0.5 mm with appropriate optical instrumentation. The overexpression of the folate receptor in 90–95% of epithelial ovarian cancers prompted investigation into the use of this probe in intraoperative specific fluorescence navigation to improve the prognosis of patients with ovarian cancer.20 Further development of a near-infrared fluorescent probe (21) allowed identification of more deeply seated tumors based on the stronger penetration properties of near-infrared dye IR-783 compared with fluorescein.20,49,95
Low et al. also synthesized a fluorescent folate conjugate (22) to label the disulfide bond-reducing process in endosomal compartments.96 The approach adopted was to directly link folate to two fluorescent dyes via a disulfide bond, which engaged in fluorescence resonance energy transfer. Reduction of the disulfide bond resulted in a change in fluorescence from red to green. It was observed that disulfide reduction began in the endosomes and occurred with a half-time of 6 h after folate-FRET endocytosis.
Choi et al. developed a folate receptor-specific probe (23) for NIR fluorescence imaging of ovarian tumors in vivo.97 The probe was synthesized by conjugating folate to the ATTO655 fluorophore via a cathepsin B-cleavable peptide spacer. The fluorescence emission from the fluorophore was effectively quenched by folate. After specific uptake into SKOV3 cells, the tumor-associated cathepsin B enzymes subsequently activated NIR fluorescence emission by cleaving the spacer. Assessment of the tumor targeting of probe 23 in a xenograft mouse model of ovarian cancer showed that fluorescence images could be obtained after 3 h post-injection.
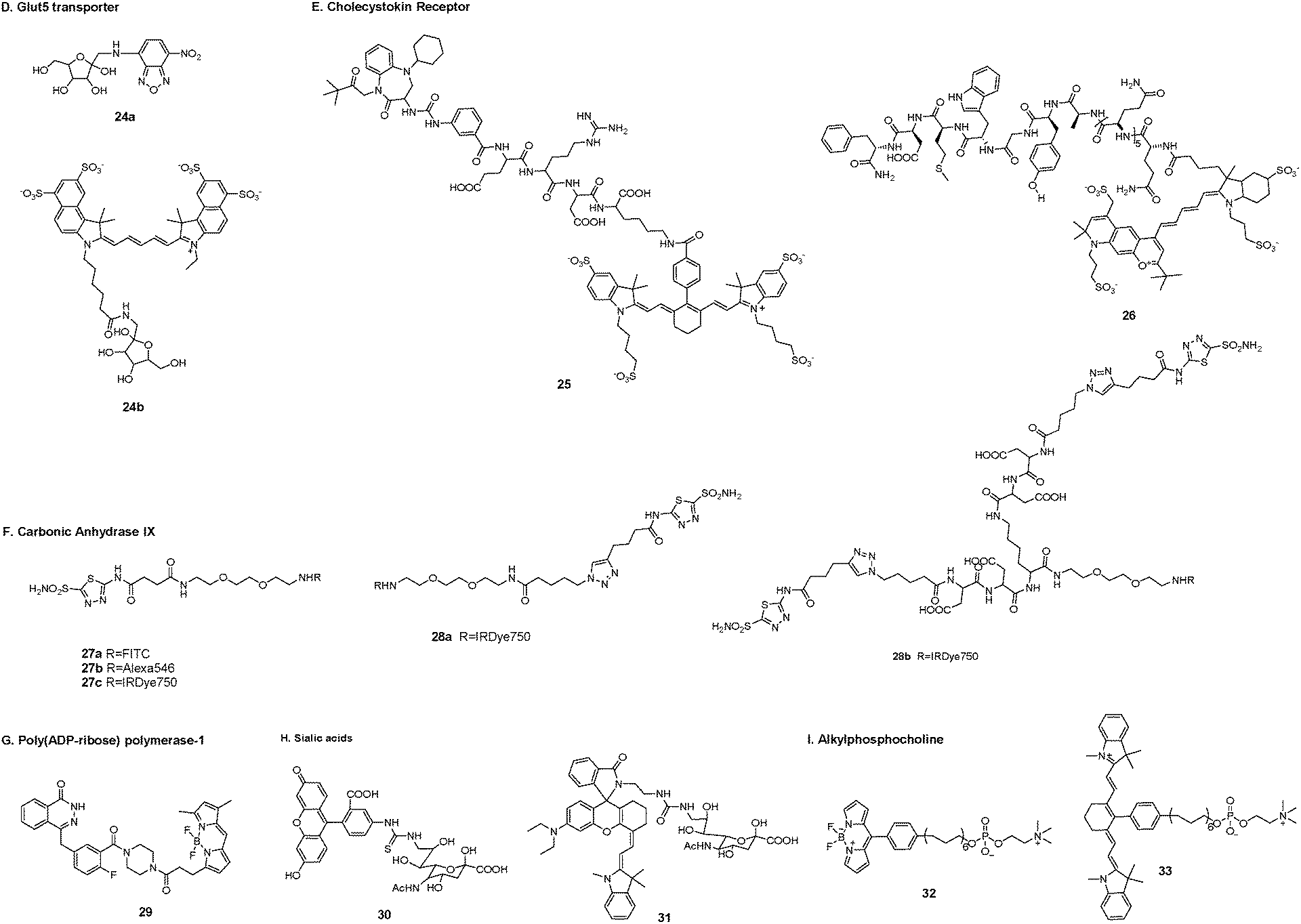
D. Glut5 transporter. Glut transporters are a family of transmembrane proteins that transport sugars such as glucose, fructose, and galactose across the cell membrane. They are expressed in different types of tumor tissues but have distinct substrates.98 Tumor cells need to overexpress at least one Glut transporter to meet their high levels of energy consumption. Glut5 is a fructose-specific transporter that is expressed at high levels in breast cancer cells but is not dominantly expressed in normal breast cells.99
To achieve molecular differentiation between cancerous and normal breast cells, Gambhir et al. synthesized two fructose-based fluorescent probes using 7-nitro-1,2,3-benzoxadiazole (24a) and cyanine5.5 dye (24b) as fluorophores, respectively.98 Evaluation of the first metabolic pathway of fructose showed the involvement of hexokinases, which could phosphorylate fructose only in the breast cancer cell lines. On the basis of this result, the fluorophore labeling site was chosen in the C-1 position of fructose. The uptake of probe 24a was dependent on the presence of the fructose-specific transporter Glut5. Probe 24b with the very bulky fluorophore cyanine5.5 resulted in nonspecific accumulation in breast cancer cells (Fig. 5).
 | ||
| Fig. 5 Small molecules as tumor targeting ligands: D–I. | ||
E. Cholecystokin receptor. The cholecystokin 2 receptor (CCK2R) belongs to the family of membrane G protein-coupled receptors. The CCK2R and its tumor-specific splice variant CCK2i4svR are overexpressed in tumors of the pancreas, medullary thyroid, lung, breast, ovary, gastrointestinal tract, and colon.100–106 The CCK2R has normal organ distributions in the central nervous system and gastrointestinal tract.
Low et al. reported a tumor-specific nonpeptidic conjugate (25) that could target the CCK2R and its splice variants for use in fluorescence imaging.107 The conjugate was composed of a sulfonated NIR fluorophore LS-288, a hydrophilic tetrapeptide spacer, and a targeting ligand Z-360 (a benzodiazepine-derived antagonist). The nonpeptidic ligand could bind in a similar manner to the natural ligand and access both the inactive and active states of the receptor. Additionally, the nonpeptidic ligand could not be taken up by a peptide scavenging receptor, thus avoiding unwanted accumulation in the liver and kidneys. Conjugate 25 accumulated in tumor tissue within 2 h following tail vein injection in mice. The results showed that 25 specifically localized to CCK2R-expressing HEK 293 murine tumor cells in both primary tumors and xenografts.
Hilger et al. synthesized a CCK2R targeted probe (26) by linking a NIR fluorophore DY-754 and a high-affinity minigastrin peptide analog with the sequence H2N-(DGln)6-Ala-Tyr-Gly-Trp-Met-Asp-Phe-amid via a hydrophilic linker.108 The binding assay demonstrated a high binding affinity for CCK2R-expressing A431 cells. After intravenous injection of the probe, a fast and long-lasting accumulation in tumors was observed. CCK2R expression was clearly identified by fluorescence imaging in the A431/CCK2R xenografts at 8 h post-injection.
F. Carbonic anhydrase IX. Carbonic anhydrases are a family of transmembrane zinc metalloenzymes that exist in 16 different isoforms. These isoforms are distributed in different subcellular organelles where they play catalytic roles and are susceptible to their own specific inhibitors. Carbonic anhydrase IX (CAIX) is normally expressed in the stomach and gallbladder epithelia. However, CAIX can be used as a marker of hypoxia because of its overexpression in solid tumors such as glioblastoma, colorectal, and breast109–111 and is therefore a potential tumor target.
Neri et al. designed and characterized a series of small-molecule fluorescence conjugates (27a–c) targeting CAIX.112 These acetazolamide-based fluorophore conjugates were found to preferentially target CAIX-expressing tumor cells. Compound 27c was used to evaluate biodistribution in a mouse model. The results showed that accumulation of 27c in the tumor was rapid and efficient at 1 h post-injection; however, the short retention time in the tumor (residence half-life t1/2 ≈ 1 h) suggested that the bivalent inhibitor acetazolamide against CAIX might result in improved CAIX binding affinity compared with its corresponding monovalent counterpart. Neri et al. synthesized monovalent and bivalent acetazolamide fluorescence conjugates (28a–b) and tested their binding to tumor cells.113 Their results showed that the bivalent conjugate 28b efficiently accumulated in CAIX-expressing SKRC52 kidney xenograft tumors with a retention time (>24 h) longer than that of the corresponding monovalent conjugate 28a.
G. Poly(ADP-ribose) polymerase-1. Poly(ADP-ribose) polymerase-1 (PARP-1), a well-known DNA-binding enzyme, is expressed in human tumors due to a deficiency in DNA repair enzymes, rendering the tumor cells susceptible to the small-molecule PARP1 inhibitor olaparib. Weissleder et al. synthesized a fluorescent olaparib derivative (29) for single-cell and subcellular pharmacokinetic analysis in murine cancer models.114,115 They performed high-resolution, temporal in vivo imaging of single cells in tumors to measure drug distribution and pharmaceutics in real time, and utilized a quantitative framework to extract and extrapolate single-cell data to be used in predictive models.114 Compound 29 reached the cellular target compartment, the nucleus, within 6 min in vivo. At the whole-body level, 29 had a weighted blood half-life of 18 min. The authors also used these data to validate predictive finite element modeling. Overexpression of PARP-1 was particularly apparent in the nuclei of glioblastoma, but not normal brain tissue. The high expression of PARP-1 in tumors and low expression in healthy tissue made 29 a valuable candidate for the detection and staging of tumors, especially glioblastoma. 29 could also be used to exploit the overexpression of PARP-1 in various other forms of cancer, including breast cancer, melanoma, and brain malignancies.116
H. Sialic acids. Sialic acids are anionic monosaccharides located at the termini of cell surface glycans. These derivatives of N-acetylneuraminic acid are involved in host–pathogen interactions and cell–cell adhesions. Overexpression of sialic acids on the cell surface has been associated with metastatic potential in a broad spectrum of tumors, suggesting an enhanced metabolic demand for sialic acids by these tumor cells.117,118
Han et al. reported a 9-fluoresccinylthioureido-9-deoxy-N-acetylneuraminic acid (30) for high-performance detection of tumors through labeling of sialic acid.119 Probe 30 could be transferred into the Golgi and metabolically incorporated into glycoproteins via a cellular sialylation pathway. The fluorescence signal was observed in tumors within 20 min after administration. The off-target probe was quickly eliminated from the circulation. The tumor-to-organ fluorescence ratios remained high up to 10 h post-injection. Implanted H22 hepatocellular carcinoma in the liver could be clearly discerned for tumors with a diameter of 0.2–5 mm. However, the active imaging probe 30 suffered from “always-on” green fluorescence, which limited deep tissue penetration. Han et al. next presented a sialylated pH-activatable NIR probe 31 for fluorescence targeted tumor detection.120 The sialic acid ligand provided effective tumor targeting in H22 hepatocellular carcinoma lesions and the NIR fluorophore underwent lysosomal pH-triggered isomerization to emit fluorescence. The micromilieu of solid tumors is hallmarked by acidic microenvironments due to the accumulation of lactic acid by glycolysis. Therefore, probe 31 displayed a high tumor-to-normal tissue signal contrast. The NIR signal was observed in nude mice 30 min after administration via the tail vein, and the signal could be retained for 144 h after injection. Additionally, 31 could effectively convert NIR light into cytotoxic heat to kill tumor cells, suggesting tumor-activatable photothermal therapy.
I. Alkylphosphocholine. Alkylphosphocholine derivatives (APCs) selectively accumulate in many solid tumor cells compared with normal cells.121 APCs also show suppressive effects on tumor growth and extend survival. APCs target cellular and intracellular membranes, inhibit phosphatidylcholine biosynthesis, interfere with lipid transduction pathways, block the endoplasmic reticular transport of cholesterol, and ultimately disrupt cholesterol homeostasis and membrane lipid raft function.122,123 Kuo et al. developed two APC-based cancer-selective fluorescent probes CLR1501 (32) and CLR1502 (33) for discriminating tumors from normal brain tissue in fluorescence-guided glioma surgery.123,124 These two probes provided high tumor-to-normal brain contrast in fluorescence discrimination with glioblastoma multiforme and glioblastoma stem cell-derived xenografts in mouse models. Moreover, 33 showed a superior tumor-to-brain fluorescence ratio compared with 5-aminolevulinic acid, which is the current standard for fluorescence-guided neurosurgery. The tumor fluorescence was readily detected with the indocyanine green fluorescence microscopy technique currently used in the clinic.
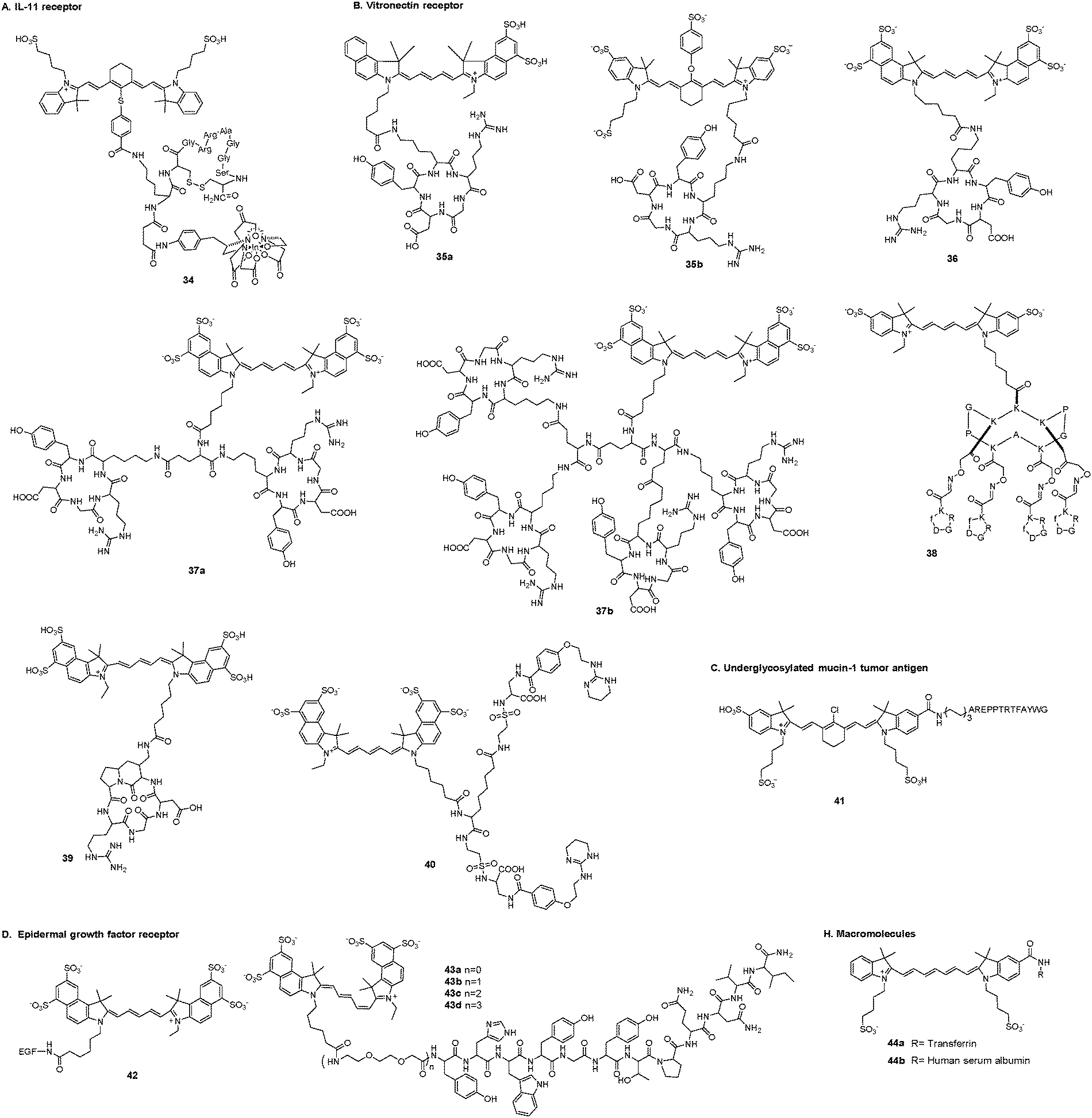
A. IL-11 receptor. Interleukin-11 (IL-11) is a multifunctional cytokine in blood cells. The IL-11 receptor alpha-chain (IL-11Rα) is a member of the gp130-dependent receptor group (Fig. 6). IL-11 and IL-11Rα are related to breast cancer development and progression and play crucial roles in bone metastasis of human breast cancer, which indicates poor prognosis.126 Targeting of IL-11Rα-positive tumors has potential as a useful strategy for noninvasive imaging. Wang et al. developed a NIR fluorescence and single photon emission tomography (SPECT) dual-modality imaging probe (34) to improve the diagnosis and management of tumors.127 The targeting moiety was the cyclic nonapeptide c(CGRRAGGSC), which showed a particular affinity for IL-11Rα, the radiotracer was radioisotope 111In complexes, and the fluorescence signal generator was the cyanine dye IR-783. Dual-modality imaging of mice bearing subcutaneous MDA-MB-231 tumors demonstrated uptake of the imaging probe in the tumor 24 h after administration.
 | ||
| Fig. 6 Peptides, proteins and antibodies as tumor targeting ligands. | ||
B. Vitronectin receptor. Integrins are a family of heterodimeric transmembrane proteins that behave as receptors for specific steric-configuration constrained extracellular ligands.128,129 Among these integrins, αvβ3, the vitronectin receptor, plays important roles in tumor progression, angiogenesis, and metastasis.129–132 Vitronectin, which contains the arginine–glycine–aspartic peptide sequence (Arg–Gly–Asp, RGD), exhibits a high affinity for αvβ3, providing a promising ligand for diagnostic imaging and a therapeutic target in tumors.
Li et al. reported the synthesis and characterization of integrin αvβ3-targeted peptide cyclo(Lys–Arg–Gly–Asp–Phe) [c(KRGDf)] labeled with NIR cyanine dyes Cy5.5 (35a) and IRDye800 (35b) for tumor imaging.133,134 These peptide–dye conjugates showed specificity for human Kaposi's sarcoma (KS1767) cells that overexpress αvβ3. Tumors were clearly visualized after intravenous injection at a dose of 6 nmol per mouse. Dynamic fluorescence images obtained over a period of 20 min revealed that 35a was rapidly taken up by KS1767 tumors after intravenous injection.
Blasberg et al. reported an IRDye 800CW-conjugated c(KRGDf) peptide that specifically targeted the overexpressed integrin receptors in murine RCAS-PDGF-driven/tv-a glioblastomas and two human orthotopic glioblastomas (U-87 MG and TS543).135 The three tumor models exhibited different levels of integrin receptor upregulation. The conjugate could specifically bind the overexpressed integrin receptors in these glioblastomas and visualization of the tumor tissue and tumor margins could be achieved by NIR imaging.
Chen et al. used the RGD peptide cyclo(Lys–Arg–Gly–Asp–D-Tyr) [c(RGDyK)] as a targeting ligand to produce a sensitive and semiquantitative NIR fluorescence imaging probe 36.136 The conjugate could function as a specific ligand toward the αvβ3 integrin receptor in the human glioblastoma cell line (U87MG) and primary human brain capillary endothelial cells (HBCECs) that are known to overexpress αvβ3 integrin. In vivo three-dimensional imaging showed typical NIR fluorescence images of athymic nude mice bearing subcutaneous U87MG glioblastoma tumors after intravenous injection of optimized doses of 36. The tumor could be clearly discriminated from the surrounding tissue from 30 minutes to 24 h post-injection, with maximum contrast occurring about 4 h post-injection. Additionally, Cy5.5-conjugated monomeric, dimeric, and tetrameric c(RGDyK) peptides (37a, 37b) were evaluated in subcutaneous U87MG glioblastoma xenograft models and were all suitable for integrin expression imaging.137
Coll et al. reported the in vitro and in vivo characteristics of conjugate 38 for targeting and imaging of tumors.138–140 Four c(RGDyK) peptides were assembled onto the functionalized RAFT scaffold via chemoselective oxime ligation. The fluorophore was Cy5. RAFTc(RGDyK)4 could be used as a therapeutic agent in nude mice. RAFTc(RGDyK)4 significantly improved the targeting specificity for subcutaneous tumor masses. Compound 38 could target αvβ3-positive human IGROV1 ovarian nodules scattered in the peritoneal cavity of nude mice after intravenous administration. The affinity between the targeting ligand RAFTc(RGDyK)4 and αvβ3 was also verified in Swiss nude mice bearing HEK293 (β1) and HEK293 (β3).140
Forni et al. reported a NIR fluorescence RGD cyclic probe 39 for noninvasive detection of αvβ3 integrin-overexpressing tumors.141 The targeting moiety was a functionalized cRGD peptide that preferentially bound the Mn-activated form of integrin, mediating rapid and extensive cell adhesion. The conjugate displayed a high binding affinity for αvβ3 integrin on human U-87 MG glioblastoma cells and their xenografts in nude mice. Probe 39 showed broad diffusion in most organs after injection, followed by a progressive increase up to 24 h with specific accumulation in tumors. The low expression of αvβ3 integrin on the C6 glioma cell line led to reduced specific accumulation in the tumor. The expression and retention of 39 in the tumor area was low compared with that observed in U-87 MG transplantable tumors.
Li et al. developed a NIR imaging probe (40) based on a bivalent nonpeptide small molecule intergrin αvβ3 antagonist for tumor imaging.142 The αvβ3 antagonist 4-[2-(3,4,5,6-tetrahydropyrimidine-2-lamino)-ethyloxy]benzoyl-2-(S)-aminoethylsulfonyl-amino-h-alanine (IA) and the Cy5.5 fluorophore were conjugated via an 8-carbon linker that had the lowest conformational energy for construction of 40. The bivalent IA contributed to a strong and specific affinity for the detection of tumors and micrometastatic lesions. Whole-animal fluorescence imaging of subcutaneously implanted U-87 xenograft tumors indicated that 40 was specific for the tumor tissue through binding to its integrin receptor. The target-to-background ratio was sufficient to clearly discern the tumor lesions at 7 h post-contrast injection. The intense signal detected at the tumor site could be retained for more than 48 h.
C. Underglycosylated mucin-1 tumor antigen. Underglycosylated mucin-1 tumor antigen (uMUC-1) is one of the early hallmarks of tumorigenesis in a wide variety of tumors.143 The EPPT peptide with the sequence AREPPTRTFAYWG targeting the uMUC-1 antigen was derived from the CDR3Vβ region of a monoclonal antibody.144 Moore et al. developed a conjugate cyanine 7-EPPT peptide (41) for tumor imaging.143 The hydrophilicity of the cyanine fluorophore was increased by introduction of a sulfonic moiety. Removal of the carboxylic acid group would eliminate the possibility of cross-linking with peptides/proteins during conjugation. NIR fluorescence imaging was obtained 24 h after injection of 41. The results showed specific accumulation in mice bearing human pancreatic adenocarcinoma CAPAN-2.
D. Epidermal growth factor receptor. The epidermal growth factor receptor (EGFR) is a transmembrane glycoprotein. Activation of the EGFR triggers the signal transduction pathways involved in regulating cellular proliferation, differentiation, and survival.145–147 The EGFR is overexpressed in many tumor cell lines, and is associated with poor prognosis and high mortality. The epidermal growth factor (EGF) is one of the most important ligands for the EGFR.
Li et al. reported an EGF–Cy5.5 fluorescent probe (42) for the detection of the EGFR in human breast tumor xenografts.148 The conjugate was conveniently synthesized from EGF and an NHS ester of Cy5.5. The selectivity of EGF–Cy5.5 for the EGFR was established by comparing the binding capability between EGFR-overexpressing MDA-MB-468 tumor cells and EGFR-negative MDAMB-435 tumor cells. NIR fluorescence in mice bearing MDA-MB-468 tumors was still detectable at 96 h after injection of 42 and the signal intensity returned to the background level at 192 h.
Basilion et al. developed a series of NIR probes 43a–d targeting the EGFR for imaging glioblastoma brain tumors in vivo.149 The probes were synthesized by varying the length of the polyethylene glycol (PEG) spacer between the peptide GE11 (YHWYGYTPQNVI) and the fluorophore Cy5.5. The length of the linker critically affected the affinity of the probes for tumors. Probe 43b, which had one unit of PEG, had the highest apparent affinity for the EGFR in glioblastoma cells and could selectively localize at glioblastoma-derived orthotopic brain tumors. When tested in cultured cell lines that expressed different levels of the EGFR and in orthotopic brain tumors using fluorescence-mediated molecular tomography, the probe could distinguish different tumors expressing various levels of the EGFR.
E. Tyrosine phosphatase μ. Receptor protein tyrosine phosphatase μ (PTPμ) protein transduces signals in response to cell adhesion and is highly expressed in the normal brain. Expression of full-length PTPμ is downregulated in the most aggressive glial tumor, glioblastoma multiforme.150 Brady-Kalnay et al. verified that the extracellular fragment of PTPμ is expressed in the human glioblastoma multiforme tumor margin but not in normal brain tissue. A series of peptides (SBK1-4) were linked to Texas Red, Cy5, and Alexa 750 fluorophores to recognize protein fragments of PTPμ.150–152 The SBK2 conjugate labeled the main tumor mass of an intracranial animal model of glioma as well as tumor cells at the margin. SBK2 could navigate the margin and recognize dispersing tumor cells up to 4 mm from the main tumor mass. The SBK2 conjugate rapidly accumulated in tumors within 10 minutes, and the fluorescent signal was retained for more than 180 min.
F. Chlorotoxin. Chlorotoxin (CTX) is a 36-amino acid peptide with four disulfide bridges that preferentially binds to glioma cells.153 The molecular targets of CTX include a lipid raft-anchored complex that contains matrix metalloproteinase-2 (MMP-2), membrane type-1 MMP, the transmembrane inhibitor of MMP-2, the glioma-specific chloride ion channel, and the ClC-3 chloride ion channel.44
For intraoperative differentiation of tumor margins or small foci of cancer cells, Olson et al. developed a molecular imaging conjugate composed of CTX and Cy5.5.154 The probe could delineate malignant glioma, medulloblastoma, prostate cancer, intestinal cancer, and sarcoma from adjacent non-neoplastic tissue. Specific binding to cancer cells was facilitated by MMP-2. The fluorescent signal was still higher in xenografts than in non-neoplastic tissue at 14 days after injection. The conjugate was concentrated in the kidneys and was excreted in urine. The Cy5.5 conjugation resulted in a mixture of mono-, di-, and tri-labeled probes because CTX contains three lysine residues.155 Substitution of the lysines at positions 15 and 23 with either alanine or arginine would result in a Lys27 monolabeled peptide that retained the stability and in vivo half-life properties of CTX. The half-life of the Cy5.5-labeled linear peptidic probe was 14 h, whereas that of Cy5.5-labeled cyclized chlorotoxin was 11 h.
Kovar et al. developed and tested a targeted NIR probe CLTX-800CW for brain tumors in the mouse model ND2:SmoA1, which spontaneously develops medulloblastoma tumors.156 Colocalization of hematoxylin and CLTX-800CW in cells at the tumor margin was distinctly visible. Specificity and functionality of the targeted probe for MMP-2-dependent tumor cell targeting were evaluated by examining cultured medulloblastoma, glioblastoma, lung carcinoma, and prostate carcinoma cell lines in a microplate assay.
Butte et al. presented a NIR imaging probe BLZ-100, using a standard charge-coupled device (CCD) camera to visualize low levels of BLZ-100 binding to the tumors.157 BLZ-100 exhibited a high affinity for glioma. After injection of BLZ-100 for 48 h, the orthotopic glioma tumors in mice were effectively discriminated from the surrounding normal brain tissue.
G. Endostatin. Endostatin, a 20 kDa C-terminal fragment of type XVIII collagen, is a potent inhibitor of angiogenesis, lymphangiogenesis, and cancer metastasis.158 Endostatin can bind to a variety of receptors including the VEGF receptor, integrin, and glypicans. All of these receptors are associated with tumors. Camphausen et al. reported an endostatin–Cy5.5 conjugate for imaging tumor in vivo.159 After injection into mice bearing Lewis lung carcinoma tumors, the conjugate selectively migrated to the site of the tumor. Injection of endostatin–Cy5.5 produced a near-infrared fluorescent image within the tumors at 18 h and reached a maximum at 42 h after injection. Intravenous injection provided a peak emission at 3 h, and the fluorescent signal persisted until 72 h. Immunofluorescence imaging of tumor specimens confirmed the intratumoral binding site for endostatin at the tumor vasculature.
H. Macromolecules. Larger particles (usually with diameters <600 nm) can passively accumulate in tumor tissues because of the enhanced permeability and retention (EPR) effect caused by defects or gaps in the tumor vasculature.160 Licha et al. reported serum protein–dye conjugates consisting of transferrin (44a) or human serum albumin (44b) and the cyanine derivative for tumor imaging.161 In contrast to 44b, the transmembrane mechanism of 44a in HT29 human colon carcinoma cells was mediated by receptor endocytosis. After intravenous injection of both compounds into HT29 tumor-bearing nude mice, maximum fluorescence intensities were reached after 6 h in tumor tissue. However, the contrast between tumor and normal tissue was significantly higher for 44a than for 44b at 24 h after injection. 44a produced fluorescence in viable tumor cells, whereas 44b fluorescence was observed along connective tissue.
I. Antibodies. Monoclonal antibodies (mAbs) usually have excellent antigen specificity and a higher binding affinity than peptides.162 Fluorescently-labeled antibodies provide potential strategies to specifically target tumor cell-specific surface epitopes in vivo while avoiding off-target effects.163 Thus, antibody-based fluorescence probes show great promise for broad applications in clinical medicine, such as cancer diagnosis, staging, and therapy response assessment.
Cetuximab is an anti-EGFR antibody that selectively binds to the external domain of the EGFR with a high affinity.164 Rosenthal et al. conjugated cetuximab with a fluorophore Cy5.5 for imaging HNSCC xenografts.165 Fluorescence images were obtained by time-domain fluorescence imaging and fluorescence stereomicroscopy. Compared with control isotype-matched IgG1–Cy5.5, Cy5.5–cetuximab was specifically accumulated in HNSCC xenografts with significantly higher fluorescence. Tumor xenograft fluorescence was retained for up to 72 h. Fluorescence was detected in multiple HNSCC tumor cell lines with different EGFR expression levels. Moreover, Cy5.5–cetuximab could be detected in small specimens (2 mm).
Warram et al. evaluated a fluorescent conjugate cetuximab–IRDye 800CW in both subcutaneous and orthotopic animal models of glioblastoma multiforme.166 Fluorescence intensities of cetuximab–IRDye 800CW correlated with EGFR expression and vessel density. Fluorescence intensity of the conjugate within the luciferase-positive tumor cell lines was retained for 3 days and offered sufficient fluorescent contrast for surgical resection. Immunohistochemistry was performed to confirm tumor fluorescence, EGFR expression, and vessel density.
Cai et al. combined an anti-EGFR Fab and an anti-CD105 Fab (Bs-F(ab)2) by bioorthogonal “click” ligation of trans-cyclooctene and tetrazine.167 Next, the NIR fluorophore ZW800-1 was conjugated to Bs-F(ab)2 for NIRF imaging of mice bearing U87MG subcutaneous xenografts. ZW800-Bs-F(ab)2 showed excellent uptake in tumors and low background in non-target tissues. The probe could delineate the tumor contours and was useful for locating the tumor and guiding removal of the tumor foci and surgical margins.
The fluorescence of indocyanine green (ICG) will be quenched upon binding to mAbs. After endocytosis and internalization, ICG dissociates from the targeting mAbs and the fluorescence emission is recovered. This property can be used to generate activatable probes. Kobayashi et al.168 developed fluorescence probes by combining indocyanine green (ICG) or Cy5.5 with monoclonal antibodies directed at cell surface markers that are overexpressed on tumors (anti-CD25, anti-EGFR1, and anti-HER2). ICG or Cy5.5 was conjugated to the antibodies daclizumab (Dac), trastuzumab (Tra), and panitumumab (Pan). ICG–mAb conjugates performed as improved activatable imaging probes in vivo in mice with tumor retention times of over 4 days. Dac–ICG could locate at CD25-expressing tumors, while tumors overexpressing HER1 and HER2 could be traced by Pan–ICG and Tra–ICG, respectively. The same group developed antibody-bound fluorescent probes using humanized anti-PSMA antibody (J591)–ICG in prostate cancer;169 Pan–Alexa680 and Tra–ICG in breast cancer;170 Pan–ICG, Pan–PEG4–ICG and Pan–PEG8–ICG in EGFR-positive tumors (MDA-MB-468);171 PSMA–MB–ICG, PSMA–MB–PEG4–ICG, PSMA–MB–PEG8–ICG, and PSMA–MB–IR700 in prostate cancer;172 and panitumumab–FNIR-774 and panitumumab–FNIR-Z-759 in MDA-MB-468 tumors.173
Inappropriate activation of the mucosal immune system can lead to production of the proinflammatory cytokine TNF-α, which plays a pivotal role in the immunopathogenesis of Crohn's disease. The monoclonal antibody adalimumab exhibits a high affinity to human mTNF. Neurath et al. labeled the adalimumab antibody with an FITC fluorophore for in vivo imaging of mucosal immune cells in Crohn's disease during colonoscopy.163 The response time of the probe was 10 min at room temperature. Ex vivo confocal imaging revealed specific fluorescence signals for the identification of mTNF-expressing mucosal cells in the inflamed tissue. They quantified the number of immune cells expressing mTNF and found that patients with high or low numbers of mTNF+ immune cells showed high or low response rates to adalimumab therapy, respectively.
2.3 Hypoxia-mediated tumor imaging
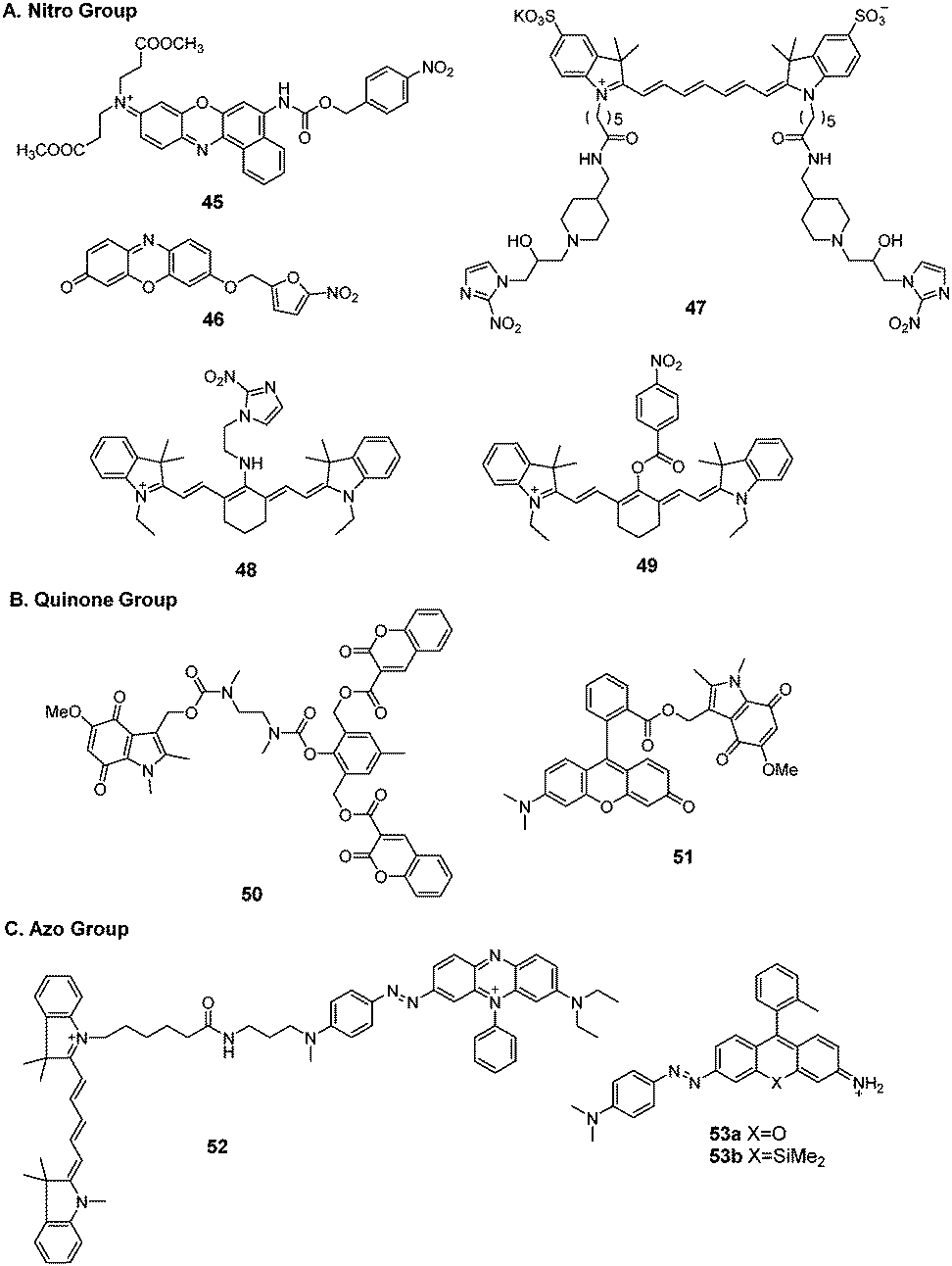
Hypoxia is one of the features typically observed in malignant solid tumors in vivo. Two different biological responses, stabilization of hypoxia-inducible factor-1 (HIF-1) and accelerated bioreductive reaction, are induced by hypoxia. Tumor cells display an elevated level of reductive enzymes such as nitroreductase, DT-diaphorase, and azoreductase. Therefore, tumors can accelerate enzyme-catalyzed one-electron reduction to reduce specific functional compounds containing a nitro group, quinone group, and azo group. This renders these enzymes ideal targets for tumor cells in vivo. Several hypoxia-sensitive fluorescent probes have been developed.174 Here, we summarize the fluorescent probes for potential tumor imaging.Qian et al. synthesized a series of nitro fluorescent probes for targeting hypoxia environments in solid tumors.175 The NIR fluorescence probe (45) was linked to a p-nitrobenzyl moiety and the fluorophore Nile Blue via a carbamate linkage.176 When activated by NTR and NADH, the p-nitrobenzyl moiety of the probe was reduced to form the unstable intermediate p-aminobenzyl derivative, which spontaneously released the Nile Blue fluorophore with fluorescence emission at 658 nm. The fluorescence response was completed within 20 min and 2 h when tested in PBS buffer and in hypoxic A549 cells, respectively.
Ma et al. reported a series of fluorescent probes for nitroreductase and tumor hypoxia.177–179 The resorufin fluorophore was decorated with 5-nitrofuran via an ether bond (46) for imaging the hypoxic status of tumor cells.177 Upon reduction by NTR and NADH, the nitro group in the probe could be selectively transformed into an amino group, followed by 1,6-rearrangement-elimination to release resorufin. The reduction reaction could be completed within 10 min in buffer. The probe was successfully applied in HeLa and A549 cells with different extents of hypoxia. The response time in cells was 8 h.
Nagasawa et al. developed a NIR fluorescent probe (47) composed of 2-nitroimidazole and Cy7 dye for in vivo imaging of tumor hypoxia.180 The probe was accumulated to a greater extent in SUIT-2/HRE-Luc pancreatic tumor cells cultured under hypoxic conditions than in cells under normoxic conditions. The fluorescence resonance time was 30 min. In vivo imaging of hypoxic cells was performed in tumor-bearing mice inoculated with SUIT-2/HRE-Luc cells. HIF-1 activity in xenografts was monitored by bioluminescence imaging (Fig. 7). After administration to the mouse xenograft model, probe 47 rapidly accumulated in tumors and NIR fluorescence could last for more than 24 h.
 | ||
| Fig. 7 Hypoxia-mediated tumor imaging. | ||
Tang et al. reported a NIR fluorescent probe (48) composed of nitroimidazole conjugated to Cy7 fluorophores for monitoring the hypoxia status via the detection of NTR.181 The probe was used to detect intracellular hypoxic levels in HepG2 cells under different oxygen concentrations. The reaction time in the presence of liver microsomes was 15 min. The probe was applied to investigation of the relationship between epithelial–mesenchymal transition in tumor progression and intracellular hypoxic level. The results showed that HepG2 cells displayed downregulation of E-cadherin expression and upregulation of α-SMA expression at low oxygen concentrations.
Li et al. reported five NIR fluorescent probes for rapid NTR imaging in vivo.182 The experimental screening results showed that only the para-nitrobenzoate group modified Cy7 fluorophore (49) probe showed rapidly increased NIR fluorescence for monitoring and bioimaging of NTR. Time-dependent fluorescence emission intensity gave a reaction kinetic time within 200 s in the presence of 1.5 μg mL−1 NTR. Probe 49 could be used to monitor NTR in A549 cells under normoxic conditions and different hypoxic conditions. 49 was also transferred into hypoxic A549 tumor-bearing mouse models via intratumoral injection. PET imaging confirmed that the A549 tumor in the murine model was a hypoxic tumor. Strong fluorescence in the hypoxic A549 tumor could be observed within 60 s, and this fluorescent signal lasted for more than 30 min. Probe 49 could distinguish the hypoxic tumor from inflamed tissue in vivo.
B. Quinone group. The quinone group can also quench the fluorescence of the conjugate system. Under hypoxic conditions, the quinone group undergoes a one-electron reduction process to trigger fluorescence emission. Nishimoto et al. synthesized two hypoxia-sensitive fluorescent probes (50 and 51) for hypoxia tumor cell imaging.183,184 Both probes possessed the hypoxia-sensitive indolequinone unit. Probe 50 was composed of two coumarin chromophores and an indolequinone unit, conjugated via a 2,6-bis(hydroxymethyl)-p-cresol linker. Upon activation by a reduction enzyme, the coumarin chromophore was released with an increase in fluorescence. The response time was 45 min in solution. Probe 50 also worked in cell lysates of the human fibrosarcoma cell line HT-1080 under hypoxia conditions with a response time of 4 h. Probe 51 consisted of an indolequinone unit and a rhodol fluorophore. The indolequinone unit functioned not only as a quenching group on the fluorophore, but also as a hypoxia-sensitive reduction group. The probe emitted fluorescence after enzymatic reduction for 30 min in solution and could produce strong fluorescence in human lung adenocarcinoma A549 cells at 6 h.
C. Azo group. The azo group is a fluorescence quenching group because of its ultrafast conformational change around the N
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond after photoexcitation; when reduced by reductases under hypoxia this quenching process will be inhibited. Nagano et al. exploited azobenzene derivatives as hypoxia-sensitive moieties and developed a NIR fluorescent probe (52) for hypoxia detection.185 The fluorescence increase was triggered by cleavage of the azo bond. The fluorescence regulation mechanism was Förster resonance energy transfer (FRET). After enzymatic reactions under hypoxic conditions, the fluorescence intensities of these probes reached a plateau within 10 min. These probes could distinguish hypoxia in MCF-7 breast cancer cells. Probe 52 could detect hypoxia in mice using an ischemia model of the mouse liver with a rapid in vivo fluorescence response within 1 min after vessel ligation. Hanaoka et al. conceived a further improvement to azo-based hypoxia sensors.186 They directly conjugated the azo group to per-rhodamine fluorophores and obtained two probes (53a and 53b). Once reduced under hypoxia, the two probes resulted in free fluorophores. The resultant colorimetric and fluorescent changes could be utilized to indicate different levels of hypoxia. Cellular hypoxia was observed under hypoxic conditions with a time-dependent course. Probe 53a was localized in the mitochondria, whereas 53b was localized inside the lysosomes. In vivo imaging studies showed that 53a could visualize retinal hypoxia in a rat model of retinal artery occlusion.
N bond after photoexcitation; when reduced by reductases under hypoxia this quenching process will be inhibited. Nagano et al. exploited azobenzene derivatives as hypoxia-sensitive moieties and developed a NIR fluorescent probe (52) for hypoxia detection.185 The fluorescence increase was triggered by cleavage of the azo bond. The fluorescence regulation mechanism was Förster resonance energy transfer (FRET). After enzymatic reactions under hypoxic conditions, the fluorescence intensities of these probes reached a plateau within 10 min. These probes could distinguish hypoxia in MCF-7 breast cancer cells. Probe 52 could detect hypoxia in mice using an ischemia model of the mouse liver with a rapid in vivo fluorescence response within 1 min after vessel ligation. Hanaoka et al. conceived a further improvement to azo-based hypoxia sensors.186 They directly conjugated the azo group to per-rhodamine fluorophores and obtained two probes (53a and 53b). Once reduced under hypoxia, the two probes resulted in free fluorophores. The resultant colorimetric and fluorescent changes could be utilized to indicate different levels of hypoxia. Cellular hypoxia was observed under hypoxic conditions with a time-dependent course. Probe 53a was localized in the mitochondria, whereas 53b was localized inside the lysosomes. In vivo imaging studies showed that 53a could visualize retinal hypoxia in a rat model of retinal artery occlusion.
3. Applications of theranostics in tumors
3.1 General strategies and activation mechanism
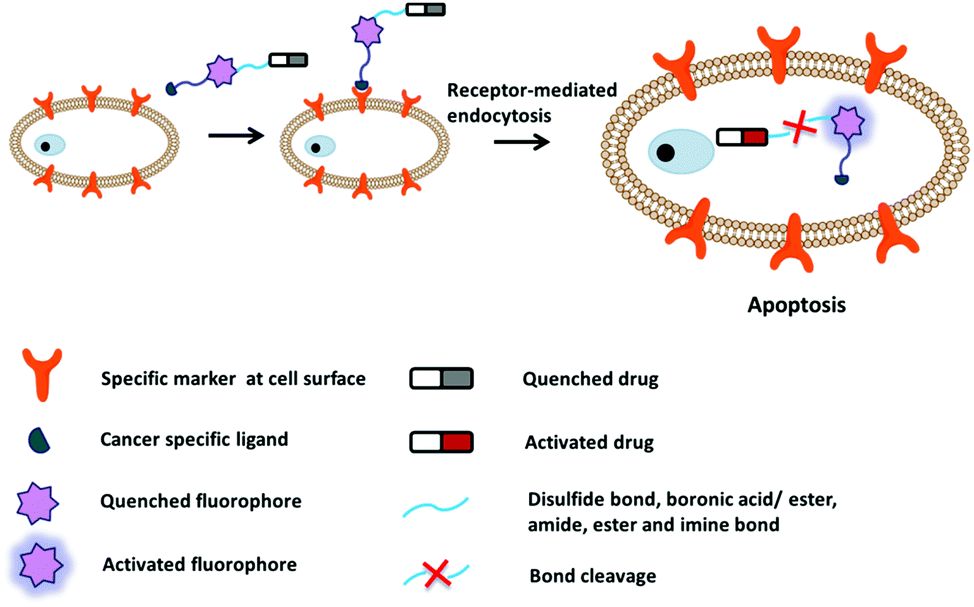
Fluorescent probes that integrate tumor-targeting ligands have successfully been used to identify and localize tumor masses in tumor diagnosis trials. The next focus is the enhancement of the drug therapeutic efficacy by improving the targeting function of small molecular drugs to achieve accumulation in tumor lesions. Conjugation of these diagnostic tools with small cytotoxic drugs generates a new synergistic system with simultaneous diagnostic and therapeutic capabilities; this field has been termed theranostics. Theranostic agents have the promise of regulating precisely when, where, and how a pharmaceutically active artificial compound is delivered to a single cell. The major features of small conjugate theranostics include targeted accumulation, improved pharmacokinetics, imaging ability, real-time information on treatment, and intraoperative guidance. Such agents can not only increase therapeutic efficacy, but also elucidate underlying disease mechanisms and allow pre- and post-treatment assessment.50The critical features of small conjugate theranostics design include the fluorescence manipulated mechanism, cytotoxic drug selection, regulation of prodrug release, and localization capability. Overall, theranostics aims to improve biodistribution, increase tumor targeting ability, reduce biotoxicity, and minimize side effects. Generally, a small molecular fluorescent conjugate therapeutic should be composed of four moieties: an imaging fluorophore, a chemotherapy drug, a targeting ligand, and an appropriate linker (Fig. 8). These theranostic agents can show increased localization and therapeutic efficacy at the tumor site via the specific recognition of receptors that are overexpressed on the surface of tumor cells. After reaching the targeting site, the cleavable linkers are broken and the masked drugs are directly released for therapy. The cleavable linkers are required to behave as stable chemical bonds unless they are selectively activated by a variety of external stimuli signals, such as temperature, light, magnetic field, ultrasound, and electric current. More importantly, the ability to selectively cleave the linkers by endogenous disorder factors, such as biothiols, reactive oxygen and nitrogen species, acidic pH, intracellular redox potential, enzymes, and glucose is even more favorable for the treatment of disease. The requirements of fluorophores and targeting ligands are the same as those of the fluorescent probes used for tumor diagnosis. The fluorescence changes of the imaging fluorophores should illustrate the biodistribution of the therapeutics, tumor lesions, and pharmacokinetics (Fig. 8). Recently, small fluorescent conjugate therapeutics have been developed for tumor diagnosis and therapy. In this section, we discuss the latest developments in these theranostics according to the cleavable mode of the linkers, which essentially form the sluice for drug release and fluorescence emission.
 | ||
| Fig. 8 Design strategy and drug release mechanism. | ||
3.2 Thiol-mediated modes
Glutathione (GSH) is the most abundant endogenous biothiol in cells. It participates in various cellular processes, including cell differentiation, cellular metabolism, antioxidant defense, and apoptosis.187 Normal concentrations of GSH range from 1 to 15 mM depending on the cell type.188 However, the intracellular GSH level is much higher in tumor cells than in normal cells.189 The disulfide bond (–S–S–) can be cleaved via the reduction of free biothiols.190 Therefore, GSH seems to be an ideal bioactivator for triggering therapeutics. A cytotoxic drug can be conjugated with a fluorophore via a disulfide linker to form a desired therapeutic agent with the specific capability to accumulate in tumor lesions. The therapeutic agent does not release the antitumor drug before it reaches the tumor cells because of the relatively low concentration of GSH in blood circulation and in normal cells (Fig. 9).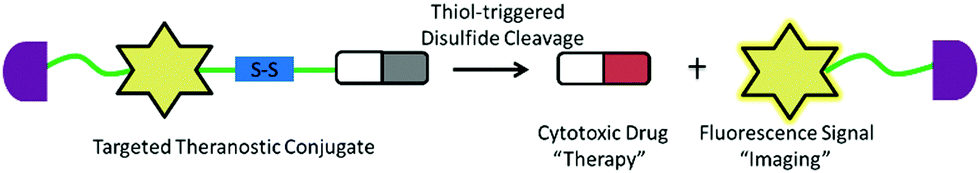 | ||
| Fig. 9 Thiol-mediated drug release. | ||
Gemcitabine, a commercial antitumor drug, principally functions as a strong inhibitor of a pyrimidine base in DNA replication or as an apoptosis inducer via an interaction with ribonucleotide reductase. Kim et al. reported the prodrug 55 as an NIR targeted drug delivery system. The theranostic agent included the targeting ligand folate, modified gemcitabine, and a Cy7 fluorophore conjugated with a cleavable disulfide linker.192 Upon reacting with GSH, the prodrug 55 decomposed into an amino fluorophore derivative and released gemcitabine. The absorption and fluorescence changes could be completed within 2 h in the presence of 0.2 mM GSH. The prodrug 55 exhibited higher cytotoxicity in FR-overexpressing cells than in FR-negative cells, indicating its targeting capability. The biothiol-induced disulfide cleavage occurred in the endoplasmic reticulum and the released gemcitabine diffused into the cell nucleus to form a faulty nucleoside and induce apoptosis. In vivo fluorescence imaging confirmed that the prodrug 55 could be selectively taken up by KB tumor tissue that overexpressed the folate receptor.
Some drugs have been directly connected to folate via a disulfide linker to strengthen their targeting abilities as therapeutic agents. Low et al. reported a folate–peptide–camptothecin prodrug 56.193 The hydrophilic peptide was introduced to improve the water solubility of the prodrug. Release of camptothecin from the folate peptide linker took 1 h in the presence of a 10-fold molar excess of dithiothreitol. 56 showed a high binding affinity for FR-overexpressing KB cells and efficiently released camptothecin to inhibit cell proliferation with an IC50 of 10 nM. Leamon et al. synthesized a folate–mitomycin C conjugate 57.194 The water-soluble conjugate exhibited a high affinity for FR-positive cells. Evaluation of cytotoxicity showed an IC50 of 5 nM in FR-positive cells. Goldmacher et al. reported the targeting prodrugs folate–maytansinoids (58a, 58b).195 These prodrugs exhibited a high binding affinity for the folate receptor and folate receptor-mediated internalization. They also showed high cytotoxicity and selectivity for folate receptor-positive KB cells with an IC50 of 50 pM.
Biotin is a B-group vitamin with an extreme affinity for avidin proteins. Biotin is a key micronutrient for cellular function and cell growth. Biotin uptake is much higher in rapidly growing tumor cells than in normal cells. Ojima et al. used biotin as a tumor targeting ligand in the design of theranostic agents utilizing different anticancer drugs and fluorophores.196–198 The biotinylated fluorescein coupled with SB-T-1214 via the self-immolative disulfide linker formed theranostic agent 59. Cleavage of the disulfide bond by endogenous GSH would release the anticancer drug and fluorescein. Fluorescent imaging demonstrated that theranostic agent 59 localized at the microtubules. This group developed a versatile platform that consisted of 1,3,5-triazine as the tripod splitter module, both SB-T-1214 and camptothecin as antitumor drugs, biotin as the tumor-targeting moiety, and the disulfide linker.199 The prodrugs 60a and 60b were delivered to cell lines that overexpressed the biotin receptor, such as MX-1, MCF-7, ID8, and L1210FR cells. After activation by GSH, 60a exhibited IC50 values of 3.22–9.80 nM against all biotin receptor-positive cell lines, and 705 nM against biotin receptor-negative WI38 cells. Next, they adopted this strategy to report two tumor-targeting theranostic conjugates 61a and 61b.200 The cytotoxic drug taxoid 3 was conjugated with a fluorine-labeled prosthetic (61a) or a fluorophore fluorescein (61b) via a disulfide bond. Once internalized into tumor cells, the disulfide bond was cleaved by endogenous biothiols and drug release was monitored by PET and fluorescence imaging, respectively. Conjugate 61b exhibited higher specificity for biotin receptor-overexpressing tumor cells than biotin receptor-negative cells. The recycling of biotin receptors varies among different cell lines, resulting in different rates of conjugate 61b internalization into cells.
Kim et al. developed a tumor-targeting theranostic agent 62 composed of the chemotherapeutic drug gemcitabine, a disulfide linker, a coumarin fluorophore, and a targeting unit biotin.201 The fluorescence intensity increased after cleavage of the disulfide linker by intracellular thiols. Temporal release of gemcitabine from the prodrug 62 was measured via fluorescence changes in the presence of GSH. The function time was 10 min. The prodrug was selectively internalized by biotin receptor-positive A549 cells compared with biotin receptor-negative WI38 cells. Colocalization experiments demonstrated that thiol induced the release of active gemcitabine in the lysosomes. In another study, Kim et al.202 designed a theranostic agent 63 based on a NIR azo-BODIPY fluorophore conjugated with biotin and linked with gemcitabine via a disulfide linker. Time-dependent fluorescence changes showed a response time of 30 min in the presence of 5.0 mM dithiothreitol. The time-dependent drug release depended on the available biothiol reactivities (Cys > Hcy > GSH > Trx). The theranostic agent 63 was easily internalized by A549 cells compared with WI38 cells. Co-localization experiments demonstrated that thiol-induced disulfide cleavage of this theranostic agent occurred in the endoplasmic reticulum (ER). Theranostic agent 64 was a cancer targeting conjugate composed of biotin, a naphthalimide fluorophore, and Holliday junction (HJ) inhibitor peptide2 (KWWCRW).203 Cleavage of the disulfide bond occurred in the ER, releasing the active HJ inhibitor peptide2 drug and the fluorophore.
SN-38 is a topoisomerase I inhibitor and the active ingredient in CPT11 (camptothecin), which is used as a therapeutic agent to treat various carcinomas. The potent antitumor drug SN-38 was connected to a biotinylated rhodol fluorophore through a cleavable disulfide bond linker.204 The theranostic agent 65 displayed a 32-fold increase in the fluorescence intensity within 10 min after exposure to GSH. The prodrug was internalized effectively within biotin receptor-enriched cells by receptor-mediated endocytosis and showed targeted antiproliferative activity against biotin receptor-positive HeLa and A549 cells as a result of release of SN-38 within these cells (Fig. 10). The antitumor efficacy of 65 was evaluated in vivo using a xenograft murine model created by subcutaneous inoculation with HeLa cells. Ex vivo optical and fluorescence imaging showed increased fluorescence of the tumor region compared with other organs, confirming the in vivo tumor targeting effect of the prodrug.
 | ||
| Fig. 10 Theranostics based on thiol-mediated release. | ||
The Shiga toxin receptor Gb3 is overexpressed on the surface of certain human cancers, including colorectal carcinoma. The cytotoxic drug SN-38 belongs to the class of camptothecin derivatives that inhibit topoisomerase I. Johannes et al. designed prodrug conjugates 66a, b, c, d to deliver the cytotoxic drug SN-38.205 The disulfide linkage of these prodrugs involves two different spacers with different stabilities in the biological system. One is based on an aromatic ring (66a,b) and the other on an aliphatic chain (66c,d). Conjugate 66b was too unstable to be used in vivo; however, conjugate 66d was completely stable over extended periods of up to 48 h in all media. Enzyme-linked immunosorbent assay (ELISA) analysis of 66d indicated that cleavage of the disulfide bond became detectable between 6 and 24 h and was essentially complete at 48 h. This slow release should sustain the continued presence of the active principle in dividing tumor cells, with the prodrug being otherwise rapidly cleared from the circulation. The disulfide bond of prodrug 66c was cleaved in the ER, which functions in cellular redox homeostasis. The ER was close to the nucleus, where the molecular target of hydrophobic SN-38 resides.
In addition to folate and biotin, cyclic peptides are another type of effective tumor targeting ligand. These peptides contain an RGD (Arg–Gly–Asp) sequence, which can be recognized and internalized by a well-known tumor-associated receptor, αvβ3 integrin. αvβ3 integrin is highly expressed on several activated endothelial cells and plays a predominant role in tumor-induced angiogenesis and growth. Kim et al. reported a therapeutic agent 67 that allowed direct, fluorescence-based monitoring of targeted cellular uptake and release of the antitumor drug camptothecin.206 The results of cellular experiments indicated that conjugate 67 bearing a cyclic RGD peptide targeting subunit was selectively internalized into U87 tumor cells via αvβ3 integrin-mediated endocytosis. Disulfide bond cleavage occurred in the ER, permitting fluorescence changes of the naphthalimide moiety within 60 min.
Wender et al. presented three disulfide-based luciferin-transporter conjugates (68a–c) to establish operationally facile methods to quantify drug-conjugate delivery, linker cleavage, and drug release in real time in cellular assays.190 The disulfide-carbonate linkers of these conjugates were stable without affecting the rapid release of luciferin and drug (within minutes) in cells through disulfide cleavage. The half-lives of the conjugates (68a–c) were 3 h, 11 h, and 33 h, respectively, and depended on cyclization of the intermediate thiol with the carbonate. When the conjugates 68b and 68c were incubated with prostate tumor cells transfected with the luciferase gene, strong luminescence of luciferin could be observed within 1 min.
Zhu and Guo et al. developed novel activatable theranostics (69a and 69b) based on the dicyanomethylene-4H-pyran NIR fluorophore for in vivo and in situ monitoring of drug delivery and cancer chemotherapy in living animals.207 The biologically abundant thiols in tumor cells triggered cleavage of the disulfide linker, leading to the release of camptothecin and emission of NIR fluorescence. These theranostics showed similar cytotoxicity with significant in vitro antitumor activity against several cell lines including BCap-37, HepG2, MCF-7, HeLa, KB, and KB200. However, the cytotoxicity of 69a was slightly higher than that of camptothecin and 2-fold lower than that of 69b. The prodrug 69a and its polyethylene glycol–polylactic acid (PEG–PLA)-encapsulated nanoparticles all displayed significantly potent cancer therapy and fewer side effects compared with free camptothecin. Loading of PEG–PLA nanoparticles increased the tumor localization properties and prolonged the plasma retention time of the prodrug. The theranostic prodrug 70 contained camptothecin and NIR cyanine dye, linked by a disulfide bond.208 Cleavage of the disulfide bond resulted in generation of the active drug and induced a remarkable fluorescence shift. This change of wavelength provided dual fluorescent channels for real-time imaging of the activation and biodistribution process of the prodrug. After intravenous injection of 70 for 24 h, the prodrug predominantly accumulated and was activated in tumor lesions. Loading the theranostic prodrug in PEG–PLA nanoparticles would improve the therapeutic efficacy and reduce the side effects of comptothecin.
Chlorambucil (CLB) is a DNA alkylating agent that causes DNA damage in the nucleus. Zhou et al. developed a theranostic agent 71 composed of the potent anticancer drug CLB, a disulfide linker, and a fluorescent naphthalimide moiety.209 The prodrug 71 underwent thiol-induced disulfide bond cleavage in HeLa cells with release of the naphthalimide moiety.
Cheng et al. reported a therapeutic agent 72 containing a cyanine-amide moiety as the NIR fluorophore, camptothecin as a model antitumor drug, and a disulfide bond as the cleavable linker.210 The disulfide bond was cleaved by dithiothreitol within 120 min, with release of the active drug and NIR dye. The results of the real-time monitoring of drug release by recording NIR fluorescence changes in vitro and in vivo demonstrated potential applications in quantitative assessments in live cells and semi-quantitative measurements in live animals.
3.3 H2O2-mediated release
Reactive oxygen species (ROS) exhibit highly physiological activities and play key roles in several physiological processes including cellular redox homeostasis, cell differentiation, cell proliferation, and cell signaling.211–214 ROS include hydrogen peroxide (H2O2), hydroxyl radicals (HO˙), hypochlorous acid (HOCl), the superoxide anion (O2), and singlet oxygen (1O2). Compared with normal cells, tumor cells exhibit elevated intrinsic oxidative stress due to the dysregulated production of ROS (mainly as H2O2). Given this special feature of tumor cells, increased concentrations of ROS have been recognized as trigger switches for the development of ROS-activated theranostics. Among ROS, the chemical lifespan of H2O2 is relatively stable, which allows establishment of significant steady-state concentrations in tumor cells. These factors make H2O2 a preferred candidate for the development of ROS-activated theranostics. Such theranostics generally contain two separate functional domains: (i) a H2O2-response moiety, and (ii) a cleavable linker system. Once triggered by H2O2, the drug is released with a large increase in cytotoxic potency (Fig. 11). | ||
| Fig. 11 H2O2-mediated drug release. | ||
Nitrogen mustard, one of the DNA cross-linking agents, exhibits severe cytotoxicity but has poor selectivity for cancer cells versus normal cells. Peng et al. synthesized two prodrugs of nitrogen mustard coupled with aryl boronic acids (73a) and their pinacol esters (73b) to reduce the toxicity of cross-linking agents against normal cells.215 The aryl boronic acids and their pinacol esters can be cleaved by H2O2. These prodrugs were effectively activated in tumor cells. The ability and selectivity of these prodrugs to inhibit tumor cell growth was evaluated and both compounds inhibited various types of tumor cells at 10 μM.
Kim et al. developed a H2O2-activated theranostic agent 74.216 Compound 74 was activated by H2O2-mediated boronate oxidation, resulting in the release of the coumarin fluorophore for monitoring fluorescence and activation of the chemotherapeutic drug camptothecin for inhibition of tumor cell growth. The fluorescent signal of coumarin showed that 74 was localized in lysosomes. In vivo therapeutic activity was evaluated after intratracheal administration of 74 into mice bearing metastatic lung tumors.
Kim et al. next developed a mitochondria-targeted antitumor theranostic agent 75 that was activated by overexpressed H2O2 in mitochondria and led to self-monitored apoptosis of tumor cells to achieve precise tumor treatment.217 The theranostic agent 75 consists of four parts. The fluorophore ethidium can preferably localize in mitochondria for detection of the intrinsic apoptosis caused by the 5′-deoxy-5-fluorouridine drug moiety. Ethidium emits weak fluorescence in aqueous solution unless intercalated into double-stranded RNA or DNA. The tumor-targeting unit biotin would mediate accumulation of 75 at the tumor site. Aryl boronic acid esters were the H2O2 activator. 75 could be activated by endogenously produced mitochondrial H2O2 in tumor cells and then release the drug 5′-deoxy-5-fluorouridine to induce apoptosis. In vitro experiments showed that 75 displayed high uptake in biotin receptor-positive human lung tumor A549 cells compared with biotin receptor-negative WI-38 cells. In vivo xenografts revealed that 75 could inhibit tumor progression and cure tumor-bearing mice (Fig. 12).
 | ||
| Fig. 12 H2O2, enzyme, and pH-triggered theranostics. | ||
3.4 Enzyme-activated release
Real-time detection of drug release would enable in vivo studies of pharmacokinetics, pharmacodynamics, and cell permeation pathways. A desirable activation mechanism for drug release depends on the unique physiologic characteristics of tumor cells. As various enzymes are typically overexpressed in tumor cells, utilization of overproduced enzymes as trigger switches for the development of enzyme-activated theranostics has been considered.Shabat et al. reported the prodrug system 76 containing a 7-hydroxycoumarin with a hydroxymethyl substituent spacer, the chemotherapeutic drug melphalan as the end-unit, and phenylacetamide as the cleavable linker.218 The phenylacetamide could be cleaved by the enzyme penicillin-G-amidase (PGA) for release of drug in cells. In the presence of PGA, 76 exhibited cytotoxicity toward the cells with IC50 of 2.5 μM. The prodrug 77 achieved selective activation in tumor cells. The proteolytic enzyme cathepsin B was employed to trigger the dipeptide Phe–Lys linker. 77 was more than 7.5-fold more toxic toward starved cells (IC50 = 4 μM) than non-starved cells (IC50 = 30 μM). The increased cytotoxicity of 77 was attributed to elevated expression of proteolytic enzymes, including cathepsin B. Human umbilical vein endothelial cells incubated with 77 showed cytoplasmic accumulation of activated coumarin.
DT-diaphorase, a cytosolic flavoenzyme, plays essential roles in the cellular antioxidant system. DT-diaphorase levels are markedly higher in a number of tumor tissues than normal tissues.219 DT-diaphorase has been employed as a trigger to activate quinone antitumor drugs and acts as a valuable biomarker for efficient drug delivery.
DT-diaphorase was developed to produce therapeutic NO by metabolizing a nitric oxide (NO) prodrug.220 In addition, Wu et al. reported a DT-diaphorase-activatable theranostic prodrug 78.22178 was composed of an antitumor drug camptothecin (CPT), a self-immolative linker, and a quinone propionic acid trigger group. As a result of a photoinduced electron transfer (PET) process between CPT and the quinone propionic acid moiety, the fluorescence of CPT was almost quenched. After reduction by DT-diaphorase, the active CPT recovered its fluorescence emission. CPT release could be monitored by real-time detection of fluorescence. The prodrug exerted high cytotoxicity towards DT-diaphorase overexpressing cell lines.
Indoleamine-2,3-dioxygenase (IDO) is an immunosuppressive enzyme present in human tumors that can be used as a tumor immunotherapeutic target. Lippard et al.222 presented tumor immunochemotherapy Pt(IV) prodrugs 79a and 79b for combining immunomodulation and DNA cross-link-triggered apoptosis. The conjugates include the IDO inhibitor (D)-1-methyltryptophan ((D)-1-MT) and the Pt agent cisplatin. After being preferentially targeted to IDO, (D)-1-MT and cisplatin were released inside tumor cells. The release of (D)-1-MT would enhance T-cell proliferation and the generation of cisplatin would concomitantly induce DNA damage in tumor cells. Dynamic blood stability experiments of 79b in Balb/c mice revealed t1/2 of 1 h, and the human blood stability study showed t1/2 of 2.2 h.
3.5 pH-triggered theranostics
The tumor cell micromilieu is highly acidic (pH 6.0–7.0), mainly as a result of glycolysis and hypoxia.223 Growth of solid tumors requires large amounts of nutrients such as glucose; however, highly efficient glycolysis will induce intracellular acidification due to the generation of lactic acid. In addition, the extracellular diffusion effect of CO2 is discharged by tumor tissue hypoxia, producing carbonic acid with H2O.Zhang et al. designed a pH-responsive prodrug (80) for real-time monitoring of drug release in the tumor mass.224 The prodrug contains three parts: a coumarin fluorophore, a targeting moiety GRGDS oligo-peptide that can be recognized by αvβ3 integrin, and doxorubicin, which functioned not only as an antitumor drug but also as a fluorescent quencher for coumarin. The GRGDS moiety promoted selective uptake of the prodrug into tumor cells that overexpressed αvβ3 integrin. The hydrazone bond in the prodrug could be cleaved in the endosomes/lysosomes (pH 5–6) of tumor cells and the activated doxorubicin induced cell apoptosis. In addition, the fluorescence of coumarin was recovered. In vitro studies showed approximately 93.8% release of drug at pH 5.0 and only 40.7% release at pH 7.4. Merged fluorescence images indicated that this prodrug was internalized and cleaved in endo/lysosomes.
Hanson et al. synthesized a therapeutic agent of steroidal anti-estrogen and doxorubicin (81).225 This prodrug reduced the side effects of doxorubicin and the targeted drug delivery of steroidal anti-estrogen improved its efficacy. Fluorescence microscopy studies in MCF-7 cells that overexpress the estrogen receptor (ER) suggested that the uptake process was controlled via a membrane ER-mediated mechanism, resulting in cellular accumulation of doxorubicin. Once the prodrug entered the acidic cytoplasm of tumor cells, hydrolysis of the pH-sensitive hydrazone linker released free doxorubicin to kill the tumor cells.
3.6 Light-activated theranostics
As an alternative to chemotherapy, a new modality termed photodynamic therapy (PDT) has been successfully applied in the clinic for the treatment of a variety of tumors and other diseases.226 Photoresponsive theranostics allows precise control of drug release, including location, timing and dosage, through an active “phototrigger”.227 The primary components of photoresponsive theranostics include a photosensitizer, a masked drug, a photo-switched linker, light, and molecular oxygen. All of these elements are required in PDT to produce reactive oxygen species and release cytotoxic drugs for killing of tumor cells (Fig. 13).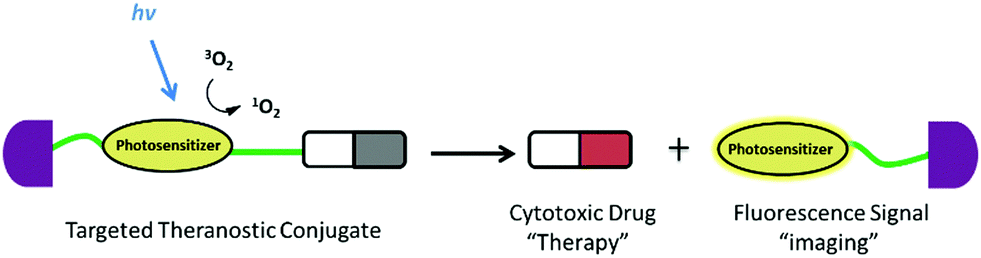 | ||
| Fig. 13 Light-activated drug release. | ||
Berkman et al. reported PSMA inhibitor conjugates of pyropheophorbide-a (82 and 83) for targeted photodynamic therapy of prostate tumors.228,229 Compared with PSMA-negative PC-3 cells, the prodrug 82 selectively induced apoptosis in PSMA-positive LNCaP prostate tumor cells via in vitro targeted photodynamic therapy. Fluorescence labeling showed that conjugate 83 was specifically localized in PSMA-positive LNCaP cells, but not PSMA-negative PC-3 cells. Studies with specific caspase inhibitors revealed that conjugate 83 mediated apoptosis through the caspase-8/-3 pathway in PSMA-positive LNCaP cells; after 4 h of PDT treatment with 83 the activities of caspases-8, -9, and -3 were increased.
Ju et al. reported a pH-activatable aniline-substituted aza-boron-dipyrromethene as a photosensitizer for efficient photodynamic therapy and therapeutic monitoring (84).230 To achieve high accumulation in tumors, compound 84 was encapsulated in a cyclic RGD peptide-functionalized poly(ethylene glycol)-block-poly(lactic acid) and methoxyl poly(ethylene glycol)-block-poly(lactic acid) nanomicelle (Fig. 14). After activation by the physiologically acidic pH in the tumor, 84 exhibited strong phototoxicity by producing 1O2 and emitted fluorescence. The fluorescence at the tumor increased gradually and reached a maximum at 8 h post-injection. The high fluorescence level could be maintained for more than 16 h after injection.
 | ||
| Fig. 14 Light-activated theranostics. | ||
Choi et al. developed a folate–photosensitizer conjugate (85) via a protein linker for photodynamic therapy.231 The NIR fluorescence emission of the photosensitizer was quenched when folate was removed by the tumor-associated lysosomal enzyme cathepsin B. KB cells were implanted into mice as a xenograft model to assess the utility of conjugate 85 for in vivo fluorescence imaging and photodynamic therapy. Strong fluorescence signals were observed at the tumor site at 3 h post-injection and could be maintained for up to 24 h.
You et al. synthesized three double activatable prodrugs of the CA-4, SN-38, and coumarin system (86a–c) that were first activated by intracellular esterase and then drug release was induced by light irradiation.232 A photo-cleavable aminoacrylate-linker and a deactivated photosensitizer allowed the spatiotemporally controlled release of drugs by visible light irradiation. When the prodrug 86a was irradiated with very low intensity light (540 nm, 8 mW cm−2) for 30 min up to 99% of the coumarin was released.
NIR light has great potential for spatiotemporally controlled release of therapeutic agents due to its deep tissue penetration. You et al. demonstrated that the aminoacrylate group could be cleaved to release parent drugs after oxidation by 1O2 when activated by NIR light.233 The prodrugs of combretastatin A-4 (87a–c) contain core-modified porphyrin as a NIR photosensitizer to generate 1O2. The aminoacrylate linker of 87a was broken by tissue-penetrable NIR light (690 nm), releasing the antitumor drug. The prodrug 87a exhibited a significantly better antitumor effect after irradiation than 87b. The prodrug 87c was designed for in vivo fluorescence imaging after cleavage of the aminoacrylate group in mice. The prodrug 88a was developed to achieve multifunction activity, including the release of drug, fluorescence imaging, and photodynamic therapy.234 The compound 88b was designed as a pseudo-prodrug to assess the effects of photodynamic therapy and confirm the potential application of the prodrug 88ain vivo. The time-dependent distribution of 88a was investigated using Balb/c mice with SC tumors (colon-26 cells, 4–6 mm in length).
You et al. expanded their NIR light activatable strategy by integrating a tumor-targeting folic acid group into the prodrug systems (89a–e).235 The photosensitizer and fluorophore was phthalocyanine. The light switch was aminoacrylate with variable polyethylene glycol chains. Compared with the lipophilic prodrugs 89a and 89b, the hydrophilic prodrugs 89c and 89d had higher cellular uptake in SC colon-26 tumors. The prodrug 89e without folic acid indicated the effectiveness of tumor targeting.
Singh et al.236 developed a targeted photoresponsive prodrug 90 containing biotinylated coumarin and chlorambucil, in which the phototrigger was directly attached to the drug. The prodrug 90 could be selectively taken up by MDA-MB-231 cells. The release time of active chlorambucil was 20 min after light irradiation. Biotin-mediated accumulation could significantly increase the efficiency of intracellular drug delivery in the MDA-MB-231 cell line.
The tropomyosin receptor kinase C (TrkC) receptor is normally expressed in neurons, but is overexpressed in highly metastatic tumor cells such as neuroblastoma, glioblastoma, thyroid cancer, melanoma, and breast cancer.237–244 The TrkC receptor could be used as a potential molecular target for chemotherapeutics. Burgess et al. reported fluorescence conjugates for PDT based on a synthetic peptidomimetic (including isoleucine and tyrosine side chains) targeting ligand.245 Conjugate 91a composed of a peptidomimetic and a boron dipyrromethene (BODIPY) fluorophore for tumor imaging was selectively accumulated in TrkC receptor-expressing tumor tissues. Intracellular imaging studies showed that 91a was internalized into lysosomes, and behaved as the natural TrkC ligand neurotrophin-3 when internalized via the TrkC receptor. The diiodo-BODIPY could perform as a photosensitizer to produce triplet oxygen for PDT. Conjugate 91a induced significant photocytotoxicity in TrkC-expressing NIH-3T3 fibroblasts and SY5Y neuroblastoma cells compared with TrkC-negative cells. The biodistribution of conjugate 91a was tested in mice bearing 4T1 tumors. Fluorescence was maintained at a high level for up to 72 h after intravenous administration. The isomeric conjugate 91b that could not bind TrkC was used as a control probe.
The application of PDT to lesions in deep tissue is often hampered by light penetration, as well as absorption and scattering by biological tissues. Wang et al. developed a PDT system in which the photosensitizer was activated by bioluminescence instead of an external light source.246 In this PDT system, luminol (92a), hydrogen peroxide, and horseradish peroxidase were used as bioluminescent molecules. Cationic oligo (p-phenylene vinylene) (OPV, 92b) was used as the photosensitizer. The activation mechanism was bioluminescence resonance energy transfer (BRET). The blue bioluminescence generated from luminol excited OPV to produce reactive oxygen species to kill cancer cells and pathogenic microbes. The cationic 92b could bind to the HeLa cell surface. This BRET system could function in deeper tissues.
Schnermann et al. reported NIR light-mediated cleavage of antibody-drug conjugate 93 based on cyanine photocages.247 The conjugate includes a heptamethine cyanine fluorophore as the caging component, the drug combretastatin A4, and panitumumab (Pan), a clinical monoclonal antibody to the human epidermal growth factor receptor (EGFR). The fluorescence signal offered a useful marker for accumulation of conjugate 93. Loss of the fluorescence signal after excitation by light at 690 nm indicated drug release. The consequence of irradiation with NIR light was evaluated in mice bearing A431 tumors.
Tumor-associated M2-type macrophages are correlated with tumor invasion and metastasis. The biomarker CD206 is specifically expressed in M2 macrophages. Liu et al. reported a CD206-targeting photoimmunotherapy agent generated by conjugating a monoclonal anti-CD206 antibody with the near-infrared phthalocyanine dye IRDye700.248 Upon light irradiation, this agent suppressed proliferation of sorafenib-resistant 4T1 tumors and prevented lung metastasis.
Zhang et al. developed a photosensitive prodrug 94 for fluorescence imaging-guided photodynamic therapy and chemotherapy of tumors.249 The prodrug included a fluorescent photosensitizer meso-tetraphenylporphyrin (TPP) and an antitumor drug gemcitabine. Upon irradiation with low-energy red light, TPP generated 1O2. Subsequently, 1O2 mediated cleavage of the thioketal linker, which resulted in the cascaded release of gemcitabine and combination therapy against tumor cells. The prodrug was formulated in PEG–PLA to form uniform micelles to enhance the plasma stability and endow tumor-targeting activity. Strong fluorescence was detected in tumor lesions after 48 h post-injection, displaying significantly enhanced tumor accumulation and good tumor retention.
Zhang et al. developed an anticancer theranostic prodrug (95) based on hypoxia and a photo dual activation process.250 The photo-activated group (o-hydroxyl E-cinnamic ester) of the prodrug was modified using a hypoxic trigger 4-nitrobenzyl group, which was directly attached to the anticancer drug gemcitabine. In hypoxic MCF-7 cells, the hypoxic trigger was reduced by nitroreductase and underwent a 1,6-rearrangement-elimination reaction to remove the masked group. Subsequent UV irradiation would induce isomerization and intramolecular esterification processes leading to the formation of a coumarin fluorophore and the release of gemcitabine. The theranostic prodrug exhibited significant cytotoxicity against MCF-7 cells.
3.7 Aggregation-induced emission-based theranostics
Aggregation-induced emission (AIE) is a unique photophysical process that is induced by a series of silole or tetraphenylethene derivatives that are non-emissive in dilute solutions but become highly emissive in solid or aggregated states.251 Fluorescent probes with AIE characteristics possess unparalleled advantages in biological detection compared with traditional fluorescent probes. On one hand, they can achieve brighter fluorescence as a result of more AIE active molecules binding to the target analytes without aggregation-induced fluorescence quenching. On the other hand, the feature of a drastic enhancement in the fluorescence intensity associated with an aggregation event can be used as a method of quantitative detection (Fig. 15). | ||
| Fig. 15 AIE-based theranostics. | ||
Tang et al. presented a series of theranostic agents based on AIE characteristics (Fig. 16).252–254 The theranostic prodrug 96 was a targetable theranostic Pt(IV) prodrug for monitoring and assessing drug-induced cell apoptosis.252 The targeting ligand was a cyclic arginine–glycine–aspartic acid (cRGD) tripeptide for intergrin αvβ3 that was overexpressed on the surface of tumor cells. The prodrug was a nontoxic Pt(IV) complex that was reduced to toxic Pt(II) in tumor cells. Asp–Glu–Val–Asp (DEVD)-conjugated tetraphenylsilole (TPS) that possessed AIE characteristics was used as an apoptosis sensor. The prodrug 96 selectively accumulated in tumor cells that overexpressed αvβ3 integrin and then released the toxic drug Pt(II) to induce apoptosis. Subsequently, caspase-3 cleaved the apoptosis unit TPS-DEVD and triggered the AIE fluorescence.
 | ||
| Fig. 16 Theranostics based on aggregation-induced emission. | ||
A targeted theranostic Pt(IV) prodrug 97 based on an AIE luminogen for in situ monitoring activation of the platinum(IV) prodrug was developed.253 The theranostic prodrug consisted of nontoxic Pt(IV) complexes, a tetraphenylethene pyridinium unit with AIE properties, a short hydrophilic peptide with five aspartic acid (D5) units to ensure its water solubility, and a cRGD tripeptide as the targeting ligand. Prodrug 97 could be selectively taken up by MDA-MB-231 cells that overexpressed αvβ3 integrin. The theranostic prodrug 98 contained a targeted cRGD moiety, a tetraphenylene (TPE) derivative, a fluorescent antitumor drug doxorubicin (DOX), and a chemotherapeutic Pt(IV) prodrug. 98 was selectively taken up by tumor cells that overexpressed αvβ3 integrin accompanied by the release of two drugs: Pt(II) and DOX. The fluorescence of TPE recovered, followed by the separation of TPE and DOX.254
The outer membrane of mitochondria has a more negative potential in tumor cells.255 Triphenyl phosphonium cation (TPP) facilitates penetration of the phospholipid bilayers in mitochondria due to the positive charge on the phosphonium and three lipophilic phenyl groups.256 Liu et al. presented a mitochondrial targeting probe (99) formed by conjugation of a salicyladazine fluorophore with the mitochondria-targeting TPP.257 The fluorescence manipulation mechanism involved both AIE and excited-state intramolecular proton transfer processes, which could enhance the imaging contrast and the signal-to-noise ratio. The probe exhibited no fluorescence in culture media but fluorescence was activated after its accumulation in the mitochondria of tumor cells. The probe could induce the generation of reactive oxygen species and affect essential cancer cell progression, leading to tumor cell apoptosis.
3.8 NIR dye-based theranostics
NIR hemicyanine dyes are desirable candidates for the development of fluorescence imaging probes due to their photophysical properties, biocompatibility, and low toxicity to living systems.258 Heptamethine indocyanine dyes are often utilized for lesion targeting and imaging. However, these dyes are lipophilic cationic molecules that can preferentially accumulate in the mitochondria of tumor cells, therefore some cyanine probes show activity in photodynamic therapy. These types of dyes have promise as candidate theranostic agents.Shi et al. developed a class of near infrared (NIR) heptamethine indocyanine dyes that preferentially accumulated in tumor cells for in vivo imaging.45,259–263 The probe 100 not only preferentially accumulated in a broad spectrum of tumor cells for in vivo tumor targeting and imaging, but also had unique photodynamic therapeutic properties.261 This probe avoided the problems of chemical conjugation with tumor-specific ligands and photosensitizers. Changes in the lipophilicity of heptamethine indocyanine dyes would increase their mitochondrial toxicity. A series of IR-808 analogs (101a–c) was developed for simultaneous cancer-targeted NIR imaging and potent anticancer activities.262 The theranostic agent 101a, a butyl ester derivative of IR-808, showed preferential accumulation in the mitochondria of A549 cells and PDT activity. The tumor inhibition effect of 101a was better than that of cyclophosphamide. The tumor selective NIR dye IR-780 (1) also exhibited efficient anti-tumor activity by targeting drug-resistant cell populations.263 IR-780 preferentially accumulated in the mitochondria of drug-resistant human lung tumor cells (A549/DR) and blocked the self-renewal and migration ability of A549/DR cells. The IR-780 dye showed tumor targeting ability and inhibited tumor recurrence in a mouse syngeneic Lewis lung carcinoma xenograft model.
Shi et al. synthesized a mitochondria-targeting theranostic agent (102) for synchronous photodynamic therapy and photothermal therapy.264 The theranostic agent 102 was screened from a panel of heptamethine cyanine dyes modified with various N-alkyl side chains. This NIR fluorescent dye could clearly visualize the margins of tumors, which is greatly helpful for imaging-guided surgical operations. 102 exhibited synchronous photodynamic and photothermal therapy effects on multiple cancer cells by targeting mitochondria (Fig. 17). The theranostic agent preferentially accumulated in tumor lesions via organic-anion transporting polypeptide (OATP)-mediated active transport and was retained in mitochondria because of its lipophilic cationic properties.
 | ||
| Fig. 17 Theranostics based on NIR dyes. | ||
Shi et al. developed a NIR therapeutic autophagy-enhancer (103) to kill tumor cells.265103 preferentially accumulated in the mitochondria of tumor cells in a glycolysis-dependent and organic anion transporter polypeptide-dependent manner. 103 killed tumor cells via inducing excessive autophagy both in vitro and in vivo, which is mediated through the reactive oxygen species (ROS)-Akt-mammalian target of the rapamycin (mTOR) pathway. Translocase of inner mitochondrial membrane 44 (TIM44), a novel autophagy-related gene, correlated positively with CRC development and prognosis. Downregulation of TIM44 resulted in the induction of autophagy and cell death through the TIM44-SOD2-ROS-mTOR pathway.
Mitochondria-bound monoamine oxidase A (MAOA) can catalyze the degradation of monoamine neurotransmitters and dietary amines. High MAOA levels are associated with prostate cancer progression and poor prognosis for patients.266 Shih et al. demonstrated a tumor-targeting NIR hemicyanine–MAOA inhibitor conjugate (104) as a theranostic agent for the treatment and diagnosis of prostate cancer.266 The fluorophore of this theranostic agent could be taken up and localized in tumor cells. The MAOA-targeting moiety was derived from the MAOA inhibitor clorgyline. The theranostic agent 104 accumulated in human PCa LNCaP cells and localized in the mitochondria to inhibit the activity of overexpressed MAOA. In vivo and ex vivo imaging of tumor xenografted mice demonstrated that 104 possessed tumor-targeting properties and antitumor efficacy.
The delivery of diagnostic and therapeutic agents for detection and therapy of brain tumors is hindered by the blood brain barrier (BBB) and the blood tumor barrier (BTB) between brain tumor cells and microvessels. Chung et al. synthesized an IR-783–gemcitabine conjugate 105 as an imaging and therapeutic agent for the detection and treatment of human brain tumors and brain metastases.267 The targeting mechanism was mediated by activation of the tumor hypoxia-inducible factor 1α/OATP signaling pathway. Conjugate 105 could penetrate the BBB/BTB and accumulate in intracranial human tumor and brain tumor metastases of a human prostate tumor model within 24 h.
3.9 Metal complex-based theranostics
Metal complexes are widely used in antitumor theranostic agents because of their favorable pharmacological profiles in vitro and in vivo. Ruthenium (Ru) (III) complexes have been identified as leading candidates due to their good photochemical and pharmacological properties. Several Ru(III) complexes, such as NAMI-A, ([Him][trans-RuCl4(DMSO)(im)], im = imidazole), KP1019, (146-[trans-RuCl4(ind)2], ind = indazole), and its Na+ analogue (KP1339), have been developed. NAMI-A and KP1339 have successfully entered clinical trials.268Spiccia et al. demonstrated the use of coordinatively saturated and substitutionally inert polypyridyl Ru(II) compounds 106a–d as theranostic drugs.269 The derivatives 106c and 106d exhibited enhanced cytotoxicity toward all six cell lines tested: human cervical tumor HeLa, breast carcinoma MCF7, osteosarcoma U2OS, ovarian carcinoma A2780, and cisplatin-resistant ovarian carcinoma A2780–CP70 cells. The complex 106c was mainly targeted to mitochondria, whereas 106d was primarily located in the outer cellular membrane. The anticancer activity of 106c was similar to that of cisplatin. 106c exerted its toxicity through a mitochondria-related pathway. The mitochondrial membrane potential of tumor cells was impaired as early as 2 h after the introduction of 106c.
Mao et al. designed three fluorescent Ru(II)–polypyridyl complexes 107a–c targeting histone deacetylase inhibitors (HDACs) as antitumor agents.270 These antitumor agents showed relatively high quantum yields, large stokes shifts, and long emission life times. The fluorescence regulation mechanism was a metal-to-ligand charge transfer process. Treatment of HeLa cells with complex 107c significantly increased the acetylation of histone H3 in a dose- and time-dependent manner. Confocal microscopy images illustrated that 107c accumulated in the nuclei. Cell apoptosis induced by 107c was determined to involve mitochondrial dysfunction and the production of reactive oxygen species. The compounds 108a–c based on cyclometalated iridium(III) complexes, which included bis-N-heterocyclic carbene ligands as photodynamic and mitochondrial targeting groups, were also reported as theranostic agents.271 All complexes were effectively localized in mitochondria and displayed higher cytotoxicity than cisplatin against all tested human tumor cell lines. The order of complexes for active transport with uptake was 108c > 108b > 108a. The mechanism for apoptosis was mitochondrial damage caused by overproduction of reactive oxygen species (Fig. 18).
 | ||
| Fig. 18 Theranostics based on metal complexes and aptamers. | ||
Farrell et al. presented Ru complexes 109a–c and 110a,b, which showed high cellular and uptake antitumor activity.272 Ru complex 109a showed high cytotoxicity to HCT116 p53+/+ cells and HCT116 p53−/− cells with IC50 values of 0.1 and 0.7 μM, respectively, which indicated that the hydrogen bond of the imidazole units strongly affected antitumor activity. Fluorescence imaging indicated that 109a and 109b localized in the cytoplasm and in the nucleus, respectively. Flow cytometry and western blotting indicated that complex 109a inhibited the G1 and S phases of the cell cycle.
Che et al. developed a class of antitumor Au(III) complexes containing N-heterocyclic carbene and 2,6-bis(imidazol-2-yl)pyridine or 2,6-bis(benzimidazol-2-yl)pyridine ligands (111a–g, 112a–c).273 The biothiols reduced the Au(III) complexes to the Au(I) state with release of the fluorescent ligand. These Au(III) complexes could suppress tumor growth in mice bearing HeLa xenografts. 112a was easily taken up by cells and localized in the mitochondria of HeLa cells. 111e exhibited significant suppression of tumor volume in vivo.
3.10 Aptamer-based theranostics
Aptamers are structured single-stranded DNA or RNA molecules that have a high affinity for small molecules, peptides, proteins, and oligosaccharides. Therefore, aptamers have attracted great attention because of their potential application for therapeutic or diagnostic targeted-delivery.274–279Jon et al. reported an A10 RNA aptamer–doxorubicin conjugate (113) for the targeted delivery of doxorubicin to tumor cells.280 The conjugate could bind to the PSMA protein on the surface of prostate cancer cells. The anticancer drug intercalated into the CG sequence of the A10 aptamer following maximal quenching of the fluorescence of doxorubicin at an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 molar equivalence of doxorubicin to the aptamer. Microscopy and flow cytometry studies showed that conjugate 113 could preferentially accumulate in PSMA-expressing LNCaP prostate epithelial cells compared with PSMA-negative PC3 prostate epithelial cells.
:1.2 molar equivalence of doxorubicin to the aptamer. Microscopy and flow cytometry studies showed that conjugate 113 could preferentially accumulate in PSMA-expressing LNCaP prostate epithelial cells compared with PSMA-negative PC3 prostate epithelial cells.
4. Challenges and perspectives for optical chemical probes
This review focuses on small molecular ligand-targeted fluorescent imaging probes and fluorescent theranostics for tumor diagnosis and therapy. It does not include nanoparticles, polymers, or liposomes, even though small molecular targeting groups and/or cytotoxic drugs have been attached and loaded to these transport carriers. The general tumor treatment methods include surgery, radiotherapy and chemotherapy. However, surgery is the preferred modality for the treatment of most solid tumors. The primary challenge for surgical intervention is how to distinguish the surgical margin between the lesion and its healthy surrounding tissue at the cellular level. Fluorescence bioimaging technologies for delimiting the surgical margin are able to precisely discern diseased tissues in patients. With current fluorescence imaging instruments, fluorescent imaging probes can be exploited for intraoperative decisions concerning which tissues need to be resected and which tissues need to be preserved during surgery. Successful surgical fluorescent navigation for tumor lesions is achieved by significantly minimizing the background and maximizing the target-to-background ratio to increase contrast. The general requirements and objectives of imaging probes are designed to achieve optimal visualization.In general, there are two approaches for targeting imaging probes to tumor lesions (Fig. 1 and 19). Passive targeting benefits from the enhanced permeability and retention (EPR) effect, in which the defects or gaps in the tumor vasculature are typically 200 to 2000 nm in diameter. The EPR effect allows the extravasation and accumulation of both small molecules and larger particles (mainly with diameters <600 nm). Small molecules can easily diffuse back into the blood circulation by way of the defects or gaps, but larger particles (>4 nm or 30 kDa) are retained in solid tumors. This effect results in the preferential accumulation of larger particles around tumors. However, deep penetration of larger particles is hampered by the dense tumor interstitial space. This tumor backpressure strongly misrepresents diffusion patterns of larger particles in solid tumors. Obstacles to tumor penetration originating from the tumor backpressure should be overcome, as small molecules can be released and integrated into the tumor independently. This strategy belongs to the second category of active targeting, which requires the involvement of targeting moieties such as ligands, peptides, proteins, and antibodies. These targeting moieties are used to implement the identifying features (i.e., tumor-specific receptors) that uniquely exist on the surface of tumor cells, such as proteins, sugars, and lipids. That is why the design focus for active targeting is seeking the right targeting moiety. These imaging probes are often represented as targeting moiety–spacer–fluorophore conjugates (Fig. 20). As the overall design approach, we first investigate the tumor-specific receptor that is the binding site of the imaging probes. To effectively capture the targeting moiety, the desirable receptor must meet the following criteria. First, the receptor is overexpressed on cancer cells. Relative to normal cells, the absolute level of the expressed receptor on cancer cells must be at least 3-fold higher. Second, the candidate receptor is fixed on the cell surface and should not be cleaved or shed into the circulation. Here, receptor-mediated endocytosis does not need to be considered. The critical need is a fluorescent imaging probe that accurately labels the tumor lesions. The candidate receptor is required to have a high affinity for the imaging probe, as this is a major contributing factor to the target-to-background ratio, tumor accumulation, and residence time. Any unbound imaging probe must be rapidly cleared by the circulation. If the targeted receptor is overexpressed within the cytoplasm or nucleus, the imaging conjugate should be designed to be nonspecifically permeable across cell membranes. The design of the targeting moiety must sufficiently take into account the affinity, specificity, molecular size, and functional-group modifiability. The dissociation constant (Kd) of the targeting moiety for its receptor is suggested to be lower than 10 nM. The spacer between the targeting moiety and the fluorophore via the derivatizable functional group must not severely reduce the affinity. The derivatizable functional group of the spacer is required to be chemically stable. Removal of the unbound imaging probe is controlled by the diffusion and clearance rate from the circulation. A NIR fluorophore is preferred for the selection of the fluorophore, in part because the low biological background is conducive to bioimaging. To date, fluorescent imaging probes that are applied to assist intraoperative dissection have been untargeted fluorescent dyes including methylene blue, indocyanine green, fluorescein, and 5-aminolevulinic acid. Although numerous targeting moiety–fluorophore conjugates have been reported only a few are in the process of clinical translation, such as IRDye 800CW,281 ZW800-1,282 EC17,283 and OTL-0038.95 In terms of increasing the target-to-background ratio, an activatable imaging probe is better than an active imaging probe due to the controllable change in fluorescence.
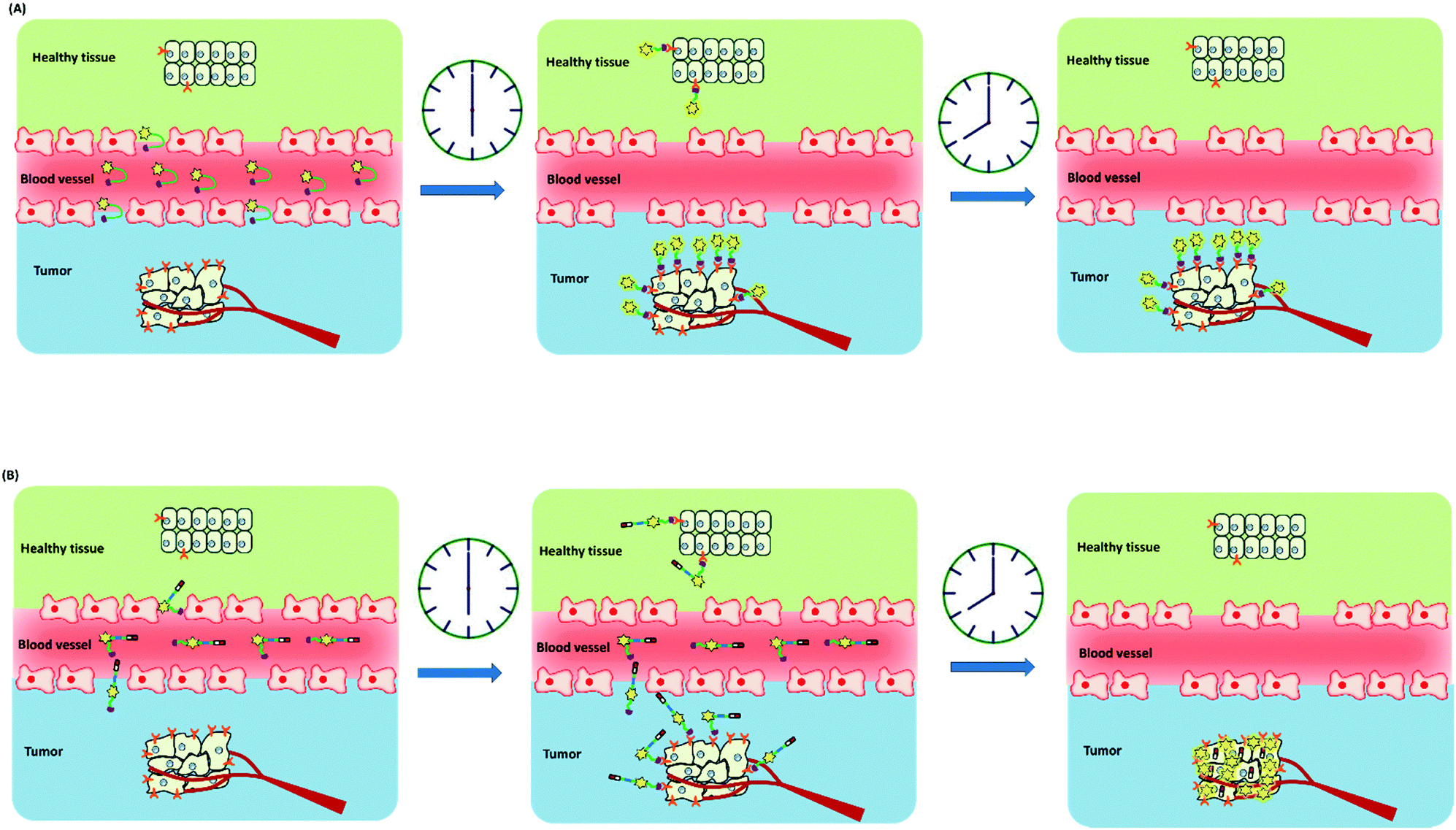 | ||
| Fig. 19 Schematic of the in vivo imaging process using an imaging fluorescent probe (A) and fluorescent theranostics (B). | ||
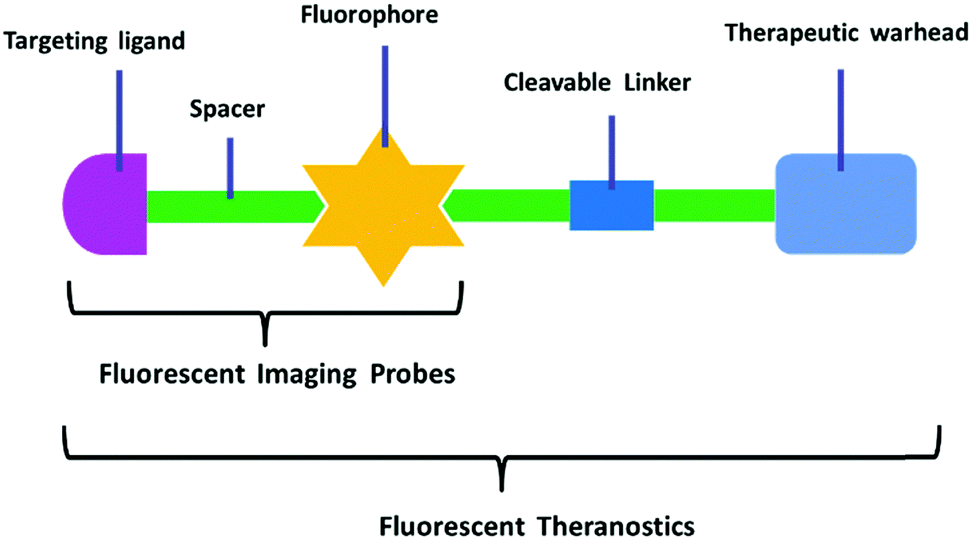 | ||
| Fig. 20 Schematic of the design strategy for fluorescent imaging probes and fluorescent theranostics. | ||
Anticancer drugs have made chemotherapy an indispensable therapeutic intervention for inhibiting the rapid growth of tumor cells. Chemotherapeutic drugs have achieved great advances, but their non-specific distribution in normal and tumor cells leads to low efficacy and severe side effects. Additionally, drug resistance is another factor that should be considered. The mechanisms of drug resistance involve sub-therapeutic concentrations and upregulation of efflux transporters and metabolizing enzymes. In order to improve the effectiveness of cancer treatment, it is essential to develop new strategies for the effective delivery of chemotherapeutic drugs to cancer cells. It seems that the targeted delivery of drugs can improve pharmacokinetics and increase efficacy. Passive targeting employs polymeric structures as carriers, such as nanoparticles, polymers, or liposomes.284–291 This strategy is highly dependent on the enhanced permeability and retention effect. The direct conjugation of a chemotherapeutic drug to a certain tumor cell targeting ligand represents active targeting. The structure of this kind of drug delivery system generally includes a targeting moiety linked to a potent chemotherapeutic drug via a linker that embodies a cleavable bond.292 The above-mentioned targeted delivery strategies have provided great advantages over traditional non-targeted chemotherapeutic drugs.292 However, the progression and treatment of tumors involves many complicated stages including initiation, progression, metastasis, recurrence, and resistance to therapy. A precise diagnosis is indispensable for tumor staging and early therapy. To meet this demand, diagnostics and therapeutics are being integrated to provide promising tools for tumor diagnosis and therapy (Fig. 19). This newly emerging combined system has been termed theranostics. Small-molecule fluorescent theranostics are conceived and synthesized as three or four moieties. A targeting moiety is covalently labeled with a fluorophore via a spacer, and then a chemotherapeutic drug is introduced into the molecular structure through a cleavable linker. This can be represented as a targeting moiety–spacer–fluorophore–linker–drug (Fig. 20). For the design of small-molecule fluorescent theranostics, the issues to be considered combine the requirements of both diagnostics and therapeutics. However, one must also pay attention to receptor-mediated endocytosis. The candidate receptors on the cell surface must be sufficient to meet the precise delivery of theranostics into tumor tissues. The theranostics are intended to be retained within the tumor cells and achieve the desired steady-state concentrations. Another focal point is the manipulation of drug release at tumor lesions because non-targeted drug release will result in systemic toxicity. Structural constraints on the introduction of chemotherapeutic drugs are often imposed by the linkers therefore the design of cleavable linkers using tumor factors is a key element for drug triggering release. The tumor physiological microenvironment is different from that of normal tissues. Insufficient and defective vascular structures of the tumors result in hypoxia, low pH, and high interstitial fluid pressure. Some other unique characteristics of tumors, such as high levels of reactive oxygen species, high concentrations of glutathione, as well as overexpressed enzymes, also act as advantageous stimulants. External stimulators, such as light and heat, are good releasing triggers. It is worth noting that no residues of the linker can remain in the released chemotherapeutic drug. In terms of the design requirements of the overall small-molecule fluorescent theranostic agents, they are expected to have low cytotoxicity, excellent pharmacokinetics, and chemical stability before reaching their destination. At the same time, we require the linker to play a role in regulation of the fluorescence changes that function as non-invasive, real-time, in situ, and direct detection signals.
Theranostics have been developed to target the primary tumor and metastases for early diagnosis and therapy. After being specifically activated by tumor factors, the cytotoxic drugs are released to inhibit the growth of tumor cells. High accumulation in tumor lesions allows cytotoxic drugs to act as effectively as possible. These expected effects allow patients to be administered lower doses, thus reducing overall systemic toxicity. Fluorescence signals provide an imaging modality that can both address the tumor lesions (by overexpressed receptors) and directly indicate the administered drugs (for distribution and accumulation). Fluorescence imaging of theranostics can allow visual evaluation and normalization of the efficacies of targeted drugs at tumor lesions. The results facilitate drug development by offering more information on the tumor stage, targeting ability, pharmacokinetics, and dosage. To date, there are no small-molecule fluorescent theranostics in clinical trials.
Imaging technologies are essential for the detection and visualization of tumor lesions. Current clinical imaging modalities include positron emission tomography (PET), single photon emission computed tomography (SPECT), magnetic resonance imaging/spectroscopy (MRI/MRS), computed tomography (CT), and ultrasound (US). Most of these have no depth sensitivity limit, but they cannot provide a real-time response for intraoperative diagnosis. Fluorescence imaging technology is expected to overcome some of the limitations of conventional therapeutics and improve tumor treatments. For fluorescence imaging tools, excellent selectivity and high sensitivity are the key parameters for visualizing physiological and pathological factors at a molecular level. These tools are also required to have sufficient spatial resolution for in vivo imaging. Unfortunately, the narrow tissue penetration of fluorescence has become the main obstacle to deep body scanning. Therefore, fluorescence imaging is performed at different spatial and temporal resolutions, ranging from micrometers (<5 mm) to centimeters (<10 cm).48 Which imaging modality is chosen depends on the purpose of the examination and the site of the tumor lesion. To better understand the comprehensive nature of the tumor, development of this field has advanced toward combining fluorescence imaging with other radioactive imaging modalities, as well as intraoperative ultrasonography (termed as photoacoustic imaging). This strategy is expected to coordinate the individual benefits of each imaging technology, and thus overcome the limitations. In addition, attention must be paid to the cost and regulatory requirements for imaging reagents.
5. Conclusions
Cancer has been identified as a dominant cause of death. Surgery and chemotherapy drugs play vital roles in the treatment of tumors. Fluorescence imaging provides an exceptional approach to imaging and quantification of cellular dynamic events that have an enormous impact on the tumor. Fluorescence imaging is achieved by maximizing the target-to-background ratio. The availability of fluorescently labeled probes for targeting tumor lesions, including tumor margins, nerves, lymph nodes, and lymphatics, greatly benefit intraoperative diagnosis. Such small molecular fluorescent probe-based therapeutic systems, composed of an antitumor drug, an imaging unit, and a tumor targeting ligand linked by a cleavable bond, show high accumulation at tumor cells and low toxicity to normal cells. The therapeutic agents are responsive to physiological factors, such as GSH, H2O2, enzymes, and pH, and to external stimulators such as phototriggers to specifically release the incorporated prodrug. Such agents are not only used to examine the necessary dose and timing and the pharmacokinetics of chemotherapy drugs, but are also applied in real time to indicate the biodistribution and efficacy of chemotherapy drugs. Future generations of smart fluorescent imaging and theranostic agents should incorporate a specific tumor-targeting unit and cleavable bond to achieve enhanced cellular selectivity and targeted drug release. The ultimate goal of tumor therapy is to improve the quality of life and prolong the survival time.Acknowledgements
We thank the National Nature Science Foundation of China (grant numbers: 21405172, 21575159, 31470415, 81670064), the National Research Foundation of Korea (grant numbers: 2008-0061891, 2009-00426), the Korea Health Industry Development Institute (grant number: HI16C2129), the program of Youth Innovation Promotion Association, CAS (Grant 2015170), and State Key Laboratory of Environmental Chemistry and Eco-toxicology, Research Center for Eco-Environmental Scienc-es, CAS (Grant KF2016-22).References
- R. Siegel, J. M. Ma, Z. H. Zou and A. Jemal, Ca-Cancer J. Clin., 2014, 64, 9–29 CrossRef PubMed.
- J. Ferlay, H. R. Shin, F. Bray, D. Forman, C. Mathers and D. M. Parkin, Int. J. Cancer, 2010, 127, 2893–2917 CrossRef CAS PubMed.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed.
- C. A. Boswell and M. W. Brechbiel, Nucl. Med. Biol., 2007, 34, 757–778 CrossRef CAS PubMed.
- S. Salipalli, P. K. Singh and J. Borlak, BMC Cell Biol., 2014, 15, 26 CrossRef PubMed.
- I. R. Correa, Curr. Opin. Chem. Biol., 2014, 20, 36–45 CrossRef CAS PubMed.
- K. M. Dean and A. E. Palmer, Nat. Chem. Biol., 2014, 10, 512–523 CrossRef CAS PubMed.
- M. Baker, Nature, 2010, 466, 1137–1142 CrossRef CAS PubMed.
- X. Q. Chen, K. A. Lee, X. T. Ren, J. C. Ryu, G. Kim, J. H. Ryu, W. J. Lee and J. Yoon, Nat. Protoc., 2016, 11, 1219–1228 CrossRef CAS PubMed.
- F. Yu, P. Li, B. Wang and K. Han, J. Am. Chem. Soc., 2013, 135, 7674–7680 CrossRef CAS PubMed.
- M. de Jong, J. Essers and W. M. van Weerden, Nat. Rev. Cancer, 2014, 14, 481–493 CrossRef CAS PubMed.
- M. Olivo, C. J. H. Ho and C. Y. Fu, Laser Photonics Rev., 2013, 7, 646–662 CrossRef CAS.
- X. Chen, F. Wang, J. Y. Hyun, T. Wei, J. Qiang, X. Ren, I. Shin and J. Yoon, Chem. Soc. Rev., 2016, 45, 2976–3016 RSC.
- J. Li, D. Yim, W. D. Jang and J. Yoon, Chem. Soc. Rev., 2016 10.1039/c6cs00619a.
- J. Yin, Y. Hu and J. Yoon, Chem. Soc. Rev., 2015, 44, 4619–4644 RSC.
- Y. Tang, D. Lee, J. Wang, G. Li, J. Yu, W. Lin and J. Yoon, Chem. Soc. Rev., 2015, 44, 5003–5015 RSC.
- A. L. Vahrmeijer, M. Hutteman, J. R. van der Vorst, C. J. H. van de Velde and J. V. Frangioni, Nat. Rev. Clin. Oncol., 2013, 10, 507–518 CrossRef CAS PubMed.
- Y. Liu, R. Njuguna, T. Matthews, W. J. Akers, G. P. Sudlow, S. Mondal, R. Tang, V. Gruev and S. Achilefu, J. Biomed. Opt., 2013, 18, 101303 CrossRef PubMed.
- E. M. Sevick-Muraca, Annu. Rev. Med., 2012, 63, 217–231 CrossRef CAS PubMed.
- G. M. van Dam, G. Themelis, L. M. A. Crane, N. J. Harlaar, R. G. Pleijhuis, W. Kelder, A. Sarantopoulos, J. S. de Jong, H. J. G. Arts, A. G. J. van der Zee, J. Bart, P. S. Low and V. Ntziachristos, Nat. Med., 2011, 17, 1315–1319 CrossRef CAS PubMed.
- Q. T. Nguyen, E. S. Olson, T. A. Aguilera, T. Jiang, M. Scadeng, L. G. Ellies and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4317–4322 CrossRef CAS PubMed.
- M. A. Pysz, S. S. Gambhir and J. K. Willmann, Clin. Radiol., 2010, 65, 500–516 CrossRef CAS PubMed.
- J. V. Frangioni, Curr. Opin. Chem. Biol., 2003, 7, 626–634 CrossRef CAS PubMed.
- S. A. Hilderbrand and R. Weissleder, Curr. Opin. Chem. Biol., 2010, 14, 71–79 CrossRef CAS PubMed.
- M. Gao, R. Wang, F. B. Yu, J. M. You and L. X. Chen, Analyst, 2015, 140, 3766–3772 RSC.
- P. Liu, X. T. Jing, F. B. Yu, C. J. Lv and L. X. Chen, Analyst, 2015, 140, 4576–4583 RSC.
- X. Jing, F. Yu and L. Chen, Chem. Commun., 2014, 50, 14253–14256 RSC.
- K. Yin, F. Yu, W. Zhang and L. Chen, Biosens. Bioelectron., 2015, 74, 156–164 CrossRef CAS PubMed.
- X. Han, F. Yu, X. Song and L. Chen, Chem. Sci., 2016, 7, 5098–5107 RSC.
- J. H. Park, S. Lee, J. H. Kim, K. Park, K. Kim and I. C. Kwon, Prog. Polym. Sci., 2008, 33, 113–137 CrossRef.
- J. A. Hubbell and A. Chilkoti, Science, 2012, 337, 303–305 CrossRef PubMed.
- R. Tong and J. J. Cheng, Polym. Rev., 2007, 47, 345–381 CrossRef CAS.
- K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., Int. Ed., 2009, 48, 872–897 CrossRef CAS PubMed.
- J. V. Jokerst and S. S. Gambhir, Acc. Chem. Res., 2011, 44, 1050–1060 CrossRef CAS PubMed.
- W. Zhang, Y. Wang, X. Sun, W. Wang and L. Chen, Nanoscale, 2014, 6, 14514–14522 RSC.
- D. Lin, T. Qin, Y. Wang, X. Sun and L. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 1320–1329 CAS.
- X. Niu, H. Chen, Y. Wang, W. Wang, X. Sun and L. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 5152–5160 CAS.
- C. Vu Thanh, E. O. Ganbold, J. K. Saha, J. Jang, J. Min, J. Choo, S. Kim, N. W. Song, S. J. Son, S. B. Lee and S.-W. Joo, J. Am. Chem. Soc., 2014, 136, 3833–3841 CrossRef PubMed.
- J. W. Singer, R. Bhatt, J. Tulinsky, K. R. Buhler, E. Heasley, P. Klein and P. de Vries, J. Controlled Release, 2001, 74, 243–247 CrossRef CAS PubMed.
- P. V. Paranjpe, S. Stein and P. J. Sinko, Anti-Cancer Drugs, 2005, 16, 763–775 CrossRef CAS PubMed.
- J. J. Khandare, P. Chandna, Y. Wang, V. P. Pozharov and T. Minko, J. Pharmacol. Exp. Ther., 2006, 317, 929–937 CrossRef CAS PubMed.
- A. Nel, T. Xia, L. Mädler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- M. Garland, J. J. Yim and M. Bogyo, Cell Chem. Biol., 2016, 23, 122–136 CrossRef CAS PubMed.
- S. Luo, E. Zhang, Y. Su, T. Cheng and C. Shi, Biomaterials, 2011, 32, 7127–7138 CrossRef CAS PubMed.
- M. H. Lee, J. L. Sessler and J. S. Kim, Acc. Chem. Res., 2015, 48, 2935–2946 CrossRef CAS PubMed.
- J. H. Rao, A. Dragulescu-Andrasi, H. Q. Yao and H. Q. Yao, Curr. Opin. Biotechnol., 2007, 18, 17–25 CrossRef CAS PubMed.
- M. Rudin and R. Weissleder, Nat. Rev. Drug Discovery, 2003, 2, 123–131 CrossRef CAS PubMed.
- M. Srinivasarao, C. V. Galliford and P. S. Low, Nat. Rev. Drug Discovery, 2015, 14, 203–219 CrossRef CAS PubMed.
- R. Kumar, W. S. Shin, K. Sunwoo, W. Y. Kim, S. Koo, S. Bhuniya and J. S. Kim, Chem. Soc. Rev., 2015, 44, 6670–6683 RSC.
- J. O. Escobedo, O. Rusin, S. Lim and R. M. Strongin, Curr. Opin. Chem. Biol., 2010, 14, 64–70 CrossRef CAS PubMed.
- F. Yu, X. Han and L. Chen, Chem. Commun., 2014, 50, 12234–12249 RSC.
- J. R. van der Vorst, B. E. Schaafsma, F. P. R. Verbeek, M. Hutteman, J. S. D. Mieog, C. Lowik, G. J. Liefers, J. V. Frangioni, C. J. H. van de Velde and A. L. Vahrmeijer, Ann. Surg. Oncol., 2012, 19, 4104–4111 CrossRef PubMed.
- Y. Ashitate, C. S. Vooght, M. Hutteman, R. Oketokoun, H. S. Choi and J. V. Frangioni, Mol. Imaging, 2012, 11, 301–308 CAS.
- W. Stummer, A. Novotny, H. Stepp, C. Goetz, K. Bise and H. J. Reulen, J. Neurosurg., 2000, 93, 1003–1013 CrossRef CAS PubMed.
- G. C. Gurtner, G. E. Jones, P. C. Neligan, M. I. Newman, B. T. Phillips, J. M. Sacks and M. R. Zenn, Ann. Surg. Innov. Res., 2013, 7, 1–14 CrossRef PubMed.
- M. Gao, F. Yu, H. Chen and L. Chen, Anal. Chem., 2015, 87, 3631–3638 CrossRef CAS PubMed.
- F. Yu, M. Gao, M. Li and L. Chen, Biomaterials, 2015, 63, 93–101 CrossRef CAS PubMed.
- Y. Huang, F. Yu, J. Wang and L. Chen, Anal. Chem., 2016, 88, 4122–4129 CrossRef CAS PubMed.
- C. Zhang, S. Wang, J. Xiao, X. Tan, Y. Zhu, Y. Su, T. Cheng and C. Shi, Biomaterials, 2010, 31, 1911–1917 CrossRef CAS PubMed.
- X. Yang, C. Shi, R. Tong, W. Qian, H. E. Zhau, R. Wang, G. Zhu, J. Cheng, V. W. Yang and T. Cheng, Clin. Cancer Res., 2010, 16, 2833–2844 CrossRef CAS PubMed.
- C. Shi, J. B. Wu, G. C. Chu, Q. L. Li, R. Wang, C. Zhang, Y. Zhang, H. L. Kim, J. Wang, H. E. Zhau, D. Pan and L. W. Chung, Oncotarget, 2014, 5, 10114–10126 CrossRef PubMed.
- G. S. Hong, J. C. Lee, J. T. Robinson, U. Raaz, L. M. Xie, N. F. Huang, J. P. Cooke and H. J. Dai, Nat. Med., 2012, 18, 1841–1846 CrossRef CAS PubMed.
- G. S. Hong, J. T. Robinson, Y. J. Zhang, S. Diao, A. L. Antaris, Q. B. Wang and H. J. Dai, Angew. Chem., Int. Ed., 2012, 51, 9818–9821 CrossRef CAS PubMed.
- S. Diao, G. S. Hong, J. T. Robinson, L. Y. Jiao, A. L. Antaris, J. Z. Wu, C. L. Choi and H. J. Dai, J. Am. Chem. Soc., 2012, 134, 16971–16974 CrossRef CAS PubMed.
- A. L. Antaris, H. Chen, K. Cheng, Y. Sun, G. S. Hong, C. R. Qu, S. Diao, Z. X. Deng, X. M. Hu, B. Zhang, X. D. Zhang, O. K. Yaghi, Z. R. Alamparambil, X. C. Hong, Z. Cheng and H. J. Dai, Nat. Mater., 2016, 15, 235–242 CrossRef CAS PubMed.
- E. R. Trivedi, A. S. Harney, M. B. Olive, I. Podgorski, K. Moin, B. F. Sloane, A. G. M. Barrett, T. J. Meade and B. M. Hoffman, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1284–1288 CrossRef CAS PubMed.
- H. S. Choi, S. L. Gibbs, J. H. Lee, S. H. Kim, Y. Ashitate, F. B. Liu, H. Hyun, G. Park, Y. Xie, S. Bae, M. Henary and J. V. Frangioni, Nat. Biotechnol., 2013, 31, 148–153 CrossRef CAS PubMed.
- M. H. Park, H. Hyun, Y. Ashitate, H. Wada, G. Park, J. H. Lee, C. Njiojob, M. Henary, J. V. Frangioni and H. S. Choi, Theranostics, 2014, 4, 823–833 CrossRef PubMed.
- M. Kawakami and J. Nakayama, Cancer Res., 1997, 57, 2321–2324 CAS.
- R. E. Carter, A. R. Feldman and J. T. Coyle, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 749–753 CrossRef CAS.
- W. T. Tino, M. J. Huber, T. P. Lake, T. G. Greene, G. P. Murphy and E. H. Holmes, Hybridoma, 2000, 19, 249–257 CrossRef CAS PubMed.
- S. E. Lupold, B. J. Hicke, Y. Lin and D. S. Coffey, Cancer Res., 2002, 62, 4029–4033 CAS.
- S. E. Lupold and R. Rodriguez, Mol. Cancer Ther., 2004, 3, 597–603 CAS.
- C. Liu, H. N. Huang, F. Donate, C. Dickinson, R. Santucci, A. El-Sheikh, R. Vessella and T. S. Edgington, Cancer Res., 2002, 62, 5470–5475 CAS.
- M. G. Pomper, J. L. Musachio, J. Zhang, U. Scheffel, Y. Zhou, J. Hilton, A. Maini, R. F. Dannals, D. F. Wong and A. P. Kozikowski, Mol. Imaging, 2002, 1, 96–101 CrossRef CAS PubMed.
- H. Tang, M. Brown, Y. Ye, G. Huang, Y. Zhang, Y. Wang, H. Zhai, X. Chen, T. Y. Shen and M. Tenniswood, Biochem. Biophys. Res. Commun., 2003, 307, 8–14 CrossRef CAS PubMed.
- V. Humblet, R. Lapidus, L. R. Williams, T. Tsukamoto, C. Rojas, P. Majer, B. Hin, S. Ohnishi, A. M. De Grand and A. Zaheer, Mol. Imaging, 2005, 4, 448 Search PubMed.
- T. Liu, L. Wu, M. Kazak and C. E. Berkman, Prostate, 2008, 68, 955–964 CrossRef CAS PubMed.
- T. Liu, L. Wu, M. R. Hopkins, J. K. Choi and C. E. Berkman, Bioorg. Med. Chem. Lett., 2010, 20, 7124–7126 CrossRef CAS PubMed.
- Y. Chen, S. Dhara, S. R. Banerjee, Y. Byun, M. Pullambhatla, R. C. Mease and M. G. Pomper, Biochem. Biophys. Res. Commun., 2009, 390, 624–629 CrossRef CAS PubMed.
- S. Jayaprakash, X. Wang, W. D. Heston and A. P. Kozikowski, ChemMedChem, 2006, 1, 299–302 CrossRef CAS PubMed.
- C. E. Eberhart, R. J. Coffey, A. Radhika, F. M. Giardiello, S. Ferrenbach and R. N. Dubois, Gastroenterology, 1994, 107, 1183–1188 CrossRef CAS.
- M. J. Uddin, B. C. Crews, A. L. Blobaum, P. J. Kingsley, D. L. Gorden, J. O. McIntyre, L. M. Matrisian, K. Subbaramaiah, A. J. Dannenberg, D. W. Piston and L. J. Marnett, Cancer Res., 2010, 70, 3618–3627 CrossRef CAS PubMed.
- M. J. Uddin, B. C. Crews, K. Ghebreselasie and L. J. Marnett, Bioconjugate Chem., 2013, 24, 712–723 CrossRef CAS PubMed.
- M. J. Uddin, B. C. Crews, I. Huda, K. Ghebreselasie, C. K. Daniel and L. J. Marnett, ACS Med. Chem. Lett., 2014, 5, 446–450 CrossRef CAS PubMed.
- M. J. Uddin, B. C. Crews, K. Ghebreselasie, C. K. Daniel, P. J. Kingsley, S. Xu and L. J. Marnett, J. Biomed. Opt., 2015, 20, 50502 CrossRef PubMed.
- H. Zhang, J. Fan, J. Wang, S. Zhang, B. Dou and X. Peng, J. Am. Chem. Soc., 2013, 135, 11663–11669 CrossRef CAS PubMed.
- H. Zhang, J. Fan, J. Wang, B. Dou, F. Zhou, J. Cao, J. Qu, Z. Cao, W. Zhao and X. Peng, J. Am. Chem. Soc., 2013, 135, 17469–17475 CrossRef CAS PubMed.
- H. Zhang, J. L. Fan, K. Wang, J. Li, C. X. Wang, Y. M. Nie, T. Jiang, H. Y. Mu, X. J. Peng and K. Jiang, Anal. Chem., 2014, 86, 9131–9138 CrossRef CAS PubMed.
- B. Wang, J. Fan, X. Wang, H. Zhu, J. Wang, H. Mu and X. Peng, Chem. Commun., 2015, 51, 792–795 RSC.
- W. Xia and P. S. Low, J. Med. Chem., 2010, 53, 6811–6824 CrossRef CAS PubMed.
- M. D. Kennedy, K. N. Jallad, D. H. Thompson, D. Ben-Amotz and P. S. Low, J. Biomed. Opt., 2003, 8, 636–641 CrossRef PubMed.
- M. D. Kennedy, K. N. Jallad, J. Lu, P. S. Low and D. Ben-Amotz, Pharm. Res., 2003, 20, 714–719 CrossRef CAS.
- S. M. Mahalingam, S. A. Kularatne, J. Roy and P. S. Low, Abstracts of Papers of the American Chemical Society, 2013, vol. 246 Search PubMed.
- J. Yang, H. Chen, I. R. Vlahov, J. X. Cheng and P. S. Low, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13872–13877 CrossRef CAS PubMed.
- H. Lee, J. Kim, H. Kim, Y. Kim and Y. Choi, Chem. Commun., 2014, 50, 7507–7510 RSC.
- J. Levi, Z. Cheng, O. Gheysens, M. Patel, C. T. Chan, Y. B. Wang, M. Namavari and S. S. Gambhir, Bioconjugate Chem., 2007, 18, 628–634 CrossRef CAS PubMed.
- S. P. ZamoraLeon, D. W. Golde, I. I. Concha, C. I. Rivas, F. DelgadoLopez, J. Baselga, F. Nualart and J. C. Vera, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 15522 Search PubMed.
- K. Savage, H. A. Waller, M. Stubbs, K. Khan, S. A. Watson, P. A. Clarke, S. Grimes, D. Michaeli, A. P. Dhillon and M. E. Caplin, Int. J. Oncol., 2006, 29, 1429–1435 CAS.
- D. S. Weinberg, B. Ruggeri, M. T. Barber, S. Biswas, S. Miknyocki and S. A. Waldman, J. Clin. Invest., 1997, 100, 597–603 CrossRef CAS PubMed.
- J. C. Reubi and B. Waser, Int. J. Cancer, 1996, 67, 644–647 CrossRef CAS PubMed.
- T. Sethi, T. Herget, S. V. Wu, J. H. Walsh and E. Rozengurt, Cancer Res., 1993, 53, 5208–5213 CAS.
- J. C. Reubi, J. C. Schaer and B. Waser, Cancer Res., 1997, 57, 1377–1386 CAS.
- K. Hur, M. K. Kwak, H. J. Lee, D. J. Park, H. Lee, H. S. Lee, W. H. Kim, D. Michaeli and H. K. Yang, J. Cancer Res. Clin. Oncol., 2006, 132, 85–91 CrossRef CAS PubMed.
- J. Zhou, M. Chen, Q. Zhang, J. Hu and W. Wang, Recept. Channels, 2004, 10, 185–188 CrossRef CAS PubMed.
- C. Wayua and P. S. Low, Mol. Pharmaceutics, 2014, 11, 468–476 CrossRef CAS PubMed.
- S. Kossatz, M. Behe, R. Mansi, D. Saur, P. Czerney, W. A. Kaiser and I. Hilger, Biomaterials, 2013, 34, 5172–5180 CrossRef CAS PubMed.
- M. A. Proescholdt, M. J. Merrill, E. M. Stoerr, A. Lohmeier, F. Pohl and A. Brawanski, Neuro-Oncology, 2012, 14, 1357–1366 CrossRef CAS PubMed.
- A. M. Niemela, P. Hynninen, J. P. Mecklin, T. Kuopio, A. Kokko, L. Aaltonen, A. K. Parkkila, S. Pastorekova, J. Pastorek, A. Waheed, W. S. Sly, T. F. Orntoft, M. Kruhoffer, H. Haapasalo, S. Parkkila and A. J. Kivella, Cancer Epidemiol., Biomarkers Prev., 2007, 16, 1760–1766 CAS.
- S. A. Hussain, R. Ganesan, G. Reynolds, L. Gross, A. Stevens, J. Pastorek, P. G. Murray, B. Perunovic, M. S. Anwar, L. Billingham, N. D. James, D. Spooner, C. J. Poole, D. W. Rea and D. H. Palmer, Br. J. Cancer, 2007, 96, 104–109 CrossRef CAS PubMed.
- N. Krall, F. Pretto, W. Decurtins, G. J. L. Bernardes, C. T. Supuran and D. Neri, Angew. Chem., Int. Ed., 2014, 53, 4231–4235 CrossRef CAS PubMed.
- N. Krall, F. Pretto and D. Neri, Chem. Sci., 2014, 5, 3640–3644 RSC.
- G. M. Thurber, K. S. Yang, T. Reiner, R. H. Kohler, P. Sorger, T. Mitchison and R. Weissleder, Nat. Commun., 2013, 4, 1504 CrossRef PubMed.
- G. M. Thurber, T. Reiner, K. S. Yang, R. H. Kohler and R. Weissleder, Mol. Cancer Ther., 2014, 13, 986–995 CrossRef CAS PubMed.
- C. P. Irwin, Y. Portorreal, C. Brand, Y. C. Zhang, P. Desai, B. Salinas, W. A. Weber and T. Reiner, Neoplasia, 2014, 16, 432–440 CrossRef CAS PubMed.
- R. J. Bernacki and U. Kim, Science, 1977, 195, 577–580 CAS.
- J. Dennis, C. Waller, R. Timpl and V. Schirrmacher, Nature, 1982, 300, 274–276 CrossRef CAS PubMed.
- X. Wu, Y. Tian, M. Yu, B. Lin, J. Han and S. Han, Biomater. Sci., 2014, 2, 1120–1127 RSC.
- X. Wu, M. Yu, B. Lin, H. Xing, J. Han and S. Han, Chem. Sci., 2015, 6, 798–803 RSC.
- R. Andreesen, M. Modolell, H. U. Weltzien, H. Eibl, H. H. Common, G. W. Lohr and P. G. Munder, Cancer Res., 1978, 38, 3894–3899 CAS.
- W. J. van Blitterswijk and M. Verheij, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2013, 1831, 663–674 CrossRef CAS PubMed.
- J. P. Weichert, P. A. Clark, I. K. Kandela, A. M. Vaccaro, W. Clarke, M. A. Longino, A. N. Pinchuk, M. Farhoud, K. I. Swanson, J. M. Floberg, J. Grudzinski, B. Titz, A. M. Traynor, H. E. Chen, L. T. Hall, C. J. Pazoles, P. J. Pickhardt and J. S. Kuo, Sci. Transl. Med., 2014, 6, 240ra75 CrossRef PubMed.
- K. I. Swanson, P. A. Clark, R. R. Zhang, I. K. Kandela, M. Farhoud, J. P. Weichert and J. S. Kuo, Neurosurgery, 2015, 76, 115–123 CrossRef PubMed.
- M. Stefanidakis and E. Koivunen, Curr. Pharm. Des., 2004, 10, 3033–3044 CrossRef CAS PubMed.
- Y. B. Kang, W. He, S. Tulley, G. P. Gupta, I. Serganova, C. R. Chen, K. Manova-Todorova, R. Blasberg, W. L. Gerald and J. Massague, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13909–13914 CrossRef CAS PubMed.
- W. Wang, S. Ke, S. Kwon, S. Yallampalli, A. G. Cameron, K. E. Adams, M. E. Mawad and E. M. Sevick-Muraca, Bioconjugate Chem., 2007, 18, 397–402 CrossRef CAS PubMed.
- R. O. Hynes, Cell, 1992, 69, 11–25 CrossRef CAS PubMed.
- R. Seftor, E. A. Seftor, K. R. Gehlsen, W. G. Stetler-Stevenson, P. D. Brown, E. Ruoslahti and M. Hendrix, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 1557–1561 CrossRef CAS.
- C. L. Gladson and D. Cheresh, J. Clin. Invest., 1991, 88, 1924 CrossRef CAS PubMed.
- K. R. Gehlsen, G. E. Davis and P. Sriramarao, Clin. Exp. Metastasis, 1992, 10, 111–120 CrossRef CAS PubMed.
- E. J. Filardo, P. C. Brooks, S. L. Deming, C. Damsky and D. A. Cheresh, J. Cell Biol., 1995, 130, 441–450 CrossRef CAS PubMed.
- W. Wang, S. Ke, Q. Wu, C. Charnsangavej, M. Gurfinkel, J. G. Gelovani, J. L. Abbruzzese, E. M. Sevick-Muraca and C. Li, Mol. Imaging, 2004, 3, 343–351 CrossRef CAS PubMed.
- S. Kwon, S. Ke, J. P. Houston, W. Wang, Q. Wu, C. Li and E. M. Sevick-Muraca, Mol. Imaging, 2005, 4, 75–87 Search PubMed.
- R. M. Huang, J. Vider, J. L. Kovar, D. M. Olive, I. K. Mellinghoff, P. Mayer-Kuckuk, M. F. Kircher and R. G. Blasberg, Clin. Cancer Res., 2012, 18, 5731–5740 CrossRef CAS PubMed.
- X. Chen, P. S. Conti and R. A. Moats, Cancer Res., 2004, 64, 8009–8014 CrossRef CAS PubMed.
- Z. Cheng, Y. Wu, Z. Xiong, S. S. Gambhir and X. Chen, Bioconjugate Chem., 2005, 16, 1433–1441 CrossRef CAS PubMed.
- E. Garanger, D. Boturyn, Z. H. Jin, P. Dumy, M. C. Favrot and J. L. Coll, Mol. Ther., 2005, 12, 1168–1175 CrossRef CAS PubMed.
- Z. H. Jin, V. Josserand, J. Razkin, E. Garanger, D. Boturyn, M. C. Favrot, P. Dumy and J. L. Coll, Mol. Imaging, 2006, 5, 188–197 Search PubMed.
- Z. H. Jin, V. Josserand, S. Foillard, D. Boturyn, P. Dumy, M. C. Favrot and J. L. Coll, Mol. Cancer, 2007, 6, 41 CrossRef PubMed.
- S. Lanzardo, L. Conti, C. Brioschi, M. P. Bartolomeo, D. Arosio, L. Belvisi, L. Manzoni, A. Maiocchi, F. Maisano and G. Forni, Contrast Media Mol. Imaging, 2011, 6, 449–458 CrossRef CAS PubMed.
- F. Li, J. Liu, G. S. Jas, J. Zhang, G. Qin, J. Xing, C. Cotes, H. Zhao, X. Wang, L. A. Diaz, Z. Shi, D. Y. Lee, K. C. Li and Z. Li, Bioconjugate Chem., 2010, 21, 270–278 CrossRef CAS PubMed.
- W. Pham, Z. Medarova and A. Moore, Bioconjugate Chem., 2005, 16, 735–740 CrossRef CAS PubMed.
- R. Hussain, N. Courtenay-Luck and G. Siligardi, Biomed. Pept., Proteins Nucleic Acids, 1995, 2, 67–70 Search PubMed.
- R. S. Herbst, Int. J. Radiat. Oncol., Biol., Phys., 2004, 59, 21–26 CrossRef CAS PubMed.
- H. Modjtahedi and C. Dean, Int. J. Oncol., 1994, 4, 277–296 CAS.
- E. Chung, J. Lee, J. Yu, S. Lee, J. H. Kang, I. Y. Chung and J. Choo, Biosens. Bioelectron., 2014, 60, 358–365 CrossRef CAS PubMed.
- S. Ke, X. Wen, M. Gurfinkel, C. Charnsangavej, S. Wallace, E. M. Sevick-Muraca and C. Li, Cancer Res., 2003, 63, 7870–7875 CAS.
- R. S. Agnes, A. M. Broome, J. Wang, A. Verma, K. Lavik and J. P. Basilion, Mol. Cancer Ther., 2012, 11, 2202–2211 CrossRef CAS PubMed.
- A. M. Burgoyne, J. M. Palomo, P. J. Phillips-Mason, S. M. Burden-Gulley, D. L. Major, A. Zaremba, S. Robinson, A. E. Sloan, M. A. Vogelbaum, R. H. Miller and S. M. Brady-Kalnay, Neuro-Oncology, 2009, 11, 767–778 CrossRef CAS PubMed.
- S. M. Burden-Gulley, M. Q. Qutaish, K. E. Sullivant, M. Q. Tan, S. E. L. Craig, J. P. Basilion, Z. R. Lu, D. L. Wilson and S. M. Brady-Kalnay, Int. J. Cancer, 2013, 132, 1624–1632 CrossRef CAS PubMed.
- S. E. L. Craig, J. Wright, A. E. Sloan and S. M. Brady-Kalnay, World Neurosurg., 2016, 90, 154–163 CrossRef PubMed.
- L. Soroceanu, Y. Gillespie, M. B. Khazaeli and H. Sontheimer, Cancer Res., 1998, 58, 4871–4879 CAS.
- M. Veiseh, P. Gabikian, S. B. Bahrami, O. Veiseh, M. Zhang, R. C. Hackman, A. C. Ravanpay, M. R. Stroud, Y. Kusuma, S. J. Hansen, D. Kwok, N. M. Munoz, R. W. Sze, W. M. Grady, N. M. Greenberg, R. G. Ellenbogen and J. M. Olson, Cancer Res., 2007, 67, 6882–6888 CrossRef CAS PubMed.
- M. Akcan, M. R. Stroud, S. J. Hansen, R. J. Clark, N. L. Daly, D. J. Craik and J. M. Olson, J. Med. Chem., 2011, 54, 782–787 CrossRef CAS PubMed.
- J. L. Kovar, E. Curtis, S. F. Othman, M. A. Simpson and D. M. Olive, Anal. Biochem., 2013, 440, 212–219 CrossRef CAS PubMed.
- P. V. Butte, A. Mamelak, J. Parrish-Novak, D. Drazin, F. Shweikeh, P. R. Gangalum, A. Chesnokova, J. Y. Ljubimova and K. Black, Neurosurg. Focus, 2014, 36, E1 CrossRef PubMed.
- A. Walia, J. F. Yang, Y. H. Huang, M. I. Rosenblatt, J. H. Chang and D. T. Azar, Biochim. Biophys. Acta, Gen. Subj., 2015, 1850, 2422–2438 CrossRef CAS PubMed.
- D. Citrin, A. K. Lee, T. Scott, M. Sproull, C. Menard, P. J. Tofilon and K. Camphausen, Mol. Cancer Ther., 2004, 3, 481–488 CAS.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- A. Becker, B. Riefke, B. Ebert, U. Sukowski, H. Rinneberg, W. Semmler and K. Licha, Photochem. Photobiol., 2000, 72, 234–241 CrossRef CAS PubMed.
- H. I. Kim, D. Hwang, S. J. Jeon, S. Lee, J. H. Park, D. Yim, J. K. Yang, H. Kang, J. Choo, Y. S. Lee, J. Chung and J. H. Kim, Nanoscale, 2015, 7, 6363–6373 RSC.
- R. Atreya, H. Neumann, C. Neufert, M. J. Waldner, U. Billmeier, Y. Zopf, M. Willma, C. App, T. Munster, H. Kessler, S. Maas, B. Gebhardt, R. Heimke-Brinck, E. Reuter, F. Dorje, T. T. Rau, W. Uter, T. D. Wang, R. Kiesslich, M. Vieth, E. Hannappel and M. F. Neurath, Nat. Med., 2014, 20, 313–318 CrossRef CAS PubMed.
- J. A. Bonner, K. P. Raisch, H. Q. Trummell, F. Robert, R. F. Meredith, S. A. Spencer, D. J. Buchsbaum, M. N. Saleh, M. A. Stackhouse, A. F. LoBuglio, G. E. Peters, W. R. Carroll and H. W. Waksal, J. Clin. Oncol., 2000, 18, 47–53 Search PubMed.
- E. L. Rosenthal, B. D. Kulbersh, T. King, T. R. Chaudhuri and K. R. Zinn, Mol. Cancer Ther., 2007, 6, 1230–1238 CrossRef CAS PubMed.
- J. M. Warram, E. de Boer, M. Korb, Y. Hartman, J. Kovar, J. M. Markert, G. Y. Gillespie and E. L. Rosenthal, Br. J. Neurosurg., 2015, 29, 850–858 CrossRef PubMed.
- H. M. Luo, R. Hernandez, H. Hong, S. A. Graves, Y. N. Yang, C. G. England, C. P. Theuer, R. J. Nickles and W. B. Cai, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12806–12811 CrossRef CAS PubMed.
- M. Ogawa, N. Kosaka, P. L. Choyke and H. Kobayashi, Cancer Res., 2009, 69, 1268–1272 CrossRef CAS PubMed.
- T. Nakajima, M. Mitsunaga, N. H. Bander, W. D. Heston, P. L. Choyke and H. Kobayashi, Bioconjugate Chem., 2011, 22, 1700–1705 CrossRef CAS PubMed.
- K. Sano, M. Mitsunaga, T. Nakajima, P. L. Choyke and H. Kobayashi, Breast Cancer Res., 2012, 14, R61 CrossRef CAS PubMed.
- K. Sano, T. Nakajima, K. Miyazaki, Y. Ohuchi, T. Ikegami, P. L. Choyke and H. Kobayashi, Bioconjugate Chem., 2013, 24, 811–816 CrossRef CAS PubMed.
- R. Watanabe, K. Sato, H. Hanaoka, T. Harada, T. Nakajima, I. Kim, C. H. Paik, A. M. Wu, P. L. Choyke and H. Kobayashi, ACS Med. Chem. Lett., 2014, 5, 411–415 CrossRef CAS PubMed.
- K. Sato, A. P. Gorka, T. Nagaya, M. S. Michie, R. R. Nani, Y. Nakamura, V. L. Coble, O. V. Vasalatiy, R. E. Swenson, P. L. Choyke, M. J. Schnermann and H. Kobayashi, Bioconjugate Chem., 2016, 27, 404–413 CrossRef CAS PubMed.
- Z. Yang, J. Cao, Y. He, J. Yang, T. Kim, X. Peng and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4563–4601 RSC.
- Y. Liu, Y. Xu, X. Qian, Y. Xiao, J. Liu, L. Shen, J. Li and Y. Zhang, Bioorg. Med. Chem. Lett., 2006, 16, 1562–1566 CrossRef CAS PubMed.
- T. Guo, L. Cui, J. Shen, W. Zhu, Y. Xu and X. Qian, Chem. Commun., 2013, 49, 10820–10822 RSC.
- Z. Li, X. Li, X. Gao, Y. Zhang, W. Shi and H. Ma, Anal. Chem., 2013, 85, 3926–3932 CrossRef CAS PubMed.
- Z. Li, X. Gao, W. Shi, X. Li and H. Ma, Chem. Commun., 2013, 49, 5859–5861 RSC.
- Q. Wan, X. Gao, X. He, S. Chen, Y. Song, Q. Gong, X. Li and H. Ma, Chem. – Asian J., 2014, 9, 2058–2062 CrossRef CAS PubMed.
- K. Okuda, Y. Okabe, T. Kadonosono, T. Ueno, B. G. M. Youssif, S. Kizaka-Kondoh and H. Nagasawa, Bioconjugate Chem., 2012, 23, 324–329 CrossRef CAS PubMed.
- K. Xu, F. Wang, X. Pan, R. Liu, J. Ma, F. Kong and B. Tang, Chem. Commun., 2013, 49, 2554–2556 RSC.
- Y. Li, Y. Sun, J. Li, Q. Su, W. Yuan, Y. Dai, C. Han, Q. Wang, W. Feng and F. Li, J. Am. Chem. Soc., 2015, 137, 6407–6416 CrossRef CAS PubMed.
- K. Tanabe, N. Hirata, H. Harada, M. Hiraoka and S. I. Nishimoto, ChemBioChem, 2008, 9, 426–432 CrossRef CAS PubMed.
- H. Komatsu, H. Harada, K. Tanabe, M. Hiraoka and S. i. Nishimoto, MedChemComm, 2010, 1, 50–53 RSC.
- K. Kiyose, K. Hanaoka, D. Oushiki, T. Nakamura, M. Kajimura, M. Suematsu, H. Nishimatsu, T. Yamane, T. Terai and Y. Hirata, J. Am. Chem. Soc., 2010, 132, 15846–15848 CrossRef CAS PubMed.
- W. Piao, S. Tsuda, Y. Tanaka, S. Maeda, F. Liu, S. Takahashi, Y. Kushida, T. Komatsu, T. Ueno and T. Terai, Angew. Chem., Int. Ed., 2013, 52, 13028–13032 CrossRef CAS PubMed.
- H. Sies, Free Radical Biol. Med., 1999, 27, 916–921 CrossRef CAS PubMed.
- F. Yu, P. Li, P. Song, B. Wang, J. Zhao and K. Han, Chem. Commun., 2012, 48, 4980–4982 RSC.
- Z. Zheng, G. Zhu, H. Tak, E. Joseph, J. L. Eiseman and D. J. Creighton, Bioconjugate Chem., 2005, 16, 598–607 CrossRef CAS PubMed.
- L. R. Jones, E. A. Goun, R. Shinde, J. B. Rothbard, C. H. Contag and P. A. Wender, J. Am. Chem. Soc., 2006, 128, 6526–6527 CrossRef CAS PubMed.
- S. Santra, C. Kaittanis, O. J. Santiesteban and J. M. Perez, J. Am. Chem. Soc., 2011, 133, 16680–16688 CrossRef CAS PubMed.
- Z. Yang, J. H. Lee, H. M. Jeon, J. H. Han, N. Park, Y. He, H. Lee, K. S. Hong, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 11657–11662 CrossRef CAS PubMed.
- W. A. Henne, D. D. Doorneweerd, A. R. Hilgenbrink, S. A. Kularatne and P. S. Low, Bioorg. Med. Chem. Lett., 2006, 16, 5350–5355 CrossRef CAS PubMed.
- C. P. Leamon, J. A. Reddy, I. R. Vlahov, M. Vetzel, N. Parker, J. S. Nicoson, L.-C. Xu and E. Westrick, Bioconjugate Chem., 2005, 16, 803–811 CrossRef CAS PubMed.
- C. A. Ladino, R. V. Chari, L. A. Bourret, N. L. Kedersha and V. S. Goldmacher, Int. J. Cancer, 1997, 73, 859–864 CrossRef CAS PubMed.
- I. Ojima, Acc. Chem. Res., 2007, 41, 108–119 CrossRef PubMed.
- J. Chen, S. Chen, X. Zhao, L. V. Kuznetsova, S. S. Wong and I. Ojima, J. Am. Chem. Soc., 2008, 130, 16778–16785 CrossRef CAS PubMed.
- S. Chen, X. Zhao, J. Chen, J. Chen, L. Kuznetsova, S. S. Wong and I. Ojima, Bioconjugate Chem., 2010, 21, 979–987 CrossRef CAS PubMed.
- J. G. Vineberg, E. S. Zuniga, A. Kamath, Y.-J. Chen, J. D. Seitz and I. Ojima, J. Med. Chem., 2014, 57, 5777–5791 CrossRef CAS PubMed.
- J. G. Vineberg, T. Wang, E. S. Zuniga and I. Ojima, J. Med. Chem., 2015, 58, 2406–2416 CrossRef CAS PubMed.
- S. Maiti, N. Park, J. H. Han, H. M. Jeon, J. H. Lee, S. Bhuniya, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 4567–4572 CrossRef CAS PubMed.
- S. Bhuniya, M. H. Lee, H. M. Jeon, J. H. Han, J. H. Lee, N. Park, S. Maiti, C. Kang and J. S. Kim, Chem. Commun., 2013, 49, 7141–7143 RSC.
- H. MiáJeon, H. ThiáLe, T. WooáKim and J. SeungáKim, Chem. Commun., 2014, 50, 7690–7693 RSC.
- S. Bhuniya, S. Maiti, E. J. Kim, H. Lee, J. L. Sessler, K. S. Hong and J. S. Kim, Angew. Chem., 2014, 126, 4558–4563 CrossRef.
- A. El Alaoui, F. Schmidt, M. Amessou, M. Sarr, D. Decaudin, J. C. Florent and L. Johannes, Angew. Chem., Int. Ed., 2007, 46, 6469–6472 CrossRef CAS PubMed.
- M. H. Lee, J. Y. Kim, J. H. Han, S. Bhuniya, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2012, 134, 12668–12674 CrossRef CAS PubMed.
- X. Wu, X. Sun, Z. Guo, J. Tang, Y. Shen, T. D. James, H. Tian and W. Zhu, J. Am. Chem. Soc., 2014, 136, 3579–3588 CrossRef CAS PubMed.
- M. Ye, X. Wang, J. Tang, Z. Guo, Y. Shen, H. Tian and W. Zhu, Chem. Sci., 2016, 7, 4958–4965 RSC.
- J. Wu, R. Huang, C. Wang, W. Liu, J. Wang, X. Weng, T. Tian and X. Zhou, Org. Biomol. Chem., 2013, 11, 580–585 CAS.
- Y. Zhang, Q. Yin, J. Yen, J. Li, H. Ying, H. Wang, Y. Hua, E. J. Chaney, S. A. Boppart and J. Cheng, Chem. Commun., 2015, 51, 6948–6951 RSC.
- O. Oktyabrsky and G. Smirnova, Biochemistry, 2007, 72, 132–145 CAS.
- M. López-Lázaro, Cancer Lett., 2007, 252, 1–8 CrossRef PubMed.
- S. Reuter, S. C. Gupta, M. M. Chaturvedi and B. B. Aggarwal, Free Radical Biol. Med., 2010, 49, 1603–1616 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and A. Iyer, J. Drug Targeting, 2007, 15, 475–486 CrossRef CAS PubMed.
- Y. Kuang, K. Balakrishnan, V. Gandhi and X. Peng, J. Am. Chem. Soc., 2011, 133, 19278–19281 CrossRef CAS PubMed.
- E. J. Kim, S. Bhuniya, H. Lee, H. M. Kim, C. Cheong, S. Maiti, K. S. Hong and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 13888–13894 CrossRef CAS PubMed.
- R. Kumar, J. Han, H. J. Lim, W. X. Ren, J. Y. Lim, J. H. Kim and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 17836–17843 CrossRef CAS PubMed.
- R. Weinstain, E. Segal, R. Satchi-Fainaro and D. Shabat, Chem. Commun., 2010, 46, 553–555 RSC.
- S. Danson, T. H. Ward, J. Butler and M. Ranson, Cancer Treat. Rev., 2004, 30, 437–449 CrossRef CAS PubMed.
- K. Sharma, A. Iyer, K. Sengupta and H. Chakrapani, Org. Lett., 2013, 15, 2636–2639 CrossRef CAS PubMed.
- P. Liu, J. Xu, D. Yan, P. Zhang, F. Zeng, B. Li and S. Wu, Chem. Commun., 2015, 51, 9567–9570 RSC.
- S. G. Awuah, Y.-R. Zheng, P. M. Bruno, M. T. Hemann and S. J. Lippard, J. Am. Chem. Soc., 2015, 137, 14854–14857 CrossRef CAS PubMed.
- J. Y. Winum, M. Rami, A. Scozzafava, J. L. Montero and C. Supuran, Med. Res. Rev., 2008, 28, 445–463 CrossRef CAS PubMed.
- S. Y. Li, L. H. Liu, H. Z. Jia, W. X. Qiu, L. Rong, H. Cheng and X. Z. Zhang, Chem. Commun., 2014, 50, 11852–11855 RSC.
- K. L. Dao, R. R. Sawant, J. A. Hendricks, V. Ronga, V. P. Torchilin and R. N. Hanson, Bioconjugate Chem., 2012, 23, 785–795 CrossRef CAS PubMed.
- D. E. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- J. Lai, B. P. Shah, E. Garfunkel and K.-B. Lee, ACS Nano, 2013, 7, 2741–2750 CrossRef CAS PubMed.
- T. Liu, L. Y. Wu, J. K. Choi and C. E. Berkman, Prostate, 2009, 69, 585 CrossRef CAS PubMed.
- T. Liu, L. Y. Wu, J. K. Choi and C. E. Berkman, Int. J. Oncol., 2010, 36, 777–784 CrossRef CAS PubMed.
- J. Tian, J. Zhou, Z. Shen, L. Ding, J. Yu and H. Ju, Chem. Sci., 2015, 6, 5969–5977 RSC.
- J. Kim, C. H. Tung and Y. Choi, Chem. Commun., 2014, 50, 10600–10603 RSC.
- A. M. Hossion, M. Bio, G. Nkepang, S. G. Awuah and Y. You, ACS Med. Chem. Lett., 2012, 4, 124–127 CrossRef PubMed.
- M. Bio, P. Rajaputra, G. Nkepang, S. G. Awuah, A. M. Hossion and Y. You, J. Med. Chem., 2013, 56, 3936–3942 CrossRef CAS PubMed.
- M. Bio, P. Rajaputra, G. Nkepang and Y. You, J. Med. Chem., 2014, 57, 3401–3409 CrossRef CAS PubMed.
- G. Nkepang, M. Bio, P. Rajaputra, S. G. Awuah and Y. You, Bioconjugate Chem., 2014, 25, 2175–2188 CrossRef CAS PubMed.
- S. Karthik, B. P. Kumar, M. Gangopadhyay, M. Mandal and N. P. Singh, J. Mater. Chem. B, 2015, 3, 728–732 RSC.
- D. J. Yamashiro, X. G. Liu, C. P. Lee, A. Nakagawara, N. Ikegaki, L. M. McGregor, S. B. Baylin and G. M. Brodeur, Eur. J. Cancer, 1997, 33, 2054–2057 CrossRef CAS PubMed.
- G. M. Brodeur, A. Nakagawara, D. J. Yamashiro, N. Ikegaki, X. G. Liu, C. G. Azar, C. P. Lee and A. E. Evans, J. Neuro-Oncol., 1997, 31, 49–55 CrossRef CAS PubMed.
- Y. Wang, C. Hagel, W. Hamel, S. Muller, L. Kluwe and M. Westphal, Acta Neuropathol., 1998, 96, 357–364 CrossRef CAS PubMed.
- S. Kumar and J. deVellis, J. Neurosci. Res., 1996, 44, 490–498 CrossRef CAS PubMed.
- L. M. McGregor, B. K. McCune, J. R. Graff, P. R. McDowell, K. E. Romans, G. D. Yancopoulos, D. W. Ball, S. B. Baylin and B. D. Nelkin, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 4540–4545 CrossRef CAS.
- X. W. Xu, S. R. Tahan, T. L. Pasha and P. J. Zhang, J. Cutaneous Pathol., 2003, 30, 318–322 CrossRef.
- M. J. Blasco-Gutierrez, I. J. S. Jose-Crespo, E. Zozaya-Alvarez, R. Ramos-Sanchez and N. Garcia-Atares, Cancer Invest., 2007, 25, 405–410 CrossRef CAS PubMed.
- W. Jin, G. M. Kim, M. S. Kim, M. H. Lim, C. Yun, J. Jeong, J. S. Nam and S. J. Kim, Carcinogenesis, 2010, 31, 1939–1947 CrossRef CAS PubMed.
- A. Kamkaew and K. Burgess, J. Med. Chem., 2013, 56, 7608–7614 CrossRef CAS PubMed.
- H. Yuan, H. Chong, B. Wang, C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, J. Am. Chem. Soc., 2012, 134, 13184–13187 CrossRef CAS PubMed.
- R. R. Nani, A. P. Gorka, T. Nagaya, H. Kobayashi and M. J. Schnermann, Angew. Chem., Int. Ed., 2015, 54, 13635–13638 CrossRef CAS PubMed.
- C. Zhang, L. Gao, Y. Cai, H. Liu, D. Gao, J. Lai, B. Jia, F. Wang and Z. Liu, Biomaterials, 2016, 84, 1–12 CrossRef CAS PubMed.
- L. Liu, W. Qiu, B. Li, C. Zhang, L. Sun, S. Wan, L. Rong and X. Zhang, Adv. Funct. Mater., 2016, 26, 6257–6269 CrossRef CAS.
- W. Feng, C. Gao, W. Liu, H. Ren, C. Wang, K. Ge, S. Li, G. Zhou, H. Li, S. Wang, G. Jia, Z. Li and J. Zhang, Chem. Commun., 2016, 52, 9434–9437 RSC.
- Y. Hong, J. W. Lam and B. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- Y. Yuan, R. T. Kwok, B. Tang and B. Liu, J. Am. Chem. Soc., 2014, 136, 2546–2554 CrossRef CAS PubMed.
- Y. Yuan, Y. Chen, B. Tang and B. Liu, Chem. Commun., 2014, 50, 3868–3870 RSC.
- Y. Yuan, R. T. Kwok, R. Zhang, B. Tang and B. Liu, Chem. Commun., 2014, 50, 11465–11468 RSC.
- D. R. Green, L. Galluzzi and G. Kroemer, Science, 2011, 333, 1109–1112 CrossRef CAS PubMed.
- M. Ross, G. Kelso, F. Blaikie, A. James, H. Cocheme, A. Filipovska, T. Da Ros, T. Hurd, R. Smith and M. Murphy, Biochemistry, 2005, 70, 222–230 CAS.
- Q. Hu, M. Gao, G. Feng and B. Liu, Angew. Chem., Int. Ed., 2014, 53, 14225–14229 CrossRef CAS PubMed.
- W. Sun, S. G. Guo, C. Hu, J. L. Fan and X. J. Peng, Chem. Rev., 2016, 116, 7768–7817 CrossRef CAS PubMed.
- C. Zhang, T. Liu, Y. Su, S. Luo, Y. Zhu, X. Tan, S. Fan, L. Zhang, Y. Zhou and T. Cheng, Biomaterials, 2010, 31, 6612–6617 CrossRef CAS PubMed.
- E. Zhang, C. Zhang, Y. Su, T. Cheng and C. Shi, Drug Discovery Today, 2011, 16, 140–146 CrossRef CAS PubMed.
- X. Tan, S. Luo, D. Wang, Y. Su, T. Cheng and C. Shi, Biomaterials, 2012, 33, 2230–2239 CrossRef CAS PubMed.
- S. Luo, X. Tan, Q. Qi, Q. Guo, X. Ran, L. Zhang, E. Zhang, Y. Liang, L. Weng and H. Zheng, Biomaterials, 2013, 34, 2244–2251 CrossRef CAS PubMed.
- Y. Wang, T. Liu, E. Zhang, S. Luo, X. Tan and C. Shi, Biomaterials, 2014, 35, 4116–4124 CrossRef CAS PubMed.
- S. Luo, X. Tan, S. Fang, Y. Wang, T. Liu, X. Wang, Y. Yuan, H. Q. Sun, Q. R. Qi and C. M. Shi, Adv. Funct. Mater., 2016, 26, 2826–2835 CrossRef CAS.
- Y. Huang, J. Zhou, S. Luo, Y. Wang, J. He, P. Luo, Z. Chen, T. Liu, X. Tan, J. Ou, H. Miao, H. Liang and C. Shi, Gut, 2016 DOI:10.1136/gutjnl-2016-311909.
- J. B. Wu, T. P. Lin, J. D. Gallagher, S. Kushal, L. W. Chung, H. E. Zhau, B. Z. Olenyuk and J. C. Shih, J. Am. Chem. Soc., 2015, 137, 2366–2374 CrossRef CAS PubMed.
- J. B. Wu, C. Shi, G. C. Y. Chu, Q. Xu, Y. Zhang, Q. Li, S. Y. John, H. E. Zhau and L. W. Chung, Biomaterials, 2015, 67, 1–10 CrossRef CAS PubMed.
- I. Bratsos, S. Jedner, T. Gianferrara and E. Alessio, Chimia, 2007, 61, 692–697 CrossRef CAS.
- V. Pierroz, T. Joshi, A. Leonidova, C. Mari, J. Schur, I. Ott, L. Spiccia, S. Ferrari and G. Gasser, J. Am. Chem. Soc., 2012, 134, 20376–20387 CrossRef CAS PubMed.
- R. Ye, Z. Ke, C. Tan, L. He, L. Ji and Z. Mao, Chem. – Eur. J., 2013, 19, 10160–10169 CrossRef CAS PubMed.
- Y. Li, C. P. Tan, W. Zhang, L. He, L. N. Ji and Z. W. Mao, Biomaterials, 2015, 39, 95–104 CrossRef CAS PubMed.
- C. R. Cardoso, M. V. Lima, J. Cheleski, E. J. Peterson, T. Venancio, N. P. Farrell and R. M. Carlos, J. Med. Chem., 2014, 57, 4906–4915 CrossRef CAS PubMed.
- T. Zou, C. T. Lum, S. S. Y. Chui and C. M. Che, Angew. Chem., 2013, 125, 3002–3005 CrossRef.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537–550 CrossRef CAS PubMed.
- O. C. Farokhzad, J. Cheng, B. A. Teply, I. Sherifi, S. Jon, P. W. Kantoff, J. P. Richie and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6315–6320 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, Adv. Drug Delivery Rev., 2006, 58, 1456–1459 CrossRef CAS PubMed.
- D. Shangguan, Y. Li, Z. Tang, Z. C. Cao, H. W. Chen, P. Mallikaratchy, K. Sefah, C. J. Yang and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11838–11843 CrossRef CAS PubMed.
- X. Fang and W. Tan, Acc. Chem. Res., 2009, 43, 48–57 CrossRef PubMed.
- Z. Tang, D. Shangguan, K. Wang, H. Shi, K. Sefah, P. Mallikratchy, H. W. Chen, Y. Li and W. Tan, Anal. Chem., 2007, 79, 4900–4907 CrossRef CAS PubMed.
- V. Bagalkot, O. C. Farokhzad, R. Langer and S. Jon, Angew. Chem., Int. Ed., 2006, 45, 8149–8152 CrossRef CAS PubMed.
- H. S. Choi, K. Nasr, S. Alyabyev, D. Feith, J. H. Lee, S. H. Kim, Y. Ashitate, H. Hyun, G. Patonay and L. Strekowski, Angew. Chem., Int. Ed., 2011, 50, 6258–6263 CrossRef CAS PubMed.
- M. V. Marshall, D. Draney, E. M. Sevick-Muraca and D. M. Olive, Mol. Imaging Biol., 2010, 12, 583–594 CrossRef PubMed.
- R. J. Amato, A. Shetty, Y. Lu, R. Ellis and P. S. Low, J. Immunother., 2013, 36, 268–275 CrossRef CAS PubMed.
- K. L. Kiick, Science, 2007, 317, 1182 CrossRef CAS PubMed.
- C. C. Lee, J. A. MacKay, J. M. Fréchet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef CAS PubMed.
- Y. Zhou, W. Huang, J. Liu, X. Zhu and D. Yan, Adv. Mater., 2010, 22, 4567–4590 CrossRef CAS PubMed.
- S. M. Lee, H. Chen, C. M. Dettmer, T. V. O'Halloran and S. T. Nguyen, J. Am. Chem. Soc., 2007, 129, 15096–15097 CrossRef CAS PubMed.
- L. Linderoth, P. Fristrup, M. Hansen, F. Melander, R. Madsen, T. L. Andresen and G. n. H. Peters, J. Am. Chem. Soc., 2009, 131, 12193–12200 CrossRef CAS PubMed.
- R. Mo, T. Jiang, R. DiSanto, W. Tai and Z. Gu, Nat. Commun., 2014, 5, 3364 Search PubMed.
- R. Mo, T. Jiang, J. Di, W. Tai and Z. Gu, Chem. Soc. Rev., 2014, 43, 3595–3629 RSC.
- R. Mo and Z. Gu, Mater. Today, 2015, 19, 276–284 Search PubMed.
- C. S. Kue, A. Kamkaew, K. Burgess, L. V. Kiew, L. Y. Chung and H. B. Lee, Med. Res. Rev., 2016, 36, 494–575 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |