
Methods for the detection and identification of pathogenic bacteria: past, present, and future
Linda
Váradi
ab,
Jia Lin
Luo
a,
David E.
Hibbs
 a,
John D.
Perry
c,
Rosaleen J.
Anderson
d,
Sylvain
Orenga
e and
Paul W.
Groundwater
*a
a,
John D.
Perry
c,
Rosaleen J.
Anderson
d,
Sylvain
Orenga
e and
Paul W.
Groundwater
*a
aFaculty of Pharmacy, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: paul.groundwater@sydney.edu.au
bCSIRO Manufacturing, Research Way, Clayton, VIC 3168, Australia
cMicrobiology Department, Freeman Hospital, Newcastle upon Tyne, UK
dSunderland Pharmacy School, University of Sunderland, Sunderland, UK
ebioMérieux Inc., Durham, NC, USA
First published on 23rd June 2017
Abstract
In order to retard the rate of development of antibacterial resistance, the causative agent must be identified as rapidly as possible, so that directed patient treatment and/or contact precautions can be initiated. This review highlights the challenges associated with the detection and identification of pathogenic bacteria, by providing an introduction to the techniques currently used, as well as newer techniques that are in development. Focusing on the chemical basis for these techniques, the review also provides a comparison of their advantages and disadvantages.
Key learning points(1) Why rapid identification of pathogenic bacterial is necessary.(2) The requirements of a bacterial detection and identification method. (3) A comparison of the attributes of culture-based and molecular methods for the detection of bacteria. (4) The chemical basis of the techniques used in the identification of pathogenic bacteria. (5) Chromogenic and fluorogenic enzyme substrates for bacterial detection. |
1. Introduction
1.1. The increasing prevalence of antibiotic resistance and the need for rapid identification of pathogenic bacteria
As a result of the increasing prevalence of antimicrobial resistance (AMR), in both the community and hospital setting, the UK Chief Medical Officer (CMO), Prof. Dame Sally Davies, suggested that AMR be included on the National Risk Register, as its potential impact on health is as significant as that of global warming.1 Among the challenges highlighted by the 2011 Annual Report of the CMO was the need to improve diagnostic testing in order to ensure more tailored (or directed) therapeutic interventions and to take advantage of the opportunities available through point-of-care (POC) testing.2 In 2014, the UK Government re-instituted the Longitude Prize and asked the UK public to vote on which global problem should be the subject of the £10m prize (https://longitudeprize.org/). The successful challenge was ‘to create a cost-effective, accurate, rapid and easy-to-use test for bacterial infections that will allow health professionals worldwide to administer the right antibiotics at the right time’, in other words, to develop a POC diagnostic test that either helps identify the effective antibiotic for a patient's infection or rules out antibiotic use. The 5 year period for teams to submit their applications began in November 2014, with assessments of submissions made every 4 months; the first team to meet all the criteria will win the prize, which currently remains unclaimed. The $20m Antimicrobial Resistance Diagnostic Challenge, which is funded by the US National Institutes of Health (NIH), National Institute of Allergy and Infectious Disease (NIAID) and the Biomedical Advanced Research and Development Authority (BARDA), also seeks innovative and rapid POC diagnostic tests for use in combating the development and spread of drug resistant bacteria (https://dpcpsi.nih.gov/AMRChallenge). This review outlines why the rapid identification of pathogenic bacteria is necessary along with the requirements and challenges associated with the development of a bacterial detection/identification method. It also provides a comparison of methods that are currently employed for the detection of bacteria and highlights the chemical basis of these techniques.While a natural phenomenon, AMR has been accelerated by the injudicious use of antibacterial agents. As the pipeline for new agents dries up (only 2 antibacterials with new targets, daptomycin and linezolid, have been introduced into practice this century) due to reduced economic incentives and increased regulatory barriers,3 global healthcare systems require methods that can be employed as part of antibacterial stewardship programmes to help maintain the efficacy of current antibacterials.
In the hospital setting, methods for the detection of multi-drug resistant (MDR) organisms are required as part of an organism-specific approach for the prevention and control of infection. An effective risk management strategy for the prevention of cross-infection between patients relies upon rapid and reliable analysis of specimens and the subsequent introduction of contact precautions (e.g. patient isolation).4
Rapid identification of bacterial pathogens would also inform a more effective, pathogen-directed clinical treatment of the infection. In developed countries, prescription in general practice accounts for approximately 90% of all human antibiotic use;5 a POC test would enable optimal patient treatment outcomes and also reduce the probability of developing further resistance by informing the choice of the right antibacterial agent for the right patient using pathogen-directed therapy. One of the specific steps that is recommended (in order to reduce demand for antibiotics) in the final report of the UK review on antimicrobial resistance, chaired by Lord O’Neill (https://amr-review.org/), is the promotion of rapid new diagnostics, and the proposal that, by 2020, it should be mandatory for antibiotic prescription to be informed by data and testing technology. This review team has also projected that, by 2050, AMR will be the cause of more annual global deaths (10 million) than cancer (8.2 million), and that by then $60–100 trillion of global economic output will have been lost due to lost productivity if AMR is not tackled.
It is difficult to assess the effect AMR has on the disease burden due to community acquired infections, but the data for nosocomial (hospital-based) infections are both alarming and indicative of the magnitude of the problem healthcare systems face. In the US, healthcare-associated infections (HAI) are estimated to be implicated in 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths p.a.; in Europe the average prevalence of HAI is 7.1 per 100 patients, while in low income countries this burden has been estimated to be as high as 15.5 per 100 patients.5 These infections are responsible for prolonged hospital stays, increasing antimicrobial resistance rates, huge financial burdens on healthcare systems and society, and avoidable patient morbidity and mortality. For example, in Europe it has been estimated that infections due to resistant organisms result in an excess 25000 deaths p.a., with associated healthcare costs and productivity losses equalling €1.5 billion. In the US, it has been estimated that there are at least 2 million illnesses and 23000 deaths associated with antibacterial resistance,6 and the combined cost of medical bills and extended hospital care has been estimated at $30 billion per annum.5
000 deaths p.a.; in Europe the average prevalence of HAI is 7.1 per 100 patients, while in low income countries this burden has been estimated to be as high as 15.5 per 100 patients.5 These infections are responsible for prolonged hospital stays, increasing antimicrobial resistance rates, huge financial burdens on healthcare systems and society, and avoidable patient morbidity and mortality. For example, in Europe it has been estimated that infections due to resistant organisms result in an excess 25000 deaths p.a., with associated healthcare costs and productivity losses equalling €1.5 billion. In the US, it has been estimated that there are at least 2 million illnesses and 23000 deaths associated with antibacterial resistance,6 and the combined cost of medical bills and extended hospital care has been estimated at $30 billion per annum.5
The majority of nosocomial infections are caused by the ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) organisms, so called because, as a result of their MDR, they have the potential to escape the actions of antibacterial agents.3 Other organisms that the Centers for Disease Control and Prevention (CDC) has classified at its top level of urgent threat in the U.S.A. are Clostridium difficile (250000 infections p.a., 14000 deaths, $1 billion in excess medical costs), and carbapenem-resistant Enterobacteriaceae (CRE; 9000 drug-resistant infections p.a.), which have developed resistance to virtually all antibiotics and are frequently susceptible only to last line antibacterials, such as aminoglycosides, tigecycline, fosfomycin, and colistin (polymyxin E).
A method that could identify the causative bacterial agent and its susceptibility to antibacterials would have significant advantages in terms of directed (rather than empirical) antibacterial therapy. As outlined previously, the ideal detection method for pathogenic bacteria would be easy to use (with no requirement for expensive instrumentation and easily interpreted), rapid (capable of delivering results within a timeframe which would enable POC testing), reliable, and cost effective (simple, so that it can be employed in any global setting).
2. The use of bacterial surveillance techniques in infection control
Surveillance is most effective when it is inclusive, comprehensive, and communicated. Organised programmes were set up in the USA (Active Bacterial Core surveillance, ABCs, as part of the CDC: http://www.cdc.gov/abcs/) in 1995 and in the EU (European Centre for Disease Prevention and Control, ECDC: http://ecdc.europa.eu) in 1996. Each brings together hospital infection incidence, screening, epidemiological data, academic research and government-enabled networks to provide accurate and up-to-date information that is sufficiently broad to capture wide trends and sufficiently detailed to identify pockets of new infections and resistance. Despite these long-standing surveillance programmes, a World Health Organisation (WHO) report on global antibacterial surveillance in 2014 found large gaps in surveillance across many parts of the World. Regardless of the international efforts, a high incidence of 3rd generation cephalosporin and fluoroquinolone resistant Escherichia coli and 3rd generation cephalosporin resistant K. pneumoniae, with a significant incidence of resistance to carbapenems in K. pneumoniae, was observed globally.7 Additional surveillance programmes have been initiated but many countries still lack surveillance programmes and agreed standards for surveillance methods and international coordination. The Center for Disease Dynamics Economics and Policy (CDDEP) supports data gathering from across the globe and, through ResistanceMap, provides information in a variety of forms for interrogation [http://www.resistancemap.org]; their report ‘The State of the World's Antibiotics 2015’ reviews the data and brings together the conclusions of CDDEP's Global Antibiotic Resistance Partnership (GARP) into six essential strategies for national antibiotic policies, encouraging countries without antibiotic stewardship and surveillance programmes to initiate national policies.8Two key aspects of surveillance are the ability to correctly detect and identify pathogenic bacteria, requiring consideration of sensitivity,† specificity, cost, time to detection, the ability to identify bacteria directly from clinical samples and to evaluate their antibiotic susceptibilities, so that the correct antibiotic treatment can be chosen, if one is needed, for each specific incidence of infection.
2.1. Detection and identification
The first objective for detection and identification‡ in a clinical setting is to answer the question ‘Is it a bacterial infection or not?’ and, if it is, to provide bacterial identification with confidence.When identifying bacteria in a clinical sample after culture (see Section 3.1), the genus (e.g. Staphylococcus or Streptococcus) is assigned using a combination of morphological features (e.g. colony size and colour), microscopy (e.g. by Gram stain) and rapid biochemical tests (e.g. for catalase and/or oxidase activity) using simple reagents. Identification to species level (e.g. S. aureus) is then performed using targeted biochemical or serological tests (e.g. latex agglutination tests), because the different species within a genus can have very different pathogenicities and resistance profiles, causing a range of infections of varying severity and requiring different antibiotic treatment regimens. The issue of resistance adds further complexity, as each incidence of a particular species can carry different resistance genes, giving even bacteria within a specific genus different antibiotic susceptibility profiles. There must be high levels of confidence in the results, so that correct diagnoses and treatment decisions can be made. Test methods therefore require high specificity, identifying accurately the bacterial genus and species, along with the susceptibility profile whenever possible.
One approach to high specificity is, for example, to use DNA detection to provide genomic data (in Section 4, we will consider molecular diagnostic techniques). The use of PCR (polymerase chain reaction) to amplify the DNA present in a sample has transformed gene-based assays and overcomes the major challenge of rapid testing of samples in a POC healthcare setting, with the potential for the question ‘is it bacterial or not?’ to be answered quickly. However, the main disadvantage of using PCR-based techniques for bacterial identification is their sensitivity. Genomic testing, which was once prohibitively expensive for routine use is now approaching a cost-effective level. Although genomic methods are rapid and provide confident identity of any bacteria present in a sample, they are so sensitive that they can detect bacteria present in very small numbers, below the infection threshold, and so could result in unnecessary antibiotic therapy being initiated. Detection methods must therefore be suitably analytically sensitive for each suspected pathogen.
As we shall see in Section 3, the more commonly employed identification methods are phenotypically based (the observable characteristics of an organism that result from the interaction of its genotype with its environment); if overnight culture with a suitable broth or agar medium allows bacterial growth the characteristic protein expression profile may be obtained. This requirement for culture of a clinical sample before bacterial identification is a major hurdle to the development of rapid phenotypic POC tests.
2.2. Susceptibility
Having detected and confidently identified the genus and species of bacteria causing an infection, the likely susceptibility profiles can be searched on a database or directly evaluated. It is not good practice merely to assume antibiotic susceptibility from the bacterial identity, due to increasing and unpredictable levels of resistance. Susceptibility profiles for bacteria without resistance mechanisms are established regularly and curated by the European Committee for Antimicrobial Susceptibility Testing (EUCAST: http://www.eucast.org) for the EU countries and the Clinical and Laboratory Standards Institute (CLSI: http://www.clsi.org) for North America. Each clinical sample under investigation requires its susceptibility profile to be established, as it may contain bacteria that have acquired resistance to antibiotics and do not have the susceptibility typical for their genus and species. The EUCAST and CLSI data provide the standards against which the observed susceptibilities can be compared.The only reliable method for determining whether bacteria are susceptible or resistant to a particular antimicrobial is to perform antimicrobial susceptibility testing (AST), i.e. by challenging the bacterial isolate with the antimicrobial at clinically-relevant concentrations to see if growth is inhibited. There are a wide variety of AST methods, including determination of the minimum inhibitory concentration (e.g. by broth or agar dilution) and disc susceptibility testing (e.g. EUCAST method: http://www.eucast.org). These require overnight incubation, whereas automated methods may generate results in a few hours. Enzymes that hydrolyze antimicrobials (e.g. β-lactamases) may be detected more rapidly by phenotypic methods to infer likely antimicrobial resistance and genotypic methods (e.g. PCR) may be used to rapidly detect genes encoding antimicrobial resistance mechanisms, as discussed in subsequent sections.
3. Phenotypic methods for bacterial identification
Each genus of bacteria has a characteristic protein expression ‘fingerprint’. Although many proteins, including enzymes, are common to most bacteria, a range of unique biochemical pathways define each bacterial genus and the proteins expressed can even differ between species within a genus. These differences in biochemical processes can be explained by the vast range of environments in which bacteria thrive, and their need to survive by gaining an advantage over other microorganisms. These differences in protein expression between bacteria can be exploited in tests for specific bacteria, providing identities with relatively good certainty.3.1. Biochemical testing
The majority of clinical microbiology laboratories still rely on culture for the detection of most bacterial pathogens from clinical samples. Traditionally, culture is performed using general purpose agar-based media (e.g. blood agar) that will support the growth of a wide range of pathogens. Each type of colony that is recovered is then investigated to identify likely pathogens. For the detection of certain pathogens, it is essential to use more specific culture media. For example, ‘differential’ media target differences in the metabolic activity of bacteria utilising biochemical indicator systems (e.g. the incorporation of a sugar (nutrition)) plus a pH indicator (to sense metabolites/digested nutrient) to indicate the likely presence of a specific pathogen. Culture media may also be ‘selective’ (by incorporation of antimicrobials) to reduce the amount of commensal§ flora able to grow and thereby increase the likelihood of isolating a particular target pathogen. For example, to isolate Salmonella from a stool sample that may contain dozens of other species, it is essential to use a culture medium that is both selective (e.g. by the use of antimicrobials such as cefsulodin and novobiocin) and differential (by detecting hydrogen sulfide production or specific enzymes – see Table 1).| Pathogen | Enzyme targeted |
|---|---|
| Gram positive pathogens | |
| Staphylococcus aureus (including MRSA) | Phosphatase or α-glucosidase |
| Clostridium difficile | β-Glucosidase |
| Group B streptococcus | Phosphatase |
| Gram negative pathogens | |
| Salmonella species | C8-Esterase or α-galactosidase |
| Shigella species | β-Ribosidase |
| Escherichia coli | β-Glucuronidase or β-galactosidase |
| Pseudomonas aeruginosa | β-Alanyl aminopeptidase |
After isolation, genus and species level identification is carried out, for which commercially available panels of biochemical tests are frequently employed; these typically include sugars to detect acidification (via oxidation or fermentation) using a pH indicator. Other tests included in such panels may target enzymes involved in amino acid metabolism (e.g. decarboxylases, deaminase, tryptophanase) or hydrolase enzymes, such as urease and β-galactosidase. The inoculation and reading of biochemical panels can be performed manually using commercial kits such as Analytical Profile Index kits. Increasingly, such testing is automated and there are a variety of commercially available instruments that perform automated inoculation and reading of biochemical panels such as the BD Phoenix or the Vitek 2 instruments. For some species, such automated systems can achieve bacterial identification in 2–3 h, as well as performing automated antimicrobial susceptibility testing.9
3.2. Chromogenic media
Due to the ease of use, relatively low cost, and minimal expertise required, one commonly used bacterial identification approach employed in clinical laboratories involves the use of chromogenic media. These media require the culture of small amounts of clinical samples from suspected infection sites, using optimal broth or agar media in the presence of chromogenic or fluorogenic substrates, which are hydrolyzed and develop colour in the presence of unique enzymes that are expressed by the bacterium of interest. The substrates added to the media have muted/no colour or fluorescence, but when acted upon by enzymes specific to the suspected infectious bacterium, they are subject to structural changes, resulting in the release of a coloured or fluorescent marker that indicates the presence of bacteria in the sample under investigation, Fig. 1.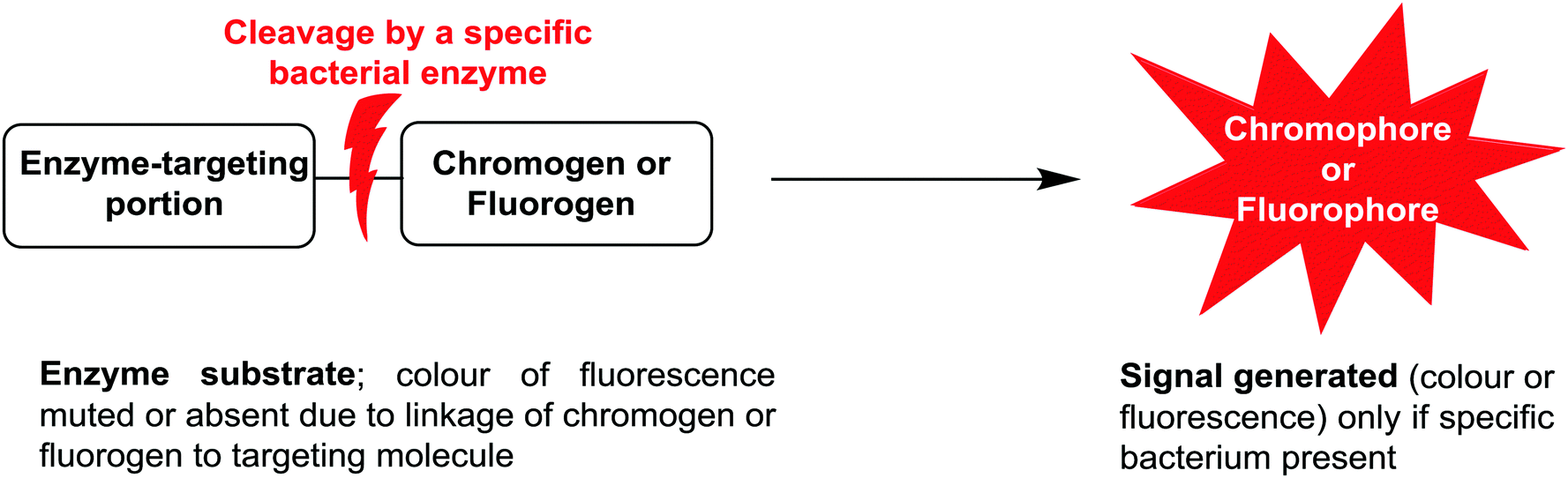 | ||
| Fig. 1 Schematic representation of phenotypic bacterial identification using chromogenic/fluorogenic substrates in culture media. | ||
The application of chromogenic culture media is an attempt to perform isolation and identification of specific bacterial pathogens in a single step. Such media incorporate one or more chromogenic enzyme substrates that target specific bacterial enzymes, normally in conjunction with selective antimicrobial agents to inhibit non-target species. This allows the targeting of bacterial pathogens with high specificity and their immediate identification based on their colony colour. The chromogenic substrates utilized in such media typically target bacterial hydrolases – most commonly glycosidases, such as β-galactosidase or β-glucosidase.10 Cleavage of the sugar residue from these substrates by a bacterial enzyme releases a brightly coloured, most often insoluble, chromogen that remains highly localized on bacterial colonies, thus differentiating targeted colonies possessing the enzyme from those that do not, Fig. 2. Other, less commonly targeted, hydrolases are esterases or peptidases. For example, a chromogen linked to octanoic acid can be utilized to detect the C8-esterase activity of Salmonella species, Fig. 3 and Table 1, whereas the detection of β-alanine aminopeptidase has been exploited for the detection of P. aeruginosa, Fig. 4.10
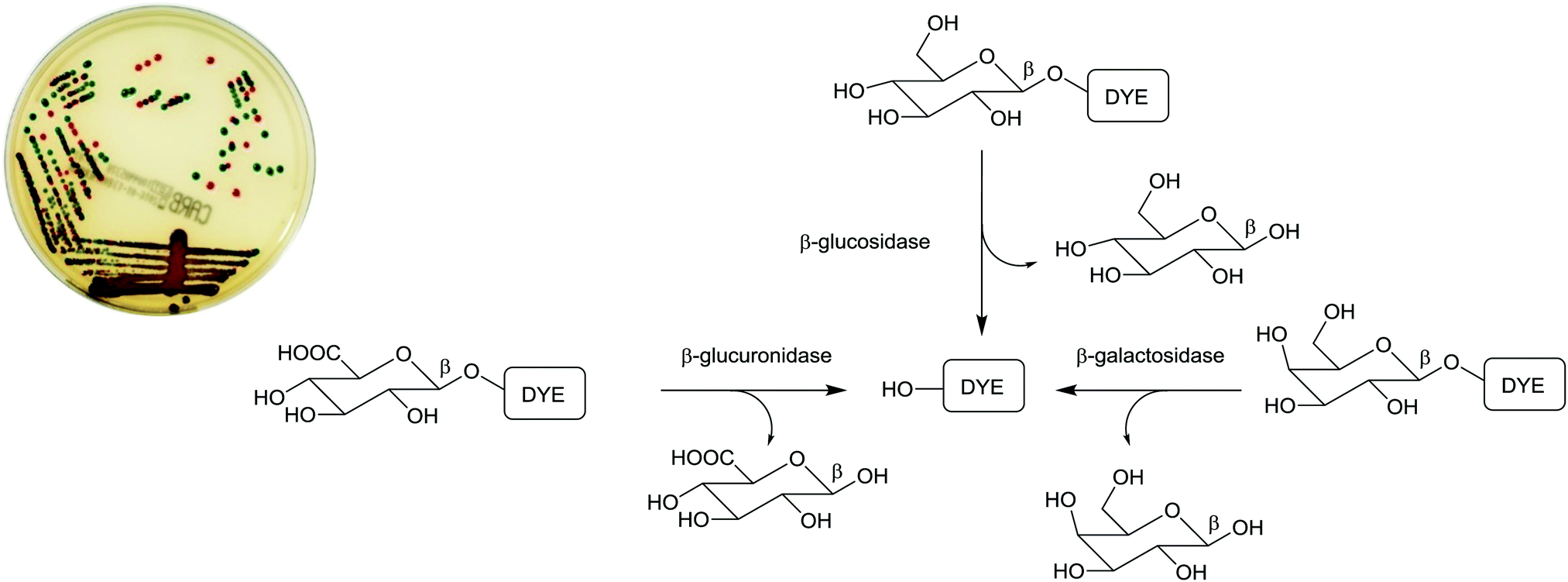 | ||
| Fig. 2 Chromogenic medium (chromID® CARBA) employing a combination of chromogenic substrates for the detection of carbapenemase-producing Enterobacteriaceae (CPE). The culture reveals E. coli as red colonies (due to β-glucuronidase/β-galactosidase activity) and K. pneumoniae as green-blue colonies (as a result of β-glucosidase activity). (Image courtesy of bioMérieux). | ||
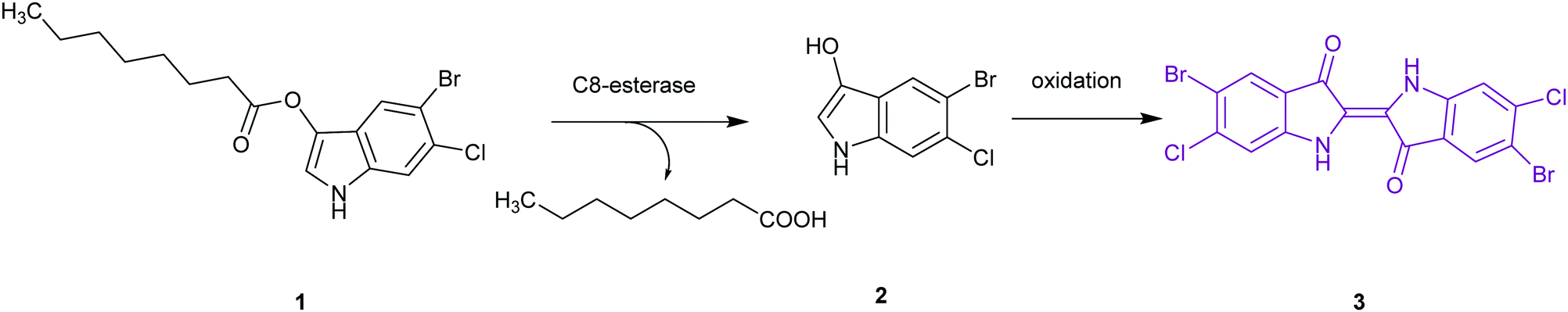 | ||
| Fig. 3 Chromogenic basis (Brilliance™ Salmonella agar) for the detection of Salmonella as a result of C8-esterase activity on 5-bromo-6-chloro-3-indolyl caprylate 1, giving indoxyl 2, which undergoes oxidization to the insoluble magenta indigo 3. | ||
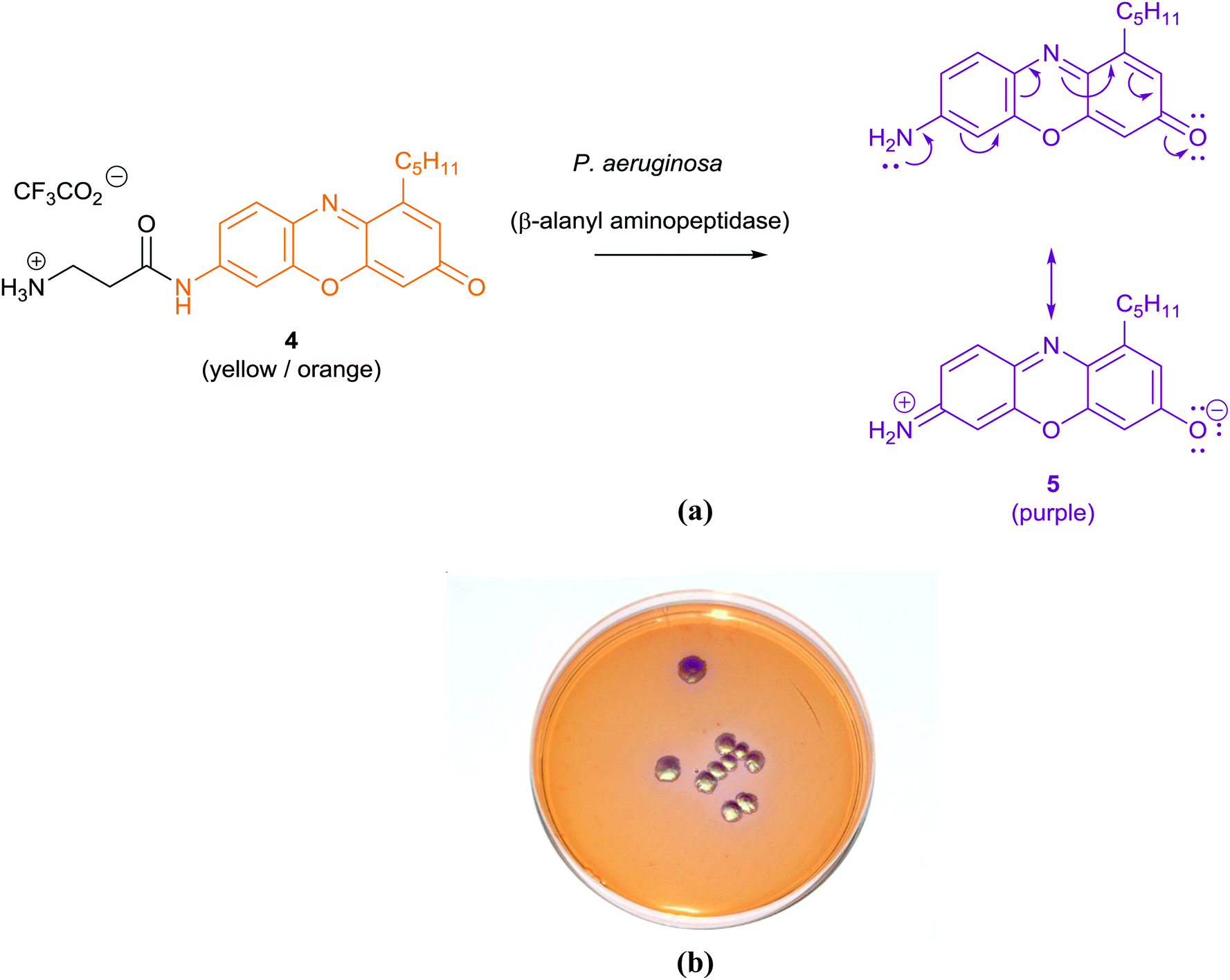 | ||
| Fig. 4 Diagram illustrating the principle behind the origin of the purple colour in the chromogenic detection of Pseudomonas aeruginosa colonies through the hydrolysis of β-alanyl pentylresorufamine 4 by β-alanyl aminopeptidase to give resorufamine 5 (a), using chromID™ P. aeruginosa (b) (image courtesy of Larissa Laine, Freeman Hospital, Newcastle upon Tyne, UK). | ||
In practice, chromogenic media are not completely specific and further confirmation of species identity is required, e.g. using MALDI-TOF MS (see Section 3.3). Nevertheless, chromogenic media have been widely adopted in clinical laboratories, as they contribute to a reduced workload (as only coloured colonies require processing) and they may enhance detection, because coloured colonies are less likely to be overlooked, particularly when a complex mixture of species is present in a sample. Table 1 contains examples of the enzymatic activity that is targeted by chromogenic media for specific pathogens. Chromogenic media are extensively used to screen for colonization of patients with antibiotic resistant bacteria, such as methicillin-resistant S. aureus (MRSA), vancomycin-resistant enterococci (VRE) and Enterobacteriaceae with extended-spectrum β-lactamases or carbapenemases.11
A major drawback with chromogenic media is the time taken to detection, which is usually 16–48 h, as this relies upon the incubation producing sufficient bacterial colonies (and thus enzyme) for the development of colour. This disadvantage could be addressed by the use of fluorogenic substrates or dyes, as fluorescence is more easily detected than perception of colour/absorption of light. This could potentially allow the detection of microcolonies within 2 h of incubation using sensitive instrumentation, but this is a relatively unexplored area in clinical microbiology.12
3.3. Matrix assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS)
Since 2010, the identification of bacterial pathogens has been revolutionized by the introduction of MALDI-TOF MS and this is now the method of choice for bacterial identification in most advanced clinical laboratories.13 In MALDI-TOF MS the sample is treated with a matrix, which absorbs energy from a laser, resulting in rapid heating, vaporization, and the ionization of the analytes; the ions are then separated on the basis of the time they take to reach the detector, as all ions of the same charge are given the same kinetic energy. In practice, the majority of bacterial molecules observed by MALDI-TOF MS are ribosomal proteins, Fig. 5, with characteristic masses for the proteins of particular bacteria giving rise to ‘fingerprint’ MS spectra (a peptide mass fingerprint [PMF]) that can be compared to an online MS database to identify specific genera and species. The identification of subspecies and strains depends upon the availability of extensive databases, while the use of whole/intact cells has resulted in improvements to reproducibility in comparison to earlier methods, which required protein extraction and analysis to be performed under standardized conditions.14 Despite its success, there are some limitations to the use of MALDI-TOF MS, such as its inability to differentiate taxonomically related bacteria, e.g. the highly pathogenic Shigella species (a cause of dysentery) from commensal E. coli, and its inability to differentiate Streptococcus pneumoniae from some commensal species of oral streptococci. Currently, MALDI-TOF MS is used mainly with culture methods to confirm the identification of bacteria that have been amplified by selective culture, meaning that confirmation of identity requires more than one day. MALDI-TOF MS is, however, rapid and allows for species-level identification within minutes rather than hours and, despite the high capital investment necessary for acquiring the instrumentation, the method is very economical when performing high throughput testing (e.g. at least 100 strains per day).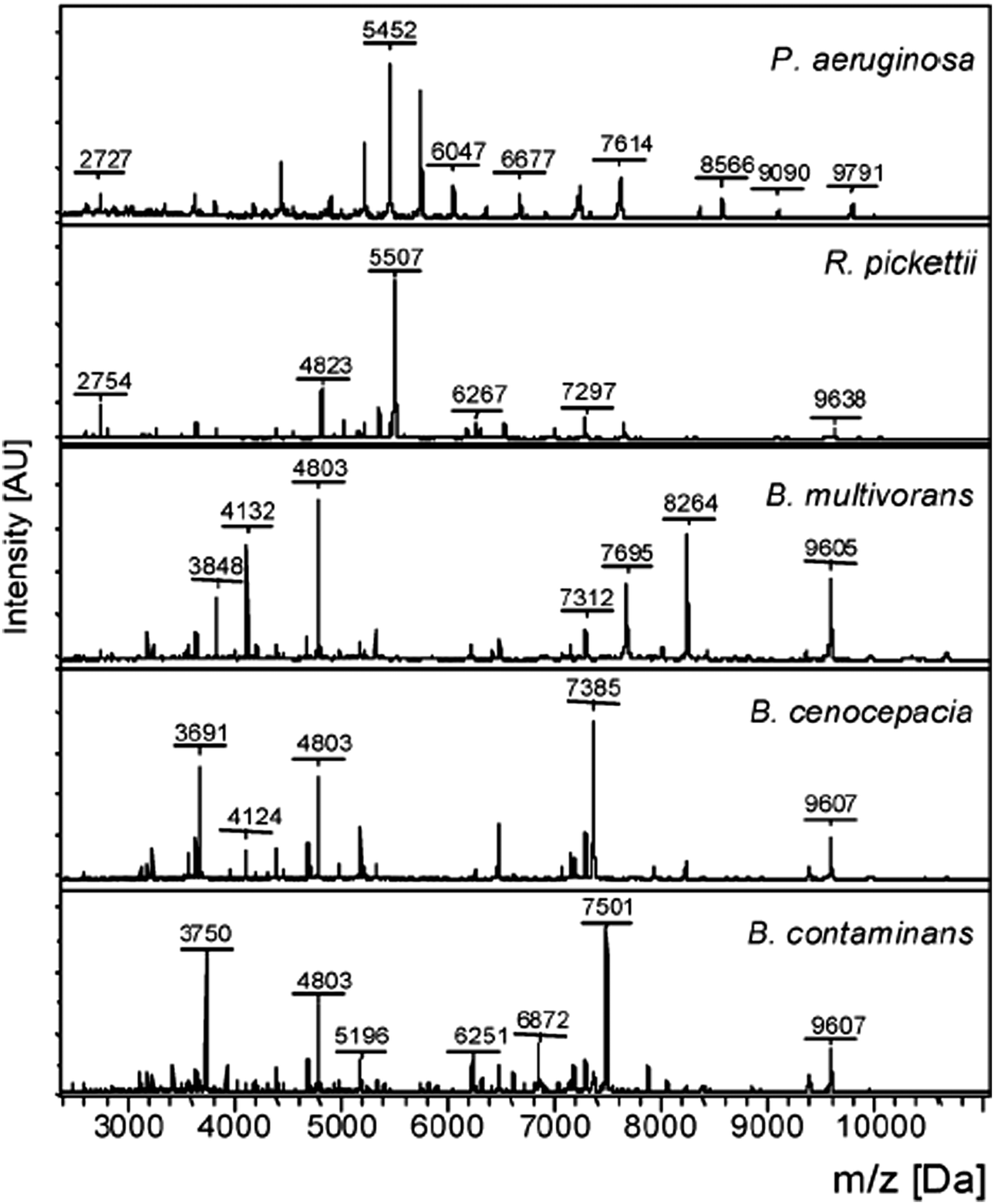 | ||
| Fig. 5 Intact cell MALDI-TOF MS spectra of Pseudomonas aeruginosa, Ralstonia pickettii, Burkholderia multivorans, Burkholderia cenocepacia, and Burkholderia contaminans (Reprinted from Minan et al.50 with permission from The Royal Society of Chemistry. Copyright 2009.). | ||
Applications of MALDI-TOF MS in clinical microbiology extend beyond microbial identification and this technique can also be used for rapid detection of antibacterial resistance. For example, β-lactamase activity in isolated bacteria can be detected using MALDI-TOF MS and used as a means of quickly inferring resistance to β-lactam antibiotics in a 2 h assay, Fig. 6.15
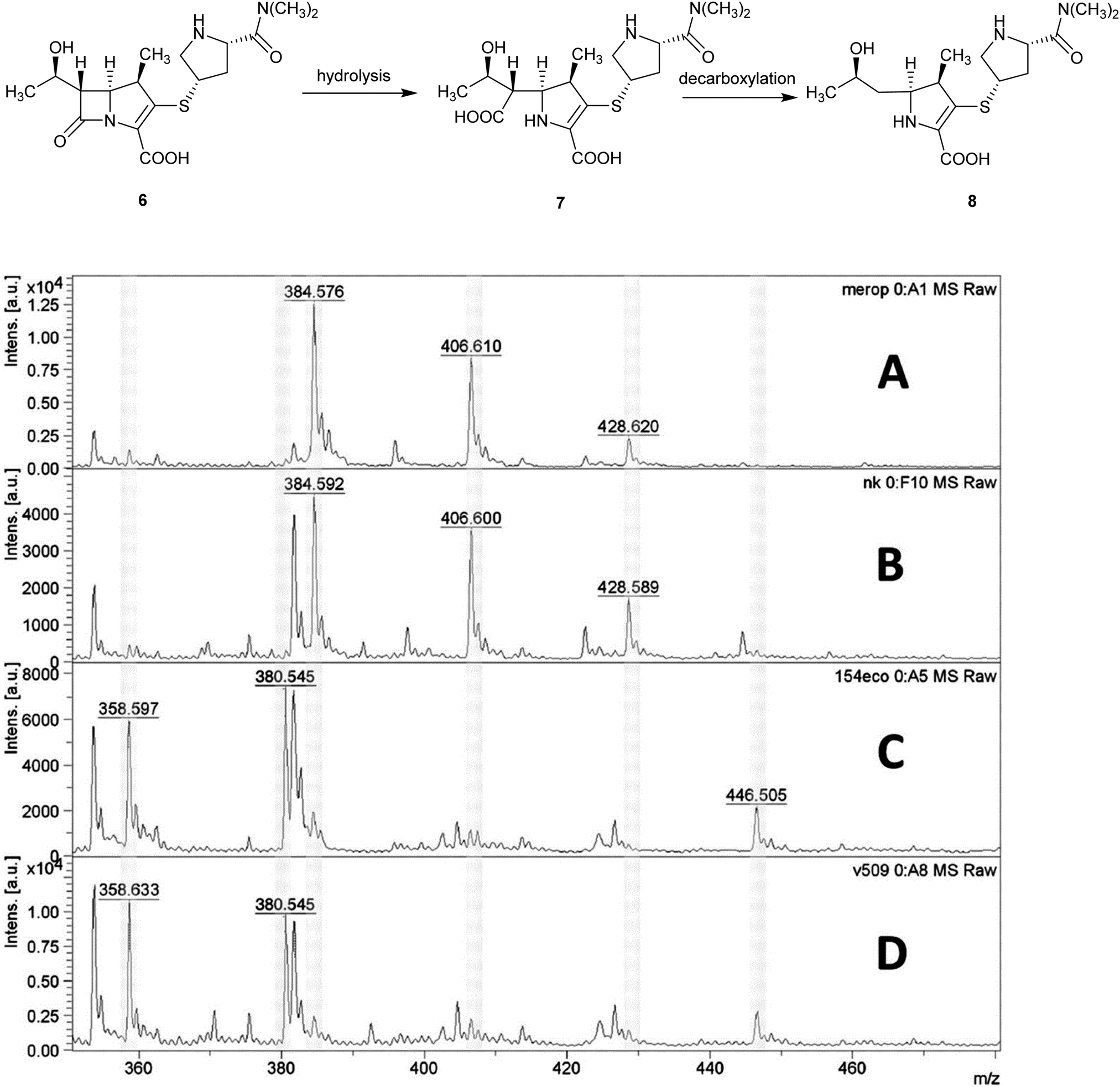 | ||
| Fig. 6 Detection of carbapenemase activity using MALDI-TOF analysis of meropenem hydrolysis; (A) spectrum of meropenem solution; (B) spectrum of non-carbapenemase-producing isolate of Klebsiella pneumoniae; (C) Escherichia coli isolate producing the New Delhi metallo-β-lactamase (NDM-1) carbapenemase; (D) NDM-1-producing Acinetobacter baumannii. MS peaks at m/z 358.6 (meropenem decarboxylation product after carbapenemase hydrolysis, [8 + H]+); 380.5 ([8 + Na]+), 384.5 (meropenem, [6 + H]+), and 406.6 ([6 + Na]+) (Reprinted from Hrabák et al.15 with permission from The American Society of Microbiology. Copyright 2012.). | ||
The role of culture in the clinical microbiology laboratory remains important and has been re-invigorated by complementary automated methods, such as MALDI-TOF MS, as well as automated methods of plate inoculation and automated reading devices that are particularly suited to interpretation of chromogenic media.16 Unlike other methods, culture has the advantage of delivering a bacterial isolate that can be subjected to further analyses – in particular antimicrobial susceptibility testing – but also to test for virulence determinants (e.g. toxins) and to perform typing to support investigation of outbreaks. It is important to remember that culture is not appropriate for a number of important bacterial pathogens because they are difficult or impossible to grow under laboratory conditions. There are many examples of such pathogens, such as the spirochetes, e.g. Borrelia burgdorferi (the primary causative agent of Lyme disease) and Treponema pallidum (the cause of syphilis). Such pathogens are usually diagnosed by the detection of specific antibodies in conjunction with the assessment of clinical symptoms or the molecular detection of specific DNA sequences. The inherent disadvantage of culture is that it is relatively slow compared to direct detection of antigens (e.g. using immunoassays) or molecular methods (e.g. using PCR). Most culture methods require at least 18 h incubation before colonies can be reliably detected and culture for slower-growing pathogens (e.g. Mycobacterium tuberculosis) may take several weeks.
4. Molecular diagnostic methods for bacterial detection
Molecular diagnostic methods rely on the analysis of genomic markers corresponding to nucleic acid sequences. The taxonomy and phylogeny of bacteria is based on the sequences of conserved genes, especially those coding for ribosomal ribonucleic acids (rRNA). As they are ubiquitous and comprise portions with various mutation rates (some highly conserved, others highly variable), they are exploited as universal molecular chronometers, similar to the hands of a clock.17 Molecular techniques, emerging from the 1960s onwards, allow for the fingerprinting and characterisation of microorganisms providing clinically essential information on their genus, species, antibiotic susceptibility, and viability. Diagnostic information is most often gained via (i) the direct characterisation of the rRNA, (ii) the amplification of the genes and characterisation of the amplicons, or (iii) the direct sequencing of the ribosomal genes.4.1. Hybridization-based detection
Hybridization-based detection provides a means of revealing the presence or absence of genes of interest. The probes are single or double stranded synthetic DNA fragments labelled with fluorescent dyes (e.g. Cy3 9 or FITC 10), Fig. 7, which, due to their complementarity to the target nucleic acid, allow for hybridization. A fluorescent signal thus indicates the presence of the analyte. For example, Fluorescence In Situ Hybridization (FISH), using bacterial and yeast universal probes, was able to identify 96.5% of the microorganisms present in 115 bacteraemia cases down to the family, genus or species level within 2.5 h.18 Although the assessment of the samples required microscopic observation and had a detection limit of 103 CFU per mL blood, the time saving achieved (in comparison to conventional methods) can be lifesaving in cases such as septicaemia. This approach was adapted using peptide nucleic acid fluorescence in situ hybridisation (PNA-FISH, AdvanDx) for the identification of Gram positive and Gram negative bacteria, as well as Candida species, from blood cultures.19 By targeting around 10 groups of the most commonly reported pathogens implicated in nosocomial bloodstream infections, the accuracy of this method was shown to be 100% for bacteria and 91% for yeast in samples with at least 105 CFU per mL present.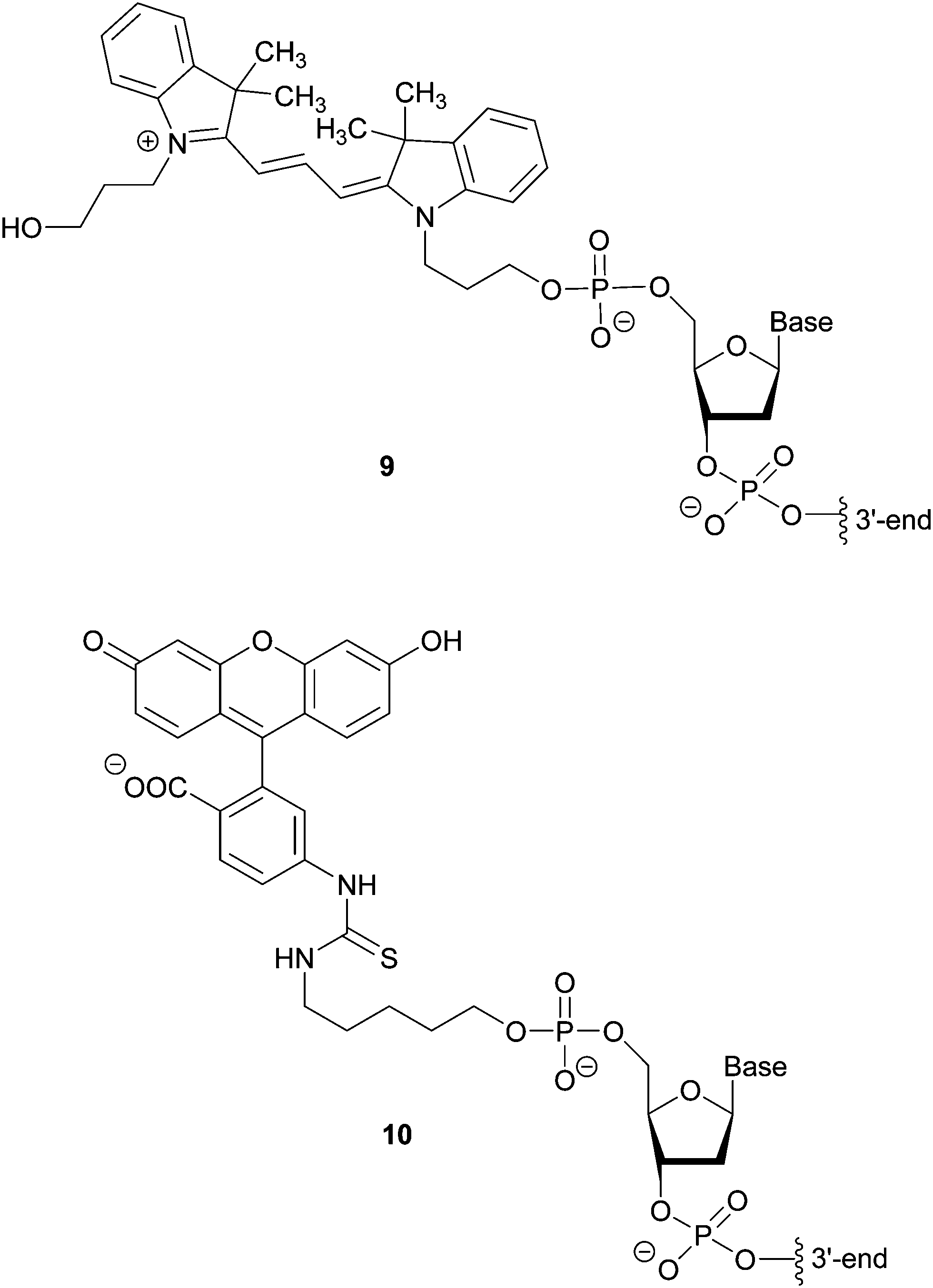 | ||
| Fig. 7 Structures of fluorescent oligonucleotide-linked labels Cy3 9 and FITC 10. | ||
4.2. Amplification methods
Amplification methods for the enhancement of the detection signal generated were designed for complex specimens comprising potentially billions of cells from other organisms along with the low numbers of bacteria of interest present. These methods allow for the targeting of sequences other than ribosomal nucleic acids, which is often necessary when looking for specific virulence or resistance mechanisms.Three of these amplification techniques, developed for the detection of Salmonella in food, were compared by targeting the invA gene or its corresponding messenger RNA, as they code for an essential virulence factor of salmonellae.20 Quantitative real-time polymerase chain reaction (qPCR) and reverse transcriptase real-time PCR (RT-qPCR) are both in vitro methods relying on precise thermal steps for nucleic acid denaturation and polymerisation (extension) with specific added, and often labelled, oligonucleotide primers to form new analogue double stranded DNAs, which then provide new targets for the next cycle, Fig. 8. The million-fold amplification achieved within 2 h allows for detection times (from sampling to result) of 5–24 h or less. By measuring the changes in fluorescence signal from the beginning of the chain reaction process, qPCR allows for minimal sample manipulation pre- and post-amplification, thereby reducing the risk of contamination and the time required. Moreover, the key advantage of RT-qPCR is its ability to differentiate and measure only viable organisms. The third method, Loop Mediated Isothermal Amplification (LAMP) has several added advantages over PCR techniques, including its moderate-temperature amplification procedure, thus allowing for simple and cost-effective equipment. High specificity is achieved in a rapid and robust single step amplification (avoiding high temperature denaturation), which provides direct detection of DNA at 60–65 °C within 15–60 min from nearly raw samples by using 4 primers and the strand-displacement activity of Bst DNA polymerase.
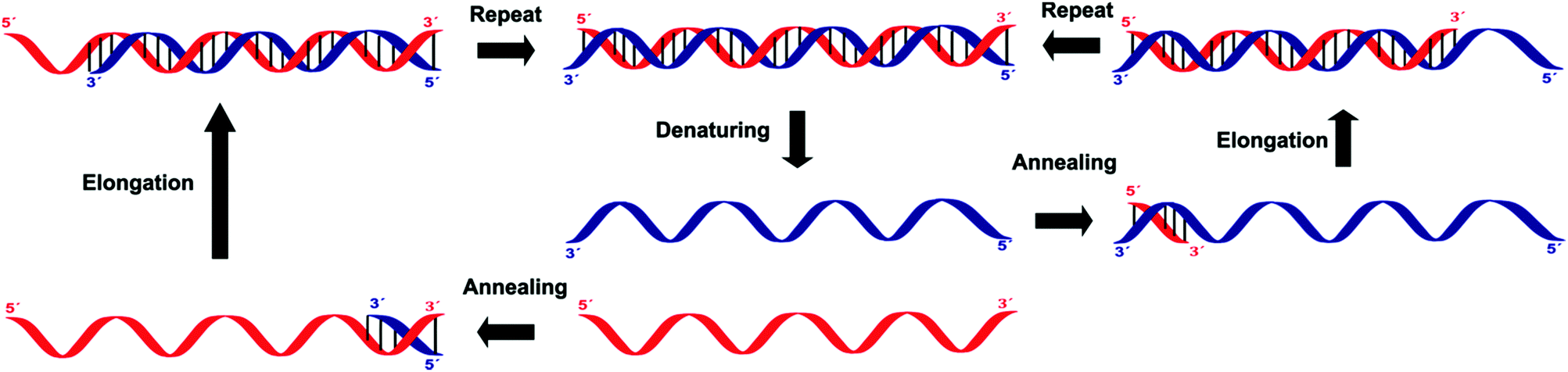 | ||
| Fig. 8 Outline of the processes involved in the polymerase chain reaction (PCR). | ||
In staphylococci, the resistance to methicillin and cephalosporins is linked to an altered penicillin-binding protein 2a (PBP2a). Methicillin resistant Staphylococcus aureus (MRSA) is detected with excellent specificity and sensitivity by IDI-MRSA (Infectio Diagnostic, Inc.) via simultaneous targeting of the staphylococcal chromosomal cassettes (SCCmec) and a S. aureus highly conserved open reading frame, ofrX, within 1.5 h from having the specimen in hand.21 This approach utilizes a specific PCR primer coupled with a molecular beacon probe, Fig. 9. The complex design of such probes is balanced out by their extreme specificity, their success in multiple target assays and the fact that they allow for detection directly from a swab specimen, without the need for initial culture.
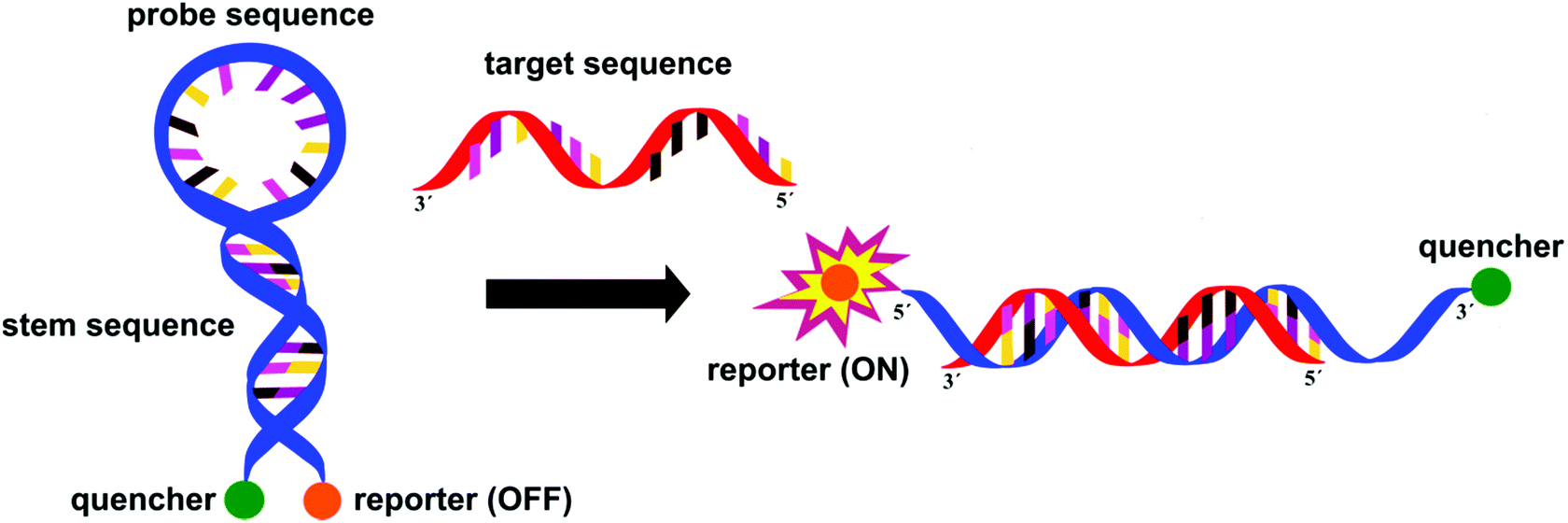 | ||
| Fig. 9 Molecular beacons are hairpin shaped probes consisting of a sequence specific oligonucleotide enclosed by 5–6 complimentary nucleotides (resulting in the hairpin shape) and two fluorescent dyes – a quencher and a reporter – on the 3′- and 5′-ends. When the target sequence is present, the hairpin selectively opens and displays the fluorescence of the reporter dye. | ||
With molecular methods, the risk of cross-contamination is linked to their intrinsic sensitivity and is exacerbated by the generation of billions of copies of the targeted sequences. Consequently, daily use of these in vitro diagnostic products has been significantly enhanced by the rise of self-contained, microfluidic lab-on-a-chip devices combining the various stages, such as cell lysis, nucleic acid purification, sequence amplification and target detection.22 To provide detection on sub-micron samples within 30 min to 2 h, most recently, a self-contained microfluidic in-gel LAMP (gLAMP) has been reported.23 This multiplexed pathogen detection device allowed for the detection of E. coli, Proteus hauseri, Vibrio parahaemolyticus and Salmonella enterica from serum samples. The extremely low detection limit of 3 copies per μL is partly facilitated by the incorporation of a non-fluorescent calcein–manganese complex, Fig. 10.
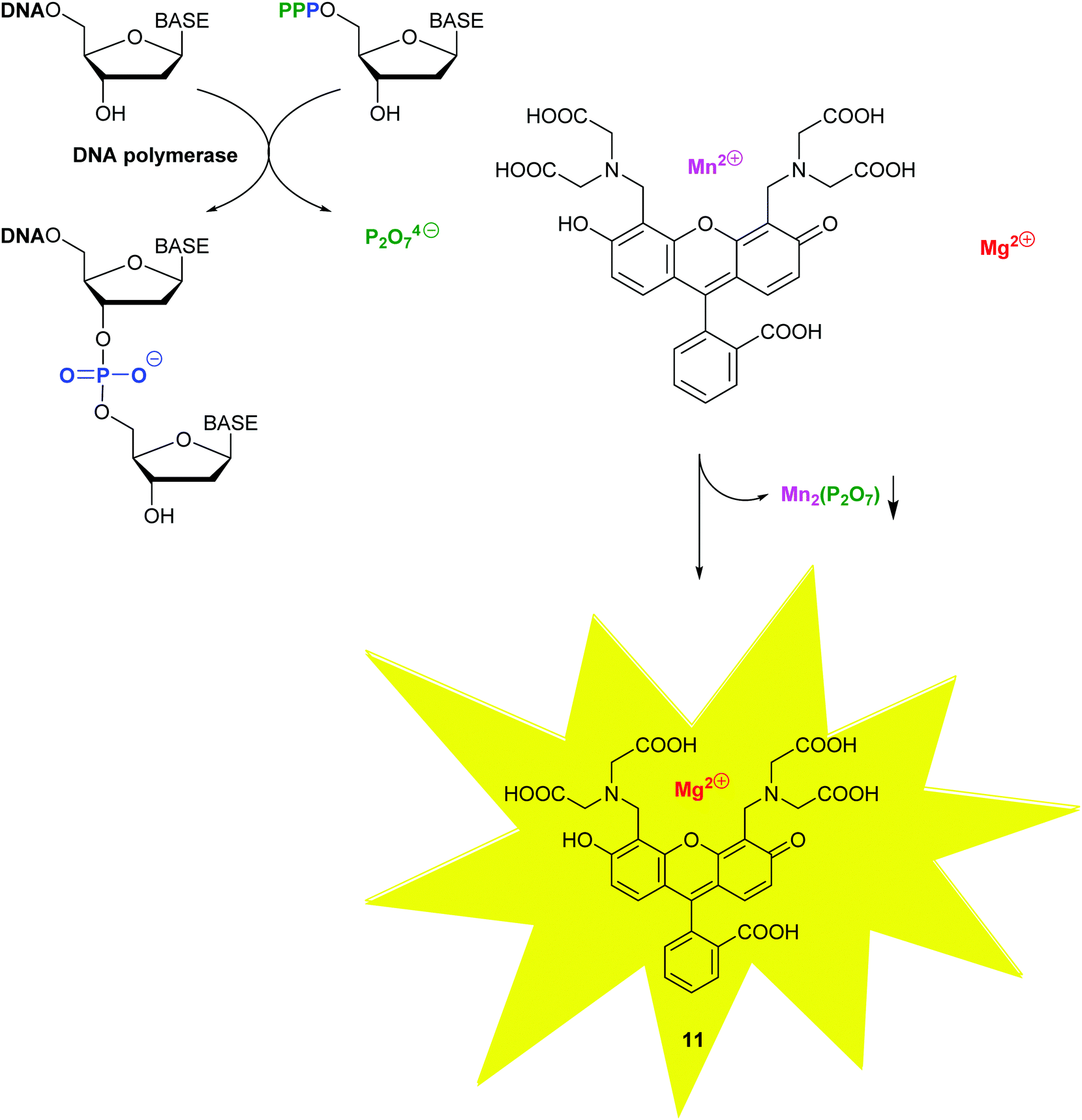 | ||
| Fig. 10 The fluorescence of calcein 8 is quenched in the presence of manganese(II), which precipitates out with the pyrophosphate by-product of the DNA polymerase-catalyzed addition of a nucleotide monophosphate at the 3-OH of the growing DNA chain, thus re-activating the fluorescence of calcein. | ||
The high analytical sensitivity and specificity of molecular diagnostics are especially powerful in the detection of slow growing microorganisms and ones that are difficult to isolate on culture due to their special growth requirements, such as Mycoplasma pneumoniae and Legionella spp.24 The Xpert MTB/RIF assay (Cepheid) incorporates a combination of PCR amplifications resulting in the simultaneous detection of M. tuberculosis complex and rifampin–resistance profiling within 2 h.25
4.3. DNA microarrays (gene chip technology)
Large-scale multiplex analysis, utilising a variety of probes in order to target a wide range of organisms, along with their resistance mechanisms, allows for a deeper differentiation between closely related species, and facilitates the identification of multiple organisms within the same specimen. Due to the publically available large scale whole genome sequencing data, genes and combinations of genes can be specifically targeted by universal or consensus primers and capturing probes. In recent years there has been an exponential development in these multiplex syndromic platforms, where compact design allows for the enclosed and automated extraction of genetic analyte, amplification, hybridisation, and even endpoint melting curve analysis within a single instrument (on sample sizes of 2–400 μL).The FilmArray® Blood Culture (FA-BC, bioMérieux) panel offers a potential tool for the management of bloodstream infections through its ability to identify more than 25 pathogens and 4 antibiotic resistance genes in 1 h with excellent specificity.26
In a comparative study for the identification of gastroenteritis-causing bacteria, parasites, and viruses Verigene® enteric pathogens, Biofire FilmArray™ gastrointestinal and Luminex xTAG® gastrointestinal pathogen panels were evaluated on 152 stool specimens.27 Not only can these platforms achieve 100% sensitivity for most of the pathogens, but they can also identify co-infections not detected by conventional methods. Although these systems increase laboratory expense, the total hospital costs were shown to be reduced with the introduction of the above techniques.
Nosochip, a low density array for nosocomial pneumonia-causing bacteria (5 Gram-positive, 18 Gram-negative) and fungi (4) targets bacterial gyrA, fus/rps and fungal COX-2 genes with a variety of universal and consensus primers. The use of immobilised capture probes allows for detection limits of 10–1000 DNA copies and the identification of multi-pathogen infections make this method competitive when compared to culture.28
Resistance profiling relies on the simultaneous detection of a vast diversity of genes and mutations. Using LAMP assays in 64 and 384-well card designs, Gram-positive cocci were screened and antibiotic resistance was identified in only 30 min starting from isolated colonies.29
4.4. Whole genome sequencing
Whole Genome Sequencing (WGS) techniques provide comprehensive information allowing for the (i) identification of pathogens, (ii) exact profiling of resistance genes, (iii) recognition of outbreaks, (iv) non-species specific targeting without the requirement for continuous development of probes and primers (to follow organism mutations and evolution), and (v) the immediate design of PCR probes based on the generated genetic data in the event of outbreaks. Since the publication of the first generation sequencing techniques in the late 1970s, a variety of rapid and cost-effective technologies have become available. The most popular of the first generation techniques is the Sanger method, which relies on the replication of one predefined 15–200 nucleotide long DNA target sequence and the use of the 32P-labelled dideoxy analogues of the natural deoxynucleoside triphosphates to terminate the replicated chains by inhibiting the DNA polymerase. Second generation techniques (reversible termination, pyrosequencing, and sequencing by ligation) have the advantage of generating massively parallel sequences (instead of the multiplication of a single predefined target). Further developments, including the use of capillary instead of gel electrophoresis, have resulted in less laborious, faster sequencing methods, Fig. 11.30 Detection of the analytes varies across the different methods and can be based on fluorescence, label-free electronic (e.g. change of pH, or translocation of analyte through nanopores), or atomic signals.31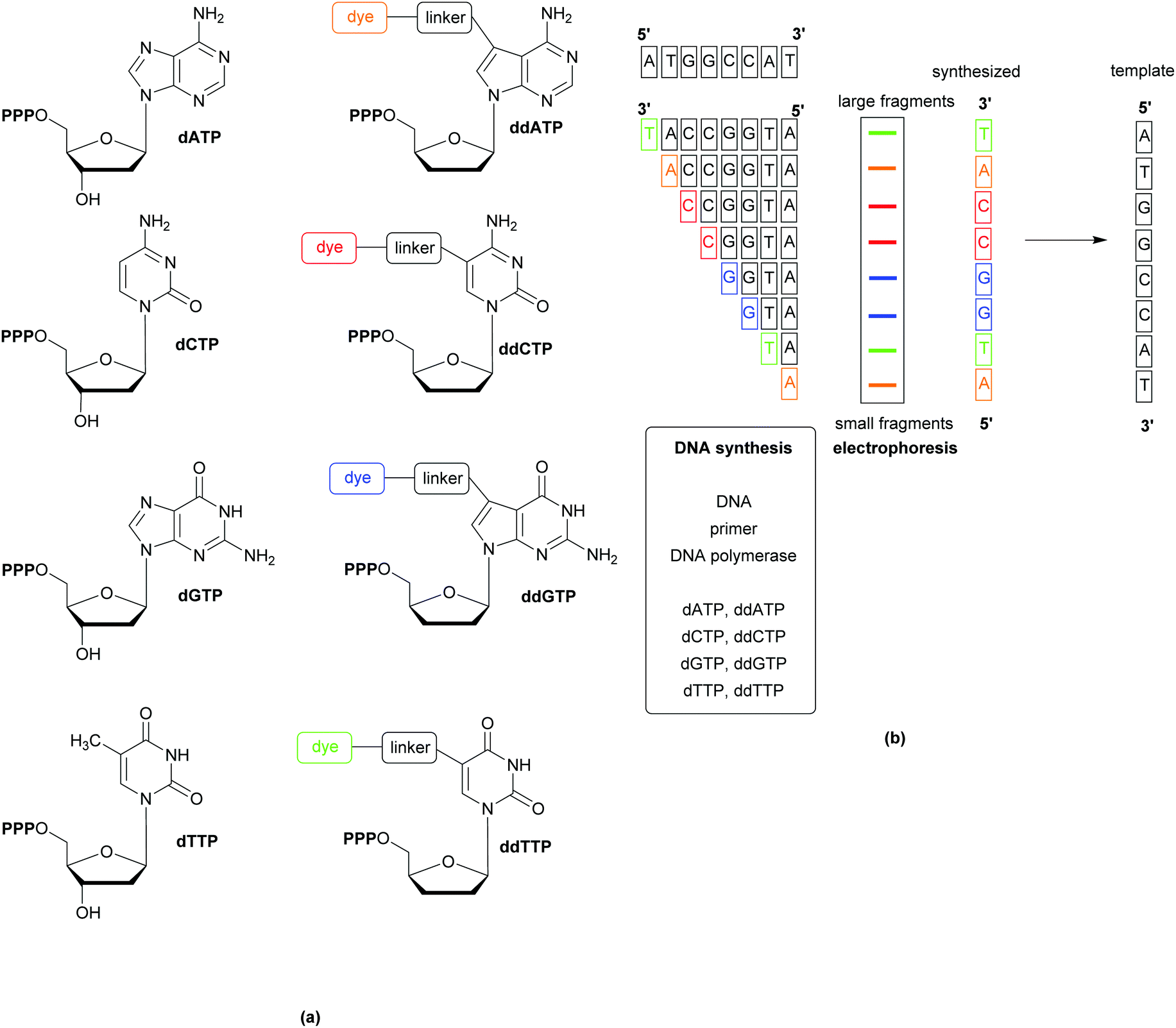 | ||
| Fig. 11 Schematic showing Sanger sequencing using coloured/fluorescent dyes linked to the chain terminating dideoxynucleotide triphosphates. A mixture of all four deoxynucleotide triphosphates (dNTP) and minor amounts of the dye-labelled dideoxynucleotide triphosphates (ddNTP) (a) is incubated with the DNA to be sequenced, a primer, and DNA polymerase (b). After separation of the fragments by electrophoresis, the sequence is indicated by the coloured bands, denoting where the sequence has been terminated by the inclusion of a dideoxynucleotide triphosphate. | ||
The wealth of information gained from WGS has facilitated improvements in current diagnostic methods, such as the discovery of the mecC gene (a homologue of the mecA gene,that is responsible for methicillin resistance in MRSA), which has prompted the redesign of PCR assays to improve sensitivity and avoid false negatives.32 Comparisons of predictions based on WGS data against phenotypic drug susceptibility have shown WGS to be highly sensitive and specific, as well as displaying high concordance with phenotypic antibacterial susceptibility/resistance techniques.33 These promising results indicate that WGS could be developed into a useful tool for informing clinical decision making; analysis of the entire genome for the presence of mobile genetic elements or point mutations known to confer resistance can be achieved with little additional cost (or effort) to that of the initial sequencing. WGS is also important when the elucidation of the mechanisms of resistance is important, such as when carbapenem resistance is detected during screening; using WGS it would be possible to determine if the resistance is due to carbapenemase or by virtue of other mechanisms.34 The source of an outbreak, tracking of its onward transmission, and whether separate cases are linked can also be determined using WGS.
For the evaluation of the performance indicators and financial viability of WGS methods, a multi-centre study was conducted with 8 laboratories across Europe and North America. Identification, typing, and resistance profiling against 7 classes of antibiotics on culture positive M. tuberculosis specimens were performed via sequencing using the MiSeq (Illumina) platform and a semi-automated bioinformatics pipeline. The sequencing here is based on the second generation technique, reversible termination. The consequent steps to generate massively parallel sequences include: the capture of high-density primers, the clonal amplification applying surface PCR (resulting in signal enhancement), flow cell array facilitation of the DNA polymerase incorporation of one of the 4 different fluorescently labelled terminators (which allow for detection and sequence-reading), followed by the cleavage of these terminators to allow for hundreds of repeat loops in order to generate sufficient data, which then can be computationally evaluated. Of 672 drug/specimen combinations, WGS provided 93% accuracy and shortened the time to result from 31 days to 9 days (median). In addition, this method was able to link 15 out of the 91 UK patients to nine outbreak clusters. Moreover, under the conditions of this study, a 7% overall cost-reduction was achieved by WGS.35
Similarly, WGS analysis of MRSA, VRE, MDR E. coli and MDR P. aeruginosa species in clinical isolates brought a total saving of €200k during a 6-month study conducted in German hospitals. According to this evaluation, the cost-reduction was a result of the rapid (4–5 days) turnover, thus avoiding the unnecessary isolation of patients.36
For its direct application to clinical specimens, WGS needs to meet the requirements of providing results (that correlate with the phenotypical evaluation) within a few hours via autonomous handling of mixed-flora specimens. In a preliminary study of 35 urine samples – a low complexity human matrix – direct sequencing from urine samples was proven to be superior to phenotypic characterisation in resolving some otherwise uncharacterized mixed-cultures, which were unable to grow on the culture medium. When only the abundant resistance genes were included, in almost all cases direct sequencing detected the exact same resistance genes and predicted susceptibility profiles accurately within 24 h when compared to the evaluation of single isolates.37 This turnaround time was reduced when third generation MinION (Oxford Nanopore) nanopore sequencing system was used on pre-handled urine samples. Digested DNA was translocated through a cyclodextrin-modified nanopore system consisting of the natural channel protein, α-haemolysin. The sequencing results were routinely compared via a BLAST search and the CARD (Comprehensive Antibiotic Resistance Database);38 51 of the 55 acquired resistance genes (identified by reference sequencing of the cultivated bacteria) were found by MinION directly from urine. When combined with Metrichor WIMP and ARMA software and rapid (15 min) library preparation kit, the total turnaround time was further reduced to 4 h. However, for routine application, improvements in sensitivity (105 CFU per mL), in performance on polymicrobial urines, in characterisation of various resistance mechanisms and in reduction of analysis cost need to be initiated.
Despite the encouraging developments reached by the end of 2016,39 a subcommittee of EUCAST has requested a further expansion of knowledge in order to consider WGS as an accurate tool for antimicrobial susceptibility screening.40 Establishment of harmonized analytical approaches, defined quality control metrics and performance standards still remain a challenge. Currently, the multiple short read lengths that are generated from WGS sequencing must be mapped out against reference genomes or assembled de novo in order for the information to be useful. Mapping-based assembly may leave gaps if the attained sequence fails to match reference genome sequences, while de novo methods can produce incomplete sequences.41 The development of automated/semi-automated sequence assembly, analysis and presentation of results would transform the generated information into an easily interpretable format.41 While phenotypic methods face the challenge of accounting for the strain diversity within a taxon, the accuracy of molecular methods will always be dependent on the diversity and quality of the sequences, as baseline measures in the design of the applied reagents (primers and probes) and data bases, which – as proven by WGS – varies from strain to strain within species.42
4.5. Multi-omics approaches
Multi-omics approaches combining metagenomics and metabolomics are able to give an overall picture of the microbial genomes and their metabolic activities (including expressed proteins and produced metabolites). This exhaustive microbial analysis of specimens allows for the determination of all expressed metabolic activities by the community of microorganisms and the direct impact thereof on the host. Generation and disclosure of such information allows for appropriate, targeted therapeutic decisions in order to inactivate detrimental microbial activities with minimal side-effects and to avoid the transition and emergence of multi-drug resistant organisms. To meet therapeutic time constraints, these multi-omics platforms require further development of integrated analysis pipelines associating highly powerful analysis tools and extensive data bases. As proof-of-concept, human and food samples were evaluated in an integrated pipeline for their 16S rRNA gene sequencing, inferred gene function profiles, and metabolomics, with the latter involving liquid chromatography-tandem mass spectrometry (LC-MS/MS).43 The combination of standardized protocols, dedicated analysis infrastructure and connected supercomputers allowed for the determination of relationship between consumption of fermented nutrients, gut bacterial diversity and metabolites in stool within 48 h from receiving the specimens. These results are encouraging for the identification of microbiome dysbiosis or metabolome changes that indicate disease.5. Conclusions
As can be seen from Table 2, current techniques each have their advantages and disadvantages, with their suitability for bacterial detection/identification being determined by a combination of their specificity/sensitivity, financial considerations, and the availability of the necessary instrumentation. The need for better bacterial detection and identification methods is, however, driving change.| Characteristic | Techniquea | |||||
|---|---|---|---|---|---|---|
| Biochemical testing | Chromogenic media | MALDI-TOF | PCR | DNA microarrays | WGS | |
| a Within each row, + represents the least and ++++ the most. b From previously isolated colonies. | ||||||
| Sensitivity | ++ | ++ | +++ | +++ | +++ | +++ |
| Specificity | ++ | + | +++ | +++ | +++ | +++ |
| Cost | ++ | + | ++ | +++ | ++++ | ++++ |
| Complexity | ++ | + | ++ | ++ | + | ++++ |
| Direct detection from clinical samples | No | Yes | No | Yes | Yes | No |
| Time to result (h) | 2–3b | >16 | 0.2–3b | 2 | 1 | >24 |
MALDI-TOF MS techniques are being further refined and developed to allow direct application of some clinical samples to the MS analysis plate, removing the need for prior overnight culture and speeding up the bacterial identification process. These new methods are reliable, with urine samples in which the level of infection is between 1.5 × 105 and 5 × 106 bacterial cells per mL, determined by flow cytometry;44,45 however, although the metrics are promising, with >85% of bacteria identified, and the time from sample receipt to bacterial ID as low as 1 h, some bacteria are not easily identified and susceptibility testing still requires 18–24 h. In the future, direct bacterial identification using MS from a wider range of samples may become a welcome reality; however, such direct techniques will not be suitable for all bacteria and clinical samples, so the need for overnight culture will remain for some applications.
An application with potential for use in a point-of-care device is the classification of bacteria based upon the volatile organic compounds (VOCs), which are unique to each genus (or species), using pH sensor impregnated cellulose acetate membranes and a software App for a smart phone. Using this method, it was possible to discriminate between isolated strains of four pathogenic Enterobacteriaceae; K. pneumoniae, Proteus vulgaris, Proteus mirabilis, and E. coli.46
A colorimetric sensor array (CSA) has also been utilised for the detection of VOCs: 15 bacterial pathogens cultured on blood agar were identified, with 91% sensitivity and 99.4% specificity, on average 1.9 h before colonies were detected by visual inspection. As with the previous method, testing on cultures containing more than one pathogen will help to determine the extent of the utility of this method; even if the identification of individual pathogens in a polymicrobial mixture is not possible, the detection of bacterial growth before colonies are visible would be a very useful indicator of infection.47
‘Lab-on-a-chip’ developments are beginning to overcome some of the technical, biological and chemical obstacles, offering a future promise of USB-accessed microelectronic and microfluidic technology for clinical applications, including bacterial identification and susceptibility.48
In a comprehensive evaluation of ten alternative technologies, which are predicted to have significant impact on the future use of antibiotics, point-of-care diagnostics was proven to have the potential to most profoundly affect the demand of antimicrobial agents.49 Improvement of community treatment regimes, rational prescribing habits and the reduction of R&D trial expenditures could all benefit from POC diagnostics and, due to their high sensitivity and specificity, molecular methods are probably the technology of choice for such POC.
Eventually, multidisciplinary methods, including responsive revolutionary materials, electronics, big data, autonomous systems, machine learning, and supercomputing will combine to provide lifesaving diagnostic solutions for the future's most concerning healthcare threat.
References
- S. C. Davies, T. Fowler, J. Watson, D. M. Livermore and D. Walker, Lancet, 2013, 381, 1606–1609 CrossRef.
- S. C. Davies, Annual Report of the Chief Medical Officer, Volume Two, 2011. Infections and the rise of antimicrobial resistance, London, 2013 Search PubMed.
- M. Bassetti, M. Merelli, C. Temperoni and A. Astilean, Ann. Clin. Microbiol. Antimicrob., 2013, 12, 22 CrossRef PubMed.
- National Health and Medical Research Council (NHMRC), Australian Guidelines for the Prevention and Control of Infection in Healthcare, Canberra, 2010, ISBN 1864965282 Search PubMed.
- World Health Organization (WHO), The evolving threat of antimicrobial resistance. Options for action, Geneva, 2012, ISBN 9789241503181 Search PubMed.
- Centers for Disease Control and Prevention, Antibiotic resistance threats in the United States, 2013, CDC, Atlanta, US, 2013 Search PubMed.
- World Health Organization (WHO), Antimicrobial resistance: global report on surveillance, 2014 Search PubMed.
- Center for Disease Dynamics, Economics and Policy, State of the World's Antibiotics, 2015, http://cddep.org/publications/state_worlds_antibiotics_2015#sthash.ztACc9UO.dpbs.
- J. H. Jorgensen and M. J. Ferraro, Clin. Infect. Dis., 2009, 49, 1749–1755 CrossRef CAS PubMed.
- S. Orenga, A. L. James, M. Manafi, J. D. Perry and D. H. Pincus, J. Microbiol. Methods, 2009, 79, 139–155 CrossRef CAS PubMed ; and references therein.
- J. D. Perry, Clin. Microbiol. Rev., 2017, 30, 449–479 CrossRef PubMed ; and references therein.
- C. J. Ingham, S. Boonstra, S. Levels, M. de Lange, J. F. Meis and P. M. Schneeberger, PLoS One, 2012, 7, e33818 CAS.
- C. D. Doern and S. M. Butler-Wu, J. Mol. Diagn., 2016, 18, 789–802 CrossRef CAS PubMed.
- N. Singhal, M. Kumar, P. K. Kanaujia and J. S. Virdi, Front. Microbiol., 2015, 6, 791 Search PubMed.
- J. Hrabák, V. Studentova, R. Walkova, H. Zemlickova, V. Jakubu, E. Chudackova, M. Gniadkowski, Y. Pfeifer, J. D. Perry, K. Wilkinson and T. Bergerova, J. Clin. Microbiol., 2012, 50, 2441–2443 CrossRef PubMed.
- T. J. Kirn, J. Clin. Microbiol., 2016, 54, 2424–2426 CrossRef PubMed.
- C. R. Woese, Microbiol. Rev., 1987, 51, 221–271 CAS.
- V. A. J. Kempf, K. Trebesius and I. B. Autenrieth, J. Clin. Microbiol., 2000, 38, 830–838 CAS.
- D. M. Harris and D. J. Hata, Ann. Clin. Microbiol. Antimicrob., 2013, 12, 2 CrossRef PubMed.
- G. Zhang, E. W. Brown and N. Gonzalez-Escalona, Appl. Environ. Microbiol., 2011, 77, 6495–6501 CrossRef CAS PubMed.
- D. K. Warren, R. S. Liao, L. R. Merz, M. Eveland and W. M. Dunne, J. Clin. Microbiol., 2004, 42, 5578–5581 CrossRef CAS PubMed.
- R. H. Liu, J. Yang, R. Lenigk, J. Bonanno and P. Grodzinski, Anal. Chem., 2004, 76, 1824–1831 CrossRef CAS PubMed.
- C. Chen, P. Liu, X. Zhao, W. Du, X. Feng and B.-F. Liu, Sens. Actuators, B, 2017, 239, 1–8 CrossRef CAS.
- M. J. Espy, J. R. Uhl, L. M. Sloan, S. P. Buckwalter, M. F. Jones, E. A. Vetter, J. D. Yao, N. L. Wengenack, J. E. Rosenblatt, F. R. Cockerill, 3rd and T. F. Smith, Clin. Microbiol. Rev., 2006, 19, 165–256 CrossRef CAS PubMed.
- R. Blakemore, E. Story, D. Helb, J. Kop, P. Banada, M. R. Owens, S. Chakravorty, M. Jones and D. Alland, J. Clin. Microbiol., 2010, 48, 2495–2501 CrossRef CAS PubMed.
- A. J. Blaschke, C. Heyrend, C. L. Byington, M. A. Fisher, E. Barker, N. F. Garrone, S. A. Thatcher, A. T. Pavia, T. Barney, G. D. Alger, J. A. Daly, K. M. Ririe, I. Ota and M. A. Poritz, Diagn. Microbiol. Infect. Dis., 2012, 74, 349–355 CrossRef CAS PubMed.
- R. S. P. Huang, C. L. Johnson, L. Pritchard, R. Hepler, T. T. Ton and J. J. Dunn, Diagn. Microbiol. Infect. Dis., 2016, 86, 336–339 CrossRef CAS PubMed.
- S. Burteau, P. Bogaerts, R. de Mendonca, L. Irenge, C. Berhin, J. Hiffe, N. de San, P. Beyne, S. Hamels, Y. Glupczynski, M. Struelens, J. L. Gala and J. Remacle, Eur. J. Clin. Microbiol. Infect. Dis., 2008, 27, 17–27 CrossRef CAS PubMed.
- T. Kostić, M. Ellis, M. R. Williams, T. M. Stedtfeld, J. B. Kaneene, R. D. Stedtfeld and S. A. Hashsham, Appl. Microbiol. Biotechnol., 2015, 99, 7711–7722 CrossRef PubMed.
- S. Ambardar, R. Gupta, D. Trakroo, R. Lal and J. Vakhlu, Indian J. Microbiol., 2016, 56, 394–404 CrossRef CAS PubMed ; and references therein.
- S. Moorthie, C. J. Mattocks and C. F. Wright, HUGO J., 2011, 5, 1–12 CrossRef PubMed.
- G. K. Paterson, E. M. Harrison and M. A. Holmes, Trends Microbiol., 2014, 22, 42–47 CrossRef CAS PubMed.
- N. C. Gordon, J. R. Price, K. Cole, R. Everitt, M. Morgan, J. Finney, A. M. Kearns, B. Pichon, B. Young, D. J. Wilson, M. J. Llewelyn, J. Paul, T. E. A. Peto, D. W. Crook, A. S. Walker and T. Golubchik, J. Clin. Microbiol., 2014, 52, 1182–1191 CrossRef CAS PubMed.
- C. U. Köser, L. J. Fraser, A. Ioannou, J. Becq, M. J. Ellington, M. T. G. Holden, S. Reuter, M. E. Török, S. D. Bentley, J. Parkhill, N. A. Gormley, G. P. Smith and S. J. Peacock, J. Antimicrob. Chemother., 2014, 69, 1275–1281 CrossRef PubMed.
- L. J. Pankhurst, C. del Ojo Elias, A. A. Votintseva, T. M. Walker, K. Cole, J. Davies, J. M. Fermont, D. M. Gascoyne-Binzi, T. A. Kohl, C. Kong, N. Lemaitre, S. Niemann, J. Paul, T. R. Rogers, E. Roycroft, E. G. Smith, P. Supply, P. Tang, M. H. Wilcox, S. Wordsworth, D. Wyllie, L. Xu and D. W. Crook, Lancet Respir. Med., 2016, 4, 49–58 CrossRef CAS PubMed.
- A. Mellmann, S. Bletz, T. Böking, F. Kipp, K. Becker, A. Schultes, K. Prior and D. Harmsen, J. Clin. Microbiol., 2016, 54, 2874–2881 CrossRef PubMed.
- H. Hasman, D. Saputra, T. Sicheritz-Ponten, O. Lund, C. A. Svendsen, N. Frimodt-Møller and F. M. Aarestrup, J. Clin. Microbiol., 2014, 52, 139–146 CrossRef PubMed.
- K. Schmidt, S. Mwaigwisya, L. C. Crossman, M. Doumith, D. Munroe, C. Pires, A. M. Khan, N. Woodford, N. J. Saunders, J. Wain, J. O'Grady and D. M. Livermore, J. Antimicrob. Chemother., 2017, 72, 104–114 CrossRef CAS PubMed.
- R. H. Deurenberg, E. Bathoorn, M. A. Chlebowicz, N. Couto, M. Ferdous, S. García-Cobos, A. M. D. Kooistra-Smid, E. C. Raangs, S. Rosema, A. C. M. Veloo, K. Zhou, A. W. Friedrich and J. W. A. Rossen, J. Biotechnol., 2017, 243, 16–24 CrossRef CAS PubMed.
- M. J. Ellington, O. Ekelund, F. M. Aarestrup, R. Canton, M. Doumith, C. Giske, H. Grundman, H. Hasman, M. T. G. Holden, K. L. Hopkins, J. Iredell, G. Kahlmeter, C. U. Köser, A. MacGowan, D. Mevius, M. Mulvey, T. Naas, T. Peto, J. M. Rolain, Ø. Samuelsen and N. Woodford, Clin. Microbiol. Infect., 2017, 23, 2–22 CrossRef CAS PubMed.
- J. Price, N. Claire Gordon, D. Crook, M. Llewelyn and J. Paul, Clin. Microbiol. Infect., 2013, 19, 784–789 CrossRef CAS PubMed.
- H. Tettelin, V. Masignani, M. J. Cieslewicz, C. Donati, D. Medini, N. L. Ward, S. V. Angiuoli, J. Crabtree, A. L. Jones, A. S. Durkin, R. T. DeBoy, T. M. Davidsen, M. Mora, M. Scarselli, I. Margarit y Ros, J. D. Peterson, C. R. Hauser, J. P. Sundaram, W. C. Nelson, R. Madupu, L. M. Brinkac, R. J. Dodson, M. J. Rosovitz, S. A. Sullivan, S. C. Daugherty, D. H. Haft, J. Selengut, M. L. Gwinn, L. Zhou, N. Zafar, H. Khouri, D. Radune, G. Dimitrov, K. Watkins, K. J. B. O'Connor, S. Smith, T. R. Utterback, O. White, C. E. Rubens, G. Grandi, L. C. Madoff, D. L. Kasper, J. L. Telford, M. R. Wessels, R. Rappuoli and C. M. Fraser, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13950–13955 CrossRef CAS PubMed.
- R. A. Quinn, J. A. Navas-Molina, E. R. Hyde, S. J. Song, Y. Vázquez-Baeza, G. Humphrey, J. Gaffney, J. J. Minich, A. V. Melnik, J. Herschend, J. DeReus, A. Durant, R. J. Dutton, M. Khosroheidari, C. Green, R. da Silva, P. C. Dorrestein and R. Knight, mSystems, 2016, 1 DOI:10.1128/mSystems.00038-16.
- M. Íñigo, A. Coello, G. Fernández-Rivas, B. Rivaya, J. Hidalgo, M. D. Quesada and V. Ausina, J. Clin. Microbiol., 2016, 54, 988–993 CrossRef PubMed.
- Y. Zboromyrska, E. Rubio, I. Alejo, A. Vergara, A. Mons, I. Campo, J. Bosch, F. Marco and J. Vila, Clin. Microbiol. Infect., 2016, 22, 561 Search PubMed.
- L. Bueno, A. Cottell, S. M. Reddy and T. Paixao, RSC Adv., 2015, 5, 97962–97965 RSC.
- S. H. Lim, S. Mix, V. Anikst, I. Budvytiene, M. Eiden, Y. Churi, N. Queralto, A. Berliner, R. A. Martino, P. A. Rhodes and N. Banaei, Analyst, 2016, 141, 918–925 RSC.
- J. D. Besant, E. H. Sargent and S. O. Kelley, Lab Chip, 2015, 15, 2799–2807 RSC.
- E. Nwokoro, R. Leach, C. Årdal, E. Baraldi, K. Ryan and J. Plahte, J. Pharm. Policy Pract., 2016, 9, 34 CrossRef PubMed.
- A. Minan, A. Bosch, P. Lasch, M. Stammler, D. O. Serra, J. Degrossi, B. Gatti, C. Vay, M. D'Aquino, O. Yantorno and D. Naumann, Analyst, 2009, 134, 1138–1148 RSC.
Footnotes |
| † The sensitivity of these clinical tests refers to their ability to correctly identify the presence of the bacterium of interest in all cases where it is present (true positives, with no false negatives); specificity refers to their ability to detect only the microorganism of interest (i.e. no false positives). |
| ‡ The focus in this review is on the detection and identification of clinically relevant bacteria, but the principles are essentially the same for bacteria and other microorganisms in food and water. |
| § Microorganisms that gain benefit from their human host and cause no harm, providing they remain in a specific location. |
| This journal is © The Royal Society of Chemistry 2017 |