
Hypercrosslinked porous polymer materials: design, synthesis, and applications
Liangxiao
Tan
and
Bien
Tan
 *
*
Key Laboratory for Large-Format Battery Materials and System Ministry of Education, Hubei Key Laboratory of Material Chemistry and Service Failure, School of Chemistry and Chemical Engineering Huazhong University of Science and Technology, Wuhan 430074, China. E-mail: bien.tan@hust.edu.cn
First published on 22nd February 2017
Abstract
Hypercrosslinked polymers (HCPs) are a series of permanent microporous polymer materials initially reported by Davankov, and have received an increasing level of research interest. In recent years, HCPs have experienced rapid growth due to their remarkable advantages such as diverse synthetic methods, easy functionalization, high surface area, low cost reagents and mild operating conditions. Judicious selection of monomers, appropriate length crosslinkers and optimized reaction conditions yielded a well-developed polymer framework with an adjusted porous topology. Post fabrication of the as developed network facilitates the incorporation of various chemical functionalities that may lead to interesting properties and enhance the selection toward a specific application. To date, numerous HCPs have been prepared by post-crosslinking polystyrene-based precursors, one-step self-polycondensation or external crosslinking strategies. The advent of these methodologies has prompted researchers to construct well-defined porous polymer networks with customized micromorphology and functionalities. In this review, we describe not only the basic synthetic principles and strategies of HCPs, but also the advancements in the structural and morphological study as well as the frontiers of potential applications in energy and environmental fields such as gas storage, carbon capture, removal of pollutants, molecular separation, catalysis, drug delivery, sensing etc.
 Liangxiao Tan | Liangxiao Tan received his bachelor's degree in chemical engineering and technology in 2012 from Huazhong University of Science and Technology. Then, he joined Professor Bien Tan's group and studied for his PhD degree in polymer chemistry and physics from Huazhong University of Science and Technology. His current scientific interests include the design, synthesis and morphology study of hypercrosslinked polymers. |
 Bien Tan | Bien Tan is a professor at the School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology (HUST). He received his PhD in 1999 from the College of Materials at South China University of Technology. He then joined Beijing Institute of Aeronautical Materials for postdoctoral research in National Laboratory of Advanced Composites (1999–2001). He worked as a PDRA at the University of Liverpool (2001–2007). He then returned to China and joined HUST in September 2007 as a professor. In December 2009, Tan was awarded New Century Excellent Talents in University by the Ministry of Education, and in the same year he was awarded Chutian Scholar Distinguished Professor by Hubei province. His main research interests include polymeric materials, supercritical fluids, microporous materials, hydrogen storage, metal nanoparticles, emulsion-templated materials, and high-throughput materials methodology. |
1. Introduction
Design, synthesis and utilization of advanced functional materials with a special porous architecture in the micro- and nanoscale range is always an important subject in many scientific fields.1 Porous materials have undergone evolution from the inorganic skeleton of zeolites,2 activated carbon,3 silica,4 to hybrid metal–organic frameworks (MOFs),5 porous coordination polymers (PCPs),6 pure organic networks of porous cages7 and porous organic polymers (POPs).8,9 Recently, POPs have received an increased level of research interest due to their special properties achieved by combining the advanced properties of both porous materials and polymers. For example, porous polymers can be designed and prepared at the molecular level with a controlled surface area and a well refined pore topology.10,11–14 A wide range of optional organic building blocks as well as multitudinous synthetic routes have been discovered, which facilitate the facile incorporation of multiple chemical functionalities into the resulting porous architecture.15–17 Moreover, porous polymers have easy processability based on their essential polymeric character and can be synthesized with controlled micromorphologies such as quasi-zero-dimensional nanoparticles,18–20 hollow capsules,21–23 and two-dimensional (2D) membranes,24–26 as well as three-dimensional (3D) monolithic blocks.27,28 Among these, a few of the porous polymers can even be dissolved in common organic solvents for further solution-processing without significantly affecting their porous architecture.29,30 In order to maintain inner cavities, highly rigid polymer networks must be constructed to prevent the collapse of polymer chains into a nonporous dense state.31 The key strategy for this purpose is the use of rigid building units fixed with strong covalent bonds. In addition, a wide range of synthetic methods are available to produce porous organic polymer networks with different structures including covalent organic frameworks (COFs),32,33 hypercrosslinked polymers (HCPs),34,35 conjugated microporous polymers (CMPs),36–38 polymers of intrinsic microporosity (PIM),39,40 covalent triazine frameworks (CTFs),41,42 and porous aromatic frameworks (PAFs)43,44etc.Among the above mentioned polymer networks, HCPs, strictly speaking, are not much novel porous polymers but have still undergone rapid development in recent years. The synthesis of HCPs is mainly based on Friedel–Crafts chemistry which provides a fast kinetics to form strong linkages resulting in a highly crosslinked network with predominant porosity.45,46 Due to the simple and versatile synthetic approach, a great diversity of aromatic monomers can be employed for the development of polymer networks with various pore architectures or may utilize particular functionalities which boost the high surface area and unique features.47 Moreover, the conventional synthetic methodologies of HCPs require low-cost reagents (monomers, reaction media, and catalysts), and easy to handle and control reaction conditions result in production of high yield products and are thus competing with conventional inexpensive microporous materials such as activated carbon. From a synthetic perspective, HCPs are predominantly prepared by the following three approaches: (1) post-crosslinking polymer precursors, (2) direct one-step polycondensation of functional monomers, and (3) knitting rigid aromatic building blocks with external crosslinkers. The diversity of building blocks, coupled with the extended synthetic approaches, makes HCPs invaluable platforms for exploring new organic porous materials with enormous potential to solve challenging energy and environmental issues.
2. Synthetic strategies/methodologies
2.1 Post-crosslinking
The first examples of HCPs (also called Davankov resins) were developed by Tsyurupa and Davankov in the early 1970s48 in which polystyrene-based precursors such as linear solvated polystyrene or gel-type swollen polystyrene-co-divinylbenzene (polySt–DVB) were post-crosslinked by external crosslinking agents (external electrophiles) in the presence of a Lewis acid catalyst and an appropriate solvent.49–51 Generally speaking, this simple process involves two crucial steps: (1) the complete dissolution or swelling of polymer precursors and (2) the intensive post-crosslinking. The detailed overview of the synthesis is illustrated in Fig. 1, prior to the crosslinking, pre-synthesized polystyrene chains are dispersed homogeneously throughout the entire solvent along with a stoichiometric bifunctional crosslinker and catalyst. Afterwards, the reaction proceeds very quickly which causes strong linking of neighboring phenyl rings with rigid bridges in numerous chain segments. These rigid linkages eventually keep the resulting network in a confined conformation and prevent the chains from mobilizing or collapsing after the removal of the solvent.31 As a result, previously solvated polystyrene was transformed into a one-phase material with a low packing density and a highly porous framework.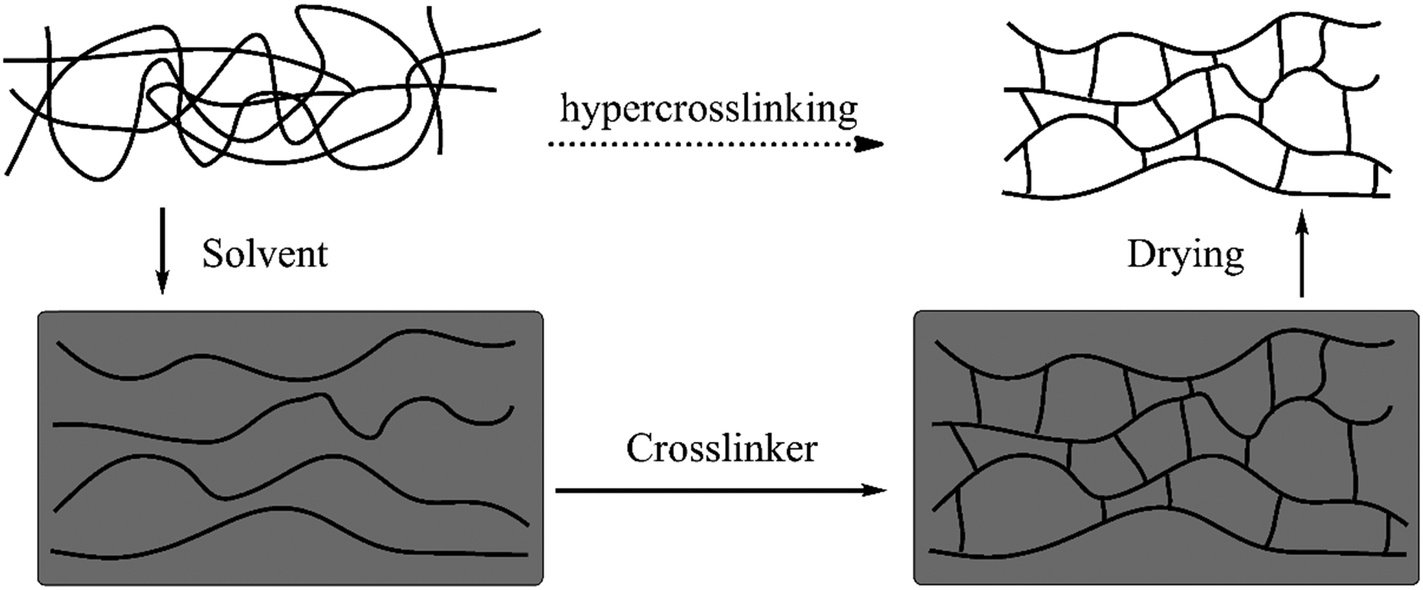 | ||
| Fig. 1 Schematic representation of the hypercrosslinking process. Reproduced with permission from ref. 31. Copyright 2007, The Royal Society of Chemistry. | ||
Previously, commonly used crosslinkers were halogen-containing compounds such as bifunctional monochlorodimethyl ether (MCDE),52–54 tetrachloromethane (CCl4),55,56 dichloroxylene (DCX),49 4,4′-bis(chloromethyl)biphenyl (BCMBP),49p,p′-bis-chloromethyl-l,4-diphenylbutane (DPB),57 and trifunctional tris-(chloromethyl)-mesitylene (TCMM)57 as well as di or triiodoalkanes31 affording network containing electron-donating moieties. By applying various crosslinkers with different linking units (for example changed length and rigidity), the interior skeleton construction of the final products can be easily adjusted (showing a difference in the surface area and porosity).57 For instance, compared with bifunctional crosslinkers, TCMM can join three polymer chains simultaneously thus creating enhanced rigidity in the final network and a much higher apparent surface area at a relatively low crosslinking degree. Moreover, all of the above bifunctional crosslinkers are able to construct a confined network with limited conformational mobility except for DPB crosslinkers. The long flexible alkyl units from DPB facilitate the conformational rearrangements in the final network with almost no surface area obtained at any crosslinking degree.
Usually, the hypercrosslinking process of linear polystyrene or gel-type polySt–DVB polymers with a low divinylbenzene content (0.3–2%) generates intrinsic microporosity. However, when the DVB contents are increased, a macroporous polymer network is initially formed. After hypercrosslinking, a bimodal porous distribution is expected to be obtained with micropores derived from post-crosslinking while retaining the macropores.51,58 In addition, the crosslinking degree achieved from a macroporous precursor is much lower compared to the solvated or gel-type swollen precursors owing to the intense steric impediment.
Hypercrosslinking of polystyrene-based precursors with external crosslinkers is usually described as a one-pot process where chloromethyl of the phenyl ring is converted into methylene linkages. Veverka has proposed a two-step process using commercial chloromethylated polySt–DVB resins and then crosslinking them via the internal condensation of chloromethyl groups.51 Despite the apparent complexity of the process, the produced results have enlightened the plausible mechanism of the polymer-analogical Friedel–Crafts reaction which seems to be ever-expanding.
In order to gain further insights into the effect of the chloromethyl content, Sherrington and co-workers have prepared a series of vinylbenzyl chloride–divinylbenzene copolymers (polyVBC–DVB) with well-defined functional compositions (with 2% and 20% DVB content respectively).59 Afterwards, hypercrosslinking was performed under variable conditions including different monomer ratios, solvents, Lewis acid catalysts and reaction times. Based on the surface area results, FeCl3 exhibits the best catalytic efficiency compared with AlCl3 and SnCl4 in dichloroethane (DCE), which can be ascribed to a better compromise between solubility and the molecular size. In this regard, optimized conditions have resulted in the production of materials with the highest surface area of up to 2090 m2 g−1 using a gel-type precursor with only 2% DVB. One possible explanation for this ultrahigh surface area is ascribed to a more efficient crosslinking produced by a doubly bridged structure between the initial swellable polystyrene chains (Fig. 2a). This might yield a more regular microporous structure than that from the porous precursor with 20% DVB. Meanwhile, a preferential decrease in the surface area was attributed to a low chloromethyl content. The effect of the reaction time was also investigated using a 2% DVB gel-type precursor under the same reaction conditions which exhibited an uncontrollable rapid cooperative phenomenon. The crosslinking degree can be enhanced quite fast within the initial 15 min, and relatively high surface area polymers (1200 m2 g−1) were obtained which increased up to >1800 m2 g−1 after 2 h. However, beyond this time, only a very small increment in the surface area was observed. In this regard, precursor resins with a higher DVB content (20%) may present an inherent limitation for the solvent and catalyst accessibility, resulting in a significantly lower surface area even when matching the functional degree of chloromethyl groups with gel-type precursors (1055 m2 g−1vs. 1706 m2 g−1). A drastic decrease in the surface area was, however, observed using precursors of low functionality. For instance, a hypercrosslinked network from the gel-type 2% DVB precursor with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 monomer ratio (St:VBC) resulted in extremely low surface area polymers of only 2 m2 g−1. Interestingly, a much higher surface area (474 m2 g−1) was obtained using the 20% DVB precursor with the same monomer ratio. These results imply that pre-crosslinked DVB plays an important role and can influence the porous structure and surface area, however, more meticulous experiments are required to validate these conclusions.
:1 monomer ratio (St:VBC) resulted in extremely low surface area polymers of only 2 m2 g−1. Interestingly, a much higher surface area (474 m2 g−1) was obtained using the 20% DVB precursor with the same monomer ratio. These results imply that pre-crosslinked DVB plays an important role and can influence the porous structure and surface area, however, more meticulous experiments are required to validate these conclusions.
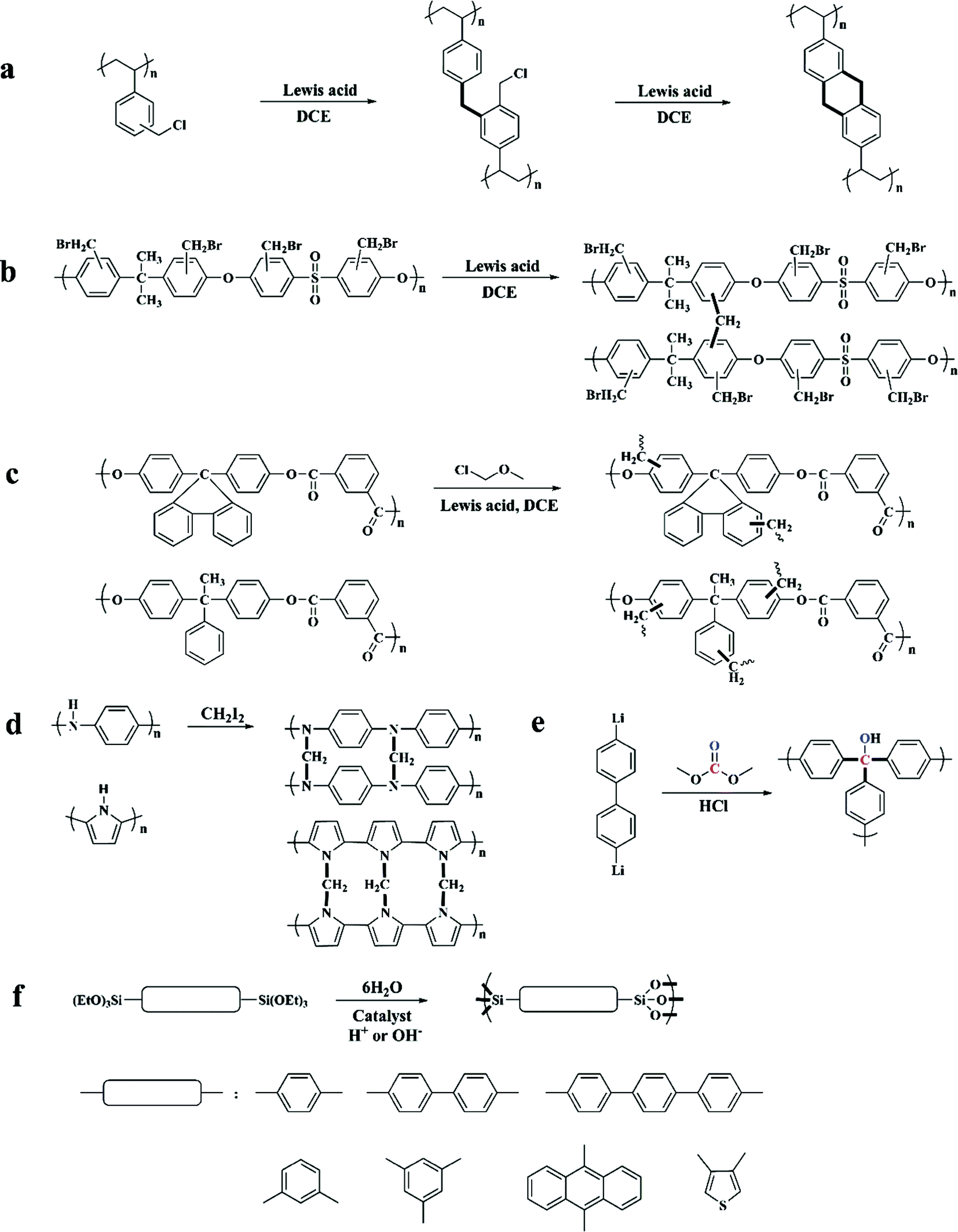 | ||
| Fig. 2 Several examples of hypercrosslinked polymers by the post-crosslinking of polyVBC-based precursors from ref. 59, (a) polysulfone (b) and polyarylate (c) from ref. 45, polyaniline from ref. 31 and polypyrrole from ref. 62 (d) as well as hypercrosslinked condensation networks of dilithio-biphenyl with dimethylcarbonate from ref. 65 (e), hybrid arylene-bridged polysilsesquioxane networks from ref. 67 (f). | ||
Following this, Tan and co-workers demonstrated a particular control over the pore structure from macroporosity to microporosity in the resulting hypercrosslinked divinylbenzene–vinylbenzyl chloride polymers (HCP-DVB–VBC) by increasing the DVB content from 0–10%.60 By increasing the DVB content, initially the surface area of HCP-DVB–VBC showed an obvious increase but it decreased when further increasing the DVB content. The highest surface area of 2060 m2 g−1 was obtained with a 2% DVB content which is consistent with the previous results. Meanwhile, a bimodal porous architecture containing both macropores and micropores was efficiently transformed into a more uniform and narrowly distributed microporous structure. Additionally, polymer networks become purely microporous when the DVB concentration is higher than 7%. These results were ascribed to the solubility of polymer precursors without DVB pre-crosslinking in DCE, therefore, the macromolecular chains extended into the whole solution and resulted in a loose and disordered twisted network with flexible neighboring chains. During the hypercrosslinking process, linking these neighboring twisted polymer chains caused a random packing density which thus produced a mixed macro- and microporosity. In the case of slightly pre-crosslinked conditions (DVB less than 2%), the chains are still very loose so a similar porous framework is achieved. Upon increasing the DVB content, chains become fixed and compact with the distribution changing from random to quite even, resulting in more uniform hypercrosslinking being achieved throughout the networks.
Although the control over microporosity can be successfully achieved by varying the DVB content, the adjustment of the mesopores is usually difficult. Seo et al. reported the synthesis of hierarchical porous polymers which contain permanent micropores and well-defined 3D continuous mesopores by combination of block copolymer self-assembly and post-hypercrosslinking.61 Copolymerization of VBC and DVB was conducted in the presence of a polylactide (PLA) macro chain transfer agent to produce slightly crosslinked block copolymer precursors (PLA-b-polyVBC–DVB). Polymerization induced microphase separation was observed to form a bicontinuous morphology containing PLA and polyVBC–DVB microdomains. The post-crosslinking approach for the precursors not only generates micropores in the polyVBC–DVB microdomain but also selectively removes PLA microdomains at the same time to yield reticulated mesoporous channels. Moreover, by controlling the molecular weight of the PLA macro agent, the mesopores of the resulting porous polymers can be precisely tuned from 6 to 15 nm.61
Except for these polystyrene-based resins, some other hetero-chain polymers were also hypercrosslinked to form highly porous networks such as polysulfone,45 polyarylates,45 polyaniline31 and polypyrrole.62
The hypercrosslinking of polysulfone was carried out via a two-step reaction in the presence of a Friedel–Crafts catalyst (Fig. 2b). Firstly, a safe method for halomethylation of polysulfone was conducted by Warshavsky et al. permitting the introduction of bromomethyl into every benzene ring.63 Then, a hypercrosslinked structure was obtained by a simple self-crosslinking reaction similar to that of DVB–VBC precursors. These precursors have the inherent polymeric chain flexibility and low activity to achieve a limited crosslinking degree with a very low apparent surface area. Polyarylate precursors composed of isophthalic acid and two kinds of bis-phenol were directly employed to prepare hypercrosslinked networks in a typical procedure using MCDE as a crosslinker (Fig. 2c) yielding much lower surface areas compared to the hypercrosslined polystyrene i.e. 380 and 108 m2 g−1 respectively. In addition, polyarylates with more rigid side substituents resulted in a relatively high porosity.
Hypercrosslinked polyaniline and polypyrrole (Fig. 2d) were developed by Fréchet and Svec. Polyaniline and polypyrrole precursors without further modification can be directly crosslinked using the more reactive iodoalkane crosslinker, which compensated for the higher reactivity of the precursors reduced by electron-donating groups. The resulting networks exhibited apparent surface areas of up to 630 and 720 m2 g−1 respectively. Recently, Sharma et al. also reported the synthesis of nanoporous hypercrosslinked polyaniline however by applying a facile microwave assisted process which resulted in a much high surface area of up to 1059 m2 g−1.64 With a high surface area, ultramicroporous structure and nitrogen moieties, this material showed good CO2 and H2 storage properties.
Some other types of porous networks can also be categorized into hypercrosslinked species, although they were not synthesized via the typical Friedel–Crafts reaction. Webster et al. prepared a highly porous organic material by treating 4,4′-dilithio-biphenyl with dimethylcarbonate at an extremely low temperature of −80 °C, which resulted in a relatively high surface area of 1167 m2 g−1 (Fig. 2e).65,66 The synthesis of hybrid arylene-bridged polysilsesquioxane networks was introduced by Lloy and Shea (Fig. 2f).67,68 In their experiments, triethoxysilyl containing aromatic hydrocarbons were used as the starting monomers and then hydrolyzed to yield siloxane bonds in the presence of an acidic or a basic catalyst. Resulting surface areas ranging from 600 to 1800 m2 g−1 have been achieved by simply varying the bridge length. Recently, Li and co-workers proposed the synthesis of a novel series of hypercrosslinked organic microporous polymers based on the radical copolymerization from rigid bi-vinyl containing monomers, bismaleimides and divinylbenzene.69 Due to the intrinsic alternating copolymerization properties, the obtained HCPs revealed a defined molecular structure with a highly crosslinked density, which resulted in high specific surface areas ranging from 627 to 841 m2 g−1. Meanwhile, this method prevents the employment of Lewis acid catalysts as well as eliminates the release of corrosive, and thus can be considered more environmentally friendly for large-scale industrial production.
2.2 Direct polycondensation or one-step condensation
Since the discovery of Davankov resins, a rapid explosion has been witnessed for hypercrosslinked polystyrene in their design and synthesis as well as finding their potential applications. The most notable advantage is the direct utilization of commercially available polymeric products as precursors for post-crosslinking. However, the synthesis of polymer precursors is time-consuming and limited functional monomers can be selected to satisfy the combined conditions from reactions of radical polymerization and Friedel–Crafts alkylation. The most commonly used precursors are still polystyrene-based polymers. Even though self-condensation of small rigid molecules with bifunctionality has already been carried out to produce porous polymers, the resulting surface areas are not very high.45 The self-condensation method was not much explored until further investigation was made by Cooper and co-workers.70,71 The results indicated a promising synthetic strategy and the optional building units may be extended to a wide range. In their experiment, three bis(chloromethyl) aromatic building blocks including DCX, BCMBP, and bis(chloromethyl) anthracene (BCMA) (as shown in Fig. 3a) were employed to obtain a series of hypercorsslinked polymers using a simple one-pot condensation polymerization process. With Lewis acid catalysts, chloromethyl groups are easy to react with neighboring phenyl rings to form highly rigid methylene linkages between small molecule building blocks by eliminating hydrogen chloride molecules. The resulting networks revealed permanent microporous structures and comparable surface areas. Among these three monomers, a much higher surface area of up to 1874 m2 g−1 was obtained for BCMBP via homo-polymerization. In contrast, p-DCX and BCMA monomers generated surface areas of 1391 and 921 m2 g−1 only under the same conditions. By varying the monomer ratios and reaction conditions, apparent surface areas of the hypercrosslinked networks can be simply controlled in the range of 700 to 2000 m2 g−1 as well as their pore size. In addition, a higher surface area of 1904 m2 g−1 was achieved from p-DCX and BCMBP copolymerization with a molar ratio of 1:3.
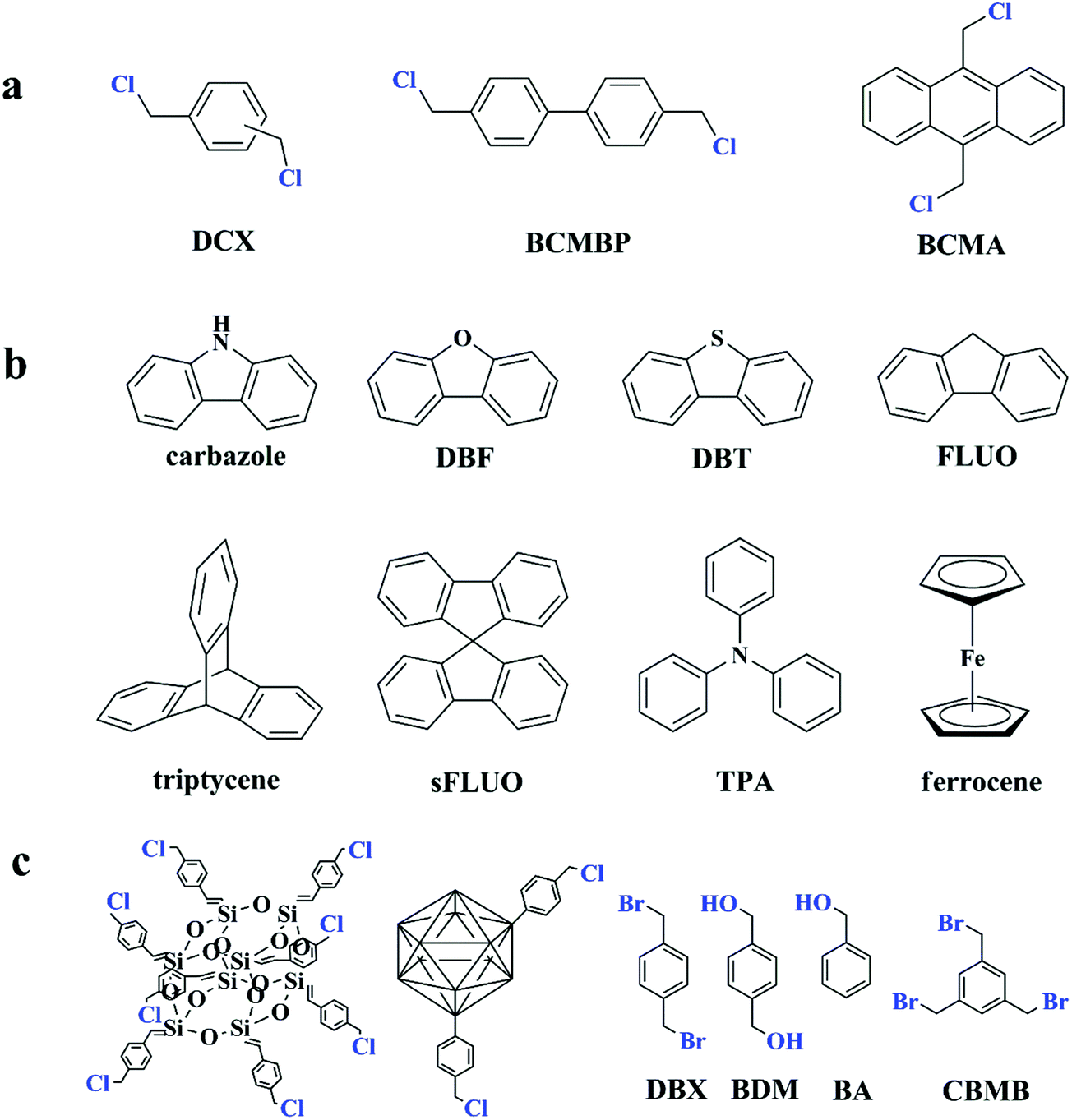 | ||
| Fig. 3 (a) Functional monomers used for the self-polycondensation synthesis of HCPs from ref. 70, (b) some typical examples of co-monomers from ref. 10, 72 and 73, (c) other types of functional building blocks for self-condensation polymerization from ref. 77–81. Atoms labeled in blue are functional moieties. | ||
Except for the self-condensation reaction, bis(chloromethyl) aromatic monomers can also act as external crosslinkers to link other substances. The molecular structure of typical co-monomers is displayed in Fig. 3b. The synthesis of a series of fluorene-based hypercrosslinked copolymers was carried out by Schwab et al. through condensation of BCMBP crosslinkers with four monomers including fluorene (FLUO), 9,9′-spirobi(fluorene) (sFLUO), dibenzofuran (DBF) and dibenzothiophene (DBT).72 The molar fraction of fluorene-based monomers was fixed to 10 and 25% and the resulting porous materials exhibited type II nitrogen sorption isotherms with combined micro- and macroporous structures. The highest surface area of up to 1800 m2 g−1 was obtained from DBF monomers with 10% molar fraction. However, even with various spatial structures and electrophilic properties, no big difference in the surface area was observed among all monomers. Microporous polytriphenylamine networks were prepared from triphenylamine (TPA) and DCX crosslinkers using a FeCl3 promoted oxidative polymerization and Friedel–Crafts alkylation process.73,74 By increasing the DCX content in the starting monomer ratio, specific surface areas of the resulting materials can be controlled from 318 to 1530 m2 g−1 with the pore width maximized around 0.55 to 1.8 nm. Chen et al. prepared a series of cost-effective nanoporous organic polymers with hierarchical pores by utilizing DCX and BCMBP crosslinkers to connect heterocyclic(carbazole), metal-doped (ferrocene) and highly rigid (triptycene) building blocks.10 By controlling the length of crosslinking molecules, the porosity of the resultant polymers can also be well-tailored. More specifically, shorter crosslinker DCX provides the polymers with greater microporosity, whereas longer crosslinker BCMCP generates a larger pore diameter. However, employing BCMBP as a crosslinker resulted in a much higher surface area of 1650 m2 g−1 for the ferrocene-based polymer. Dai and co-workers developed a soft chemistry synthetic strategy to construct a bimodal micro-mesoporous architecture in the textural engineering of phenolic resin based on the Friedel–Crafts alkylation reaction.75 A dense nonporous phenolic resin was synthesized around F127 triblock copolymer templates to form a composite matrix in which hypercrosslinking occurred to introduce microporosity in phenolic resin nanodomains. Moreover, the formation of rigid molecular bridges between neighboring aromatic units in a highly crosslinked state also strengthened the soft organic system for robust mesoporous generation, preventing pore collapse during template removal. As a result, the resultant sample maintained a bimodal micro-mesoporous architecture with a high surface area of up to 782 m2 g−1. Compared with the other pore formation strategies such as carbonization, this soft chemistry approach works under milder conditions revealing significant advantages in preserving the mesoporous architecture as well as functional moieties, which may be beneficial for further gas adsorption applications. Following this investigation, an improved one-pot strategy was proposed by Huo et al., which combined the hypercrosslinking process and solvent extraction of the triblock copolymer template into one step.76 It is demonstrated from the results that the released HCl in Friedel–Crafts alkylation is beneficial and highly demanded to remove the templates from the phenolic matrix for the preparation of ordered bimodal micro-mesoporous phenolic polymers. As a consequence, additional treatment for template removal is no longer required. However, the obtained surface area was much lower and not higher than 398 m2 g−1.
Migration of functional chloromethyl to other building units leads to a successful development of novel HCP synthesis with a distinctive topological architecture. Some types of functional building blocks are summarized in Fig. 3c. Dibromo-p-xylene (DBX) was also confirmed as an efficient Friedel–Crafts alkylation crosslinker by Bhunia et al.77 Therefore a direct crosslinking of carbazole building units has afforded a hypercrosslinked supermicroporous polymer which revealed an apparent BET surface area of 913 m2 g−1. As the resultant network possessed unsubstituted carbon atoms on benzene rings, a post synthetic functionalization with sulphonic moieties yielded an efficient solid state catalyst (acid) for the production of biodiesel via esterification of free fatty acids and esters at room temperature.
Liu et al. reported a series of porous HCPs through a facile AlCl3 or FeCl3 catalyzed template-free Friedel–Crafts alkylation of rigid aromatic building blocks with tribromomethylbenzene crosslinkers (CBMBs).78 A comparative study showed that anhydrous AlCl3 is a much more effective Lewis acid catalyst than FeCl3 and is more beneficial in terms of porosity formation and gas sorption ability. The highest surface area of 1783 m2 g−1 was achieved for triphenylbenzene (TPB)-based polymers using AlCl3 catalysts.
Hypercrosslinked hybrid materials can be achieved by self-condensation of benzyl chloride containing building blocks. Chaikittisilp et al. described a simple approach to produce highly porous hypercrosslinked siloxane–organic hybrids via a Friedel–Crafts self-condensation reaction of benzyl chloride-terminated double-four-ring cubic siloxane cages.79 The resulting porous siloxane-based organic–inorganic hybrid materials exhibited an ultrahigh surface area of 2500 m2 g−1 and a large pore volume of 3.3 cm3 g−1 exceedingly high as compared to all the other siloxane-based materials reported to date. POPs containing carborane units were successfully prepared by Yuan and co-workers.80 Carboranes were firstly functionalized by a simple grafting of two benzyl chloride groups and then directly self-polymerized or copolymerized with DCX crosslinkers. The resulting hybrid materials possessed high surface areas of up to 864 and 1037 m2 g−1, respectively, and showed relatively high H2 adsorption due to their highly electron-deficient structure that enhances H2 and polymer interactions.
Except for the chloromethyl groups, hydroxymethyl can also act as functional moieties to form reticular networks via self-condensation. Luo et al. chose two kinds of aromatic hydroxymethyl monomers i.e. 1,4-benzenedimethanol (BDM) and benzyl alcohol (BA) as the starting building blocks.81 Under reaction conditions similar to chloromethyl groups, FeCl3 was also used as a catalyst and provided high activity. A high surface area of up to 847 m2 g−1 was obtained using the bifunctional monomer BDM with a predominant microporous architecture. Interestingly, self-condensation of monofunctional BA monomers still generated a moderate surface area of 742 m2 g−1. These findings stipulate that multifunctional groups are not crucial for hypercrosslinking and open up more possibilities for constructing novel porous networks.
Following these achievements, Tan and co-workers proposed a cost-effective approach for the synthesis of microporous polymers from a wide variety of low functional aromatic building blocks (as illustrated in Fig. 4).82 This approach is based on the Scholl coupling reaction83 that directly links adjacent phenyl rings by eliminating two aryl-bound hydrogen atoms and forming a new aryl–aryl bond in the presence of an anhydrous AlCl3 catalyst (sometimes Brønsted acid such as hydrogen chloride is also needed). Interestingly, conjugation from monomers is easily extended to a long-range scale because no external bridges are formed. Moreover, due to the high catalytic activity, a wide range of optional monomers involved in the reaction including those with high electron density (anisole, benzyl amine) or low electron density (tetraphenylporphyrin), acidic (benzoic acid) or alkaline (bipyridine) functional groups, an aryl ring (TPB), a fused ring (naphthalene, pyrene) or even a heterocyclic ring (pyrrole). These monomers with various functionalities provided the resulting materials with multifunctional properties for diverse potential applications including gas storage, heterogeneous catalysis, luminescence and semiconductors. The highest surface area of 1421 m2 g−1 was obtained by co-condensation of TPB and pyrrole, while homo-polymerization of aryl or fused monomers produced ultra-microporous polymer networks with relatively lower surface areas of 636 to 1254 m2 g−1. Embedding small molecular ligands of tetraphenylporphyrin and bipyridine as a part of the resulting networks provided a bottom-up strategy to design heterogeneous catalytic scaffolds. Organometallic catalysts coordinating Pd(II) and Cu(II) were successfully synthesized which revealed catalytic activities in Suzuki–Miyaura crosscoupling and alcohol oxidation reactions. Due to the extended π-conjugation and 3D π–π stacking, colorful luminescence and p-type semiconductor character were observed for all of the Scholl coupling microporous networks.
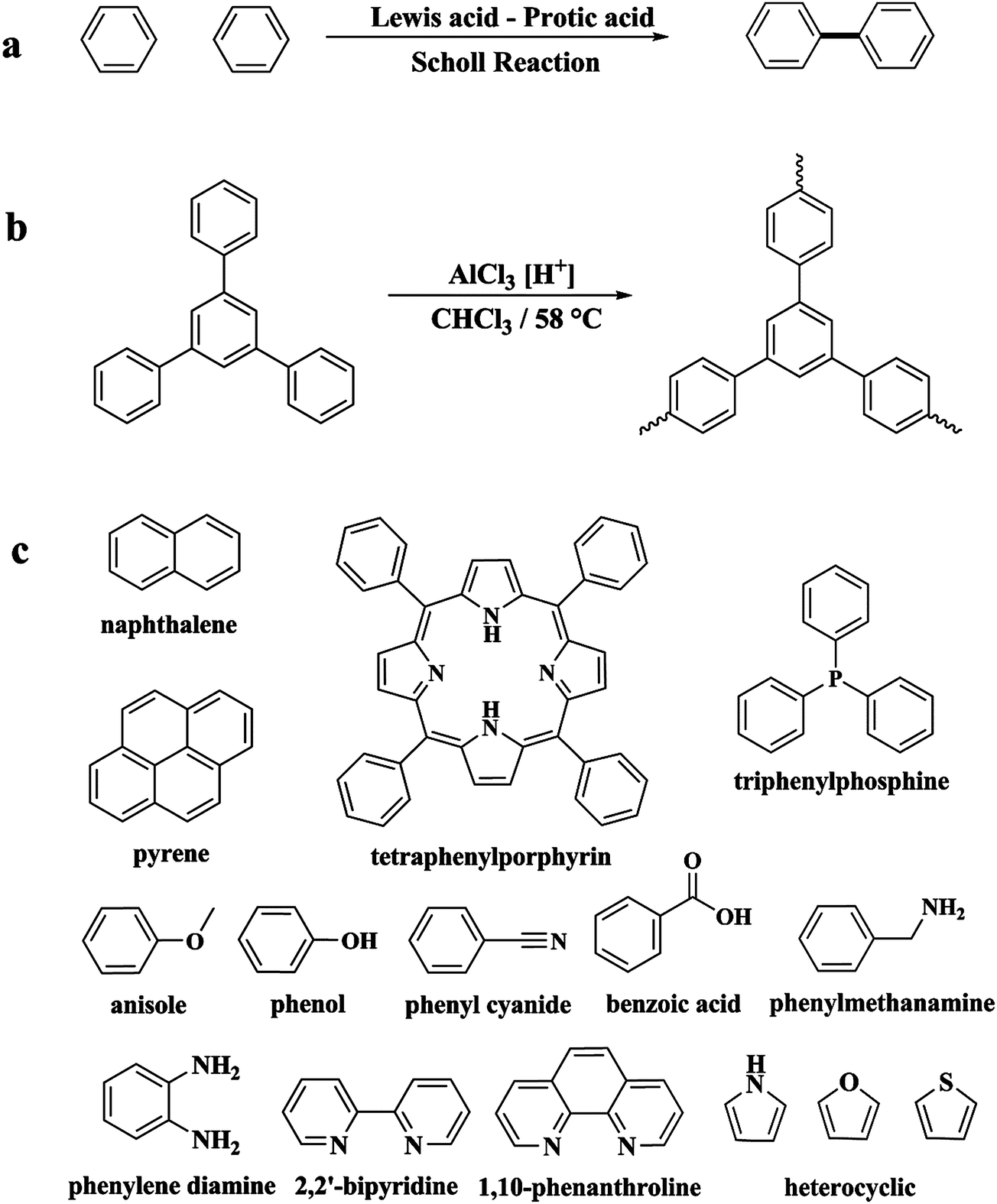 | ||
| Fig. 4 (a) The typical Scholl coupling reaction, (b) a model structure of the Scholl coupling polymer network from TPB monomers, (c) other monomers or co-monomers used for synthesizing polymer networks with multifunctionality. Reproduced with permission from ref. 82. Copyright 2014, The Royal Society of Chemistry. | ||
Li et al. chose several monomers such as TPA, tetraphenylmethane, tetraphenylsilane, and tetraphenylgermane for constructing spatially hypercrossslinked networks via the Scholl coupling reaction.84 The resulting networks displayed moderate surface areas ranging from 515 to 1119 m2 g−1 with a uniform pore size distribution mainly centered at 0.5 nm.
Very recently, McKeown et al. prepared a series of amorphous porous polymers with a very high surface area via a AlCl3-mediated coupling polymerization from a wide variety of readily available and inexpensive aromatic hydrocarbon building blocks.85 Herein DCE was used as a solvent but it also played a role of the crosslinker in providing methylene bridges which were afterwards confirmed by 13C solid-state NMR. Greatly enhanced microporosity was obtained in the resulting amorphous porous polymers and the highest surface area of 2435 m2 g−1 was achieved from TPB reproducibly. This work opened up a new perspective in synthesizing microporous network polymers with high surface areas even in excess of commercially activated carbons.
2.3 External crosslinking
Several strategies including self or co-condensation of chloromethyl and hydroxymethyl containing monomers or the Scholl coupling reaction have been proposed for the synthesis of hypercrosslinked polymer materials, but all of these approaches have certain limitations. For example, the preparation of building blocks containing special functional groups always needs multistep organic synthesis that requires large volumes of organic reagents and solvents associated with energy cost for further purification steps. The condensation of chloromethyl groups normally generates hydrogen chloride which is harmful to the environmental and industrial facilities. In Scholl coupling, the complex and strict reaction conditions usually prolong the production period. Therefore, a facile strategy is still desired for preparing microporous polymers with a high surface area from a wide variety of easily available simple aromatic building blocks under mild conditions.A breakthrough approach was proposed by Tan and co-workers known as the knitting strategy in which quite active formaldehyde dimethyl acetal (FDA) was employed as an external crosslinker to combine simple aromatic compounds like benzene or biphenyl with rigid methylene bridges via the anhydrous FeCl3 catalyzed Friedel–Crafts reaction (Fig. 5a).86 The possible mechanism was proposed as follows: a Lewis acid catalyst first complexes with a crosslinker molecule which reduces its binding strength between the methoxyl group and the central carbon atom and then produces a large amount of intermediate carbocations in the DCE solvent. As the reaction proceeds, carbocations react with phenyl rings to add multi-methoxymethyl groups to aromatic molecules with the release of methanol. These highly active methoxymethy groups are further converted to methylene linkages when reacted with other phenyl rings thus forming highly rigid crosslinked networks. Several aromatic building blocks, such as benzene, biphenyl, TPB, methylbenzene, chlorobenzene, and phenol (as shown in Fig. 5b), were directly knitted resulting in networks with predominant microporosity and a high surface area. Moreover, porous structures and surface areas are adjustable by varying the molar ratio of crosslinkers to monomers which eventually affect the crosslinking degree. The benzene knitting networks (with a 3:1 crosslinker to benzene ratio) displayed the highest apparent surface area of up to 1391 m2 g−1 which nevertheless dropped to 897 m2 g−1 with an equal amount of crosslinker and benzene. In addition, the functionality of polymer networks can be changed simply by employing aromatic building blocks which contain different functional groups. For example, phenolic hydroxyl group containing polymer networks were successfully synthesized using phenol as the starting monomer, which displayed a 50% increase in CO2 adsorption compared with the biphenyl knitting polymers with a similar surface area owing to the enhanced CO2 binding affinity. In summary, this strategy showed outstanding characteristics: versatile and flexible (extended building blocks and tunable porous structures); facile (low cost reagents and mild conditions) and highly efficient (resulting in an abundant micropore structure and a high surface area). Ever since the knitting strategy was developed, it has been employed extensively in synthesizing hypercrosslinked microporous materials from a wide variety of rigid building units with different structures and special functionalities.
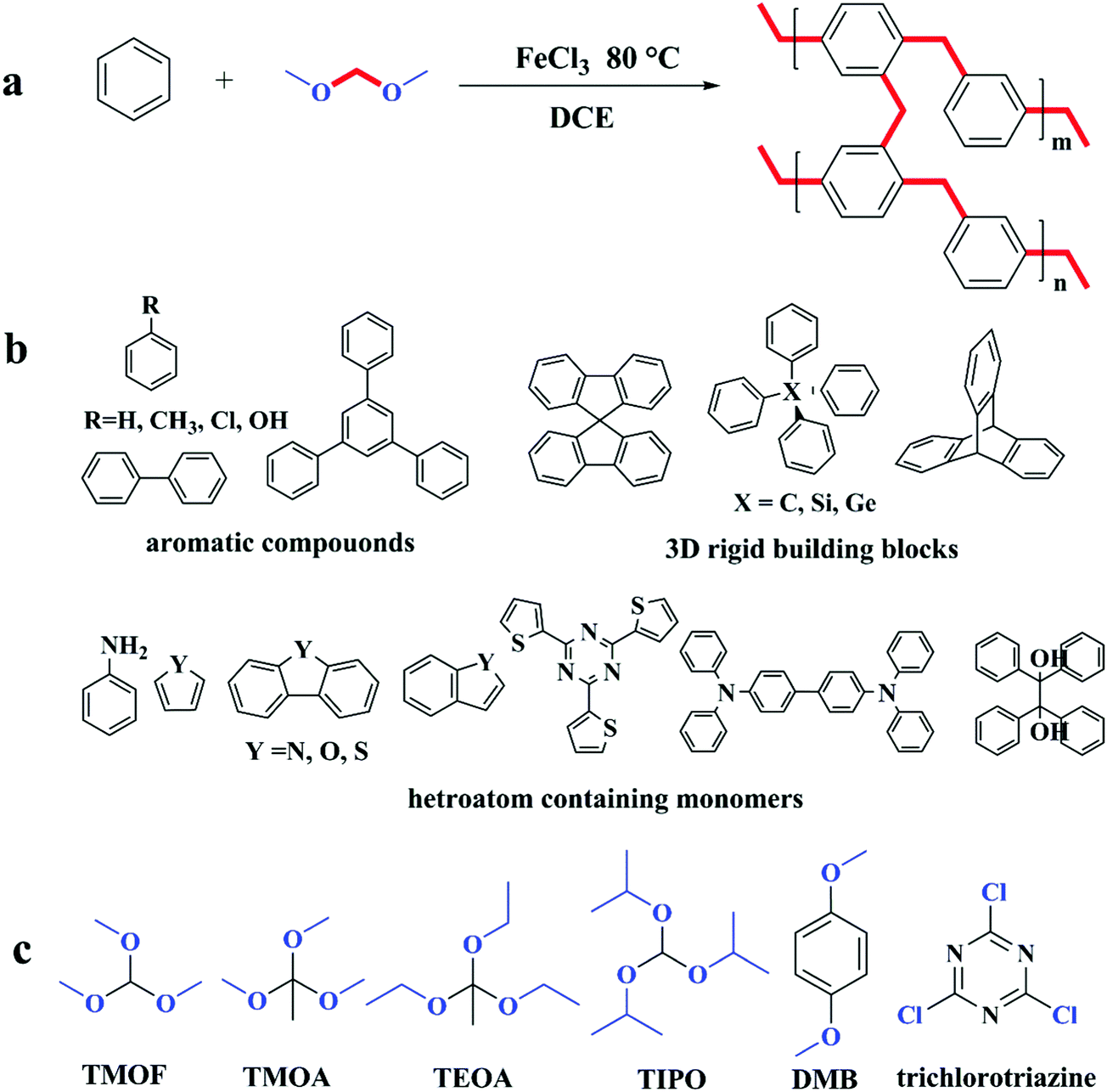 | ||
| Fig. 5 (a) The typical knitting aromatic polymer network from benzene monomers from ref. 86, (b) molecular structure of optional building blocks from ref. 86–90, 93, 98, 100, 181 and 182, (c) other types of crosslinkers from ref. 119, 120 and 122. Blue parts are functional groups to be eliminated in the hypercrosslinking process. | ||
Using the low-cost and effective knitting strategy, three dimensional spatial networks were successfully constructed from corresponding aromatic compounds including sFLUO, tetraphenyl-X (X represents C, Si, Ge center atoms), triptycene etc. Hypercrosslinked networks obtained by using tetraphenylmethane building blocks revealed varied surface areas from 948 to 1314 m2 g−1 with an increasing amount of FDA crosslinker which, however, decreased to 1162 m2 g−1 when the highest molar ratio of 30:1 (crosslinker to monomer) was chosen.87 It may be ascribed to the excessive linking between the monomer and the un-consumed crosslinker which eventually decreased the crosslinking degree. Compared to the carbon centered monomer, larger center atoms of Si and Ge produced comparable materials but with slightly lower surface areas of 1137 and 1059 m2 g−1, respectively.88,89 The synthesis of a series of sFLUO-based conjugated organic polymers was reported by Modak et al. starting from unsubstituted sFLUO monomers through FeCl3 mediated oxidative polymerization, Friedel–Crafts alkylation and a combination of both.90 The highest surface area of 1980 m2 g−1 was obtained using mixed reaction conditions. In contrast, isolated oxidative coupling only generated a much lower surface area of 940 m2 g−1 indicating the high efficiency in preparing high surface area materials via the knitting strategy.
Triptycene and its derivatives with a 3D rigid structure are widely applied in supramolecular chemistry and materials science fields and have already been used in the synthesis of PIMs.91,92 Zhang and co-workers prepared a series of triptycene-based knitting microporous polymers from triptycene,93 hexaphenylbenzene based triptycene94 and tricarbazolyltriptycene monomers.95 According to the N2 adsorption/desorption analysis, the resulting materials exhibited abundant micropores with surface areas of up to 1469, 569 and 1426 m2 g−1, respectively, indicating the high efficiency of triptycene based monomers in the synthesis of hypercrosslinked microporous networks. However, hexaphenylbenzene based triptycenes produced a relatively low surface area which may be due to the steric effect of the big molecules that reduced the crosslinking degree. Jin et al. also reported the synthesis of triptycene-based polymers by directly crosslinking triptycene monomers with FDA crosslinkers which revealed a comparable value of the surface area up to 1340 m2 g−1.96 Starting from four differently functionalized triptycene monomers, including amino (–NH2), formyl (–CHO), acetyl (–COCH3) and nitro (–NO2) functional groups, Peng and co-workers achieved a series of pre-functionalized triptycene-based 3D rigid polymer networks.97 Owing to the different activities of the side groups, materials with a wide range of surface areas from 863 m2 g−1 (with amino group) to 140 m2 g−1 (with nitro groups) were obtained. Moreover, alkyl-substituted amino groups were further incorporated into the amino containing network by post-synthetic modification leading to an enhanced CO2 adsorption capacity.
Except for rigid monomer units with pendant functional groups, the knitting strategy can also be directly used for crosslinking heterocyclic aromatic building blocks. Luo et al. synthesized heteroatom decorated knitting polymers using three typical heterocyclic molecules: pyrrole, furan and thiophene.98 Owing to a low crosslinking density, the obtained BET surface areas were lower compared to the benzene knitting polymer network displaying different values of 437 (Py-1), 514 (Fu-1) and 726 m2 g−1 (Th-1). However, the introduction of heteroatoms increased the availability of lone electron pairs in the network skeleton which enhanced the binding affinity to CO2 molecules by dipole–dipole interactions. Very recently, Cooper and co-workers reported the synthesis of ultra-high porous carbon materials using Py-1, Th-1 and benzene knitting polymers as carbon sources via the KOH activation strategy.99 Under optimized carbonization conditions, a specific surface area of 4334 m2 g−1 was obtained after anaerobic pyrolysis of Py-1 polymers. With such a high surface area and a large pore volume, these carbon materials could be promising candidates in adsorption application fields. Saleh et al. explored more heterocyclic monomers including indole, benzothiophene, benzofuran, carbazole, DBF and DBT for the synthesis of heteroatom functionalized knitting polymers.100 Based on the N2 sorption analysis, the synthesized polymer networks showed diversified surface areas in the range of 391–1022 m2 g−1.
Recently, the knitting strategy was also confirmed to be highly efficient for hypercrosslinking aromatic polymer precursors such as styrene-based polymers, linear polyphenylenes and block copolymers. Cooper and co-workers synthesized a series of hypercrosslinked polystyrenes by directly knitting linear monodisperse polystyrene with different degrees of polymerization.101 At a lower degree of 5, limited porosity was obtained with an extremely low surface area of 9 m2 g−1 which, however, increased to the highest value of 974 m2 g−1 with 45 degree polymerization. Moreover, the surface areas of the resulting networks increase gradually with the degree of polymerization below 20. Tan and co-workers further investigated the synthesis of soluble HCPs by intramolecular knitting of commercially available polystyrene.30 At very low concentration, the polystyrene precursors are highly dispersed and expanded which prevent the crosslinking of two neighboring chains. On the other hand, linkage bridges are more likely formed between two phenyl rings from the same chain. As a result, the single chain crosslinked polystyrene showed a solution-processing property and can be produced as thin membranes using a simple filtration technique.
Zhang et al. prepared a series of HCPs by directly linking polyphenylenes of different molecular weights by using CCl4 and FDA as the crosslinkers.56 The surface area and pore volume can be tuned by changing the molecular weight of the linear polyphenylene precursors which exhibited an increasing trend at first and then a decreasing one with increasing molecular weight. Both crosslinkers revealed the same trend; however, FDA crosslinkers played a more significant role in producing a microporous architecture. Therefore, the surface areas obtained by employing FDA crosslinkers were higher especially using large molecular weight polymer precursors.
Huang and co-workers did some research on synthesizing microporous organic nanotube networks (MONNs) by combining the formation of molecular templating precursors of core–shell bottlebrush copolymers and in situ hypercrosslinking with FDA crosslinkers.102–107 A typical synthetic scheme of amino-containing MONNs is shown in Fig. 6. Firstly, a bottlebrush like copolymer with aromatic side polymeric chains is synthesized by a combination of atom transfer radical polymerization, ring-opening polymerization and reversible addition–fragmentation chain transfer polymerization (RAFT) which also results in a multi-layer structure consisting of a poly(glycidyl methacrylate) backbone (shown in Fig. 6 as black color), an intermediate PLA layer (blue color) and a functional amino layer (green color) as well as an outer PS shell (red color). The pendant PS shell was then knitted with FDA crosslinkers to form a permanent microporous hypercrosslinked network which was simultaneously retained after removing the PLA inner core. Finally, the hollow nanotube network is obtained with a hierarchical trimodal micro, meso- and macroporous architecture. Furthermore, by incorporating an additional polymer layer with special functional groups such as thiol,103 amino,104,105 carboxyl,106 and sulfonic acid,104 MONNs with multi functionality were produced which were further extended for more applications in catalysis, adsorption and fast separation.
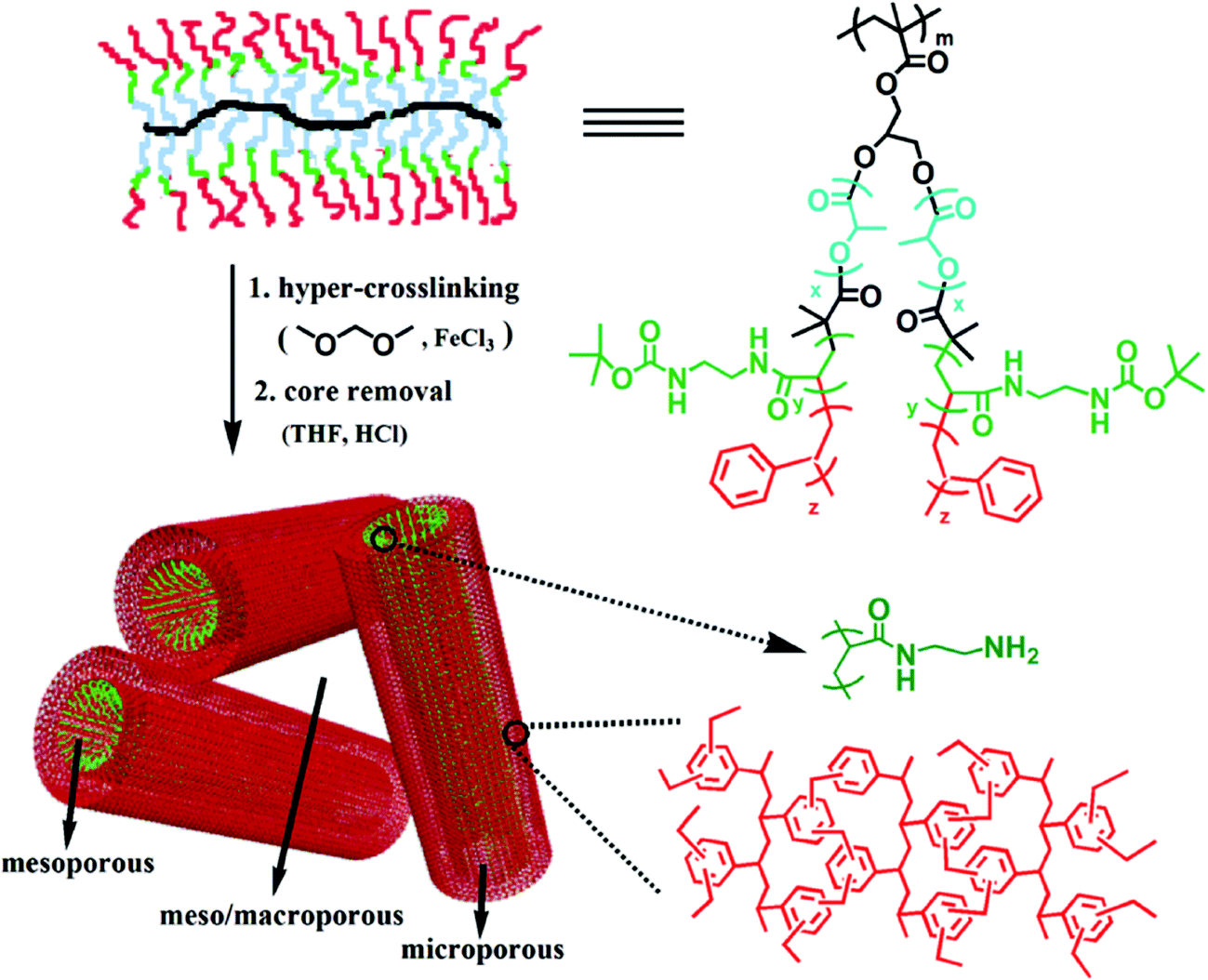 | ||
| Fig. 6 Preparation of the microporous organic nanotube networks with amino functionality. Reproduced with permission from ref. 105. Copyright 2016, The Royal Society of Chemistry. | ||
Except for the above mentioned factitious building units from industrial chemicals, the knitting strategy also presents a versatile performance in synthesizing microporous materials from natural products or raw sources such as lignin or pitch. Renewable organosolv lignin (OL) building blocks were employed to prepare a novel kind of solid absorbent by Weber et al. via FDA knitting.108 According to the N2 sorption analysis, the hypercrosslinked OL (OL-HC) showed a rather low surface area i.e. less than 5 m2 g−1, however with an extremely high CO2 adsorption capacity of about 6.0 wt%. It may be ascribed to the ultra-small pore size that cannot be probed by N2 molecules as well as abundant hydroxyl groups. The specific surface area of OL-HC increased to 253 m2 g−1 as calculated by the GCMC method from CO2 adsorption at 0 °C. After carbonization at 550 °C, much higher CO2 adsorption was obtained i.e. almost two times the initial value. Being very abundant, OL can be considered as a promising candidate to construct microporous materials for gas storage applications. Recently Li and co-workers demonstrated the large-scale preparation of high surface area pitch-based microporous polymers via Friedel–Crafts chemistry.109,110 Pitch is a mixture of various small molecules produced from petro- and coal-chemical industries. However, the aromatic compounds in pitch such as phenols, polycyclic aromatic hydrocarbons (PAHs) and heterocyclic compounds are efficient building blocks in constructing microporous networks which resulted in surface areas of 758 and 1337 m2 g−1via the knitting and Scholl coupling methods respectively. In addition, these materials revealed comparable gas uptake abilities. The utilization of sustainable and inexpensive raw precursors such as pitch will open up a way to scale-up the preparation as well as practical applications of HCPs.
Microporous hybrid materials combining the specific features from inorganic materials as well as microporous polymers normally exhibit enhanced properties such as thermal stability and mechanical strength compared to pure organic porous materials. The knitting strategy was simultaneously confirmed as an efficient method for the fabrication of polyhedral oligomeric silsesquioxne (POSS)-based hybrids and magnetic hybrids. Wang et al. synthesized POSS-based microporous materials from commercial octaphenylsilsesquioxane building blocks via FDA knitting as well as the Scholl coupling reaction.111 The resulting networks showed predominantly microporous structures with surface areas of 795 and 472 m2 g−1 respectively. Zhang et al. selected a double-decker-shaped silsesquioxane as the building block which was directly crosslinked by FDA crosslinkers using the Friedel–Crafts reaction.112 The molar ratio of crosslinkers to monomers was varied from 4 to 20 resulting in a surface area increase from 411 to 923 m2 g−1 at first which decreased to 868 m2 g−1 afterwards. Tian et al. reported a novel series of hybrid microporous polymers prepared from octaphenylcyclotetrasiloxane building blocks.113 Scholl coupling and Friedel–Crafts reactions using FDA and DCX as crosslinkers were adopted providing the resulting materials with excellent thermal stability, high surface areas and controllable pore structures. Moreover, these composites can serve as desirable carbon precursors to fabricate microporous carbons. After carbonization and etching, the surface area revealed a large enhancement from 599 to 782 m2 g−1. Liu and co-workers have prepared a series of hybrid porous polymers based on the Friedel–Crafts reactions of the vinyl group functionalized POSS building block with various aromatic co-monomer units such as benzene, tetraphenylsilane, commercial polystyrene, triphenylphosphine (TPP) and triphenylphosphine oxide.114–118 The resulting hybrid materials displayed a variation in the surface area and pore volume by adjusting the mole ratio of POSS and co-monomers. The highest surface area of 1105 m2 g−1 was obtained from copolymerization of cubic octavinylsilsesquioxane and TPP monomers in a molar ratio of 1.5:0.8. Moreover, due to the high surface area and narrow pore size, these hybrid materials exhibited an excellent size-selective adsorption performance for different dyes and could rapidly remove rhodamine B.118
In summary, the knitting strategy which has been widely used for the creation of novel microporous materials not only provides versatile routes towards the molecular structure construction and incorporation of multitudinous special functionality but also inspires the rapid development of HCP materials prepared via other related synthetic strategies. Novel crosslinkers with methoxyl or chloride groups were developed for the construction of HCPs from aromatic compounds, which are summarized in Fig. 5c.
Zhang et al. reported the synthesis of TPA-based microporous networks accomplished by the FeCl3-promoted one-step oxidative coupling reaction and Friedel–Crafts alkylation in one-pot.119 Four kinds of common reagents including trimethyl orthoformate (TMOF), trimethyl orthoacetate (TMOA), triethyl orthoacetate (TEOA), triisopropyl orthoformate (TIPO) were used as the crosslinkers to adjust the porosity in the resultant networks. Meanwhile, a TPA knitting network using FDA as an external crosslinker was synthesized for comparison which showed a relatively low surface area of 841 m2 g−1. The highest specific surface area of 1543 m2 g−1 was obtained using TEOA crosslinkers.
Tan et al. carried out a further exploration to construct a long-range conjugated network by using a phenyl bridged crosslinker, dimethoxybenzene (DMB), to directly knit aromatic monomers.120 Varying the building units leads to the pore size change as well as the surface area increase from 424 to 800 m2 g−1. With rigid phenyl groups as the linkage, the resulting networks revealed a conjugated structure confirmed by the solid fluorescence excitation and emission spectra. Moreover, a bright blue fluorescence can be observed when exposing to 365 nm UV light indicating great potential in the optics field.
Schute et al. proposed a novel metal-free route for the synthesis of HCP materials based on the typical self-condensation polymerization of bis(chloromethyl) aromatics DCX, BCMBP, BCMA and knitting benzene polymers via the Brønsted acid catalysts such as trifluoromethanesulfonic acid as well as H2SO4.121 The same experiments were also conducted under conventional conditions using stoichiometric amounts of metal-based Lewis acids for comparison. The traditional FeCl3 catalyst provided much higher surface areas compared to that obtained from metal-free routes. However, the self-condensation of BCMBP monomers using trifluoromethanesulfonic acid as the catalyst resulted in a comparable surface area of 1842 m2 g−1 (vs. 1874 m2 g−1) indicating the possibility of synthesizing HCP materials in a more “green” way.
Recently, new kinds of triazine-based organic polymers were synthesized via a mild AlCl3-catalyzed Friedel–Crafts reaction in which 2,4,6-trichloro-1,3,5-triazine was used as a trifunctional crosslinker to react with various aromatic compounds including benzene, biphenyl, terphenyl, TPB, trans-stilbene, triptycene, FLUO and DBF.122–125 This resulted in polymer networks with different surface areas depending on the building units and displayed the highest value of up to 1668 m2 g−1 using triptycene monomers.
In general, the utilization of this simple and efficient knitting strategy has greatly expanded the variety of HCPs as well as their diversified applications by incorporating different functional monomers. Moreover, this strategy was also adopted for the preparation of microporous materials with a precisely controlled micro-morphology.
3. Structure and morphology
Normally, hypercrosslinking is a rapid and random process due to the essence of Friedel–Crafts alkylation. As the resulting highly crosslinked networks are very rigid and strongly fixed in an amorphous skeleton topology, normal analytical tools are not suitable for characterization such as X-ray diffraction (for inner structure investigation) or gel permeation chromatography (for molecular weight determination). Gas adsorption/desorption analysis is the most widely used technique to determine the inner porous structure of the materials, however in an indirect way, which may cause errors because polymer networks tend to swell in liquid or gaseous media.46 Although molecular construction in the nanoscale range is still a mystery, some assumptions or simulations have been proposed to have a better understanding of the porous structure.For polystyrene precursors crosslinked by methylene groups, the main structural unit is a spatially nonplanar cycle formed via linking the neighboring phenyl residues in two or more chain segments.57 The contour length is predominantly determined by the degree of crosslinking. At a relatively low crosslinking degree, the two pre-crosslinked chains are flexible and a third chain is allowed to insert between them by vibrational motion thus forming a large unstrained cycle from three chains. In another case of a high crosslinking degree, a second bridge may be formed between two phenyl residues near the first bridge increasing a small strained cycle from two chains. In both situations, a large number of interconnected cycles are produced and mutually condensed with other cycles thus providing high rigidity and helping to preserve the unstressed conformation of the final network.
Molecular simulations have already been employed in the investigation of the skeleton structure and the pore formation mechanism of various hypercrosslinked materials which are treated as complementary pieces of evidence to the experimental results. In addition, an important advantage of using such a simulated approach is the ability to explore the evolution of the porosity at each stage of hypercrosslinking via virtual synthesis.126 Molecular simulations of hypercrosslinked poly(styrene-co-vinylbenzyl chloride) were carried out by Colina and co-workers utilizing a Polymatic code to study the effect of the crosslinking degree on the formation of porosity and the evolution of a simulated pore structure is shown in Fig. 7.127,128 The crosslinking degree was controlled by varying the VBC content in the range of 25–100% of pre-formed copolymer precursors. The simulated progression of the pore formation throughout virtual synthesis, both during the crosslinking in the swollen state and moving from the swollen to dried states, indicated a gradual formation of pores with increased VBC content in the swollen state and a higher degree of crosslinking was required to prevent collapse of the pores when moving from the swollen to the dried states. The trends in the surface areas as a function of the VBC content were consistent with available experimental data. These simulation results have provided a better understanding of the structure and porosity of HCPs which are currently unobtainable experimentally, and allow for a more purposeful approach to adjust the properties of multitudinous materials for given applications.
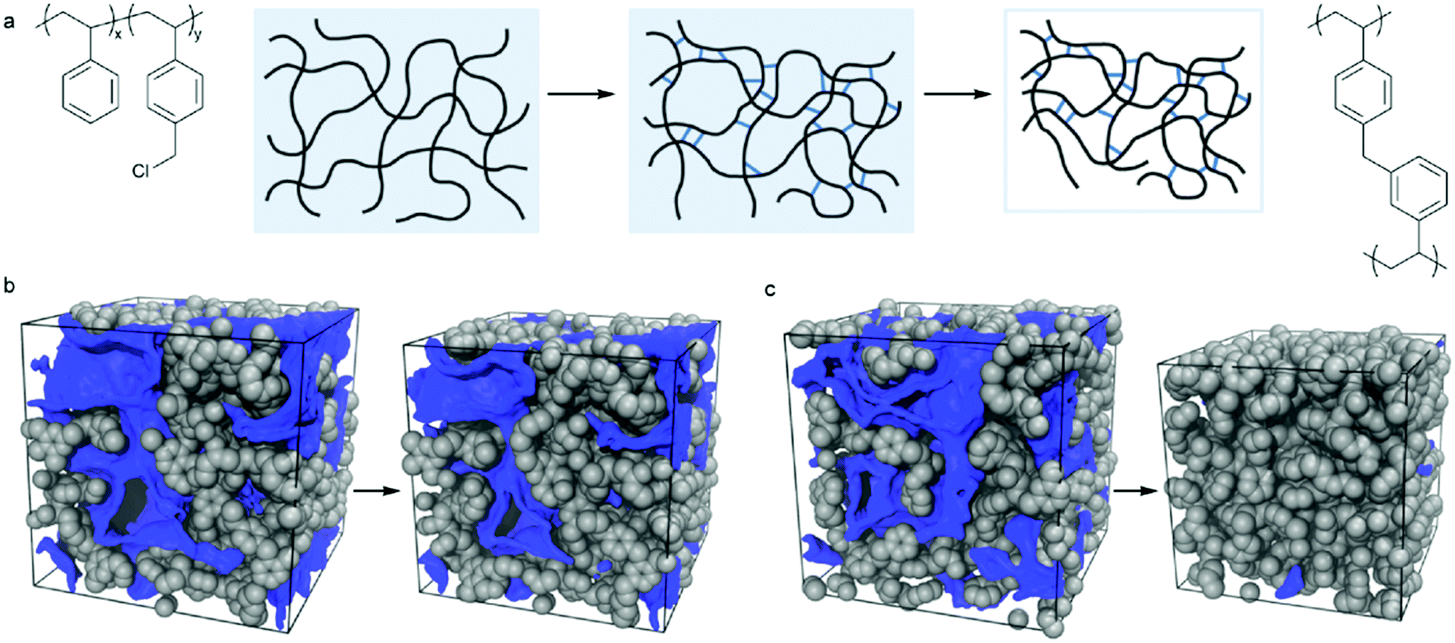 | ||
| Fig. 7 (a) Scheme showing the hypercrosslinked process and chemical structures of the poly(styrene-co-vinylbenzyl chloride) (St–VBC) polymer before and after hypercrosslinking, example of snapshots from the simulations for (b) VBC and (c) St–VBC (with 25% VBC) with pore surfaces mapped out by the center of a nitrogen-sized probe in blue. Reproduced with permission from ref. 127. Copyright 2014, American Chemical Society. | ||
In another study, the hypercrosslinking of bifunctional monomers DCX was simulated to mimic the atomistic structure constructed during self-condensation polymerization.129,130 The simulated structures in terms of inner porosity and surface area were compared to the experimental data as well as gas adsorption of hydrogen and methane which were found to be in agreement. Detailed investigation of the crosslinking procedure determined two important factors for producing microporosity in HCPs i.e. the degree of crosslinking and the density of the system. Specifically, a low density during crosslinking seems to be crucial for allowing pores to form within the network, while a higher number of crosslinks within the system are needed to lock the chains in the expanded state.
Even though the inner precise molecular structure plays a decisive role in determination of the essential physical properties of the hypercrosslinked materials such as packing density, thermal stability, porosity and surface area, the apparent morphology and state affect their potential application fields in reality.8 For instance, the regular aggregated nanoparticles show an accelerated adsorption rate for small gas molecules and enhanced gas storage capacity compared to their amorphous analogues.19 Compared to the numerous synthetic methods for developing materials with tunable porous structures or special functionality, the investigation of the polymer properties and applications affected by the material morphology is rare.
3.1 Nanoparticles
Hypercrosslinked polystyrene particles prepared via suspension polymerization are usually polydisperse with particle sizes in the micron range which is inconvenient for their practical applications in certain fields.34 Therefore, alternate polymerization methods have been developed to prepare hypercrosslinked materials with a more uniform particle size distribution such as emulsion polymerization (with a particle size of ∼420 nm),131 non-aqueous dispersion polymerization (with diameters in the range 4–10 μm)132 and precipitation polymerization (spherical particles with ∼4 μm).132 However, the synthesis of uniform hypercrosslinked nanoparticles with particle sizes less than 100 nm and the investigation of the particle size effect have not yet been realized.A series of monodispersed microporous polymer nanoparticles with a tunable nanoscale particle size were successfully synthesized by Tan and co-workers through post-crosslinking emulsion polymerized polyVBC–DVB particle precursors.19 The particle sizes varied from 131 to 36 nm by controlling the emulsifier amount, which were much smaller compared to the sizes of polydispersed hypercrosslinked suspension polymerized particles (with a typical particle size of 10–500 μm). Although hypercrosslinked nanoparticles have a higher surface area of up to 1500 m2 g−1, the smaller nanoparticles have a shorter gas diffusion path which could accelerate the rate of adsorption of hydrogen as well as enhance the heats of adsorption and adsorption capacity.
Wu and co-workers reported the preparation of monodisperse microporous polystyrene nanospheres via a versatile post-crosslinking strategy of nonporous spherical precursors.133 Initially, polystyrene nanospheres with diameters as small as 190 nm were obtained from traditional emulsion polymerization and were directly crosslinked by CCl4 in the presence of an AlCl3 catalyst. With a small particle size, an unreactive crosslinked outer skin was pre-formed as a protective layer at the very beginning of hypercrosslinking which minimized the undesired inter-sphere crosslinking thus achieving a monodisperse morphology. Recently, they proposed a facile method for the fabrication of multifunctional microporous spheres with water dispersible and pH/temperature responsive properties based on the combination of the hypercrosslinking chemistry via surface-initiated atom transfer radical polymerization (SI-ATRP).20 Monodisperse polyVBC–DVB nanoparticles were firstly synthesized using the surfactant-free emulsion polymerization and then hypercrosslinked to provide microporosity with a high surface area of up to 1323 m2 g−1. The as-prepared hypercrosslinked spheres were then utilized as macro transfer agents to graft hydrophilic poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) and poly(N-isopropylacrylamide) (PNIPAM) polymer chains which introduced additional water-soluble and stimuli-responsive properties. The stimuli-responsive phenomenon is described in Fig. 8. At low pH, = 2, PDMAEMA chains show a swollen state with a large diameter of 1364 nm due to the inter-chain electrostatic repulsion from protonated cationic polyelectrolytes. With an increase in pH (up to 10), PDMAEMA chains become deprotonated and the inter-chain electrostatic repulsion gets weaker leading to the contraction of the outer polymer shell resulting in the formation of much smaller particles i.e. 837 nm. The particle size was further reduced to 661 nm upon increasing the temperature to 50 °C. In a reverse stimulation order, the hairy spheres can display the same results indicating the efficient pH and temperature stimuli-responsive properties. In addition, the hairy thermo-decomposable polymer shell can protect the inner microporous structure during the carbonization process thus producing well defined microporous carbon materials after carbonization.
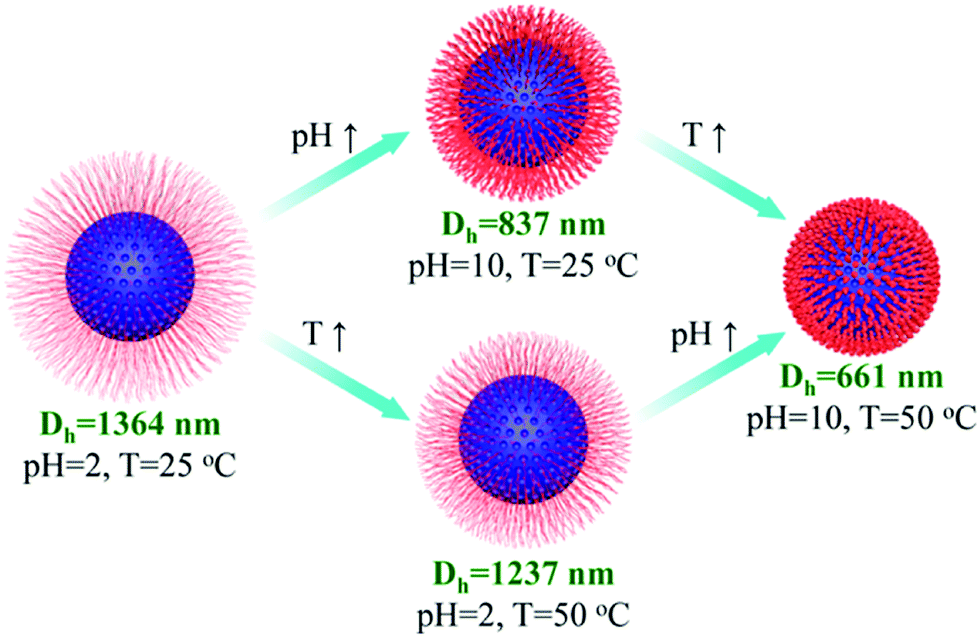 | ||
| Fig. 8 Illustration of hydrodynamic diameter change for the hairy microporous nanospheres at different pH and temperature stimulations. Reproduced with permission from ref. 20. Copyright 2015, American Chemical Society. | ||
The combination of porous polymers with inorganic magnetic materials increases the functionality of the resulting composite with easy separation properties. Tan and co-workers reported a synthetic route for the fabrication of magnetic microporous polymer nanoparticles (MMPNs) with a high surface area and superparamagnetic properties via mini-emulsion polymerization followed by a FDA knitting procedure.134 The obtained MMPNs were used as adsorbents for the removal of organic pollutants with an adsorption capacity of 53.3 mg g−1 for phenol and MMPNs can be easily recovered by applying an external magnetic field and thus can be recycled too. Ma et al. synthesized a hypercrosslinked magnetic polymer composite with improved acid resistance by coating Fe3O4 with vinyltriethoxysilane followed by polymerization.135 The resulting hybrid materials were highly stable at pH ≥ 2 and showed superparamagnetic properties with a specific surface area of 1022 m2 g−1. As a result, these kinds of materials surpassed commercial absorbents in p-nitrophenol adsorption and separation under optimal conditions at pH ≤ 4. Xu et al. demonstrated the preparation of nanoscale porous organic polymer composite microspheres consisting of Fe3O4 nanoparticles as the core and micro-/mesoporous polymer as the shell (Fig. 9).136 In this approach, the core–shell Fe3O4@PS microspheres were first synthesized, and then subjected to seeded swelling polymerization forming a blended shell of polyVBC–DVB and PS. Upon Friedel–Crafts type hypercrosslinking treatment, distinct phase segregation occurred with the extraction of non-crosslinked PS chains which resulted in the formation of mesopores. Furthermore, the mesopore size can be tuned from 4 nm to 30 nm by varying the amount of VBC and DVB. Based on their rapid magnetic responsiveness, hierarchical pore structure and high surface area, these composite nanospheres can easily be impregnated with Pt nanoparticles which showed high catalytic activity and specificity towards the enantioselective hydrogenation of ethyl pyruvate to ethyl lactate. Pan et al. proposed a novel one-step strategy to directly produce Fe3O4@HCP composite microspheres via a FeCl3-promoted self-polycondensation reaction between DCX and chloropropyltriethoxysilane functionalized Fe3O4 nanoparticles.137 Due to the high surface area of 770 m2 g−1, fast adsorption kinetics and large adsorption capacities were obtained for organic dyes such as methyl orange and basic fuchsine.
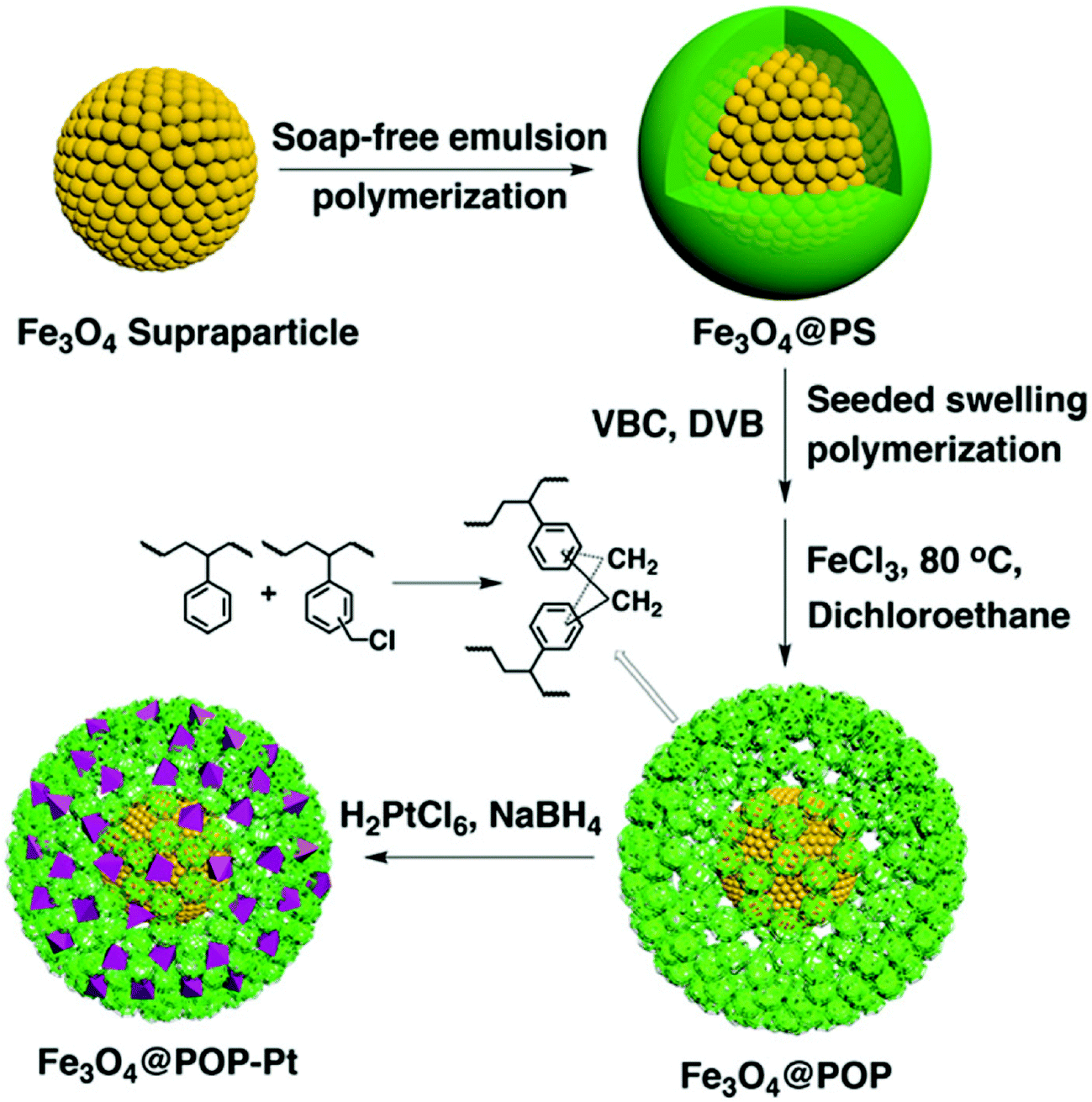 | ||
| Fig. 9 Schematic of the preparation of core–shell-structured Fe3O4@HCP microspheres. Reproduced with permission from ref. 136. Copyright 2015, The Royal Society of Chemistry. | ||
3.2 Hollow capsules
Hollow capsules have recently received much attention due to their inner hollow cavity showing great potential for the storage and controlled release of drugs/chemicals and use as nanoreactors.138 Normally, the formation of a core–shell structure is the prerequisite to get the hollow cavities after removing the core part. The templating strategy is the most frequently used technique in which both hard and soft templates are widely employed including silica, block copolymers and small molecular surfactants.Hollow microporous organic capsules (HMOCs) with a tunable particle size and a precisely controlled porous structure were fabricated by Tan and co-workers via a four-step templating method in which SiO2 nanoparticles were used as the core template.23,139,140 Nano sized SiO2 was firstly prepared using the Stöber method with vinyl functional groups decorated on the surface and was then used as seeds in the emulsion polymerization to form SiO2@PS–DVB core–shell spheres. Following the knitting and etching approach, the final HMOCs were obtained with a specific hollow cavity occupied previously by the SiO2 core and revealed a comparable surface area of up to 1129 m2 g−1. By varying the reaction conditions of emulsion polymerization, the particle size and the shell thickness can be easily tuned simultaneously. With increasing DVB content, the surface area of the HMOCs decreased and the micropore size became much narrower. Moreover, pure microporous networks were obtained when the DVB content was increased to more than 10% as confirmed by the nitrogen sorption analysis. HMOCs further acted as precursors to produce porous hollow carbon spheres (HCSs) with precise control over the pore morphology, hollow cavity, and the shell thickness via a direct carbonization strategy.140 Showing a distinct behavior compared with HMOCs, the surface area of the HCSs was initially decreased and then increased with increasing DVB content. The highest surface area of 816 m2 g−1 was obtained from the 15% DVB–HMOCs precursor with the lowest surface area of 478 m2 g−1. It can be ascribed to a decrease in the pore size owing to the shrinkage of the polymer network skeleton during carbonization. It resulted in the reduction of the particle size, a decrease in the pore size from 0.68 nm to 0.59 nm followed by a reduction in the shell thickness.
Following this research, nanorod-shaped hollow capsules with a tunable aspect ratio were also prepared employing a similar templating procedure but using silica nanorods as the template.141 The resulting hollow microporous organic nanorods with a comparable surface area revealed potential applications in gas storage and drug delivery systems.
Wu and co-workers developed various synthetic routes for the preparation of a series of porous materials with a controlled morphology of hollow capsules.22,142–146 Hairy nanoparticles were prepared by grafting polystyrene chains onto the surface of Br-functionalized silica nanoparticles via a SI-ATRP.142 The outer polymer shell was followed by an intra-/inter-particle crosslinking with CCl4 using the AlCl3 catalyst which resulted in a hollow structure with an average size of about 18 nm and a surface area of 417 m2 g−1. However, the hollow nanoparticles revealed an aggregated morphology due to inter-particle crosslinking. After a direct carbonization process, nanoporous carbon materials were obtained without destroying their hollow structure showing an increased surface area of 525 m2 g−1. Highly monodisperse hollow microporous polystyrene nanospheres were prepared using vinyl modified nano-sized SiO2 as a template followed by a combination of the emulsion polymerization and hypercrosslinking processes.143,146 With a highly rigid crosslinked polymer shell, these hollow nanospheres were confirmed to be efficient precursors for carbon material fabrication and exhibited a much higher surface area of 1166 m2 g−1. Recently, novel hollow structured periodic mesoporous polymer nanoparticles were designed by a conceptually different approach using reactive interface-guided co-assembly.22 In a model study, aldehyde group containing SiO2 nanoparticles were employed as the core template and reacted with resole to in situ form stable covalent bonds at the interface which also assisted the successful self-assembly of resole and triblock copolymer F127. After removing F127 porogen and the inner core template, monodisperse hollow polymer nanoparticles with highly periodic mesoporosity were generated. The PMMA-b-PS diblock copolymers can be easily self-assembled into uniform core–shell nanospheres without any other special treatment (Fig. 10).144 These pre-synthesized polymers were then used as precursors to construct microporous carbon materials by a combined hypercrosslinking process with a CCl4 crosslinker and carbonization while retaining their porous structure and morphology. In another study, a non-ionic surface active agent (Triton X-100) was used as the soft template in the chemical oxidation of pyrrole and aniline co-monomers to produce hollow capsules with ultra-small particles smaller than 100 nm.145 By a simple carbonization procedure with carefully selected carbon precursors and carbonization conditions, these nonporous polymer precursors were converted to highly porous hollow carbon capsules with a high surface area of up to 3022 m2 g−1.
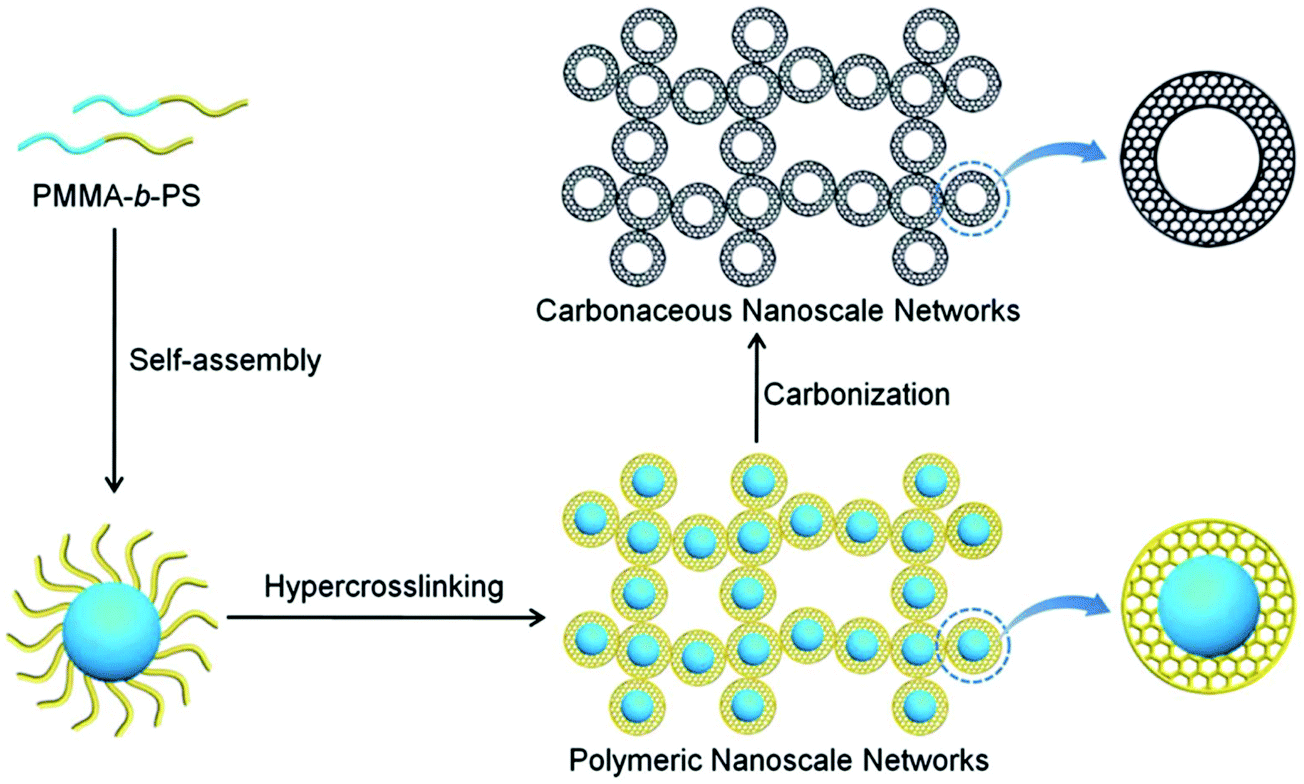 | ||
| Fig. 10 Schematic illustration of preparation of hollow microspheres from block copolymer self-assembly, hypercrosslinking and further carbonization. Reproduced with permission from ref. 144. Copyright 2014, The Royal Society of Chemistry. | ||
Hu and co-workers developed a bottom-up strategy for the synthesis of carbon hollow spheres through the Friedel–Crafts crosslinking of phenyl self-assembled monolayers on silica nanospheres followed by high temperature pyrolysis and template removal.147,148 However, the unavoidable inter-particle crosslinking caused large aggregation of the HCSs.
3.3 2D membranes
High-performance polymeric membranes have attracted enormous attention due to their superior molecular level separation for diverse applications including industrial-scale chemical, energy and environmental processes.149 However, most of the polymeric molecular sieve membranes are based on a few solution-processable polymers such as PIMs which limit their production on a large scale.39,40 Dai and co-workers developed a facile and low-cost approach for the synthesis of polymeric molecular sieve membranes in which nonporous PS membranes were fabricated by a simple coating method using the precursors which were then readily hypercrosslinked via the FDA knitting process (Fig. 11).26 The resulting knitting polymeric membrane showed a sandwich porous structure comprising a dense microporous layer with an inner macroporous core and an outer mesoporous surface formed by the crosslinking of small polymer particles. With hierarchical and tailorable porous structures, these membranes demonstrated exceptional performance in terms of both good gas permeability and selectivity for smaller gas molecules such as CO2 and O2, but relatively poor performance for larger N2 molecules. Anyway, this method provided a milder route for the synthesis of microporous polymer membranes and because of the low-cost commercial raw materials and easy handling reaction conditions, these membranes may have promising potential in large-scale gas separation.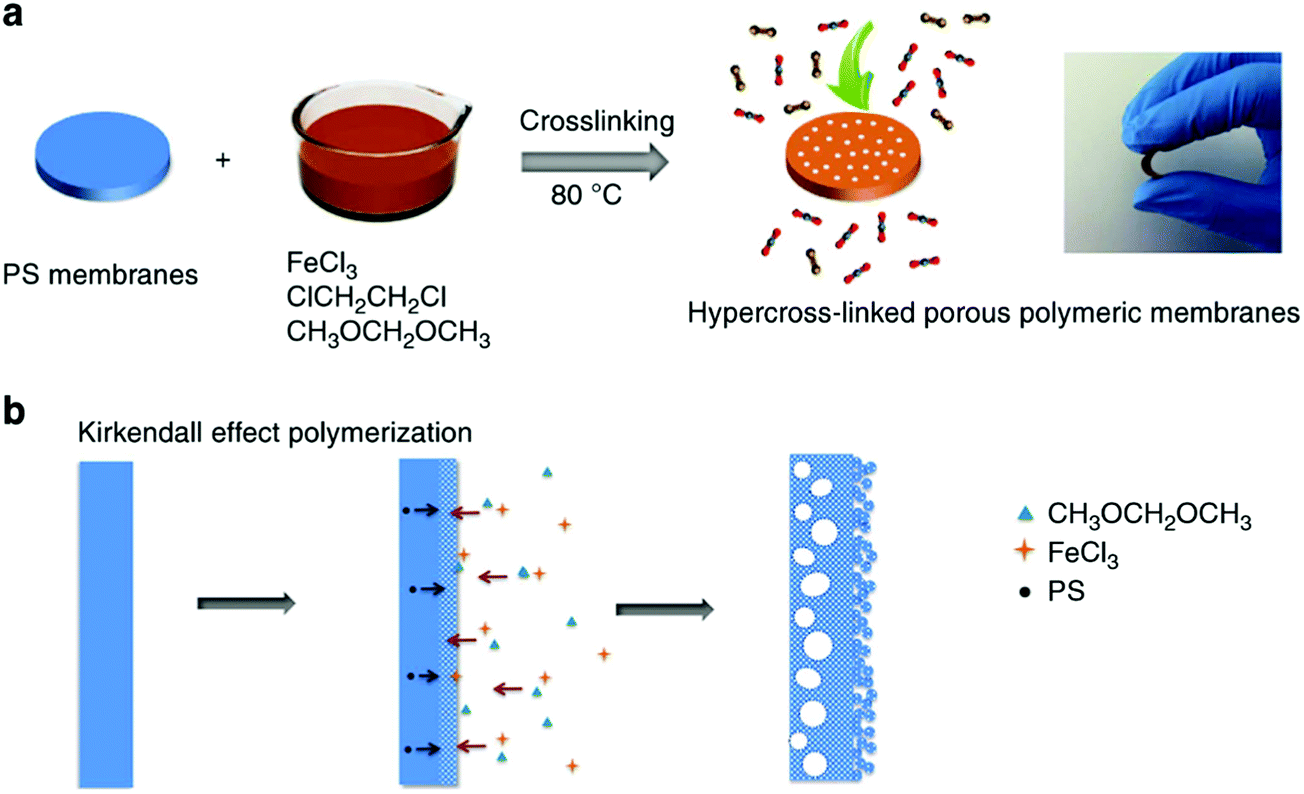 | ||
| Fig. 11 (a) Synthesis procedure of the in situ hypercrosslinked porous polymeric membranes, (b) the non-equilibrium diffusion at the interface in membranes by Kirkendall effect polymerization (a photo inserted to show the flexibility of the hypercrosslinked polymeric membranes). Reproduced with permission from ref. 26. Copyright 2014, Macmillan Publishers Limited. | ||
Based on this novel strategy, Chinnappan et al. synthesized knitting microporous polystyrene/ionic liquid membranes via an in situ crosslinking of polystyrene/ionic liquid membrane precursors.150,151 The ionic liquid was incorporated into the polystyrene matrix directly by dissolving in the DMF solvent. Followed by a casting and knitting process, the hypercrosslinked polystyrene ionic liquid membranes were obtained. The mixed ionic liquid molecules in the matrix can serve as ligands to coordinate other metal ions resulting in the production of effective catalysts for applications in H2 generation and 4-nitrophenol reduction.
Li and co-workers further extended this approach to the preparation of hierarchical porous polystyrene membranes in which the CO2-expanded liquid selective swelling and the static breath figure (BF) process were applied to produce the macroporous membrane precursors and subsequently the knitting process was adopted to fabricate micro-mesoporous membranes.152,153 The surface areas of the resulting porous membranes were not too different from those of Dai's work due to the same inherent hypercrosslinked polystyrene networks. However, the BF process generated a uniform macropore distribution of ∼2.5 μm on the surface of the membrane precursor which can be retained after the knitting process.
Zhao et al. reported another example for the synthesis of 2D microporous polymers via a RAFT emulsion polymerization approach and FeCl3-promoted Friedel–Crafts hypercrosslinking (Fig. 12).154 The synthetic route involves the use of a highly soluble trithiocarbonate functionalized graphene oxide as the micro-CTA for RAFT emulsion polymerization which provided a 2D template in this approach. The resulting nanosheets showed a multilayer structure with an inner graphene oxide core and an outer polyVBC–DVB shell. After hypercrosslinking, a high specific surface area of up to 1224 m2 g−1 was obtained with enhanced H2 and CO2 capacities which might come from the contribution of the 2D graphene template and the 2D morphology.
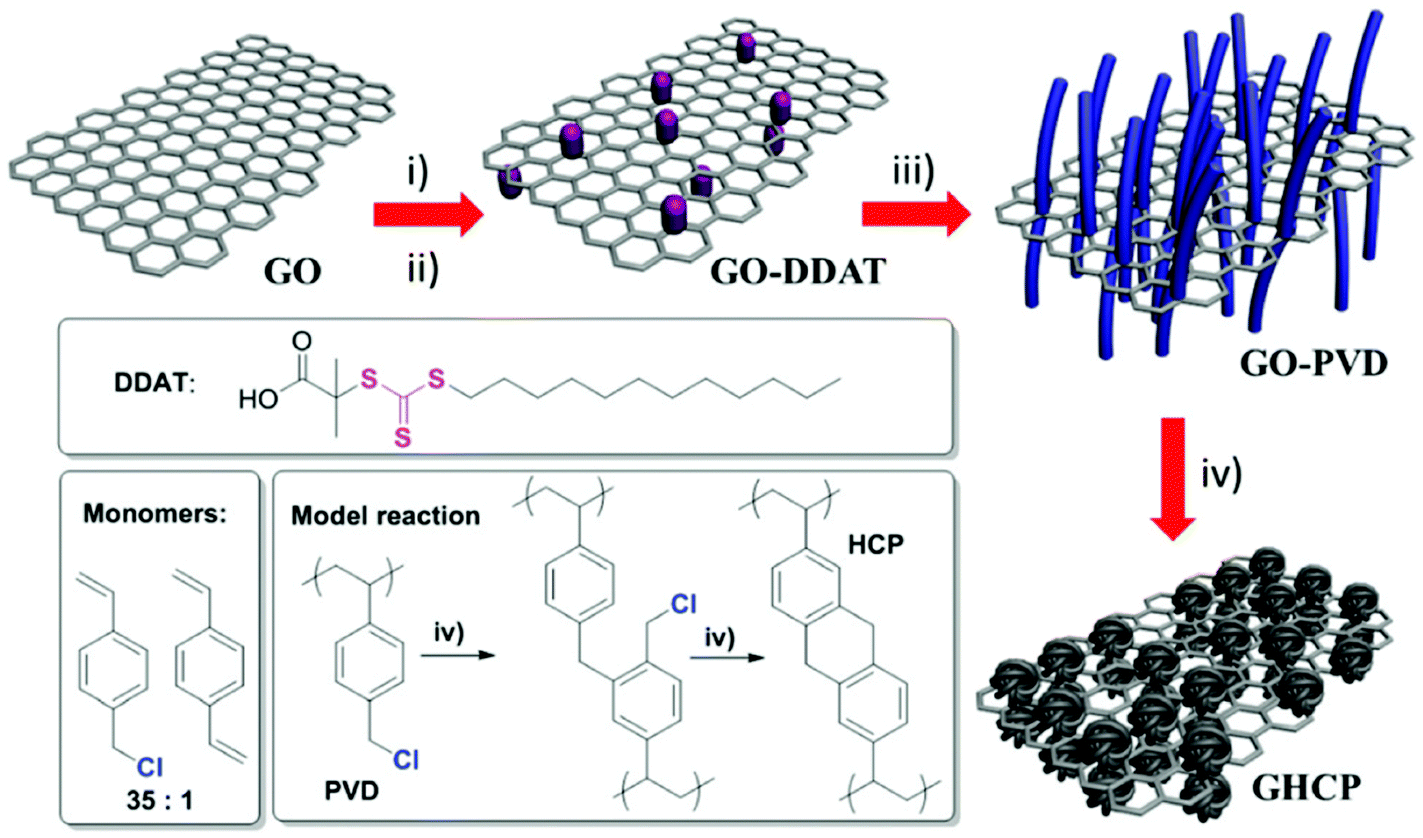 | ||
| Fig. 12 The synthetic pathway of 2D nanosheet hypercrosslinked polymers using graphene oxide templates. Reproduced with permission from ref. 154. Copyright 2015, The Royal Society of Chemistry. | ||
A novel study has been carried out recently by Cooper and Budd for investigating the permeability and aging property change of a composite membrane.155 Firstly, hypercrosslinked polyVBC particles with controlled particle sizes were synthesized via the traditional emulsion polymerization and then served as fillers to be incorporated into the platform PIM-1 forming a composite membrane. With an extremely small particle size of 55 nm, HCP nanoparticles were uniformly distributed into PIM membranes. In addition, due to the high surface area of approximately 1700 m2 g−1 as well as rigid structure from the HCP filler, the composite membrane not only showed a higher permeability but also a significant arrest in polymer aging and permeability loss. Wood and co-workers further investigated the physical aging behavior by adding a highly dispersible and scalable hypercrosslinked polymer additive based on the self-polycondensation of DCX monomers.156 The results revealed that these additives are capable of reducing physical aging in super glassy membranes which can also provide a significant processability advantage. In short, the use of HCPs as additives may open up a way to tailor physical aging thus commercializing high-performance, selective-aging membranes for future applications such as carbon capture.
3.4 3D monoliths
Adding a top third layer to the previous mentioned 2D planar materials in the vertical direction, a key macroscopic 3D morphology of monolithicity was obtained.157 Hierarchical porous monolithic materials which possess well-defined macropores, interconnected mesopores, and micropores are generally required in energy and environmental applications.158,159 The inter-connected macropores are beneficial for rapid mass transport and micropores act as active sites for many interfacial reactions. Moreover, the shape of a monolith can be easily adapted to any tank or filter geometry or other shapes and allows safe handling since dust formation is prevented.In the pioneering hypercrosslinked monolith synthesis, a monolith precursor was initially produced by either high internal phase emulsion polymerization (polyHIPE) or normal radical polymerization in capillary columns using aromatic monomers such as styrene, vinylbenzyl chloride, and divinylbenzene. The hypercrosslinking process was then conducted by internal post-crosslinking or crosslinking with external crosslinkers. Svec and co-workers reported the preparation of porous poly(styrene-co-divinylbenzene) monoliths with three kinds of external crosslinkers including DCX, BCMBP and FDA via an FeCl3 catalyzed Friedel–Crafts reaction in a capillary column.160 The reaction conditions were optimized to yield the highest surface area of around 900 m2 g−1 in a 2.5 h hypercrosslinking process with BCMBP crosslinkers. Owing to its high surface area and interconnecting hierarchical porous structure, the hypercrosslinked capillary monolith was used as the stationary phase for liquid chromatography and demonstrated a remarkable efficiency in the reversed phase separation of small molecules. Monolithic thin layers were also prepared between two surface modified glass plates filled with a 4-methylstyrene, chloromethylstyrene and divinylbenzene monomer mixture through a thermally initiated polymerization and post-crosslinking process.161 These monolithic layers revealed a well-defined porous structure with a globular morphology and were used for thin-layer chromatography of peptides and proteins.
Tan and co-workers further investigated the control of the morphology and porous structure of the hierarchical porous polystyrene monoliths and extended their potential application to oil cleaning and organic solvent separation from water.162 Polystyrene precursors with an interconnecting macroporous structure were obtained by polyHIPE from styrene and divinylbenzene co-monomers and then knitted with FDA crosslinkers forming a hierarchical porous structure. By increasing the DVB content in polyHIPE precursors, surface areas of the resulting monoliths showed a decrease trend from 595 to 196 m2 g−1 as demonstrated by N2 sorption analysis.
Microporous monoliths can also be prepared from the self-condensation of bifunctional monomers71 or by knitting aromatic building blocks86 in a tube directly. The monolithic materials were brown in color and had similar surface areas to that of the powder state. Pore size distribution by N2 sorption analysis showed a minor difference in the meso-macropore range that microporous polymers synthesized in bulk solution showed a broad distribution containing a portion of macropores. In contrast, monolithic materials exhibited a much purer microporous structure which may be due to the high concentration of the monomer resulting in a higher crosslinking degree and rigidity in the final network.
4. Applications
During the last few decades, hypercrosslinked polymers have undergone fast development in terms of both synthetic strategy and morphology control. HCP materials with a tunable porous structure or special functionalities can now be designed at the molecular level and successfully prepared by post functionalization as well as copolymerization with multifunctional building blocks. With adjustable physical and chemical properties, these HCP materials are promising candidates for potential applications in gas storage, carbon capture, removal of pollutants, molecular separation, catalysis, drug delivery, sensing.4.1 Gas storage
The hydrogen adsorption properties of hypercrosslinked polyVBC–DVB materials at low pressure were investigated by Fréchet and Svec.164 With an extremely high surface area of up to 1930 m2 g−1, this material can undoubtedly absorb 1.6 wt% hydrogen at a pressure of 0.12 MPa which is almost twice that adsorbed with commercially available macroporous polymer sorbents. Cooper and co-workers studied high pressure hydrogen adsorption using another “Davankov-type” resin prepared by the post-crosslinking of polyVBC precursors from suspension polymerization.165 A relatively high surface area of 1466 m2 g−1 was achieved with 3.0 wt% hydrogen adsorption at 77 K/15 bar. This is 60% higher than that achieved with the best-performing organic polymer reported at that time, however far from meeting the US Department of Energy (DOE) target (6.5 wt%). A series of self-condensed HCPs obtained from bis(chloromethyl) aromatic monomers were also studied because of their hydrogen sorption properties.70 The pure p-DCX condensation network with a moderate surface area showed higher hydrogen adsorption capacity compared with other networks that even showed a higher surface area at low pressure. This can be ascribed to its narrower pore size compared to other polymer networks which is helpful for the adsorption of small molecules. However, at a high pressure of 15 bar, surface area plays a more important role.
The phenomenon of “hydrogen spillover” observed over supported metal catalysts provides a possible design/synthesis strategy to produce materials with enhanced hydrogen adsorption capacity. Tan and co-workers proposed a new method for the synthesis of microporous HCPs with highly dispersed Pt nanoparticles.166 By incorporating 2.0 wt% Pt nanoparticles, the hydrogen storage capacity of HCPs was enhanced to 0.2 wt% at 298.15 K and 19 bar with an enhanced factor of 1.75. The higher hydrogen adsorption can be explained by the dissociation of hydrogen molecules on the Pt surface and the subsequent surface diffusion and adsorption of atomic hydrogen on the microporous polymer surface.
| HCPs | Monomer units | Synthetic strategy | BET surface area (m2 g−1) | CO2 uptakea (wt%) | Ref. |
|---|---|---|---|---|---|
| a CO2 adsorption was measured at 273 K, 1 bar (a) and 1.13 bar (b). | |||||
| KAPs-1 |
|
FDA knitting | 1391 | 13.5a | 86 |
| KAP-3 |
|
FDA knitting | 1059 | 15.9a | 86 |
| HCPs |
|
FDA knitting | 1470 | 13.0a | 172 |
| THPS |
|
FDA knitting | 1426 | 15.7a | 93 |
| HCPs |
|
FDA knitting | 1980 | 16.0a | 182 |
| HCPs |
|
FDA knitting | 1015 | 17.4a | 174 |
| PAF-32-OH |

|
FDA knitting | 1608 | 10.0a | 176 |
| PAF-32-NH2 |
|
FDA knitting | 1230 | 7.1a | 176 |
| TSP-1 |
|
FDA knitting | 563 | 13.2a | 177 |
| TSP-1 |
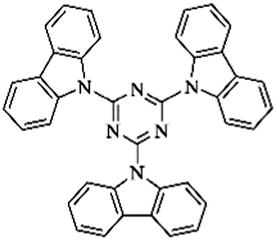
|
FDA knitting | 913 | 18.0a | 177 |
| CPOP-13 |
|
FDA knitting | 890 | 16.8a | 179 |
| CPOP-14 |
|
FDA knitting | 820 | 15.6a | 179 |
| CPOP-15 |
|
FDA knitting | 1190 | 14.2a | 179 |
| FCBCz |
|
FDA knitting | 1067 | 15.9b | 183 |
| FCTCz |
|
FDA knitting | 1845 | 20.4b | 183 |
| InCz-HCP1 |
|
FDA knitting | 750 | 9.9b | 185 |
| InCz-HCP2 |
|
FDA knitting | 1421 | 15.8b | 185 |
| FCDTPA |
|
FDA knitting | 871 | 12.5b | 181 |
| TCP-B |
|
FDA knitting | 1469 | 16.1a | 95 |
| HCPs-1 |
|
FDA knitting | 1236 | 12.9b | 184 |
| HCPs-2 |

|
FDA knitting | 1175 | 12.7b | 184 |
| HCPs-3 |
|
FDA knitting | 1147 | 12.3b | 184 |
| Py-1 |
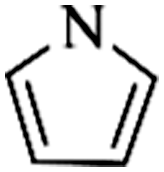
|
FDA knitting | 437 | 11.9b | 98 |
| Fu-1 |
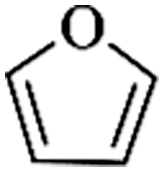
|
FDA knitting | 514 | 9.7b | 98 |
| Th-1 |
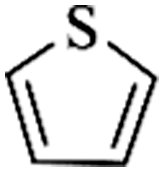
|
FDA knitting | 726 | 12.7b | 98 |
| HCPs | Pitch | FDA knitting | 695 | 11.8a | 109 |
| SMPs |
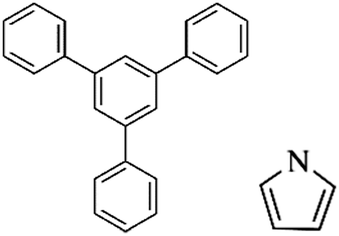
|
Scholl coupling | 1421 | 20.4a | 82 |
| PAF-41 |
|
Scholl coupling | 1119 | 15.5a | 82 |
| TCP-A |
|
Scholl coupling | 893 | 12.5a | 95 |
| PHCP-3 | Pitch | Scholl coupling | 1337 | 17.7a | 110 |
| HCP–BDM |
|
Self-condensation | 847 | 12.6a | 81 |
| HCP–BA |
|
Self-condensation | 742 | 8.5a | 73 |
| PP-N-25 |
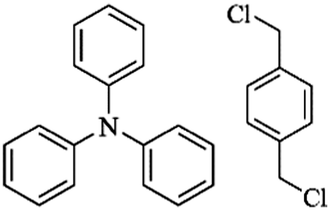
|
Self-condensation | 1257 | 20.2a | 73 |
| DMB-B |
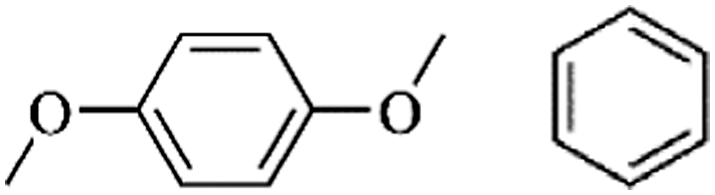
|
DMB knitting | 800 | 12.7b | 120 |
| HCPs |
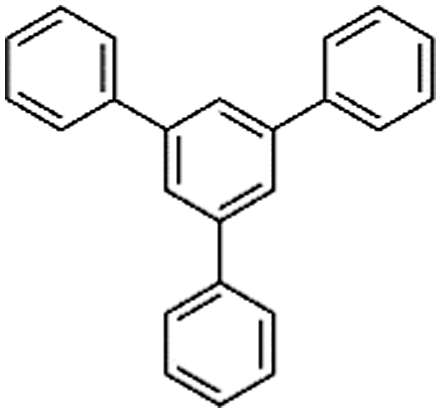
|
Solvent knitting | 2435 | 26.0a | 85 |
| HUST-1 |
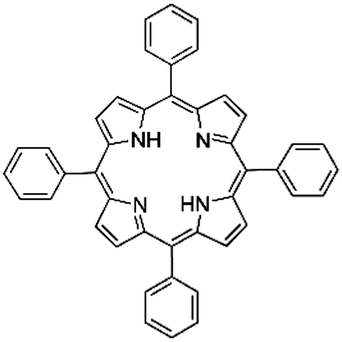
|
Solvent knitting | 1410 | 20.1a | 189 |
| FCDTPA-700 |
|
FDA knitting and carbonization | 417 | 12.8b | 181 |
| FCDTPA-K-700 |
|
FDA knitting and carbonization | 2065 | 28.6b | 181 |
| Ben750 |
|
FDA knitting and carbonization | 3105 | 14.5a | 99 |
| Th850 |
|
FDA knitting and carbonization | 2682 | 10.6a | 99 |
| Py800 |
|
FDA knitting and carbonization | 4334 | 15.4a | 99 |
Recent research has shown that the introduction of functional groups such as carboxyl and amine may enhance CO2 adsorption and selectivity for CO2/N2 by increasing the interactions between the adsorbent and the adsorbate.170,171 Based on this knowledge, Cooper and co-workers have developed a number of polymer series with various special functionality to investigate their performance for CO2 adsorption.172 The knitting strategy was initially adopted for the synthesis of a rigid 3D polymer network from tetraphenylmethane monomers (network E). A surface area of 1470 m2 g−1 was obtained with 13.0 wt% CO2 adsorption at 273 K/1 bar which is slightly lower than the triazole containing clicked network (network C, 17.0 wt% at 273 K/1 bar). This can be ascribed to the contribution of electron-rich triazole units in the networks to enhance the interaction between CO2 molecules and polymer networks. However, compared with a non-functional network (network A), even with a much higher surface area of 4077 m2 g−1, the network E exhibited a higher adsorption value.
Amino containing polymer networks were also obtained by copolymerization of aniline and benzene via FDA knitting.173 Owing to the strong electron donating effect, the incorporation of aniline into benzene led to a decrease in the surface areas from 1289 to 7 m2 g−1 as aniline concentration was increased from 0 to 100%. The CO2 adsorption capacity also decreased as the surface area was decreased. However, an improved CO2/N2 selectivity was obtained indicating the important role of functional amino groups in CO2 selectivity.
Complex adsorption conditions for post combustion CO2 capture such as water vapor and acidic impurities in gas streams were considered and investigated using several kinds of microporous materials.174 Alcohol containing polymer networks were synthesized by a Friedel–Crafts assisted knitting process with surface areas of up to 1015 m2 g−1 (for a binaphthol network). With the highest surface area and abundant hydroxyl groups, the binaphthol network showed the highest CO2 uptake ability among those tested under dry conditions which was substantially decreased by 50% under more realistic “wet” conditions due to the more polar structure. However, another network with more hydrophobicity showed a much smaller drop in CO2 uptake capacity i.e. 5%. CO2 adsorption behavior at high pressure was also studied among several different kinds of microporous materials including carbon, zeolite 13×, ZIF-8, HKUST-1 and benzene knitting polymers.175 By comparing CO2 adsorption capacities under various conditions, it was concluded that the hydrophobic benzene knitting polymers with strong hydrophobicity maintained their adsorption ability even under wet conditions and obtained a higher CO2 capacity and selectivity at high pressure due to physical swelling just like a sponge adsorbing water (Fig. 13). As a result, these knitting polymers prepared from very simple organic feedstocks have superior function as selective gas adsorbents as would be required for materials preparation on large industrial scales required for carbon capture.
 | ||
| Fig. 13 The swelling of knitting microporous polymers from benzene monomers in high pressure CO2. Reproduced with permission from ref. 175. Copyright 2014, American Chemical Society. | ||
Three kinds of knitting aromatic heterocyclic microporous polymers (Py-1, Fu-1, Th-1) were selected to investigate their CO2 adsorption and selectivity performance which revealed different behaviors at different pressure regions.98 At a high pressure of more than 1 bar, the adsorption capacity was very dependent on the surface area. As a result, Th-1 (726 m2 g−1) possessed the highest CO2 uptake i.e. 12.7 wt% at 273 K/1.13 bar. At a pressure lower than 0.2 bar, even with the lowest surface area of 437 m2 g−1, Py-1 showed a much higher adsorption capacity and rate in comparison with other polymer networks. Moreover, extraordinarily high selectivity of CO2/N2 about 117 was also obtained for Py-1 at 273 K, which was the highest value among all of the microporous materials reported at that time. Some other researchers also carried out related work on the design and synthesis of microporous polymer materials with special functionality for CO2 storage using the knitting strategy.173–185
Zhu et al. reported the preparation of amino and hydroxyl containing polymer networks based on knitting tetrahedral building blocks which showed apparent surface areas of up to 1230 m2 g−1 and 1608 m2 g−1 respectively.176 Owing to their functionality, the corresponding polymer networks revealed enhanced CO2 adsorption capacities and heats of adsorption.
Dai and co-workers also produced triazine and carbazole bifunctionalized porous polymers with surface areas of 563 and 913 m2 g−1 respectively.177 Although the surface areas were not very high, a comparable CO2 uptake (18.0 wt%, 273 K/1 bar) and CO2/N2 selectivity (38) were obtained. In addition, a series of novel carbohydrate-based microporous polymers were synthesized by knitting various hydroxyl-functionalized carbohydrate monomers.178 Several factors including the quantity and reactivity of hydroxyl groups and the structure of the carbohydrate monomers that contributed to CO2 adsorption were discussed indicating promising applications of these microporous polymers in carbon capture.
Han and co-workers selected two carbazole-based building blocks to construct porous polymer networks via FeCl3-promoted one-step oxidative coupling and FDA knitting reactions.179 N2 adsorption analysis confirmed that these networks were permanently microporous with average surface areas of around 1190 m2 g−1. Gas adsorption analysis showed that their H2 storage is up to 1.3 wt% at 1 bar and 77 K, and the uptake capacity for CO2 reached a comparable value of 16.8 wt% at 1 bar and 273 K. Another class of hypercrosslinked carbazole-based POPs was prepared via a FeCl3-promoted one-step oxidative coupling reaction and Friedel–Crafts alkylation from the vinyl or hydroxymethyl functionalized carbazole monomers.180 The specific surface area and CO2 adsorption performance showed similar results which can be up to 1130 m2 g−1 and 16.7 wt% (at 1 bar and 273 K), respectively. However, the hydrogen uptake capacity was enhanced up to 2.4 wt% at 1 bar and 77 K.
Jiang and co-workers adopted the knitting strategy to synthesize different kinds of microporous polymer networks containing various functional groups including amine,181 hydroxyl,182 carbazole,183 silole184 and indolo[3,2-b]carbazole.185 All of the resulting polymers showed enhanced CO2 capture ability due to their electron rich nature that is a strong binding affinity between CO2 molecules and pore channels. The polymer networks based on tetraphenylethylene and/or 1,1′,2,2′-tetraphenylethane-1,2-diol (TPD) displayed moderate surface areas ranging from1980 to 618 m2 g−1 with a decrease in CO2 adsorption capacities. However, the CO2/N2 selectivity was enhanced to the highest value of 119 with the TPD content increase. Another activated carbon material was obtained by activated carbonization of a nitrogen-rich polymer based on the knitting N,N,N′,N′-tetraphenylbiphenyl-4,4′-diamine (DTPA) monomer. The resulting carbon material with a high surface area of up to 2065 m2 g−1 was obtained exhibiting an exceptionally high CO2 uptake up to 28.6 wt% and a comparable CO2/N2 selectivity of 57.
Wang et al. prepared a series of nitro-functionalized HCPs from 4-nitrobenzyl chloride and styrene copolymerization with a chloromethyl methyl ether crosslinker using the Friedel–Crafts alkylation reaction.186 With an increase in the molar ratio of 4-nitrobenzyl chloride to styrene, the surface area initially increased and then decreased finally approaching the highest value of 1275 m2 g−1. CO2 adsorption analysis demonstrated that the pore texture and the N-content in the porous polymers were equally important for adsorbing CO2. Polymer networks with a moderate surface area and nitrogen content revealed the highest CO2 adsorption capacity. However, the equilibrium selectivity for CO2/N2 separation was well correlated with the N-content.
Recently, the CO2 adsorption and conversion technique has attracted a lot of attention as it combines the adsorption of CO2 molecules in the absorbent network as well as the conversion of CO2 into organic compounds. Zhang and co-workers designed two kinds of hypercrosslinked porous polymers incorporated by phosphonium salt and imidazolium salt within the polymer networks which showed a high surface area of 1168 and 926 m2 g−1 respectively.187,188 With incorporated salts as catalytic sites, these polymers revealed high catalytic activity even after several cycle reactions for the conversion of CO2 by converting propylene oxide into propylene carbonate. Tan and co-workers reported the preparation of metalloporphyrin-based microporous organic polymers which showed extremely high CO2 capture (21.4 wt%) as well as efficient chemical conversion of CO2 under ambient conditions.189 This polymer was synthesized by a novel strategy called the solvent knitting method using tetraphenylporphyrins as building blocks in which dichloromethane acted as both the solvent and crosslinker in the presence of an AlCl3 catalyst. A high surface area of 1360 m2 g−1 was obtained with a hierarchical porous architecture comprising abundant ultra-micropores and continuous meso-macropores which not only enhanced the interaction between the pore walls and CO2, but also was favorable for the catalysis process. After incorporating with Co(II) ions, this material revealed efficient catalytic (yields >93%) and recycling performance (more than 15 times) for the coupling of CO2 with substituted epoxides with various functional groups at room temperature and atmospheric pressure.
756 barrer when the thickness was changed to 86 μm indicating a much faster gas passing. Unfortunately, the N2 permeability was also enhanced which led to a decrease of CO2/N2 selectivity from 27 to 7 only. These results provide a clear direction for future investigation of polymeric sieve membranes that a balance needs to be considered in real industrial applications within gas permeability and selectivity. Cooper and Budd investigated the gas permeability variation of a composite membrane by adding HCP fillers into the PIM membrane matrix.155 After filling, the CO2 permeability increased 250% compared to the pristine PIM membrane which showed a 39.9% decrease after aging for 150 days. These results opened up a useful strategy for cost-effective and scalable synthesis of mixed matrix membranes for gas separations.
4.2 Removal of micropollutants
HCPs have a long history of being used as solid absorbents and being widely employed in solid phase extraction (SPE),190–195 treatment of waste waters,196,197 adsorption of organic vapors198 and chromatographic analysis199–201etc. Due to their abundant narrow micropores, the adsorption in HCPs is not only a classical surface phenomenon but also the adsorbate molecules are transferred from the surface to inner micropores through the channels.202 As a result, the adsorption capacity is greatly enhanced as well as the adsorption rate. In addition, HCP materials with a hydrophobic skeleton have a stronger affinity for organic molecules showing promising potential in water treatment compared with traditional inorganic materials such as activated carbon, zeolite or silica.Hypercrosslinked styrene-based polymers with strong ion-exchange character are well-suited for their use as SPE sorbents for the extraction of ionic species from real water samples.203,204 Novel polymers with hydrophilic character arising from 2-hydroxyethyl methacrylate monomers were prepared in the form of microspheres and then applied to an off-line SPE.190 With a large specific surface area and a moderate degree of hydrophilicity, the polymer absorbent showed a high recovery of ∼90% for various polar compounds such as oxamyl, methomyl, phenolic compounds as well as some pharmaceuticals from water, and out-performed the commercially available sorbents.
Huang and co-workers synthesized a series of polar modified hypercrosslinked resins by simple copolymerization of VBC with various functional building units and DVB.205–208 By adjusting the initial crosslinking degree of the precursor copolymers, the porosity and polarity of these resins could be effectively tuned. The post-crosslinked resin with the initial lower crosslinking degree possessed a much greater BET surface area and narrower pore size distribution, however less polarity; while that with a higher initial crosslinking degree had a smaller BET surface area as well as less micro/mesopores and considerably less macropores but more polarity. Owing to strong hydrogen bonding, hydrophobic and π–π stacking interactions, these resins revealed efficient adsorption and selectivity for aromatic compounds such as phenol, salicylic acid and aniline.
Ouyang et al. investigated the adsorption performance by means of the SPE process using both Davankov-type HCP nanoparticles as well as knitting aromatic polymers (KAPs) as solid absorbents.209,210 Owing to their high surface area, a narrow pore size and hydrophobicity, the as-synthesized HCP coated fibers exhibited a rapid and selective adsorption ability towards a series of aromatic compounds (including benzene, toluene, ethylbenzene and m-xylene generally referred to as BTEX), PAHs and highly hydrophobic long-chain n-alkanes. Based on the efficient adsorption performance, the HCP coated fibers were further adopted for the determination of BTEX and PAHs in environmental water samples revealing sensitive detection with low limits of detection of 0.10–1.13 ng L−1 and 0.05–0.49 ng L−1, respectively. Recently Li et al. synthesized a hypercrosslinked β-cyclodextrin porous polymer (BnCD-HCPP) by directly knitting benzylated β-cyclodextrin monomers via a Friedel–Crafts alkylation route.211 Due to a specific bifunctional structure of β-cyclodextrin comprising hydrophobic core and a hydrophilic shell as well as a high surface area, the obtained polymer network showed a high efficiency for the removal of aromatic pollutants from water. The adsorption efficiency which was defined as the ratio of adsorption capacity to the equilibrium adsorbate concentration ranged from 103 to 106 mL g−1 which was one order of magnitude higher than those of other β-cyclodextrin-based adsorbents reported previously. Moreover, BnCD-HCPP can be further functionalized by incorporation of gold nanoparticles for the catalytic transformation of adsorbed phenolic compounds such as 4-nitrophenol into less toxic 4-aminophenol.
The removal of toxic metal ions from wastewater was also investigated using HCP materials as solid absorbents. It is believed that the atoms in the ligand with lone pair electrons, such as N and S in the functional groups, may significantly enhance metal ion removal. The sulfonic acid-modified microporous HCPs were prepared by Li et al. via a sulfonation process of traditional hypercrosslinked ‘‘Davankov Resins”.212 After sulfonic acid modification, the original microporous structure and the spherical morphology were retained completely with a high surface area of 1025 m2 g−1 and the HCP was subsequently investigated as a highly efficient adsorbent for the removal of toxic metal ions. The permanent microporosity as well as massive sulfonic active sites rendered these materials with good adsorption capacity for metal ions such as Pb(II), Cu(II), Cr(III), Ni(II). Moreover, these modified resins can be recycled several times with minimal loss of their adsorption capacity and thus may have potential industrial applications. He et al. synthesized a triazine and thiophene bifunctionalized microporous polymer by a one-step FDA knitting process of 2,4,6-trithiophen-1,3,5-triazine monomers.213 A relatively low surface area of 255 m2 g−1 was obtained due to the electron rich properties of the monomer which resulted in low activity in the Friedel–Crafts reaction. However, on the other hand, the abundant N and S heteroatoms within the network have a stronger affinity for the metal ions, forming coordination complexes that led to high adsorption capacity for Cu(II) reaching a maximum of 98.33 mg g−1.
Desulfurization is a vitally important operation in petroleum refining since the combustion of sulfur containing fuels can produce toxic SOX compounds which cause serious environmental pollution. Hu and co-workers investigated the desulfurization efficiency of the methylbenzene knitting polymer network.214 A surface area of 803 m2 g−1 was obtained with a sulphur adsorption capacity of 9.68 mg S g−1. In addition, the large surface area as well as suitable mesoporosity ensured that this material was a good substrate for loading metal species and provided an improved desulfurization performance. Among all of the incorporated metals, Pd(II) performed the best reaching a saturated adsorption as high as 25.97 mg S g−1. Following this research, another task-specific triptycene-based polymer was synthesized and subsequently utilized as a desulfurization absorbent which exhibited an efficient saturated adsorption of DBT as high as 111.1 mg g−1.96 In addition, incorporation of uniform Ag(I) species inside the network was found to achieve a significantly higher uptake of 203.7 mg g−1 for DBT molecules. It can be ascribed to the multiple-site interaction of π–π stacking between DBT and phenyl rings as well as additional π-complexation adsorption with Ag(I) ions.
Nowadays, the emission of volatile organic compounds (VOCs) has also gained more and more attention not only because they cause damage to human health and the environment but also because of economic interests.215 Wang et al. prepared a series of well-developed micro-mesoporous hypercrosslinked polystyrene-based adsorbents and investigated their adsorption and desorption efficiency of VOCs from gas streams.216 Dichloromethane and 2-butanone were considered as two model molecules and achieved much higher uptake capacities of 1345.3 mg g−1 and 853.5 mg g−1, respectively, at 308 K, which were about 1.78 and 1.88 times higher than that of commercial porous absorbents under the same conditions. Hydrophobic benzyl chloride knitting polymers with a high surface area and a large pore volume were synthesized by Liu and co-workers as novel absorbents for the removal of benzene vapor from humid streams.198 The highest adsorption capacity of 1480 mg g−1 was achieved at 298 K for a polymer network with the largest surface area of 1394 m2 g−1. Dynamic adsorption experiments of benzene under dry and humid conditions (relative humidity = 80%) revealed that the existence of water vapor had little effect on the adsorption of benzene due to the hydrophobic network skeleton. Han and co-workers investigated the uptake capacity for poisonous and harmful organic vapors such as toluene and formaldehyde using carbazole-based microporous polymers.179,180 The high adsorption capacity for toluene can be up to 1470 mg g−1 at the saturated vapor pressure owing to the hydrophobicity, high porosity, and the π–π interactions between toluene molecules and the polymer networks. However, the formaldehyde adsorption capacity was much lower i.e. 11.2 mg g−1.
The permanent microporous structure of HCP absorbents plays an important role in absorbing small organic compounds or metal ions, which however is not suitable for large scale removal of oil and organic solvents. Hierarchically porous monoliths with an interconnecting macroporous architecture were synthesized by knitting polystyrene HIPE precursors and investigated for their oil sorption ability.162 Due to their hydrophobic and superoleophilicity properties, the obtained monoliths showed fast adsorption kinetics to various organic solvents with comparable uptake capacities in the range of 800–1900% depending on the density of the organic solvent. Moreover, the monoliths can also efficiently adsorb floating oil and organic solvents on water surface indicating their promising applications in oil spill clean-up.
4.3 Chromatographic separation
Hypercrosslinked monolithic capillary columns containing an array of small pores are ideally suited for high efficiency isocratic separation.217 During the last few decades, significant effort has been made to improve the adsorption efficiencies of capillary columns for various small organic molecules as well as large biological molecules such as alkylbenzenes and their derivatives,218 uracil,219 proteins161 and phosphopeptides.220For example, hierarchical porous poly(styrene-divinylbenzene) monoliths were prepared by hypercrosslinked monolithic precursors via a FeCl3 catalyzed Friedel–Crafts reaction using three external crosslinkers including BCMBP, DCX and FDA.160 The highest surface area of 900 m2 g−1 was obtained by using a BCMBP crosslinker. The capillary columns containing hypercrosslinked monoliths were used as the stationary phase for liquid chromatography and provided a significant improvement in efficiency in the reversed phase separation of a mixture of acetone and six alkylbenzenes (Fig. 14). In addition, a comparable high column efficiency over 70000 plates m−1 was achieved. Another approach for the preparation of porous polymer monoliths possessing both a large surface area and functional groups was also developed, in which acetoxystyrene was used as a co-monomer and finally deprotected to produce networks decorated with reactive phenolic hydroxyl functionalities.221 The percentage of hydroxyl groups in the monoliths enabled the modulation of polarity of the stationary phase. As a result, capillary columns filled with HCPs showed good column efficiencies for the separation of small molecules using both reversed phase and normal phase chromatographic modes.
 | ||
| Fig. 14 Separation of acetone and six alkylbenzenes using monolithic poly(styrene-co-divinylbenzene) capillary column before (a) and after hypercrosslinking (b). Peaks: acetone (1), benzene (2), toluene (3), ethylbenzene (4), propylbenzene (5), butylbenzene (6), n-pentylbenzene (7). Reproduced with permission from ref. 160. Copyright 2014, Elsevier Ltd. | ||
Lu et al. reported the gas chromatographic separation performance using the benzene knitting polymer coated capillary column for the first time.222 The fabricated column exhibited a nonpolar nature that resulted in high separation performance for a series of VOCs even for the challenging ethylbenzene and xylene isomers which cannot be resolved even by the commercial 5% phenyl polysiloxane stationary phase. In addition, high column efficiency for n-dodecane was also obtained i.e. up to 7769 plates m−1. The relative standard deviations for five repeated determinations of the targets were 0.0–0.6%, 0.9–3.2%, 1.1–5.9%, 0.8–3.7% for the retention time, peak area, peak height and peak width, respectively, indicating a good repeatability for KAP coated capillary columns.
4.4 Catalysis
Tan and co-workers reported the synthesis of organometallic catalysts by incorporating Pd(II) ions into three HCP networks containing PPh3 as small molecular ligands.224,225 The synthetic routes showed three different pathways for the preparation of catalyst supports (Fig. 15). In detail, one platform was formed by directly knitting PPh3 and TPB (named as KAPs(Ph-PPh3) as shown in Fig. 15a) while the other two were prepared by grafting PPh3 onto pre-formed polymer networks (denoted PS-PPh2 in Fig. 15b and KAPs(Ph)-PPh3 in Fig. 15c). After incorporating Pd(II) ions, the resulting material KAPs(Ph-PPh3)-Pd showed the highest surface area of 1036 m2 g−1 indicating that the porous channels were communicating and not blocked by Pd(II) ions. The PPh3 ligands were uniformly distributed throughout the entire polymer matrix due to the similar molecular structure which resulted in a high dispersion of Pd active sites in the network. As a result, excellent catalytic performance such as high activity and selectivity can be achieved in the Suzuki–Miyaura cross-coupling reaction of aryl chlorides even under milder reaction conditions and in aqueous media. This work also highlights that the microporous polymers can not only play the role of support materials, but can also protect the catalyst and improve the catalytic activity. Following this, N-heterocyclic carbine-based porous polymers obtained by knitting N-heterocyclic carbine with benzene monomers were further adopted to successfully incorporate other metals such as palladium227 and copper.228 With substantial porosity and individual pore structures, the resulting catalysts doped with Pd afforded a rapid conversion of aryl halides and arylboronic acids in aqueous media. The copper catalysts displayed outstanding catalytic performance in a wide range of organic synthetic reactions including the oxidative condensation reaction of indole, 1,3-cyclohexandione and phenylglyoxal monohydrate as well as three-component click, Ullmann C–N coupling, and Glaser coupling reactions.
 | ||
| Fig. 15 The synthetic routes towards three different types of Pd loading heterogeneous catalysts. Reproduced with permission from ref. 224. Copyright 2012, Wiley-VCH Verlag GmbH & Co. | ||
KAPs were successfully synthesized by Ding and co-workers from copolymerization of TPP and aryl compounds and were directly used as the platform to support Rh catalysts.233 The resulting heterogeneous catalyst revealed higher activity and selectivity for the hydroformylation of higher olefins to aldehydes compared to silica based catalysts. Furthermore, a facile preparation of heterogeneous asymmetric hydrogenation catalysts was carried out by coordinating Ru into knitting aryl networks.234 The uniform incorporation of BINAP monomers into the polymer backbones led to a high activity and enantioselectivity for the asymmetric hydrogenation of β-keto esters.
Song et al. immobilized phosphomolybdic acid as the active sites onto the knitting aryl networks based on phosphomolybdic acid and benzene which can be used for the selective oxidation of olefins to epoxide with aqueous hydrogen peroxide (H2O2) as an oxidant.235 This heterogeneous catalyst revealed a solvent selective property i.e. higher activity and selectivity were obtained by using ethyl acetate as a reaction medium which, however, was reduced when the reaction was carried out in acetonitrile medium.
Another knitting aryl polymer from benzene and benzylamine copolymerization was also used as a catalyst support to load Ru nanoparticles.236 The RuCl3 precursor was reduced using NaBH4, ethylene glycol, and hydrothermal reduction to produce Ru(0) nanoparticles. Homogeneous dispersion of Ru nanoparticles was achieved due to the confinement of Ru precursors in the micropores resulting in excellent catalytic performance for the hydrogenation of nitroarenes at room temperature with high conversion, selectivity and stability.
Han and co-workers reported an in situ strategy for the synthesis of a gold nanoparticle incorporated carbazole-based polymer catalyst in which the surface-functionalized gold nanoparticles were pre-synthesized and further subjected to FeCl3-promoted oxidative coupling copolymerization.237 Thiol groups containing carbazole monomers not only acted as small molecular ligands to stabilize gold nanoparticles but also participated in the formation of microporous networks. Through this preparative approach, the gold nanoparticles did not occupy the cavities of the polymer thus avoiding the decrease of the surface area and pore volume. The catalytic activities were studied by the model reaction of reduction of 4-nitrophenol to 4-aminophenol which displayed an activity factor of up to 17.57 s−1 g−1.
Confining metal nanoparticles in the hollow spheres to make a yolk–shell structure is an effective way to introduce stable active sites which not only avoid aggregation to protect active metal sites but also offer tiny nanoreactors to be used in shape selective catalysis. Shi et al. developed a novel method for the preparation of gold encapsulated yolk–shell type polymer composites.238 The phenyl modified SiO2 template with a gold nanoparticle inside was further subjected to the FDA knitting process with methylbenzene co-monomers which produced a microporous polymer outer shell on the template surface. After etching, the SiO2 core was removed leaving gold nanoparticles trapped inside the cavity separately. The obtained material served as an efficient nanoreactor for the catalytic decomposition of cyclohexyl hydroperoxide. By applying a similar procedure, Liang et al. also prepared a hollow and yolk–shell structured polymer network encapsulating gold nanoparticles (Fig. 16).22 The surface modified SiO2 encapsulating gold nanoparticles were used as the core template onto which the periodic mesoporous polymer shell was grown using reactive interface-guided co-assembly. Having a mesopore channel for fast reactant diffusion, the obtained catalyst revealed good performance for the catalytic reduction of nitrobenzene and 4-nitrophenol. Yang et al. proposed a ship-in-bottle strategy for the fabrication of Pt nanoparticle embedding HMOCs.139 During the process, the initially prepared HMOCs were used as nanoreactors for the synthesis of polymer dot supports as well as for embedding Pt nanoparticles. Due to the high dispersion of Pt active sites, the obtained Pt catalyst exhibited excellent activity and selectivity for the nitroaromatic hydrogenation under mild conditions and the possibility of using alcoholic reaction media.
 | ||
| Fig. 16 The synthesis of yolk–shell structured Au@periodic mesoporous polymer nanoparticles by developing the approach of reactive interface-guided co-assembly. Reproduced with permission from ref. 22. Copyright 2015, The Royal Society of Chemistry. | ||
A series of MONNs synthesized by Huang and co-workers have been confirmed to be useful platforms to support Au101 and Pd104 nanoparticles or to directly serve as efficient metal-free organic catalysts102,103 for various heterogeneous catalysis. For instance, well-dispersed Au nanoparticles with an average size of 3.0 nm, synthesized by in situ reduction of HAuCl4, were anchored into a thiol-functionalized MONNs which showed remarkable catalytic performances for the reduction reaction of 4-nitrophenol.103 Carboxyl-containing MONNs with highly dispersed palladium nanoparticles were achieved by a thermal decomposition method.106 The obtained catalyst exhibited high activities for the Suzuki–Miyaura cross-coupling reactions and can be easily recovered and reused for several cycles. Amino-functionalized and sulfonic/amino-bifunctionalized MONNs with a large surface area and interconnected porosity showed highly efficient activity, selectivity and excellent reusability in the Knoevenagel condensation/Henry reaction and deacetalization–Knoevenagel cascade reactions, respectively.104,105
4.5 Drug delivery
HMOCs with special morphological features containing a hollow structure and microporosity have great potential to be used as storage materials or reaction chambers while supplying the necessary path for the design of controlled uptake/release systems. The investigation of drug loading and controlled release properties of HMOCs was carried out by Tan and co-workers using ibuprofen (IBU) as a model drug.23 The efficient loading was achieved in IBU hexane solution with a loading amount of 1.68 to 2.04 (IBU/HMOCs) calculated from the data obtained with UV-vis spectrophotometric and thermogravimetric analysis. While the solid HCP spheres showed a much lower loading of 0.80 indicating the entrapment of the drug inside the hollow cavity of HMOCs. The drug release was achieved by soaking the drug-loaded HMOCs in simulated body fluid (PBS, pH = 7.4, buffer solution). Comparing the drug release behavior of a series of HMOCs (as shown in Fig. 17a), it was concluded that the porous structure of the shell deeply affects the drug release kinetics. HMOCs with a combined meso- and microporous structure showed a regular first order kinetic model indicating that the release mechanism is mainly controlled by simple diffusion and the release rate is proportional to the amount of drug remaining in the cavity. However, the pure microporous HMOCs resulted in a constant drug release rate. It may be ascribed to the restricted diffusion of drug molecules in small micropore channels. (The different diffusion process for IBU release is illustrated in Fig. 17b.) In addition, the constant release rate stabilizes the drug concentration in blood which is highly desired in clinical therapy.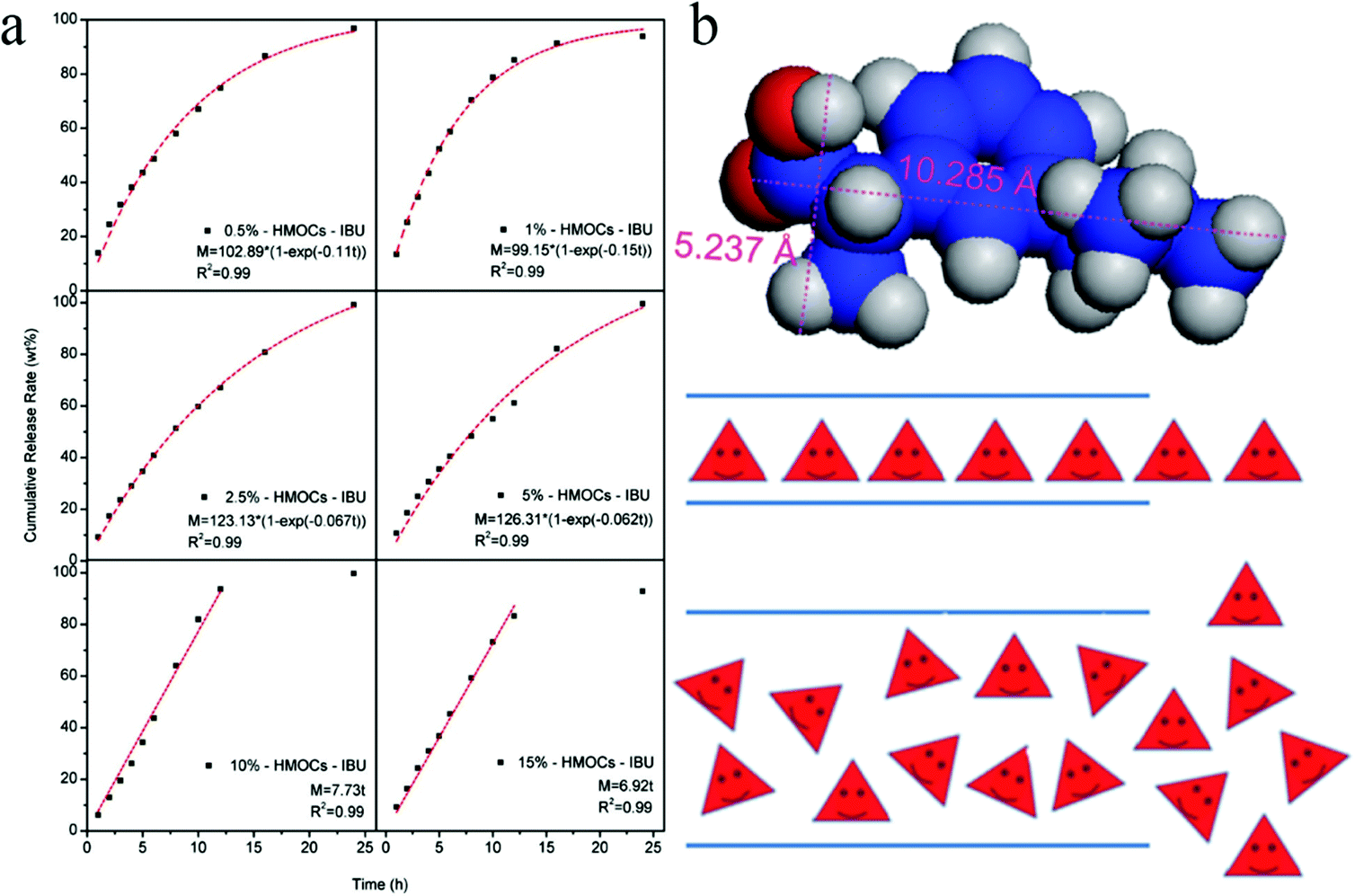 | ||
| Fig. 17 (a) Drug release profile of HMOCs with different DVB contents. Red line is the fitting line. (b) Simulated IBU molecules and different diffusion processes of release in micropores and mesopores. Reproduced with permission from ref. 23. Copyright 2013, Macmillan Publishers Limited. | ||
4.6 Sensing
Electrochemical humidity sensors were prepared successfully by Zhang et al. using the KAPs from 3-hydroxybenzoic acid building blocks.240,241 Even with rich hydroxyl and carboxyl groups, the pure polymer sensor did not show a visible change of impedance until the relative humidity (RH) reached as high as 54% RH. In order to enhance the hydrophilic properties of porous polymers, lithium modification was carried out using LiCl salt which enhanced the interaction between materials and water molecules. After loading LiCl salt, the modified sensor resulted in a decrease of impedance by three orders of magnitude in the whole humidity range of 11–95% RH indicating a much higher sensitivity. In another experiment, using lithium hydroxide instead of LiCl salt as a Li source significantly enhanced the sensitivity of electrochemical humidity sensors.5. Conclusions and future perspectives
Considering the design, versatility and conditions employed for the synthesis of HCPs, currently there are three main strategies that have provided firm control over the pore topology, and allowed introduction of diverse functionalities as well as unprecedented control over the material morphology, and are classified as post-crosslinking, one-step self-polycondensation and external crosslinking strategies. Furthermore, considerable progress in the development of strategies has provided great innovation in materials science by employing a wide range of optional building blocks. Thus, the use of a well-defined porous framework has been dramatically expanded to high value applications in terms of energy, environment, and health. For instance, hydrogen and methane storage, CO2 capture, removal of organic pollutants and metal ions, chromatographic separation, heterogeneous and photo catalysis, humidity sensing and drug delivery are the customized applications that can be easily accessed by employing the HCP architecture. In spite of these outstanding achievements, a number of challenges still exist that need to be addressed. For example, the polymer networks of HCPs are highly irregular due to the fast kinetics of the Lewis acid catalytic reaction. The synthesis of HCPs with precise control over their pore structure from a molecular design point of view is still a formidable challenge. At present, it is possible to produce benzene-based knitting polymers on the kilogram scale but further enhancing the production/yield is still a significant challenge. The main obstacle associated with hypercrosslinking is the huge amount of heat generated along with the liberation of corrosive hydrogen chloride by hydrolysis of the catalysts. Moreover, a huge disparity always exists between HCPs and ultrahigh surface area materials such as PAFs with a surface area of 5640 m2 g−1 that keeps inspiring scientists and researchers worldwide to look for new materials with desired properties. Meanwhile, except for the existing synthetic strategies, more and more polymerization methods can be explored for the synthesis of HCPs with special properties. Further approaches are being expected in the near future to produce HCPs with new functionalities that will keep up the research interest in this area to produce new effective materials for diverse applications.List of abbreviations
| MOFs | Metal–organic frameworks |
| PCPs | Porous coordination polymers |
| POPs | Porous organic polymers |
| COFs | Covalent organic frameworks |
| HCPs | Hypercrosslinked polymers |
| CMPs | Conjugated microporous polymers |
| PIMs | Polymers of intrinsic microporosity |
| CTFs | Covalent triazine frameworks |
| PAFs | Porous aromatic frameworks |
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| MCDE | Monochlorodimethyl ether |
| CCl4 | Tetrachloromethane |
| DCX | Dichloroxylene |
| BCMBP | 4,4′-Bis(chloromethyl)biphenyl |
| DPB | p,p′-Bis-chloromethyl-l,4-diphenylbutane |
| TCMM | Trifunctional tris-(chloromethyl)-mesitylene |
| BCMA | Bis(chloromethyl) anthracene |
| polySt–DVB | Polystyrene-co-divinylbenzene |
| polyVBC–DVB | Vinylbenzyl chloride–divinylbenzene copolymer |
| HCP-DVB–VBC | Hypercrosslinked divinylbenzene–vinylbenzyl chloride polymer |
| DCE | Dichloroethane |
| PLA | Polylactide |
| DBT | Dibenzothiophene |
| DBF | Dibenzofuran |
| FLUO | Fluorene |
| sFLUO | 9,9′-Spirobi(fluorene) |
| DBX | Dibromo-p-xylene |
| CBMB | Tribromomethylbenzene |
| TPB | Triphenylbenzene |
| TPA | Triphenylamine |
| BDM | 1,4-Benzenedimethanol |
| BA | Benzyl alcohol |
| FDA | Formaldehyde dimethyl acetal |
| MONNs | Microporous organic nanotube networks |
| RAFT | Reversible addition-fragmentation chain transfer polymerization |
| OL | Organosolv lignin |
| OL-HC | Hypercrosslinked organosolv lignin |
| POSS | Polyhedral oligomeric silsesquioxne |
| TPP | Triphenylphosphine |
| TMOF | Trimethyl orthoformate |
| TMOA | Trimethyl orthoacetate |
| TEOA | Triethyl orthoacetate |
| TIPO | Triisopropyl orthoformate |
| DMB | Dimethoxybenzene |
| SI-ATRP | Surface-initiated atom transfer radical polymerization |
| PDMAEMA | Poly(2-(dimethylamino)ethyl methacrylate) |
| PNIPAM | Poly(N-isopropylacrylamide) |
| MMPNs | Magnetic microporous polymer nanoparticles |
| HMOCs | Hollow microporous organic capsules |
| HCS | Hollow carbon spheres |
| BF | Breath figure |
| polyHIPE | High internal phase emulsion polymerization |
| DOE | Department of energy |
| CCS | Carbon capture and storage |
| TPD | 1,1′,2,2′-Tetraphenylethane-1,2-diol |
| SPE | Solid phase extraction |
| KAPs | Knitting aromatic polymers |
| BTEX | Benzene compounds |
| PAHs | Polycyclic aromatic hydrocarbons |
| VOCs | Volatile organic compounds |
| IBU | Ibuprofen |
Acknowledgements
We are grateful for the financial support given by the International S&T Cooperation Program of China (2016YFE0124400), the Program for HUST Interdisciplinary Innovation Team and the Fundamental Research Funds for the Central University (2016JCTD104), the Program for New Century Excellent Talents in University (NCET-10-0389) and National Natural Science Foundation of China (No. 21474033/51273074/51173058).Notes and references
- A. G. Slater and A. I. Cooper, Science, 2015, 348, 8075 CrossRef PubMed.
- J. Li, A. Corma and J. Yu, Chem. Soc. Rev., 2015, 44, 7112–7127 RSC.
- M.-M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. del Monte, J. H. Clark and M. J. MacLachlan, Chem. Soc. Rev., 2015, 44, 250–290 RSC.
- Y. Zhang, B. Y. W. Hsu, C. Ren, X. Li and J. Wang, Chem. Soc. Rev., 2015, 44, 315–335 RSC.
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 RSC.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed.
- D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959–4015 CrossRef CAS PubMed.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS.
- D. Chen, S. Gu, Y. Fu, Y. Zhu, C. Liu, G. Li, G. Yu and C. Pan, Polym. Chem., 2016, 7, 3416–3422 RSC.
- Y. Zhu, S. Wan, Y. Jin and W. Zhang, J. Am. Chem. Soc., 2015, 137, 13772–13775 CrossRef CAS PubMed.
- T.-Y. Zhou, S.-Q. Xu, Q. Wen, Z.-F. Pang and X. Zhao, J. Am. Chem. Soc., 2014, 136, 15885–15888 CrossRef CAS PubMed.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS PubMed.
- R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Macromolecules, 2010, 43, 8524–8530 CrossRef CAS.
- R. Dawson, A. I. Cooper and D. J. Adams, Polym. Int., 2013, 62, 345–352 CrossRef CAS.
- R. Dawson, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1173–1177 RSC.
- Y. Xu and D. Jiang, Chem. Commun., 2014, 50, 2781–2783 RSC.
- P. A. G. Cormack, A. Davies and N. Fontanals, React. Funct. Polym., 2012, 72, 939–946 CrossRef CAS.
- B. Li, X. Huang, L. Liang and B. Tan, J. Mater. Chem., 2010, 20, 7444–7450 RSC.
- W. Mai, B. Sun, L. Chen, F. Xu, H. Liu, Y. Liang, R. Fu, D. Wu and K. Matyjaszewski, J. Am. Chem. Soc., 2015, 137, 13256–13259 CrossRef CAS PubMed.
- J. Han, M. Wang, R. Chen, N. Han and R. Guo, Chem. Commun., 2014, 50, 8295–8298 RSC.
- Y. Liang, W. Mai, J. Huang, Z. Huang, R. Fu, M. Zhang, D. Wu and K. Matyjaszewski, Chem. Commun., 2016, 52, 2489–2492 RSC.
- B. Li, X. Yang, L. Xia, M. I. Majeed and B. Tan, Sci. Rep., 2013, 3, 2128 Search PubMed.
- Y. Shin, E. Prestat, K.-G. Zhou, P. Gorgojo, K. Althumayri, W. Harrison, P. M. Budd, S. J. Haigh and C. Casiraghi, Carbon, 2016, 102, 357–366 CrossRef CAS.
- P. M. Budd, K. J. Msayib, C. E. Tattershall, B. S. Ghanem, K. J. Reynolds, N. B. McKeown and D. Fritsch, J. Membr. Sci., 2005, 251, 263–269 CrossRef CAS.
- Z. Qiao, S. Chai, K. Nelson, Z. Bi, J. Chen, S. M. Mahurin, X. Zhu and S. Dai, Nat. Commun., 2014, 5, 3705 Search PubMed.
- F. Svec, J. Chromatogr. A, 2010, 1217, 902–924 CrossRef CAS PubMed.
- B. Beiler, Á. Vincze, F. Svec and Á. Sáfrány, Polymer, 2007, 48, 3033–3040 CrossRef CAS.
- P. M. Budd, E. S. Elabas, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib, C. E. Tattershall and D. Wang, Adv. Mater., 2004, 16, 456–459 CrossRef CAS.
- Y. Yang, B. Tan and C. D. Wood, J. Mater. Chem. A, 2016, 4, 15072–15080 CAS.
- J. Germain, J. M. J. Fréchet and F. Svec, J. Mater. Chem., 2007, 17, 4989–4997 RSC.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- N. Fontanals, R. M. Marce, F. Borrull and P. A. G. Cormack, Polym. Chem., 2015, 6, 7231–7244 RSC.
- S. Xu, Y. Luo and B. Tan, Macromol. Rapid Commun., 2013, 34, 471–484 CrossRef CAS PubMed.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230–231 RSC.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- S. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Adv. Mater., 2012, 24, 2357–2361 CrossRef CAS PubMed.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- T. Ben and S. Qiu, CrystEngComm, 2013, 15, 17–26 RSC.
- M. P. Tsyurupa and V. A. Davankov, React. Funct. Polym., 2002, 53, 193–203 CrossRef CAS.
- M. P. Tsyurupa and V. A. Davankov, React. Funct. Polym., 2006, 66, 768–779 CrossRef CAS.
- L. Tan and B. Tan, Acta Chim. Sin., 2015, 73, 530–540 CrossRef CAS.
- V. A. Davankov, S. V. Rogozhin and M. P. Tsyurupa, US. Pat., 3729457, 1971.
- V. A. Davankov, S. V. Rogoshin and M. P. Tsyurupa, J. Polym. Sci., Polym. Symp., 1974, 47, 95–101 CrossRef CAS.
- V. A. Davankov, G. I. Timofeeva, M. M. Ilyin and M. P. Tsyurupa, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 3847–3852 CrossRef CAS.
- P. Veverka and K. Jeřábek, React. Funct. Polym., 1999, 41, 21–25 CrossRef CAS.
- V. A. Davankov, M. M. Ilyin, M. P. Tsyurupa, G. I. Timofeeva and L. V. Dubrovina, Macromolecules, 1996, 29, 8398–8403 CrossRef CAS.
- A. V. Pastukhov, M. P. Tsyurupa and V. A. Davankov, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 2324–2333 CrossRef CAS.
- M. P. Tsyurupa, Z. K. Blinnikova, Y. A. Davidovich, S. E. Lyubimov, A. V. Naumkin and V. A. Davankov, React. Funct. Polym., 2012, 72, 973–982 CrossRef CAS.
- J. Hradil and E. Králová, Polymer, 1998, 39, 6041–6048 CrossRef CAS.
- D. Zhang, L. Tao, J. Ju, Y. Wang, Q. Wang and T. Wang, Polymer, 2015, 60, 234–240 CrossRef CAS.
- V. A. Davankov and M. P. Tsyurupa, React. Polym., 1990, 13, 27–42 CrossRef CAS.
- M. G. Schwab, I. Senkovska, M. Rose, N. Klein, M. Koch, J. Pahnke, G. Jonschker, B. Schmitz, M. Hirscher and S. Kaskel, Soft Matter, 2009, 5, 1055–1059 RSC.
- J.-H. Ahn, J.-E. Jang, C.-G. Oh, S.-K. Ihm, J. Cortez and D. C. Sherrington, Macromolecules, 2006, 39, 627–632 CrossRef CAS.
- B. Li, R. Gong, Y. Luo and B. Tan, Soft Matter, 2011, 7, 10910–10916 RSC.
- M. Seo, S. Kim, J. Oh, S.-J. Kim and M. A. Hillmyer, J. Am. Chem. Soc., 2015, 137, 600–603 CrossRef CAS PubMed.
- J. Germain, J. M. J. Frechet and F. Svec, Chem. Commun., 2009, 1526–1528 RSC.
- A. Warshawsky, A. Deshe and R. Gutman, Br. Polym. J., 1984, 16, 234–238 CrossRef CAS.
- V. Sharma, A. Sahoo, Y. Sharma and P. Mohanty, RSC Adv., 2015, 5, 45749–45754 RSC.
- O. W. Webster, F. P. Gentry, R. D. Farlee and B. E. Smart, Makromol. Chem., Macromol. Symp., 1992, 54–55, 477–482 CrossRef.
- C. Urban, E. F. McCord, O. W. Webster, L. Abrams, H. W. Long, H. Gaede, P. Tang and A. Pines, Chem. Mater., 1995, 7, 1325–1332 CrossRef CAS.
- K. J. Shea, D. A. Loy and O. W. Webster, Chem. Mater., 1989, 1, 572–574 CrossRef CAS.
- D. A. Loy and K. J. Shea, Chem. Rev., 1995, 95, 1431–1442 CrossRef CAS.
- H. Gao, L. Ding, W. Li, G. Ma, H. Bai and L. Li, ACS Macro Lett., 2016, 5, 377–381 CrossRef CAS.
- C. D. Wood, B. Tan, A. Trewin, H. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Khimyak, N. L. Campbell, R. Kirk, E. Stöckel and A. I. Cooper, Chem. Mater., 2007, 19, 2034–2048 CrossRef CAS.
- C. D. Wood, B. Tan, A. Trewin, F. Su, M. J. Rosseinsky, D. Bradshaw, Y. Sun, L. Zhou and A. I. Cooper, Adv. Mater., 2008, 20, 1916–1921 CrossRef CAS.
- M. G. Schwab, A. Lennert, J. Pahnke, G. Jonschker, M. Koch, I. Senkovska, M. Rehahn and S. Kaskel, J. Mater. Chem., 2011, 21, 2131–2135 RSC.
- Y. Yang, Q. Zhang, S. Zhang and S. Li, Polymer, 2013, 54, 5698–5702 CrossRef CAS.
- Y. Yang, Q. Zhang, S. Zhang and S. Li, RSC Adv., 2014, 4, 5568–5574 RSC.
- J. Zhang, Z.-A. Qiao, S. M. Mahurin, X. Jiang, S.-H. Chai, H. Lu, K. Nelson and S. Dai, Angew. Chem., Int. Ed., 2015, 127, 4665–4669 CrossRef.
- X. Huo, Q. Lan and Y. Wang, Ind. Eng. Chem. Res., 2016, 55, 6398–6404 CrossRef CAS.
- S. Bhunia, B. Banerjee and A. Bhaumik, Chem. Commun., 2015, 51, 5020–5023 RSC.
- G. Liu, Y. Wang, C. Shen, Z. Ju and D. Yuan, J. Mater. Chem. A, 2015, 3, 3051–3058 CAS.
- W. Chaikittisilp, M. Kubo, T. Moteki, A. Sugawara-Narutaki, A. Shimojima and T. Okubo, J. Am. Chem. Soc., 2011, 133, 13832–13835 CrossRef CAS PubMed.
- S. Yuan, D. White, A. Mason and D.-J. Liu, Int. J. Energy Res., 2013, 37, 732–740 CrossRef CAS.
- Y. Luo, S. Zhang, Y. Ma, W. Wang and B. Tan, Polym. Chem., 2013, 4, 1126–1131 RSC.
- B. Li, Z. Guan, X. Yang, W. D. Wang, W. Wang, I. Hussain, K. Song, B. Tan and T. Li, J. Mater. Chem. A, 2014, 2, 11930–11939 CAS.
- M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900–9930 CrossRef CAS PubMed.
- L. Li, H. Ren, Y. Yuan, G. Yu and G. Zhu, J. Mater. Chem. A, 2014, 2, 11091–11098 CAS.
- K. J. Msayib and N. B. McKeown, J. Mater. Chem. A, 2016, 4, 10110–10113 CAS.
- B. Li, R. Gong, W. Wang, X. Huang, W. Zhang, H. Li, C. Hu and B. Tan, Macromolecules, 2011, 44, 2410–2414 CrossRef CAS.
- M. Errahali, G. Gatti, L. Tei, G. Paul, G. A. Rolla, L. Canti, A. Fraccarollo, M. Cossi, A. Comotti, P. Sozzani and L. Marchese, J. Phys. Chem. C, 2014, 118, 28699–28710 CAS.
- P. Cui, X.-F. Jing, Y. Yuan and G.-S. Zhu, Chin. Chem. Lett., 2016, 27, 1479–1484 CrossRef CAS.
- P. Puthiaraj and W.-S. Ahn, Ind. Eng. Chem. Res., 2016, 55, 7917–7923 CrossRef CAS.
- A. Modak, Y. Maegawa, Y. Goto and S. Inagaki, Polym. Chem., 2016, 7, 1290–1296 RSC.
- B. S. Ghanem, K. J. Msayib, N. B. McKeown, K. D. M. Harris, Z. Pan, P. M. Budd, A. Butler, J. Selbie, D. Book and A. Walton, Chem. Commun., 2007, 67–69 RSC.
- B. S. Ghanem, M. Hashem, K. D. M. Harris, K. J. Msayib, M. Xu, P. M. Budd, N. Chaukura, D. Book, S. Tedds, A. Walton and N. B. McKeown, Macromolecules, 2010, 43, 5287–5294 CrossRef CAS.
- C. Zhang, P.-C. Zhu, L. Tan, J.-M. Liu, B. Tan, X.-L. Yang and H.-B. Xu, Macromolecules, 2015, 48, 8509–8514 CrossRef CAS.
- C. Zhang, P.-C. Zhu, L. Tan, L.-N. Luo, Y. Liu, J.-M. Liu, S.-Y. Ding, B. Tan, X.-L. Yang and H.-B. Xu, Polymer, 2016, 82, 100–104 CrossRef CAS.
- T.-L. Zhai, L. Tan, Y. Luo, J.-M. Liu, B. Tan, X.-L. Yang, H.-B. Xu and C. Zhang, Chem. – Asian J., 2016, 11, 294–298 CrossRef CAS PubMed.
- T. Jin, S. An, X. Yang, J. Hu, H. Wang, H. Liu, Z. Tian, D.-e. Jiang, N. Mehio and X. Zhu, AIChE J., 2016, 62, 1740–1746 CrossRef CAS.
- Y. He, X. Zhu, Y. Li, C. Peng, J. Hu and H. Liu, Microporous Mesoporous Mater., 2015, 214, 181–187 CrossRef CAS.
- Y. Luo, B. Li, W. Wang, K. Wu and B. Tan, Adv. Mater., 2012, 24, 5703–5707 CrossRef CAS PubMed.
- J.-S. M. Lee, M. E. Briggs, T. Hasell and A. I. Cooper, Adv. Mater., 2016, 28, 9804–9810 CrossRef CAS PubMed.
- M. Saleh, H. M. Lee, K. C. Kemp and K. S. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 7325–7333 CAS.
- T. Ratvijitvech, M. Barrow, A. I. Cooper and D. J. Adams, Polym. Chem., 2015, 6, 7280–7285 RSC.
- Z. He, A. Zhong, H. Zhang, L. Xiong, Y. Xu, T. Wang, M. Zhou and K. Huang, Macromol. Rapid Commun., 2016, 37, 1566–1572 CrossRef CAS PubMed.
- Y. Xu, T. Wang, Z. He, A. Zhong and K. Huang, Microporous Mesoporous Mater., 2016, 229, 1–7 CrossRef CAS.
- H. Zhang, L. Xiong, Z. He, A. Zhong, T. Wang, Y. Xu and K. Huang, New J. Chem., 2016, 40, 7282–7285 RSC.
- H. Zhang, L. Xiong, Z. He, A. Zhong, T. Wang, Y. Xu, M. Zhou and K. Huang, Polym. Chem., 2016, 7, 4975–4982 RSC.
- Y. Xu, T. Wang, Z. He, A. Zhong and K. Huang, RSC Adv., 2016, 6, 39933–39939 RSC.
- M. Zhou, H. Zhang, L. Xiong, Z. He, A. Zhong, T. Wang, Y. Xu and K. Huang, RSC Adv., 2016, 6, 87745–87752 RSC.
- Q. B. Meng and J. Weber, ChemSusChem, 2014, 7, 3312–3318 CrossRef CAS PubMed.
- W. Li, A. Zhang, H. Gao, M. Chen, A. Liu, H. Bai and L. Li, Chem. Commun., 2016, 52, 2780–2783 RSC.
- H. Gao, L. Ding, H. Bai, A. Liu, S. Li and L. Li, J. Mater. Chem. A, 2016, 4, 16490–16498 CAS.
- S. Wang, L. Tan, C. Zhang, I. Hussain and B. Tan, J. Mater. Chem. A, 2015, 3, 6542–6548 CAS.
- A. Zhang, H. Gao, W. Li, H. Bai, S. Wu, Y. Zeng, W. Cui, X. Zhou and L. Li, Polymer, 2016, 101, 388–394 CrossRef CAS.
- Z. Tian, J. Huang, Z. Zhang, G. Shao, A. Liu and S. Yuan, Microporous Mesoporous Mater., 2016, 234, 130–136 CrossRef CAS.
- L. Li and H. Liu, RSC Adv., 2014, 4, 46710–46717 RSC.
- Y. Wu, D. Wang, L. Li, W. Yang, S. Feng and H. Liu, J. Mater. Chem. A, 2014, 2, 2160–2167 CAS.
- W. Yang, D. Wang, L. Li and H. Liu, Eur. J. Inorg. Chem., 2014, 2976–2982 CrossRef CAS.
- Y. Wu, L. Li, W. Yang, S. Feng and H. Liu, RSC Adv., 2015, 5, 12987–12993 RSC.
- R. Shen and H. Liu, RSC Adv., 2016, 6, 37731–37739 RSC.
- D. Zhang, L. Tao, Q. Wang and T. Wang, Polymer, 2016, 82, 114–120 CrossRef CAS.
- L. Tan, B. Li, X. Yang, W. Wang and B. Tan, Polymer, 2015, 70, 336–342 CrossRef CAS.
- K. Schute and M. Rose, ChemSusChem, 2015, 8, 3419–3423 CrossRef CAS PubMed.
- H. Lim, M. C. Cha and J. Y. Chang, Macromol. Chem. Phys., 2012, 213, 1385–1390 CrossRef CAS.
- P. Puthiaraj, S.-M. Cho, Y.-R. Lee and W.-S. Ahn, J. Mater. Chem. A, 2015, 3, 6792–6797 CAS.
- L. Xiang, Y. Zhu, S. Gu, D. Chen, X. Fu, Y. Zhang, G. Yu, C. Pan and Y. Hu, Macromol. Rapid Commun., 2015, 36, 1566–1571 CrossRef CAS PubMed.
- S. Dey, A. Bhunia, D. Esquivel and C. Janiak, J. Mater. Chem. A, 2016, 4, 6259–6263 CAS.
- L. J. Abbott, J. E. Hughes and C. M. Colina, J. Phys. Chem. B, 2014, 118, 1916–1924 CrossRef CAS PubMed.
- L. J. Abbott and C. M. Colina, Macromolecules, 2014, 47, 5409–5415 CrossRef CAS.
- G. Kupgan, T. P. Liyana-Arachchi and C. M. Colina, Polymer, 2016, 99, 173–184 CrossRef CAS.
- A. Trewin, D. J. Willock and A. I. Cooper, J. Phys. Chem. C, 2008, 112, 20549–20559 CAS.
- L. J. Abbott and C. M. Colina, Macromolecules, 2011, 44, 4511–4519 CrossRef CAS.
- N. Fontanals, P. Manesiotis, D. C. Sherrington and P. A. G. Cormack, Adv. Mater., 2008, 20, 1298–1302 CrossRef CAS.
- F. S. Macintyre, D. C. Sherrington and L. Tetley, Macromolecules, 2006, 39, 5381–5384 CrossRef CAS.
- Y. Ouyang, H. Shi, R. Fu and D. Wu, Sci. Rep., 2013, 3, 1430 Search PubMed.
- X. Yang, B. Li, I. Majeed, L. Liang, X. Long and B. Tan, Polym. Chem., 2013, 4, 1425–1429 RSC.
- Y. Ma, Q. Zhou, A. Li, C. Shuang, Q. Shi and M. Zhang, J. Hazard. Mater., 2014, 266, 84–93 CrossRef CAS PubMed.
- S. Xu, Z. Weng, J. Tan, J. Guo and C. Wang, Polym. Chem., 2015, 6, 2892–2899 RSC.
- L. Pan, M.-Y. Xu, Z.-L. Liu, B.-B. Du, K.-H. Yang, L. Wu, P. He and Y.-J. He, RSC Adv., 2016, 6, 47530–47535 RSC.
- X. Wang, J. Feng, Y. Bai, Q. Zhang and Y. Yin, Chem. Rev., 2016, 116, 10983–11060 CrossRef CAS PubMed.
- X. Yang, K. Song, L. Tan, I. Hussain, T. Li and B. Tan, Macromol. Chem. Phys., 2014, 215, 1257–1263 CrossRef CAS.
- K. Wang, L. Huang, S. Razzaque, S. Jin and B. Tan, Small, 2016, 12, 3134–3142 CrossRef CAS PubMed.
- Q. Li, S. Jin and B. Tan, Sci. Rep., 2016, 6, 31359 CrossRef CAS PubMed.
- D. Wu, C. M. Hui, H. Dong, J. Pietrasik, H. J. Ryu, Z. Li, M. Zhong, H. He, E. K. Kim, M. Jaroniec, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2011, 44, 5846–5849 CrossRef CAS.
- X. Kang, Y. Liang, L. Chen, W. Mai, Z. Lin, R. Fu and D. Wu, RSC Adv., 2014, 4, 26166–26170 RSC.
- Z. Li, D. Wu, X. Huang, J. Ma, H. Liu, Y. Liang, R. Fu and K. Matyjaszewski, Energy Environ. Sci., 2014, 7, 3006–3012 CAS.
- F. Xu, Z. Tang, S. Huang, L. Chen, Y. Liang, W. Mai, H. Zhong, R. Fu and D. Wu, Nat. Commun., 2015, 6, 7221 CrossRef PubMed.
- L. Chen, Y. Liang, H. Liu, W. Mai, Z. Lin, H. Xu, R. Fu and D. Wu, RSC Adv., 2016, 6, 49661–49667 RSC.
- Y. Wang, R. Xiong, L. Dong and A. Hu, J. Mater. Chem. A, 2014, 2, 5212–5217 CAS.
- Y. Zhang, Y. Wang and A. Hu, RSC Adv., 2015, 5, 70297–70301 RSC.
- S. Kim and Y. M. Lee, Prog. Polym. Sci., 2015, 43, 1–32 CrossRef CAS.
- A. Chinnappan, W.-J. Chung and H. Kim, J. Mater. Chem. A, 2015, 3, 22960–22968 CAS.
- A. Chinnappan, A. H. Tamboli, W.-J. Chung and H. Kim, Chem. Eng. J., 2016, 285, 554–561 CrossRef CAS.
- H. Wang, H. Bai and L. Li, RSC Adv., 2015, 5, 68639–68645 RSC.
- L. Ding, A. Zhang, W. Li, H. Bai and L. Li, J. Colloid Interface Sci., 2016, 461, 179–184 CrossRef CAS PubMed.
- W. Zhao, Z. Hou, Z. Yao, X. Zhuang, F. Zhang and X. Feng, Polym. Chem., 2015, 6, 7171–7178 RSC.
- T. Mitra, R. S. Bhavsar, D. J. Adams, P. M. Budd and A. I. Cooper, Chem. Commun., 2016, 52, 5581–5584 RSC.
- C. H. Lau, X. Mulet, K. Konstas, C. M. Doherty, M.-A. Sani, F. Separovic, M. R. Hill and C. D. Wood, Angew. Chem., Int. Ed., 2016, 55, 1998–2001 CrossRef CAS PubMed.
- A. M. Saeed, C. A. Wisner, S. Donthula, H. Majedi Far, C. Sotiriou-Leventis and N. Leventis, Chem. Mater., 2016, 28, 4867–4877 CrossRef CAS.
- S. J. Yang, T. Kim, J. H. Im, Y. S. Kim, K. Lee, H. Jung and C. R. Park, Chem. Mater., 2012, 24, 464–470 CrossRef CAS.
- D.-C. Guo, J. Mi, G.-P. Hao, W. Dong, G. Xiong, W.-C. Li and A.-H. Lu, Energy Environ. Sci., 2013, 6, 652–659 CAS.
- F. Maya and F. Svec, Polymer, 2014, 55, 340–346 CrossRef CAS.
- Y. Lv, Z. Lin, T. Tan and F. Svec, J. Chromatogr. A, 2013, 1316, 154–159 CrossRef CAS PubMed.
- X. Yang, L. Tan, L. Xia, C. D. Wood and B. Tan, Macromol. Rapid Commun., 2015, 36, 1553–1558 CrossRef CAS PubMed.
- J. Germain, J. M. J. Fréchet and F. Svec, Small, 2009, 5, 1098–1111 CrossRef CAS PubMed.
- J. Germain, J. Hradil, J. M. J. Fréchet and F. Svec, Chem. Mater., 2006, 18, 4430–4435 CrossRef CAS.
- J.-Y. Lee, C. D. Wood, D. Bradshaw, M. J. Rosseinsky and A. I. Cooper, Chem. Commun., 2006, 2670–2672 RSC.
- B. Li, X. Huang, R. Gong, M. Ma, X. Yang, L. Liang and B. Tan, Int. J. Hydrogen Energy, 2012, 37, 12813–12820 CrossRef CAS.
- C. J. Nielsen, H. Herrmann and C. Weller, Chem. Soc. Rev., 2012, 41, 6684–6704 RSC.
- A. I. Cooper, Nature, 2015, 519, 294–295 CrossRef CAS PubMed.
- C. F. Martín, E. Stöckel, R. Clowes, D. J. Adams, A. I. Cooper, J. J. Pis, F. Rubiera and C. Pevida, J. Mater. Chem., 2011, 21, 5475–5483 RSC.
- S. Chen, J. Zhang, T. Wu, P. Feng and X. Bu, J. Am. Chem. Soc., 2009, 131, 16027–16029 CrossRef CAS PubMed.
- S. Couck, J. F. M. Denayer, G. V. Baron, T. Rémy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326–6327 CrossRef CAS PubMed.
- R. Dawson, E. Stoeckel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239–4245 CAS.
- R. Dawson, T. Ratvijitvech, M. Corker, A. Laybourn, Y. Z. Khimyak, A. I. Cooper and D. J. Adams, Polym. Chem., 2012, 3, 2034–2038 RSC.
- R. Dawson, L. A. Stevens, T. C. Drage, C. E. Snape, M. W. Smith, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2012, 134, 10741–10744 CrossRef CAS PubMed.
- R. T. Woodward, L. A. Stevens, R. Dawson, M. Vijayaraghavan, T. Hasell, I. P. Silverwood, A. V. Ewing, T. Ratvijitvech, J. D. Exley, S. Y. Chong, F. Blanc, D. J. Adams, S. G. Kazarian, C. E. Snape, T. C. Drage and A. I. Cooper, J. Am. Chem. Soc., 2014, 136, 9028–9035 CrossRef CAS PubMed.
- X. Jing, D. Zou, P. Cui, H. Ren and G. Zhu, J. Mater. Chem. A, 2013, 1, 13926–13931 CAS.
- X. Zhu, S. M. Mahurin, S.-H. An, D.-T. Chi-Linh, C. Tian, Y. Li, L. W. Gill, E. W. Hagaman, Z. Bian, J.-H. Zhou, J. Hu, H. Liu and S. Dai, Chem. Commun., 2014, 50, 7933–7936 RSC.
- H. Li, B. Meng, S. M. Mahurin, S.-H. Chai, K. M. Nelson, D. C. Baker, H. Liu and S. Dai, J. Mater. Chem. A, 2015, 3, 20913–20918 CAS.
- J.-H. Zhu, Q. Chen, Z.-Y. Sui, L. Pan, J. Yu and B.-H. Han, J. Mater. Chem. A, 2014, 2, 16181–16189 CAS.
- L. Pan, Q. Chen, J.-H. Zhu, J.-G. Yu, Y.-J. He and B.-H. Han, Polym. Chem., 2015, 6, 2478–2487 RSC.
- X. Yang, M. Yu, Y. Zhao, C. Zhang, X. Wang and J.-X. Jiang, J. Mater. Chem. A, 2014, 2, 15139–15145 CAS.
- S. Yao, X. Yang, M. Yu, Y. Zhang and J.-X. Jiang, J. Mater. Chem. A, 2014, 2, 8054–8059 CAS.
- X. Yang, M. Yu, Y. Zhao, C. Zhang, X. Wang and J.-X. Jiang, RSC Adv., 2014, 4, 61051–61055 RSC.
- Y. Zhang, Y. Li, F. Wang, Y. Zhao, C. Zhang, X. Wang and J.-X. Jiang, Polymer, 2014, 55, 5746–5750 CrossRef CAS.
- D. Chang, M. Yu, C. Zhang, Y. Zhao, R. Kong, F. Xie and J.-X. Jiang, Microporous Mesoporous Mater., 2016, 228, 231–236 CrossRef CAS.
- J. Wang, J. Huang, X. Wu, B. Yuan, Y. Sun, Z. Zeng and S. Deng, Chem. Eng. J., 2014, 256, 390–397 CrossRef CAS.
- J. Wang, W. Sng, G. Yi and Y. Zhang, Chem. Commun., 2015, 51, 12076–12079 RSC.
- J. Wang, J. G. W. Yang, G. Yi and Y. Zhang, Chem. Commun., 2015, 51, 15708–15711 RSC.
- S. Wang, K. Song, C. Zhang, Y. Shu, T. Li and B. Tan, J. Mater. Chem. A, 2017, 5, 1509–1515 CAS.
- D. Bratkowska, N. Fontanals, F. Borrull, P. A. G. Cormack, D. C. Sherrington and R. M. Marcé, J. Chromatogr. A, 2010, 1217, 3238–3243 CrossRef CAS PubMed.
- N. Fontanals, P. A. G. Cormack, D. C. Sherrington, R. M. Marce and F. Borrull, J. Chromatogr. A, 2010, 1217, 2855–2861 CrossRef CAS PubMed.
- N. Fontanals, M. Galia, P. A. G. Cormack, R. M. Marce, D. C. Sherrington and F. Borrull, J. Chromatogr. A, 2005, 1075, 51–56 CrossRef CAS PubMed.
- N. Fontanals, R. M. Marce, P. A. G. Cormack, D. C. Sherrington and F. Borrull, J. Chromatogr. A, 2008, 1191, 118–124 CrossRef CAS PubMed.
- E. Candish, H.-J. Wirth, A. A. Gooley, R. A. Shellie and E. F. Hilder, J. Chromatogr. A, 2015, 1410, 9–18 CrossRef CAS PubMed.
- V. V. Tolmacheva, V. V. Apyari, A. A. Furletov, S. G. Dmitrienko and Y. A. Zolotov, Talanta, 2016, 152, 203–210 CrossRef CAS PubMed.
- D. Bratkowska, A. Davies, N. Fontanals, P. A. G. Cormack, F. Borrull, D. C. Sherrington and R. M. Marce, J. Sep. Sci., 2012, 35, 2621–2628 CrossRef CAS PubMed.
- M. P. Tsyurupa, Z. K. Blinnikova, Y. A. Borisov, M. M. Ilyin, T. P. Klimova, K. V. Mitsen and V. A. Davankov, J. Sep. Sci., 2014, 37, 803–810 CrossRef CAS PubMed.
- W.-Q. Wang, J. Wang, J.-G. Chen, X.-S. Fan, Z.-T. Liu, Z.-W. Liu, J. Jiang and Z. Hao, Chem. Eng. J., 2015, 281, 34–41 CrossRef CAS.
- V. Davankov, M. Tsyurupa, M. Ilyin and L. Pavlova, J. Chromatogr. A, 2002, 965, 65–73 CrossRef CAS PubMed.
- F. Svec, J. Sep. Sci., 2004, 27, 747–766 CrossRef CAS PubMed.
- F. Svec and Y. Lv, Anal. Chem., 2015, 87, 250–273 CrossRef CAS PubMed.
- P. Veverka and K. Jeřábek, React. Funct. Polym., 2004, 59, 71–79 CrossRef CAS.
- D. Bratkowska, R. M. Marce, P. A. G. Cormack, D. C. Sherrington, F. Borrull and N. Fontanals, J. Chromatogr. A, 2010, 1217, 1575–1582 CrossRef CAS PubMed.
- N. Fontanals, P. A. G. Cormack and D. C. Sherrington, J. Chromatogr. A, 2008, 1215, 21–29 CrossRef CAS PubMed.
- J. Huang, X. Wang, P. D. Patil, J. Tang, L. Chen and Y.-N. Liu, RSC Adv., 2014, 4, 41172–41178 RSC.
- X. Ling, H. Li, H. Zha, C. He and J. Huang, Chem. Eng. J., 2016, 286, 400–407 CrossRef CAS.
- W. Kuang, Y.-N. Liu and J. Huang, J. Colloid Interface Sci., 2017, 487, 31–37 CrossRef CAS PubMed.
- X. Wang, T. Zhang, J. Huo, J. Huang and Y.-N. Liu, J. Colloid Interface Sci., 2017, 487, 231–238 CrossRef CAS PubMed.
- S. Liu, D. Chen, J. Zheng, L. Zeng, J. Jiang, R. Jiang, F. Zhu, Y. Shen, D. Wu and G. Ouyang, Nanoscale, 2015, 7, 16943–16951 RSC.
- S. Liu, Q. Hu, J. Zheng, L. Xie, S. Wei, R. Jiang, F. Zhu, Y. Liu and G. Ouyang, J. Chromatogr. A, 2016, 1450, 9–16 CrossRef CAS PubMed.
- H. Li, B. Meng, S.-H. Chai, H. Liu and S. Dai, Chem. Sci., 2016, 7, 905–909 RSC.
- B. Li, F. Su, H.-K. Luo, L. Liang and B. Tan, Microporous Mesoporous Mater., 2011, 138, 207–214 CrossRef CAS.
- Y. He, Q. Liu, F. Liu, C. Huang, C. Peng, Q. Yang, H. Wang, J. Hu and H. Liu, Microporous Mesoporous Mater., 2016, 233, 10–15 CrossRef CAS.
- Y. Xia, Y. Li, Y. Gu, T. Jin, Q. Yang, J. Hu, H. Liu and H. Wang, Fuel, 2016, 170, 100–106 CrossRef CAS.
- J. Wu, L. Jia, L. Wu, C. Long, W. Deng and Q. Zhang, RSC Adv., 2016, 6, 28986–28993 RSC.
- S. Wang, L. Zhang, C. Long and A. Li, J. Colloid Interface Sci., 2014, 428, 185–190 CrossRef CAS PubMed.
- F. Svec, J. Chromatogr. A, 2012, 1228, 250–262 CrossRef CAS PubMed.
- J. Urban, F. Svec and J. M. J. Fréchet, J. Chromatogr. A, 2010, 1217, 8212–8221 CrossRef CAS PubMed.
- J. Urban, F. Svec and J. M. J. Fréchet, Anal. Chem., 2010, 82, 1621–1623 CrossRef CAS PubMed.
- A. Saeed, F. Maya, D. J. Xiao, M. Najam-ul-Haq, F. Svec and D. K. Britt, Adv. Funct. Mater., 2014, 24, 5790–5797 CrossRef CAS.
- F. Maya and F. Svec, J. Chromatogr. A, 2013, 1317, 32–38 CrossRef CAS PubMed.
- C. Lu, S. Liu, J. Xu, Y. Ding and G. Ouyang, Anal. Chim. Acta, 2016, 902, 205–211 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- B. Li, Z. Guan, W. Wang, X. Yang, J. Hu, B. Tan and T. Li, Adv. Mater., 2012, 24, 3390–3395 CrossRef CAS PubMed.
- Z. Guan, B. Li, G. Hai, X. Yang, T. Li and B. Tan, RSC Adv., 2014, 4, 36437–36443 RSC.
- K. Song, P. Liu, J. Wang, L. Pang, J. Chen, I. Hussain, B. Tan and T. Li, Dalton Trans., 2015, 44, 13906–13913 RSC.
- S. Xu, K. Song, T. Li and B. Tan, J. Mater. Chem. A, 2015, 3, 1272–1278 CAS.
- Z. Jia, K. Wang, T. Li, B. Tan and Y. Gu, Catal. Sci. Technol., 2016, 6, 4345–4355 CAS.
- K. Song, P. Liu, J. Wang, B. Tan and T. Li, J. Porous Mater., 2016, 23, 725–731 CrossRef CAS.
- K. Song, Z. Zou, D. Wang, B. Tan, J. Wang, J. Chen and T. Li, J. Phys. Chem. C, 2016, 120, 2187–2197 CAS.
- Z. Dou, L. Xu, Y. Zhi, Y. Zhang, H. Xia, Y. Mu and X. Liu, Chem. – Eur. J., 2016, 22, 9919–9922 CrossRef CAS PubMed.
- A. Mouradzadegun and M. Alsadat Mostafavi, RSC Adv., 2016, 6, 42522–42531 RSC.
- M. Jiang, Y. Ding, L. Yan, X. Song and R. Lin, Chin. J. Catal., 2014, 35, 1456–1464 CrossRef CAS.
- T. Wang, Y. Lyu, X. Chen, C. Li, M. Jiang, X. Song and Y. Ding, RSC Adv., 2016, 6, 28447–28450 RSC.
- X. Song, W. Zhu, Y. Yan, H. Gao, W. Gao, W. Zhang and M. Jia, J. Mol. Catal. A: Chem., 2016, 413, 32–39 CrossRef CAS.
- J. Mondal, S. K. Kundu, W. K. Hung Ng, R. Singuru, P. Borah, H. Hirao, Y. Zhao and A. Bhaumik, Chem. – Eur. J., 2015, 21, 19016–19027 CrossRef CAS PubMed.
- J. Liu, Q. Chen, Y.-N. Sun, M.-Y. Xu, W. Liu and B.-H. Han, RSC Adv., 2016, 6, 48543–48549 RSC.
- S. Shi, C. Chen, M. Wang, J. Ma, H. Ma and J. Xu, Chem. Commun., 2014, 50, 9079–9082 RSC.
- R. Li, Z. J. Wang, L. Wang, B. C. Ma, S. Ghasimi, H. Lu, K. Landfester and K. A. I. Zhang, ACS Catal., 2016, 6, 1113–1121 CrossRef CAS.
- K. Jiang, T. Fei and T. Zhang, Sens. Actuators, B, 2014, 199, 1–6 CrossRef CAS.
- K. Jiang, D. Kuang, T. Fei and T. Zhang, Sens. Actuators, B, 2014, 203, 752–758 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |