
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorine-18 labelled building blocks for PET tracer synthesis
Dion
van der Born†
 a,
Anna
Pees†
a,
Alex J.
Poot
a,
Romano V. A.
Orru
b,
Albert D.
Windhorst
a and
Danielle J.
Vugts
*a
a,
Anna
Pees†
a,
Alex J.
Poot
a,
Romano V. A.
Orru
b,
Albert D.
Windhorst
a and
Danielle J.
Vugts
*a
aDepartment of Radiology & Nuclear Medicine, VU University Medical Center, Amsterdam, De Boelelaan 1085c, 1081 HV Amsterdam, The Netherlands. E-mail: d.vugts@vumc.nl
bDepartment of Chemistry and Pharmaceutical Sciences and Amsterdam Institute for Molecules, Medicines & Systems (AIMMS), VU University Amsterdam, De Boelelaan 1083, 1081 HV, Amsterdam, The Netherlands
First published on 13th June 2017
Abstract
Positron emission tomography (PET) is an important driver for present day healthcare. Fluorine-18 is the most widely used radioisotope for PET imaging and a thorough overview of the available radiochemistry methodology is a prerequisite for selection of a synthetic approach for new fluorine-18 labelled PET tracers. These PET tracers can be synthesised either by late-stage radiofluorination, introducing fluorine-18 in the last step of the synthesis, or by a building block approach (also called modular build-up approach), introducing fluorine-18 in a fast and efficient manner in a building block, which is reacted further in one or multiple reaction steps to form the PET tracer. This review presents a comprehensive overview of the synthesis and application of fluorine-18 labelled building blocks since 2010.
 Dion van der Born | Dion van der Born received his MSc in chemistry in 2016 at the VU University in Amsterdam, where he is also currently finishing his PhD thesis under the supervision of Dr D. J. Vugts, Prof. Dr ir. R. V. A. Orru and Prof. Dr A. D. Windhorst on the development of novel radiochemical methodology for the incorporation of the fluorine-18 labelled tri-fluoromethyl group directly onto arenes. This methodology enables radiolabelling and biological evaluation of active pharmaceutical ingredients containing the trifluoromethyl group. Dion is currently employed as a radiochemist at FutureChemistry, where he is involved in the development of radiolabelled compounds and their biological evaluation. |
 Anna Pees | Anna Pees studied biomedical chemistry at the University of Mainz, Germany. She performed her Master's thesis in the group of Prof. Dr F. Rösch focusing on the synthesis of lipophilic gallium-68 chelators. Since 2016, she is working as a doctoral student under supervision of Prof. Dr A. D. Windhorst and Dr D. J. Vugts in the field of radiopharmaceutical chemistry at the VU University Medical Center Amsterdam, The Netherlands. Her research interest is the development of new radiofluorination methods which enable the synthesis of new PET tracers for imaging cancer and neurological disorders. |
 Alex J. Poot | In 2008 Alex J. Poot obtained his PhD in Medicinal Chemistry from Utrecht University under supervision of Prof. Liskamp. As postdoctoral researcher at the VU University Medical Center (VUmc), Amsterdam, he developed radiolabelled anticancer drugs for PET imaging. In 2014, he accepted a research fellowship from Memorial Sloan Kettering Cancer Center New York to develop carbon-13 labelled probes for tumour metabolism imaging with MRI. In 2015 he was appointed as assistant professor at the VUmc, where his research focusses on novel radiolabelling methods with carbon-11 and fluorine-18, and the application of radiolabelled drugs for oncology PET imaging. |
 Romano V. A. Orru | Romano V. A. Orru studied molecular sciences at the Agricultural University in Wageningen, the Netherlands, where he obtained his PhD in 1994. From 1996–2000 he worked in the group of K. Faber at the Technical and Karl-Franszens Universities (Graz, Austria). In 2000 he was appointed assistant professor and later associate professor at the VU University Amsterdam. In 2007 he became a full professor of synthetic and bioorganic chemistry. His current research focuses on the development of novel diversity-oriented synthetic methodology for the synthesis of pharmaceutically relevant compounds and natural products. |
 Albert D. Windhorst | Albert D. Windhorst received his PhD in radiopharmaceutical chemistry in 1998 and was appointed as chair of radiopharmaceutical chemistry in 2014 at the VU University Medical Center Amsterdam, The Netherlands. His research interests are the development of new radio-labelling methods for fluorine-18 and carbon-11 and application of these new techniques in the synthesis of PET tracers for oncology, neurology and cardiology and the translation of these new PET tracers to clinical research. He is immediate past president of the Society of Radiopharmaceutical Sciences, editor of Journal of Labelled Compounds & Radio-pharmaceuticals and associate editor of Nuclear Medicine & Biology. |
 Danielle J. Vugts | Danielle J. Vugts received her PhD in organic chemistry from the VU University Amsterdam, The Netherlands under the supervision of Prof. Orru. During a post-doctoral period at the VU University Medical Center (VUmc), Amsterdam, The Netherlands she worked on (pre-targeted) immuno-PET. She was appointed assistant professor in 2012 at VUmc. Her research interests are immuno-PET as well as development of new radiolabelling methods for fluorine-18, carbon-11 and zirconium-89 and application of these new techniques in the synthesis of PET tracers for oncology and neurology. |
1 Introduction
Positron emission tomography (PET) is a powerful molecular imaging technique with a broad range of applications including diagnosis of disease, monitoring of treatment and early phase determination of pharmacokinetics and pharmacodynamics of novel drug candidates.1–6Via detection of the γ-radiation formed by annihilation of positrons (β+) emitted by radionuclides, such as carbon-11 (t1/2 = 20 min), nitrogen-13 (t1/2 = 10 min), oxygen-15 (t1/2 = 2 min), fluorine-18 (t1/2 = 110 min), gallium-68 (t1/2 = 68 min) and zirconium-89 (t1/2 = 78 h), PET is able to provide well-defined, three-dimensional quantitative images of the distribution of biologically active compounds labelled with these radionuclides.2,7,8 The sensitivity of PET is superior to other molecular imaging techniques, since only picomolar concentrations of the labelled compounds have to be used. At these concentrations, biological targets of interest can be visualised without causing a biological effect by the radiolabelled compound, thus truly meeting the tracer principle of Hevesy.1The principal application of PET is to diagnose disease in patients by administering a PET tracer which visualises the biological pathway or the therapeutic target which is involved with the disease. Such clear visualization techniques greatly facilitate physicians to establish the correct diagnosis and decide on an effective treatment strategy. The most popular PET tracer is 2-[18F]fluoro-2-deoxy-D-glucose ([18F]FDG), which allows visualisation of glucose metabolism. Therefore [18F]FDG is widely used for the diagnosis of cancer and monitoring of cancerous lesions that often show increased glucose metabolism (Fig. 1).9,10
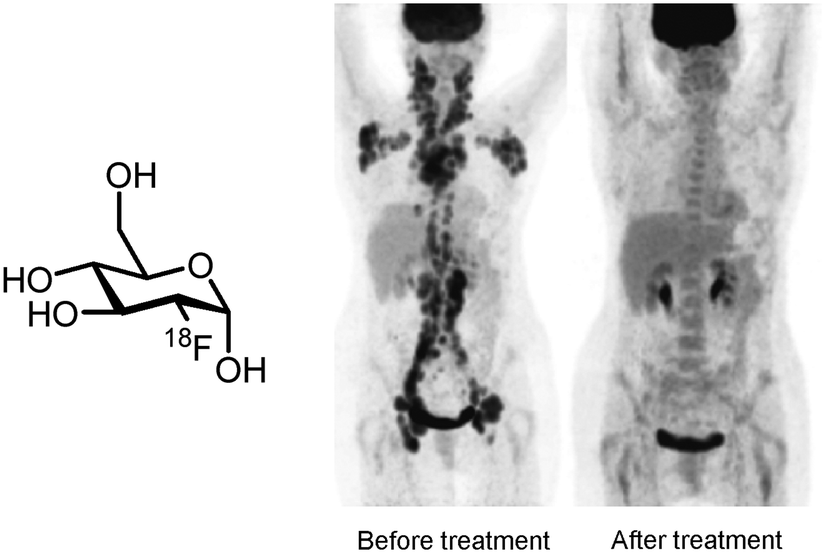 | ||
| Fig. 1 PET imaging with [18F]FDG.10 | ||
PET is also a useful asset in the development of drugs. PET can be used to investigate the effect of the drug on a biological target or pathway by visualisation with a PET tracer, probing the drug response downstream of the target, or by labelling the drug candidate itself and charting its distribution and kinetics.3,11
Of the many positron emitting radionuclides available for the production of PET tracers, fluorine-18 is amongst the most frequently used due to the unique and ideal combination of a 110 minute half-life (allowing transport to satellite PET scan facilities), a clean decay profile (97% positron emission and 3% electron capture), and a low positron energy (max. 0.635 MeV), resulting in high-resolution PET images with a maximum positron range of 2.4 mm in water.7
To enable radiochemists to label a wide range of compounds, including naturally occurring compounds and pharmaceuticals, the availability of a large toolkit of fluorine-18 labelling methods is important. Two major strategies can be identified to access fluorine-18 labelled tracers: (1) late-stage radiofluorination, introducing fluorine-18 in the last step of PET tracer synthesis by direct labelling of the precursor with [18F]fluoride and (2) the building block approach (also called modular build-up approach), where fast and efficient introduction of fluorine-18 into the building block by radiolabelling with [18F]fluoride occurs prior to one or more additional reaction steps to arrive at the actual PET tracer.
In recent years, new and very promising methodologies for late-stage aromatic radiofluorination reactions have been developed and excellently reviewed by Preshlock et al.12 and Brooks et al.13 The overview of the different late-stage aromatic radiofluorination reactions given in Table 1, nicely demonstrates the progress in this area.12–35 For several reasons however, we believe that application of fluorine-18 labelled building blocks for radiolabelling of biologically active molecules as an alternative for late-stage fluorination, is still of high value. In the first place the building block approach allows a modular build-up of fluorine-18 labelled PET tracers which cannot be made by direct late-stage radiofluorination methods. Second, using a labelling strategy that employs fluorine-18 labelled building blocks, the desired PET tracers can be obtained in higher radiochemical yields and radiochemical purity compared to application of late-stage radiofluorination techniques. Finally, once a building block is developed, the same generic labelling methodology can easily be applied to other compounds, e.g. N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB) for peptides or a library of analogues of a lead compound. This in contrast to late-stage radiofluorination techniques that only allow the synthesis of a dedicated precursor and where labelling conditions always need to be optimised for every new compound.
| Method | Highlights | Tested on complex molecules? | Ref. | |
|---|---|---|---|---|
| a Yields: low = 0–30%; moderate = 0–70%; good = 70–90%; excellent = 90–100%. | ||||
|
01 Radiofluorination of (hetero)aryl iodonium salts
|
• Low to moderate radiochemical yields on electron-rich and electron-neutral precursors.
• Reasonable selectivity towards the less electron rich arene. • Precursors can be challenging to prepare. |
• Precursors have modest shelf lives.
• Harsh reaction conditions, temperatures of >150 °C. • Limited tolerance to common functional groups. |
✓ | 13, 14, 34 and 35 |
|
02 Radiofluorination of (hetero)aryl iodonium ylides
|
• Low to good radiochemical yields on electron rich and electron-deficient precursors.
• Precursors are stable crystalline materials. |
• Method has been successfully applied to highly functionalised molecules and existing PET radiopharmaceuticals. | ✓ | 13, 15 and 16 |
|
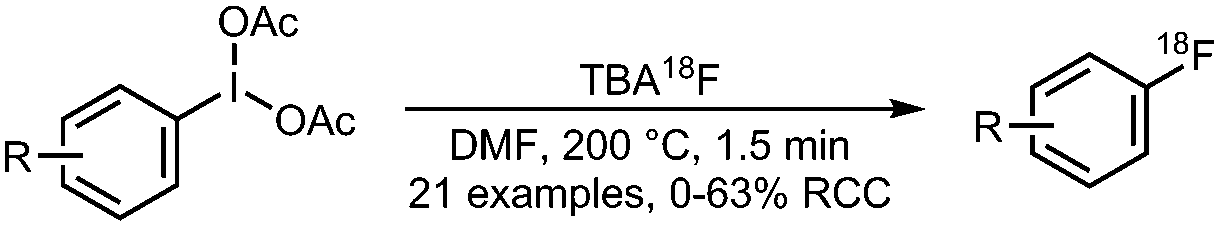
03 Radiofluorination of (diacetoxyiodo)arenes
|
• Low to moderate radiochemical conversions on electron-neutral to electron-deficient precursors.
• Limited scope concerning arene electron density. |
• Synthesis of precursors can be challenging.
• Method has not yet been tested on highly functionalised molecules. |
17 | |
|
04 Cu-Mediated radiofluorination of (mesityl)(aryl) iodonium salts
|
• Low to good radiochemical yields on electron-rich and electron-deficient precursors.
• High reproducibility of radiochemical yields. • Relatively mild reaction conditions. |
• No reduced specific activity due to isotopic exchange on BF4 anion.
• Precursors can be challenging to prepare. • Copper catalyst is air stable and commercially available. |
✓ | 12, 13, 18 and 19 |
05 Radiofluorination of triarylsulfonium salts 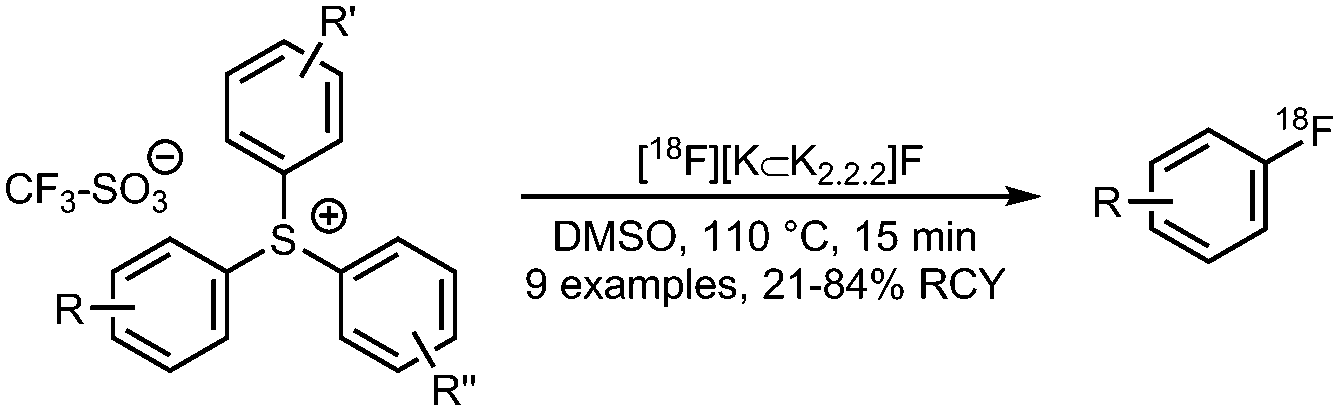
|
• Low to good radiochemical yields on electron-neutral and electron-deficient precursors.
• Precursors can be challenging to prepare. • Precursors show high thermal and chemical stability. |
• 18F-Fluorination proceeds in presence of basic functional groups and heterocyclic moieties. | ✓ | 12, 20 and 21 |
|
06 Radiofluorination of diaryl sulfoxides
|
• Moderate to excellent radiochemical yields on electron-deficient precursors.
• No or very low radiochemical yield on electron-rich or electron-neutral precursors. • Precursors can be challenging to prepare. |
• Good regioselectivity towards more electron-deficient arenes.
• No results yet available on the reaction with complex substrates. |
12 and 22 | |
|
07 Pd-Catalysed radiofluorination of Pd-precursors
|
• Low to moderate radiochemical yields on electron-rich precursors.
• Two-step procedure, synthesis of [Pd]–18F complex and subsequent radiofluorination of a [Pd]-arene. |
• [Pd]–18F complex is sensitive to air and moisture. | ✓ | 13 and 23–25 |
|
08 Radiofluorination of arylnickel complexes
|
• Low to moderate radiochemical yields on a wide scope of precursors.
• Room temperature reaction and short reaction times (<1 min). • The volume of aqueous [18F]fluoride must be kept <1% to prevent degradation of Ni-precursor. |
• Basicity of the [18F]fluoride must be reduced/tuned, when [18F]fluoride is dried by classic azeotropic distillation.
• Synthesis of Ni-precursors may be challenging. |
✓ | 13, 26 and 27 |
|
09 Copper mediated radiofluorination of (hetero)aryl boronic acid pinacolesters
|
• Low to good radiochemical yields on electron-rich and electron-deficient precursors.
• Precursors are stable, however challenging to synthesise. • Reasonable functional group tolerance. |
• Challenging to reproduce.
• Products are difficult to purify due to the presence of aryl-H, formed from aryl-BPin during the reaction. • Copper catalyst is air stable and commercially available, but sensitive for basic conditions. |
✓ | 13 and 28 |
|
10 Copper mediated radiofluorination of aryl boronic acids
|
• Low to good radiochemical yields on electron-rich and electron-deficient precursors.
• Precursors are stable, however challenging to prepare. • Reasonable functional group tolerance. |
• Not yet tested on heteroaryl precursors.
• Not extensively tested yet on more complex precursor structures. |
12 and 29 | |
|
11 Copper mediated radiofluorination of arylstannanes
|
• Low to good radiochemical conversions on electron deficient arenes.
• Precursors are stable, however may be challenging to prepare. |
• Method has been successfully applied to highly functionalised molecules and existing PET radiopharmaceuticals. | ✓ | 30 |
|
12 Oxidative radiofluorination of phenols
|
• Low to moderate radiochemical yields on electron-rich and electron-deficient precursors.
• Reasonable functional group tolerance. |
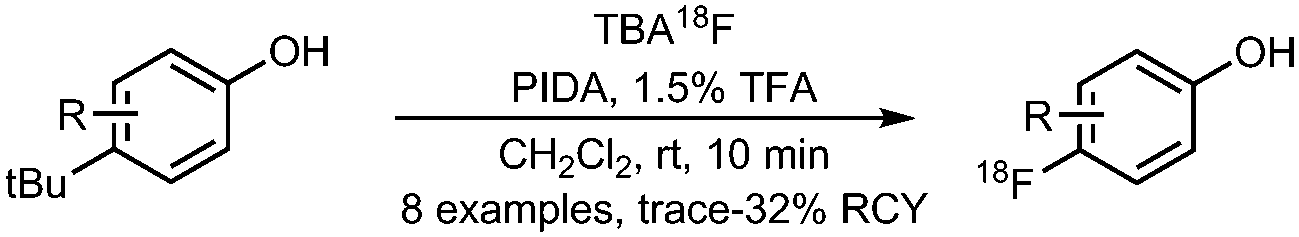
• Not yet evaluated on structural more complex precursors.
• Not yet evaluated on hetero-arenes. |
13 and 31 | |
|
13 Deoxyradiofluorination of phenols
|
• Moderate to excellent radiochemical yields on electron-neutral and electron-deficient precursors.
• Phenolic precursors are relatively easy to synthesise and stabile. • Excellent functional group tolerance. |
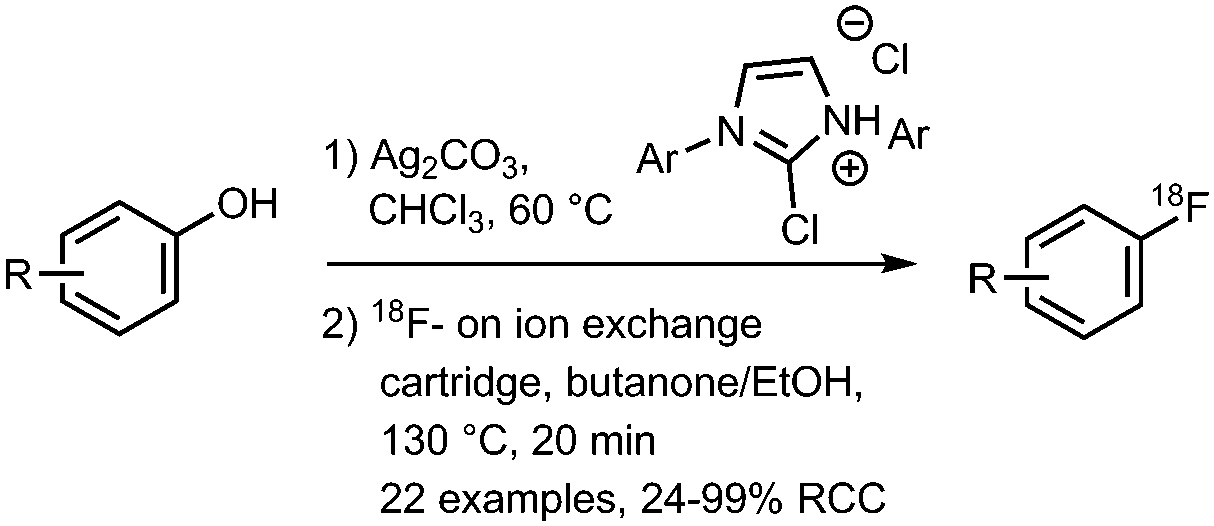
• Radiochemical conversions are based on eluted [18F]fluoride, which is 62% of the total fluoride activity. | ✓ | 32 |
|
14 TiO2 mediated radiofluorination of tosylated precursors
|
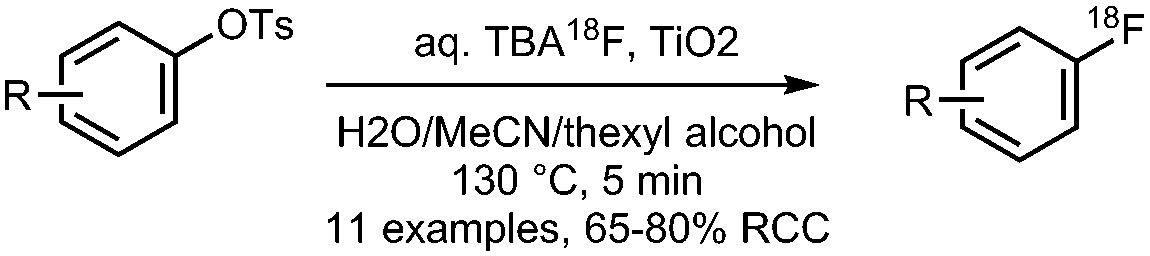
• Good yields on electron-neutral and electron-deficient precursors.
• Precursors are simple to synthesise from phenolic precursors. • No azeotropic drying of [18F]fluoride required, may be performed in up to 25% v/v water. |
• Method has been successfully applied to existing PET radiopharmaceuticals.
• Scaling up the amount of aqueous [18F]fluoride had limited success. |
✓ | 12 and 33 |
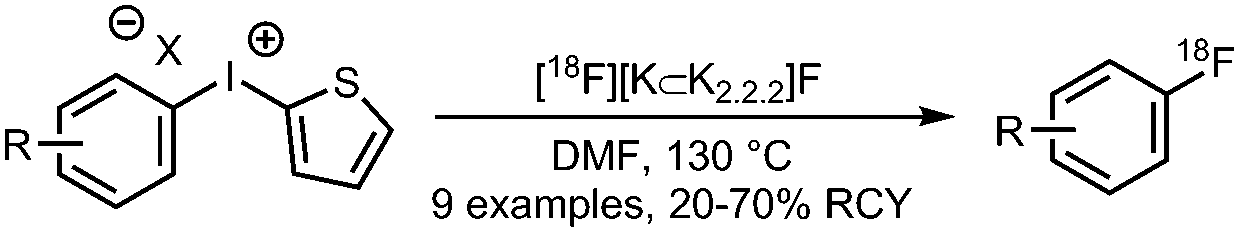
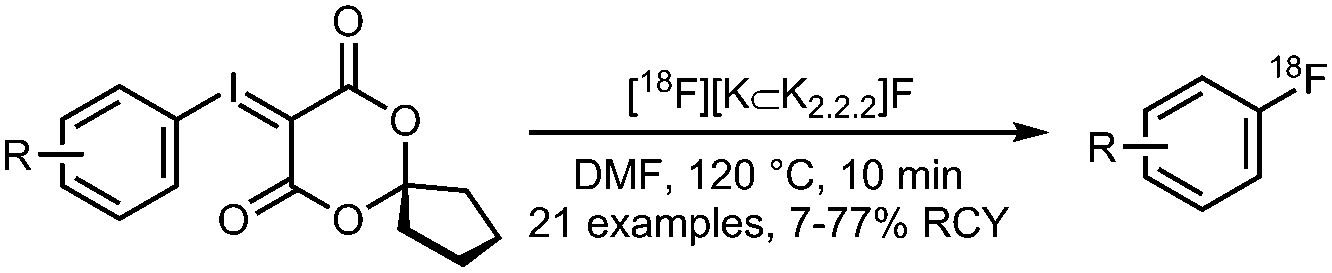
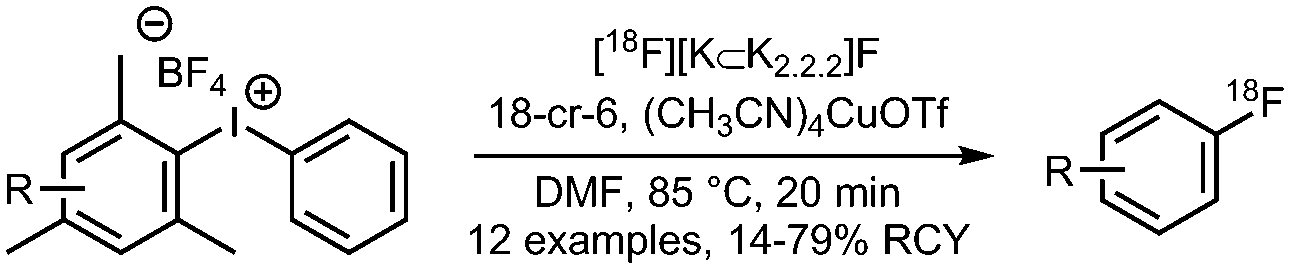
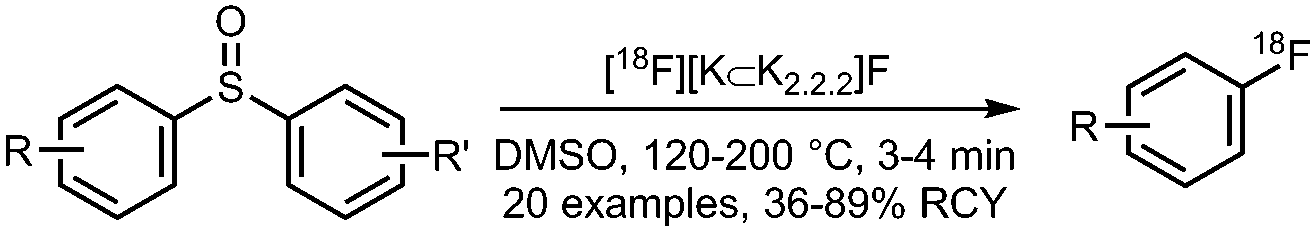
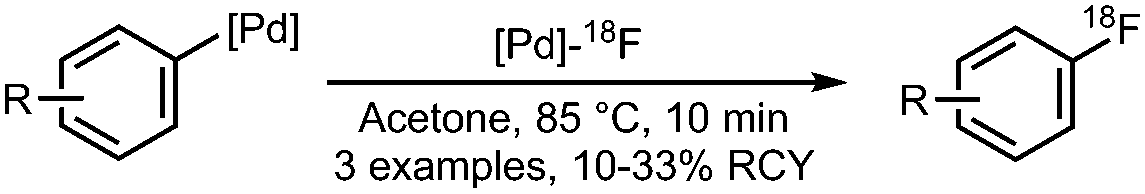
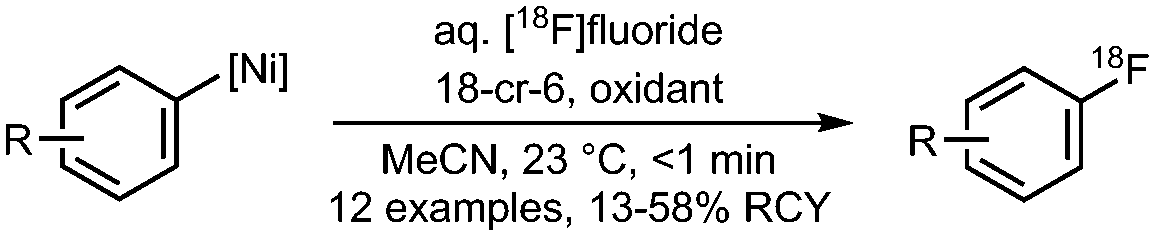
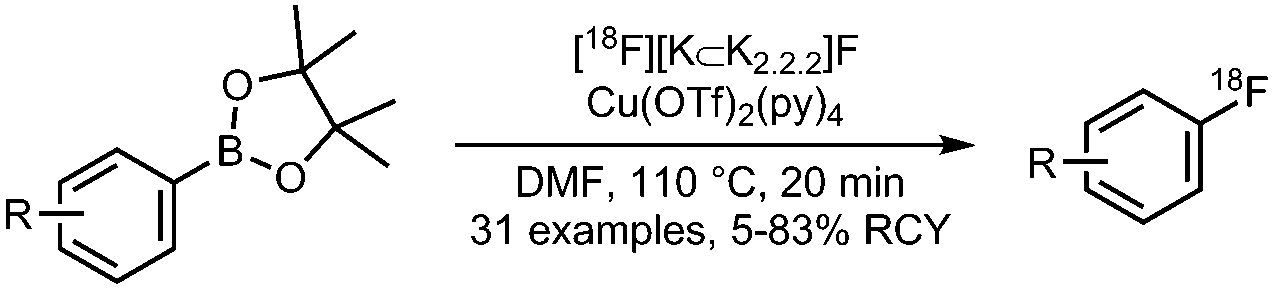
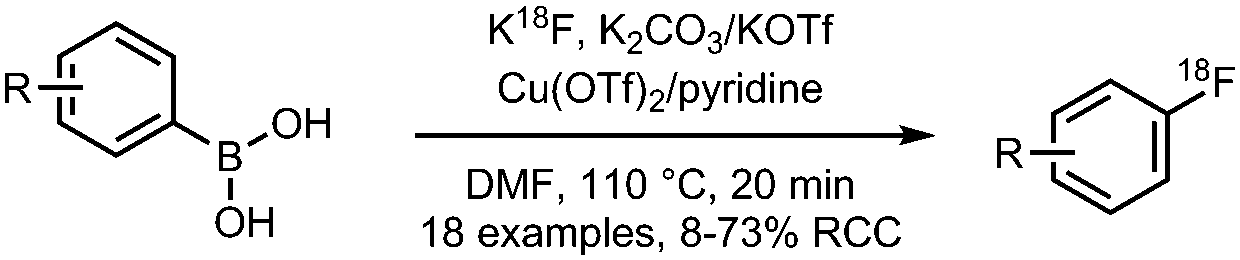
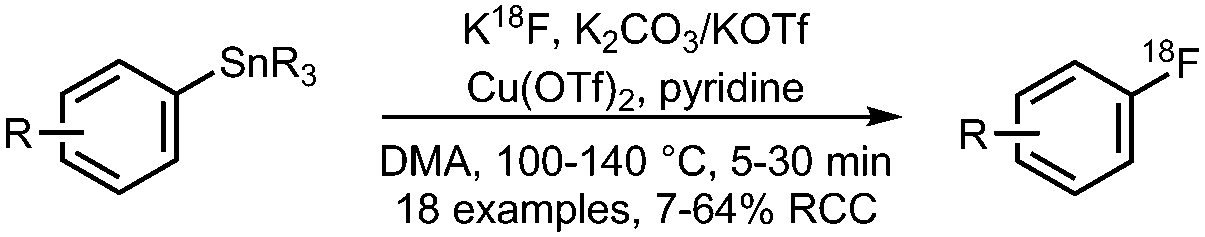
The aim of this review is to summarise the recent developments in fluorine-18 labelled building blocks containing a carbon–fluorine bond and their applications in PET tracer synthesis. A comprehensive overview of publications since 2010 that describe the synthesis and development of fluorine-18 labelled building blocks and their potential application to radiolabel low molecular weight compounds for PET tracers, is provided. To demonstrate that these building blocks are indeed a valuable asset, in each case the following characteristics will be discussed:
• Overall radiochemical yield, specific activity and radiochemical purity.
• Ease of synthesis of the labelling precursors.
• Technical ease of labelling.
• Labelling conditions (types of conditions, reaction times, and commercial availability of reagents).
• Purification method (distillation, solid phase extraction (SPE), high performance liquid chromatography (HPLC)).
2 Fluorine-18 labelled aliphatic building blocks
A broad spectrum of aliphatic fluorine-18 labelled building blocks has been utilised in PET tracer synthesis. The spectrum ranges from simple molecules such as radiofluorinated methyl and ethyl halides and sulfonates to complex structures such as [18F]FDG. Fluorine-18 labelled aliphatic building blocks have been used to synthesise either the radiolabelled lead structure or a structural derivative of the lead structure. In the following sections, the synthesis and application of aliphatic fluorine-18 labelled building blocks applied since 2010 will be discussed with respect to their ease of synthesis, stability and application in follow-up reactions.2.1 [18F]Fluoromethyl halides and sulfonates
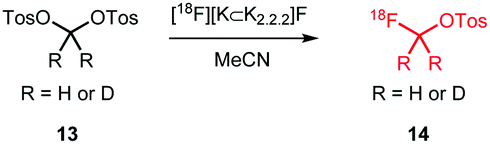
The replacement of a methyl group by a [18F]fluoromethyl group is a relatively minor modification in the structure of most small molecules and the chance of significantly influencing the physicochemical and biological properties is minimal. Therefore, [18F]fluoromethylation is often considered as labelling approach for molecules that contain no native fluorine, or where labelling in another position is less preferred. However, the [18F]fluoromethyl group is prone to metabolic instability, resulting in bone uptake of released free [18F]fluoride.36 The metabolic stability can however be enhanced by deuteration of the [18F]fluoromethyl group37 or by inhibiting enzyme reactivity with a pharmacological intervention employing disulfiram or miconazole.38,39Multiple [18F]fluoromethylation agents are described and available: [18F]fluoromethyl bromide and iodide are most established, while more recently, [18F]fluoromethyl tosylate as a reagent is of increased interest. This is especially due to the ease of handling and purification of [18F]fluoromethyl tosylate compared to its volatile bromine and iodine analogues. [18F]Fluoromethyl triflate can also be used for [18F]fluoromethylation, but it needs to be synthesised in a two-step procedure from dibromomethane, and is therefore less preferred over other reagents.40
Hereafter, synthesis and application of every [18F]fluoromethylation agent for PET tracer synthesis reported since 2010 will be discussed.
[18F]Fluoromethyl bromide is synthesised in one step by reacting dibromomethane 1 with [18F]fluoride in MeCN at 90–100 °C (Scheme 1). The main challenge is in the purification of the volatile product. Although [18F]fluoromethyl bromide has a much lower boiling point (b.p. 9 °C) than its precursor (b.p. 97 °C) and the solvent MeCN (b.p. 82 °C), no pure product could be obtained by straightforward distillation.41 Purification using gas chromatography is an alternative, however it is incompatible with automation.40 Distillation over 3 to 4 silica plus Sep-Pak cartridges however provides pure [18F]fluoromethyl bromide in an automation-compliant manner, as impurities are retained on the cartridges while most of the product passes through.41,42 Reported radiochemical yields of [18F]fluoromethyl bromide vary strongly, from 37 to 74% (dc), which is a major drawback for widespread application of this radiolabelled building block.40,43
 | ||
| Scheme 1 Synthesis of [18F]fluorobromomethane. | ||
The subsequent reactions that can be performed with [18F]fluoromethyl bromide to obtain a PET tracer can be divided into two categories; O-alkylation and N-alkylation (Scheme 2). Although O-alkylation comprises the reaction with aliphatic as well as aromatic hydroxyl groups, since 2010 only aromatic O-alkylations with [18F]fluoromethyl bromide have been described. An overview of all labelled structures is given in Scheme 3. All syntheses were conducted under comparable reaction conditions. Mostly, reactions were performed in MeCN, only Lodge and co-workers described the use of N,N-dimethylformamide (DMF).44 The reactions required high reaction temperatures (90–100 °C) and the addition of an inorganic base. The choice of base proved to have an impact on the yield as reported by Klein et al. Purification of the final 18F-fluoromethylated PET tracers was carried out by semi-preparative HPLC.42–45
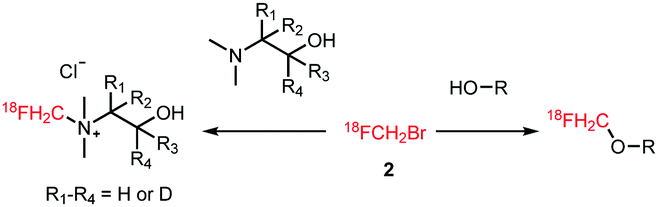 | ||
| Scheme 2 N-Alkylation and O-alkylation reactions with [18F]fluoromethyl bromide. | ||
 | ||
| Scheme 3 Recently produced tracers using O-alkylation with [18F]fluoromethyl bromide.42–45 | ||
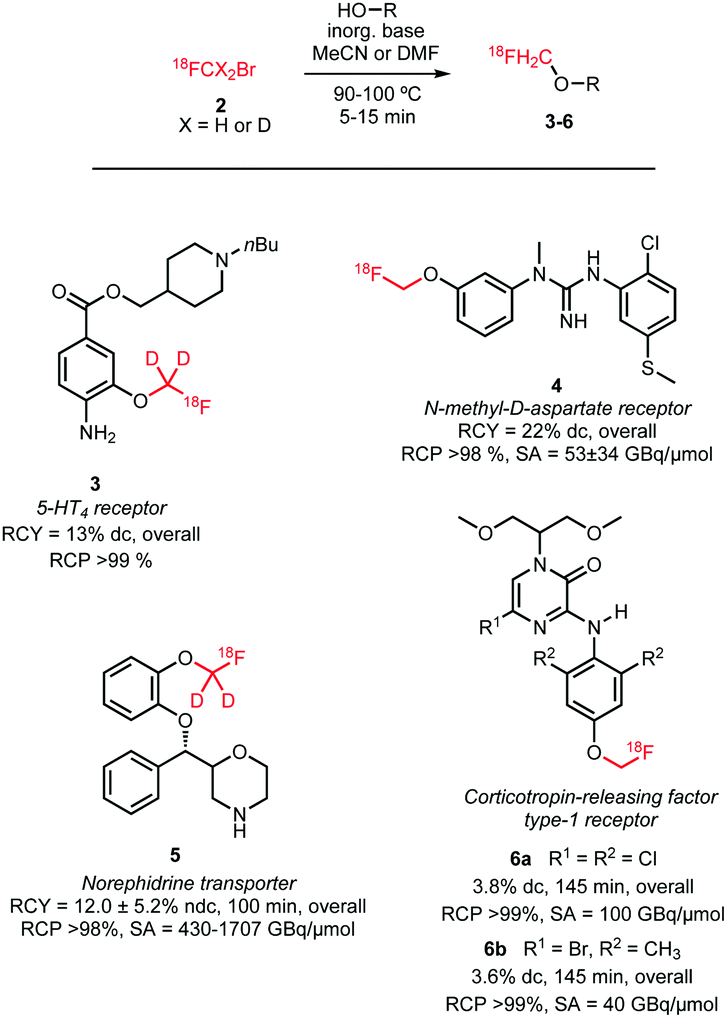
There is only one example of the application of [18F]fluoromethyl bromide in N-alkylation reported since 2010, namely, the synthesis of [18F]fluorocholine 8. This is an established oncologic PET tracer for imaging prostate cancer (Scheme 4). This tracer is routinely synthesised at numerous laboratories and in some cases even commercially available. An automated synthesis of [18F]fluorocholine has been developed by Shao and co-workers. Synthesis and purification of the alkylating reagent was carried out as described above, by distilling [18F]fluoromethyl bromide over three silica Sep-Pak cartridges. This approach proved to be the most efficient regarding yield as well as radiochemical and chemical purity. The volatile product was trapped on a C18 cartridge, where it reacted with the [18F]fluorocholine precursor, dimethylaminoethanol. The tracer was obtained after a cartridge purification procedure with a radiochemical yield of 4–6% (dc).41
 | ||
| Scheme 4 Synthesis of [18F]fluorocholine and deuterated derivatives with [18F]fluoromethyl bromide. | ||
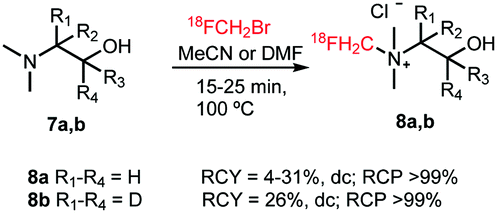
Smith and co-workers investigated the influence of different [18F]fluoromethylation agents in the [18F]fluorocholine synthesis. Next to [18F]fluoromethyl bromide, less volatile [18F]fluoromethyl tosylate was used for [18F]fluoromethylation, to simplify handling and purification. However, for both synthesis routes, a synthesis time of about 150 minutes and similar yields have been observed, both for deuterated 8b as well as non-deuterated [18F]fluorocholine 8a.40
Thus, [18F]fluoromethyl bromide 2 is a useful building block for [18F]fluoromethylation, providing comparable yields to its more popular and easy-to-handle analogue [18F]fluoromethyl tosylate. Purification and handling of the gaseous compound have been mastered and translated to automated production, making [18F]fluoromethyl bromide 2 easily accessible for novel PET tracer development.
Deuterated [18F]fluoromethyl iodide 10 was obtained via a nucleophilic substitution reaction of diiodomethane-d29 with [18F]fluoride in the presence of potassium carbonate and kryptofix K2.2.2 (Scheme 5). Separation of the volatile building block (b.p. 54–56 °C)46 from the precursor (b.p. 181 °C)47 was achieved by distillation in a stream of helium. Unfortunately the radiochemical yield of [18F]fluoromethyl iodide was not reported, which makes the comparison between the production of this reagent with other [18F]fluoromethyl alkylating agents difficult.
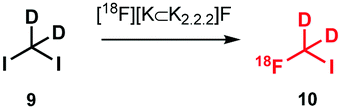 | ||
| Scheme 5 Synthesis of [18F]fluoromethyl iodide.46 | ||
For [18F]fluoroalkylation, the distilled [18F]FCD2I 10 was reacted with the radiolabelling precursor in DMF in the presence of cesium carbonate for 5 minutes at 90 °C (Scheme 6). Purification by semi-preparative HPLC resulted in only 0.3–1.6% (overall) product, which was attributed to the complexity of the [18F]fluoromethylation reagent synthesis.
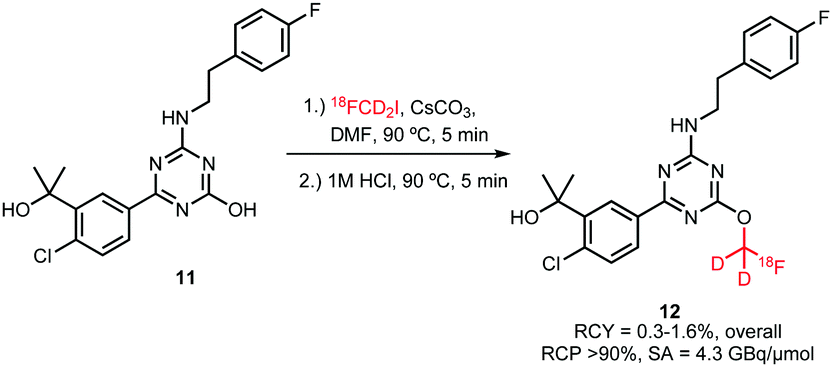 | ||
| Scheme 6 [18F]Fluoroalkylation of a CB2 cannabinoid receptor ligand.46 | ||
It remains unclear why this [18F]fluoroalkylation agent was chosen, the choice possibly depended on the availability of the deuterated precursor. This building block has the same disadvantages as the corresponding bromide, being gaseous and difficult to separate from its precursor. Unfortunately, there is not enough data available to draw a conclusion whether this building block can be synthesised in similar yields as its analogues and whether it shows comparable reactivity in [18F]fluoroalkylation reactions.36
[18F]Fluoromethyl tosylate 14 is synthesised in a one-step reaction from methylene ditosylate 13 in MeCN at temperatures between 80 and 120 °C (Scheme 7). Unfortunately, in addition to the desired [18F]fluoromethyl tosylate, [18F]tosylfluoride is formed as side product.40 Many efforts have been undertaken to reduce the amount of this side product and thus increase the yield of [18F]fluoromethyl tosylate. Neal and co-workers were first to recognise that traces of water have a positive influence on the reaction and reduce side product formation.48 Up to 20% of water can be beneficial to the reaction outcome.49
 | ||
| Scheme 7 Synthesis of [18F]fluoromethyl tosylate. | ||
Another approach, pursued by Beyerlein et al., is to replace water by the protic solvent tert-butanol. The optimal yield of 14 using this approach was achieved in a solvent mixture of 75% MeCN and 25% tert-butanol. Furthermore, it was shown that with the commonly used combination of potassium carbonate and kryptofix K2.2.2, degradation of the precursor occurred. Tetrabutylammonium bicarbonate and the combination of potassium carbonate and 18-crown-6 proved better alternatives.37,40
In contrast to the volatile analogues [18F]fluoromethyl bromide and iodide, purification of [18F]fluoromethyl tosylate can be easily carried out by semi-preparative HPLC, and even successful [18F]fluoromethylations without intermediate purification have been described.50,51 Automated procedures have been developed using conventional synthesis units as well as microfluidic devices. Both provide the radiolabelled building block in a radiochemical yield of 44% (dc).37,49
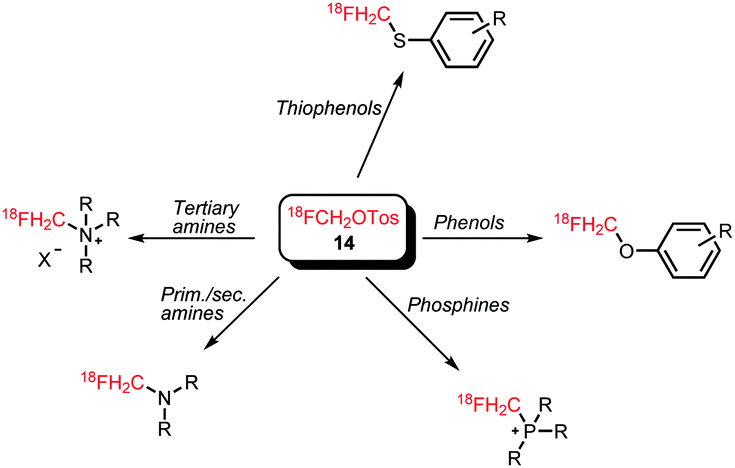
[18F]Fluoromethyl tosylate can be used for a variety of alkylation reactions, next to the common N- and O-alkylation reactions, even S- and P-alkylations have been reported with this reagent (Scheme 8).
 | ||
| Scheme 8 Reactions with [18F]fluoromethyl tosylate. | ||
O-Alkylations of phenolic hydroxyl groups form besides N-alkylations the major part of the conducted [18F]fluoromethylations with deuterated [18F]fluoromethyl tosylate. Scheme 9 shows the reported tracers obtained via [18F]fluoromethyl tosylate. O-Alkylations were all conducted under similar reaction conditions, proving that the reaction is generally applicable without the need to intensively adjust the different reaction parameters. Dimethyl sulfoxide (DMSO) served as solvent and the reactions were carried out at 110–120 °C for 10–20 minutes. Sodium hydroxide and cesium carbonate have been used as base in the nucleophilic substitution reactions. Radiochemical yields are comparable and range from 60 to 80% for the final alkylation step.37,52,53
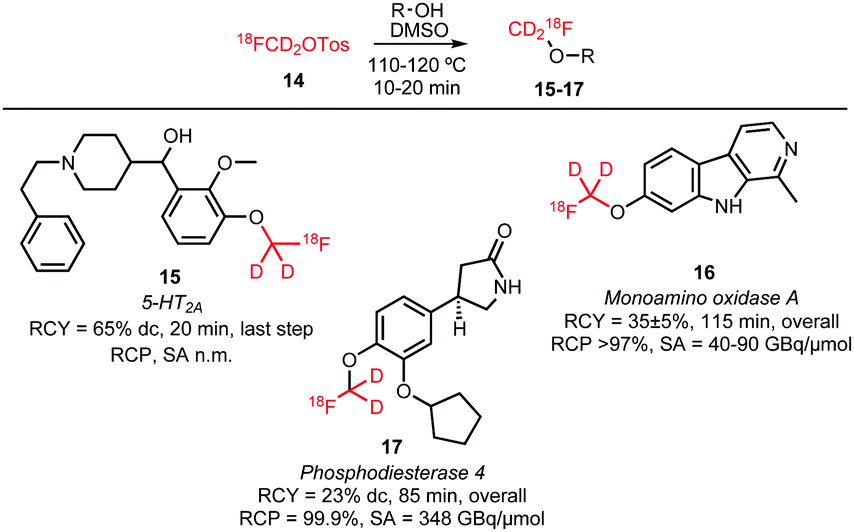 | ||
| Scheme 9 O-Alkylated tracers with [18F]fluoromethyl tosylate.37,52,53 | ||
Analogous to the O-alkylation, aromatic S-alkylation of guanidine derivative 18 has been performed (Scheme 10). The reaction was carried out in MeCN at 110 °C for 15 minutes using cesium carbonate as base. After purification by semi-preparative HPLC, the compound however decomposed to give free [18F]fluoride. Hence, no yield has been determined for the synthesis of tracer 19.54
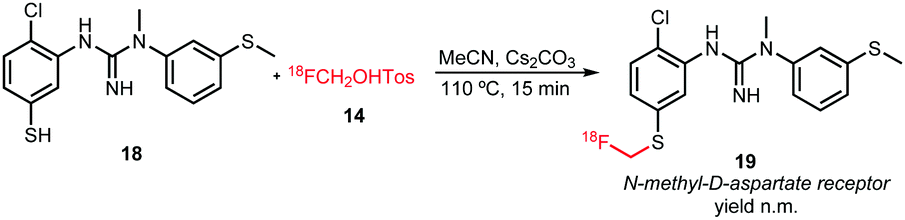 | ||
| Scheme 10 Synthesis of a S-fluoroalkyl guanidine derivative.54 | ||
Similar to [18F]fluoromethyl bromide, the main application of [18F]fluoromethyl tosylate in N-alkylation reactions is the production of the PET tracer [18F]fluorocholine for the imaging of prostate cancer (Scheme 11). Extensive studies have been performed to establish optimal labelling conditions and to develop an automated synthesis procedure. Smith and co-workers were the first to report [18F]fluoromethylation of the choline precursor using [18F]fluoromethyl tosylate. They compared [18F]fluorocholine and its d2 and d4 derivatives (Scheme 11) and showed enhanced stability of the deuterated species towards in vivo oxidation during metabolism. They reported that temperature had a strong influence on N- or O-alkylation. At 100 °C, desired N-alkylation was favoured whereas higher temperatures directed the reaction towards O-alkylation. Compared to MeCN, the use of DMF resulted in higher radiochemical yields (70 ± 5% dc, n = 5, alkylating step) that could be achieved in decreased reaction times. This was especially beneficial for the deuterated analogues, which required a longer reaction time compared to the non-deuterated compound. The optimised procedure gave the tracers in an overall radiochemical yield of about 10% (ndc) for the d4 and the non-deuterated compound and 8% (ndc) for [18F]d2-fluorocholine with a total synthesis time of 150 minutes.40
 | ||
| Scheme 11 Synthesis of [18F]fluorocholine and deuterated derivatives with [18F]fluoromethyl tosylate.40,49,50 | ||
Almost simultaneously, Pascali et al. developed a microfluidic synthesis procedure for dose-on-demand [18F]fluorocholine production. They produced [18F]fluorocholine in 13–15 minutes with a radiochemical yield of 20 ± 2% (ndc, overall) without intermediate purification of [18F]fluoromethyl tosylate.49
Further proof that intermediate purification is not required for successful [18F]fluorocholine production is provided by the fully-automated one-pot synthesis developed by Rodnick and co-workers. To the mixture of crude [18F]fluoromethyl tosylate, dimethylamino ethanol was added and heated for 10 minutes at 120 °C to yield 7% (ndc, overall) in a total synthesis time of 75 minutes.
Not only [18F]fluorocholine but also other tracers obtained by N-alkylation have been reported using [18F]fluoromethyl tosylate. As can be seen in Schemes 12 and 13, next to tertiary nitrogen atoms also primary and secondary nitrogen atoms can be alkylated. This is rarely found in literature because reactivity of the nitrogen increases in each step leading to polyalkylation. However, the huge excess of precursor compared to alkylation agent (in this case [18F]fluoromethyl tosylate) used in radiochemistry allows in this case selective monoalkylation.
 | ||
| Scheme 12 N-Alkylation reactions of primary amines.55 | ||
 | ||
| Scheme 13 N-Alkylation reactions of secondary amines.37,56 | ||
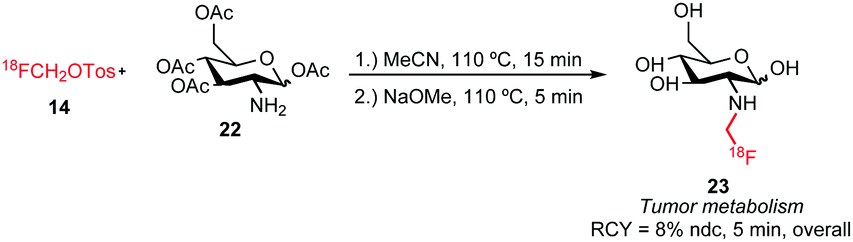
An example of [18F]fluoroalkylation of primary amines is the synthesis of the glucosamine derivative 23 (Scheme 12). Using the conditions of Smith and co-workers, [18F]fluoromethyl glucosamine derivative 23 could be obtained in a radiochemical yield of 8 ± 2% (n = 15, ndc).55
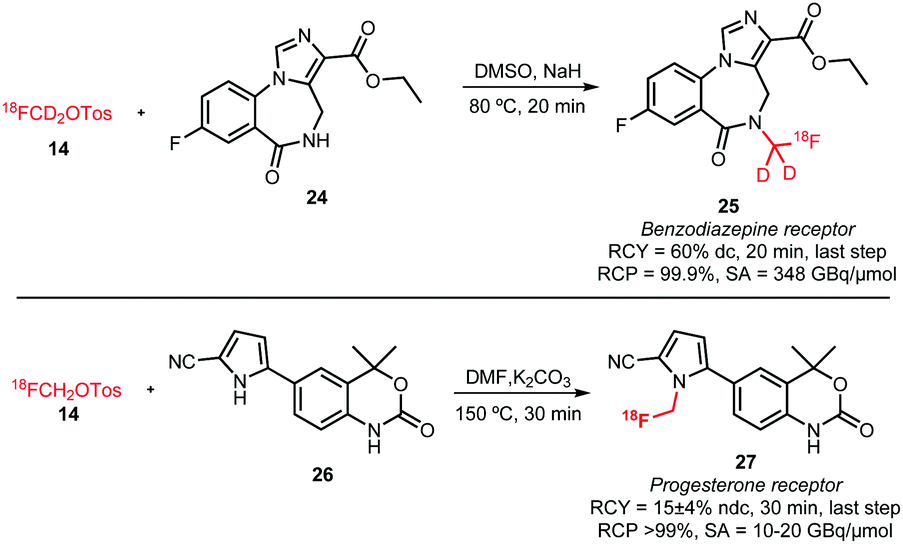
Two successful [18F]fluoromethylations of secondary amines with [18F]fluoromethyl tosylate (Scheme 13) are (1) the synthesis of [18F]fluoro-d2-methylflumazenil 25 for imaging benzodiazepine receptors and (2) the synthesis of the progesterone receptor agonist 27. Very different reaction conditions were applied in each reaction. While Beyerlein et al. reported the [18F]fluoro-d2-methyl-flumazenil 25 radiosynthesis with a radiochemical yield of 60% dc over the last step using sodium hydride at 80 °C in DMSO,37 Merchant et al. reported the synthesis of 27 in DMF at 150 °C in presence of potassium carbonate as base. Lower temperatures led in the latter case to yields lower than 1% and the addition of base was necessary for a clean reaction. Thus, 27 was synthesised from [18F]fluoromethyl tosylate in 30 minutes with a radiochemical yield of 15 ± 4% (ndc).56
Since 2010 only one case of P-alkylation with [18F]fluoromethyl tosylate has been reported. [18F]Fluoromethyl triphenylphosphonium cation 29 was synthesised in a one-pot reaction from [18F]fluoromethyl tosylate 14 and triphenylphosphine 28 with an overall radiochemical yield of 30–34% dc (Scheme 14).51
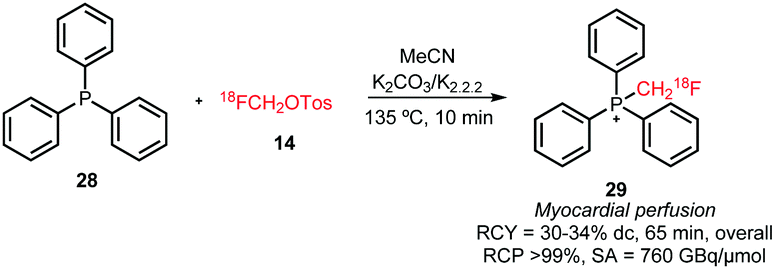 | ||
| Scheme 14 Synthesis of [18F]fluoromethyl triphenylphosphonium cation.51 | ||
In conclusion, [18F]fluoromethyl tosylate is a building block, which can be applied in many different alkylation reactions. Due to its ease of handling and purification compared to the volatile analogues [18F]fluoromethyl bromide and [18F]fluoromethyl iodide, it has gained increased interest for PET tracer development and routine production.
2.2 [18F]Fluoroethyl halides and sulfonates
Like the [18F]fluoromethyl halides and sulfonates, [18F]fluoroethyl halides and sulfonates find widespread application in fluorine-18 labelling. Especially for molecules which contain no native fluorine atom or where the fluorine atom is difficult to introduce. [18F]Fluoroethylation often allows for the radiofluorination of easier accessible precursors under milder reaction conditions and can therefore be advantageous over direct labelling. Furthermore, it facilitates separation of the labelled compound from the precursor by HPLC purification.57[18F]Fluoroethyl groups are often used as a substitute for a methyl group and in contrast to [18F]fluoromethylated tracers they offer the advantage of showing high in vivo stability.
Next to the most popular [18F]fluoroethylation agent [18F]fluoroethyl tosylate ([18]FETos), the bromide and different sulfonates have found application in PET tracer synthesis. The different building blocks and their advantages will be discussed in the following sections.
All syntheses of this building block followed established procedures by Zhang et al. (Scheme 15).58,59 Typically, 2-bromoethyl triflate served as precursor, but also the use of the corresponding tosylate or nosylate has been reported.60,61 The radiofluorination reaction was carried out in o-dichlorobenzene as solvent, reaction temperatures varied between 85–135 °C and reaction times between 2–15 minutes. [18F]Fluoroethyl bromide 31 was distilled during or after the reaction and transferred to a second reaction vial where it was trapped in a solution at around −15 °C, containing the precursor and base for the subsequent reaction. Distillation was straightforward and the product was produced in high (radio)chemical purity due to the much lower boiling point of [18F]fluoroethyl bromide (71.5 °C) compared to o-dichlorobenzene (179 °C) and the triflate precursor (230 °C). Decay-corrected radiochemical yields of 53–71% have been reported.60,62 Schmaljohann and co-workers reported a cartridge-based purification procedure instead of distillation. They obtained the building block in a radiochemical yield of 63% (dc).63
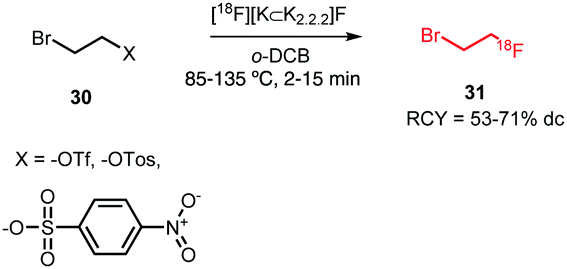 | ||
| Scheme 15 Synthesis of the building block [18F]fluoroethyl bromide. | ||
Labelling reactions with [18F]fluoroethyl bromide can be divided into two main categories, being N- and O-alkylation (Scheme 16). Whereas N-alkylation of various amines is performed, O-alkylation is almost exclusively applied and reported for phenolic precursors. Apart from phenolic O-alkylation, two ester formations with carboxyl groups have been reported.
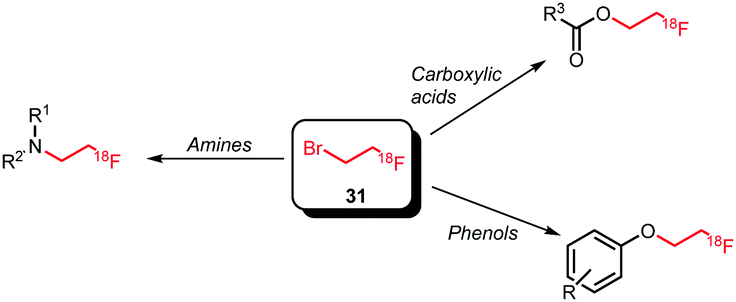 | ||
| Scheme 16 Reaction scope of [18F]fluoroethyl bromide as building block. | ||
An overview of tracers obtained by N-alkylation is given in Scheme 17.60,63–67 Typical reaction conditions for N-alkylations are the use of DMF as solvent and the addition of a base in the presence of the amine precursor. As base, either TBAOH or cesium carbonate were used. After heating to 60–100 °C for 5–15 minutes, the products were usually purified by (semi-)preparative HPLC and obtained in overall radiochemical yields of 10–40% (dc). However, some variations were reported depending on the labelling precursor. Murali and co-workers describe alkylation without any addition of base as otherwise epimerisation of the chiral centre occurs. Consequently, the coupling step required long reaction times and afforded the dopamine transporter (DAT) tracer 36 in a relatively low radiochemical yield of 34% (dc, last step). By performing the same reaction with the more reactive [18F]fluoroethyl triflate, the radiochemical yield could be increased to 84% for the coupling step and the overall reaction time was reduced from 148 to 96 minutes.60 Schmaljohann et al. succeeded in the development of a cartridge based purification procedure for fast and reliable automated synthesis. They produced [18F]fluoroethyl choline 37, an imaging agent for prostate cancer and brain tumours, on two different commercially available synthesis units obtaining comparable overall radiochemical yields of 33% and 37% (dc) in 55 minutes.63 Though, comparable synthesis times could also be achieved including an HPLC purification procedure.66
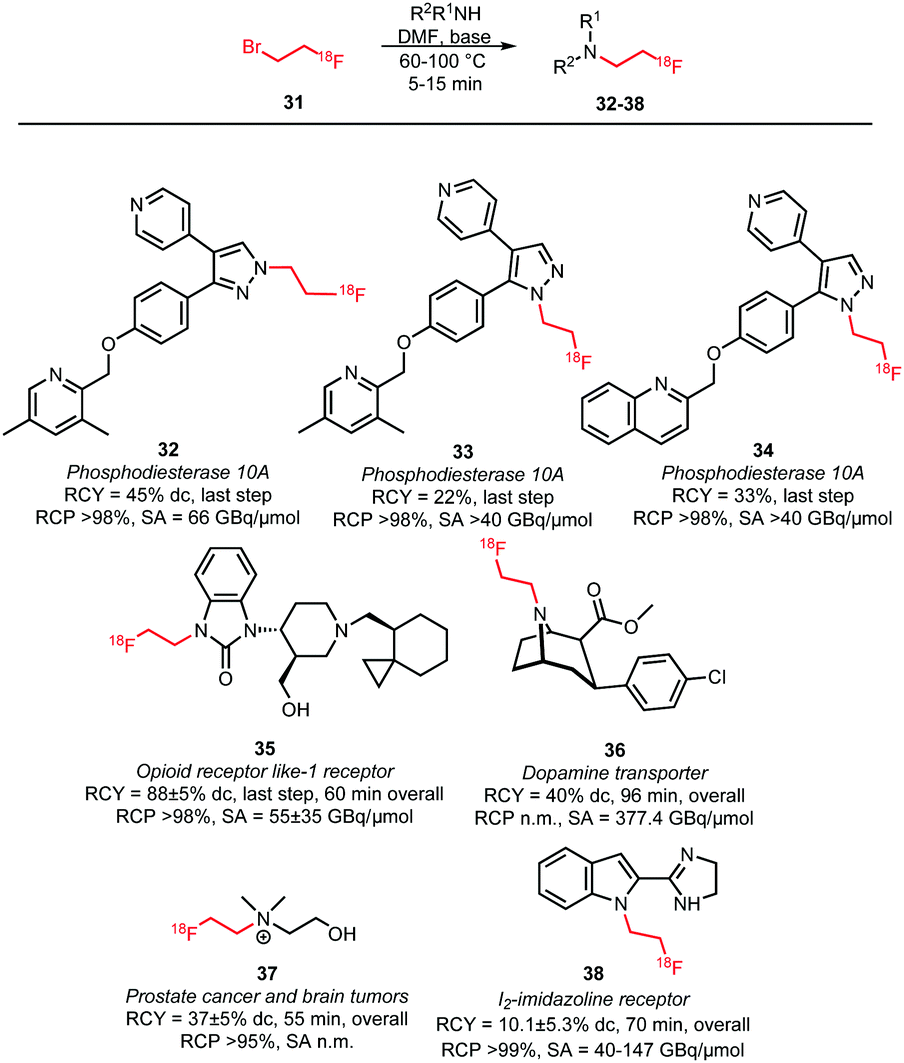 | ||
| Scheme 17 N-Alkylation with [18F]fluoroethyl bromide.60,63–67 | ||
Phenolic O-alkylation is the most popular application using [18F]fluoroethyl bromide as the building block. A variety of PET tracers have been synthesised based on this labelling strategy (Scheme 18).61,62,68–74 The general reaction conditions are similar to N-alkylation: the phenolic precursor was reacted with [18F]fluoroethyl bromide under basic conditions at elevated temperatures for 2–20 minutes in DMF or DMSO. Although sodium hydroxide was often employed as base, the use of other bases has been reported depending on the reactivity of the precursor. Liu and co-workers for example described in their synthesis of 46 a coupling reaction with the weaker base potassium carbonate, to prevent degradation of the lactone moiety.62 Furthermore, addition of sodium iodide to the coupling reaction increased the reactivity of the building block by in situ formation of the more reactive [18F]fluoroethyl iodide.59,62
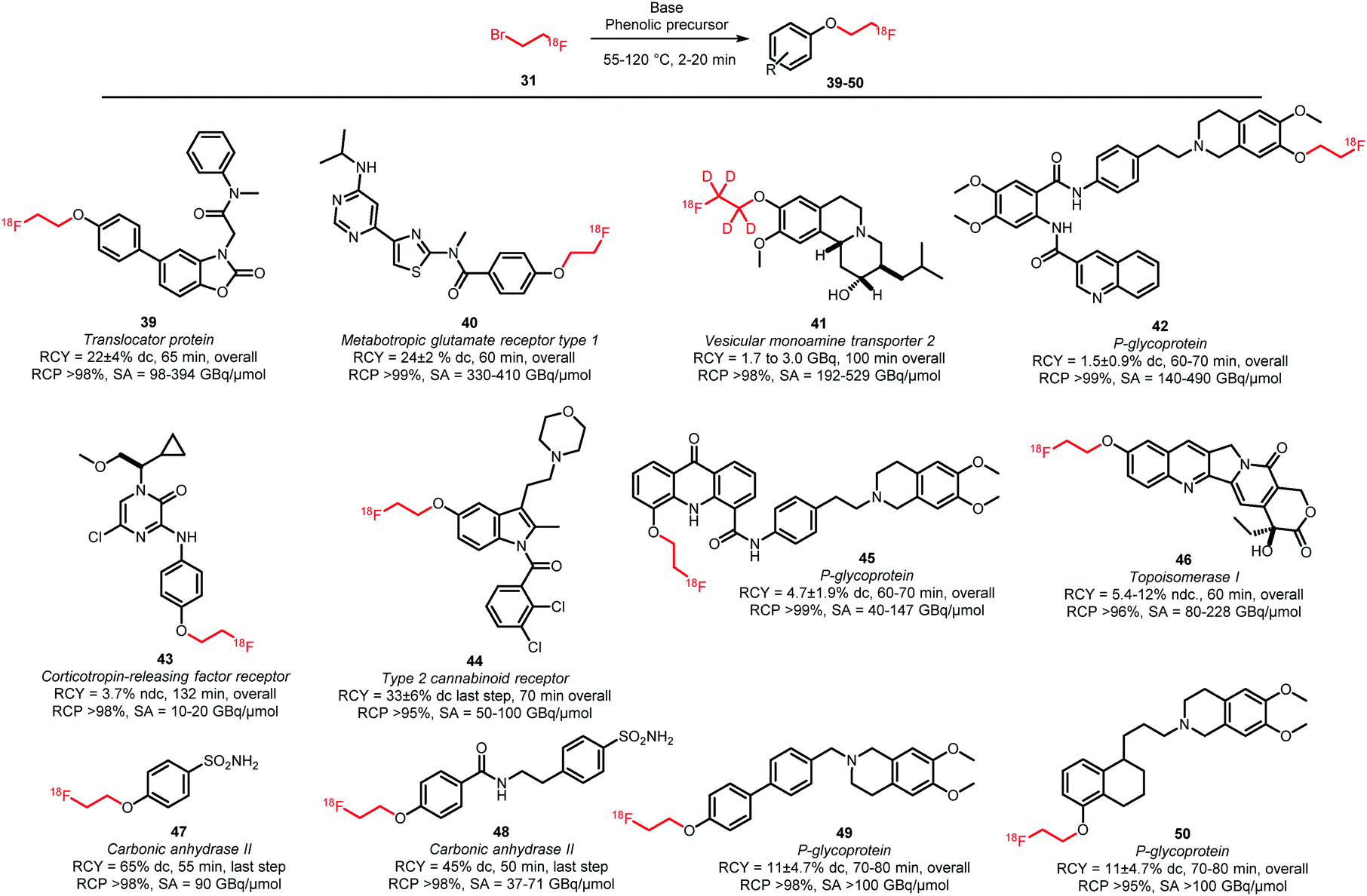 | ||
| Scheme 18 Phenolic O-alkylation with [18F]fluoroethyl bromide.61,62,68–74 | ||
Next to O-alkylation of phenolic precursors, esterification of carboxylic acid precursors has been investigated (Scheme 19). Rami-Mark and co-workers obtained radiochemical yields of 67 ± 16% (dc) in the coupling reaction forming the dopamine transporter ligand 51. The reaction was carried out under TBAOH catalysis at a reaction temperature of 100 °C. In contrast to phenolic O-alkylation, no increase in yield was observed in the presence of sodium iodide.75 Another coupling reaction with an acid precursor was conducted by Philippe et al., in order to obtain a derivative of the melanin concentrating hormone receptor 1 antagonist SNAP-7941 52. Despite screening different solvents, temperatures and reaction times, no reaction of the radiolabelled building block was observed.76
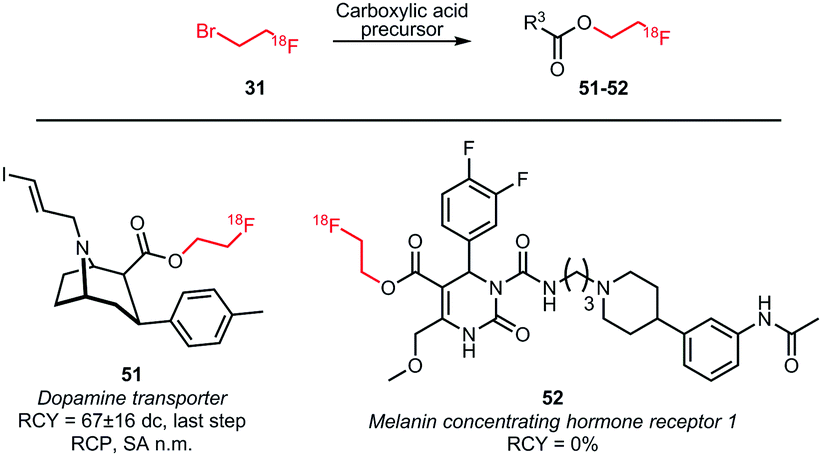 | ||
| Scheme 19 Esterification of carboxylic acids with [18F]fluoroethyl bromide.75,76 | ||
In conclusion, [18F]fluoroethyl bromide can be regarded a convenient reagent for radiofluorination of amines, phenols and carboxylic acids via alkylation. [18F]Fluoroethyl bromide can be synthesised fast and reliably and likewise the coupling reactions proceed fast, providing in one step the fluoroalkylated products in decay-corrected overall radiochemical yields of up to 40%.
The synthesis procedures of [18F]FETos 54 follow similar reaction conditions (Scheme 20). The kryptofix-potassium carbonate-[18F]fluoride complex ([18F][K⊂K2.2.2]F) reacted, after azeotropic drying, with the precursor ethylene ditosylate 53 in MeCN. Temperatures for this reaction varied between 75 °C and 130 °C and reaction times were between 3 and 15 minutes. Radiochemical yields of 20–90% were reported depending on purification and whether the production was executed manually, semi-automated, automated or by using a microfluidic system. In addition to manual synthesis of the building block, production using automated modules has frequently been carried out, either for only [18F]FETos synthesis or also for subsequent alkylation reactions.78 Pascali et al. developed a microfluidic approach providing the crude radiolabelled building block in a radiochemical yield of 67% (based on radio-TLC analysis).49
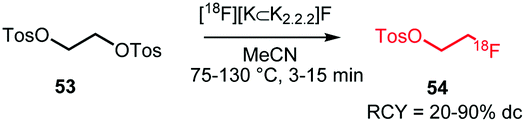 | ||
| Scheme 20 Radiosynthesis of [18F]FETos. | ||
Of the different approaches to purify [18F]FETos, semi-preparative HPLC generally provided the product with the highest chemical purity, leading to better conversions in the subsequent alkylation reaction. Furthermore, reduced formation of non-radioactive by-products was observed during the alkylation reaction which made final purification of the tracer easier.57
As HPLC purification is time-consuming, several SPE-based purification procedures have been developed. However, most of them focus on the removal of free [18F]fluoride, potassium carbonate and kryptofix only.78 Moreover, significant losses of radioactivity during cartridge purification or the subsequent drying step were observed.79,80 Schoultz et al. presented a SPE procedure including precipitation of the precursor with acetic acid, followed by filtration. The building block was obtained in high radiochemical purity (>99%) and in radiochemical yields of over 45%.78
Next to that, many successful one-pot methods have been described where [18F]FETos was used without intermediate purification before the subsequent alkylation reaction. Heinrich et al. reported an increased yield when using a one-pot strategy compared to a two-pot reaction with intermediate SPE purification, because they could avoid the activity losses on the cartridge.79 Majo et al. on the other hand obtained in a one-pot procedure only half of the radiochemical yield (20–25%) that was achieved when using a two-pot procedure, with intermediate purification by semi-preparative HPLC.81
[18F]FETos has found widespread application as a building block (Scheme 21). Besides N- and O-alkylation, reactions of [18F]FETos with phosphonates and thiols are known. Further, next to phenolic O-alkylation aliphatic hydroxyl groups and aromatic carboxylic acids can also be labelled with [18F]FETos.
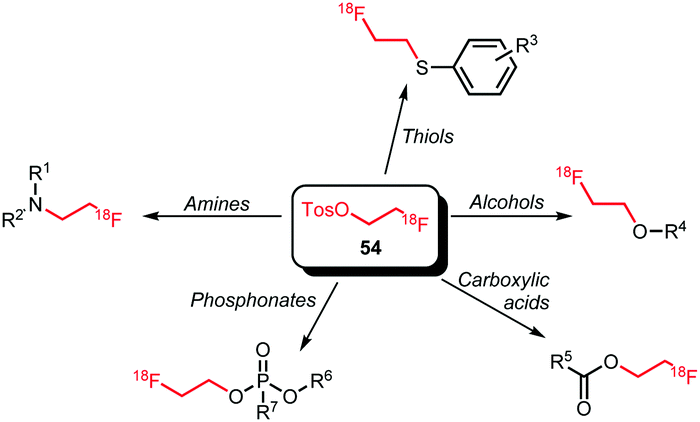 | ||
| Scheme 21 Reaction scope of [18F]FETos as building block. | ||
Numerous N-alkylations with [18F]FETos have been performed (Scheme 22).49,57,82–95 General reaction conditions involve the use of polar aprotic solvents such as DMSO, DMF or MeCN and temperatures ranging from 82 to 130 °C. Mostly, inorganic bases with a range of different pKa-values were employed depending on the reactivity of the corresponding amine reactant. Alkylation in absence of base has been reported for 56 and 58, which are potential imaging agents for matrix metalloproteinases and phosphatidylinositol 3-kinase, respectively.83,88 To achieve [18F]fluoroalkylation of 61 and 64, the amine reactants were treated with base (NaH or NaOH) prior to radiolabelling to generate the corresponding sodium salt. Radiolabelled 61 and 64 are important PET tracers for translocator protein and VEGF, respectively.85,90
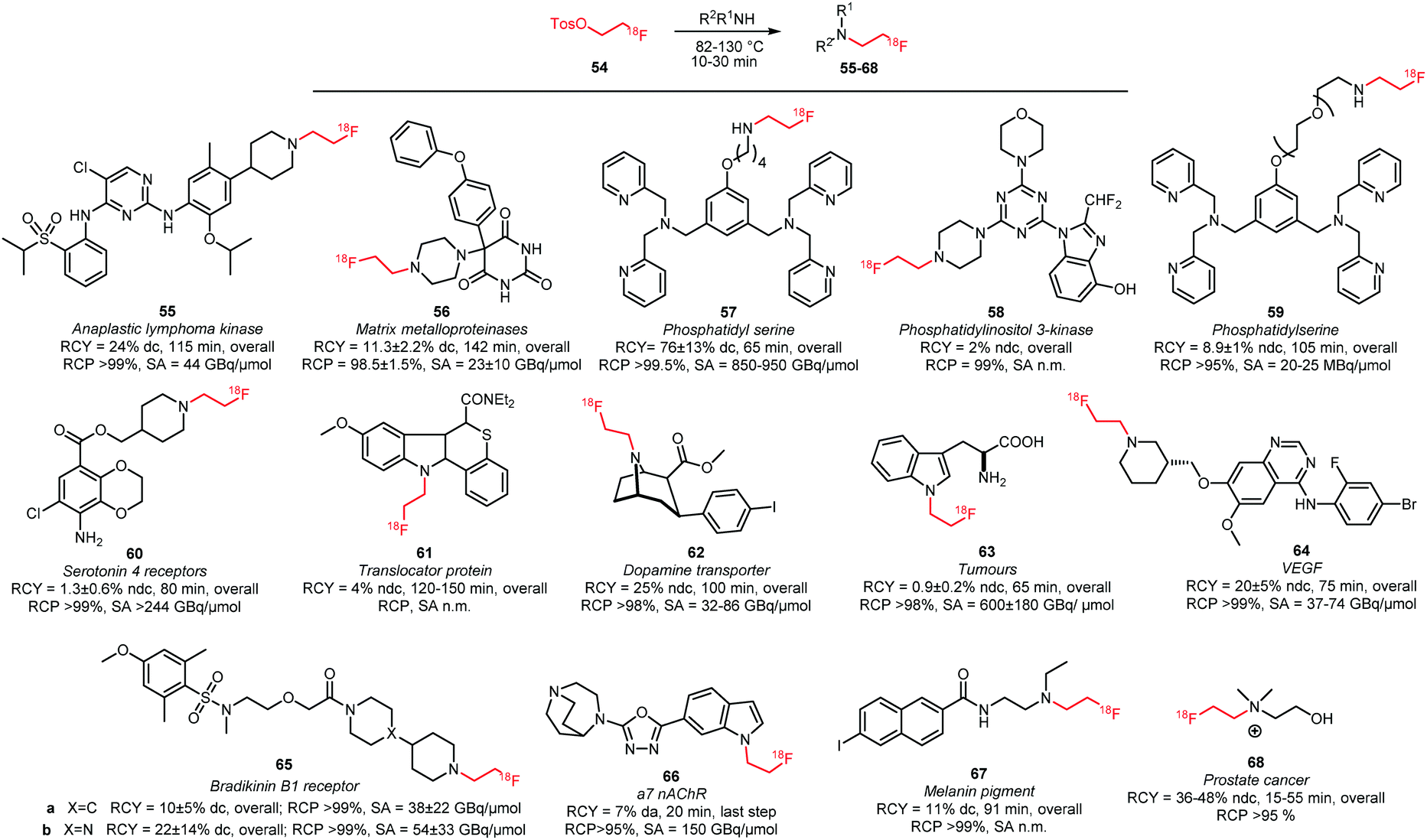 | ||
| Scheme 22 PET tracers synthesised by N-alkylation with [18F]FETos.49,57,82–95 | ||
Studies that involve Finkelstein-type alkylation reactions by addition of alkali iodides showed promising results indicating that in situ iodide exchange indeed could increase the yield of N-alkylation.96 However, this strategy has only been applied to access the serotonin 4 receptor tracer 60 and actually did not appear beneficial for the overall reaction outcome.89
The majority of tracers produced by alkylation with [18F]FETos were purified by semi-preparative HPLC. Only two SPE-based purification procedures were described in literature. They were developed for the cancer tracer fluoroethylcholine 68 and for tracer 59 that targets phosphatidyl serine to image cell death.49,93 Fluoroethyl-ceritinib 55, an imaging agent for anaplastic lymphoma kinase, was purified by normal phase flash chromatography because no HPLC conditions were found to obtain the tracer in decent purity.94
For some of the tracers in Scheme 22, direct and indirect labelling methods of the molecule were compared. In general no clear preference for either method could be concluded as in some cases (61 and 66) higher yields with direct labelling were found whereas in other cases (55 and 62) the indirect labelling strategy was more successful.91,94
Many O-alkylations with [18F]FETos have been performed and Scheme 23 shows all tracers synthesised by fluoroalkylation of phenolic precursors.52,80,81,97–120 The synthesis is overall similar to the N-alkylations and reactions were carried out in DMF, DMSO, MeCN or mixtures of these solvents with water at elevated temperatures (80–130 °C). Reaction times ranged from 10 to 20 minutes. A variety of bases have been employed: amongst others cesium carbonate, sodium hydroxide and sodium hydride have mostly been described. The choice of base and its concentration has a big influence on the yield of the alkylation reaction. Basic formation of the phenolate generates the nucleophile that will react with [18F]FETos in the radiolabelling.117 In approximately one third of all synthesis procedures, the phenolic precursor was deprotonated prior to alkylation to form the corresponding phenolate. The time of this preformation of phenolate varied from a few minutes up to several hours.104,105 The base was either filtered off after phenolate formation was complete or added together with the precursor to the reaction mixture containing [18F]FETos.98,104
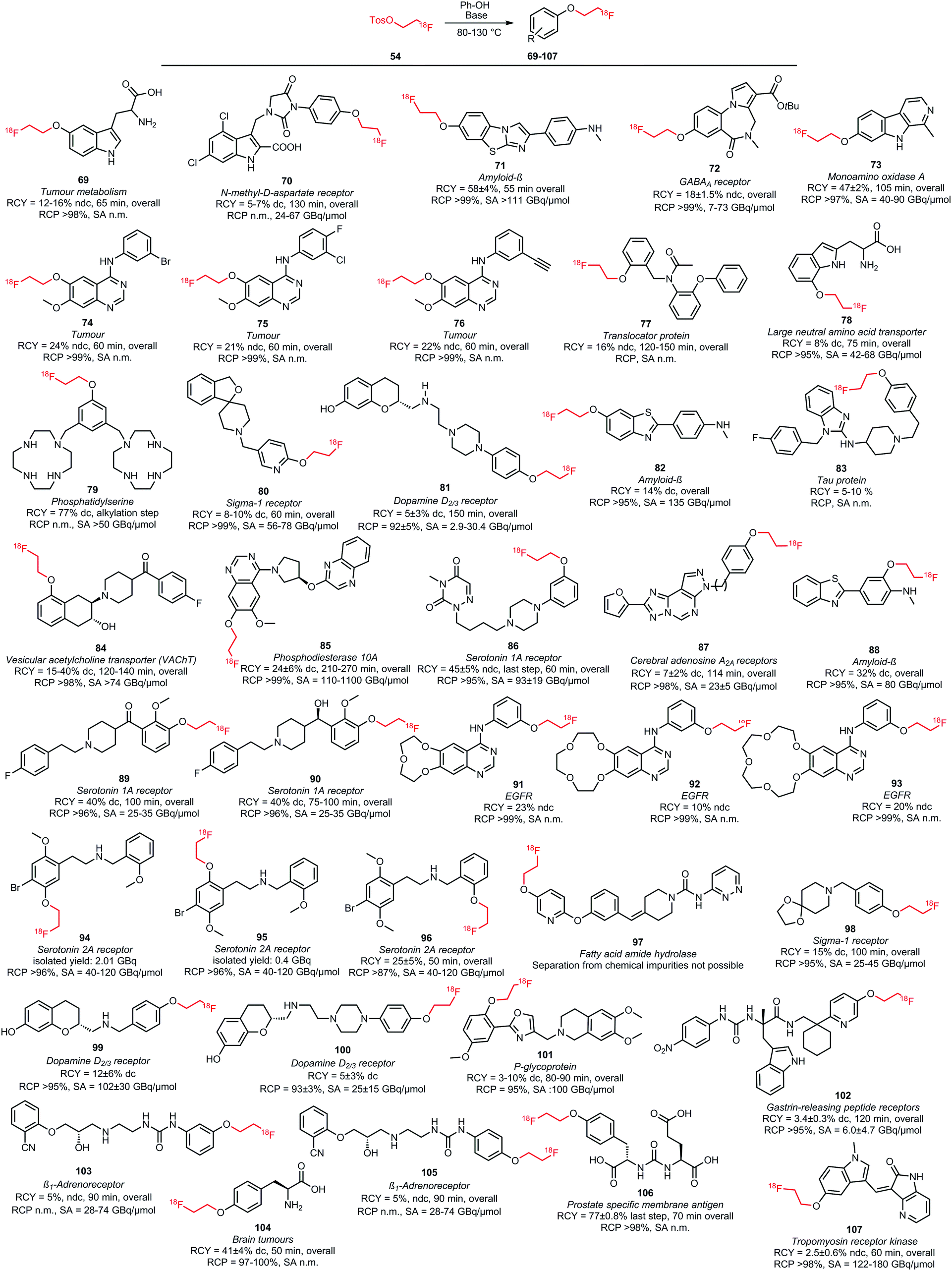 | ||
| Scheme 23 PET tracers synthesised by O-alkylation of phenolic precursors with [18F]FETos.52,80,81,97–120 | ||
For some of the synthesised PET tracers, direct and indirect radiolabelling has been compared. For the tracers 71, 73, 103 and 105, higher labelling yields for indirect labelling compared to direct labelling have been reported. For example, Schieferstein et al. obtained an overall radiochemical yield of 47 ± 2% in the synthesis of the monoamino oxidase A tracer 73 with [18F]FETos as labelling reagent, whereas the direct labelling approach in their hands only led to decomposition of the precursor.52 In another study, 73 was obtained via direct labelling in 23% decay-corrected radiochemical yield.121
For the tracers 85 and 83 however, direct labelling was superior to the indirect method. Purification of the phosphodiesterase 10A tracer 85, which was synthesised by the two-step reaction via [18F]FETos, turned out to be challenging and provided 85 in variable radiochemical purities, ranging from 92–99%. Therefore, direct labelling was performed which afforded 85 in high purity (≥99%) and comparable overall yields (25 ± 9%).102
Next to aromatic O-alkylation, alkylation of aliphatic hydroxyl groups has also been reported with [18F]FETos. Schoultz et al. studied an automated synthesis procedure for the opioid receptor tracers 109a–c. The tracers were synthesised in a two-step one-pot procedure from [18F]FETos and the trityl-protected precursor 108. The aliphatic hydroxyl group in the precursor was first deprotonated by treatment for 5 minutes with sodium hydride to generate the alkoxide. Then [18F]FETos was added and efficient alkylation occurred within 10 minutes at 100 °C. In the second step, the trityl-protected hydroxyl group was removed under acidic conditions. After HPLC purification, all derivatives of 109 were obtained in decay-corrected radiochemical yields of 26 ± 8% (Scheme 24).78,122
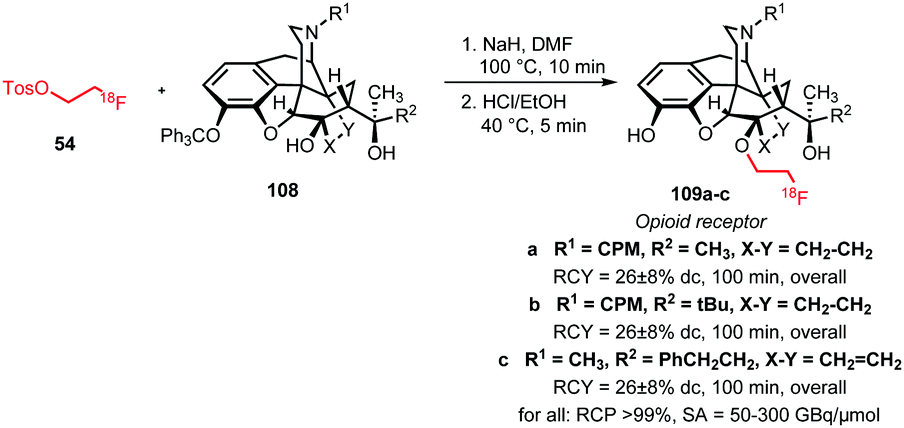 | ||
| Scheme 24 Aliphatic O-alkylation with [18F]FETos.78,122 | ||
Esterification of carboxylic acids with [18F]FETos has been studied by Heinrich et al. and Philippe et al.76,79 Effective esterification towards 111, a potential imaging agent for the melanin concentrating hormone receptor 1, was not observed when using [18F]FETos, and only direct radiofluorination in a microfluidic procedure proved successful.76 In contrast, 110, a tracer for myocardial perfusion, was synthesised successfully via esterification of the carboxylic acid precursor with [18F]FETos, performed in a one-pot procedure. After formation of [18F]FETos, the carboxylic acid was added under base catalysis in anhydrous MeCN for 30 minutes at 165 °C. The resulting product 110 was isolated by SPE or HPLC purification in an overall radiochemical yield of 36% (dc). Evaporation of the solvent during alkylation resulted in higher yields due to increased concentration of the reactants (Scheme 25).79
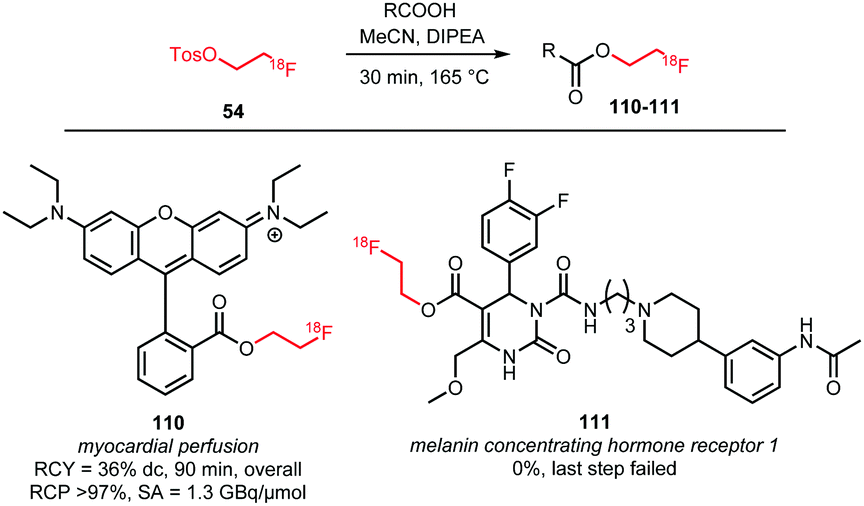 | ||
| Scheme 25 O-Alkylation of carboxylic acids with [18F]FETos.76,79 | ||
James et al. described the synthesis of an acetylcholine esterase tracer via O-alkylation of a phosphonate precursor (Scheme 26). The reaction was carried out in a microwave reactor with addition of cesium carbonate as base and with molecular sieves. Without intermediate purification of [18F]FETos and after semi-preparative HPLC, the desired tracer was obtained in a yield of 6.5% (dc) after the alkylation step.123
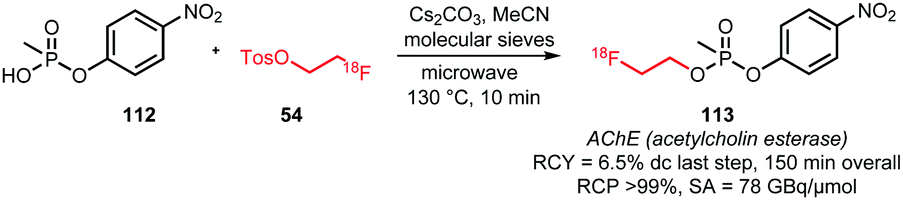 | ||
| Scheme 26 O-Alkylation of a phosphonate with [18F]FETos.123 | ||
Analogous to N- and O-alkylation, S-alkylation has been successful to obtain the NMDA receptor tracer 115 (Scheme 27). Overall radiochemical yields were 4–9% (ndc) in a synthesis time of 3 to 4 hours. In contrast, direct radiolabelling conditions led to degradation of the precursor and no radiofluorinated product was obtained.54
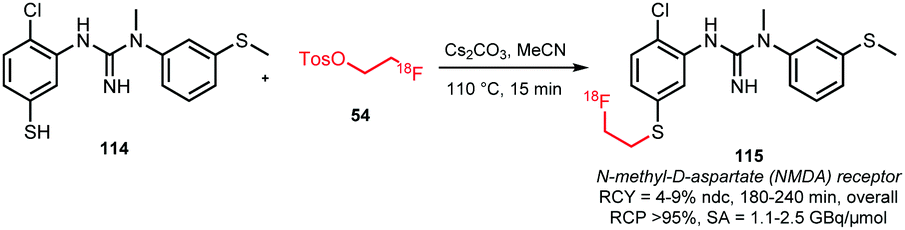 | ||
| Scheme 27 Synthesis of 115 by S-alkylation.54 | ||
In conclusion, [18F]FETos is an easy to synthesise, versatile building block which has been employed in the synthesis of many tracers because of its versatility and stability. It shows several advantages over the other [18F]fluoroethyl halides and sulfonates such as low volatility and decent reactivity. Furthermore, in many cases, [18F]FETos performs better or as good as the direct radiolabelling approach regarding conversion and purification of the PET tracer.
2.2.3.1 [18F]Fluoroethyl triflate. [18F]Fluoroethyl triflate 120 is more reactive towards alkylation than [18F]fluoroethyl bromide and therefore affords [18F]fluoroethylated products under very mild reaction conditions such as room temperature and without the need to add a base. Furthermore, it enables [18F]fluoroethylation of less nucleophilic precursors.
Three different strategies for [18F]fluoroethyl triflate synthesis have been developed (Scheme 28). Philippe et al. reported a one-step procedure starting from ethylene glycol bistriflate 117 and [18F][K⊂K2.2.2]F complex. The building block was obtained using triflic anhydride at elevated temperatures. Purification was performed with an alumina cartridge providing the product in a radiochemical yield of 19.2 ± 9.6% (dc).76
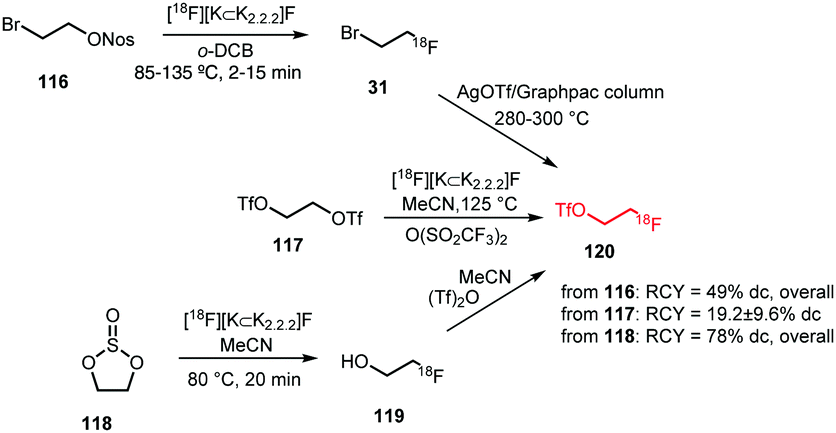 | ||
| Scheme 28 [18F]Fluoroethyl triflate synthesis from [18F]fluoroethyl bromide or ethylene glycol bistriflate.60,76,124 | ||
Murali et al. on the other hand discovered that the moisture-sensitive bistriflate precursor 117 suffers from poor stability even when stored at −20 °C. They observed low radiolabelling yields and therefore followed a two-step synthesis procedure with [18F]fluoroethyl bromide 31 as intermediate product. The bromide was distilled over an AgOTf/Graphpac column heated to 280–300 °C where it was converted to the corresponding triflate 120. They reported a total decay-corrected radiochemical yield of 49%, which is more than twice as high as the yield reported for the one-step procedure.60
A third approach was developed by Peters et al. They used [18F]fluoroethanol 119 as an intermediate that was synthesised from ethylene sulfate 118 in MeCN at 80 °C. After passing the crude reaction mixture through a QMA light cartridge, [18F]fluoroethanol was treated with triflic anhydride. The reaction proceeded smoothly (1 min) and did not require elevated temperatures. [18F]Fluoroethyl triflate was obtained after purification using an Alumina N light cartridge in a radiochemical yield of 78% (dc) starting from dried [18F]fluoride.124
Triflate building block 120 has been used in both N-alkylations and O-alkylations.60,76,125 While O-alkylation of 121, which is the acid precursor of a potential PET tracer for the melanin concentrating hormone receptor 1, proved unsuccessful under common [18F]fluoroethylation conditions (the tracer could only be synthesised using direct radiofluorination in a microfluidic system), two successful N-alkylations were described (Fig. 2).76 As the triflate is more reactive than the bromide, milder reaction conditions could be applied resulting in potential imaging agents, 122 and 123, which were obtained at room temperature without the presence of base or any other additives, after a few minutes, in good radiochemical yields.60,125 The base-free reaction conditions proved to be a big advantage particularly in the synthesis of the dopamine transporter ligand 123, avoiding epimerisation of the chiral centre at the C2-position.60
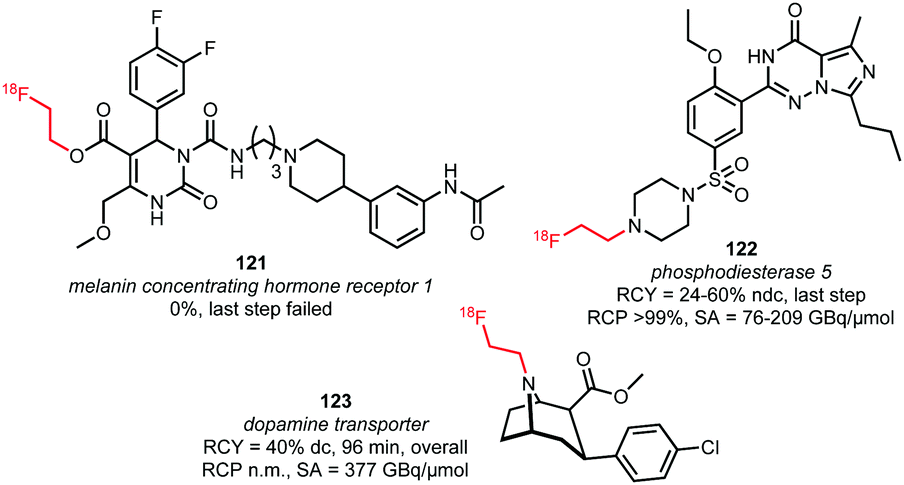 | ||
| Fig. 2 Tracers labelled with [18F]fluoroethyl triflate.60,76,125 | ||
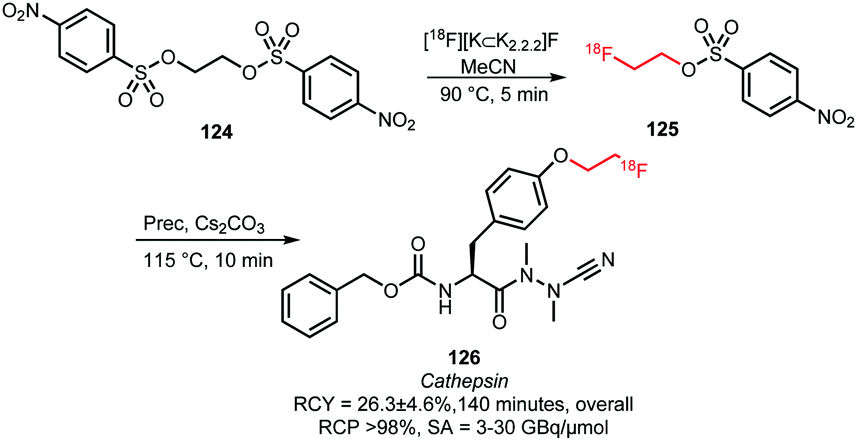
2.2.3.2 [18F]Fluoroethyl nosylate. Since 2010, [18F]fluoroethyl nosylate has only been described once as building block in a radiosynthesis. Löser and co-workers presented the synthesis of the fluorinated cathepsin inhibitor 126 in a two-step one-pot process (Scheme 29). [18F]Fluoroethyl nosylate was selected for the alkylation because the use of [18F]FETos resulted in low radiochemical yields (<26% based on radio-TLC analysis) due to degradation of the [18F]FETos under the reaction conditions. Furthermore, separation of the final product from [18F]FETos by semi-preparative HPLC was difficult and resulted in low radiochemical purity.
 | ||
| Scheme 29 [18F]Fluoroethyl nosylate synthesis and reaction to the cathepsin inhibitor 126.107 | ||
The selected [18F]fluoroethyl nosylate 125 building block could be prepared from the corresponding ethylene dinosylate 124 in MeCN at 90 °C in 5 minutes (Scheme 29). After cooling the reaction mixture to room temperature, the subsequent coupling reaction was conducted without intermediate purification. Nosylate 125 was treated with the phenolic precursor using catalytic amounts of base at 115 °C for 10 minutes. This gave product 126 in 74% radiochemical yield (based on radio-TLC analysis; 26% after HPLC purification) in an overall synthesis time of 140 minutes.107
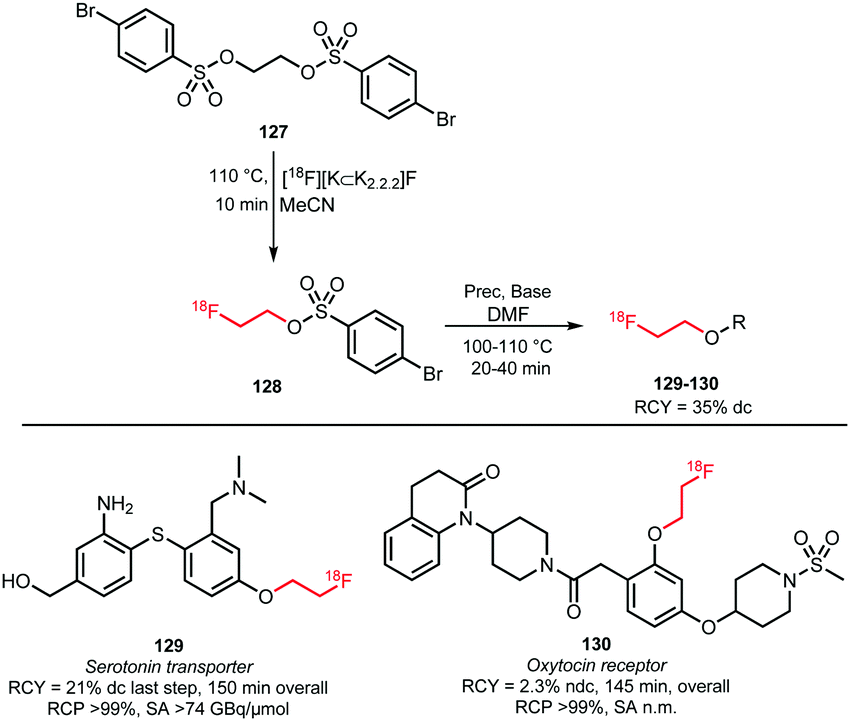
2.2.3.3 [18F]Fluoroethyl brosylate. Like the other sulfonates discussed above, 2-[18F]fluoroethyl-4-bromobenzene sulfonate ([18F]fluoroethyl brosylate) can be used as building block for the introduction of a [18F]fluoroethyl group. Its main advantage is that phenolic O-alkylation proceeds more efficiently compared to the use of the corresponding tosylate.126 Moreover, it is less volatile compared to [18F]FETos, which makes [18F]fluoroethyl brosylate more suitable for application in open vessel reactors (Scheme 30).127
 | ||
| Scheme 30 [18F]Fluoroethyl brosylate synthesis and subsequent [18F]fluoroethylation.127,129 | ||
The brosylate building block can be prepared by a procedure established by Voll and co-workers. Via a nucleophilic substitution reaction, ethylene-1,2-4-bromobenzene sulfonate precursor 127 could be radiofluorinated at elevated temperatures resulting in the desired brosylate in 35% (dc) radiochemical yield after HPLC purification.127,128
The follow-up coupling reactions were conducted analogously to those with the other ethyl sulfonate building blocks. The phenolic precursors were reacted in DMF with [18F]fluoroethyl brosylate 128 under basic conditions at 100–110 °C. Catalytic amounts of cesium carbonate or TBAOH were employed as base. In this way the serotonin transporter imaging agent 129 was obtained in an overall radiochemical yield of 21% (dc). This is quite efficient compared to the alternative preparation of fluorine-18 labelled oxytocin receptor ligand 130, which was obtained in a non-decay corrected overall radiochemical yield of only 2.3%. Total synthesis time of both tracers was about 150 minutes.
Smith and co-workers also investigated the synthesis of PET tracer 130via direct radiofluorination of the respective tosylate precursor and reported a non decay-corrected radiochemical yield of 19%, which is 8 times higher than the two-step synthesis.127,129
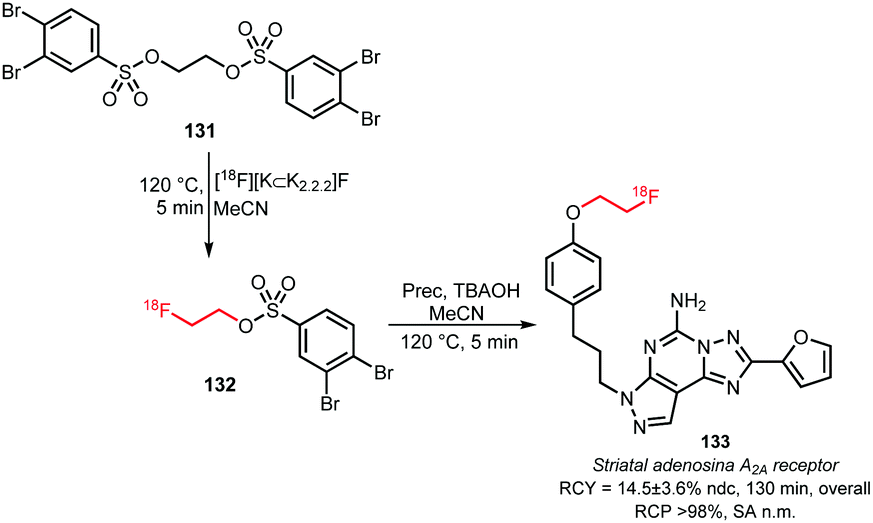
2.2.3.4 [18F]Fluoroethyl-3,4-dibromobenzenesulfonate. In 2005 Musachio et al. studied other [18F]2-fluoroethyl brosylates to mediate [18F]fluoroethylation. Their studies revealed that [18F]fluoroethyl-3,4-dibromobenzenesulfonate 132 is highly reactive and quite stable and achieves high yields in alkylating reactions (Scheme 31).126
 | ||
| Scheme 31 [18F]Fluoroethyl-3,4-dibromobenzene sulfonate synthesis and reaction to the A2A receptor tracer 133.130 | ||
Still, thusfar only one tracer synthesis based on [18F]fluoroethylation with [18F]fluoroethyl-3,4-dibromobenzene sulfonate 132 has been reported. Bhattacharjee and co-workers described the synthesis of a fluorine-18 labelled tracer for A2A receptor imaging. In the first step, ethyleneglycol-1,2-3,4-dibromobenezenesulfonate 131 was reacted with the dried [18F]fluoride at 120 °C for 5 minutes. After that, the resulting building block was purified by semi-preparative HPLC and reacted with the respective phenolic precursor in presence of TBAOH as base. Purification by semi-preparative HPLC afforded the radiotracer 133 for the striatal adenosine A2A receptor in an overall radiochemical yield of 14.5 ± 3.6% (ndc).130
2.3 [18F]Fluoroform
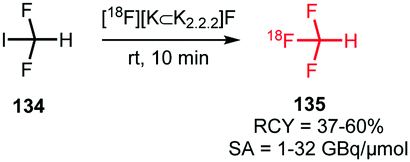
The trifluoromethyl functional group is well established for its favourable in vivo properties. Therefore it is a group incorporated in many active pharmaceutical ingredients. Consequently, the introduction of fluorine-18 using trifluoromethylation has found widespread interest. A highly effective way to achieve this is to couple [18F]fluoroform to aryl boronic acids and iodides in a copper(I) mediated reaction. However, this approach mostly gave the products in poor specific activity, which is a disadvantage for PET imaging of low density receptors in particular.Difluoroiodomethane 134 and the difluoromethylsulfonium salt 142 have been explored as alternative precursors for [18F]fluoroform synthesis (Schemes 32 and 35). Based on the precursor difluoroiodomethane 134, van der Born et al. developed two different methods to synthesise [18F]fluoroform, providing the product either in high yield or increased specific activity. In one method, [18F]fluoride was eluted with K2CO3/K2.2.2 and azeotropically dried following standard procedures. Subsequent reaction with difluoroiodomethane 134 for 10 minutes at room temperature afforded [18F]fluoroform 135 in a radiochemical yield of 60% with a specific activity of 1 GBq μmol−1. Purification was carried out by distillation over a silica Sep-Pak cartridge. The low specific activity is most probably caused by the polyfluorinated precursor. Reducing the amount of precursor 40-fold together with decreasing the amount of base for [18F]fluoride elution from the cartridge gave an average radiochemical yield of only 37%, but specific activity increased to 32 GBq μmol−1.131
 | ||
| Scheme 32 [18F]Fluoroform synthesis with difluoroiodomethane as precursor. | ||
To introduce the [18F]trifluoromethyl group into PET tracers 137 and 138 for imaging breast cancer and other tumours, copper-mediated reactions with aryl iodides and aryl boronic acids showed promising results. [18F]Trifluoromethylation of aryl iodides was carried out in the presence of copper(I) bromide and potassium tert-butoxide as a base. Further, triethylamine trihydrofluoride was employed to stabilise the resulting copper–CF3 complex. Reactions were complete after 10 minutes at 130 °C. The procedure was similar for trifluoromethylation of aryl boronic acids, but oxidation of copper(I) was required by purging the reaction solution with air. Reactions were complete within 1 min at room temperature, which is considerably faster compared to the reactions using analogous iodide precursors. In Scheme 33, the radiosynthesis of two tracers labelled by [18F]trifluoromethylation using [18F]fluoroform is depicted. Both were synthesised from the available iodide precursor as well as from the boronic acid precursor. Direct comparison of both procedures showed that use of the boronic acid precursors offered more favourable coupling conditions and ultimately higher radiochemical yields.131
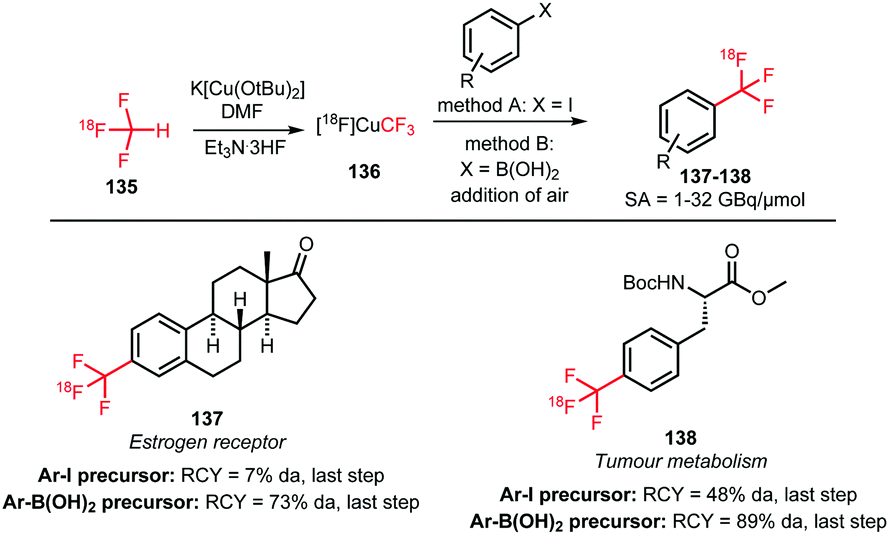 | ||
| Scheme 33 PET tracer synthesis by [18F]trifluoromethylation of iodide and boronic acid precursors.131 | ||
In addition to the above methods, a one-pot procedure to synthesise [18F]trifluoromethylated tracers in an automation-compliant manner has been developed by Rühl et al. In order to find the most efficient Cu–ligand system for the [18F]trifluoromethylation reaction in presence of the dried [18F]fluoride, different ligands and sources of reactive [18F]fluoride were screened. Optimal yields of [18F]trifluoromethylated product were obtained with a KHCO3/kryptofix/DIPEA mixture. Utilizing the optimised conditions, three potential PET tracers 139, 140 and 141 were synthesised in good radiochemical yields of 73 to 85%. A drawback is that the tracers were obtained in only a very low specific activity of 139 MBq μmol−1 (Scheme 34).132
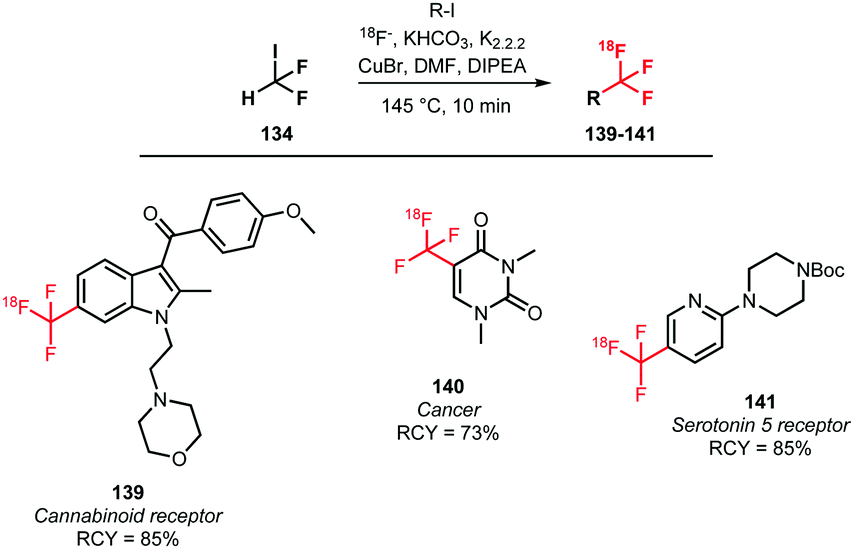 | ||
| Scheme 34 One-pot synthesis of [18F]trifluoromethylated arenes.132 | ||
Ivashkin et al. employed difluoromethylsulfonium salt 142 as precursor for the [18F]fluoroform synthesis (Scheme 35). However, [18F]fluoroform was not isolated, but distilled into a solution containing a copper(I) halide and potassium tert-butoxide. This instantaneously formed the [18F]CuCF3 complex 136, which was subsequently treated with a range of different model iodides or boronic acids. Again, the tracers were obtained in very low specific activity of 100 MBq μmol−1, comparable to the one-pot procedure described by Rühl et al.133
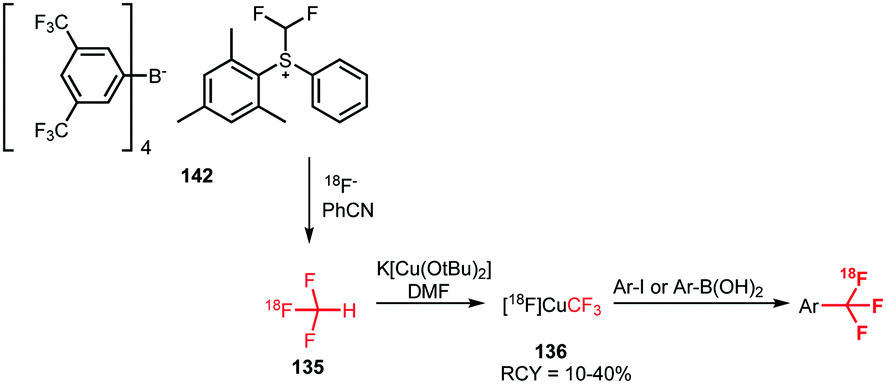 | ||
| Scheme 35 [18F]Fluoroform synthesis with the difluoromethylsulfonium salt 142 as precursor.133 | ||
In conclusion, production of [18F]fluoroform with high specific activity remains a challenge. A method has however been developed providing [18F]fluoroform with an acceptable specific activity of 32 GBq μmol−1. Aryl iodides and boronic acids have successfully been labelled with [18F]fluoroform as building block. [18F]Trifluoromethylation of boronic acids proceeds fast and under mild reaction conditions and aryl iodides have shown to be valuable precursors in one-pot syntheses of relevant tracers. Hence, [18F]trifluoromethylation with [18F]fluoroform holds great promise for fluorine-18 labelling of compounds containing native trifluoromethyl groups.
2.4 [18F]Trifluoroethyl tosylate
Application of [18F]trifluoroethyl tosylate as a building block enables the introduction of fluorine-18 via the trifluoroethyl group. Two different PET tracers have been synthesised using [18F]trifluoroethyl tosylate (Schemes 36 and 37).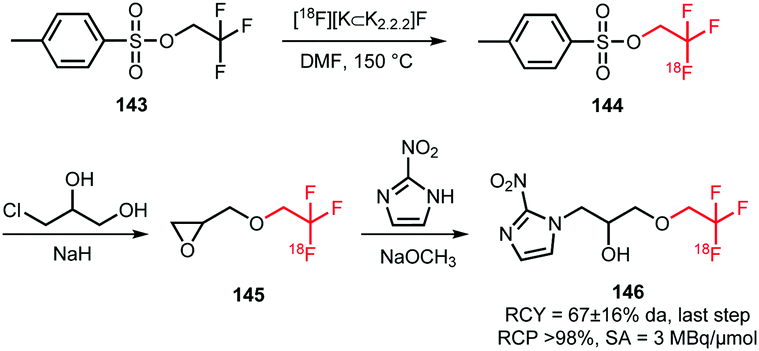 | ||
| Scheme 36 Synthesis of the hypoxia tracer [18F]TFMISO.134 | ||
Suehiro et al. developed the synthesis of [18F]trifluoromisonidazole ([18F]TFMISO) 146, a hypoxia tracer for bimodality imaging with MRI and PET. In this context, 2,2,2-[18F]trifluoroethyl tosylate 144 was found to be an excellent [18F]trifluoroethylation agent, as it reacts smoothly with alcohols to the corresponding [18F]trifluoroethyl ethers.
The building block was synthesised via18F–19F exchange from 2,2,2-trifluoroethyl tosylate 143. For this, the unlabelled compound was heated in presence of [18F][K⊂K2.2.2]F at 150 °C. After 10 minutes, the product was separated from unreacted [18F]fluoride by extraction with ether. A specific activity of this building block is not reported, but a low specific activity is expected due to the isotopic exchange methodology that was employed here.
Starting from [18F]trifluoroethyl tosylate, the hypoxia tracer was subsequently prepared via a 2-step procedure. First, [18F]trifluoroethyl tosylate was treated with deprotonated 3-chloro-1,2-propanediol, in the presence of sodium hydride. After a 45–60 minutes reaction at room temperature, the desired [18F]trifluoroethoxy intermediate 145 was obtained with good radiochemical yields of 57 ± 10% (analytically determined). In the next step, intermediate 145 was converted to [18F]TFMISO in a reaction with 2-nitroimidazole under basic conditions using NaOMe. The final product 146 was obtained in a radiochemical yield of 67 ± 16% (analytically determined) (Scheme 36).
Besides the procedure described above, other routes have been investigated to arrive at the same final compound. The analogue 2,2,2-[18F]trifluoroethyl iodide was synthesised with an excellent labelling efficiency (90–95%), but underwent nucleophilic substitution at the fluorinated carbon atom instead of substitution of the iodide. Furthermore, Suehiro et al. tried to directly label the complete precursor molecule of 146, however this led to intramolecular nucleophilic substitution of the nitro group.134
Riss et al. synthesised [18F]trifluoroethyl tosylate via nucleophilic addition of [18F]fluoride to 1,1-difluorovin-2-yl-4-toluene sulfonate 147 (Scheme 37). Extensive studies to find optimal reaction conditions were conducted and in the end 5 minutes reaction in DMSO at 85 °C proved sufficient to produce the desired compound. Trace amounts of water were crucial for product formation as in the absence of water the precursor was subject to an addition–elimination reaction resulting in fluorine-18 labelled 147. As the addition of ppm amounts of water appears to be rather cumbersome, the influence of low molecular weight alcohols on the reaction has been explored. Best results were obtained with 1 M 2-propanol in DMSO and radiochemical yields up to 67% (based on radio-HPLC analysis) were observed. Furthermore, specific activity of the fluoroethyl building block has been examined. A good specific activity (86 GBq μmol−1) was obtained even with low quantities of [18F]fluoride at the start of synthesis (5 GBq).135
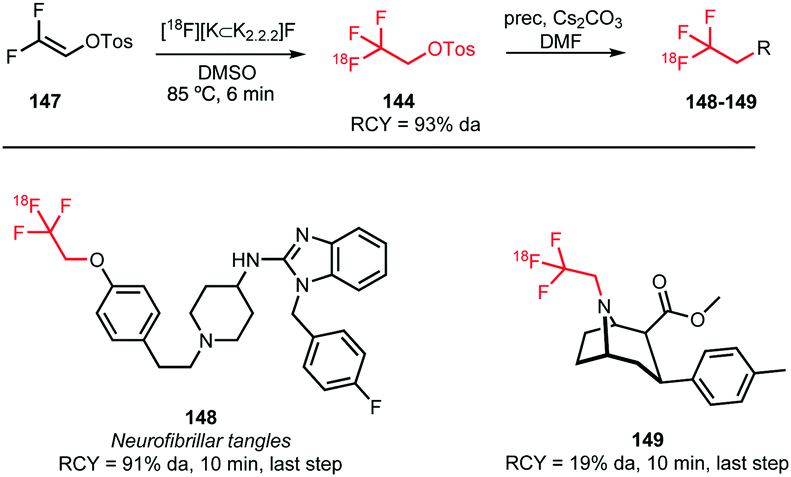 | ||
| Scheme 37 [18F]Trifluoroethyl tosylate synthesis and coupling towards 148 and 149.108,135,136 | ||
[18F]Trifluoroethyl tosylate was applied in both O- and N-alkylations resulting in two potential imaging agents, 148 and 149 (Scheme 37). The coupling reaction was conducted in DMF using cesium carbonate as base. N-Alkylation (19%) proceeded in a much lower yield than O-alkylation (91%), which was attributed to the tropane scaffold used in this specific case.136
In summary, 2,2,2-[18F]trifluoroethyl tosylate is a useful building block, forming [18F]trifluoroethyl ethers under relatively mild conditions. The possibility of native radiofluorination and the enhanced stability of the trifluoroethyl group towards metabolic degradation compared to [18F]fluoroethylates make it a promising building block for fluorine-18 labelling in the future.
2.5 Long chain (n > 2) fluorine-18 labelled aliphatic halides and sulfonates
Apart from [18F]fluoromethyl and [18F]fluoroethyl halides and sulfonates, building blocks with longer alkyl chains have been used for PET tracer synthesis. PET tracers with longer alkyl chains show enhanced in vivo stability, improved target affinity or selectivity and more favourable pharmacokinetics compared to the corresponding fluoroethylated or fluoromethylated analogues.80,100,137Scheme 38 summarises various long chain aliphatic building blocks containing a tosylate as leaving group. Especially [18F]fluoropropyl tosylate 150 is quite popular. Fluoroalkylations to access homologues with a chain length of n = 6 proved successful with this reagent. Furthermore, unsaturated and cyclic derivatives of [18F]fluorobutyl tosylate (154–156) as well as polyethyleneglycol derived building blocks (157 and 158) have been employed in PET tracer synthesis.
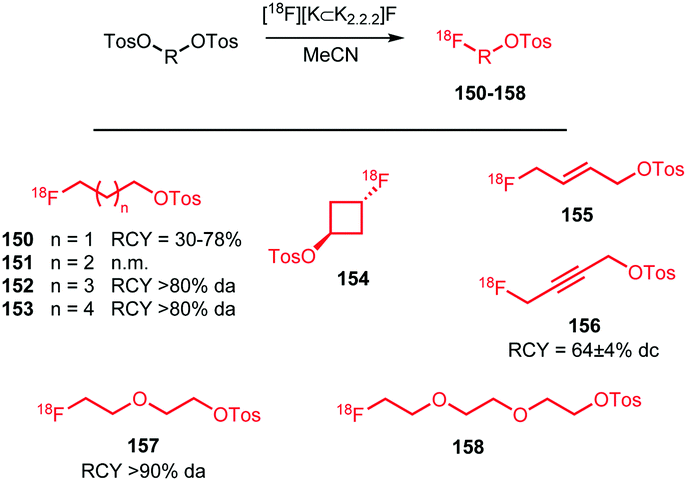 | ||
| Scheme 38 Synthesis of various long chain aliphatic building blocks from the corresponding ditosylate. | ||
The synthesis of all these tosylate fluorine-18 labelled aliphatic building blocks was similar to the synthesis of [18F]FETos 54. The ditosylate precursor was reacted with dried [18F][K⊂K2.2.2]F complex in MeCN at temperatures of 85–130 °C. The reported yields of the radiofluorination are comparable to those using [18F]FETos, which demonstrates that the procedure is generally applicable and not depending on chain length. The resulting fluorine-18 labelled aliphatic building blocks can be purified in different ways. For example, [18F]fluoropropyl tosylate 150 was purified by either HPLC or silica Sep Pak. In addition, one-pot procedures including the alkylation step were also applied. Although similar radiochemical yields for the one-pot strategy and two-step procedure including HPLC purification have been described,89 low specific activities for the products from the one-pot synthesis have been reported. The main reason for this is that the coupling product of the remaining ditosylate and the tracer precursor could not be separated from the actual PET tracer.137 For two of the butane derived fluorine-18 labelled building blocks, 151 and 156, HPLC purification has been reported. 1-[18F]Fluoro-4-tosylbut-2-ene 155, the longer chain [18F]fluoroalkyl halides (152 and 153) and the PEG derived building blocks (157 and 158) were purified by silica Sep-Pak, while 1-[18F]fluoro-3-tosyl cyclobutane 154 was purified by C18 Sep-Pak.142 For [18F]fluoropropyl tosylate 150, automated synthesis procedures have been developed including a microfluidic approach.49
As discussed, [18F]fluoropropyl tosylate 150 is used most often for longer chain alkylations. Using this building block, the compounds 160, 162 and 164 were obtained by N-alkylation of amine precursors (Fig. 3). Reactions were carried out in DMF at 130 °C for 20 to 30 minutes. When intermediate purification of the building block 150 was necessary, cesium carbonate was added as base. When a one-pot method was applied, the potassium carbonate present from the first reaction served as a base to catalyse the subsequent coupling reaction of 150. Generally, moderate to good radiochemical yields have been obtained,49,89,138 (except for the 5-HT4 receptor tracer 164). The other tracers depicted in Fig. 3 were synthesised by O-alkylation of the phenolic precursor with 150.49,68,80,89,99,100,137–140 The alkylation reactions were performed in DMSO, DMF or MeCN at around 100 °C and different bases were employed (NaH, NaOH, K2CO3 or TBAOH). The precursor of the serotonin transporter ligand 167 was pre-incubated with the base prior to [18F]fluoropropylation to form the phenolate, thereby facilitating nucleophilic substitution.140
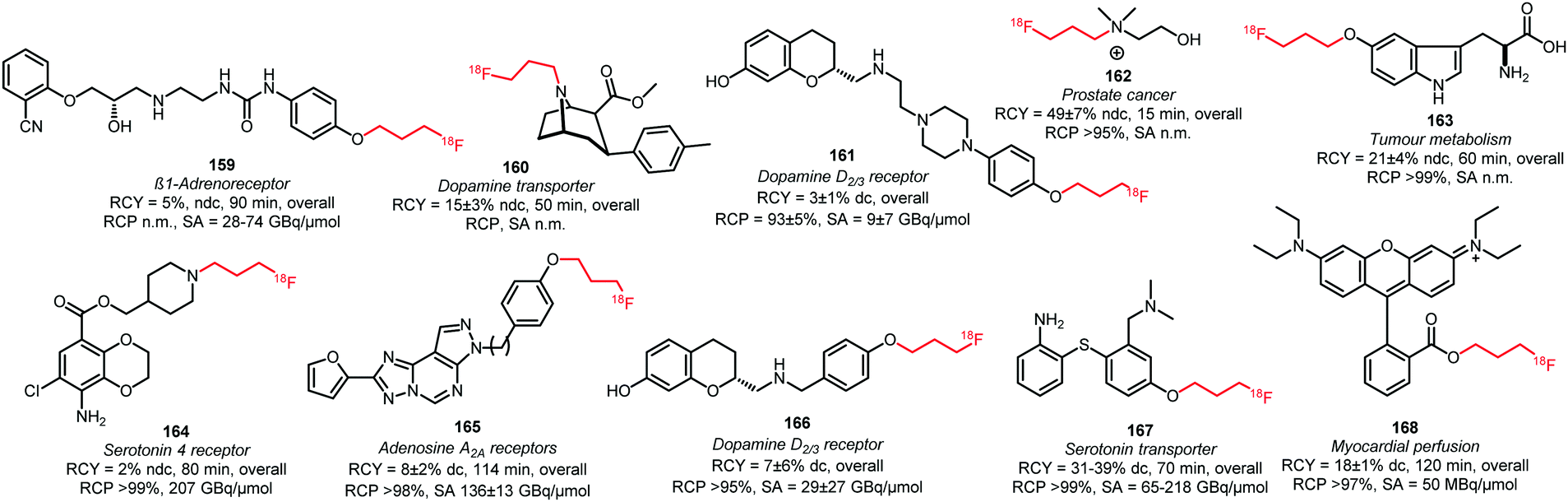 | ||
| Fig. 3 PET tracers synthesised from the building block [18F]fluoropropyl tosylate.49,68,80,89,99,100,137–140 | ||
Overall radiochemical yields of the tracers synthesised with [18F]fluoropropyl tosylate 150 were variable and ranged from low to good. Shalgunov et al. conducted a comparative study on the two dopamine D2/3 receptor tracers 161 and 166, labelled with [18F]fluoropropyl tosylate as well as [18F]fluorobutyl tosylate (169, 170) and [18F]FETos (81, 99). Similar yields were obtained for 161, 169 and 81 as well as 166, 170 and 99, showing that the chain length of the building block had no major effect on the reaction kinetics.100
Bartholomä et al. developed a synthesis of 168, which is a known imaging agent for myocardial perfusion, by esterification of the carboxyl group with [18F]fluoropropyl tosylate 150. In this case, the corresponding lactone served as precursor and the reaction was carried out in MeCN at 165 °C with DIPEA as base. They reported a decay-corrected radiochemical yield of 18 ± 1% in a total synthesis time of 120 minutes. In comparison to the [18F]fluoroethyl analogue, [18F]fluoroproyl tracer 168 showed and improves stability.137
Four different butane derived building blocks have been employed for PET tracer synthesis. Next to the parent n-butane derivative 151, also cyclobutane 154, butene 155 and butyne 156 analogues have been used in N- and O-alkylations (Fig. 4). The two imaging agents for the dopamine D2/3 receptor, 169 and 170, were synthesised by Wieringen et al. and Shalgunov et al. via O-alkylation of the phenolic precursor with [18F]fluorobutyl tosylate 151 in moderate overall radiochemical yields of 5–6% (dc) and 7–8% (dc), respectively. Using building block 151, resulted in increased lipophilicity of the tracer and thereby enhanced ability to penetrate the blood brain barrier.100,113 Also, four tropane derivatives, 171a–c and 172, were developed for imaging the dopamine transporter. The fluoroalkylation reactions could be carried out without base catalysis.57,141 Riss et al. reported a quite efficient automated alkylation of the amine present in the tropane scaffold employing [18F]fluorobutyne 156 in an overall radiochemical yield of 25% (ndc). Direct labelling of tropane 172 was also reported and proceeds in higher yield (32–36%) under microwave conditions, but this approach did not allow for automation.57 Furthermore, Franck et al. introduced the [18F]fluorocyclobutyl group the amino acid tyrosine 173 using 154, to enhance the metabolic stability of the PET tracer (cycloalkanes show in general better metabolic stability than the n-alkyl counterparts).142
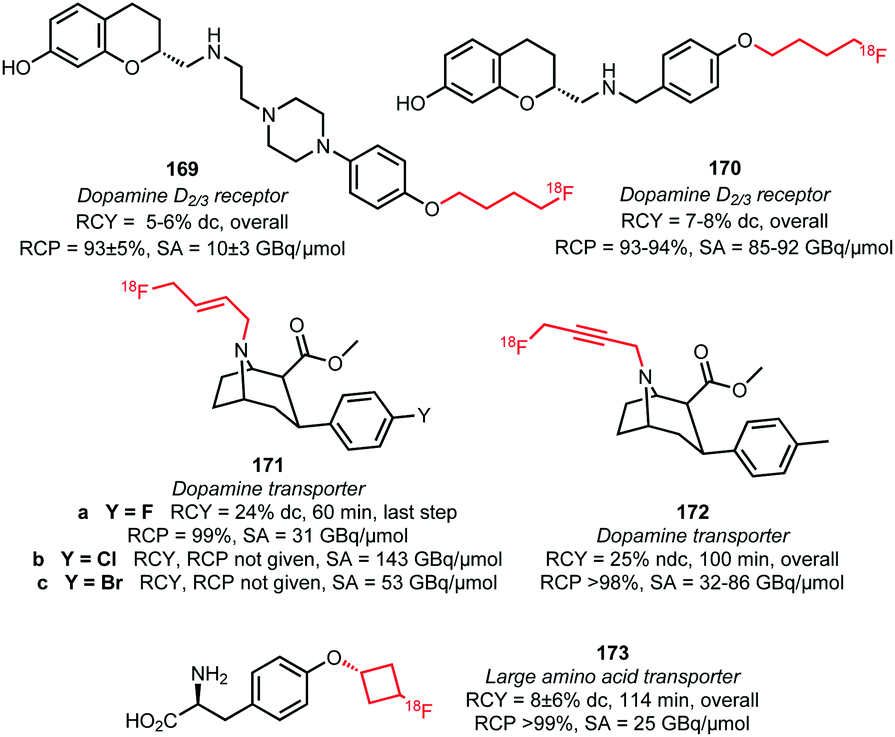 | ||
| Fig. 4 PET tracers synthesised by indirect labelling with [18F]fluorobutyl tosylate and derivatives.57,100,113,141,142 | ||
[18F]Fluoroalkyl tosylates with a carbon chain length longer than 4 were only applied in the synthesis of triphenylphosphonium salts for myocardial perfusion imaging (Fig. 5). [18F]Fluoropentyl tosylate 152 and [18F]fluorohexyl tosylate 153 were coupled to triphenyl phosphine in toluene at 220 °C. After purification by semi-preparative HPLC, both PET tracers were obtained in decay-corrected radiochemical yields of 15 to 20%, respectively.143
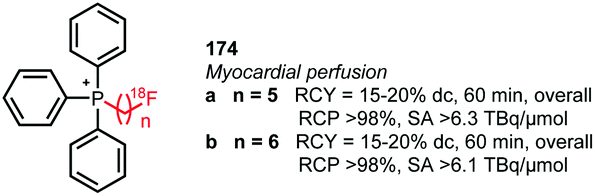 | ||
| Fig. 5 [18F]Fluoroalkyl triphenylphosphonium salts for myocardial perfusion imaging.143 | ||
The polyethylene glycol derived building blocks 2-(2-[18F]fluoroethoxy)ethyl tosylate 157 and 2-(2-(2-[18F]fluoroethoxy)ethoxy)ethyl tosylate 158 have also been used in the synthesis of myocardial perfusion imaging agents (Fig. 6). Kim et al. reported the synthesis of [18F]fluoroPEGylated phosphonium salts 175 and 177via reaction of triphenyl phosphine with 2-(2-[18F]fluoroethoxy)ethyl tosylate 158 in toluene at 220 °C, followed by purification over a small silica cartridge.143,144 Bartholomä et al. presented several 2-(2-[18F]fluoroethoxy)ethyl esters (178a–c and 179) and a (2-(2-[18F]fluoroethoxy)ethoxy)ethyl ester (176) of rhodamine B as myocardial perfusion imaging agents. For the labelling of the lactone precursors, a one-pot method was applied in which the coupling reaction was performed in MeCN at 160 to 165 °C under base-catalysis (DIPEA). All tracers were obtained in good overall radiochemical yields (∼20% (dc)) in a synthesis time of 20 minutes. In vitro and in vivo biological evaluations of 179 showed that this tracer is superior to the ethyl, propyl and triethyleneglycol analogues with respect to imaging characteristics and metabolic stability. This shows that the prosthetic group significantly influences the pharmacokinetics and metabolism of the PET tracer.137,145
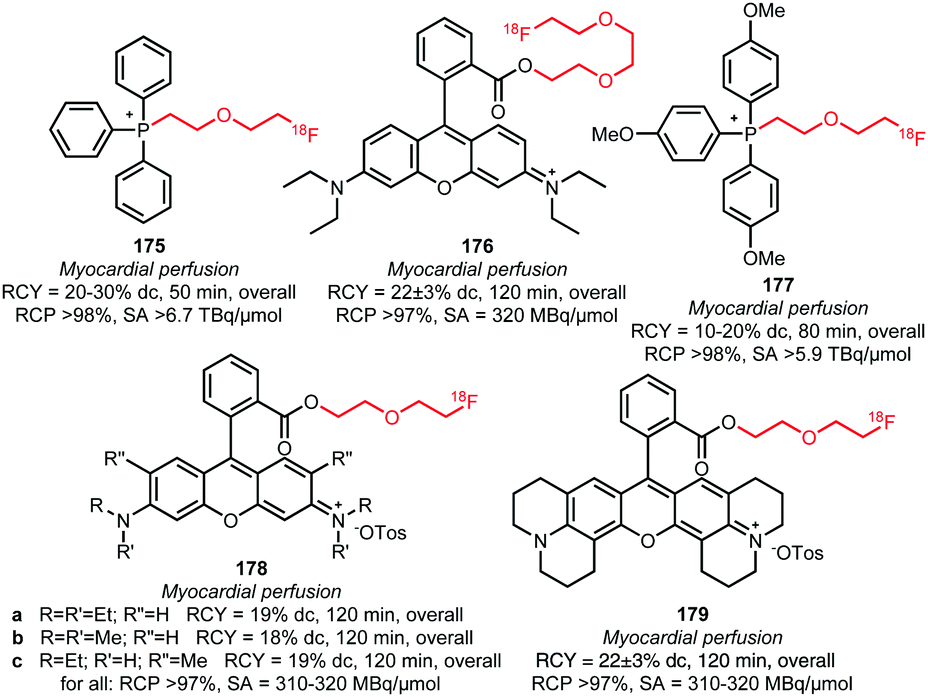 | ||
| Fig. 6 [18F]FluoroPEGylated tracers for myoardial perfusion imaging.137,143–145 | ||
Two other building blocks for indirect labelling of triphenyl phosphines have been presented by Zhao et al. in 2014. They synthesised 1-bromomethyl-3-[18F]fluoromethylbenzene 181a and 1-bromomethyl-4-[18F]fluoromethylbenzene 181b building blocks using a modified procedure of De Vries et al. (Scheme 39).146 The myocardial perfusion tracers 182a and 182b were obtained via a one-pot procedure without intermediate purification of the building blocks in decay-corrected radiochemical yields of 52 ± 9% and 51 ± 7%, respectively.147
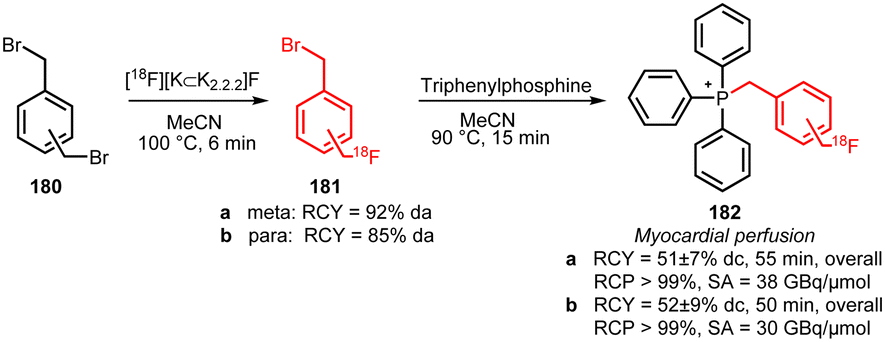 | ||
| Scheme 39 Synthesis and reaction of meta- and para-[18F]bromomethylfluoromethylbenzene.147 | ||
Not only [18F]fluoropropyl tosylate 150, but also the corresponding bromide has been employed as building block for fluoroalkylation (Scheme 40). [18F]Fluoropropyl bromide 184 was synthesised by treatment of the bromopropyl triflate with dried [18F][K⊂K2.2.2]F complex in o-dichlorobenzene and subsequent distillation at 150 to 180 °C into cooled DMF (−15 to −20 °C) containing the precursor and sodium hydroxide. After distillation, the trapping solution was heated for 10 minutes at 120 °C to react the building block with the phenolic precursors and generate the two PET tracers 185 and 186, albeit in rather low overall radiochemical yields (5% and 2%, respectively).68,69 Fujinaga et al. hypothesised that this can be explained by a decreased inductive effect of the fluorine atom further along the chain.69
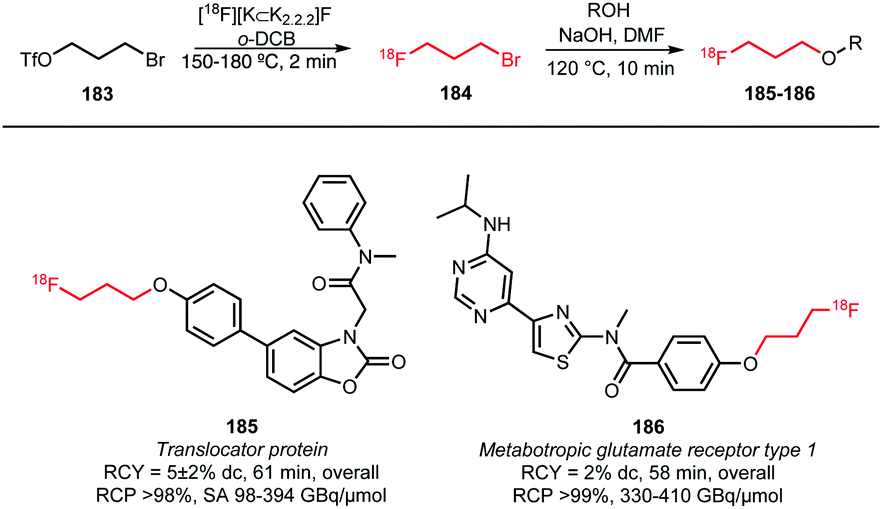 | ||
| Scheme 40 [18F]Fluoropropyl bromide as building block for [18F]fluoroalkylation.68,69 | ||
2.6 Fluorine-18 labelled azides
Many labelled azides have found widespread application in tracer synthesis. In particular, [18F]fluoroethyl azide 188 (Scheme 41) and deoxy-[18F]fluoroglucopyranosyl azides have been employed as building block. They can be coupled to PET tracers by the Huisgen 1,3-dipolar cycloaddition, also called copper(I)-catalysed azide-alkyne cycloaddition (CuAAC) or ‘click’-reaction. Alternatively, the traceless Staudinger ligation has been employed. | ||
| Scheme 41 Synthesis of [18F]fluoroethyl azide. | ||
In the CuAAC, 1,4-disubstituted triazoles are formed by reaction of an azide group with an alkyne functionality under copper(I) catalysis. Using the CuAAC protocol introduces fluorine-18 under mild aqueous conditions. Conditions that are compatible with highly functionalised polar biomolecules.148 Furthermore, these ‘click’-reactions usually show high specificity, robustness and yields.149 Many different functional groups are well tolerated, making additional protection and deprotection steps unnecessary.150 Moreover, the click reaction is an excellent method to build up libraries of compounds for screening campaigns to ultimately select the best PET tracer.151
As an alternative coupling reaction to the CuAAC, the traceless Staudinger ligation can be employed. This reaction of an azide with a phosphine-substituted thioester leads to the formation of an amide bond. Therefore, it represents a useful strategy for the labelling of amino acids and peptides. It proceeds under mild reaction conditions and no metal catalysis is required.152,153
Both the CuAAC and the Staudinger ligation methods however have also disadvantages, for example, the in vivo toxicity of the copper(I) used in the CuAAC and the instability of phosphine reagents used in the Staudinger ligation due to oxidation.154
In the following sections, the synthesis of [18F]fluoroethyl azide and deoxy-[18F]fluoroglucopyranosyl azides as well as their application in PET tracer syntheses will be discussed.
Hugenberg et al. described distillation during the reaction time to increase the non decay-corrected yield by shortening the synthesis time.160 However, none of the modifications of the distillation procedure described above led to a significant increase in radiochemical yield of 188. Kelly et al. found that addition of more MeCN to the reaction vial during distillation did increase the efficiency, but the higher MeCN content in the purified building block solution led to lower yields in the subsequent click reaction.161 This was also reported by other research groups.149
Next to the flow-and-trap-distillation method, a vacuum distillation method has been developed by Zhou et al. They reported radiochemical yields of over 80% (dc) within 10 minutes including formation of [18F]fluoroethyl azide, distillation into a dry ice cooled trapping vial and warming up to room temperature.162 However, due to co-distillation of the side-product vinyl azide, the precursor for the follow-up reaction was needed in large excess. This makes the method unsuitable for the high specific activity labelling of macromolecules due to the pseudo-carrier present.
Furthermore, two different cartridge purification procedures have been developed in order to facilitate automation. Bejot et al. and Carroll et al. employed a polyfluorinated sulfonate precursor instead of 2-azidoethyl 4-toluenesulfonate (Scheme 41), which could be separated from [18F]fluoroethyl azide 188 by fluorous solid phase extraction (FSPE).151,163 Another approach used a silica-based C18 cartridge and a Water Oasis HLB cartridge in series.149
For some tracer syntheses, successful one-pot procedures have been described.164,165 However, the one-pot method often promotes side-reactions, which necessitates elaborate purification of the PET tracer. Automated syntheses have been developed as well. Ackermann et al., for example, reported an automated synthesis on the Flex Lab module including purification by vacuum distillation.166
Scheme 42 lists all PET tracers synthesised using the [18F]fluoroethyl azide building block via the CuAAC method.55,148,149,151,156–160,163,164,166–180 In most cases, the [18F]fluoroethyl azide reacts with an alkyne precursor in the presence of a Cu(I)- or Cu(II)-catalyst and sodium ascorbate as reducing agent. Copper sulfate was used in the majority of the tracer syntheses reported. Here, Cu(II) is reduced by sodium ascorbate to the reactive Cu(I) species.158,164,180 In addition, the use of Cu(II) acetate171 as well as Cu(I) iodide has been reported. Although when Cu(I) iodide is employed the catalyst is already present in its active species, sodium ascorbate is still used since oxidation from Cu(I) to Cu(II) during the reaction is well known.167,172 Ackermann et al. reported that with a freshly prepared mixture of Cu(I) iodide and sodium ascorbate, better results in the labelling of the tumour cell proliferation imaging agent 207 were obtained than with the conventional CuSO4/Na-ascorbate system.167 In another article, Ackermann et al. investigated Cu(CH3CN)4PF6 as a catalyst system, because it is soluble in organic solvent and would be more compatible for use in an automated synthesis module. Unfortunately, a lower yield of the tracer 207 was observed with the new catalyst system.166
 | ||
| Scheme 42 PET tracers synthesised from [18F]fluoroethyl azide in a CuAAC.55,148,149,151,156–160,163,164,166–180 | ||
Different solvent systems have been used in the CuAAC. Often water or an aqueous buffer solution such as phosphate buffer is used to dissolve the copper catalyst and the sodium ascorbate.157,161 On the other hand, the alkyne precursor is mostly added in DMF, but also MeCN, DMSO and aqueous media were used.148,161,167,175 Depending on the applied purification procedure, the azide building block is added either in the distillation trapping solution or in the solvent eluted from the intermediate SPE purification cartridge.171,173 The MeCN from co-distillation or trapping led in some cases to a decrease in coupling efficiency.178 For the prostate-specific membrane antigen (PSMA) tracers 222 and 223, higher MeCN content led to a decrease in yield from 50% to <25% (measured by radio-HPLC).161 However, a few successful click reactions were also performed in MeCN.55,167 The presence of DMF has been described as a necessary condition to maintain the level of Cu(I) in the reaction solution.162
Other additives have been employed in some of the tracer syntheses too. The use of the base DIPEA172 as well as the Cu(I) stabilizing agents TBTA (tris(benzyltriazolylmethyl)amine) and BPDS (bathophenanthroline disulfonic acid disodium salt) have been reported.168,179 TBTA and BPDS served as auxiliary copper(I) chelators164 and accelerated the reaction. They were found to be especially helpful in one-pot strategies, for example in the synthesis of imaging agents 217–220 for the multidrug resistance-associated protein 1.164 However, the presence of BPDS or TBTA was not necessary for successful one-pot synthesis. Chen et al. demonstrated this in the synthesis of PSMA inhibitor 221 without ligand in an overall radiochemical yield of 14 ± 1% (ndc).
Most of the PET tracers shown in Scheme 42 were purified by semi-preparative HPLC, but purification was not always successful. In some cases, the alkyne precursor could not be separated from the product, leading to low specific activities.174,177 For the tumour proliferation imaging agents 202a and 202b, direct labelling was more successful giving increased specific activities (20–210 GBq μmol−1 instead of 0.3 GBq μmol−1) at comparable overall radiochemical yields (7% ndc).174
Based on the CuAAC with [18F]fluoroethyl azide, a “multi-click” approach has been developed for the synthesis of the apoptosis tracers 227b–d. Up to four different alkynes were synthesised in one pot from one batch of [18F]fluoroethyl azide at the same time. This makes the CuAAC a valuable tool for the screening of potential PET tracers. However, the purification of the reaction mixtures turned out to be challenging and tracers with low purity were obtained, because the alkyne precursors could not be separated from the labelled compounds.150,180
Carroll et al. presented the first examples (compounds 229–232) of fluorine-18 labelled compounds synthesised by traceless Staudinger ligation with [18F]fluoroethyl azide (Scheme 43). The reaction was carried out either in a mixture of tetrahydrofuran (THF) and water or DMF and water, at 80 °C for 30 minutes or at 120 °C for 15 minutes. The labelled compounds could be obtained in radiochemical yields of >95% (analytically determined).152 Gaeta et al. were the first to synthesise and isolate a PET tracer (228) via this method. Labelling proceeded in a mixture of DMF and MeCN within 15 minutes at 130 °C and provided the GABAA tracer 228 in an overall radiochemical yield of 7% (ndc).153
 | ||
| Scheme 43 PET tracers synthesised from [18F]fluoroethyl azide in a traceless Staudinger reaction.152,153 | ||
In conclusion, [18F]fluoroethyl azide is a useful building block for the fluorine-18 labelling of biomolecules and small molecule PET tracers under mild conditions without need of protecting groups.
2-Deoxy-2-[18F]fluoroglucopyranosyl azide can be prepared in a two-step procedure starting from the mannosyl precursor 233 (Scheme 44). In the first step, nucleophilic substitution of the triflate group with [18F][K⊂K2.2.2]F was conducted at 85 °C for 5 minutes. HPLC purification provided the acetyl-protected product 234 in a radiochemical yield of 67% (ndc). In the second step, deprotection of the hydroxyl groups was carried out by addition of aqueous sodium hydroxide at 60 °C. After complete deacetylation, the solution was neutralised with hydrogen chloride solution and directly used in the CuAAC reaction.182 Only minor alterations since the initially developed synthesis of the building block have been reported. Fischer et al. showed that a cartridge based purification procedure instead of a time consuming HPLC purification yielded 75% of protected 2-deoxy-2-[18F]fluoroglucopyranosyl azide 234.183
 | ||
| Scheme 44 Synthesis of 2-deoxy-2-[18F]fluoroglucopyranosyl azide with subsequent Cu-catalysed click reaction. | ||
Maschauer et al. described also the synthesis of 6-deoxy-6-[18F]fluoroglucopyranosyl azide, analogous to the synthesis of 2-deoxy-2-[18F]fluoroglucopyranosyl azide 235, obtained from its tosyl precursor. They found that addition of all reactants in buffered solution made neutralisation prior to the coupling reaction unnecessary.184
The coupling of building block 234 to the alkyne precursors was carried out in a CuAAC reaction, resulting in the PET tracers which are shown in Fig. 7.171,181,183,185–188 The CuAAC proceeded in a one-pot two-step reaction together with the deprotection of 234. After deacylation and neutralisation, 2-deoxy-2-[18F]fluoroglucopyranosyl azide 235 was reacted with the corresponding alkyne precursor in an aqueous solution of copper(II)acetate or sulfate and sodium ascorbate. The desired product was formed in 10 to 15 minutes at slightly elevated temperatures (50–60 °C). Typically, the products were purified by semi-preparative HPLC and obtained in overall radiochemical yields of 1–40% (dc) in a total reaction time of 70–180 minutes. Several improvements to the original reaction conditions have been published by Maschauer et al. They described a significant increase of product formation in the presence of ethanol in the aqueous reaction solution.184 Furthermore, THPTA (tris(3-hydroxypropyltriazolylmethyl)amine) and BPDS (bathophenanthroline disulfonic acid disodium salt) were presented as agents accelerating the CuAAC reaction. The use of BPDS enables click reactions in 5 minutes at room temperature.185
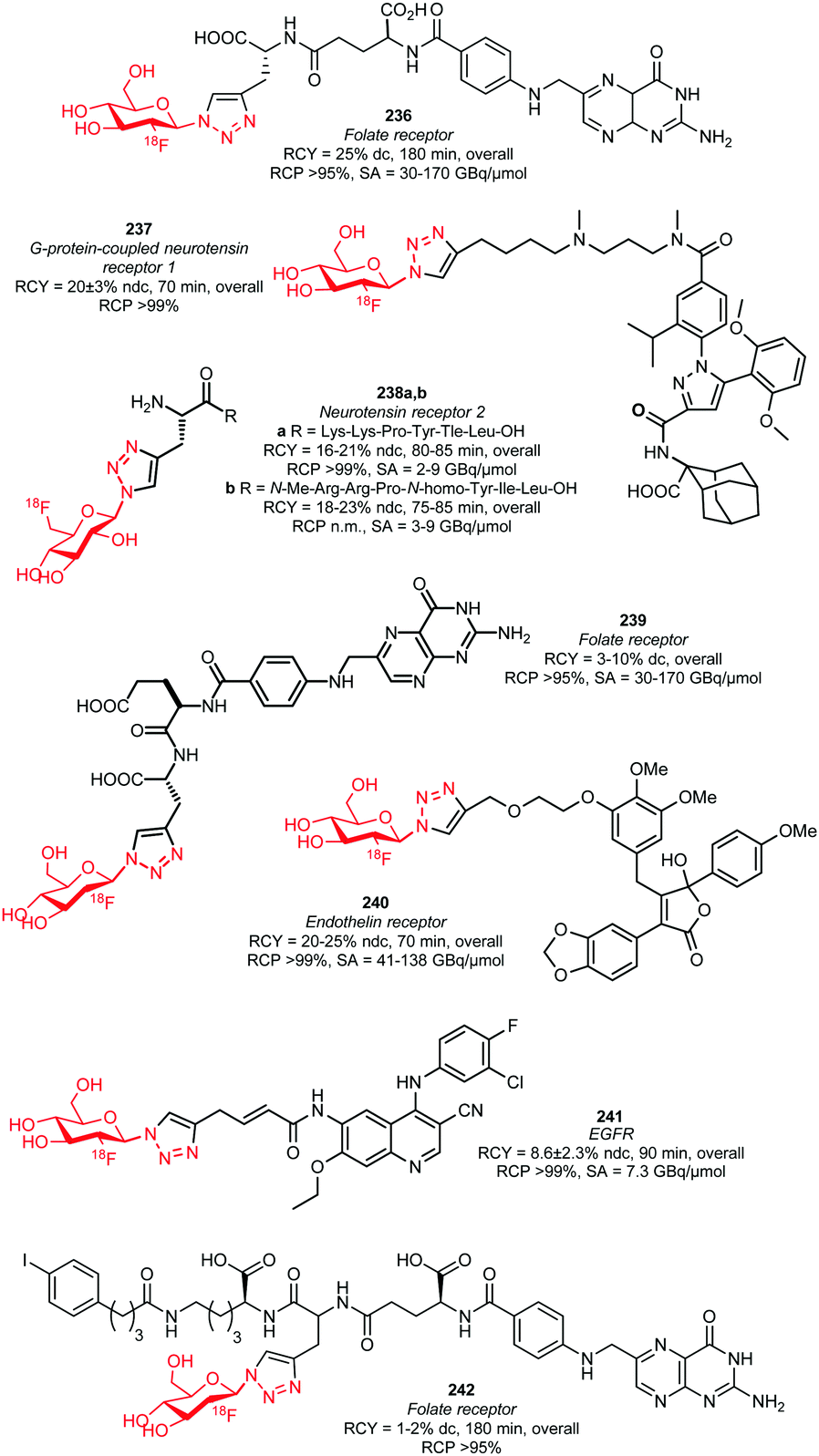 | ||
| Fig. 7 PET tracers labelled with 2-deoxy-2-[18F]fluoroglucopyranosyl azide.171,181,183–188 | ||
To conclude, 2-deoxy-2-[18F]fluoroglucopyranosyl azide 235 is a useful building block for fluorine-18 labelling and proved to be especially useful for labelling peptides. It has several advantages: the glucosyl moiety enhances, due to its hydrophilicity, the in vivo properties of the PET tracer and the CuAAC is a reliable and efficient procedure with high regioselectivity.171,183
2.7 18F-Labelled alkynes
A large range of different fluorine-18 labelled alkynes have been used in PET tracer synthesis. Scheme 45 summarises the different types of precursor used since 2010. Alkyl precursors 243 and polyethylene glycol (PEG) derived precursors 245 are popular precursors for radiofluorination and have been applied many times. By varying the chain length these precursors can be easily adapted to specific requirements without the need to change the labelling procedure significantly.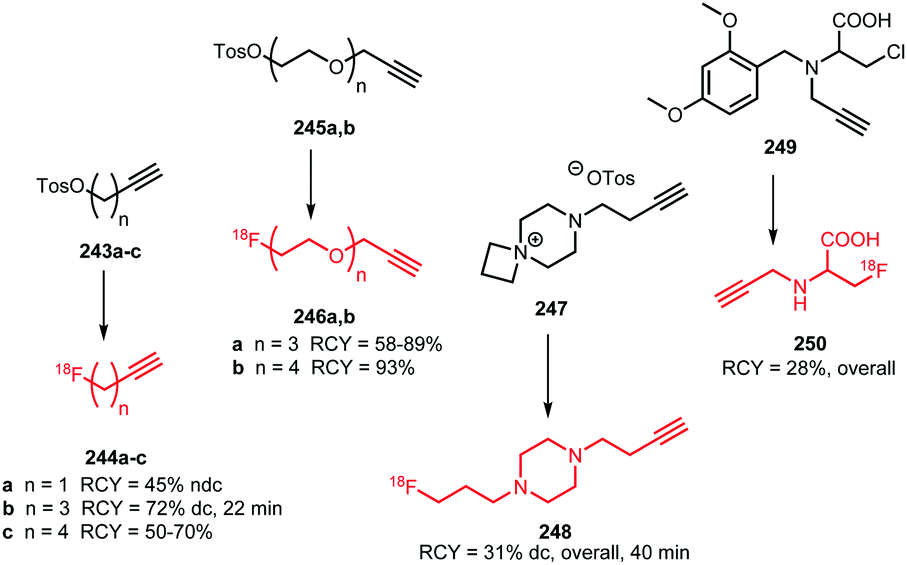 | ||
| Scheme 45 Different types of alkyne precursors. | ||
Small fluorine-18 labelled alkynes such as 244 have relatively low boiling points, which is advantageous for distillation, avoiding time-consuming preparative HPLC purification procedures. In addition, they have little influence on the (bio)chemical properties of the developed PET tracer.
Typical reaction temperatures are between 95–110 °C and distillation is quite fast (2–5 minutes). The solvent used for trapping the product after distillation may depend on the click reaction which is performed afterwards. As a leaving group, predominantly the tosylate is used, as it provides in general the best results in a nucleophilic substitution.171,189,190 The same applies for the PEG derived precursors 245. In contrast to alkyl derived fluorine-18 labelled alkynes, PEG derived fluorine-18 labelled alkynes show low volatility which simplifies handling but makes preparative HPLC purification necessary in most cases. Their amphiphilicity makes them ideal reactants in the CuAAC reaction. Nucleophilic substitution was performed at temperatures between 110 and 140 °C in MeCN or DMSO for 10 to 15 minutes. High radiochemical yields could be obtained, ranging from 58 to 93%.191–193
Other alkyne building blocks have been developed as well. The piperazine based building block 248 was chosen because of its high hydrophilicity compared to the alkyl derived fluorine-18 labelled alkynes, facilitating the radiofluorination of peptides in aqueous conditions. It was synthesised from spiro precursor 247, which could be easily separated from the resulting product 248 using reversed phase C-18 or silica gel cartridges.194 Building block 249 on the other hand is an amino acid derivative of alanine, functionalised with the alkyne moiety at the N-terminus. It is also a convenient prosthetic group for radiofluorination of biomolecules based on amino acids. The tosylated precursor however showed poor stability during purification, hence the chlorinated precursor was used yielding the product in 28 ± 5% (RCY, overall).195
Scheme 46 shows that a large set of structurally diverse PET tracers can be prepared by the application of fluorine-18 labelled alkynes in the CuAAC reaction.171,189–195 Nonetheless, labelling conditions for all click reactions are comparable. The Cu(I) catalyst in the 1,3-dipolar cycloaddition, was in every case generated in situ from a Cu(II) salt by reduction with sodium ascorbate. Attempts to directly employ the active Cu(I) species led to significant precursor degradation and poor radiochemical yields.191 Mostly, aqueous solutions of the salts were combined with polar aprotic organic solvents like MeCN or DMF containing the radiolabelled building block and/or precursor. Temperatures up to 110 °C have been reported, but in general the CuAAC proceeds under mild temperatures of 20 to 40 °C. In cases of elevated temperatures, microwave heating was shown to be more efficient than conventional heating.191,193
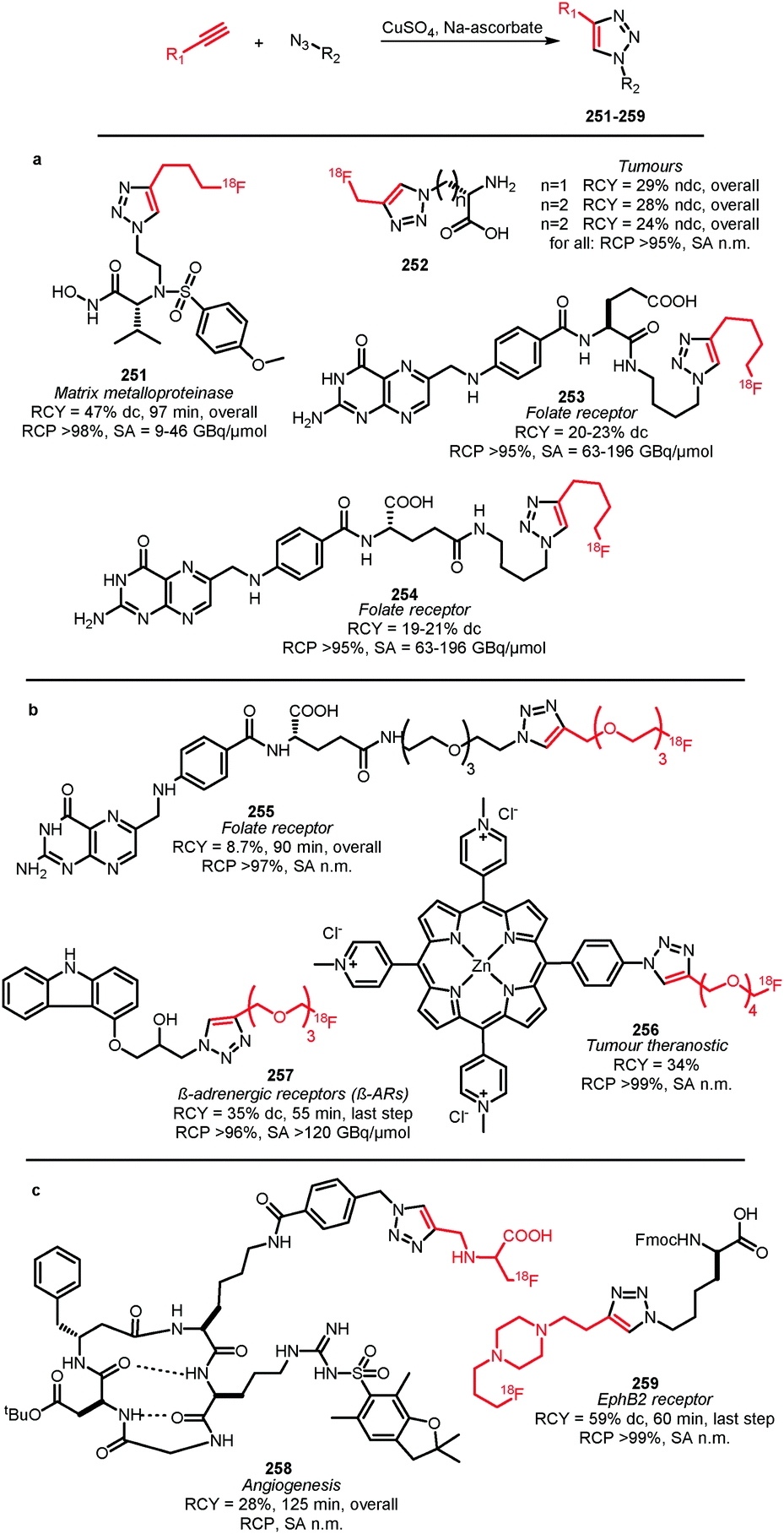 | ||
| Scheme 46 PET tracers synthesised via click reaction with different fluorine-18 labelled alkynes: (a) alkyl-derived building blocks;171,189,190 (b) PEG-derived building blocks;191–193 (c) others.194,195 | ||
Purification was usually carried out by (semi-)preparative HPLC. However, Yook and co-workers reported a synthesis procedure using an additional, more lipophilic alkyne to react with residual precursor and increase its lipophilicity to allow for separation of the tracer 252 by a SPE purification procedure.189
In summary, many different fluorine-18 labelled alkynes have been successfully employed as building blocks in CuAAC reactions for PET tracer syntheses. Due to their structural diversity, the use of fluorine-18 labelled alkynes significantly expand the spectrum of PET tracers that can be accessed.
2.8 Fluorine-18 labelled alkyl amines
[18F]Fluoroalkyl amines such as [18F]fluoroethyl amine but also quite complex molecules like 267 (Scheme 48) have been used as small versatile building blocks for radiofluorination. They can be introduced into the precursor by amide, carbamate and urea formation, functional groups that are often present in biomolecules.Two general methods for the synthesis of [18F]fluoroalkyl amines have been reported: one uses phthalimide protected alkyl amines such as 260 as precursor for [18F]fluorination (Scheme 47) whereas the other method employs Boc-protected alkyl amines 263 (Scheme 48).
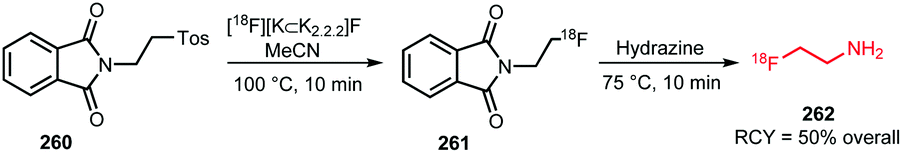 | ||
| Scheme 47 Synthesis of [18F]fluoroethylamine via the procedure of Tewson et al.195 | ||
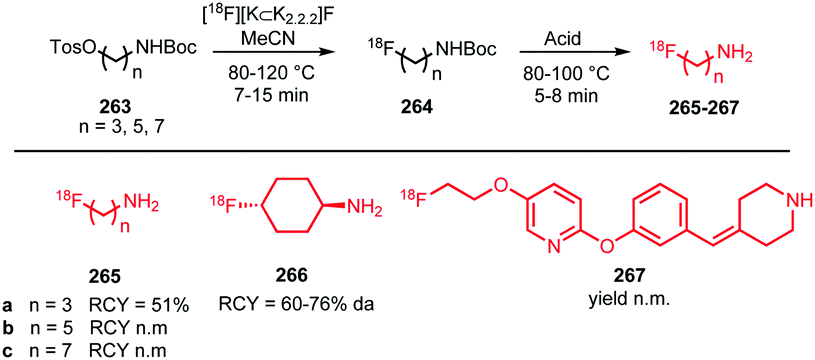 | ||
| Scheme 48 Synthesis of different types of [18F]fluoroalkyl amines via Boc-protected amines.200,202 | ||
The method using phthalimide protected amines as starting materials is based on a Gabriel reaction and was first published by Tewson and co-workers in 1997. Scheme 47 shows the 2-step synthesis of [18F]fluoroethyl amine 262. The intermediate phthalimide protected aminoethyl tosylate 260 is obtained by reaction of phthalimide with 2-bromoethanol followed by tosylation. Subsequent radiofluorination at 100 °C for 10 minutes in MeCN, followed by deprotection with hydrazine, gave [18F]fluoroethyl amine 262, which was purified by simultaneous distillation into a second reaction vessel. Although the labelling strategy itself is quite straightforward, the deprotection step proved quite complex and several reaction parameters required careful examination. Especially, the presence of water appeared mandatory for the reaction. A complicating factor was that the MeCN, which was left behind from the previous reaction step, led to azeotropic evaporation of the water. Therefore, the MeCN had to be evaporated until dryness before hydrated hydrazine was added. Furthermore, to avoid co-distillation of hydrazine, the reaction temperatures could not exceed 75 °C and the reaction time was kept between 10 and 15 minutes.196
The original procedure of Tewson et al. is still largely employed as it was first published.197,198 For example, Huang and co-workers applied it to the synthesis of [18F]fluorooctyl amine. Due to the high boiling point, purification by HPLC was explored but neither normal nor reversed phase HPLC gave satisfactory pure product. However, a radiochemical yield of 53% (dc, based on radio-HPLC analysis) was observed, which is consistent with the yields reported for [18F]fluoroethyl amine 262.199
The second method to synthesise [18F]fluoroalkyl amines starts from the corresponding Boc-protected amino tosylates (Scheme 48). [18F]Fluorination is performed in MeCN at 80–120 °C for 7–15 minutes. Radiochemical yields up to 80% (analytically determined) have been reported using this strategy. Subsequent deprotection is performed under acidic conditions at temperatures of 80 to 100 °C. Sulfuric acid as well as trifluoroacetic acid have been employed, both resulting in high conversions. After neutralisation of the sulfuric acid with phosphate buffer or evaporation of trifluoroacetic acid, the [18F]fluoroalkyl amine could be used without further purification in the next reaction.200–203 A range of structurally diverse amines have been radiofluorinated applying this method (Scheme 48). Apart from primary amines with linear alkyl chains, the cyclic primary amine 266 and the more complex secondary amine 267 were labelled in this manner with fluorine-18.201,203
In addition to the above discussed more generally established methods to access [18F]fluoroalkyl amines, Glaser et al. reported an alternative method. They described the reduction of [18F]fluoroethyl azide (188) with elemental copper under acidic conditions providing [18F]fluoroethyl amine (262) in radiochemical yields of over 90% (analytically determined).204
[18F]Fluoroalkyl amines can undergo three different types of reactions forming either carbamates, amide bonds, or carbamines. Carbamate formation is most widely applied (Fig. 8).146,198,200,201,205 The radiofluorination reaction was carried out using the purified (distilled) fluorine-18 labelled building block, but also successful one-pot three step reactions including [18F]fluorination and deprotection of the amine have been reported. In most syntheses, the carbamate formation was carried out at room temperature without additives. Antunes and co-workers prepared 268, a PET tracer for imaging glucuronidase activity, in an overall radiochemical yield of 13% (dc) including deprotection of the product with 2 M sodium hydroxide solution, while trapped on a tC18 cartridge.146 Zhang and co-workers used an additional base (triethylamine, TEA) in the synthesis of the tumour hypoxia imaging agent 269, which was obtained in a radiochemical yield of 48% (dc, last step).198 Sadovski et al. reported that a temperature of 80 °C is optimal to obtain 271, a radiotracer for fatty acid amide hydrolase imaging, with a radiochemical yield of 17–22% (ndc, overall).200 In all cases, purification of the products was conducted by semi-preparative HPLC.
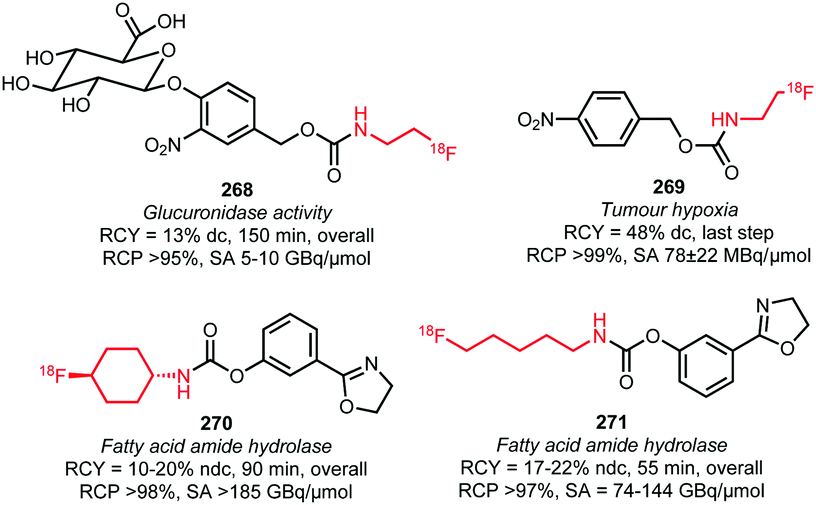 | ||
| Fig. 8 [18F]Fluorocarbamates synthesised via reaction with [18F]fluoroalkyl amines.146,198,200,201,205 | ||
Sawatzky et al. reported the use of [18F]fluoroheptyl amine 263c as building block in the multi-step synthesis of the butyrylcholinesterase tracer 274. To activate [18F]fluoroheptyl amine 263c, it was first treated with 4-nitrophenyl chloroformate in MeCN under basic conditions resulting in carbamate 273. Subsequently, 273 was coupled with the phenol to obtain the butyrylcholinesterase tracer 274 in a radiochemical yield of 18–22% (dc) (Scheme 49).207
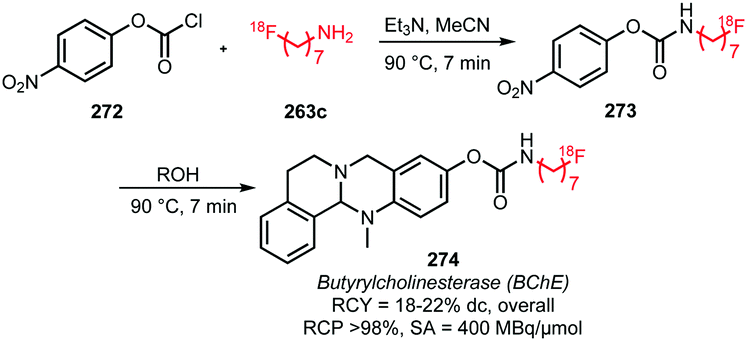 | ||
| Scheme 49 Synthesis of the butyrylcholinesterase tracer 274.206 | ||
For the synthesis of amides from [18F]fluoroalkyl amines, Silvers et al. described the use of an acyl chloride precursor, which was reacted with [18F]fluoropropylamine in MeCN under basic conditions (TEA) for 14 minutes at room temperature. This provided stearoyl-CoA desaturase-1 tracer 275 in an overall radiochemical yield of 21% (dc) (Fig. 9).203 Huang et al. reported amide formation starting from the carboxylic acid, using the coupling reagent 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) in the presence of DIPEA as base in DMF. The reaction was carried out at room temperature and yielded cyclooxygenase-2 (COX-2) inhibitor 276 in 4% (dc).199 In a third approach for amide bond formation, 9H-β-carboline pentafluorophenyl ester was used as a precursor. One-pot reaction at 80 °C with 2-[18F]fluoroethyl azide-derived [18F]fluoroethylamine under basic conditions (TEA) gave GABAA tracer 277 in 46% decay-corrected radiochemical yield (calculated from 2-[18F]fluoroethyl azide).205
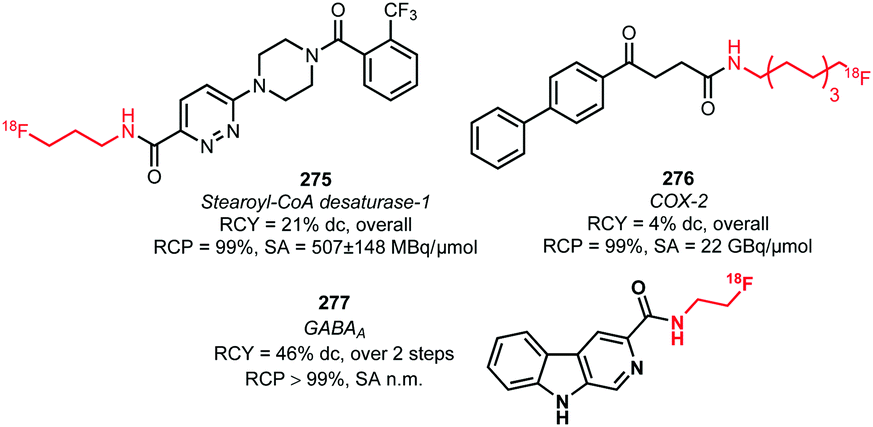 | ||
| Fig. 9 [18F]Fluoroamides.198,201,204 | ||
Since 2010 the synthesis of two fluorine-18 labelled urea derivatives has been reported. In the procedure described by Majo et al., the potential PET ligand for mTOR 278 was synthesised from an amine precursor, which was pre-treated with trisphosgene and TEA in dichloromethane. The building block [18F]fluoroethyl amine was directly distilled into the solution containing the pre-treated precursor. A radiochemical yield of 15% (dc, overall) was achieved.197 Skaddan and co-workers performed a one-pot synthesis, obtaining 279 from the corresponding carbamate precursor by reaction in MeCN under basic conditions (DIPEA) at 80 °C in a radiochemical yield of 10% (dc, overall) (Fig. 10).
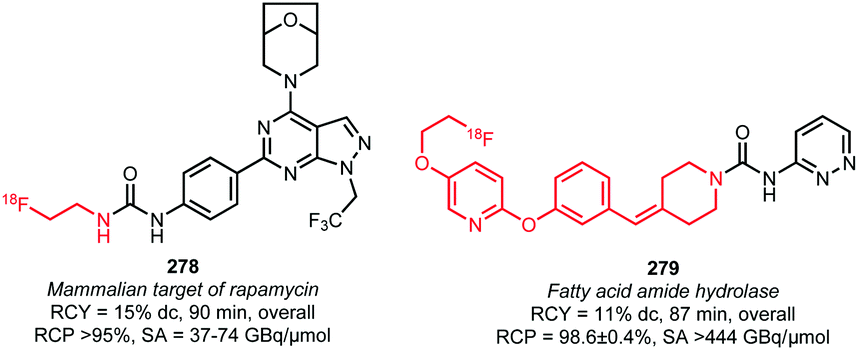 | ||
| Fig. 10 Fluorine-18 labelled urea derivatives.197,203 | ||
2.9 [18F]FDG
[18F]FDG is the most applied radiopharmaceutical for PET imaging and serves as generic tumour tracer. Because of its widespread use and availability in almost every PET centre, as well as its favourable pharmacokinetics, several attempts have been made to employ [18F]FDG not only as tracer, but also as a building block for other PET tracers. As such, it could enable convenient radiolabelling in a one-step synthesis starting from [18F]FDG.207 As [18F]FDG is readily available, the synthesis of [18F]FDG will not be discussed in this review.When amines are reacted with [18F]FDG, glycosylamines are formed, which are biochemically important for many metabolic pathways. The mechanism of the reaction between [18F]FDG as building block and an amine, is based on the Maillard reaction (Scheme 50). In a Maillard reaction, an amine reacts with the aldehyde of glucose at the 1-position to form Schiff base 282 after elimination of water. After that, the Schiff base can rearrange to the Amadori product 283 leading to ketone formation at the 2′-position. As [18F]FDG contains a fluorine atom instead of a hydroxyl group at the 2-position, it cannot undergo rearrangement to the Amadori product. Thus, in this case the Maillard reaction is blocked at the stage of Schiff base 282, which is also called a quasi-Amadori product.208
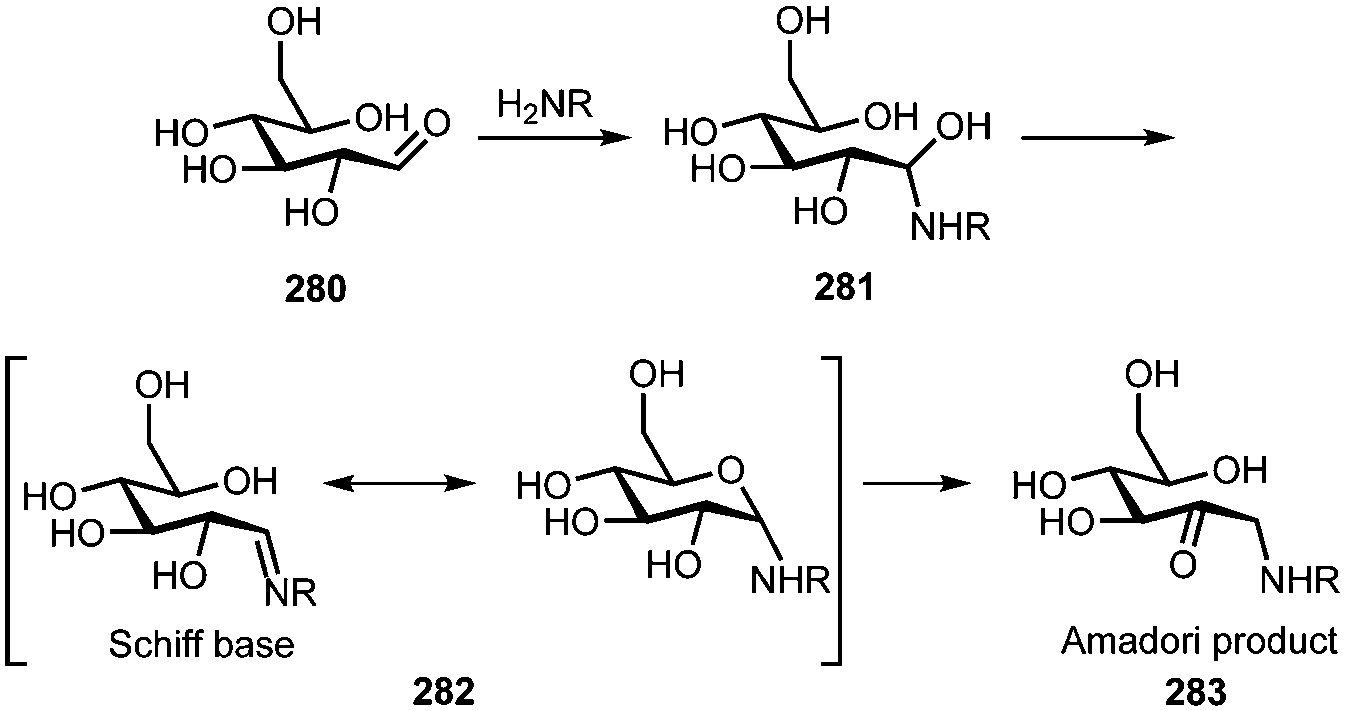 | ||
| Scheme 50 Maillard reaction.208 | ||
An overview of tracers radiolabelled with [18F]FDG is given in Fig. 11.207–212 The tracers 284–291 were synthesised from the corresponding amine precursor whereas tracers 292–296 were synthesised from oxy-amine precursors. The [18F]FDG building block was employed either in solution (saline or PBS) or azeotropically dried with MeCN prior to use.207,210
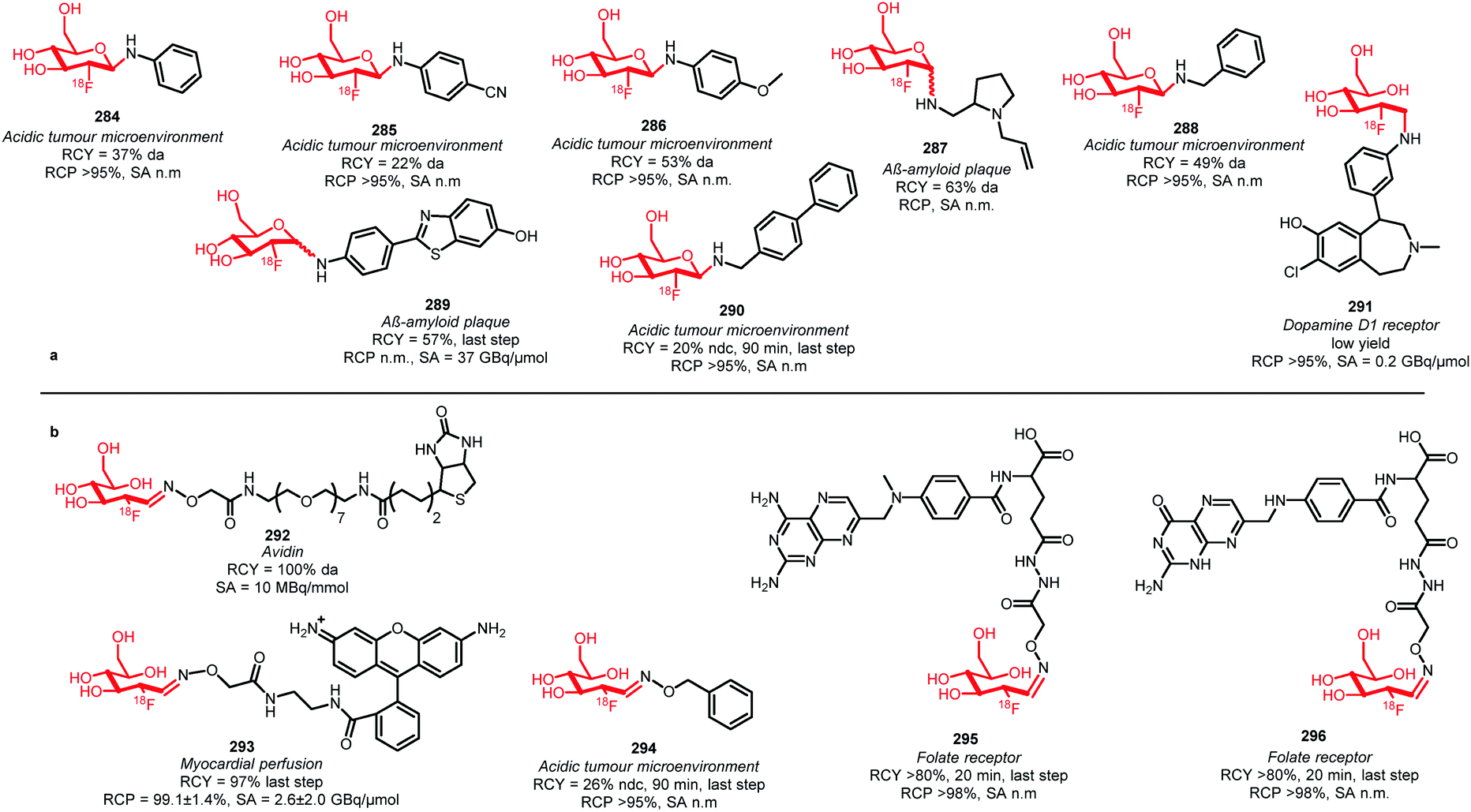 | ||
| Fig. 11 PET tracers synthesised from [18F]FDG and (a) amine precursors, (b) oxy-amine precursors.207–212 | ||
As solvent, the use of methanol, ethanol, DMSO and mixtures of them with water have been described. Furthermore, acetic acid was present in all reactions mentioned. Reaction temperatures varied between 60–99 °C with reaction times varying between 10–120 minutes.
In the reactions with [18F]FDG as labelling reagent, aniline can serve as catalyst forming [18F]FDG–aniline as an intermediate. Flavell and co-workers could shorten reaction times from 30 to 1 minute using aniline, whereas Baranwal et al. were able to perform the reactions at room temperature (instead of 99 °C) when aniline was present.207,208
Usually the tracers synthesised from [18F]FDG needed purification and a range of methods to achieve this have been applied. However, it is noteworthy that Al Jammaz and co-workers obtained the PET myocardial perfusion imaging agent 293 in a radiochemical yield of 97% by just simply passing the reaction mixture through a membrane filter, resulting in >98% radiochemical purity.212 The in vitro stability of the glycosyl amine tracers are in general high.210,211 However, at low pH, increased decomposition of the [18F]FDG amines was observed whereas the [18F]FDG oximes were stable towards hydrolysis under these conditions. Flavell and co-workers took advantage of this characteristic and designed acid-labile pro-drug tracers 284–286, 288 and 290 for imaging acidic tumour microenvironments.207
In summary, because of its commercial availability, [18F]FDG is a very easily accessible building block and allows mild radiofluorination. Valuable PET tracers have been obtained using [18F]FDG. Though, due to its size and hydrophilicity it can have a significant influence on the pharmacokinetics and the targeting of the resulting PET tracer.
2.10 5-[18F]Fluoro-5-deoxyribose
Initially developed as PET tracer for tumour imaging,213,214 5-[18F]fluoro-5-deoxyribose has also found application as a building block. Besides in peptide radiolabelling,215 it has been used to prepare tetrazine 301, a fluorine-18 labelled compound for pre-targeted PET imaging. The synthesis of the fluorinated tetrazine from an 18F-building block was necessary, because tetrazines are unstable under commonly used direct radiofluorination conditions. 5-[18F]Fluoro-5-deoxyribose can be used under mild conditions and in addition, the 5-[18F]fluoro-5-deoxyribose moiety contributes positively to the overall hydrophilicity of the PET tracer, reducing unspecific binding.2165-[18F]Fluoro-5-deoxyribose 299 could be obtained in a two-step procedure (Scheme 51). First, the protected tosylate precursor 297 was radiofluorinated by nucleophilic substitution in MeCN at 108 °C. Intermediate 298 was purified by semi-preparative HPLC and then deprotected with hydrochloric acid. After neutralisation, the obtained 5-[18F]fluoro-5-deoxyribose 299 was used without further purification.
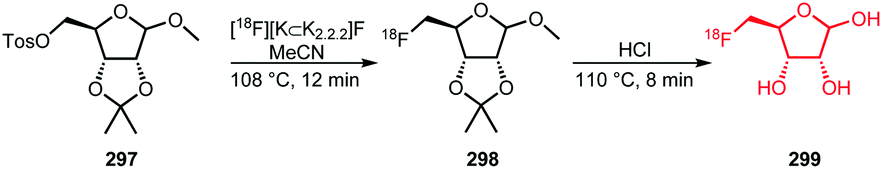 | ||
| Scheme 51 Synthesis of 5-[18F]fluoro-5-deoxyribose 299. | ||
Another elegant way to synthesise 5-[18F]fluoro-5-deoxyribose was reported by Onega and co-workers. They converted S-adenosyl-L-methionine (SAM) and [18F]fluoride in a two-step enzymatic reaction using fluorinase and adenosine hydrolase to access the product. Radiochemical yields of 75–98% (determined by radio-HPLC) were achieved and the overall synthesis time was 100–120 minutes.217
Furthermore, 5-[18F]fluoro-5-deoxyribose 299 was conjugated with the amino-oxy functionalised tetrazine 300 by oxime ether formation (Scheme 52). The reaction was carried out in anilinium acetate buffer (pH 4.6). After 10 minutes at room temperature, product 301 was purified by semi-preparative HPLC and obtained in an overall radiochemical yield of 50.5 ± 1.7% (dc).216
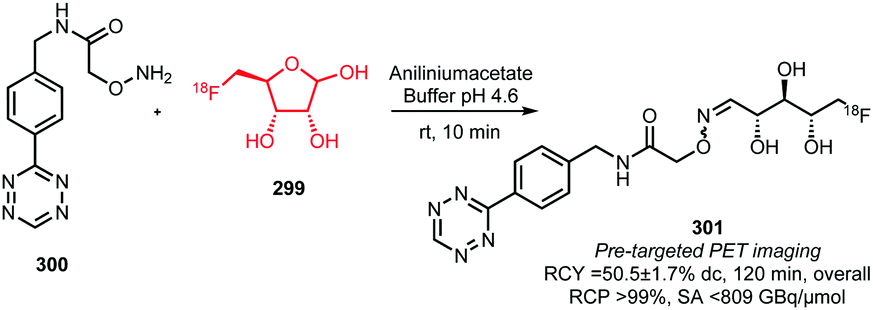 | ||
| Scheme 52 Synthesis of compound 301 by reaction of 5-[18F]fluoro-5-deoxyribose 299 with tetrazine 300.216 | ||
In conclusion, the amino-oxy functionalised tetrazine 300 could be labelled fast and in high overall yields, proving the suitability of 5-[18F]fluoro-5-deoxyribose 299 as building block for PET tracer synthesis. In its function as building block, 5-[18F]fluoro-5-deoxyribose may serve as a valuable alternative to [18F]FDG.
2.11 2-Deoxy-2-[18F]fluoroarabinofuranose
Derivatives of 2′-[18F]fluoro-2′-deoxy-1-β-D-arabinofurano-syluracil ([18F]FXAU) 307 can act as potential PET tracers to image the expression of the herpes simplex virus type-1 thymidine kinase (Scheme 53). As direct radiofluorination at the 2′-position of the sugar moiety provides extremely low yields (<1%), and therefore does not allow for routine clinical production, many efforts have been made to develop a high-yielding multi-step synthesis based on the building block 2-deoxy-2-[18F]fluoroarabinofuranose 303.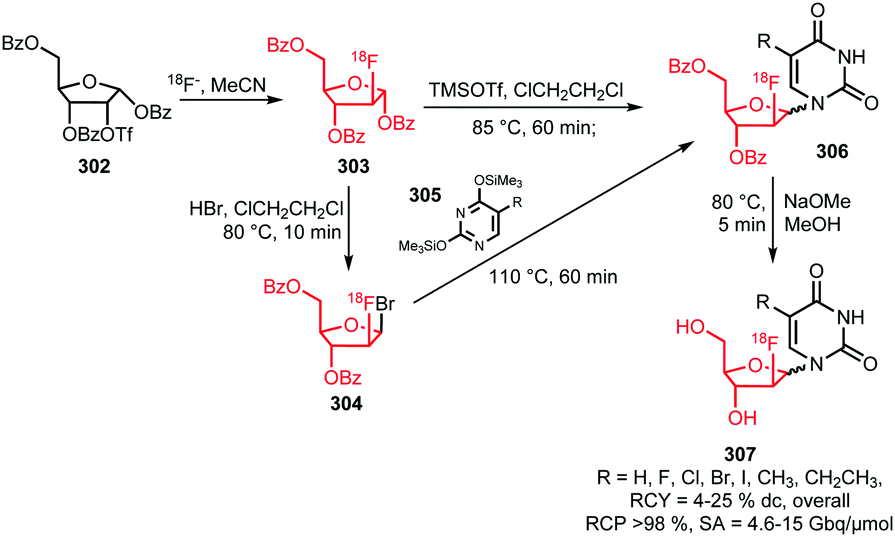 | ||
| Scheme 53 Synthesis of [18F]FXAU (307).218,220 | ||
Formation of the building block [18F]2-deoxy-2-fluoroarabinofuranose 303 is in all reported procedures based on the same strategy. In that strategy, the benzyl protected ribose triflate 302 is treated during 20 to 30 min with dried [18F]fluoride in MeCN at 80–90 °C. Alauddin and co-workers reported radiochemical yields of 58–68% for 303 and a radiochemical purity of >90% after passing the reaction mixture over a silica Sep-Pak cartridge to remove unreacted [18F]fluoride.218 However, depending on the follow-up chemistry, purification of 303 was not always required. Manual as well as automated synthesis procedures on conventional synthesis units and microfluidic devices have been performed.219,220
Two different synthesis procedures for [18F]FXAU 307 starting from building block [18F]2-deoxy-2-fluoroarabinofuranose 303 have been developed (Scheme 53). The first method, reported by Alauddin and co-workers in 2002, includes the formation of an α-bromo derivative 304 to promote β-selective coupling to the uracil moiety. For the conversion of the 1-benzoxy group of 303 to the corresponding bromide 304, hydrogen bromide in acetic acid is used. However, the highly corrosive hydrogen bromide makes this step challenging to automate. The building block was subsequently coupled to a silyl precursor 305 in a non-polar solvent which induced product formation in a favourable anomeric ratio of α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β = 1:3–1:9.218,221 The reaction time of 60 minutes was rather long, and led to low overall yields.222
:β = 1:3–1:9.218,221 The reaction time of 60 minutes was rather long, and led to low overall yields.222
Although this method is described as very reliable, major disadvantages such as the use of corrosive hydrogen bromide make it not amenable for clinical production. Therefore, an alternative method suitable for automated synthesis of [18F]FXAU has been developed, which employs trimethylsilyl trifluoromethanesulfonate (TMSOTf) as Friedel Crafts catalyst in the coupling reaction that enables direct coupling between building block 303 and silyl precursor 305. Building block 303 could be employed without purification and added to the in situ generated 305. The reaction optimally performs at 85 °C. No coupling could be observed at lower temperatures while higher temperatures led to decomposition.220
In a final step, 306 was deprotected with potassium methoxide in methanol. The product was obtained after neutralisation with HCl and subsequent purification by semi-preparative HPLC. For the 3-step reaction route, radiochemical yields of 10–12% (dc) have been reported, with synthesis times of 114–150 min. The relatively low yields can be attributed to poor α/β anomer selectivity (6:4 instead of 1:4).219,220,223 Furthermore, the product has a poor specific activity of 5 GBq μmol−1, probably caused by the excess of TMSOTf.219
To further improve the synthesis route, Chen et al. have investigated the influence of microwave heating and Lewis acid catalysis on the coupling reaction. Microwave heating can have a positive influence on the coupling reaction by reducing the reaction time from 1 hour to 10 minutes, delivering the product in a radiochemical yield of 20% (dc). However, currently this microwave approach is only suitable for manual production.224
Application of the Lewis acid catalyst SnCl4 shortens the reaction time of the coupling reaction to 15 minutes while it is compatible with automation. Though, neither microwave heating nor the use of SnCl4 influenced the anomeric ratio positively.222
In conclusion, synthesis of the building block 2-deoxy-2-[18F]fluoroarabinofuranose is straightforward, but the anomeric C-1 atom makes the follow-up chemistry challenging. So far, the scope of the building block is limited to the synthesis of 2′-[18F]fluoro-2′-deoxy-1-β-D-arabinofuranosyl-uracil and -thymidine derivatives.
2.12 2′-Deoxy-2′-[18F]fluorothymidine
2′-Deoxy-2′-[18F]fluorothymidine ([18F]FLT) has been applied only once as a building block. Carroll and co-workers reported on the synthesis of a prodrug-like tracer in 2014, PC-[18F]FLT 310a, for H2O2 detection and tumour imaging. Firstly, [18F]FLT was synthesised according to an established procedure. Thereafter it was reacted with imidazole ester precursor 309 (Scheme 54) in the presence of dimethylaminopyridine (DMAP) and TEA. After deprotection with citric acid, the product was purified by semi-preparative HPLC to afford PC-[18F]FLT 310a and CC-[18F]FLT 310b in a radiochemical yield of 41% and 44%, respectively, starting from [18F]FLT.225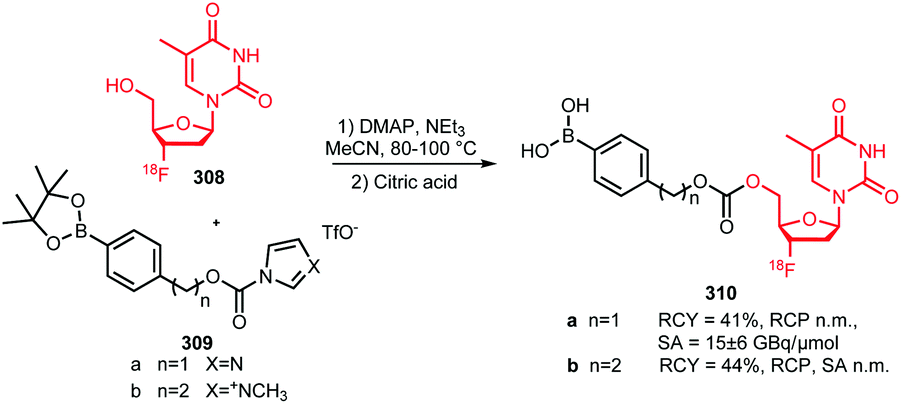 | ||
| Scheme 54 Production of PC-[18F]FLT 310a and CC-[18F]FLT 310b with [18F]FLT.225 | ||
2.13 4-(2-[18F]Fluoroethoxy)-3-methoxybenzaldehyde
4-(2-[18F]Fluoroethoxy)-3-methoxybenzaldehyde 312 has been applied as a building block in the synthesis of 1-(4-[18F]fluoroethyl)-7-(4′-methyl)curcumin 314, a tracer for β-amyloid plaque imaging. Compared to direct labelling, the 2-step synthesis shown in Scheme 55 provided higher yields and higher specific activities.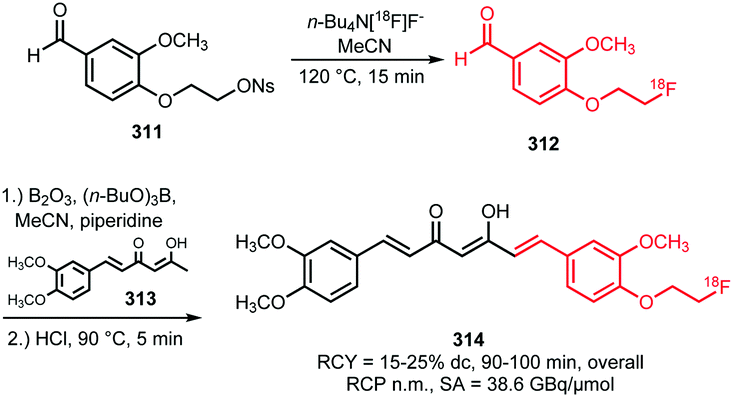 | ||
| Scheme 55 Synthesis of 1-(4-[18F]fluoroethyl)-7-(4′-methyl)curcumin.226 | ||
4-(2-[18F]Fluoroethoxy)-3-methoxybenzaldehyde 312 was synthesised from its nosylate precursor 311. [18F]Fluorination was performed with n-Bu4N[18F]F− in MeCN at 120 °C and resulted in radiochemical yields of over 70% (based on radio-TLC analysis). In the follow-up aldol condensation, 312 was reacted with a 5-hydroxy-1-phenyl-hexa-1,4-dien-3-one (313) in the presence of B2O3, (n-BuO)3B and piperidine at 120 °C. After treatment with hydrochloric acid, the final product 314 was purified by semi-preparative HPLC and obtained in a radiochemical yield of 15–25% with a specific activity of 37.6 GBq μmol−1.226
2.14 Fluorine-18 labelled phosphines
Pretze et al. explored the synthesis of fluorine-18 labelled triarylphosphine 316 and its performance in a traceless Staudinger ligation. In the traceless Staudinger ligation, triarylphosphines carrying an ester group at the ortho position of the phosphorus atom can undergo a reaction with an azide resulting in amide bond formation. The ligation usually proceeds under mild conditions, however with slower reaction kinetics than the CuAAC reaction and it suffers from the disadvantage of oxidation of the phosphine precursor. Nevertheless, it provides a “clean” alternative to the CuAAC reaction and complex biomolecules have been radiolabelled using this strategy.227Fluorine-18 labelled triarylphosphine 316 was prepared by reaction of [18F]tetrabutylammonium fluoride ([18F]TBAF) with the tosylated precursor 315 (Scheme 56). The choice of solvent to obtain 316 proved to be crucial: whereas no reaction was observed in DMF or DMSO, a mixture of MeCN and tert-butanol afforded the desired product in 65% (dc) radiochemical yield. The traceless Staudinger ligation was carried out without additional intermediate purification. Water and the azide were directly added and the reaction was stirred at different temperatures. Low temperatures required longer reaction times compared to high temperatures, but provided 317–320 in similar overall yields of 31–35%. The more complex biotin derivative 320 however required a longer reaction time at medium temperatures (60 °C) and resulted in lower yields compared to the other compounds.228
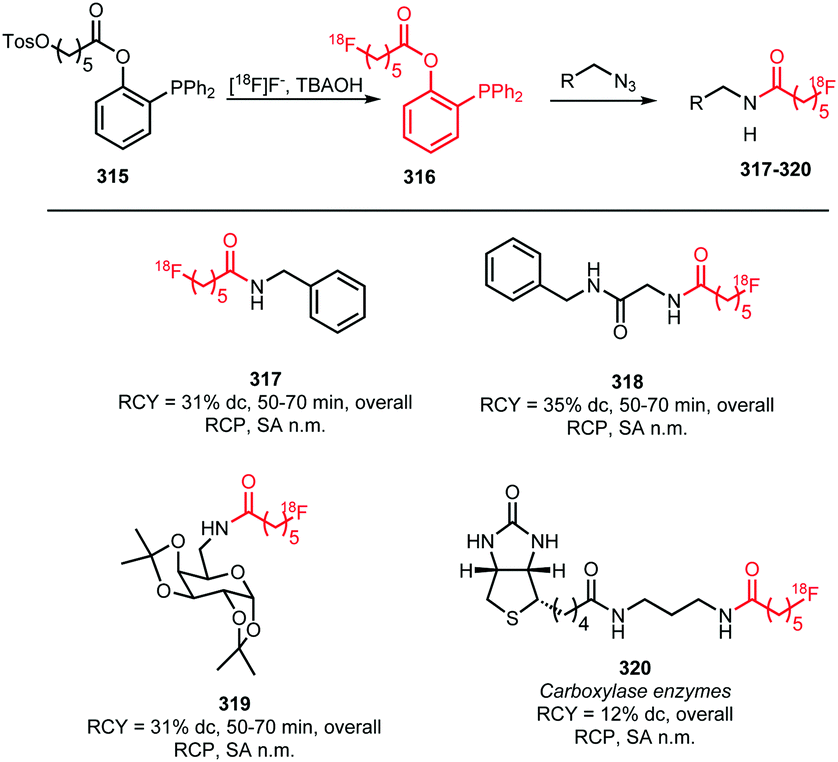 | ||
| Scheme 56 Synthesis of fluorine-18 labelled phosphine building block 316 and subsequent traceless Staudinger ligation.228 | ||
2.15 [18F]4-Nitrophenyl 2-fluoropropionate ([18F]NFP)
[18F]4-Nitrophenyl 2-fluoropropionate ([18F]NFP, 325) is commonly used for fluorine-18 labelling of amino acids, peptides and other amine derivatives. It can be coupled under very mild reaction conditions with the amine functionality of an amino acid, resulting in amide bond formation.[18F]NFP can be synthesised via a one-pot three-step procedure (Scheme 57). Via halogen exchange of ethyl-2-bromopropionate 321 with dried [18F]fluoride, ethyl-2-[18F]fluoropropanoate 322 was obtained, which was subsequently saponified under basic conditions. Usually an aqueous solution of potassium hydroxide is used, however since the subsequent step requires anhydrous conditions, Li et al. have used TBAOH which can be employed in a smaller volume, thereby shortening the time-consuming drying procedure. In the third step, bis-4-nitrophenyl carbonate 324 is added, followed by semi-preparative HPLC purification. As the [18F]NFP needs to be absolutely free from water for subsequent coupling to an amine, it was transferred via a cartridge procedure to a volatile organic solvent (e.g. ether) and then evaporated to dryness. Overall radiochemical yields of 20–50% (ndc) are reported.84,229 Reaction of 325 with amino precursors proceeds in general under mild conditions (room temperature to 60 °C) in short reaction times with good radiochemical yields. Scheme 58 summarises the molecules labelled with [18F]NFP.84,229–231 In addition to two amines, also the two amino acids, L-methionine 329 and L-arginine 327, have been radiofluorinated using this building block. Whereas Gao and co-workers described the use of the protected amino acid arginine ethyl ester dihydrochloride as precursor, Hu et al. state fast and efficient coupling of [18F]NFP to unprotected methionine. Overall radiochemical yields are comparable and approximately 15% (ndc) with reaction times of 120 minutes.229,231
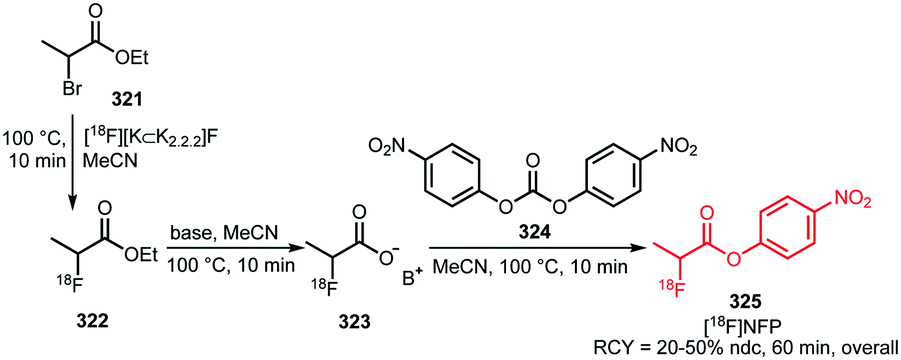 | ||
| Scheme 57 One-pot three-step synthesis of [18F]NFP.84 | ||
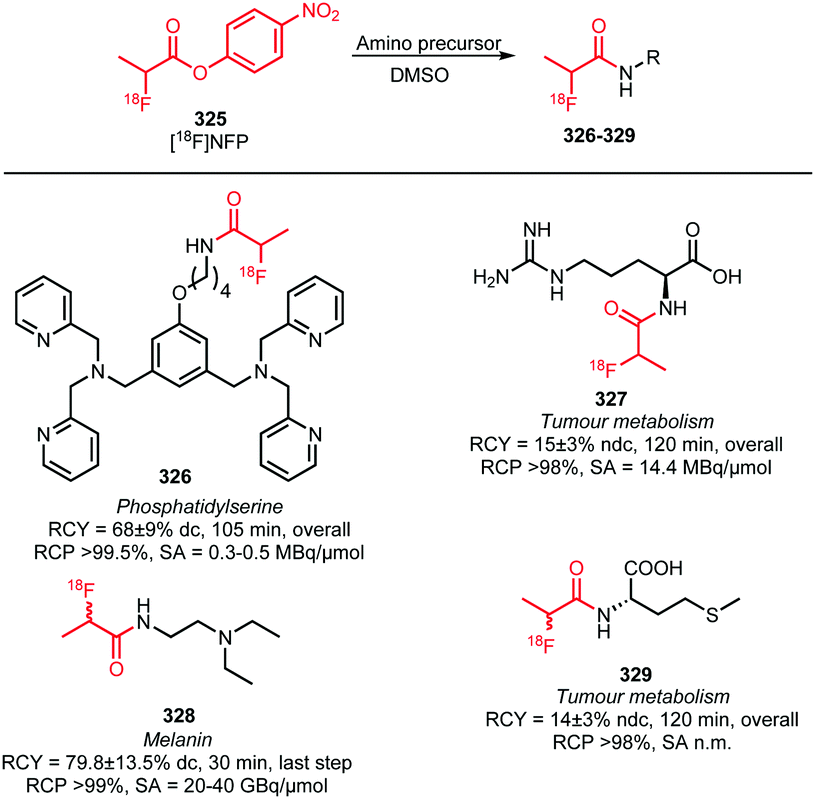 | ||
| Scheme 58 PET tracer synthesis by coupling to [18F]4-nitrophenyl 2-fluoropropionate 325.84,229–231 | ||
To summarise, [18F]NFP offers fast, mild and simple radiofluorination of amines and amino acids. The only challenge in the application of [18F]NFP is the complex and time-consuming three-step one-pot synthetic procedure of the building block itself.
2.16 Fluorine-18 labelled trans-cyclooctenes
In [4+2] inverse electron demand Diels-Alder cycloadditions, trans-cyclooctenes (TCO) and tetrazines (Tz) react very fast and selectively with each other under mild reaction conditions. Keliher et al. and Reiner et al. used this strategy to radiolabel poly-ADP-ribose-polymerase 1 inhibitor AZD2281 332 (Scheme 59). Compared to native labelling, which requires multiple steps including intermediate purifications, higher yields and reduced reaction times have been reported using this strategy.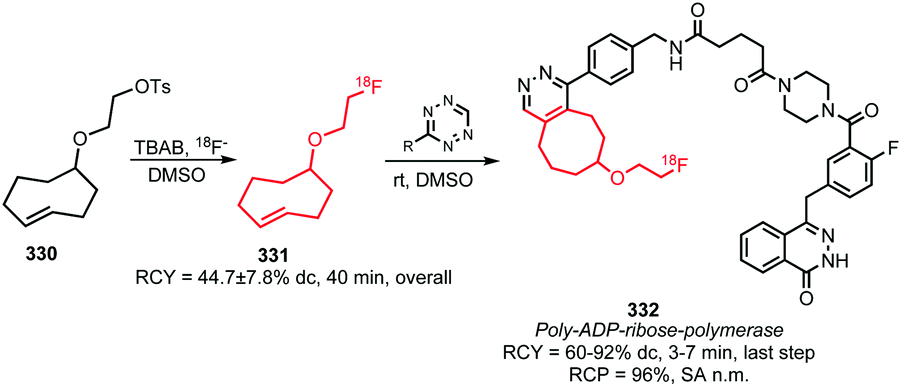 | ||
| Scheme 59 Synthesis of the fluorine-18 labelled TCO 331 and [4+2] cycloaddition with Tz precursor.233,234 | ||
As tetrazines are unstable under radiofluorination conditions,232 fluorine-18 was introduced into the trans-cyclooctene reactant 331. This building block was synthesised from its tosyl precursor in a nucleophilic substitution reaction and obtained after HPLC purification in a radiochemical yield of 44.7 ± 7.8% (dc) in 40 minutes from the start of drying the [18F]fluoride. The tetrazine moiety was integrated into the structure of the AZD2281 derivative 332 by attaching it to the piperazine unit. Reaction between the tetrazine and 331 was very fast (3 min at room temperature). HPLC purification afforded the product in a radiochemical yield of 59.6 ± 5.0% (dc).233 To allow routine production, an alternative way of purification was developed: excess amount of precursor was extracted by using magnetic beads functionalised with trans-cyclooctene. The radiochemical yield was improved to 92.1 ± 0.4% (dc).234
In conclusion, as the [4+2] cycloaddition proceeds very fast under mild reaction conditions with high selectivity, it is a very interesting alternative to the popular CuAAC reaction for radiofluorination of biomolecules.
2.17 5-(1,3-Dioxolan-2-yl)-2-(2-(2-(2-[18F]fluoroethoxy)ethoxy)ethoxy)pyridine
Carberry and co-workers introduced a novel fluorine-18 labelled building block, 5-(1,3-dioxolan-2-yl)-2-(2-(2-(2-[18F]fluoroethoxy)ethoxy)ethoxy)pyridine 334, which can react after hydrolysis with aminooxy groups, resulting in oxime formation (Scheme 60).235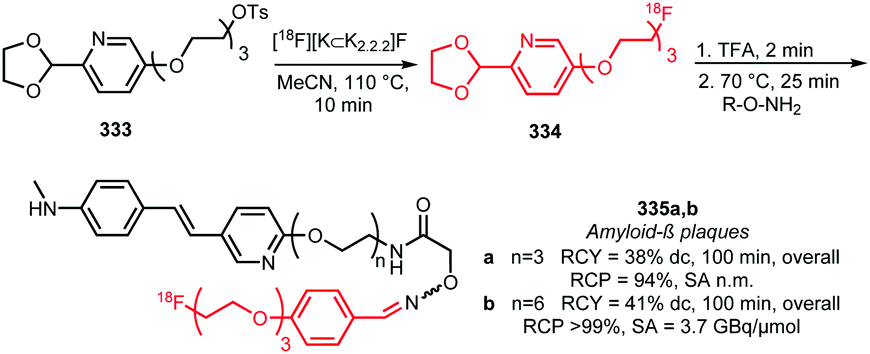 | ||
| Scheme 60 Radiosynthesis and coupling reaction of building block 334.235 | ||
Building block 334 was prepared in a 71 ± 2% radiochemical yield via nucleophilic substitution of the corresponding tosylate 333 using [18F][K⊂K2.2.2]F in MeCN at 110 °C, followed by purification using solid-phase extraction.
Next, building block 334 was converted in situ to the corresponding aldehyde under the same acidic conditions which are used in the oxime formation. Attempts to perform this step prior to radiofluorination were unsuccessful, because the presence of the free aldehyde led to formation of various side products under the radiolabelling conditions.
Two model compounds 335a and b, both potential tracers for β-amyloid plaque imaging, were synthesised by Carberry et al. using building block 334 and were obtained in comparable overall radiochemical yields (around 40% (dc)) in 100 minutes (Scheme 60).235
Successful labelling of small molecules using this building block has been demonstrated. The utility of this prosthetic group in radiofluorination of more complex biomolecules such as peptides and proteins has yet to be proven.
2.18 [18F]Fluorobutyl ethacrynic amide ([18F]FBuEA)
[18F]FBuEA 337 was used in the synthesis of the glutathione (GSH) conjugate [18F]FBuEA-GSH 338, a potential imaging agent for brain tumours targeting the lipocalin-type prostaglandin D synthase.236[18F]FBuEA was synthesised from the corresponding tosylate precursor 336 in a nucleophilic substitution reaction and obtained after Boc deprotection and HPLC purification in 20–30% radiochemical yield in 60 minutes reaction time (Scheme 61). Coupling of [18F]FBuEA with glutathione was accomplished in aqueous medium at pH 8.2, followed by semi-preparative HPLC purification, providing the racemic product in an overall radiochemical yield of 5% (dc) and an overall synthesis time of 2 hours (including the synthesis of the building block (Scheme 62)).237
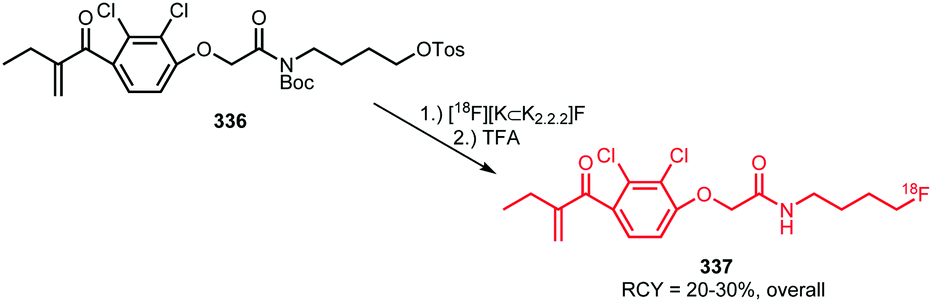 | ||
| Scheme 61 Radiosynthesis of [18F]FBuEA 337.237 | ||
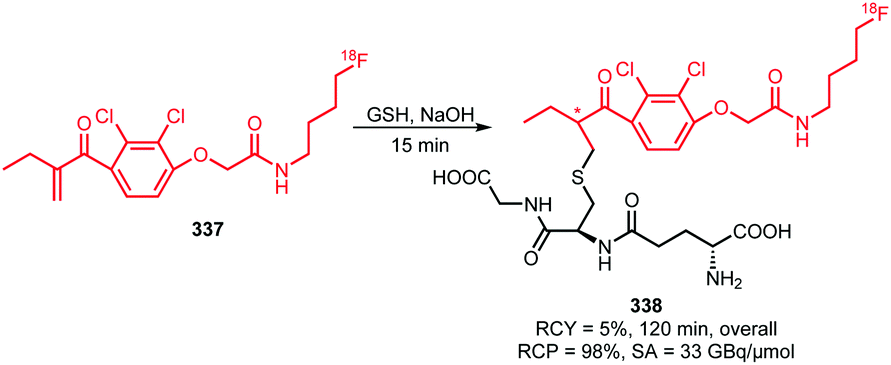 | ||
| Scheme 62 Conjugation of [18F]FBuEA with glutathione.237 | ||
2.19 New aliphatic building blocks and coupling methods with potential for PET tracer synthesis
In the following sections, new radiolabelled aliphatic building blocks as well as novel conjugation methods with aliphatic building blocks, which have not been applied yet in PET tracer synthesis, will be summarised.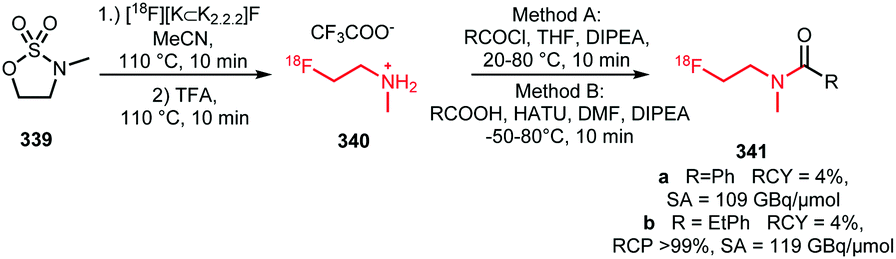 | ||
| Scheme 63 Synthesis and reaction of N-(2-[18F]fluoroethyl)-N-methylamine.238 | ||
Precursor 339 was radiofluorinated in MeCN at 110 °C within 10 minutes. Next, trifluoroacetic acid (TFA) was added and the reaction mixture was heated again at 110 °C for 10 min. After evaporation of TFA, N-(2-[18F]fluoroethyl)-N-methylamine 340 was obtained in 81% radiochemical yield and directly employed in the coupling reactions without further purification.
For the subsequent conversion to amides 341a and 341b, two methods were studied: method A employed benzyl chloride and hydrocinnamoyl chloride, respectively, in THF in presence of DIPEA as base and in method B, the corresponding carboxylic acids were reacted with N-(2-[18F]fluoroethyl)-N-methylamine 340 in presence of DIPEA and the coupling reagent 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU). Radio-TLC and HPLC analyses showed higher conversion using method B for both products. The yield of amides 341a and 341b resulting from method B were 4 and 17% in a synthesis time of 93 minutes and 134 minutes, respectively.238
An optimised synthesis procedure of building block 344 and its reaction with various amines was reported by Löser et al. in 2013. First, [18F]fluoride was introduced by nucleophilic substitution, providing thiocyanate 343 in 75–85% yield from the nosylate precursor. Compound 343 was used without intermediate purification and converted to sulfonyl chloride 344 by repetitive addition of a saturated solution of chlorine in water over a C18 SPE cartridge containing 343. 3-[18F]Fluoropropanesulfonyl chloride 344 was obtained in 40–45% decay-corrected radiochemical yield in a synthesis time of 70 minutes (Scheme 64).
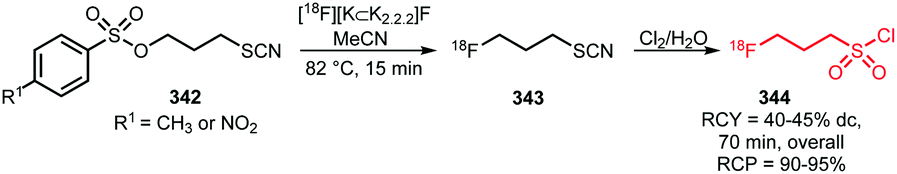 | ||
| Scheme 64 Two-step synthesis 3-[18F]fluoropropanesulfonyl chloride.240 | ||
Different amines were subjected to reaction with 3-[18F]fluoropropanesulfonyl chloride 344 to assess the usability of the building block in PET tracer synthesis (Scheme 65). Aliphatic sulfonamide 345 and 346 were formed in 2–3 minutes with high radiochemical yields of 77 to 89% (determined by radio-TLC). Addition of bases (TEA or DMAP) did not improve the yield. However, for the aniline derivatives 347, only low yields (<10%) were observed in absence of additive. Addition of TEA or DMAP improved the conversion of aniline and 4-fluoroaniline and provided radiochemical yields of 50–65% (analytically determined). For the poorly nucleophilic 4-nitroaniline, only addition of potassium trimethylsilanolate led to formation of satisfying amounts of product (RCY = 45%, analytically determined).240
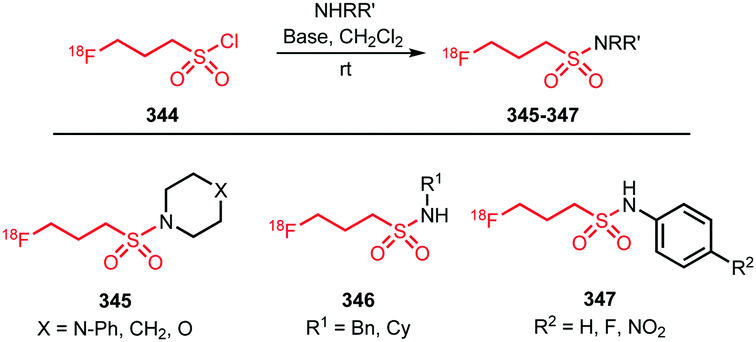 | ||
| Scheme 65 Reaction of 3-[18F]fluoropropanesulphonyl chloride with different amines.240 | ||
2-[18F]Fluoroethanol 119 was synthesised in a one-step reaction via nucleophilic substitution of ethylene carbonate 348. The reaction was carried out at 165 °C in diethylene glycol with dry [18F]TBAF as fluoride source. After 20 minutes, 2-[18F]fluoroethanol (b.p. 103.5 °C) was co-distilled with THF and trapped into a second vial containing THF in a decay-corrected radiochemical yield of 88.6 ± 2.0%. The total synthesis time including drying of [18F]fluoride was about 60 minutes.
The same procedure was applied to the synthesis of 3-[18F]fluoropropanol resulting in a decay-corrected radiochemical yield of 65.6 ± 10.2%. The lower yield in the latter case was attributed to the higher boiling point of 3-[18F]fluoropropanol (127.8 °C) and the resulting reduced distillation efficiency.
2-[18F]Fluoroethanol was reacted in two model reactions to prove its usability in the formation of [18F]fluoroalkyl aryl esters and ethers (Scheme 66). Under non-optimised reaction conditions, 2-[18F]fluoroethyl 4-fluorobenzoate 349 and 1-(2-[18F]fluoroethoxy)-4-nitrobenzene 350 were synthesised with decay-corrected radiochemical yields of 36.1 ± 5.4% and 27.7 ± 10.7%, respectively. In the synthesis of 350, the strong base tert-butoxide was employed to increase the reactivity of the building block in the nucleophilic substitution by generating 2-[18F]fluoroethoxide.
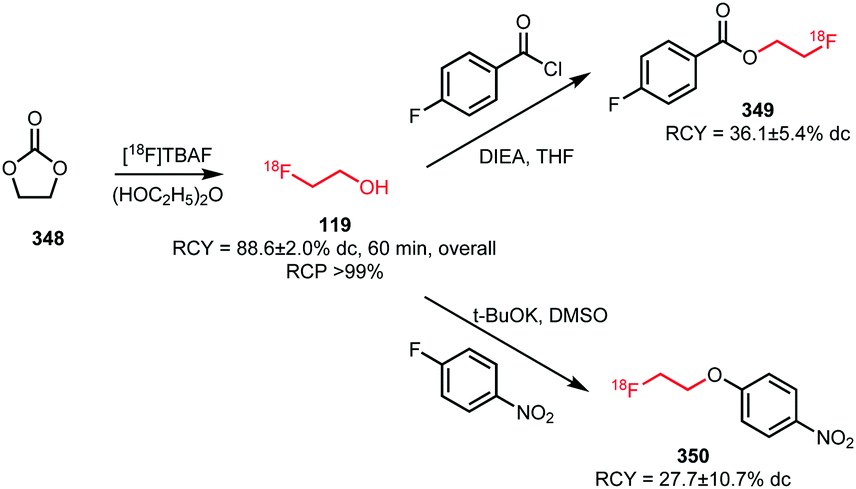 | ||
| Scheme 66 2-[18F]Fluoroethanol as building block in the synthesis of 2-[18F]fluoroethyl aryl esters and ethers.241 | ||
Due to the slightly higher nucleophilicity of 3-[18F]fluoropropanol compared to [18F]fluoroethanol, similar performance of 3-[18F]fluoropropanol was expected. But formation of 3-[18F]fluoropropyl aryl esters and ethers has not been investigated.241
[18F]Fluoromethyl iodide 10 as well as [18F]fluoromethyl bromide 2 have been investigated as building blocks in a coupling reaction with the pinacolborane-substituted benzoate 351 (Scheme 67). Initial experiments with [18F]fluoromethyl iodide 10 in DMF, using the catalyst system [Pd2(dba)3]/P(o-tolyl)3 and potassium carbonate as base, provided [18F]fluoromethylated benzoate 352 in a radiochemical yield of 23% (based on radio-HPLC analysis). Unfortunately, decomposition of building block [18F]fluoromethyl iodide 10 was observed under the coupling conditions and attempts to reduce decomposition of the precursor failed. When using [18F]fluoromethyl bromide 2, the yield of 352 could be increased up to 86% (based on radio-HPLC analysis) under optimised conditions. Due to the lower reactivity of the bromide 2 compared to the iodide 10, higher reaction temperatures of 120 °C were required and the solvent system 1,3-dimethyltetrahydropyrimidin-2(1H)-one (DMPU, N,N′-dimethyl propylene urea)/water (9:1) was found to be superior to DMF or N-methyl-2-pyrrolidone (NMP). Addition of small amounts of water suppressed side product formation, providing product 352 in 66% decay-corrected radiochemical yield in a total synthesis time of 40 minutes starting from [18F]fluoromethyl bromide.242
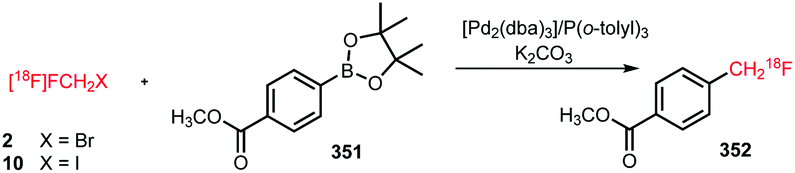 | ||
| Scheme 67 Pd(0)-Mediated C-[18F]fluoromethylation.242 | ||
3 Fluorine-18 labelled aromatic building blocks
Since 2010 a wide variety of fluorine-18 labelled aromatic building blocks have been developed and applied to synthesise PET tracers. These building blocks are mostly used for PET tracers which cannot be prepared by direct radiofluorination of the corresponding precursors, since sufficient electron withdrawing functionalities at the ortho or para position to the site of fluorination are absent, or because the precursor or product is unstable under the relatively harsh radiolabelling reaction conditions.In most cases, fluorine-18 labelled aromatic building blocks are synthesised by introduction of [18F]fluoride on phenyl precursors containing one good leaving group (–NO2 or –NMe3+) and at least one strong electron withdrawing functional group (aldehyde, ester, cyanide) positioned ortho or para from each other. Due to the electron withdrawing functional group, the benzene ring is electron deficient, allowing nucleophilic aromatic substitution with [18F]fluoride, exchanging the leaving group for fluorine-18. After radiofluorination, in the follow-up chemistry, the functional group is either (1) reacted directly in a second reaction with a precursor towards the desired PET tracer or (2) further transformed to a more useful functional group and then reacted with a precursor towards the desired PET tracer.
3.1 [18F]Fluorobenzaldehydes
The aldehyde functionality is a versatile functional group; it is therefore not surprising that [18F]fluorobenzaldehydes are often reported and used in a wide variety of subsequent chemical reactions (Scheme 68).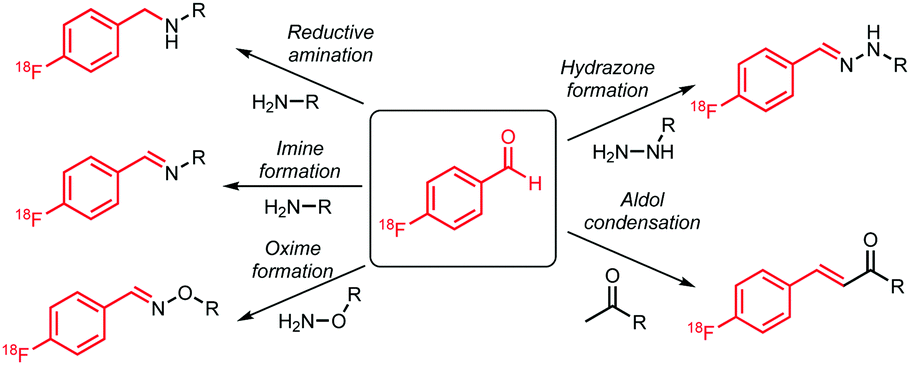 | ||
| Scheme 68 Possible applications of [18F]fluorobenzaldehydes. | ||
Of these reactions, reductive amination is the most commonly used, and will be discussed in Section 3.1.2. Furthermore, [18F]fluorobenzaldehydes are used in the condensation with various types of amines towards imines, oximes and hydrazones, which will be discussed in Section 3.1.3, 3.1.4 and 3.1.5. A well-known reaction for aldehydes is the aldol condensation, which has been used in the synthesis of PET tracers with the dibenzalacetone core structure, as can be seen in Section 3.1.6. Finally, a very innovative application of [18F]fluorobenzaldehydes is discussed in Section 3.1.7, being the use as a reagent in multi-component reactions, opening up the synthesis of a wide diversity of potential PET tracers.
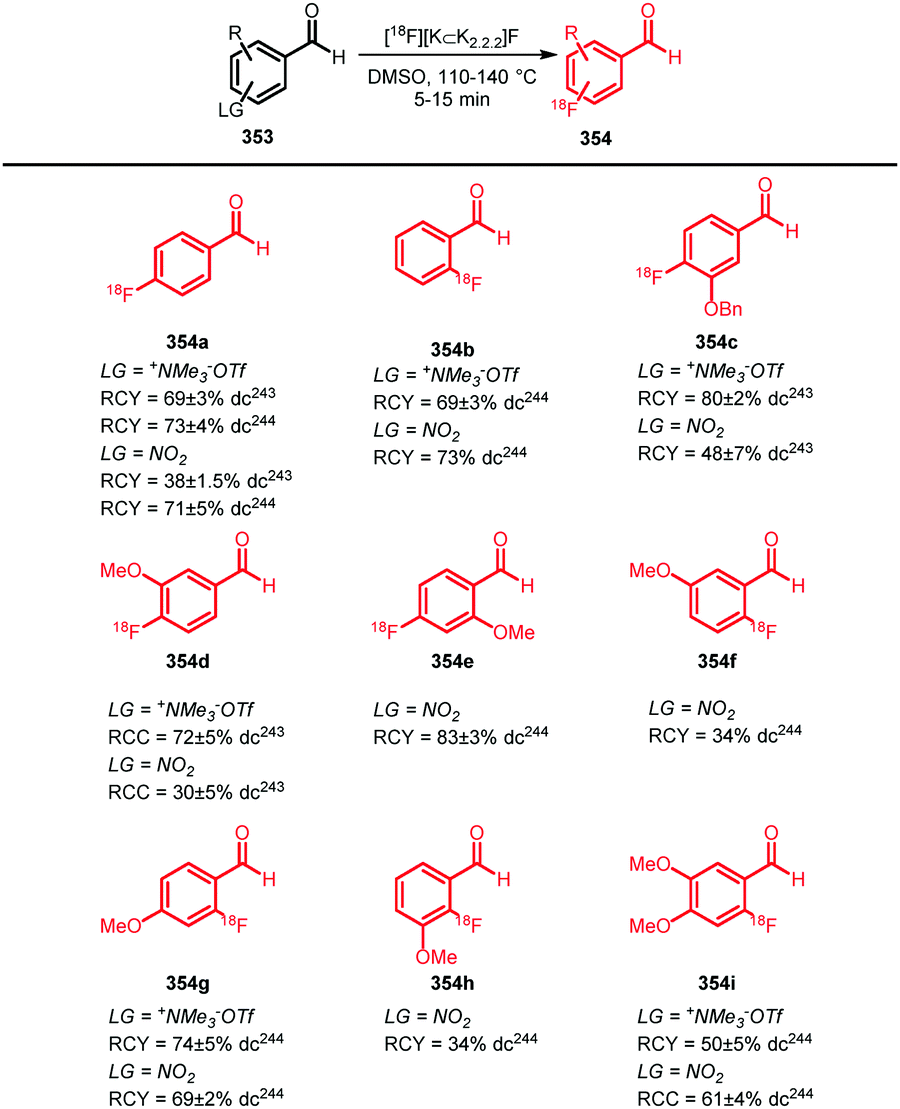 | ||
| Scheme 69 Synthesis of 2- and 4-[18F]fluorobenzaldehydes.243,244 | ||
Furthermore, in the case of the trimethylammonium precursors, purification of the 4-[18F]fluorobenzaldehydes from the trimethyl ammonium precursors can be carried out via a straightforward C18 SPE procedure. Apolar 4-[18F]fluorobenzaldehyde is retained on the C18 cartridge, while the polar, ionic, trimethylammonium precursor can be removed by washing the cartridge with aqueous media.
In case of the nitro precursors, which are also apolar molecules, such a simple purification procedure to obtain 4-[18F]fluorobenzaldehydes is not possible. As a result, trimethylammonium precursors are currently preferred for the synthesis of 4-[18F]fluorobenzaldehyde.
When a nitrogen atom is included in the aromatic ring, directly next to the fluorine-18 labelling position (thus being a pyridine derivative), the electron withdrawing effect of the nitrogen atom highly activates the labelling position, making it possible to obtain high conversions even with less reactive leaving groups. As shown by Kügler et al. (Scheme 70), a radiochemical yield of 80 ± 6% (analytically determined) was obtained when labelling the commercially available precursor 6-chloronicotinaldehyde 355.243
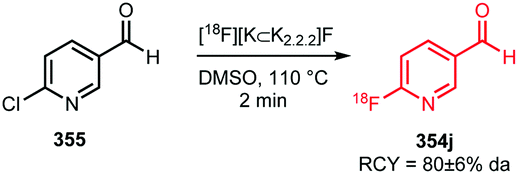 | ||
| Scheme 70 Synthesis of 6-[18F]fluoronicotinaldehyde.243 | ||
Labelling the 3-position of benzaldehyde towards 3-[18F]fluorobenzaldehyde is more challenging because this position is relatively electron rich compared to the 2- and 4-position. Attempts to prepare 3-[18F]fluorobenzaldehyde from nitro- or trimethylammonium benzaldehyde precursors resulted in very low radiochemical yields.245–247
For the synthesis of 3-[18F]fluorobenzaldehyde in higher radiochemical yields, various new radiofluorination techniques have recently been investigated (Scheme 71).18,28,34,35 These methods enable the formation of 3-[18F]fluorobenzaldehyde in moderate to good radiochemical yields (analytically determined) and thereby open up the possibility to synthesise PET tracers based on this building block. Applications of this building block have not been published yet, except in the synthesis of Lapatinib, which will be discussed in Section 3.2.3.3 (Application of [18F]fluorobenzyl halides in alkylation of phosphines and benzyl alcohols).
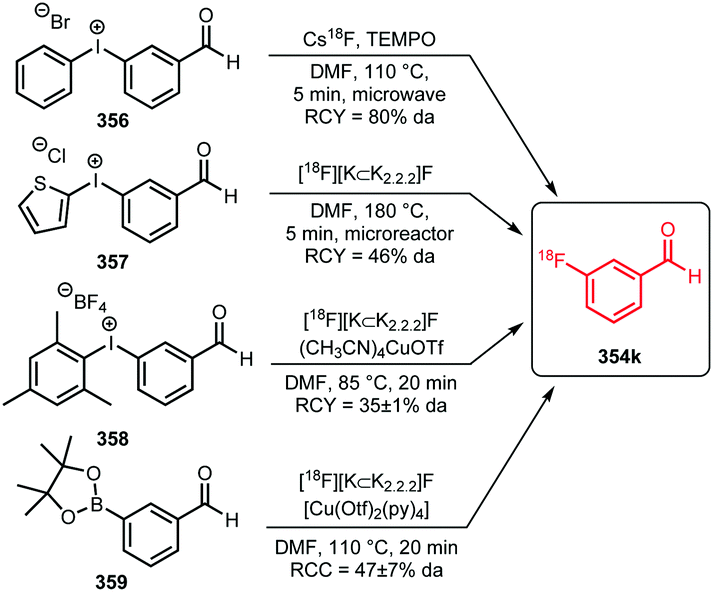 | ||
| Scheme 71 Novel fluorine-18 labelling methods for the synthesis of 3-[18F]fluorobenzaldehydes.18,28,34,35 | ||
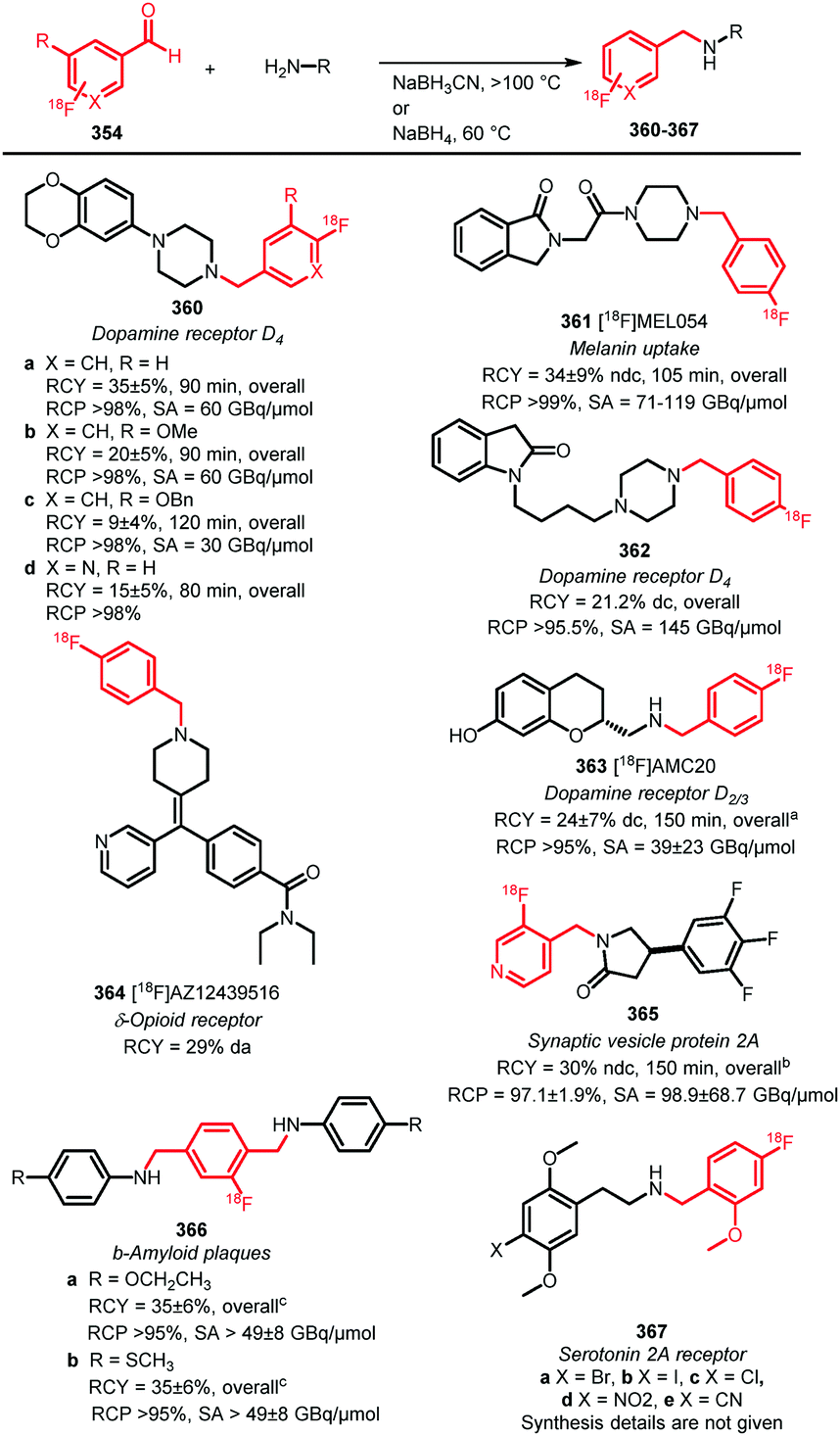 | ||
| Scheme 72 Recently produced tracers using reductive amination with [18F]fluorobenzaldehydes. aIncluding deprotection after reductive amination. bIncluding ring closure after reductive amination. cIn the synthesis of this PET tracer, the dibenzaldehyde 2-[18F]fluoroterephthalaldehyde was used, leading to double reductive amination.243,249–256 | ||
Intermediate purification of [18F]fluorobenzaldehydes as building blocks by SPE or HPLC could be omitted in some cases, resulting in a faster and easier to automate overall synthetic procedure. For example, in the case of the benzodioxin piperazines 360a–d and N-benzyl-phenethylamines 367a–e, one-pot [18F]benzaldehyde production and subsequent reductive amination was possible.243,256
Another example of a successful two-step reaction without intermediate purification is the synthesis of the delta opioid agonist [18F]AZ12439516 364. In this case, both the [18F]benzaldehyde synthesis and subsequent reductive amination could be performed using a microfluidic apparatus.250 Although a significant reduction of overall reaction time can be achieved with the one-pot procedure, it does not always yield satisfactory results. The final purification of the PET tracer can be challenging or even prove to be impossible, due to the formation of significantly more side products.
In a recent publication describing the reductive amination approach using [18F]fluorobenzaldehyde, the synthesis of fluorine-18 labelled histone deacetylase class-I tracer derivatives of [11C]Martinostat can be found. Developed originally as a carbon-11 tracer that showed excellent imaging results in preclinical studies, Strebl et al. developed a fluorine-18 derivative, which would allow multicentre clinical studies and ultimately commercialisation of the PET tracer.
Initial approaches to create a fluorine-18 derivative aimed to replace the N-methyl functionality with a [18F]fluoroethyl group showed that the [18F]fluoroethyl group led to a significant decrease in target affinity and selectivity.257 As an alternative, Strebl et al. published a fluorine-18 labelled Martinostat derivative where the aromatic ring was substituted with fluorine-18.257 Since the aromatic ring does not contain an electron withdrawing group that allows for direct nucleophilic aromatic substitution, a building block approach was used, starting from [18F]fluorobenzaldehyde 354l or 354m, followed by a multi-step procedure including a reductive amination for further functionalisation (Scheme 73). As the hydroxamate moiety is incompatible with radiofluorination conditions, two approaches were examined: (a) protecting the hydroxamic acid group with 2,2-diethoxypropane to form aprotic 5,5-dimethyl-1,4,2-dioxazole; (b) starting from the methyl ester which is converted in the last step to the hydroxamate using hydroxylamine.
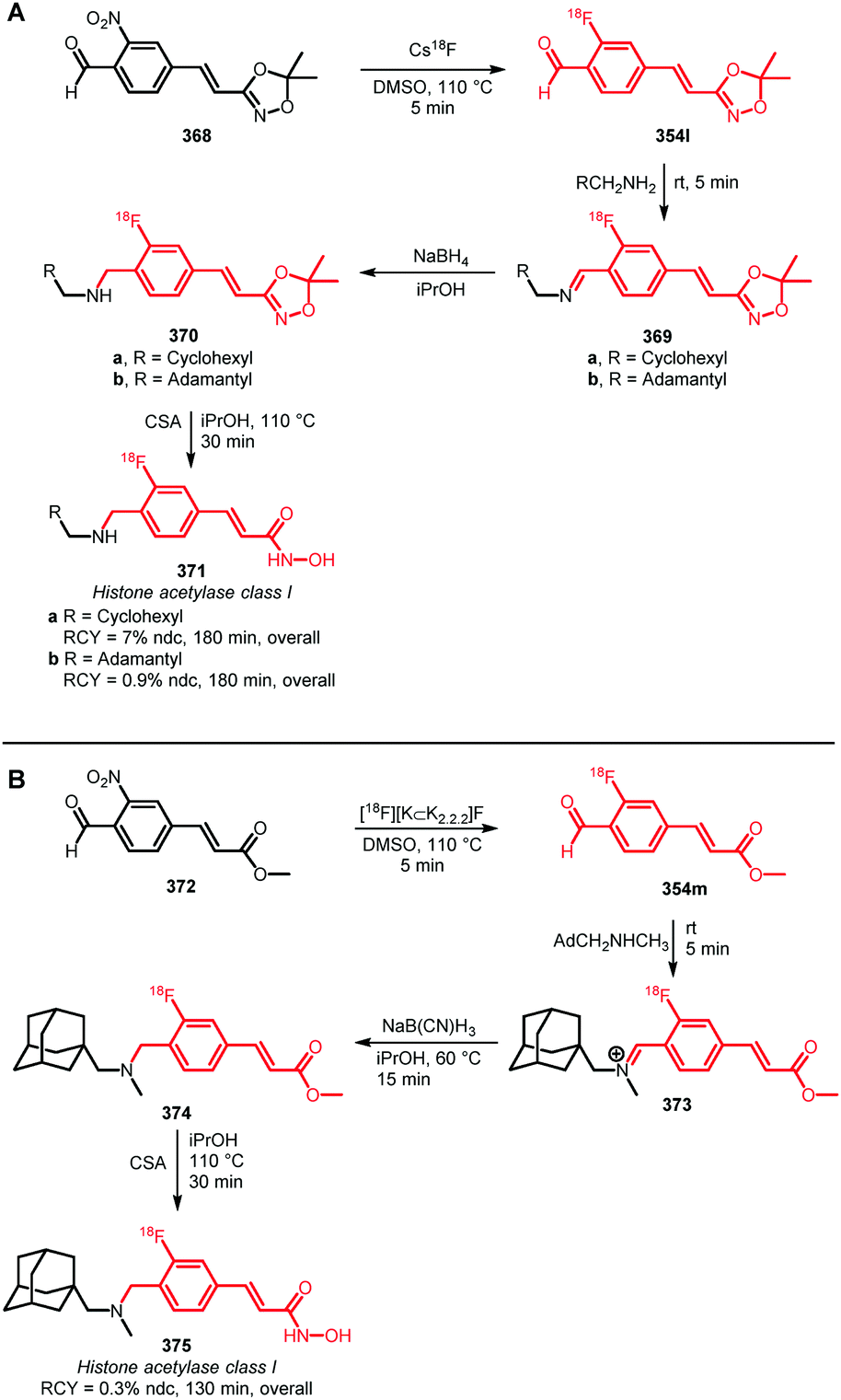 | ||
| Scheme 73 Synthesis of aromatic fluorine-18 labelled Martinostat derivatives.257 | ||
Both approaches led to the desired fluorine-18 labelled Martinostat derivative. Although the overall radiochemical yields were low, due to the multistep procedure requiring two HPLC purifications and multiple solid phase extractions, the radiochemical yields were sufficient for the preclinical evaluation of these compounds.
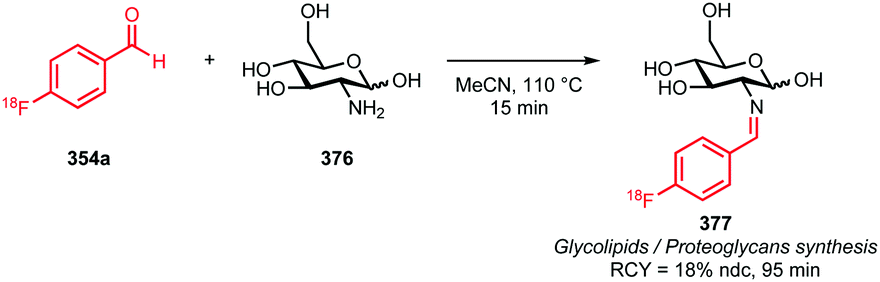 | ||
| Scheme 74 Imine formation with 4-[18F]fluorobenzaldehyde as a method to label glucosamine.55 | ||
The main advantage of this approach over the use of other fluorine-18 labelled building blocks to label glucosamine 376, is that the alcohol groups do not require protection due to the high selectivity of [18F]benzaldehydes for reaction with amines.
Abdel-Jalil et al. reported on the synthesis of a series of hypoxia tracers 379a–c by reacting [18F]4-fluorobenzaldehyde 354a with aminoxy-functionalised precursors 378a–c (Scheme 75).259 These precursors are synthesised in only 3 steps and the subsequent oxime formation proceeds in high conversions (RCY > 70% da) in 30 minutes reaction time. Since high yields and short synthesis times in general are ideal for the synthesis PET tracers, this oxime formation using [18F]4-fluorobenzaldehyde has great potential to develop fluorine-18 labelled PET tracers.
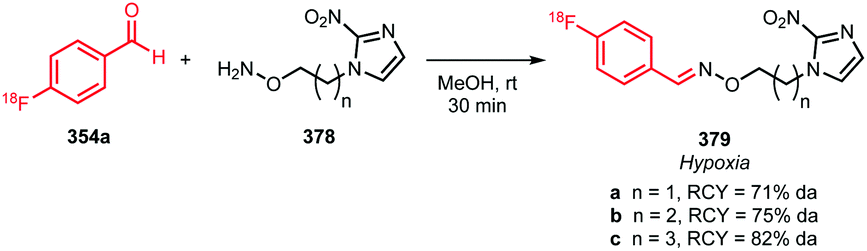 | ||
| Scheme 75 Synthesis of a hypoxia tracer by oxime formation.259 | ||
A series of derivatives was synthesised using various fluorine-18 labelled building blocks: amide formation with 4-[18F]fluorobenzoic acid, click reaction with [18F]fluoroethyl azide and imine condensation with 4-[18F]fluorobenzaldehyde. The condensation of 4-[18F]fluorobenzaldehyde with hydrazine precursor 380 resulted in 94% radiochemical yield (analytically determined) (Scheme 76). In contrast, the amide formation on the same hydrazine precursor 380 with 4-[18F]fluorobenzoic acid resulted in only 32% radiochemical yield (analytically determined).
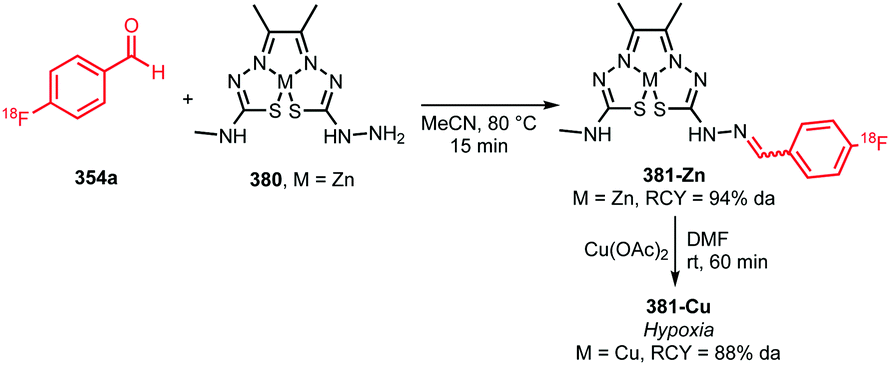 | ||
| Scheme 76 Imine formation with 4-[18F]fluorobenzaldehyde for the synthesis of a bis(thiosemicarbazonata) hypoxia tracer.163 | ||
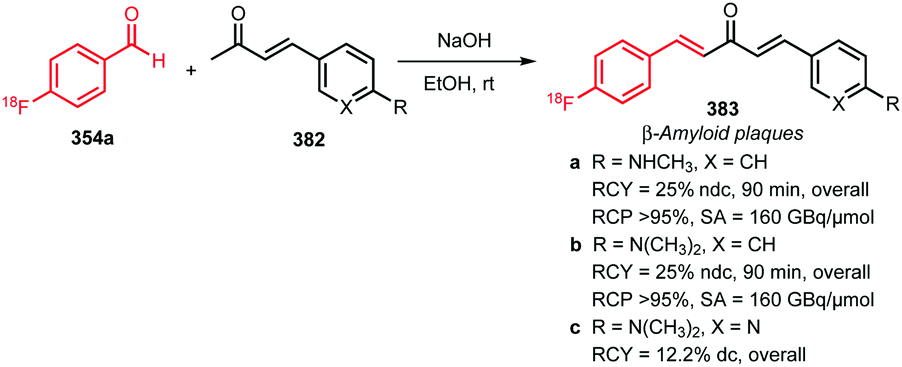 | ||
| Scheme 77 Synthesis of fluorine-18 labelled benzylidene acetones by aldol condensation with 4-[18F]fluorobenzaldehyde.261,262 | ||
Benzylideneacetones 383a–c were successfully synthesised within 90 min in moderate radiochemical yields (12–25% ndc). The aldol condensation with [18F]benzaldehydes has thereby proven to be suitable for the synthesis of PET tracers. Unfortunately the scope of the aldol condensation is limited, as an α-acidic ketone is required and the presence of other nucleophilic functional groups is not allowed.
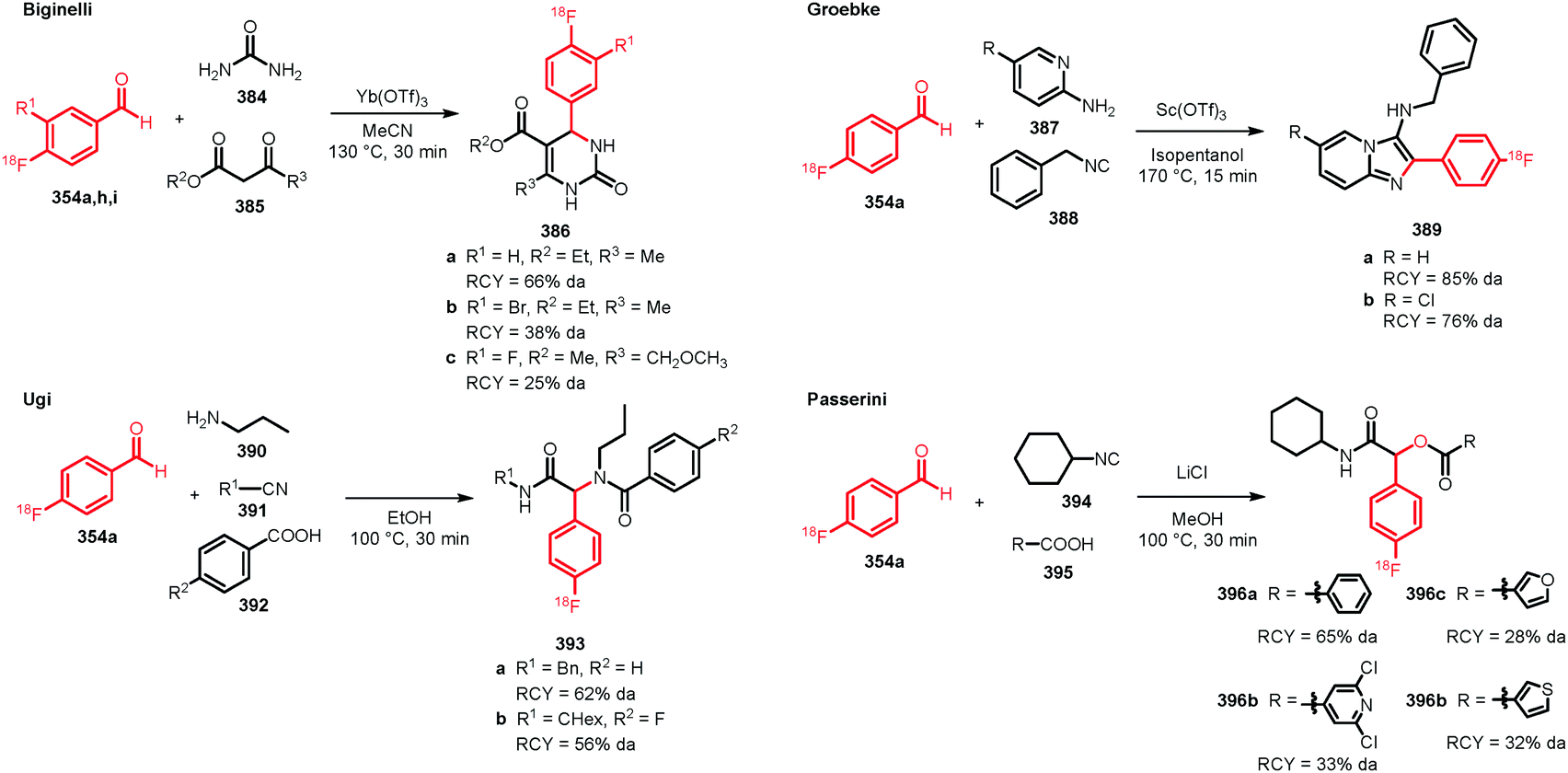 | ||
| Scheme 78 Multicomponent reactions with [18F]fluorobenzaldehydes.263 | ||
Li et al. reacted [18F]fluorobenzaldehydes in Biginelli, Groebke, Ugi or Passerini multicomponent reactions, resulting in a diversity of complex radiolabelled molecules with the fluorine-18 label on a position where direct aromatic nucleophilic substitution was not possible. By combining the Biginelli multicomponent reaction with an additional condensation reaction, α1A-selective adrenoceptor antagonist [18F]L771.668 was synthesised in a 4.4% decay-corrected overall radiochemical yield in 75 minutes (Scheme 79).
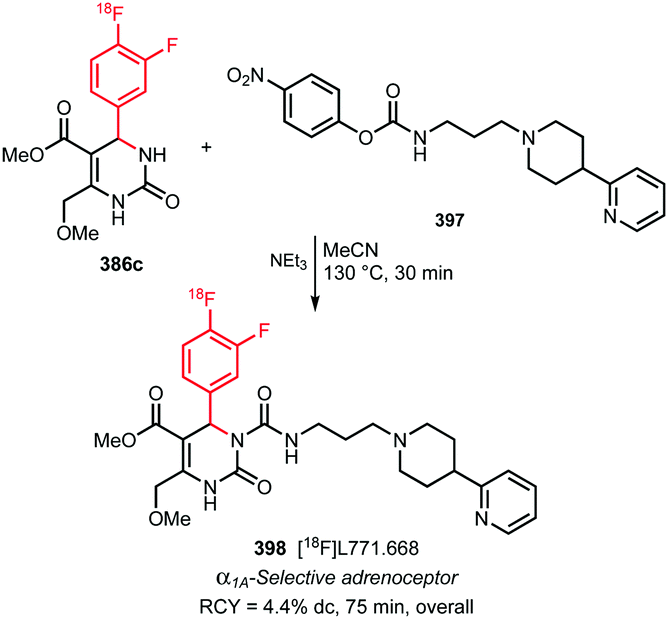 | ||
| Scheme 79 Synthesis of [18F]L771.668 using the Biginelli MCR.263 | ||
As shown in this section, [18F]4-fluorobenzaldehydes are ideal building blocks as they can be synthesised efficiently and are able to participate in a wide range of reactions in a efficient manner.
3.2 [18F]Fluoroaryl halides & [18F]fluorobenzyl halides
In recent literature, there are various building blocks described in which the fluorine-18 atom is attached to an aromatic ring and the functional group is an aromatic or aliphatic halide. The most commonly used aryl halide building block is [18F]4-fluoroiodobenzene, which is used in metal catalysed cross-coupling reactions (Section 3.2.1). Other aryl halides, which have very recently been applied are 1-bromo-3-[18F]fluorobenzene (Section 3.2.2) and 2-bromo-6-[18F]fluoropyridine (Section 3.2.3). The group of 18F-labelled benzyl halide building blocks will be shown in Section 3.2.3 and finally, the novel building block [18F]4-fluorophenetylhalide, prepared using novel fluorine-18 chemistry, will be shown in Section 3.2.5.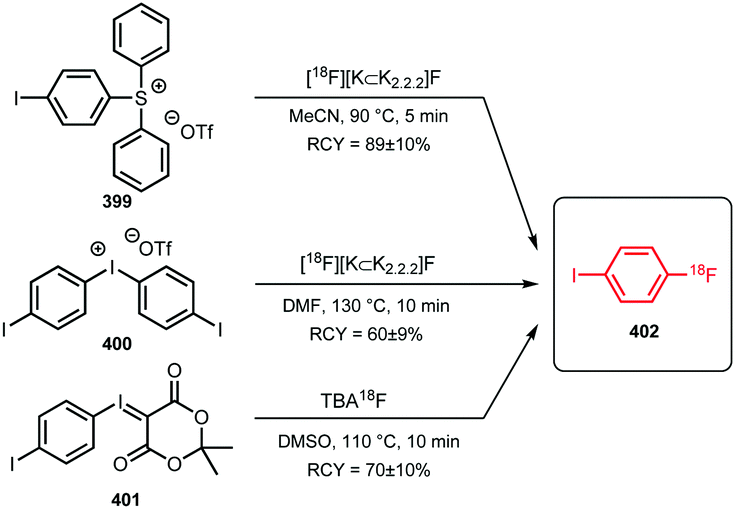 | ||
| Scheme 80 Synthesis of 4-[18F]fluoroiodobenzene.20,265–268 | ||
All three reported methods provide 4-[18F]fluoroiodobenzene 402 in moderate to excellent radiochemical yields, thereby demonstrating the advantage of novel radiofluorination technology in the synthesis of building blocks. Because these methods are rather new, applications of 4-[18F]fluoroiodobenzene are still scarce. Only 3 examples have been recently reported, each reporting a different type of metal catalysed cross-coupling (Scheme 81).
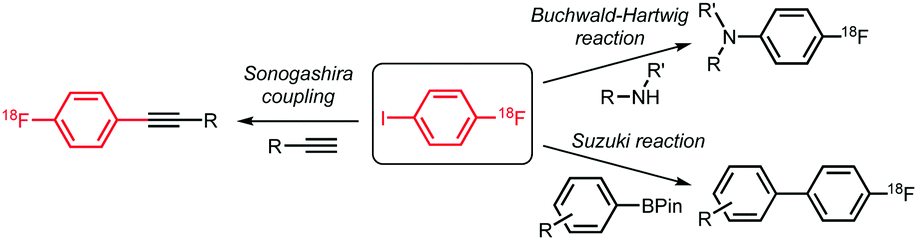 | ||
| Scheme 81 Metal catalysed coupling reactions with 4-[18F]fluoroiodobenzene. | ||
The first example is the synthesis of 2-amino-5-(4-[18F]fluorphenyl)pent-4-ynoic acid ([18F]FPhPa) 404, a novel amino acid for the PET imaging of tumours.266 The tracer was obtained via a Sonogashira coupling between alkyne 403 and 4-[18F]fluoroiodobenzene 402 (Scheme 82). This publication shows that using 4-[18F]fluoroiodobenzene as a building block, Sonogashira derived PET tracers to image amino acid transport can be produced in sufficient radiochemical yields without the use of additional protecting groups.
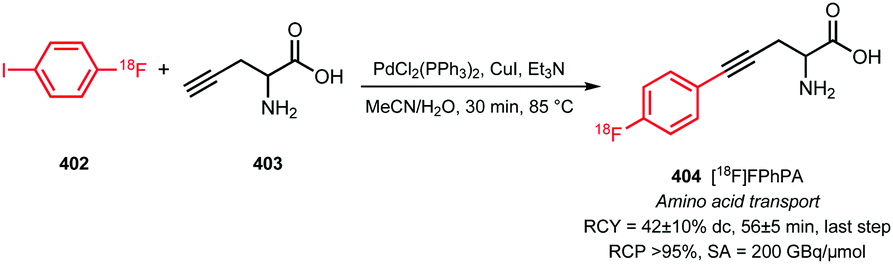 | ||
| Scheme 82 Synthesis of [18F]FPhPA by Sonogashira coupling with 4-[18F]fluoroiodobenzene.266 | ||
The second example is the synthesis of dopamine D4 ligand [18F]FAUC 316 (408). This tracer is synthesised in two steps from 4-[18F]fluoroiodobenzene 402, first by a Buchwald–Hartwig reaction of amine 405 and followed by a reductive amination with aldehyde 407 (Scheme 83).265 The low overall radiochemical yield of 10% and the long synthesis time of 80 minutes show the disadvantage and challenge of the multistep synthesis for PET tracers.
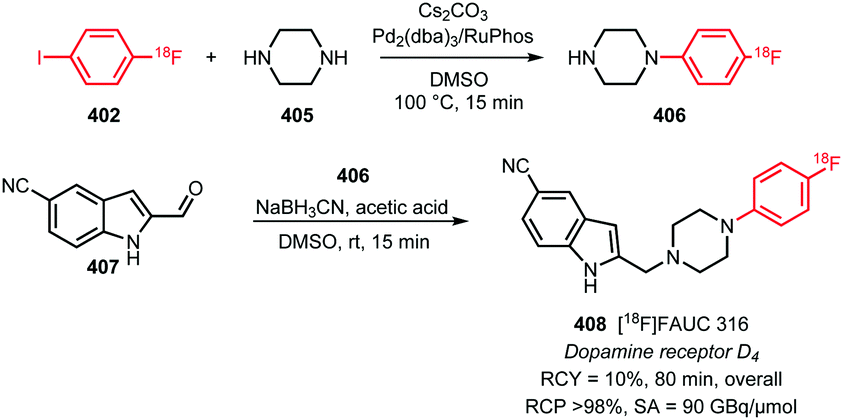 | ||
| Scheme 83 Buildup synthesis of [18F]FAUC 316.262 | ||
The last example is the synthesis of [18F]pitavastatin 410, which is prepared by Suzuki coupling between 4-[18F]fluoroiodobenzene 402 and boronic acid pinacol ester 409 (Scheme 84).268 A relatively low overall radiochemical yield (12% decay corrected) is also reported here.
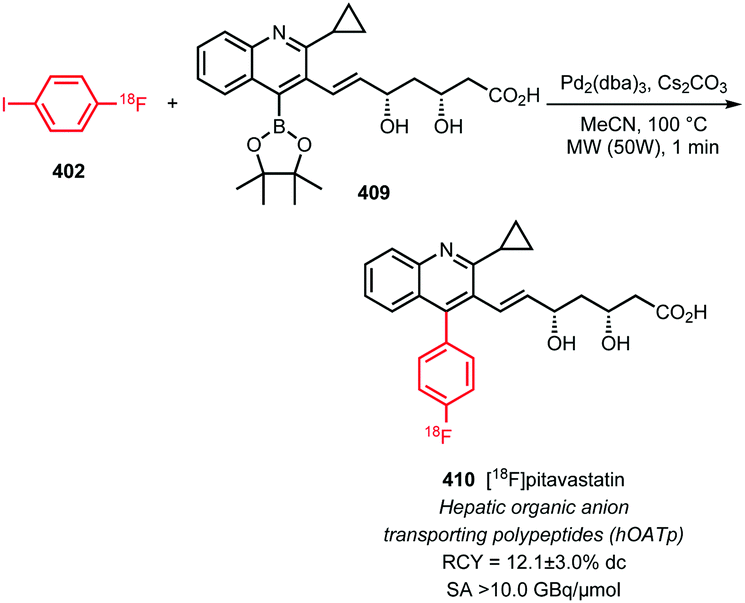 | ||
| Scheme 84 Synthesis of [18F]pitavastatin.268 | ||
In summary, the relatively low radiochemical yield of the cross-coupling reactions is most probably the main reasons that 4-[18F]fluoroiodobenzene is not very often used for PET tracer development. The building block itself however can be prepared in high radiochemical yields.
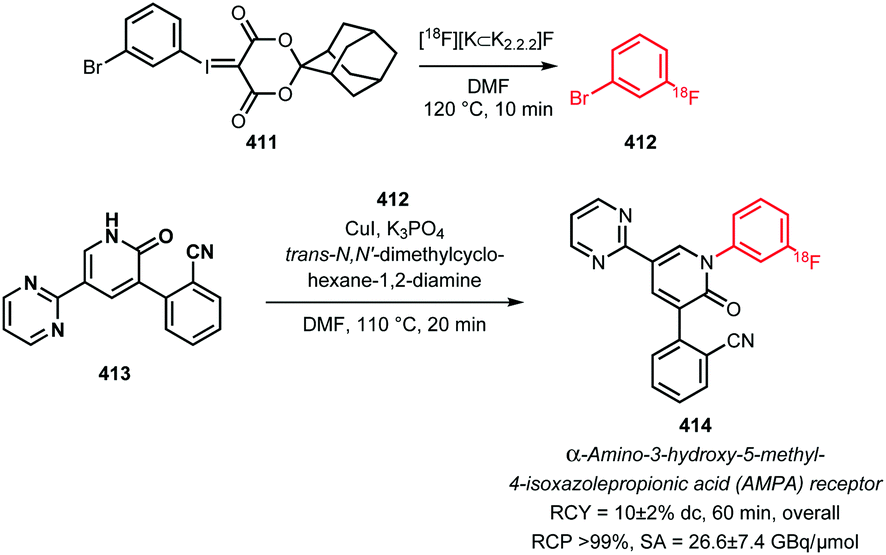 | ||
| Scheme 85 Synthesis of AMPA receptor PET tracer 414 by Cu-mediated N-arylation with 1-bromo-3-[18F]fluorobenzene.269 | ||
The fluorine-18 atom in 1-bromo-3-[18F]fluorobenzene 412 is positioned at the 3-position from the bromine atom and is thereby challenging to synthesise. Yuang et al. however did succeed in the synthesis of this building block in a radiochemical yield of 72 ± 3% (analytically determined) by radiofluorination of precursor 411.
In the same reaction vessel, 1-bromo-3-[18F]fluorobenzene 412 was further reacted by copper-mediated N-arylation resulting in the desired PET tracer 414 in an overall radiochemical yield of 10 ± 2% (dc, calculated from starting amount of [18F]fluoride) in a short 60 min synthesis time with an excellent radiochemical purity and good specific activity.
Thereby, Yuan et al. show that novel late-stage fluorination methods, in this case the fluorine-18 labelling of spirocyclic iodonium ylides, can be effectively used for the synthesis of aromatic fluorine-18 labelled building blocks which cannot be made by conventional nucleophilic aromatic substitution due to unfavourable electronic properties.
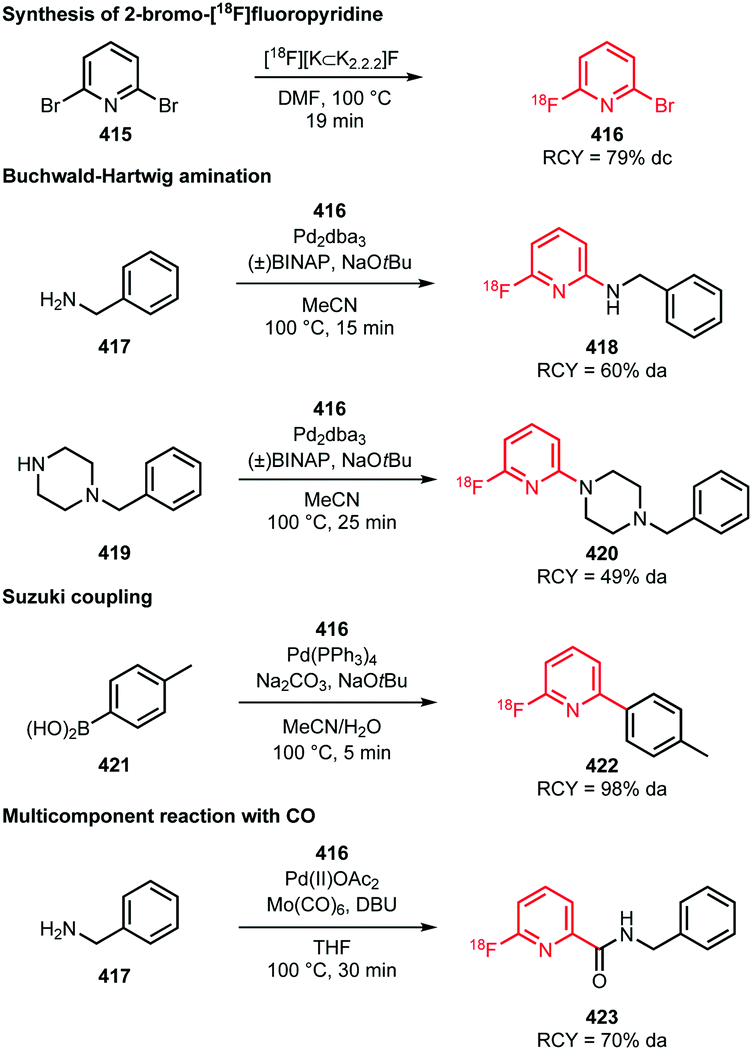 | ||
| Scheme 86 Synthesis and application of 2-bromo-6-[18F]fluoropyridine.271 | ||
Betts et al. investigated the reaction of this building block with model substrates in the Buchwald–Hartwig amination, Suzuki coupling and in a multicomponent reaction with CO and benzylamine (Scheme 86). The application in the synthesis of PET tracers has not yet been demonstrated, these first results are however promising.
3.2.4.1 Synthesis of [18F]fluorobenzyl halides. Benzyl halides are generally very useful in organic chemistry due to their versatile use in alkylation reactions at oxygen, nitrogen, sulfur, phosphor or carbon. It is therefore not surprising that multiple methods have been developed to synthesise [18F]fluorobenzyl halides.272,273
The most commonly described approach towards 4-[18F]fluorobenzyl halides and 2-[18F]fluorobenzyl halides is via a three step procedure, starting with the synthesis of [18F]fluorobenzaldehyde, followed by a reduction and finally the halogenation of the benzylalcohol. Lemaire et al. recently optimised this synthetic strategy towards various 4- and 2-[18F]fluorobenzyl halides (Scheme 87).244
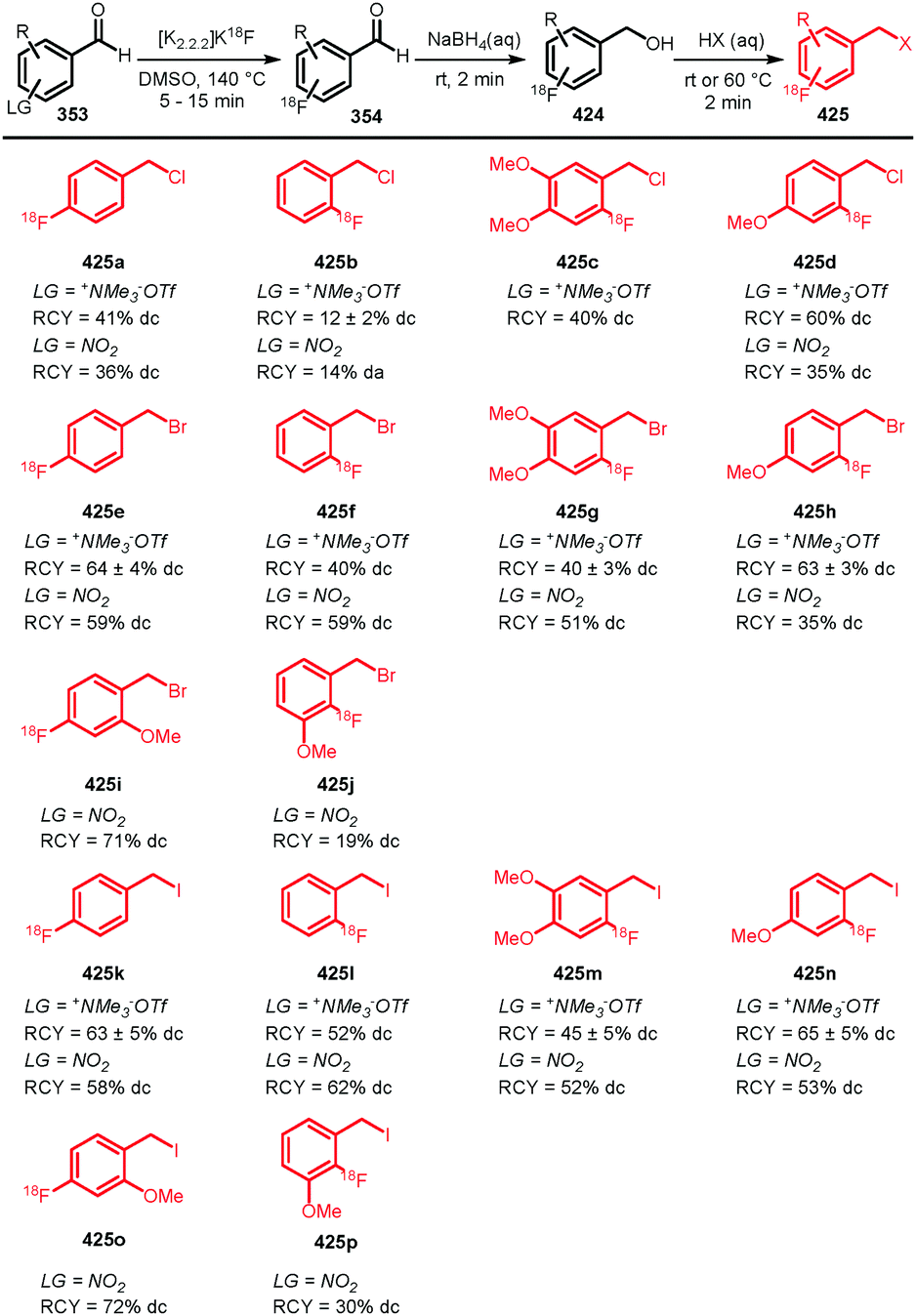 | ||
| Scheme 87 Conventional synthesis of 2- and 4-[18F]fluorobenzyl halides.244 | ||
Synthesis of 3-[18F]fluorobenzyl halides using this strategy is almost impossible due to the electron rich properties of the aromatic ring as a consequence of substitution at the 3-position. Therefore, Basuli et al. synthesised 3-[18F]fluorobenzyl bromide from iodonium salt benzaldehyde precursor 356 (Scheme 88).243
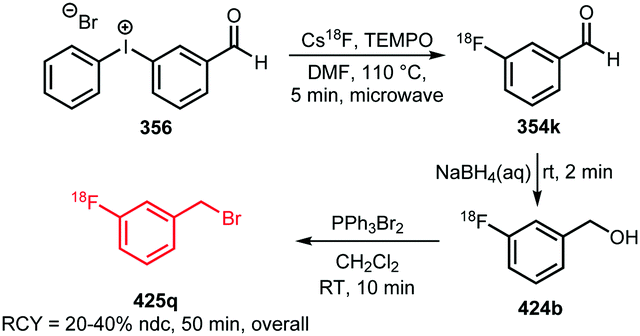 | ||
| Scheme 88 Three step strategy towards 3-[18F]fluorobenzyl bromide.240 | ||
For both three-step strategies, moderate radiochemical yields of the 2-, 3- and 4-[18F]fluorobenzyl halides could be obtained. The multi-step nature however makes this method rather complex and therefore challenging to automate and sensitive to failures.
Because not only electron deficient, but also electron neutral and electron rich iodonium salt precursors can be labelled with [18F]fluoride, Chun et al. investigated a direct one-step labelling towards 2-, 3- and 4-[18F]fluorobenzyl halides using iodonium salt precursors (Scheme 89).34 Using this strategy, the desired [18F]fluorobenzyl halides could be generated with radiochemical yields up to 55% (analytically determined) in just one synthesis step.
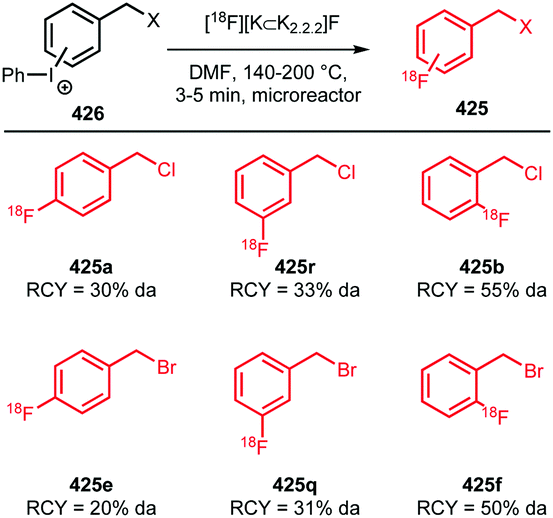 | ||
| Scheme 89 Preparation of [18F]fluorobenzylhalides in one step from diaryliodonium precursors.34 | ||
3.2.4.2 Application of [18F]fluorobenzyl halides in the synthesis of [18F]F-DOPA. Since 1991, [18F]fluorobenzyl halides have been used as useful building blocks in the synthesis of [18F]F-DOPA 427 (Scheme 90).274 Due to the electron rich properties of the aromatic ring, [18F]F-DOPA can only be synthesised by direct labelling via electrophilic fluorination using [18F]F2 gas. Drawbacks of this method is a relatively low yield (up to 5 GBq) and low specific activity, typically less than 1 GBq μmol−1.275–279 To overcome these issues, a method using a building block approach was developed, where fluorine-18 was incorporated via a nucleophilic substitution reaction in the first reaction of a five step total synthesis: (1) reaction of trimethylammonium benzaldehyde precursor 353i with [18F]fluoride towards [18F]benzaldehyde 354i, (2) reduction to [18F]fluorobenzylalcohol 424i, (3) halogenation towards benzyl halide 425g or 425m and (4) chiral C–C coupling using a chiral catalyst and (5) deprotection to yield [18F]F-DOPA 427 (Scheme 90).274,280–288
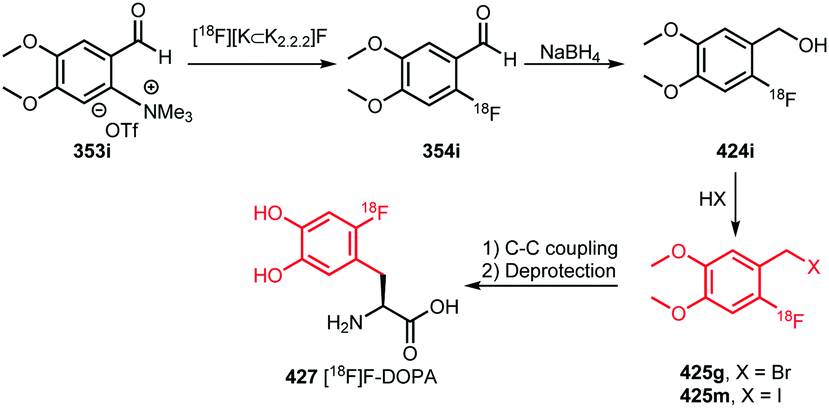 | ||
| Scheme 90 Synthesis of [18F]F-DOPA via multistep approach using a [18F]fluorobenzyl halide as a building block.274,280–288 | ||
For the enantioselective C–C coupling reaction, Lemaire et al. developed a method in 2004, where [18F]fluorobenzyl halide (bromide or iodide) is coupled with N-(diphenylmethylene)glycine tert-butyl ester 428 under basic conditions and in the presence of a phase transfer catalyst (PTC) (Scheme 91).280 This approach yielded [18F]F-DOPA 427 in an enantiomeric excess of >95%, an overall radiochemical yield of 25–30% (dc) and a specific activity of >100 GBq μmol−1 in 100 minutes.
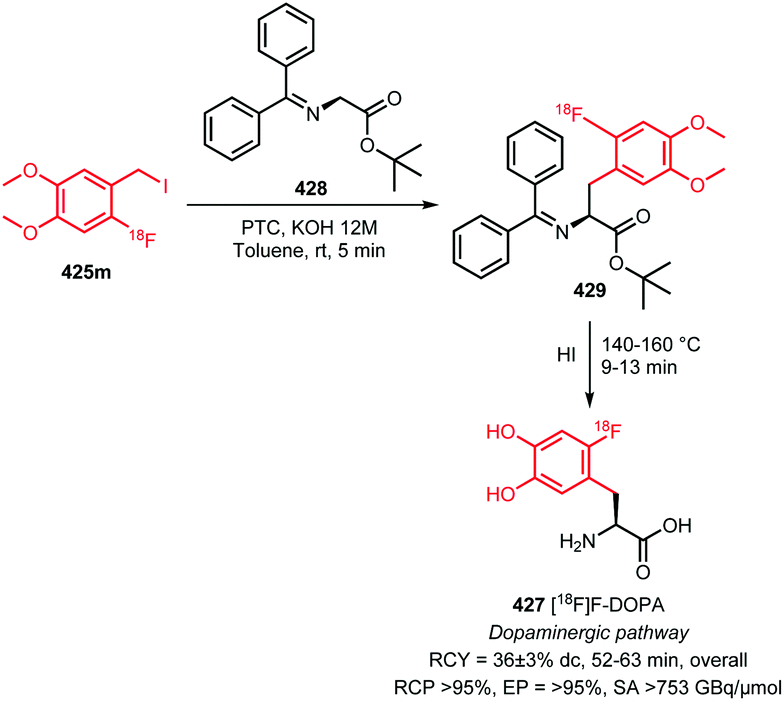 | ||
| Scheme 91 C–C coupling and hydrolysis of a [18F]fluorobenzyl halide with N-(diphenylmethylene)glycine tert-butyl ester and a phase transfer catalyst (PTC).280,287,288 | ||
Recently, Lemaire et al. optimised and automated this method by studying various reaction conditions and various new PTCs for the C–C coupling.287,288 [18F]F-DOPA was synthesised using a FASTlab synthesiser in an improved radiochemical yield of 36% ± 3% (dc) and >45 GBq at the end of synthesis, an enantiomeric excess of >95% and a synthesis time of 52–63 min (Scheme 91). In a similar fashion, 2-[18F]fluoro-L-tyrosine was synthesized by Libert et al. in an overall radiochemical yield of 50.5 ± 2.7% (dc).287,288
Pretze et al. evaluated the [18F]F-DOPA synthesis procedure, however was not able to synthesize [18F]F-DOPA in the same radiochemical yield.289 It was determined that this was caused by a combination of factors: (1) decomposition of the trimethyl ammonium triflate group of the precursor molecule into 4-aminobenzaldehyde; (2) problematic automation due to formation of precipitates during the C–C coupling reaction.
Because of these drawbacks, Pretze et al. investigated a late-stage fluorination approach, based on the radiofluorination of a nitrobenzaldehyde precursor and conversion of the aldehyde functional group to a phenol by Baeyer–Villiger oxidation. With this method, [18F]F-DOPA was synthesized in a radiochemical yield of 20 ± 1% (dc).289
3.2.4.3 Application of [18F]fluorobenzyl halides in alkylation of phospines and benzyl alcohols. Three new tracers have been synthesised using [18F]fluorobenzyl halides since 2010 (Scheme 92).290–292 Only [18F]fluorobenzyl bromides were used, probably due to a balance between a high reactivity as an alkylating agent for the alkylation of alcohols and phoshines, and a good stability.
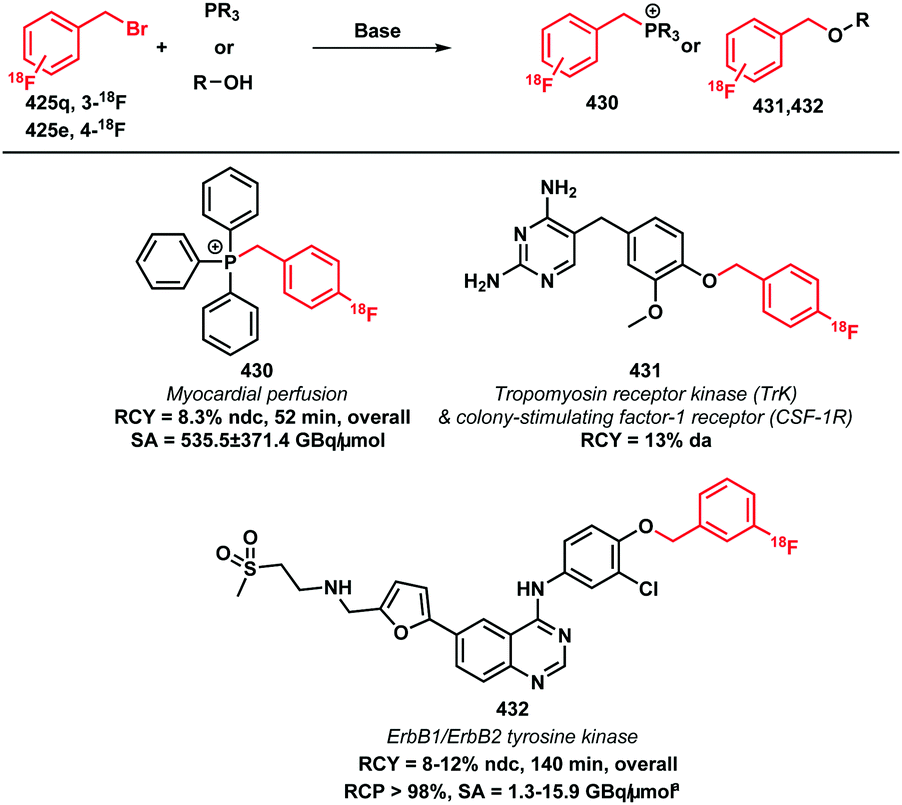 | ||
| Scheme 92 PET tracers synthesised using [18F]fluorobenzyl bromide as a building block. aThe specific activity is low due to the low amount of starting activity of [18F]fluoride.290–292 | ||
Tropomyosin receptor kinase and colony-stimulating factor-1 receptor tracer 431 (Scheme 92) was prepared via O-alkylation of the corresponding hydroxyl precursor with 4-[18F]fluorobenzyl bromide 425e.292 The yield as measured by HPLC was found to be 13%. Together with the low radiochemical yield for the three step synthesis to obtain 4-[18F]fluorobenzyl bromide of 25–30% (ndc), the main conclusion of Bernard-Gauthier et al. was that another synthesis route should be developed for this tracer. They suggested the use of diaryliodonium salts as a precursor for either 4-[18F]fluorobenzyl bromide synthesis, or even for a direct late-stage labelling approach towards 431.
The myocardial perfusion tracer 4-[18F]fluorobenzyltriphenylphosphonium ion 430, as reported by Ravert et al., was synthesised via microwave assisted alkylation using 4-[18F]fluorobenzyl bromide (Scheme 92).291 The overall radiochemical yield was 8.3% (ndc). Also in this case, a multistep procedure towards 4-[18F]fluorobenzaldehyde was used. However by performing all steps in one pot and by using microwave irradiation, the total synthesis time could be kept at 52 minutes.
In the synthesis of the ErB1/ErB2 tracer [18F]lapatinib 432, 3-[18F]fluorobenzyl bromide 425q was synthesised in a three step procedure, using an iodonium salt precursor for the synthesis of 3-[18F]fluorobenzaldehyde (Scheme 88) in an overall radiochemical yield of 12% (ndc).290
In summary, [18F]fluorobenzyl halides have proven to be successful in the synthesis of [18F]F-DOPA. In the synthesis of new PET tracers however only relatively low radiochemical yields were obtained. It is not clear yet what is causing these relatively low radiochemical yields.
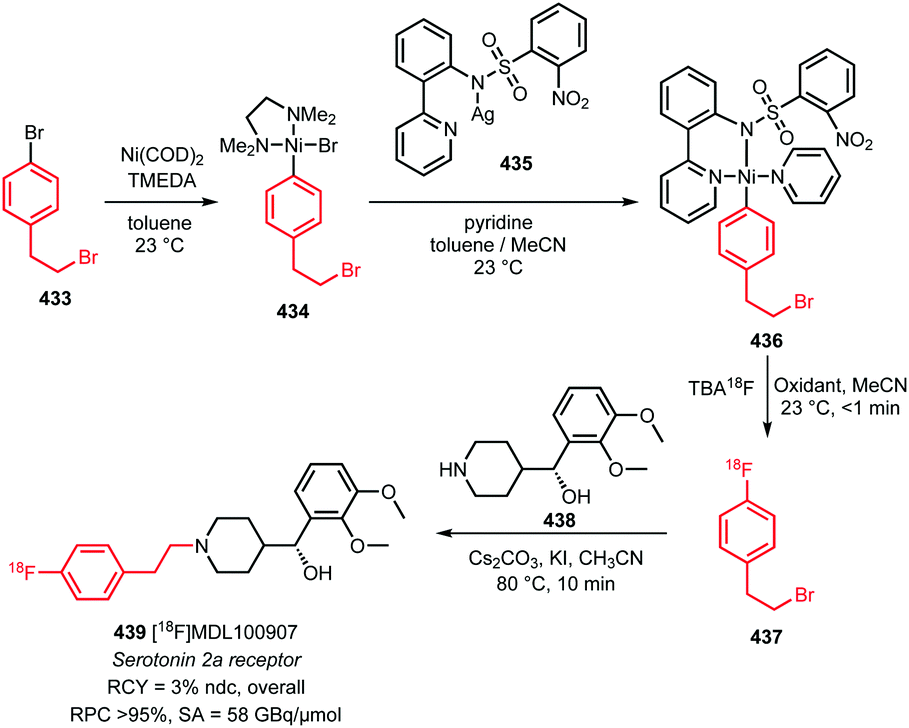 | ||
| Scheme 93 Preparation of serotonin 2a receptor tracer [18F]MDL100907 using 4-[18F]fluorophenethyl bromide.27 | ||
The overall radiochemical yield for the synthesis of [18F]MDL100907 is low (2.2%, ndc), which can be explained by a combination of a low radiofluorination yield and a low alkylation yield. Irrespective of the synthesis results, [18F]fluorophenylethyl bromide is still an attractive building block. More research is required towards the synthesis of [18F]fluorophenylethyl bromide to make this method useful for high yield tracer synthesis.
3.3 [18F]Fluorophenyl amines
Various PET tracers have been synthesised using fluorine-18 labelled aromatic amine containing building blocks since 2010. Their high versatility and selectivity in reactions with electrophiles including acid chlorides, sulfonyl halides, Michael acceptors and various others make them attractive building blocks. In this chapter, the synthesis and application of [18F]fluoroanilines (Section 2.3.1), [18F]fluorobenzylamines (Section 2.3.2), [18F]fluorophenylhydrazides (Section 2.3.3) and [18F]phenethylamines (Section 2.3.4) will be described.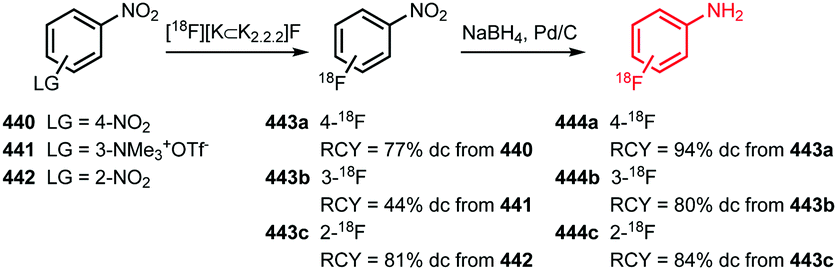 | ||
| Scheme 94 Preparation of [18F]fluoroanilines in a two-step fluorine-18 labelling and nitro reduction procedure.202,293–297 | ||
Radiofluorination of 1,3-dinitrobenzene gives 3-[18F]fluoronitrobenzene, however in only low radiochemical yields due to the increased electron density of the aromatic ring due to substitution of the 3-position. The yield can be increased if trimethylammonium precursor 441 is used as a precursor, because the trimethylammonium group is a better leaving group then the nitro group (Scheme 94).295,298,299
One group of compounds in which [18F]fluoroanilines as reagents for PET tracers have proven to be useful are the epidermal growth factor receptor (EGFR) kinase inhibitors (Scheme 95).295,298,299 The anilinoquinazoline structure can be built-up by the reaction of 4-[18F]fluoroanilines with chloro-quinazoline 445 or cyclic amide 446, in which, for the latter, a strong base (1,8-diazabicyclo[5.4.0]undec-7-ene (DBU)) and a coupling reagent (O-benzo-triazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate (BOP)) are required. Using this pathway Gefitinib 447 could be synthesised in an overall radiochemical yield of 17.2 ± 3.3% (dc) and Afatinib 449 in an overall radiochemical yield of 17.0 ± 2.5% (dc).298,299 Vasdev et al. explored this strategy by synthesizing a library of anilinoquinazolines 448a–f, showing that these tracers can be obtained in 9–55% overall radiochemical yield (dc).295
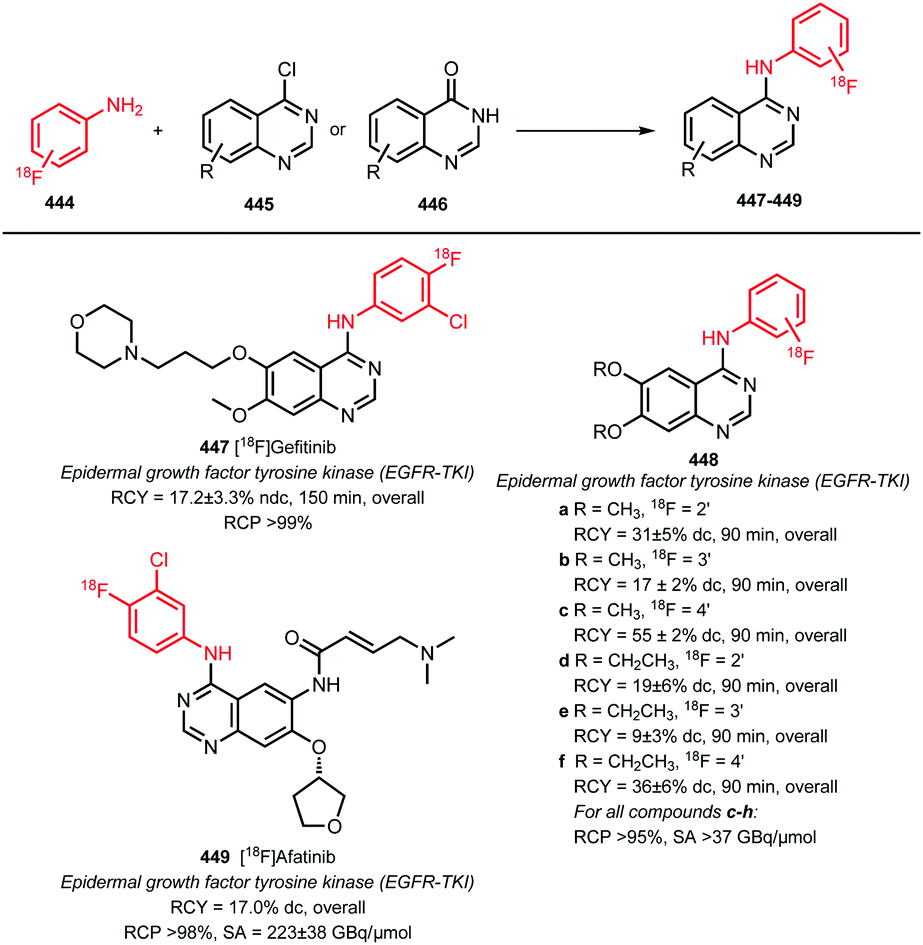 | ||
| Scheme 95 Synthesis of PET tracers for the EGFR-TKI by reaction of chloroquinazolines or cyclic amides with [18F]fluoroanilines.295,298,299 | ||
In a similar approach, Huang et al. synthesised the potential indoleamine 2,3-dioxygenase-1 tracer [18F]IDO5L 451 in a radiochemical yield comparable to the EGFR inhibitors (Scheme 96).297 A direct 18F-radiolabelling towards 451 using the corresponding trimethylammonium precursor did not yield 451, due to decomposition of the precursor under the relatively harsh (120 °C, 30 min) reaction conditions. The coupling reaction with 3-chloro-4-[18F]fluoroaniline, however, only required a temperature of 60 °C, showing a clear benefit of the building block approach.
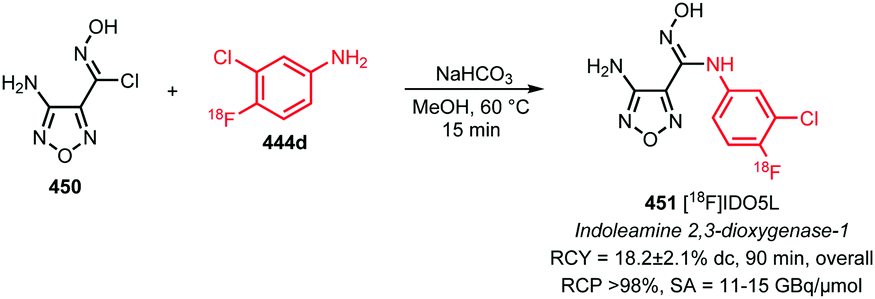 | ||
| Scheme 96 Synthesis of indolamine 2,3-dioxygenase-1 tracer [18F]IDO5L using 3-chloro-4-[18F]fluoroaniline.297 | ||
4-[18F]Fluoroaniline has been used for the synthesis of amides by reaction with acid chlorides. The first reported tracer is [18F]SAHA 453 (Scheme 97).296 The authors reported various efforts to introduce fluorine-18 by late-stage fluorination, but all approaches were unsuccessful. However, with the use of 4-[18F]fluoroaniline as building block they were successful, [18F]SAHA was obtained in a good overall 40% decay-corrected radiochemical yield. Considering this excellent yield for a 4-step synthesis, the question arises whether other strategies are actually needed at all. Stearoyl-CoA tracer 454 was also synthesised by reaction of 4-[18F]fluoroaniline with an acid chloride.202 However, this time only an overall radiochemical yield of 3% (dc) was obtained. The low yield was attributed by the authors to the poor reactivity of 4-[18F]fluoroaniline, since the same acid chloride precursor gave a radiochemical yield of 21% (dc) with the aliphatic 3-[18F]fluoropropylamine.
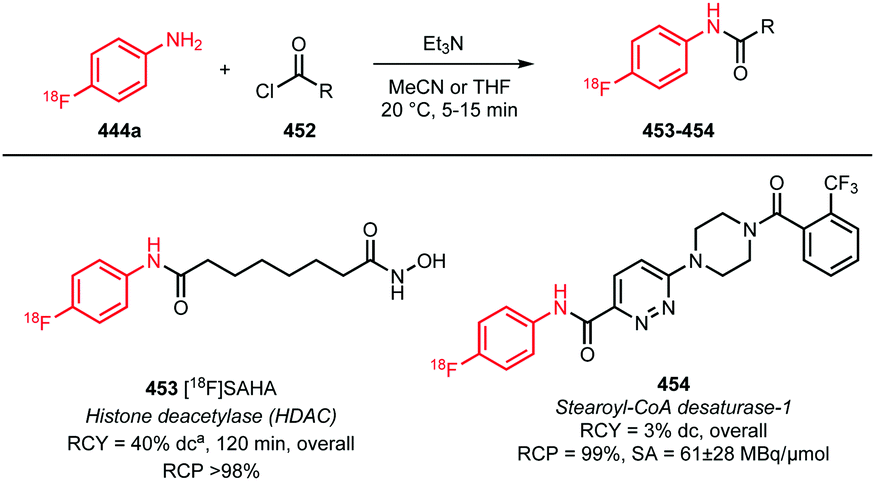 | ||
| Scheme 97 PET tracer synthesis via amide formation with 4-[18F]fluoroaniline; aIncludes formation of the hydroxamide from the methyl ester after the building block is introduced.202,296 | ||
In summary, [18F]fluoroanilines have been successfully applied in the synthesis of multiple PET tracers. The major challenges in using this building block however are in the relative low radiochemical yield caused by the required two step synthesis to produce the building block and the poor nucleophilicity of the aniline.
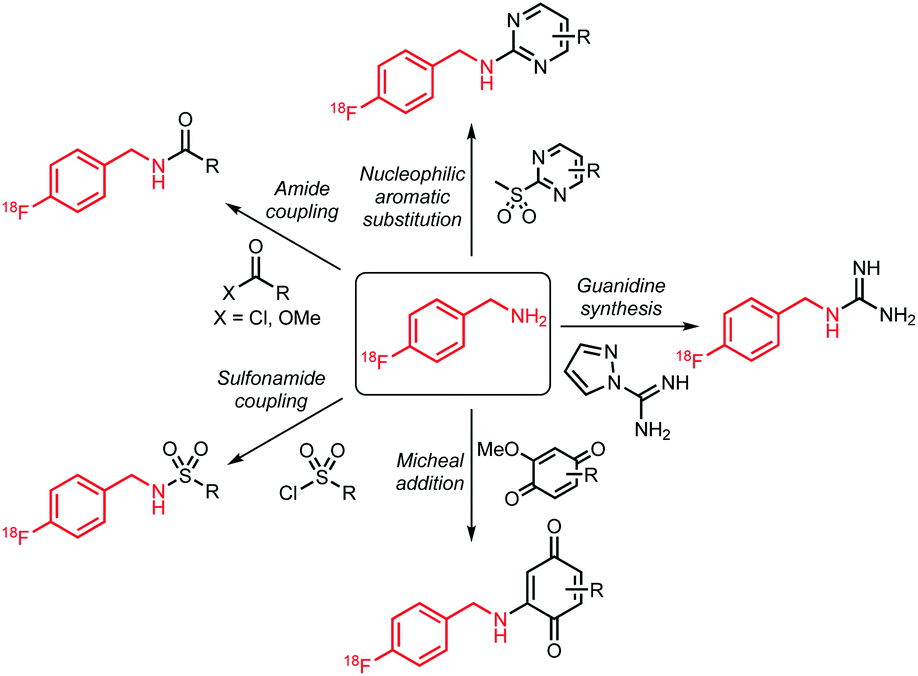 | ||
| Scheme 98 Recent examples of reactions with [18F]fluorobenzylamines. | ||
3.3.2.1 Synthesis of [18F]fluorobenzylamines. [18F]Fluorobenzylamines are generally synthesised from cyanophenyl derivatives, since the cyano functional group provides an electron withdrawing character for a nucleophilic aromatic substitution of [18F]fluoride on 4-N,N,N-trimethylammonium-benzonitrile triflate 455a (Scheme 99).300–304,306,307 After formation of [18F]fluorobenzonitrile, the cyano group is reduced to an amine. Using this synthetic pathway, 4-[18F]fluorobenzylamine 457a can be obtained in a radiochemical yield of 80–95%.
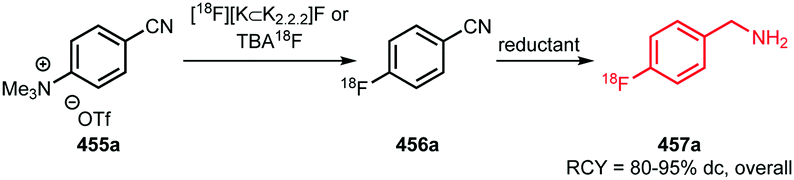 | ||
| Scheme 99 General synthesis route towards [18F]fluorobenzylamines.300–304,306,307 | ||
For the reduction, various reducing agents have been used including LiAlH4, borane dimethyl sulfide and mixtures of sodium borohydride with transition metal salts.300–304,307 Both LiAlH4 and borane dimethyl sulfide resulted in high radiochemical yields of [18F]fluorobenzylamines, but are, for practical reasons, less suited for automated synthesis procedures. Firstly, due to the highly anhydrous reaction conditions which are required and are difficult to achieve using an automated synthesis unit. Secondly, due to the formation of aluminium salts in the case of LiAlH4, which can lead to clogging of transfer lines and filters.
To prevent the issues with the automated synthesis of [18F]benzylamines, Koslowsky et al. and Way et al. developed a new method for the reduction step. By performing the reduction on a cartridge containing borohydride exchange resin (BER), 4-[18F]fluorobenzylamine could be produced in a synthesis unit in >85% decay corrected radiochemical yield in 60 minutes starting from [18F]fluoride.303,307
3.3.2.2 Application of [18F]fluorobenzylamine in (sulfon)amide coupling reactions. Both human immunodeficiency virus 1 integrase (HIV-1 IN) inhibitor 459 and CB2 receptor ligands 460a and 460b have been synthesised utilizing amide coupling reaction of a methyl ester or acid chloride precursor with 4-[18F]fluorobenzylamine (Scheme 100).300,302 In the case of the HIV-1 IN inhibitor 459, the labelled product was obtained in an overall radiochemical yield of 2% (ndc) in 90 minutes synthesis time and in the case of CB2 receptor ligands, 460a and 460b were obtained in an overall radiochemical yield of 15% (dc) in 140 minutes.
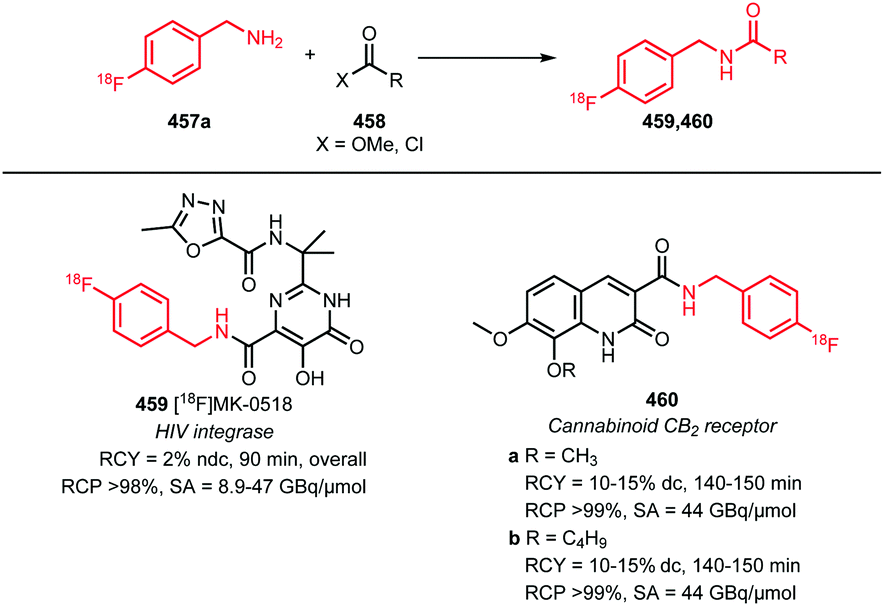 | ||
| Scheme 100 Amide coupling reactions with [18F]fluorobenzylamine.300,302 | ||
4-[18F]Fluorobenzylamine was used for the synthesis of a sulfonamide, via a reaction with sulfonyl chloride 461, to obtain COX-2 tracer 462 in a radiochemical yield of 20% (dc) in 85 min, calculated from 4-[18F]fluorobenzylamine (Scheme 101).305
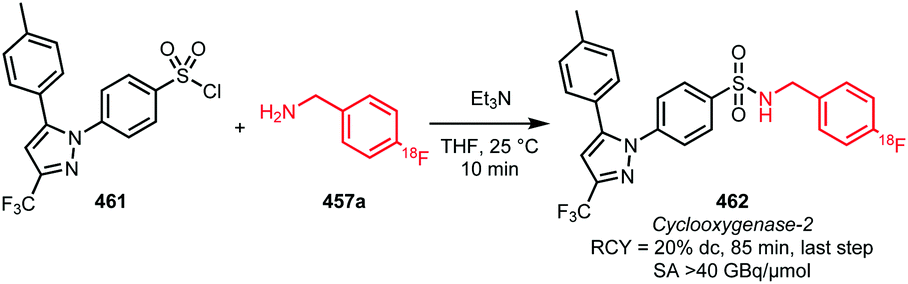 | ||
| Scheme 101 Coupling of [18F]fluorobenzylamine with sulfonyl chloride 461.305 | ||
3.3.2.3 Application of [18F]fluorobenzylamine in Michael addition reactions. The natural product geldanamycin 463 is a potent heatshock protein-90 inhibitor which contains a methoxy quinone moiety. The methoxy quinone can undergo a Michael addition reaction with various primary amines, such as [18F]fluorobenzylamine, to obtain fluorine-18 labelled derivatives (Scheme 102).
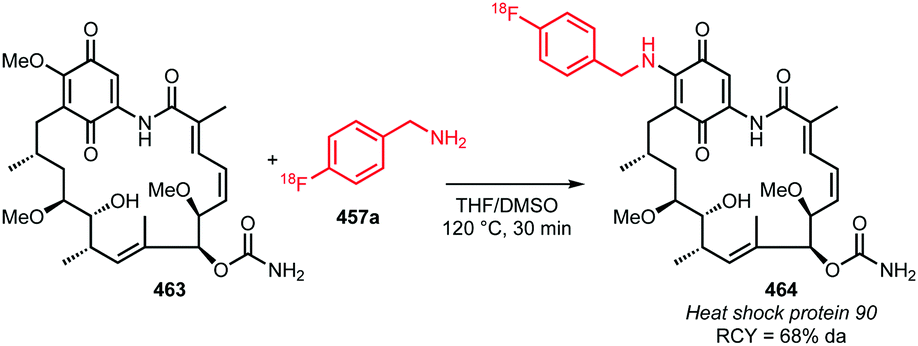 | ||
| Scheme 102 Synthesis of [18F]geldanamycin via Michael addition using [18F]4-fluorobenzylamine.303 | ||
The coupling of 4-[18F]fluorobenzylamine with geldanamycin 463 was investigated by Way et al.303 A radiochemical yield of 68% (determined by radio-TLC) after 30 minutes at 120 °C was reported. The labelled product was not isolated, thus no overall radiochemical yield and synthesis time could be given. However, since the new NaBH4/NiCl2 reduction methodology was used to synthesise 4-[18F]fluorobenzylamine, decent radiochemical yields can be expected despite the multistep procedure.
3.3.2.4 Application of [18F]fluorobenzylamine in nucleophilicaromatic substitution on (methylsulfonyl)-pyrimidines. Tietz et al. reported the synthesis of two COX-2 inhibitors by the nucleophilic aromatic substitution of 4-[18F]fluorobenzylamine on the (methylsulfonyl)pyrimidine moiety of two precursors (Scheme 103).301 Radiochemical yields of the coupling reactions were moderate, 27 ± 11% (dc) for product 466a and 23 ± 1% for product 466b.
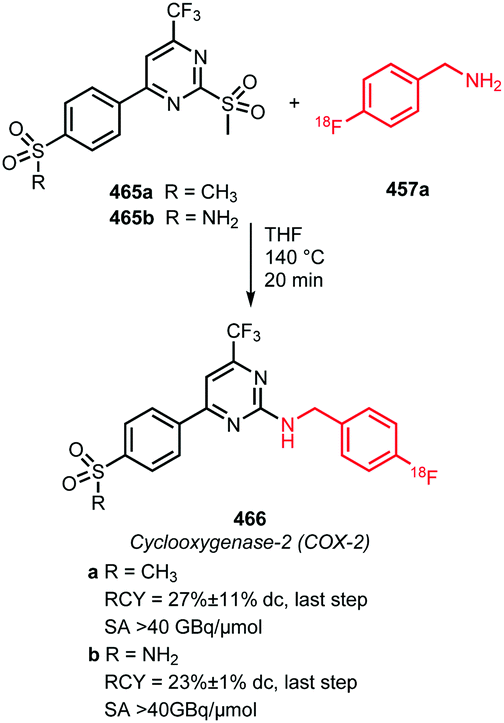 | ||
| Scheme 103 Synthesis of COX-2 inhibitors by nucleophilic aromatic substitution with 4-[18F]fluorobenzylamine.301 | ||
3.3.2.5 Application of [18F]fluorobenzylamine in guanidine synthesis. The first syntheses of the tracers para-[18F]fluorobenzylguanidine [18F]PFBG 468a and meta-[18F]fluorobenzylguanidine [18F]MFBG 468b, using [18F]fluorobenzylamines, were reported by Garg et al. in 1994 as an alternative to the commonly used cardiology and oncology tracer [123I]MIBG.306 Following this publication, two articles were published in 1996 and 2002 in which [18F]PFBG was investigated in rat and dog.308,309 It took until 2014, when Zhang et al. showed a renewed interest in [18F]MFBG and [18F]PFBG.304 For the synthesis of [18F]MFBG and [18F]PFBG, the route used was similar to that of Garg et al. (Scheme 104). A few modifications to the radiolabelling were made to improve the overall radiochemical yields, in particular by using lower reaction temperatures and shorter reaction times for the [18F]fluorobenzonitrile synthesis and by using the more reactive 1H-pyrazole-1-carboximidamide 467 instead of 2-methyl-2-thiopseudourea sulfate for the guanidine formation.
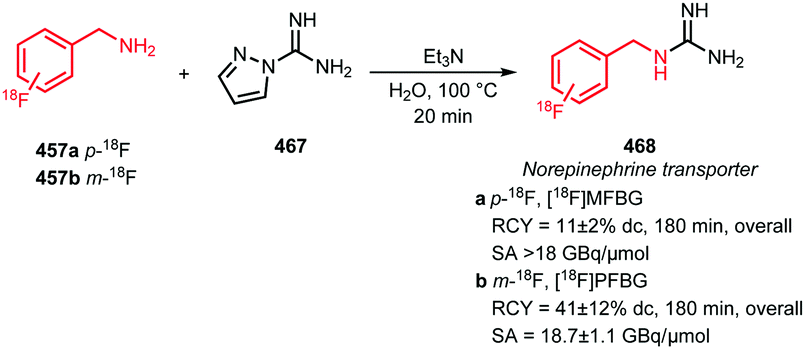 | ||
| Scheme 104 Synthesis of [18F]MFBG and [18F]PFBG.304 | ||
The overall radiochemical yields using the improved synthesis were 11 ± 2% (dc) for [18F]MFBG and 41 ± 12% (dc) for [18F]PFBG. The lower overall radiochemical yield for [18F]MFBG was mainly due to the low radiochemical yield of the synthesis of 3-[18F]fluorobenzonitrile of 21 ± 5% where 4-[18F]fluorobenzonitrile could be synthesised in 75 ± 7%.
In summary, as can be seen by the examples mentioned in Sections 3.3.2.1 to 3.3.2.4, [18F]fluorobenzylamine is a useful building block which was successfully applied in the synthesis of several PET tracers. Nevertheless, the overall radiochemical yields from PET tracer synthesis performed with [18F]fluorobenzylamine are in general moderate to low and the reactions times are quite long, which challenges the use of this methodology.
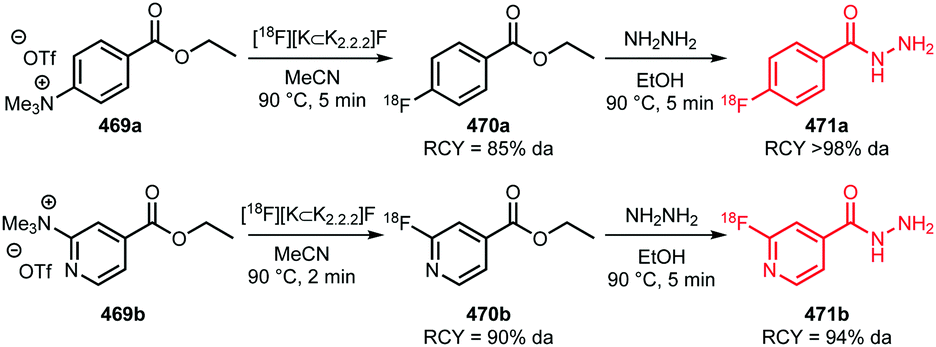 | ||
| Scheme 105 Synthesis of [18F]fluorobenzohydrazide building blocks.310 | ||
These building blocks have recently been applied in the synthesis of fluorine-18 labelled derivatives of methotrexate (Scheme 106).311 In this synthesis, the activated N-succinimidyl-methotrexate carboxylate 472 was reacted with [18F]fluorobenzohydrazide 471a or 471b under mild conditions to obtain the methotrexate derivatives 473a and 473b, labelled with fluorine-18. The overall decay corrected radiochemical yields, starting from [18F]fluoride, were >80% with a synthesis time of 40–45 minutes, which is excellent for a multistep procedure. Furthermore, the products were obtained in >97% radiochemical purity and a specific activity of 11 GBq μmol−1 without the need of a HPLC purification.
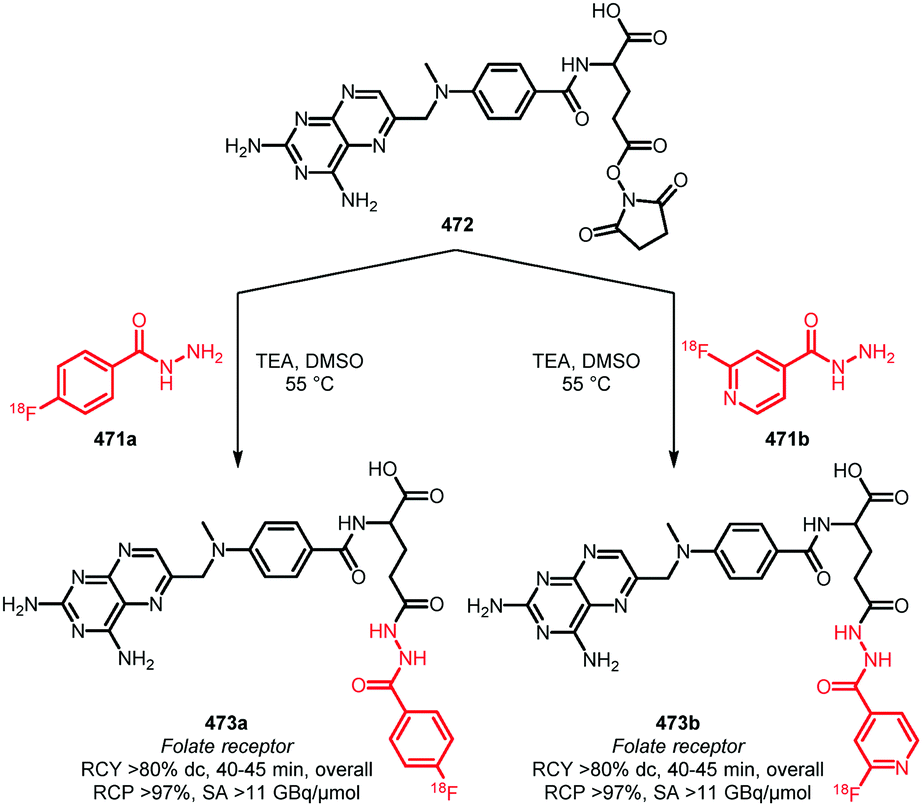 | ||
| Scheme 106 Fluorine-18 labelling of methotrexate using [18F]fluorobenzohydrazide building blocks.311 | ||
The multistep procedure towards fluorine-18 labelled methotrexate is superior to other reported methods in which the PET tracers are synthesised in one step by direct nucleophilic aromatic substitution, because these methods only provide fluorine-18 labelled methotrexate derivatives in overall radiochemical yields of less than 10%.312,313
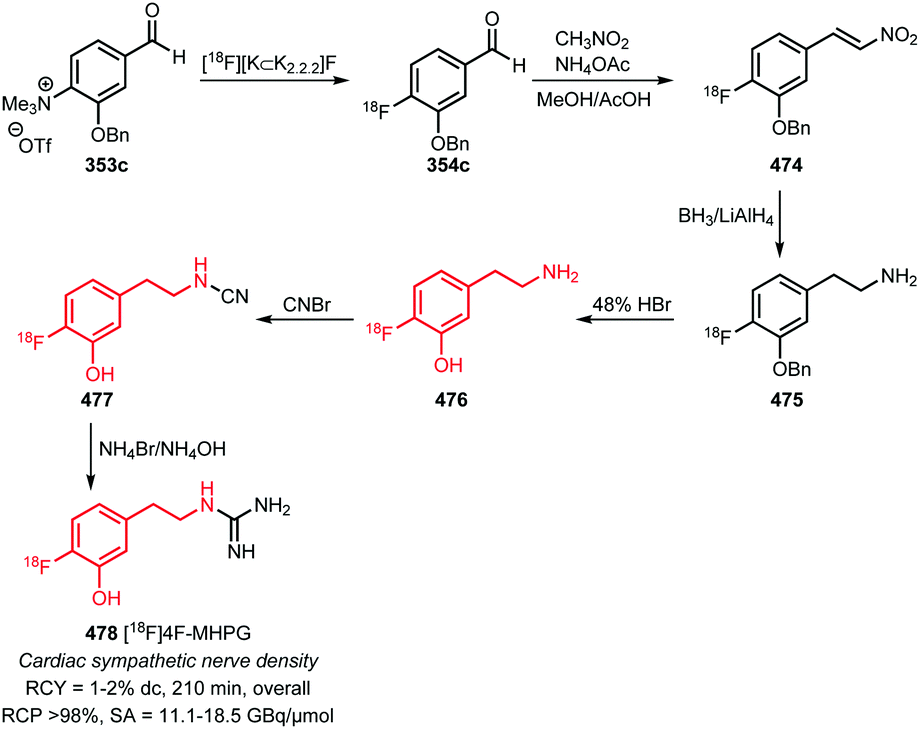 | ||
| Scheme 107 Synthesis of 4-[18F]fluoro-3-hydroxyphenylethylguaninidine in six steps.314 | ||
To improve the overall synthesis time and radiochemical yield, Jang et al. reduced the number of reaction steps to a total of four steps by first synthesizing Boc/benzyl protected phenethylamine building block 480 in one step by 18F-fluorination of iodonium salt precursor 479 (Scheme 108).315 Subsequently the Boc protecting group was removed and the primary amine reacted with reagent 481 to form guanidine 482. In the last step, all remaining protecting groups were removed to obtain PET tracer 478. This time, an increased overall radiochemical yield of 7 ± 3% (dc) was obtained and the synthesis time was decreased to 150 min.
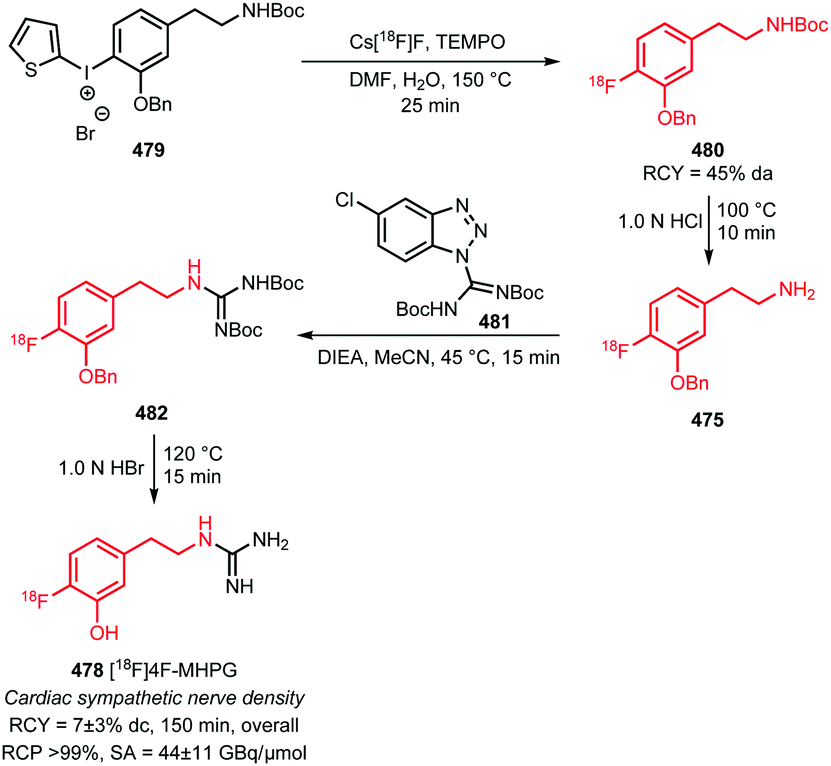 | ||
| Scheme 108 Synthesis of 4-[18F]fluoro-3-hydroxyphenylethylguaninidine in four steps.315 | ||
The second PET tracer with a fluorophenethyl moiety is neuronal nitric oxide synthase (nNOS) tracer 488. This tracer was obtained with 3-[18F]fluorophenethyl amine 485 (Scheme 109),316 which was synthesised in two steps by radiofluorination of Boc-protected iodonium ylide precursor 483 and subsequent removal of the Boc protecting group with HCl in dioxane. 3-[18F]Fluorophenethylamine was subsequently coupled to aldehyde 486 by reductive amination, followed by deprotection of the primary amine to obtain 488. Using this strategy, PET tracer 488 could be obtained in an overall radiochemical yield of 15% (dc). This procedure was not further optimised due to issues with reproducibility of the reductive amination step and because promising results were obtained with the novel late-stage radiofluorination of boronic acid pinacol esters (Table 1, entry 9).
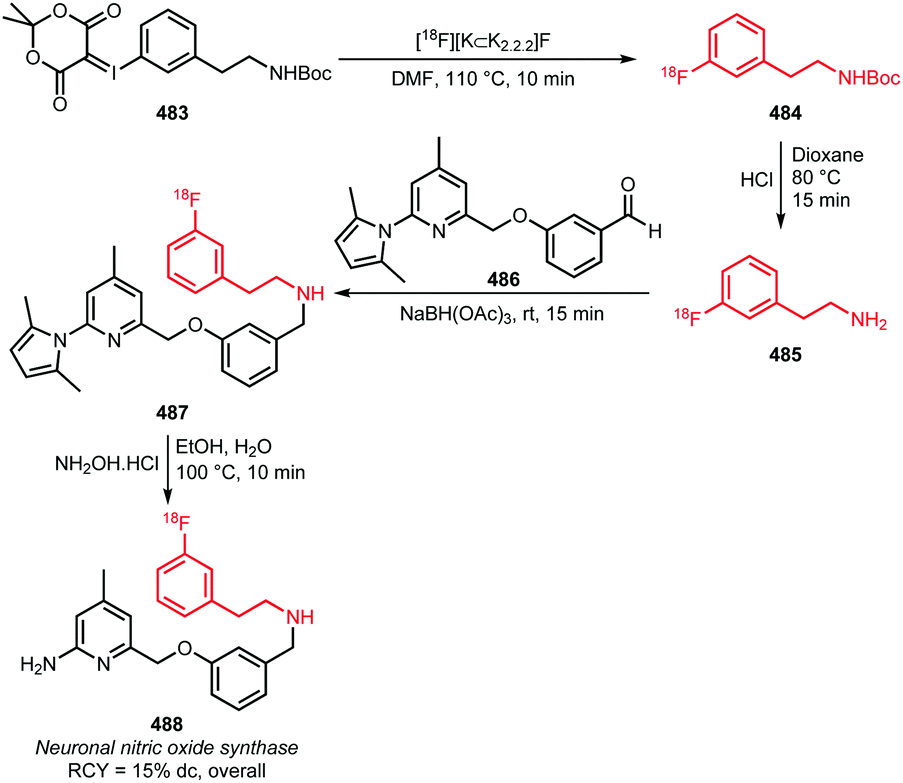 | ||
| Scheme 109 Building block approach towards neuronal nitric oxide synthase tracer 488.316 | ||
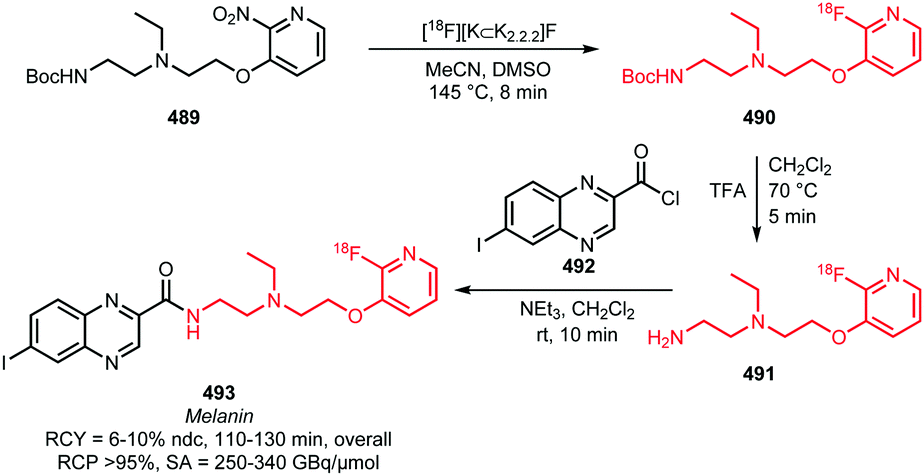 | ||
| Scheme 110 Multistep radiosynthesis of fluorine-18 labelled melanoma PET tracer 493.317 | ||
3.4 [18F]Fluorobenzoic acid & [18F]fluorobenzoic acid esters
[18F]Fluorobenzoic acids and [18F]fluorobenzoic acid esters are often applied in reactions with amines to form amides. Activated esters are either formed in situ from [18F]fluorobenzoic acid (Section 3.4.1), or the active esters are isolated before use, as is the case with [18F]SFB (Section 3.4.2) and [18F]6-fluoronicotinic acid 2,3,5,6-tetrafluorophenyl ester ([18F]FPy-TFP, Section 3.4.3).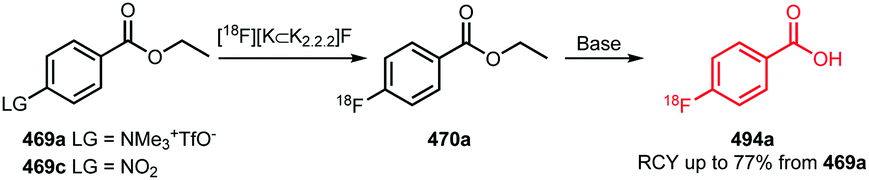 | ||
| Scheme 111 Synthesis of 4-[18F]fluorobenzoic acid.163,318,319 | ||
In these recent articles, no radiochemical yields of the obtained 4-[18F]fluorobenzoic acid are mentioned, however, in publications from before 2010, radiochemical yields up to 77% are described when started from trimethylammonium triflate precursor 469a.320
Various coupling reagents can be used to activate benzoic acid for nucleophilic substitution. Caroll et al. used BOP 495 together with N,N-diisopropylethylamine as a base to couple [18F]4-fluorobenzoic acid to hydrazine derivative 497, followed by a Cu for Zn exchange resulting in hypoxia tracer bis(thiosemicarbazonate) complex 498-Cu in 32% radiochemical yield, starting from 4-[18F]fluorobenzoic acid (Scheme 112).163
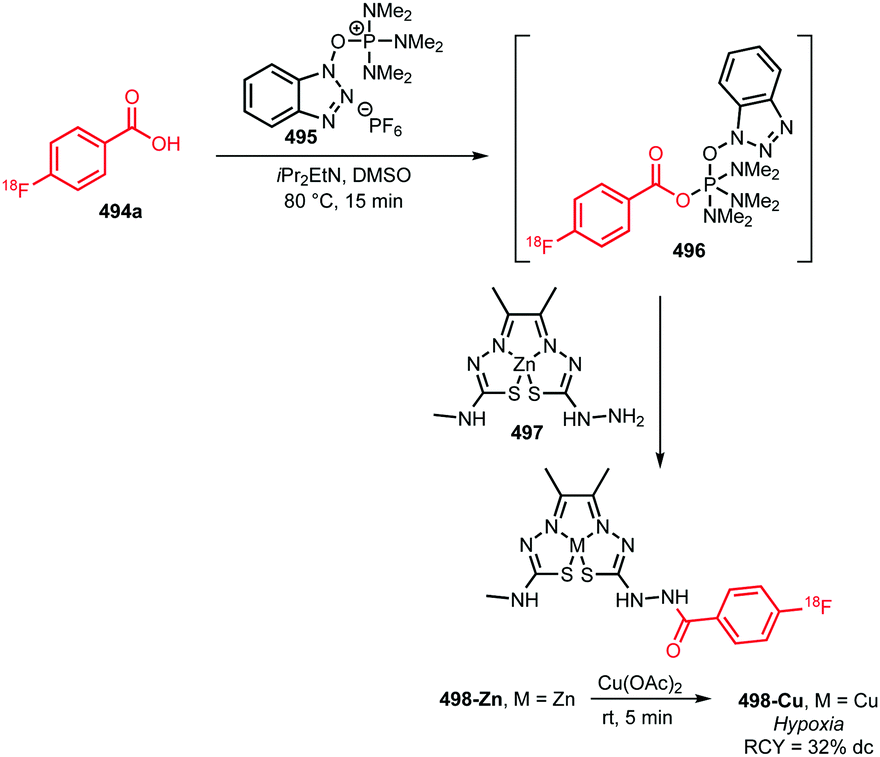 | ||
| Scheme 112 Activation of 4-[18F]fluorobenzoic acid with BOP and coupling to hydrazine derivative 497.163 | ||
Another coupling agent to activate 4-[18F]fluorobenzoic acid, N,N′-dicyclohexylcarbodiimide (DCC) 499, is applied by Ackermann et al. for the synthesis of fluorine-18 labelled naphthoquinone as a PET tracer for hypoxia (Scheme 113).318 This approach resulted in the desired labelled compound 502 in a moderate radiochemical yield of 27 ± 5% starting from 4-[18F]fluorobenzoic acid, showing that DCC can be used efficiently as reagent to couple 4-[18F]fluorobenzoic acid with primary alcohols.
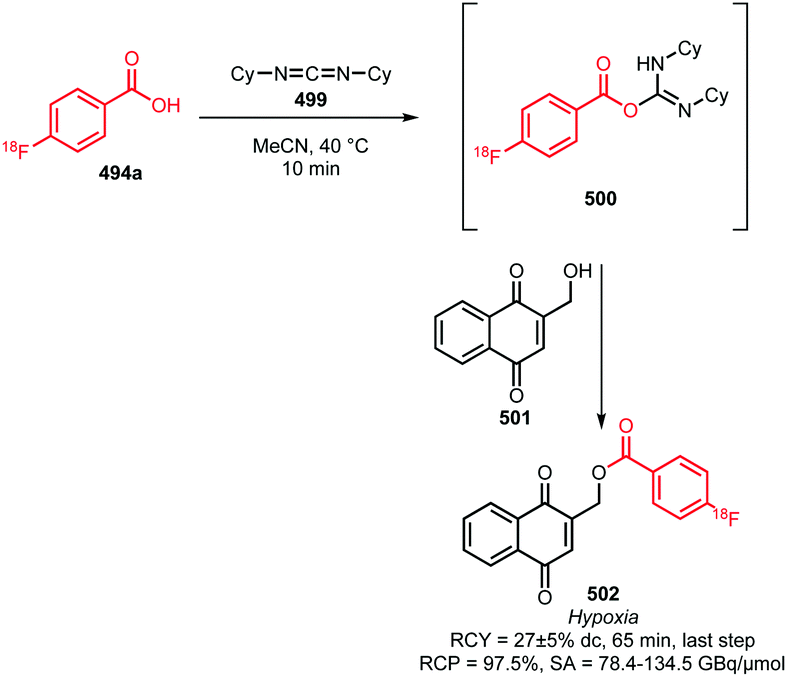 | ||
| Scheme 113 Activation of 4-[18F]fluorobenzoic acid with DCC and coupling to alcohol 501.318 | ||
The last and most recent example of the use of a coupling agent to activate 4-[18F]fluorobenzoic acid is in the synthesis of PARP1 inhibitor [18F]PARPi 506 by activation of 4-[18F]fluorobenzoic acid using HBTU and subsequent reaction with secondary amine precursor 505 in an overall radiochemical yield of 10% (ndc) (Scheme 114).319 This is a low but acceptable radiochemical yield, considering the 4-[18F]fluorobenzoic acid is reacted with a bulky secondary amine.
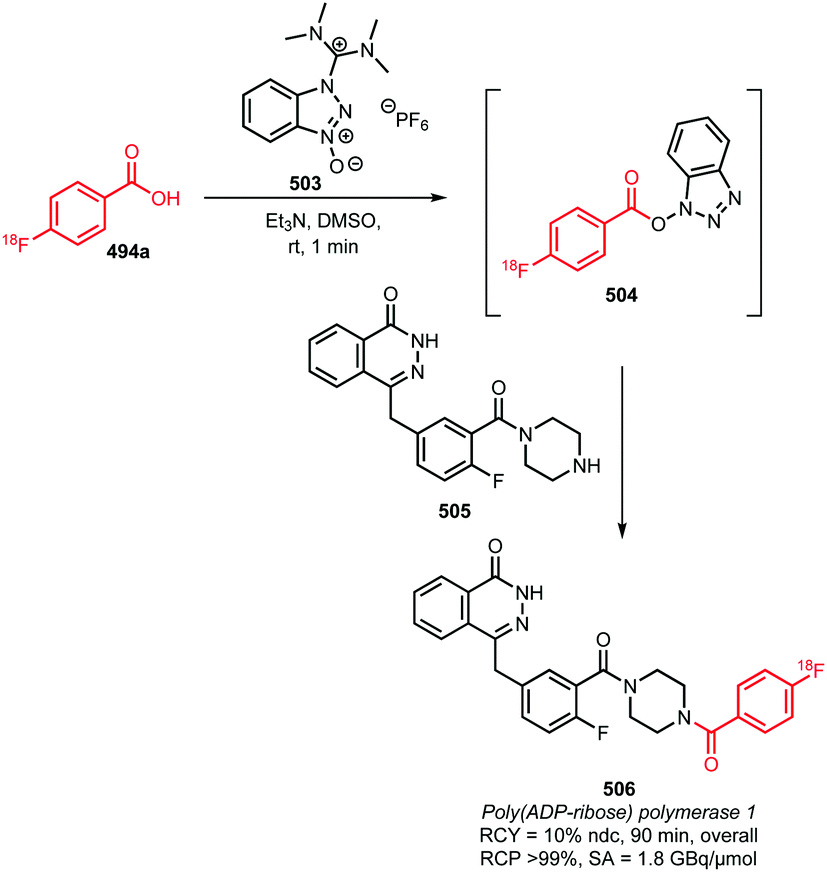 | ||
| Scheme 114 Activation of 4-[18F]fluorobenzoic acid with HBTU and coupling to secondary amine 505.319 | ||
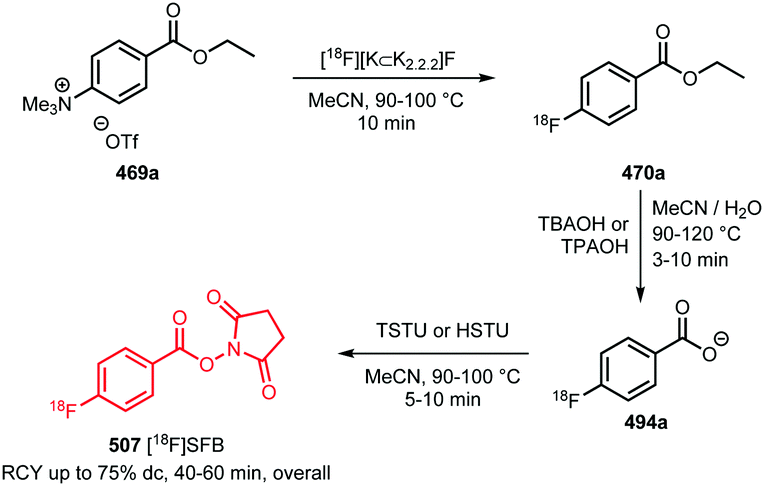 | ||
| Scheme 115 One pot, three step synthesis of [18F]SFB.84,93,322–326 | ||
Purification of [18F]SFB is performed by trapping on a C18 SPE cartridge, washing the cartridge with water and eluting the building block in an organic solvent of choice, preferably through alumina and SCX cartridges to remove any remaining [18F]fluoride and other impurities. With these methods, decay corrected radiochemical yields can be obtained up to 75%, and due to the absence of time consuming HPLC purifications and the use of just one SPE purification, the overall synthesis time can be less than 40 minutes.
The second method to synthesise [18F]SFB is a two-pot procedure, in which 4-[18F]fluorobenzoate 494a is formed by hydrolysis of ethyl 4-[18F]fluorobenzoate 470a with NaOH or HCl, which is subsequently purified by SPE before formation of [18F]SFB.327–329 This method is however not recommended, as it only results in longer synthesis times and lower radiochemical yields, without having any advantages over the one-pot procedure.
The third method to synthesise [18F]SFB is a very different, two-step approach, first reported by Glaser et al. in 2009 and which is recently used by Ganguly et al. (Scheme 116).330,331 In this method, purified 4-[18F]fluorobenzaldehyde 354a, is oxidised with (diacetoxyiodo)benzene in the presence of N-hydroxysuccinimide (NHS). HPLC purification is necessary to obtain [18F]SFB in sufficient radiochemical purities (>99%) for further reactions. Glaser et al. reported a decent overall radiochemical yield of 66 ± 6% (dc), which could however not be reproduced by Ganguly et al., reporting only a 25% (dc) overall radiochemical yield. Although this method only requires two synthetic steps, it still seems that the three-step, one-pot procedure as depicted in Scheme 115 is currently the most convenient, as it is more simple to purify [18F]SFB.
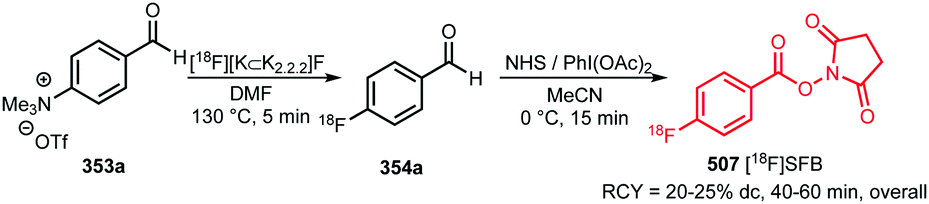 | ||
| Scheme 116 Two-step synthesis of [18F]SFB.330,331 | ||
As a building block for the synthesis of low molecular weight PET tracers, [18F]SFB is used solely in the base catalysed acylation of primary amine precursors, as shown in Scheme 117.84,93,322–329,331–336 The main advantages of labelling amine precursors with [18F]SFB over other fluorine-18 labelled building blocks such as 4-[18F]fluorobenzaldeyde and [18F]FETos is that the acylation with [18F]SFB can be performed under very mild reaction temperatures (typically 20–50 °C, mild basic) and with very high selectivity for the primary amine functional group. Due to the high selectivity of [18F]SFB towards primary amines, protection of other functional groups in the precursor molecule is generally not required (Table 2). As a consequence, the precursors are easier to synthesise and no removal of the protecting groups is necessary afterwards.
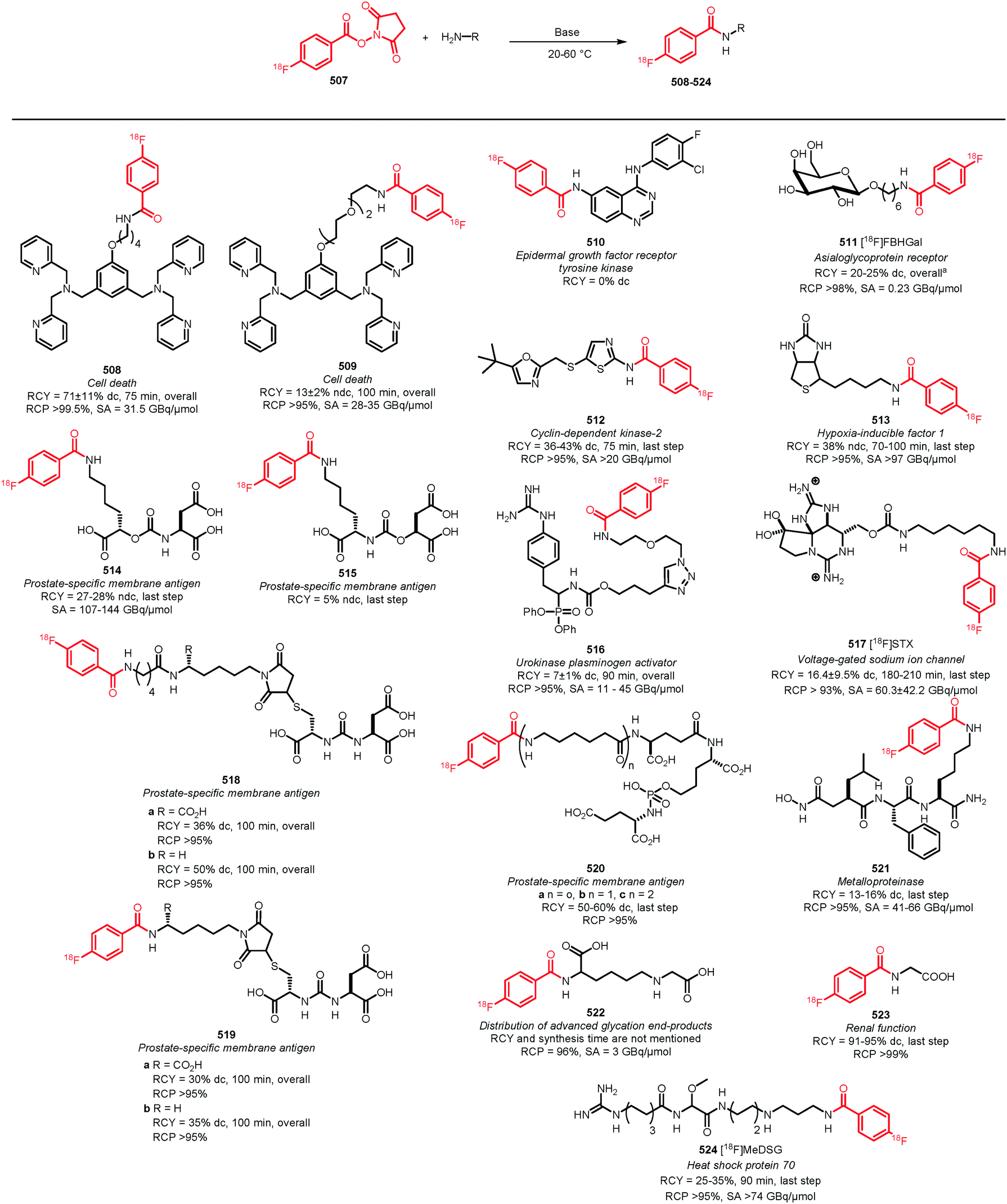 | ||
| Scheme 117 PET tracers synthesised by amide formation using [18F]SFB. aIncludes removal of acetyl protecting groups.84,93,322–329,331–336 | ||
Typically, reaction of primary amines with [18F]SFB are performed underbasic conditions. Interestingly, for this reaction not just one type of base, but a wide range of bases in various solvents are reported. Two categories for the solvents and bases can be identified:
(1) Reactions in water using water soluble bases or buffers, including borate buffer, carbonate buffer, sodium phosphate and potassium carbonate. Organic solvents, generally MeCN, can be added to increase solubility of the precursors. These conditions are used when the precursor is highly water soluble.93,323,325,327–329,331,335
(2) Reactions in organic solvents (DMSO, DMF, MeCN), using bases which readily dissolve in these solvents (DIPEA, NEt3). These conditions are used when the precursor is insoluble in water.84,322,326,332,336
Two recent publications compared the reaction of [18F]SFB, [18F]fluoroethyl tosylate and 2-[18F]fluoro-4-nitrophenyl-propionate [18F]NFP with amine precursors 525 and 526 in the synthesis of pycolylamine based cell death imaging agents (Fig. 12 and Table 3).84,93
 | ||
| Fig. 12 Alkylamine modified picolylamine derivatives.84,93 | ||
| Precursor | Building block | Labelling conditions | Overall RCY (dc) | Overall synthesis time (min) |
|---|---|---|---|---|
| 525 | [18F]SFB | DMSO, DIPEA, RT, 10 min | 71 ± 11% | 75 |
| [18F]FETos | DMSO, DIPEA, 100 °C, 10 min | 76 ± 13% | 65 | |
| [18F]NFP | RT, 10 min | 68 ± 9% | 105 | |
| 526 | [18F]SFB | Borate buffer pH 8.5, 50 °C, 10 min | 24 ± 4% | 100 |
| [18F]FETos | MeCN, K2CO3, 120 °C, 30 min | 17 ± 2% | 105 | |
Labelling of these precursors with [18F]SFB resulted in fluorine-18 labelled derivatives 508 and 509 (Scheme 117) in overall radiochemical yields of 71 ± 11% (dc) and 13 ± 2% (ndc), respectively. Comparable radiochemical yields were obtained when the precursors were reacted with [18F]FETos at 100 °C and 2-[18F]fluoro-4-nitrophenylpropionate ([18F]NFP) at room temperature. This indicates that precursors 525 and 526 are stable under high temperatures. The reason for the large difference in radiochemical yield between labelling precursor 525versus526 was not explained.
In general, [18F]SFB is used to label aliphatic amines, however there are two recent examples of labelling aniline derivatives. Neto et al. tried to synthesise a PET tracer for imaging EGFR tyrosine kinase 510 (Scheme 117).333 Unfortunately, reaction of the aniline precursor with [18F]SFB in DMSO using various buffers did not lead to the formation of 510. Because stronger alkaline conditions are probably required to facilitate labelling of aniline derivatives, the pH was increased to >9, this however resulted in undesired rapid hydrolysis of [18F]SFB. Svensson et al., however, have successfully reacted their aromatic amine precursor with [18F]SFB in the synthesis of cyclin-dependent kinase-2 inhibitor 512 in a decent radiochemical yield of 36–43% (dc).334 A major difference from the method of Neto et al. is that the reaction is performed under water-free conditions using NaH as a base. Due to the absence of water, [18F]SFB does not hydrolyse and is able to react with the amine.
Derivatives of [18F]SFB can potentially be made by changing the position of the fluorine-18 atom and by the addition of substituents on the aromatic ring. Yang et al. used this strategy to improve the properties of Prostate-Specific Membrane Antigen tracers 514 and 515.324 By changing the location of the fluorine-18 atom to the 2-position and adding a bromine or iodine atom to the 4-position, they were able to produce tracers 529a and 529b, which have increased PSMA binding, presumably due to increased interaction with a hydrophobic subpocket in the enzyme (Scheme 118).
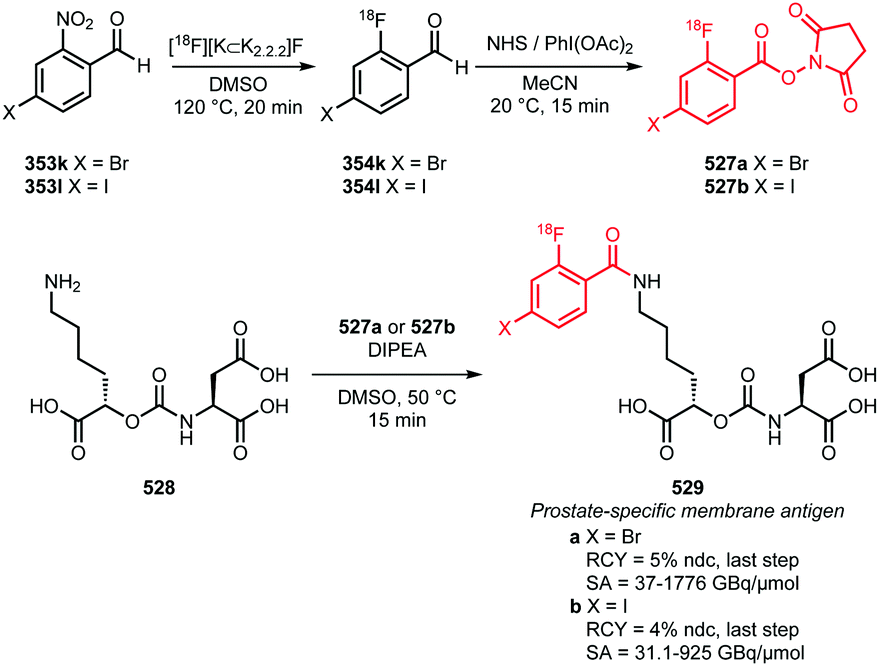 | ||
| Scheme 118 PET tracers for imaging PSMA, labelled using [18F]SFB derivatives.324 | ||
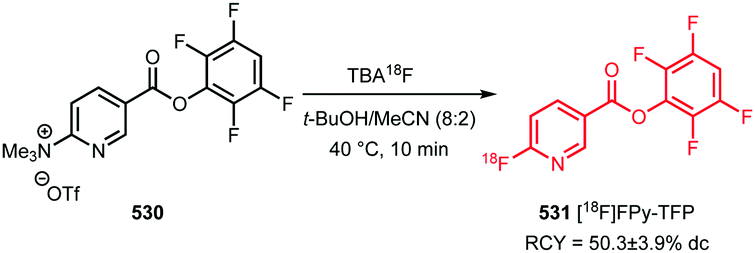 | ||
| Scheme 119 Synthesis of [18F]FPy-TFP.337 | ||
Besides labelling of large peptides, [18F]FPy-TFP is also used for the labelling of small molecules, more specifically, in the labelling of PSMA targeting tracers (Scheme 120).338,339 The carboxylic acids in the precursor were protected with 4-methoxybenzyl ether (PMB) protecting groups before reaction with [18F]FPy-TFP in case of tracer 532, therefore requiring an acidic deprotection after the coupling.338
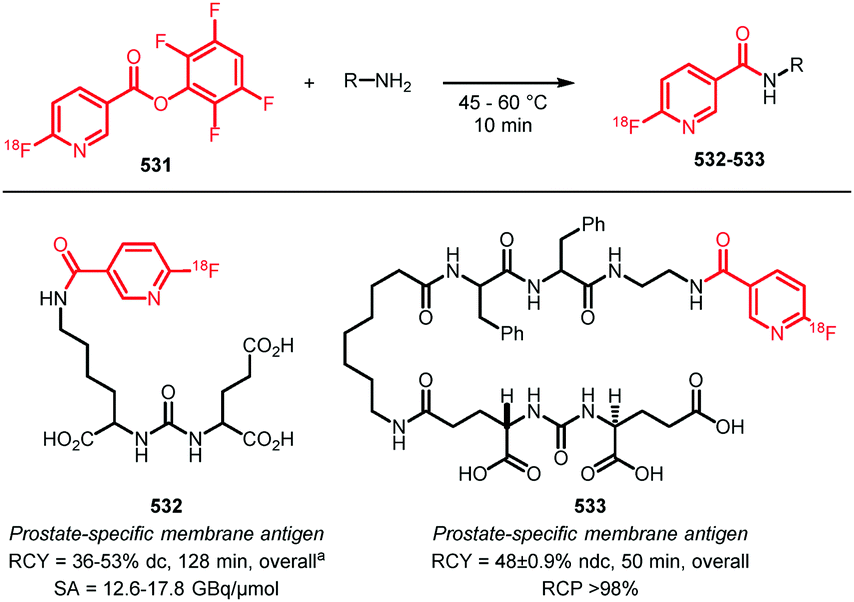 | ||
| Scheme 120 Synthesis of PSMA targeting tracers using [18F]FPy-TFP. aIncludes deprotection of the PMB protected carboxylic acids using trifluoroacetic acid.338,339 | ||
Protection of the carboxylic acids is however not required, as seen in the synthesis of tracer 533, in which the coupling went smoothly while the carboxylic acid functional groups were unprotected.339 Most probably because [18F]FPy-TFP already contains an activated ester, therefore no additional coupling reagents are needed.
In conclusion, both PSMA targeting tracers could be made in decent overall radiochemical yields, showing that [18F]FPy-TFP is also a suitable building block for the synthesis of low molecular weight PET tracers.
3.5 Other fluorine-18 labelled aromatic building blocks
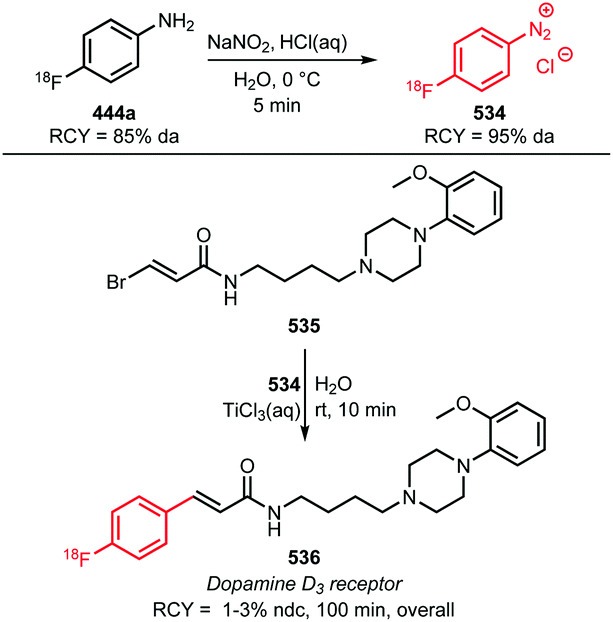 | ||
| Scheme 121 Synthesis of dopamine D3-selective ligand [18F]SH317 using 4-[18F]fluorophenyldiazonium chloride.294 | ||
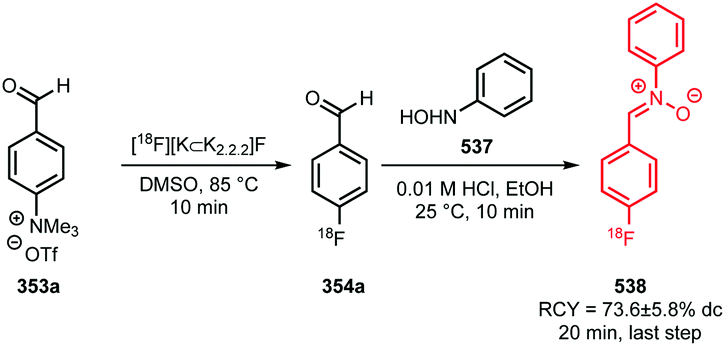 | ||
| Scheme 122 Synthesis of N-phenyl-C-(4-[18F]fluorophenyl) nitrone.340,341 | ||
Zlatopolskiy et al. investigated the reaction of N-phenyl-C-(4-[18F]fluorophenyl) nitrone 538 with a variety of N-substituted maleimides, which can be easily introduced in peptides (Scheme 123).340 The radiochemical yields towards the mixtures of endo and exo bicyclic isoxazolidines 540a–c, as measured by HPLC, were 87% for 540a, 91% for 540b and 91% for 540c. The endo/exo ratio was around 2:1 for compounds 540a–c.
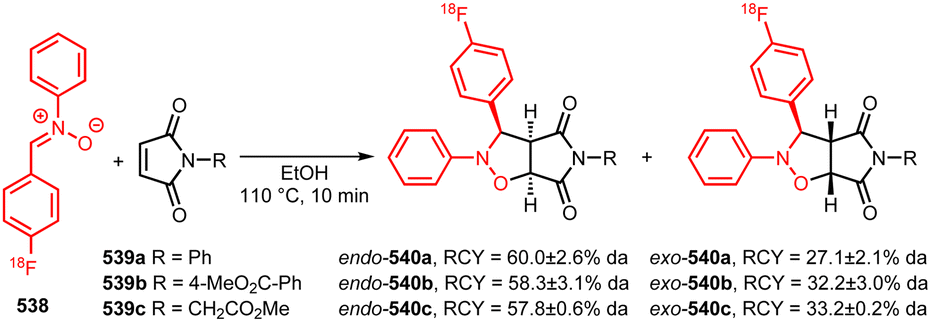 | ||
| Scheme 123 Reaction of N-phenyl-C-(4-[18F]fluorophenyl) nitrone with N-substituted maleimides.340 | ||
Furthermore Zlatopolskiy et al. investigated the Kinugasa reaction by reacting alkynes 541a–d with N-phenyl-C-(4-[18F]fluorophenyl) nitrone 538 under copper catalysis resulting in β-lactams 542a–d (Scheme 124).341 Radiochemical yields of 65–89% (analytically determined) were obtained with trans–cis ratios varying between 2:3 and 1:5 depending on the used alkyne.
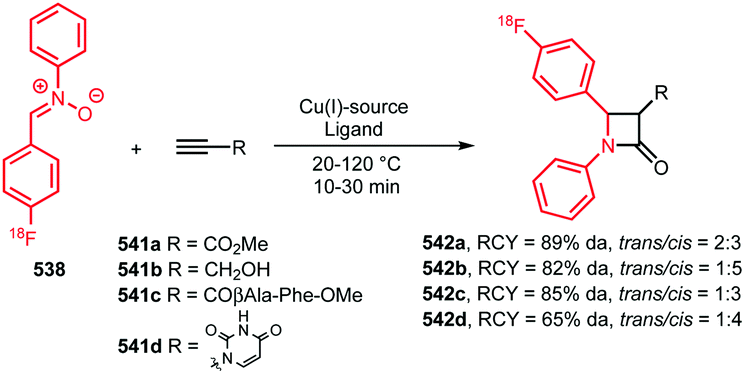 | ||
| Scheme 124 Application of N-phenyl-C-(4-[18F]fluorophenyl) nitrone in the synthesis of fluorine-18 labelled β-lactams.341 | ||
Both the reaction towards isoxazolidines as well as the Kinugasa reaction can potentially be used for the labelling of peptides equipped with maleimide or terminal alkyne functional groups, as long as the high reaction temperature of 110–120 °C would not lead to degradation of the peptides. For the synthesis of low molecular weight PET tracers these reactions could also be useful for the synthesis of PET tracers with an isoxazolidine or β-lactam core structure.
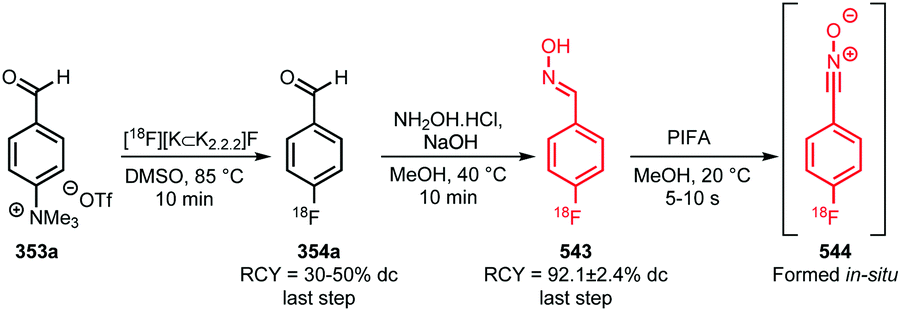 | ||
| Scheme 125 Three step synthesis of the building block 4-[18F]fluorophenyl nitrile oxide.342 | ||
4-[18F]Fluorophenyl nitrile oxide 544 can be synthesised in three steps. Firstly, 4-[18F]fluorobenzaldehyde is synthesised, (Section 3.1.1) followed by reaction with hydroxylamine and sodium hydroxide yielding benzaldoxime 543 in 92.1 ± 2.4% (ndc) in 10 minutes. Because 4-[18F]fluorophenyl nitrile oxide 544 is very reactive, it was not isolated but formed in situ and reacted in one pot with various dipolarophiles.
Initial studies in which 4-[18F]fluorophenyl nitrile oxide 543 was formed in situ from oxime 543 and reacted with various model dipolarophiles showed high radiochemical yields of 57–95% as measured by HPLC. To prove that this building block is also suitable for the synthesis of PET tracers for COX-2, it was reacted with three indomethacin derivatives which contain a dipolarophile functional group (Scheme 126). Fluorine-18 labelled indomethacin derivatives 545 and 546 could be isolated in 86% and 55% (ndc) radiochemical yield, starting from building block 543. 18F-Indomethacin derivative 547 was less easily formed and could initially only be obtained in a low radiochemical yield. However, when the oxidant in the reaction was changed from phenyliodine bis(trifluoroacetate) (PIFA) to [bis(acetoxy)iodo]-benzene (BAIB), this indomethacin derivative could also be obtained in a radiochemical yield of 35% (ndc), starting from 543. Since BAIB is a weaker oxidant, leading to the slower generation of 4-[18F]fluorophenyl nitrile oxide 544 from oxime 543, the alkene precursor has more time to react before nitrile oxide 544 decomposes by acid-promoted decomposition, solvolysis or reaction with contaminants.
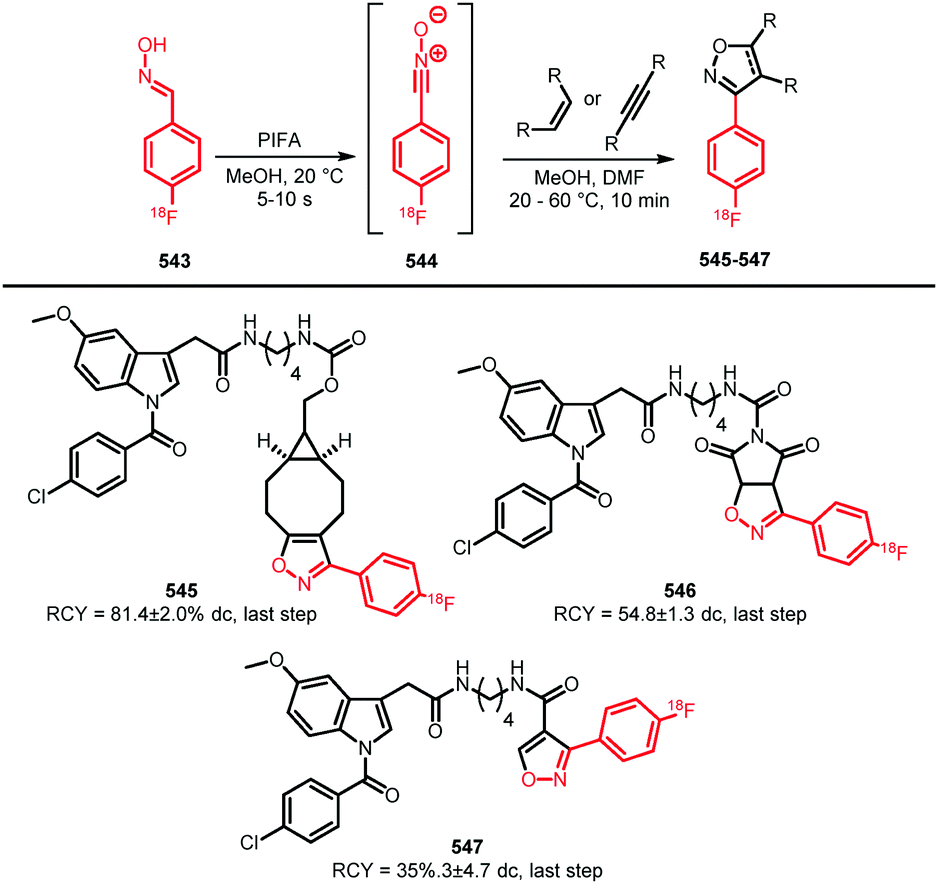 | ||
| Scheme 126 Application of 4-[18F]fluorophenyl nitrile oxide 544 in the synthesis of fluorine-18 labelled indomethacin.342 | ||
Whether this building block is useful for low molecular weight PET tracers remains unclear, as its multistep synthesis is time consuming and results in low to moderate radiochemical yields. For the labelling of biomolecules, such as peptides it can be beneficial because of the mild reaction conditions, regiospecificity and good cycloaddition yields. Zlatapolsky et al. did however notice that the amount of precursor needed for acceptable cycloaddition radiochemical yields was high, leading to low specific activities as it is generally difficult to separate a large biomolecule precursor from its labelled product. This issue was solved by in situ conversion of 4-[18F]fluorobenzaldoxime 543 to an imidoyl chloride by treatment with chloramine-T. Using this more stable derivative of 4-[18F]fluorobenzaldoxime 543, the amount of required precursor could be lowered to 5 nmol, thus making this method also useful for the labelling of biomolecules.
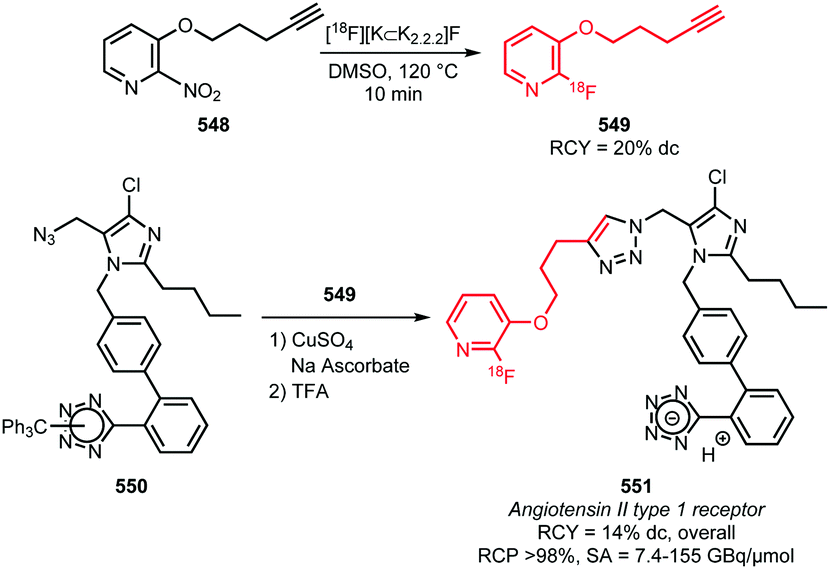 | ||
| Scheme 127 Synthesis of a fluorine-18 labelled losartan derivative using [18F]FPyKYNE as a building block.343,344 | ||
Arksey et al. showed that [18F]FPyKYNE 549 can be used as a building block in the synthesis of a fluorine-18 labelled derivative of the AT1 inhibitor losartan (Scheme 127).344 The ‘click’-reaction of [18F]FPyKYNE 549 with the azide modified losartan 550, followed by trityl deprotection, proceeded in good radiochemical yields of 44–70% (dc). Unfortunately, the overall radiochemical yield starting from [18F]fluoride was quite low (7–14% dc) due to the low yielding aromatic nucleophilic substitution on nitro precursor 548.
A recent publication by Roberts et al. describes the synthesis of another fluorine-18 labelled alkyne building block: (4-[18F]fluorophenyl)acetylene 553a, which can be obtained by direct labelling of its trimethylammonium precursor (Scheme 128).345 Although the aromatic ring is only marginally electron deficient, the (4-[18F]fluorophenyl)acetylene building block could still be obtained in a radiochemical yield of 14% (ndc). Purification of this building block was done by HPLC, because SPE methods did not lead to desired radiochemical purities of this reagent.
 | ||
| Scheme 128 Synthesis of (4-[18F]fluorophenyl)acetylene by direct nucleophilic aromatic substitution with [18F]fluoride.345 | ||
To demonstrate the application of (4-[18F]fluorophenyl)acetylene in the synthesis of PET tracers, Roberts et al. reacted this building block with a variety of azide precursors (Scheme 129).345 Compounds 554 and 555 were formed in radiochemical yields of 67% and 56% (analytically determined) respectively. Unfortunately, overall non-decay corrected radiochemical yields, based on starting [18F]fluoride were low due to the low yielding synthesis of 4-([18F]fluorophenyl)acetylene 553a.
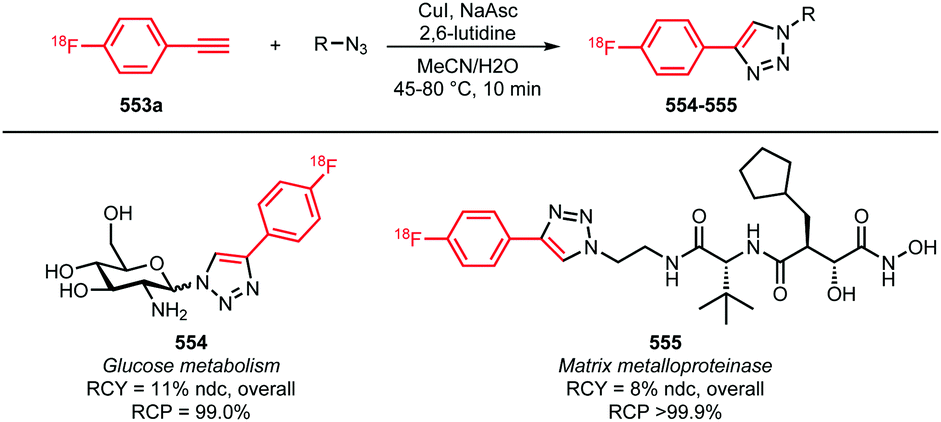 | ||
| Scheme 129 ‘Click’-reaction with (4-[18F]fluorophenyl)acetylene.345 | ||
A different approach towards the synthesis of ([18F]fluorophenyl)acetylenes was recently published by Krapf et al. (Scheme 130).346 Instead of a direct labelling approach, they reported a two-step method, consisting of first the synthesis of [18F]fluorobenzaldehydes 363a,b,k in 20–75% radiochemical yield (ndc) and subsequent Seyferth–Gilbert Homologation towards ([18F]fluorophenyl)acetylenes 553a–c using the Bestmann–Ohira reagent in 40–60% radiochemical yield (ndc). The two step approach seems more promising than the direct approach employed by Roberts et al.,345 as exemplified by the overall radiochemical yield for (4-[18F]fluorophenyl)acetylene of 26–45% instead of 14 ± 2%. For the purification of (4-[18F]fluorophenyl)acetylene, Krapf et al. discovered that radiochemical purities of >98% can be achieved by distillation, thereby avoiding cumbersome HPLC purification.346
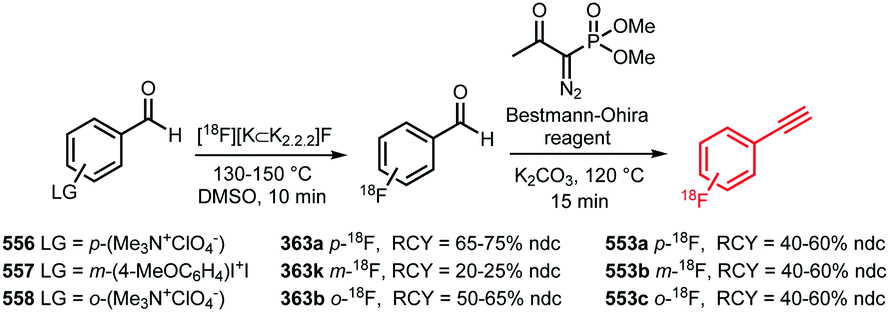 | ||
| Scheme 130 Synthesis of ([18F]fluorophenyl)acetylenes by Seyferth–Gilbert homologation.346 | ||
Using small model substrates, it was demonstrated that these alkynes can participate successfully in (1) click reactions with various dipoles (RCY = 20–53%, determined analytically), (2) the Sonogashira reaction (RCY = 83%, determined analytically) and (3) alkyne trimerisation (RCY = 18%, determined analytically). As a proof of principle, (2-[18F]fluorophenyl)acetylene was reacted with azides in the click reaction towards potential COX-2 PET tracer 559 and PSMA PET tracer 560 in a reasonable overall radiochemical yield of 30% and a short 60 min overall synthesis time (Scheme 131).346
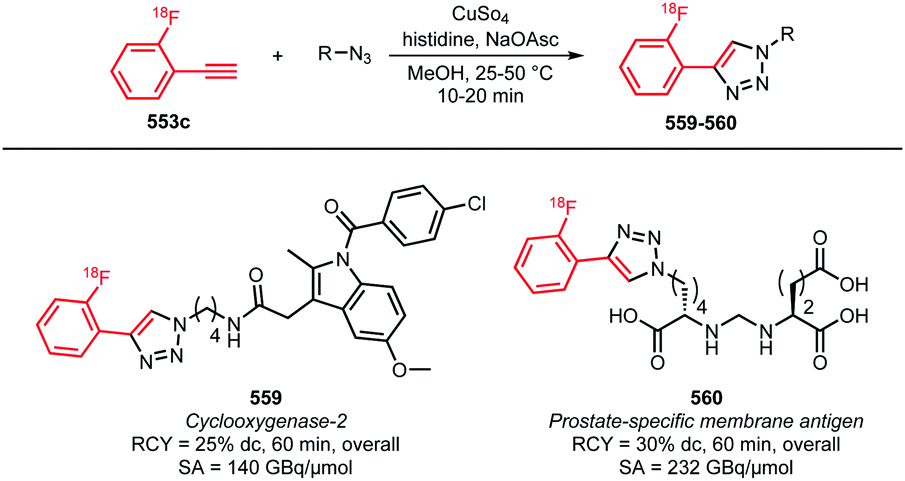 | ||
| Scheme 131 Click reaction with (2-[18F]fluorophenyl)acetylene.346 | ||
In summary, ([18F]fluorophenyl)acetylenes are a new class of building blocks with high versatility. The building blocks can be synthesised in decent radiochemical yields and reacted in cycloadditions as well as transition metal catalysed reactions.
Although [18F]fluorophenyl substituted azides have interesting properties for the use as a reagent in click reactions, there are no recent publications on the use of these building blocks in the synthesis of small molecule PET tracers. The obvious reason is the challenge to synthesise these building blocks. The azide functional group is not a strong electron withdrawing group. Therefore, the synthesis of 2- and 4-[18F]fluorophenyl azide by conventional nucleophilic aromatic substitution with [18F]fluoride leads only to very low radiochemical yields.347 When the azide functional group is attached to the aromatic ring via an aliphatic chain, as in for example [18F]fluorobenzyl azides, the electron density on the aromatic ring is too high to allow successful conventional nucleophilic aromatic substitution. Because [18F]fluorophenyl substituted azides would be a valuable addition to the radiochemist's toolkit, various late-stage fluorination methods have recently been investigated for the synthesis of these building blocks.
Chun et al. investigated the radiolabelling of diaryliodonium salt precursors towards [18F]fluorophenyl azides and [18F]fluorobenzyl azides (Scheme 132).348 For the synthesis of [18F]fluorophenyl azides 562a and 562b, the use of diaryliodonium salt precursors was unsuccessful, giving the desired building blocks only in low radiochemical yields. For the synthesis of [18F]fluorobenzyl azides 563a–c, moderate radiochemical yields were obtained for the ortho, meta and para isomers (39–45% RCY, analytically determined).
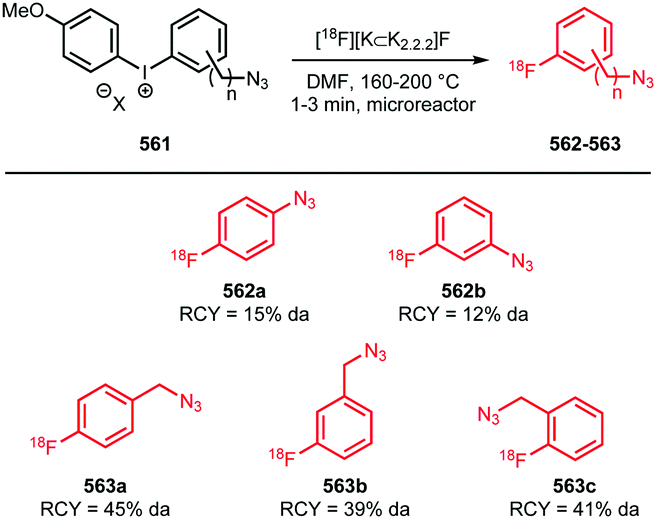 | ||
| Scheme 132 Synthesis of [18F]fluorophenyl azides and [18F]fluorobenzyl azides from iodonium salt precursors.348 | ||
Another approach towards [18F]fluorophenyl substituted azides was reported by Rotstein et al. and Wang et al. They investigated the radiofluorination of spirocyclic hypervalent iodine(III) precursors (Scheme 133).16,349 Using this novel radiofluorination method, various [18F]fluorophenyl substituted azides could be formed in moderate to excellent radiochemical yields.
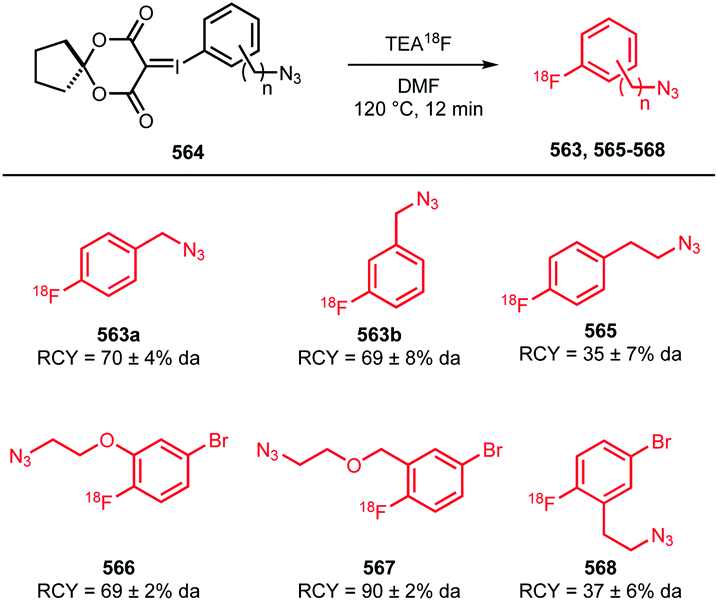 | ||
| Scheme 133 Synthesis of various [18F]fluorophenyl substituted azides from spirocyclic hypervalent iodine(III) precursors.16,349 | ||
To demonstrate the potential of these building blocks for the synthesis of PET tracers, Wang et al. showed that fluorine-18 labelled amino acid 570 could be formed in 49% radiochemical yield (analytically determined) by the ‘click’-reaction of building block 567 with alkyne modified precursor 569 (Scheme 134).344
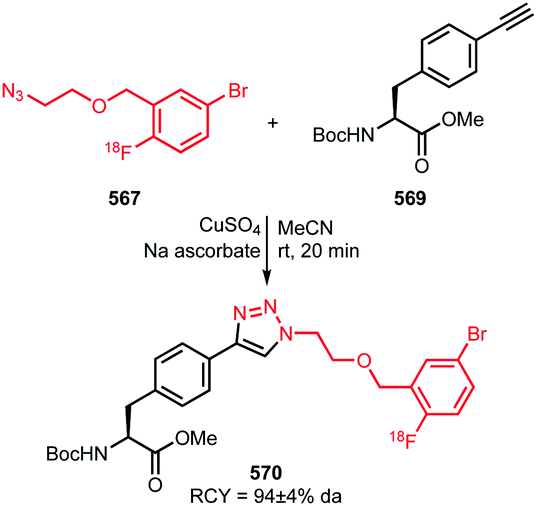 | ||
| Scheme 134 Synthesis of fluorine-18 labelled amino acid 570 using [18F]fluorophenyl substituted azide 567.349 | ||
In conclusion, novel methodologies have recently become available to synthesise [18F]fluorophenyl substituted azide building blocks in moderate to good radiochemical yields, making this group of building blocks available for the synthesis of PET tracers.
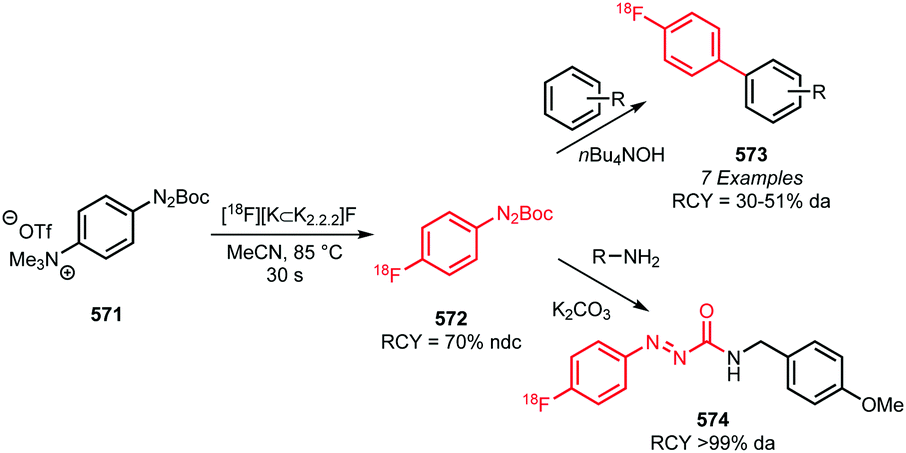 | ||
| Scheme 135 Synthesis and application of [18F]4-fluorophenylazocarboxylic tert-butyl ester.350 | ||
The application of this building block in radical arylations toward compounds 573 resulted in reasonable radiochemical yields of 30–51% (analytically determined) with simple model substrates. Examples of more complex molecules resembling PET tracers however have not yet been published. The reaction of building block 575 with amines to amides is very promising, as shown by the quantitative conversion towards compound 574 (Scheme 135) and the successful synthesis of dopamine D3 ligand 576 in an overall radiochemical yield of 20–24% (ndc) (Scheme 136).350
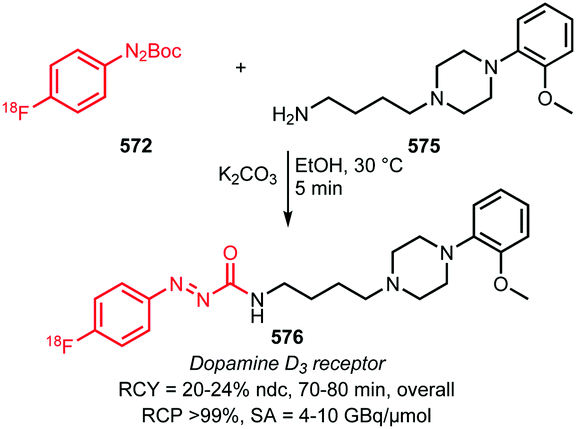 | ||
| Scheme 136 Application of [18F]4-fluorophenylazocarboxylic tert-butyl ester in the synthesis of dopamine-D3 ligand 576.350 | ||
In conclusion, this building block is a good candidate for the synthesis of various PET tracers, as it is easy to synthesise and reacts in high yields. As it is a fairly new building block, its potential needs to be further explored.
Recently, Ross et al. and Helfer et al. developed a novel two-step synthesis of 4-[18F]fluorophenol from iodonium salt precursors 577 and 578 (Scheme 137).356,357 These iodonium salt precursors are benzyl protected phenols with the reactive iodonium salt at the 4-position. After radiofluorination, benzyl protected 4-[18F]fluorophenol 579 is obtained, which is deprotected by hydrogenation to result in 4-[18F]fluorophenol. When microwave heating was used for the radiofluorination reaction, 4-[18F]fluorophenol could be obtained in an overall synthesis times of 15 minutes with radiochemical yields of 52% (dc). The only disadvantage is that complex iodonium salt precursors need to be synthesised, which are somewhat unstable.
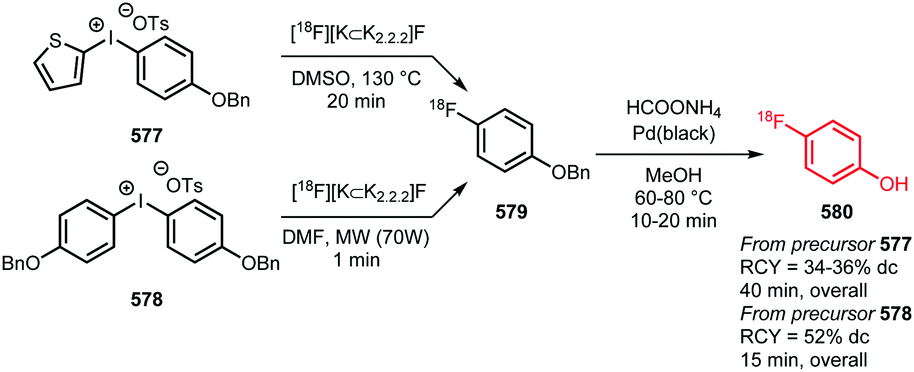 | ||
| Scheme 137 Synthesis of 4-[18F]fluorophenol from iodonium salt precursors.356,357 | ||
Gao et al. developed a novel one-pot method starting from 4-tert-butyl precursors 579.31Via oxidative radiofluorination and using phenoliodine diacetate (PIDA), various 4-[18F]fluorophenols were prepared in only 30 minutes overall synthesis time (Scheme 138). The synthesis of 4-tert-butyl precursors is simpler than of the iodonium salt precursors, some of them are even commercially available. If the radiochemical yields can be further improved, this method will be highly valuable for the synthesis of 4-[18F]fluorophenols.
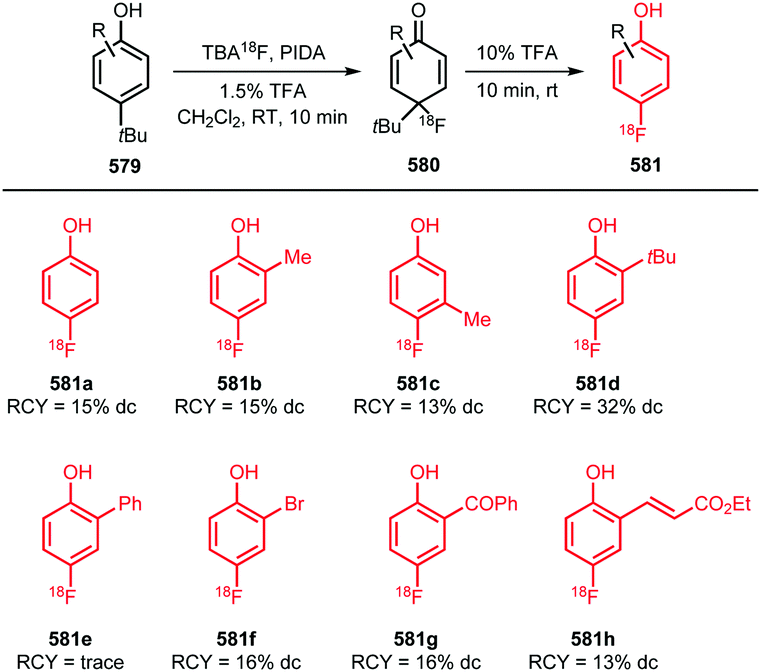 | ||
| Scheme 138 Synthesis of 4-[18F]fluorophenols by oxidative fluorination of 4-tert-butyl phenols.31 | ||
4-[18F]Fluorophenol has not been applied in the synthesis of new PET tracers. However with these novel methods, 4-[18F]fluorophenol becomes accessible as a fluorine-18 labelled building block and its application in the synthesis of PET tracers can therefore be expected in the future.
Tominaga et al. showed that [18F]FBnTP can also be synthesised by direct reaction of 4-[18F]fluorobenzyl alcohol with triphenylphosphine hydrobromide (Scheme 139) in an overall radiochemical yield of 12–14% (dc). In comparison, the radiochemical yield obtained by Ravert et al. was 8.3% (ndc), which converts to 11.5% (dc). Besides the similar radiochemical yields, both methods use SPE purifications in the building block synthesis and a final HPLC purification of the tracer. Therefore, there seems no significant preference for either method in the synthesis of [18F]FBnTP.
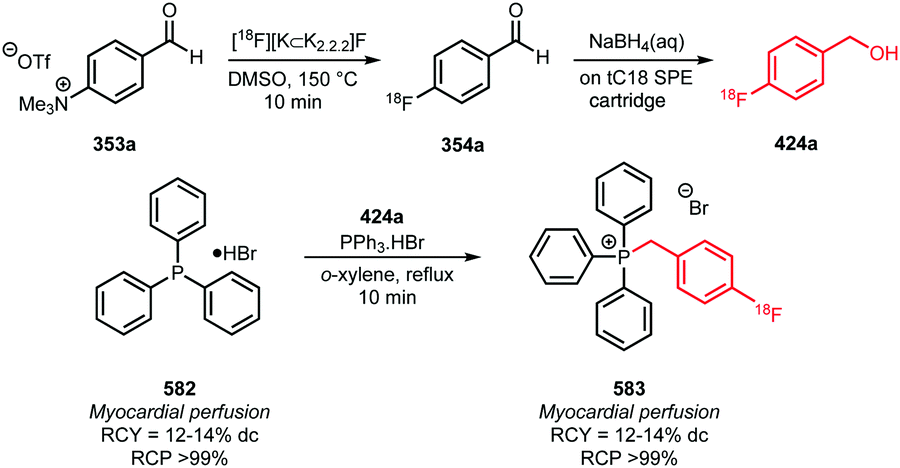 | ||
| Scheme 139 Application of [18F]4-fluorobenzyl alcohol in the synthesis of [18F]FBnTP.358 | ||
4 Conclusion and future perspectives
The interest in the development of novel fluorine-18 labelled PET tracers has increased significantly over the last two decades. For the synthesis of these tracers, radiochemists prefer late stage radiofluorination reactions for various reasons. The use of building blocks is however still of significant importance. Besides the use of fluorine-18 labelled building blocks for the modular build-up of PET tracers, which cannot be obtained via direct radiofluorination, several aromatic and aliphatic fluorine-18 labelled building blocks have been developed for generic applications. As such, fluorine-18 labelled building blocks are a good alternative to late stage radiofluorination. For example, building blocks which have been proven valuable due to their simple, easy to automate synthesis and effective reaction with precursors are the alkylating agents [18F]fluoroethyl bromide, [18F]FETos and ‘click’-reagent [18F]fluoroethyl azide. These three building blocks account, since 2010, for the synthesis of more than 120 PET tracers. A building block which has proven to be very useful due to its versatility is 4-[18F]fluorobenzaldehyde, as it has been applied as prosthetic group in at least five different types of coupling chemistry as well as in various multicomponent reactions. N-Succinimidyl-4-[18F]fluorobenzoate has proven to be valuable as one of the most selective building blocks, as it reacts rather selectively with primary amines.Other building blocks are less broadly applied, but still find applications in the synthesis of PET tracers. Many aromatic building blocks are for example used in the development of PET tracers which cannot be synthesised easily by late-stage radiofluorination. Although the overall yields via the building block approach can be low due to a challenging building block synthesis or low yielding subsequent reactions, the final PET tracer is often produced in sufficient yields for initial preclinical studies. Various aliphatic building blocks are used as an alternative to [18F]fluoroethyl halides and sulfonates to improve the biological characteristics of the PET tracer by modifying the chain length and chain structure. For the synthesis of PET tracers which are difficult to obtain by late-stage radiofluorination as well as the synthesis of PET tracer derivatives with improved biological activity, an elaborate toolkit is required which contains many different types of building blocks. With that perspective in mind, it is clear that the current set of building blocks available to the radiochemist is still rather limited and further expansion to allow the introduction of fluorine-18 at any desired position in any molecule is highly desired.
It should be noted that nowadays novel late-stage radiofluorination chemistry also provides radiochemists with opportunities to develop and access a larger variety of structurally diverse PET tracers that were previously only accessible by elaborate multistep fluorine-18 building block chemistry. These novel late-stage radiofluorination chemistry methods significantly increase the tools available for PET tracer synthesis. Nevertheless, despite these recent developments, it is still not possible to access and develop every desired PET tracer. Precursors can be difficult to synthesise and may not be very stable, the functional group tolerance and scope can still be too limited, and the reaction conditions for the fluorine-18 labelling reactions are often still harsh. It is therefore very likely that the building blocks described in this review, that have proven to be particularly successful and are widely applied, will not be replaced by late-stage radiofluorination chemistry, but will remain important in the radiochemists toolkit.
In conclusion, this review shows that building blocks are vital tools for radiochemists and will continue to be important in the future of PET tracer development, as complementary techniques to late-stage radiofluorination methods.
Abbreviations
| aq. | Aqueous |
| AMPA | α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| b.p. | Boiling point |
| BAIB | [Bis(acetoxy)iodo]-benzene |
| BOP | (Benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate |
| BuEA | Fluorobutyl ethacrynic amide |
| COX | Cyclooxygenase |
| CSA | Camphorsulfonic acid |
| CuAAC | Copper(I)-catalysed azide-alkyne cycloaddition |
| da | Determined analytically |
| dba | Dibenzylideneacetone |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| dc | Decay-corrected |
| DCC | N,N′-Dicyclohexylcarbodiimide |
| DIPEA | N,N-Diisopropylethylamine |
| DMA | Dimethylacetamide |
| DMAP | N,N-Dimethylpyridin-4-amine |
| DMF | N,N-Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| EGFR | Epidermal growth factor receptor |
| FBnTP | 4-Fluorobenzyl-triphenylphosphonium bromide |
| FBuEA | Fluorobutyl ethacrynic amide |
| FDG | 2-Deoxy-2-fluoroglucose |
| FETos | 2-Fluoroethyl tosylate |
| FPy-TFP | 6-Fluoronicotinic acid 2,3,5,6-tetrafluorophenyl ester |
| FPyKYNE | 2-Fluoro-3-pent-4-yn-1-yloxypyridine |
| FLT | 2′-Deoxy-2′-fluorothymidine |
| FXAU | 2′-Fluoro-2′-deoxy-1-β-D-arabinofuranosyl-uracil |
| GSH | Glutathione |
| HATU | 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate |
| HBTU | (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate) |
| HPLC | High-performance liquid chromatography |
| HSTU | N,N,N′,N′-Tetramethyl-O-(N-succinimidyl)uronium hexafluorophosphate |
| LG | Leaving group |
| M | Molar |
| min | Minute |
| MRP | Multi-drug resistance protein |
| MW | Microwave |
| N | Normal |
| ndc | Non decay-corrected |
| NFP | 4-Nitrophenyl 2-fluoropropionate |
| NHS | N-Hydroxysuccinimide |
| n.m. | Not mentioned |
| o-DCB | 1,2-Dichlorobenzene |
| PDE5 | Phosphodiesterase |
| PIDA | Phenoliodine diacetate |
| PIFA | Phenyliodine bis(trifluoroacetate) |
| PEG | Poly(ethylene glycol) |
| PET | Positron emission tomography |
| PMB | 4-Methoxybenzyl ether |
| PSMA | Prostate-specific membrane antigen |
| PTC | Phase-transfer catalyst |
| py | Pyridine |
| quant. | quantitative |
| RCP | Radiochemical purity |
| RCY | Radiochemical yield (isolated, if not stated otherwise) |
| rt | Room temperature |
| SA | Specific activity |
| SFB | N-Succinimidyl 4-fluorobenzoate |
| SPE | Solid-phase extraction |
| TBA | tert-Butanol |
| TEA | Triethylamine |
| TBAB | Tetrabutylammonium bromide |
| TBAF | Tetrabutylammonium fluoride |
| TBAOH | Tetrabutylammonium hydroxide |
| TCO | trans-Cyclooctenes |
| TEMPO | (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl |
| TFA | Trifluoroacetic acid |
| THF | Tetrahydrofuran |
| TMEDA | Tetramethylethylenediamine |
| TPAOH | Tetrapropylammonium hydroxide |
| TSTU | N,N,N′,N′-Tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate |
| Tz | Tetrazines |
| VEGF | Vascular endothelial growth factor |
Acknowledgements
The project is financially supported by The Netherlands Organization of Scientific Research (NWO) grant no. 731.015.413, BV Cyclotron VU and the Coops foundation.References
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed.
- J. S. Fowler and A. P. Wolf, Acc. Chem. Res., 1997, 30, 181–188 CrossRef CAS.
- P. M. Matthews, E. A. Rabiner, J. Passchier and R. N. Gunn, Br. J. Clin. Pharmacol., 2012, 73, 175–186 CrossRef CAS PubMed.
- H. Gewirtz, JACC Cardiovasc. Imaging, 2011, 4, 292–302 CrossRef PubMed.
- K.-L. Xiong, Q.-W. Yang, S.-G. Gong and W.-G. Zhang, Nucl. Med. Commun., 2010, 31, 4–11 CrossRef CAS PubMed.
- D. Papathanassiou, C. Bruna-Muraille, J.-C. Liehn, T. D. Nguyen and H. Curé, Crit. Rev. Oncol. Hematol., 2009, 72, 239–254 CrossRef PubMed.
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed.
- M. E. Phelps, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9226–9233 CrossRef CAS.
- P. F. Rambaldi, Whole-Body FDG PET Imaging in Oncology, Springer-Verlag, Mailand, 1st edn, 2013 Search PubMed.
- A. Ellmann and J. Holness, Contin. Med. Educ., 2013, 31, 279–283 Search PubMed.
- P. H. Elsinga, A. van Waarde, A. M. J. Paans and R. A. J. O. Dierckx, Trends on the Role of PET in Drug Development, World Scientific, Singapore, 1st edn, 2012 Search PubMed.
- S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719–766 CrossRef CAS PubMed.
- A. F. Brooks, J. J. Topczewski, N. Ichiishi, M. S. Sanford and P. J. H. Scott, Chem. Sci., 2014, 5, 4545–4553 RSC.
- T. L. Ross, J. Ermert, C. Hocke and H. H. Coenen, J. Am. Chem. Soc., 2007, 129, 8018–8025 CrossRef CAS PubMed.
- J. Cardinale, J. Ermert, S. Humpert and H. H. Coenen, RSC Adv., 2014, 4, 17293–17299 RSC.
- B. H. Rotstein, N. A. Stephenson, N. Vasdev and S. H. Liang, Nat. Commun., 2014, 5, 4365 CrossRef CAS PubMed.
- M. B. Haskali, S. Telu, Y.-S. Lee, C. L. Morse, S. Lu and V. W. Pike, J. Org. Chem., 2016, 81, 297–302 CrossRef CAS PubMed.
- N. Ichiishi, A. F. Brooks, J. J. Topczewski, M. E. Rodnick, M. S. Sanford and P. J. H. Scott, Org. Lett., 2014, 16, 3224–3227 CrossRef CAS PubMed.
- N. Ichiishi, A. J. Canty, B. F. Yates and M. S. Sanford, Org. Lett., 2013, 15, 5134–5137 CrossRef CAS PubMed.
- L. Mu, C. R. Fischer, J. P. Holland, J. Becaud, P. A. Schubiger, R. Schibli, S. M. Ametamey, K. Graham, T. Stellfeld, L. M. Dinkelborg and L. Lehmann, Eur. J. Org. Chem., 2012, 889–892 CrossRef CAS.
- K. Sander, T. Gendron, E. Yiannaki, K. Cybulska, T. L. Kalber, M. F. Lythgoe and E. Årstad, Sci. Rep., 2015, 5, 9941 CrossRef CAS PubMed.
- J.-H. Chun, C. L. Morse, F. T. Chin and V. W. Pike, Chem. Commun., 2013, 49, 2151–2153 RSC.
- J. R. Brandt, E. Lee, G. B. Boursalian and T. Ritter, Chem. Sci., 2014, 5, 169–179 RSC.
- A. S. Kamlet, C. N. Neumann, E. Lee, S. M. Carlin, C. K. Moseley, N. Stephenson, J. M. Hooker and T. Ritter, PLoS One, 2013, 8, e59187 CAS.
- E. Lee, A. S. Kamlet, D. C. Powers, C. N. Neumann, G. B. Boursalian, T. Furuya, D. C. Choi, J. M. Hooker and T. Ritter, Science, 2011, 334, 639–642 CrossRef CAS PubMed.
- E. Lee, J. M. Hooker and T. Ritter, J. Am. Chem. Soc., 2012, 134, 17456–17458 CrossRef CAS PubMed.
- H. Ren, H.-Y. Wey, M. Strebl, R. Neelamegam, T. Ritter and J. M. Hooker, ACS Chem. Neurosci., 2014, 5, 611–615 CrossRef CAS PubMed.
- M. Tredwell, S. M. Preshlock, N. J. Taylor, S. Gruber, M. Huiban, J. Passchier, J. Mercier, C. Génicot and V. Gouverneur, Angew. Chem., Int. Ed., 2014, 53, 7751–7755 CrossRef CAS PubMed.
- A. V. Mossine, A. F. Brooks, K. J. Makaravage, J. M. Miller, N. Ichiishi, M. S. Sanford and P. J. H. Scott, Org. Lett., 2015, 17, 5780–5783 CrossRef CAS PubMed.
- K. J. Makaravage, A. F. Brooks, A. V. Mossine, M. S. Sanford and P. J. H. Scott, Org. Lett., 2016, 18, 5440–5443 CrossRef CAS PubMed.
- Z. Gao, Y. H. Lim, M. Tredwell, L. Li, S. Verhoog, M. Hopkinson, W. Kaluza, T. L. Collier, J. Passchier, M. Huiban and V. Gouverneur, Angew. Chem., Int. Ed., 2012, 124, 6837–6841 CrossRef.
- C. N. Neumann, J. M. Hooker and T. Ritter, Nature, 2016, 534, 369–373 CrossRef CAS PubMed.
- M. E. Sergeev, F. Morgia, M. Lazari, C. Wang and R. M. Van Dam, J. Am. Chem. Soc., 2015, 137, 5686–5694 CrossRef CAS PubMed.
- J.-H. Chun and V. W. Pike, Org. Biomol. Chem., 2013, 11, 6300–6306 CAS.
- F. Basuli, H. Wu and G. L. Griffiths, J. Labelled Compd. Radiopharm., 2011, 54, 224–228 CrossRef CAS PubMed.
- L. Hortala, J. Arnaud, P. Roux, D. Oustric, L. Boulu, F. Oury-Donat, P. Avenet, T. Rooney, D. Alagille, O. Barret, G. Tamagnan and F. Barth, Bioorg. Med. Chem. Lett., 2014, 24, 283–287 CrossRef CAS PubMed.
- F. Beyerlein, M. Piel, S. Höhnemann and F. Rösch, J. Labelled Compd. Radiopharm., 2013, 56, 360–363 CrossRef CAS PubMed.
- Y. H. Ryu, J.-S. Liow, S. Zoghbi, M. Fujita, J. Collins, D. Tipre, J. Sangare, J. Hong, V. W. Pike and R. B. Innis, J. Nucl. Med., 2007, 48, 1154–1161 CrossRef CAS PubMed.
- D. N. Tipre, S. S. Zoghbi, J. S. Liow, M. V. Green, J. Seidel, M. Ichise, R. B. Innis and V. W. Pike, J. Nucl. Med., 2006, 47, 345–353 CAS.
- G. Smith, Y. Zhao, J. Leyton, B. Shan, Q.-D. Nguyen, M. Perumal, D. Turton, E. Årstad, S. K. Luthra, E. G. Robins and E. O. Aboagye, Nucl. Med. Biol., 2011, 38, 39–51 CrossRef CAS PubMed.
- X. Shao, B. G. Hockley, R. Hoareau, P. L. Schnau and P. J. H. Scott, Appl. Radiat. Isot., 2011, 69, 403–409 CrossRef CAS PubMed.
- C. Rami-Mark, M.-R. Zhang, M. Mitterhauser, R. Lanzenberger, M. Hacker and W. Wadsak, Nucl. Med. Biol., 2013, 40, 1049–1054 CrossRef CAS PubMed.
- P. J. Klein, J. A. M. Christiaans, A. Metaxas, R. C. Schuit, A. A. Lammertsma, B. N. M. van Berckel and A. D. Windhorst, Bioorg. Med. Chem., 2015, 23, 1189–1206 CrossRef CAS PubMed.
- N. J. Lodge, Y.-W. Li, F. T. Chin, D. D. Dischino, S. S. Zoghbi, J. A. Deskus, R. J. Mattson, M. Imaizumi, R. Pieschl, T. F. Molski, M. Fujita, H. Dulac, R. Zaczek, J. J. Bronson, J. E. Macor, R. B. Innis and V. W. Pike, Nucl. Med. Biol., 2014, 41, 524–535 CrossRef CAS PubMed.
- R. Xu, J. Hong, C. L. Morse and V. W. Pike, J. Med. Chem., 2010, 53, 7035–7047 CrossRef CAS PubMed.
- J. Hu, B. Gao, L. Li, C. Ni and J. Hu, Org. Lett., 2015, 17, 3086–3089 CrossRef CAS PubMed.
- P. O. Miranda, R. M. Carballo, V. S. Martín and J. I. Padrón, Org. Lett., 2009, 11, 357–360 CrossRef CAS PubMed.
- T. R. Neal, S. Apana and M. S. Berridge, J. Labelled Compd. Radiopharm., 2005, 48, 557–568 CrossRef CAS.
- G. Pascali, G. Nannavecchia, S. Pitzianti and P. A. Salvadori, Nucl. Med. Biol., 2011, 38, 637–644 CrossRef CAS PubMed.
- M. E. Rodnick, A. F. Brooks, B. G. Hockley, B. D. Henderson and P. J. H. Scott, Appl. Radiat. Isot., 2013, 78, 26–32 CrossRef CAS PubMed.
- H. Zeng, X. Wu, F. Song, C. Xu, H. Liu and W. Liu, Eur. J. Med. Chem., 2016, 118, 90–97 CrossRef CAS PubMed.
- H. Schieferstein, M. Piel, F. Beyerlein, H. Lüddens, N. Bausbacher, H.-G. Buchholz, T. L. Ross and F. Rösch, Bioorg. Med. Chem., 2015, 23, 612–623 CrossRef CAS PubMed.
- D. Thomae, T. J. Morley, H. S. Lee, O. Barret, C. Constantinescu, C. Papin, R. M. Baldwin, G. D. Tamagnan and D. Alagille, J. Labelled Compd. Radiopharm., 2016, 59, 205–213 CrossRef CAS PubMed.
- E. G. Robins, Y. Zhao, I. Khan, A. Wilson, S. K. Luthra and E. Årstad, Bioorg. Med. Chem. Lett., 2010, 20, 1749–1751 CrossRef CAS PubMed.
- L. Carroll, T. H. Witney and E. O. Aboagye, MedChemComm, 2013, 4, 653–656 RSC.
- S. Merchant, L. Allott, L. Carroll, V. Tittrea, S. Kealey, T. H. Witney, P. W. Miller, G. Smith and E. O. Aboagye, RSC Adv., 2016, 6, 57569–57579 RSC.
- P. J. Riss, S. Hoehnemann, M. Piel and F. Roesch, J. Labelled Compd. Radiopharm., 2013, 56, 356–359 CrossRef CAS PubMed.
- M.-R. Zhang, A. Tsuchiyama, T. Haradahira, Y. Yoshida, K. Furutsuka and K. Suzuki, Appl. Radiat. Isot., 2002, 57, 335–342 CrossRef CAS PubMed.
- M.-R. Zhang, K. Furutsuka, Y. Yoshida and K. Suzuki, J. Labelled Compd. Radiopharm., 2003, 46, 587–598 CrossRef CAS.
- D. Murali, T. E. Barnhart, N. T. Vandehey, B. T. Christian, R. J. Nickles, A. K. Converse, J. A. Larson, J. E. Holden, M. L. Schneider and O. T. Dejesus, Appl. Radiat. Isot., 2013, 72, 128–132 CrossRef CAS PubMed.
- H. Savolainen, M. Cantore, N. A. Colabufo, P. H. Elsinga, A. D. Windhorst and G. Luurtsema, Mol. Pharmaceutics, 2015, 12, 2265–2275 CrossRef CAS PubMed.
- Z.-F. Liu, G.-L. Wang, M.-J. Dong, J.-W. Jin, J.-J. Li, Q. Zhang, K. Zhao, S.-Y. Yang and X.-T. Lin, J. Radioanal. Nucl. Chem., 2014, 299, 1509–1515 CrossRef CAS.
- J. Schmaljohann, E. Schirrmacher, B. Wängler, C. Wängler, R. Schirrmacher and S. Guhlke, Nucl. Med. Biol., 2011, 38, 165–170 CrossRef CAS PubMed.
- J.-I. Andrés, M. De Angelis, J. Alcázar, L. Iturrino, X. Langlois, S. Dedeurwaerdere, I. Lenaerts, G. Vanhoof, S. Celen and G. Bormans, J. Med. Chem., 2011, 54, 5820–5835 CrossRef PubMed.
- M. Ooms, S. Celen, M. Koole, X. Langlois, M. Schmidt, M. De Angelis, J. I. Andrés, A. Verbruggen, K. Van Laere and G. Bormans, Nucl. Med. Biol., 2014, 41, 695–704 CrossRef CAS PubMed.
- E. D. Hostetler, S. Sanabria-Bohórquez, W. Eng, A. D. Joshi, S. Patel, R. E. Gibson, S. O’Malley, S. M. Krause, C. Ryan, K. Riffel, S. Bi, O. Okamoto, H. Kawamoto, S. Ozaki, H. Ohta, T. de Groot, G. Bormans, M. Depré, J. de Hoon, I. De Lepeleire, T. Reynders, J. J. Cook, H. D. Burns, M. Egan, W. Cho, K. van Laere and R. J. Hargreaves, Neuroimage, 2013, 68, 1–10 CrossRef CAS PubMed.
- K. Kawamura, Y. Shimoda, K. Kumata, M. Fujinaga, J. Yui, T. Yamasaki, L. Xie, A. Hatori, H. Wakizaka, Y. Kurihara, M. Ogawa, N. Nengaki and M. R. Zhang, Nucl. Med. Biol., 2015, 42, 406–412 CrossRef CAS PubMed.
- A. K. Tiwari, M. Fujinaga, J. Yui, T. Yamasaki, L. Xie, K. Kumata, A. K. Mishra, Y. Shimoda, A. Hatori, B. Ji, M. Ogawa, K. Kawamura, F. Wang and M.-R. Zhang, Org. Biomol. Chem., 2014, 12, 9621–9630 CAS.
- M. Fujinaga, T. Yamasaki, J. Yui, A. Hatori, L. Xie, K. Kawamura, C. Asagawa, K. Kumata, Y. Yoshida, M. Ogawa, N. Nengaki, T. Fukumura and M.-R. Zhang, J. Med. Chem., 2012, 55, 2342–2352 CrossRef CAS PubMed.
- N. Evens, C. Vandeputte, G. G. Muccioli, D. M. Lambert, V. Baekelandt, A. M. Verbruggen, Z. Debyser, K. Van Laere and G. M. Bormans, Bioorg. Med. Chem., 2011, 19, 4499–4505 CrossRef CAS PubMed.
- K. Kawamura, T. Yamasaki, F. Konno, J. Yui, A. Hatori, K. Yanamoto, H. Wakizaka, M. Ogawa, Y. Yoshida, N. Nengaki, T. Fukumura and M.-R. Zhang, Bioorg. Med. Chem., 2011, 19, 861–870 CrossRef CAS PubMed.
- J. A. Deskus, D. D. Dischino, R. J. Mattson, J. L. Ditta, M. F. Parker, D. J. Denhart, D. Zuev, H. Huang, R. A. Hartz, V. T. Ahuja, H. Wong, G. K. Mattson, T. F. Molski, J. E. Grace, L. Zueva, J. M. Nielsen, H. Dulac, Y.-W. Li, M. Guaraldi, M. Azure, D. Onthank, M. Hayes, E. Wexler, J. McDonald, N. J. Lodge, J. J. Bronson and J. E. Macor, Bioorg. Med. Chem. Lett., 2012, 22, 6651–6655 CrossRef CAS PubMed.
- M. Jahan, O. Eriksson, P. Johnström, O. Korsgren, A. Sundin, L. Johansson and C. Halldin, EJNMMI Res., 2011, 1, 33 CrossRef PubMed.
- O. Gheysens, V. Akurathi, R. Chekol, T. Dresselaers, S. Celen, M. Koole, D. Dauwe, B. J. Cleynhens, P. Claus, S. Janssens, A. M. Verbruggen, J. Nuyts, U. Himmelreich and G. M. Bormans, EJNMMI Res., 2013, 3, 4 CrossRef PubMed.
- C. Rami-Mark, B. Bornatowicz, C. Fink, P. Otter, J. Ungersboeck, C. Vraka, D. Haeusler, L. Nics, H. Spreitzer, M. Hacker, M. Mitterhauser and W. Wadsak, Bioorg. Med. Chem., 2013, 21, 7562–7569 CrossRef CAS PubMed.
- C. Philippe, J. Ungersboeck, E. Schirmer, M. Zdravkovic, L. Nics, M. Zeilinger, K. Shanab, R. Lanzenberger, G. Karanikas, H. Spreitzer, H. Viernstein, M. Mitterhauser and W. Wadsak, Bioorg. Med. Chem., 2012, 20, 5936–5940 CrossRef CAS PubMed.
- D. Block, H. H. Coenen and G. Stöcklin, J. Labelled Compd. Radiopharm., 1987, 24, 1029–1042 CrossRef CAS.
- B. W. Schoultz, B. J. Reed, J. Marton, F. Willoch and G. Henriksen, Molecules, 2013, 18, 7271–7278 CrossRef CAS PubMed.
- T. K. Heinrich, V. Gottumukkala, E. Snay, P. Dunning, F. H. Fahey, S. Ted Treves and A. B. Packard, Appl. Radiat. Isot., 2010, 68, 96–100 CrossRef CAS PubMed.
- S. Khanapur, S. Paul, A. Shah, S. Vatakuti, M. J. B. Koole, R. Zijlma, R. A. J. O. Dierckx, G. Luurtsema, P. Garg, A. van Waarde and P. H. Elsinga, J. Med. Chem., 2014, 57, 6765–6780 CrossRef CAS PubMed.
- V. J. Majo, V. Arango, N. R. Simpson, J. Prabhakaran, S. A. Kassir, M. D. Underwood, M. Bakalian, P. Canoll, J. John Mann and J. S. Dileep Kumar, Bioorg. Med. Chem. Lett., 2013, 23, 4191–4194 CrossRef CAS PubMed.
- M. Asti, D. Farioli, M. Iori, C. Guidotti, A. Versari and D. Salvo, Nucl. Med. Biol., 2010, 37, 309–315 CrossRef CAS PubMed.
- H. J. Breyholz, S. Wagner, A. Faust, B. Riemann, C. Höltke, S. Hermann, O. Schober, M. Schäfers and K. Kopka, ChemMedChem, 2010, 5, 777–789 CrossRef CAS PubMed.
- J. Li, B. D. Gray, K. Y. Pak and C. K. Ng, J. Labelled Compd. Radiopharm., 2012, 55, 149–154 CrossRef CAS.
- J. Prabhakaran, V. Arango, V. J. Majo, N. R. Simpson, S. A. Kassir, M. D. Underwood, H. Polavarapu, J. N. Bruce, P. Canoll, J. John Mann and J. S. Dileep Kumar, Bioorg. Med. Chem. Lett., 2012, 22, 5104–5107 CrossRef CAS PubMed.
- E. M. F. Billaud, L. Rbah-Vidal, A. Vidal, S. Besse, S. Tarrit, S. Askienazy, A. Maisonial, N. Moins, J. C. Madelmont, E. Miot-Noirault, J. M. Chezal and P. Auzeloux, J. Med. Chem., 2013, 56, 8455–8467 CrossRef CAS PubMed.
- E. Galante, T. Okamura, K. Sander, T. Kikuchi, M. Okada, M. Zhang, M. Robson, A. Badar, M. Lythgoe, M. Koepp and E. Årstad, J. Med. Chem., 2014, 57, 1023–1032 CrossRef CAS PubMed.
- A. Makino, T. Arai, M. Hirata, M. Ono, Y. Ohmomo and H. Saji, Nucl. Med. Biol., 2016, 43, 101–107 CrossRef CAS PubMed.
- F. Caillé, T. J. Morley, A. A. S. Tavares, C. Papin, N. M. Twardy, D. Alagille, H. S. Lee, R. M. Baldwin, J. P. Seibyl, O. Barret and G. D. Tamagnan, Bioorg. Med. Chem. Lett., 2013, 23, 6243–6247 CrossRef PubMed.
- D. O’Shea, R. Ahmad, E. Årstad, M. Avory, W. F. Chau, C. Durrant, E. Hirani, P. A. Jones, I. Khan, S. K. Luthra, D. Mantzilas, V. Morisson-Iveson, J. Passmore, E. G. Robins, B. Shan, H. Wadsworth, S. Walton, Y. Zhao and W. Trigg, Bioorg. Med. Chem. Lett., 2013, 23, 2368–2372 CrossRef PubMed.
- S. Rötering, M. Scheunemann, S. Fischer, A. Hiller, D. Peters, W. Deuther-Conrad and P. Brust, Bioorg. Med. Chem., 2013, 21, 2635–2642 CrossRef PubMed.
- T. Sun, G. Tang, H. Tian, X. Wang, X. Chen, Z. Chen and S. Wang, Appl. Radiat. Isot., 2012, 70, 676–680 CrossRef CAS PubMed.
- H. Wang, X. Tang, G. Tang, T. Huang, X. Liang, K. Hu, H. Deng, C. Yi, X. Shi and K. Wu, Apoptosis, 2013, 18, 1017–1027 CrossRef CAS PubMed.
- S. Perera, D. Piwnica-Worms and M. M. Alauddin, J. Labelled Compd. Radiopharm., 2016, 59, 103–108 CrossRef CAS PubMed.
- J. Henrottin, C. Lemaire, D. Egrise, A. Zervosen, B. van Den Eynde, A. Plenevaux, X. Franci, S. Goldman and A. Luxen, Nucl. Med. Biol., 2016, 43, 379–389 CrossRef CAS PubMed.
- A. Bauman, M. Piel, R. Schirrmacher and F. Rösch, Tetrahedron Lett., 2003, 44, 9165–9167 CrossRef CAS.
- R. Li, S.-C. Wu, S.-C. Wang, Z. Fu, Y. Dang and L. Huo, Appl. Radiat. Isot., 2010, 68, 303–308 CrossRef CAS PubMed.
- A. Bauman, M. Piel, S. Höhnemann, A. Krauss, M. Jansen, C. Solbach, G. Dannhardt and F. Rösch, J. Labelled Compd. Radiopharm., 2011, 54, 645–656 CrossRef CAS.
- H. S. Radeke, A. Purohit, T. D. Harris, K. Hanson, R. Jones, C. Hu, P. Yalamanchili, M. Hayes, M. Yu, M. Guaraldi, M. Kagan, M. Azure, M. Cdebaca, S. Robinson and D. Casebier, ACS Med. Chem. Lett., 2011, 2, 650–655 CrossRef CAS PubMed.
- V. Shalgunov, J.-P. van Wieringen, H. M. Janssen, P. M. Fransen, R. A. J. O. Dierckx, M. C. Michel, J. Booij and P. H. Elsinga, EJNMMI Res., 2015, 5, 41 CrossRef PubMed.
- H. Ren, H. Ning, J. Chang, M. Zhao, Y. He, Y. Chong and C. Qi, J. Radioanal. Nucl. Chem., 2016, 517–523 CAS.
- U. Funke, W. Deuther-Conrad, G. Schwan, A. Maisonial, M. Scheunemann, S. Fischer, A. Hiller, D. Briel and P. Brust, Pharmaceuticals, 2012, 5, 169–188 CrossRef CAS PubMed.
- Y. Chen, M. Feng, S. Li, J. Xu, H. Ning, Y. He, X. Wang, R. Ding and C. Qi, Bioorg. Med. Chem. Lett., 2012, 22, 4745–4749 CrossRef CAS PubMed.
- B. H. Yousefi, A. Drzezga, B. Von Reutern, A. Manook, M. Schwaiger, H.-J. Wester and G. Henriksen, ACS Med. Chem. Lett., 2011, 2, 673–677 CrossRef CAS PubMed.
- H. Wadsworth, P. A. Jones, W. F. Chau, C. Durrant, V. Morisson-Iveson, J. Passmore, D. O’Shea, D. Wynn, I. Khan, A. Black, M. Avory and W. Trigg, Bioorg. Med. Chem. Lett., 2012, 22, 5795–5800 CrossRef CAS PubMed.
- A. Jackson, B. B. Guilbert, S. D. Plant, J. Goggi, M. R. Battle, J. L. Woodcraft, A. Gaeta, C. L. Jones, D. R. Bouvet, P. A. Jones, D. M. O’Shea, P. H. Zheng, S. L. Brown, A. L. Ewan and W. Trigg, Bioorg. Med. Chem. Lett., 2013, 23, 821–826 CrossRef CAS PubMed.
- R. Löser, R. Bergmann, M. Frizler, B. Mosch, L. Dombrowski, M. Kuchar, J. Steinbach, M. Gütschow and J. Pietzsch, ChemMedChem, 2013, 8, 1330–1344 CrossRef PubMed.
- P. J. Riss, L. Brichard, V. Ferrari, D. J. Williamson, T. D. Fryer, Y. T. Hong, J.-C. Baron and F. I. Aigbirhio, MedChemComm, 2013, 4, 852–855 RSC.
- Z. Tu, X. Zhang, H. Jin, X. Yue, P. K. Padakanti, L. Yu, H. Liu, H. P. Flores, K. Kaneshige, S. M. Parsons and J. S. Perlmutter, Bioorg. Med. Chem., 2015, 23, 4699–4709 CrossRef CAS PubMed.
- X. Yue, C. Bognar, X. Zhang, G. G. Gaehle, S. M. Moerlein, J. S. Perlmutter and Z. Tu, Appl. Radiat. Isot., 2016, 107, 40–46 CrossRef CAS PubMed.
- F. Xie, R. Bergmann, T. Kniess, W. Deuther-Conrad, C. Mamat, C. Neuber, B. Liu, J. Steinbach, P. Brust, J. Pietzsch and H. Jia, J. Med. Chem., 2015, 58, 5395–5407 CrossRef CAS PubMed.
- F. Debus, M. M. Herth, M. Piel, H.-G. Buchholz, N. Bausbacher, V. Kramer, H. Lüddens and F. Rösch, Nucl. Med. Biol., 2010, 37, 487–495 CrossRef CAS PubMed.
- J. P. Van Wieringen, V. Shalgunov, H. M. Janssen, P. M. Fransen, A. G. M. Janssen, M. C. Michel, J. Booij and P. H. Elsinga, J. Med. Chem., 2014, 57, 391–410 CrossRef CAS PubMed.
- Y.-Y. Chen, X. Wang, J.-M. Zhang, W. Deuther-Conrad, X.-J. Zhang, Y. Huang, Y. Li, J.-J. Ye, M.-C. Cui, J. Steinbach, P. Brust, B.-L. Liu and H.-M. Jia, Bioorg. Med. Chem., 2014, 22, 5270–5278 CrossRef CAS PubMed.
- A. Chiotellis, A. Muller, L. Mu, C. Keller, R. Schibli, S. D. Kra and S. M. Ametamey, Mol. Pharmaceutics, 2014, 11, 3839–3851 CrossRef CAS PubMed.
- B. Neumaier, S. Deisenhofer, C. Sommer, C. Solbach, S. N. Reske and F. Mottaghy, Appl. Radiat. Isot., 2010, 68, 1066–1072 CrossRef CAS PubMed.
- E. Al-Momani, N. Malik, H.-J. Machulla, S. N. Reske and C. Solbach, J. Radioanal. Nucl. Chem., 2013, 295, 2289–2294 CrossRef CAS.
- V. Bernard-gauthier, A. Aliaga, A. Aliaga, M. Boudjemeline, R. Hopewell, A. Kostikov, P. Rosa-neto, A. Thiel and R. Schirrmacher, ACS Chem. Neurosci., 2015, 6, 260–276 CrossRef CAS PubMed.
- M. M. Herth, I. N. Petersen, H. D. Hansen, M. Hansen, A. Ettrup, A. A. Jensen, S. Lehel, A. Dyssegaard, N. Gillings, G. M. Knudsen and J. L. Kristensen, Nucl. Med. Biol., 2016, 43, 455–462 CrossRef CAS PubMed.
- P. Cumming, D. Skaper, T. Kuwert, S. Maschauer and O. Prante, Synapse, 2015, 69, 57–59 CrossRef CAS PubMed.
- E. Blom, F. Karimi, O. Eriksson, H. Hall and B. Långström, J. Labelled Compd. Radiopharm., 2008, 51, 277–282 CrossRef CAS.
- B. W. Schoultz, T. Hjørnevik, B. J. Reed, J. Marton, C. S. Coello, F. Willoch and G. Henriksen, J. Med. Chem., 2014, 57, 5464–5469 CrossRef CAS PubMed.
- S. L. James, S. K. Ahmed, S. Murphy, M. R. Braden, Y. Belabassi, H. F. VanBrocklin, C. M. Thompson and J. M. Gerdes, ACS Chem. Neurosci., 2014, 5, 519–524 CrossRef CAS PubMed.
- T. Peters, A. Vogg, I. M. Oppel and J. Schmaljohann, Appl. Radiat. Isot., 2014, 94, 141–146 CrossRef CAS PubMed.
- R. Chekol, O. Gheysens, J. Cleynhens, P. Pokreisz, G. Vanhoof, M. Ahamed, S. Janssens, A. Verbruggen and G. Bormans, Nucl. Med. Biol., 2014, 41, 155–162 CrossRef CAS PubMed.
- J. L. Musachio, J. Shah and V. W. Pike, J. Labelled Compd. Radiopharm., 2005, 48, 735–747 CrossRef CAS.
- N. Jarkas, R. J. Voll and M. M. Goodman, J. Labelled Compd. Radiopharm., 2013, 56, 539–543 CrossRef CAS PubMed.
- R. J. Voll, J. McConathy, M. S. Waldrep, R. J. Crowe and M. M. Goodman, Appl. Radiat. Isot., 2005, 63, 353–361 CrossRef CAS PubMed.
- A. L. Smith, S. M. Freeman, J. S. Stehouwer, K. Inoue, R. J. Voll, L. J. Young and M. M. Goodman, Bioorg. Med. Chem., 2012, 20, 2721–2738 CrossRef CAS PubMed.
- A. K. Bhattacharjee, L. Lang, O. Jacobson, B. Shinkre, Y. Ma, G. Niu, W. C. Trenkle, K. A. Jacobson, X. Chen and D. O. Kiesewetter, Nucl. Med. Biol., 2011, 38, 897–906 CrossRef CAS PubMed.
- D. van der Born, C. Sewing, J. D. M. Herscheid, A. D. Windhorst, R. V. A. Orru and D. J. Vugts, Angew. Chem., Int. Ed., 2014, 53, 11046–11050 CrossRef CAS PubMed.
- T. Rühl, W. Rafique, V. T. Lien and P. J. Riss, Chem. Commun., 2014, 50, 6056–6059 RSC.
- P. Ivashkin, G. Lemonnier, J. Cousin, V. Grégoire, D. Labar, P. Jubault and X. Pannecoucke, Chem. – Eur. J., 2014, 20, 9514–9518 CrossRef CAS PubMed.
- M. Suehiro, G. Yang, G. Torchon, E. Ackerstaff, J. Humm, J. Koutcher and O. Ouerfelli, Bioorg. Med. Chem., 2011, 19, 2287–2297 CrossRef CAS PubMed.
- P. J. Riss, V. Ferrari, L. Brichard, P. Burke, R. Smith and F. I. Aigbirhio, Org. Biomol. Chem., 2012, 10, 6980–6986 CAS.
- P. J. Riss and F. I. Aigbirhio, Chem. Commun., 2011, 47, 11873–11875 RSC.
- M. D. Bartholomä, V. Gottumukkala, S. Zhang, A. Baker, P. Dunning, F. H. Fahey, S. T. Treves and A. B. Packard, J. Med. Chem., 2012, 55, 11004–11012 CrossRef PubMed.
- P. Cumming, S. Maschauer, P. J. Riss, N. Tschammer, S. K. Fehler, M. R. Heinrich, T. Kuwert and O. Prante, J. Cereb. Blood Flow Metab., 2014, 34, 1148–1156 CrossRef CAS PubMed.
- S. He, G. Tang, K. Hu, H. Wang, S. Wang, T. Huang, X. Liang and X. Tang, Nucl. Med. Biol., 2013, 40, 801–807 CrossRef CAS PubMed.
- L. Zhu, G. Li, S. R. Choi, K. Plössl, P. Chan, H. Qiao, Z. Zha and H. F. Kung, Nucl. Med. Biol., 2013, 40, 974–979 CrossRef CAS PubMed.
- J. S. Stehouwer, L. M. Daniel, P. Chen, R. J. Voll, L. Williams, S. J. Plott, J. R. Votaw, M. J. Owens, L. Howell and M. M. Goodman, J. Med. Chem., 2010, 53, 5549–5557 CrossRef CAS PubMed.
- D. Franck, T. Kniess, J. Steinbach, S. Zitzmann-Kolbe, M. Friebe, L. M. Dinkelborg and K. Graham, Bioorg. Med. Chem., 2013, 21, 643–652 CrossRef CAS PubMed.
- D.-Y. Kim, H.-J. Kim, K.-H. Yu and J.-J. Min, Nucl. Med. Biol., 2012, 39, 1093–1098 CrossRef CAS PubMed.
- D.-Y. Kim, H.-J. Kim, K.-H. Yu and J.-J. Min, Bioorg. Med. Chem. Lett., 2012, 22, 319–322 CrossRef CAS PubMed.
- M. D. Bartholomä, S. Zhang, V. Akurathi, C. A. Pacak, P. Dunning, F. H. Fahey, D. B. Cowan, S. Ted Treves and A. B. Packard, Nucl. Med. Biol., 2015, 42, 796–803 CrossRef PubMed.
- I. F. Antunes, H. J. Haisma, P. H. Elsinga, R. A. Dierckx and E. F. J. de Vries, Bioconjug. Chem., 2010, 21, 911–920 CrossRef CAS PubMed.
- Z. Zhao, Q. Yu, T. Mou, C. Liu, W. Yang, W. Fang, C. Peng, J. Lu, Y. Liu and X. Zhang, Mol. Pharmaceutics, 2014, 11, 3826–3831 Search PubMed.
- L. Frullano, C. Catana, T. Benner, A. D. Sherry and P. Caravan, Angew. Chem., Int. Ed., 2010, 49, 2382–2384 CrossRef CAS PubMed.
- D. Zhou, W. Chu, X. Peng, J. McConathy, R. H. Mach and J. A. Katzenellenbogen, Tetrahedron Lett., 2015, 56, 952–954 CrossRef CAS PubMed.
- L. Jia, Z. Cheng, L. Shi, J. Li, C. Wang, D. Jiang, W. Zhou, H. Meng, Y. Qi, D. Cheng and L. Zhang, Appl. Radiat. Isot., 2013, 75, 64–70 CrossRef CAS PubMed.
- R. Bejot, L. Carroll, K. Bhakoo, J. Declerck and V. Gouverneur, Bioorg. Med. Chem., 2012, 20, 324–329 CrossRef CAS PubMed.
- L. Carroll, S. Boldon, R. Bejot, J. E. Moore, J. Declerck and V. Gouverneur, Org. Biomol. Chem., 2011, 9, 136–140 CAS.
- A. Gaeta, J. Woodcraft, S. Plant, J. Goggi, P. Jones, M. Battle, W. Trigg, S. K. Luthra and M. Glaser, Bioorg. Med. Chem. Lett., 2010, 20, 4649–4652 CrossRef CAS PubMed.
- K. Nwe and M. W. Brechbiel, Cancer Biother. Radiopharm., 2009, 24, 289–302 CrossRef CAS PubMed.
- M. Glaser and E. Årstad, Bioconjugate Chem., 2007, 18, 989–993 CrossRef CAS PubMed.
- A. Monaco, O. Michelin, J. Prior, C. Rüegg, L. Scapozza and Y. Seimbille, J. Labelled Compd. Radiopharm., 2014, 57, 365–370 CrossRef CAS PubMed.
- F. Pisaneschi, Q.-D. Nguyen, E. Shamsaei, M. Glaser, E. Robins, M. Kaliszczak, G. Smith, A. C. Spivey and E. O. Aboagye, Bioorg. Med. Chem., 2010, 18, 6634–6645 CrossRef CAS PubMed.
- E. Laurens, S. D. Yeoh, A. Rigopoulos, D. Cao, G. A. Cartwright, G. J. O'Keefe, H. J. Tochon-Danguy, J. M. White, A. M. Scott and U. Ackermann, Nucl. Med. Biol., 2014, 41, 419–425 CrossRef CAS PubMed.
- W. Chu, A. Chepetan, D. Zhou, K. I. Shoghi, J. Xu, L. L. Dugan, R. J. Gropler, M. A. Mintun and R. H. Mach, Org. Biomol. Chem., 2014, 12, 4421–4431 CAS.
- V. Hugenberg, H.-J. Breyholz, B. Riemann, S. Hermann, O. Schober, M. Schäfers, U. Gangadharmath, V. Mocharla, H. Kolb, J. Walsh, W. Zhang, K. Kopka and S. Wagner, J. Med. Chem., 2012, 55, 4714–4727 CrossRef CAS PubMed.
- J. Kelly, A. Amor-Coarasa, A. Nikolopoulou, D. Kim, C. Williams Jr, S. Ponnala and J. W. Babich, Eur. J. Nucl. Med. Mol. Imaging, 2016, 44, 647–661 CrossRef PubMed.
- D. Zhou, W. Chu, C. S. Dence, R. H. Mach and M. J. Welch, Nucl. Med. Biol., 2012, 39, 1175–1181 CrossRef CAS PubMed.
- L. Carroll, R. Bejot, R. Hueting, R. King, P. Bonnitcha, S. Bayly, M. Christlieb, J. R. Dilworth, A. D. Gee, J. Declerck and V. Gouverneur, Chem. Commun., 2010, 46, 4052–4054 RSC.
- E. Galante, B. W. Schoultz, M. Koepp and E. Årstad, Molecules, 2013, 18, 5335–5347 CrossRef CAS PubMed.
- Y. Chen, A. Lisok, S. Chatterjee, B. Wharram, M. Pullambhatla, Y. Wang, G. Sgouros, R. C. Mease and M. G. Pomper, Bioconjugate Chem., 2016, 27, 1655–1662 CrossRef CAS PubMed.
- U. Ackermann, L. Plougastel, Y. W. Goh, S. D. Yeoh and A. M. Scott, Appl. Radiat. Isot., 2014, 94, 72–76 CrossRef CAS PubMed.
- U. Ackermann, G. O’Keefe, S.-T. Lee, A. Rigopoulos, G. Cartwright, J. I. Sachinidis, A. M. Scott and H. J. Tochon-Danguy, J. Labelled Compd. Radiopharm., 2011, 54, 260–266 CrossRef CAS.
- M. Glaser, J. Goggi, G. Smith, M. Morrison, S. K. Luthra, E. Robins and E. O. Aboagye, Bioorg. Med. Chem. Lett., 2011, 21, 6945–6949 CrossRef CAS PubMed.
- R. Fortt, G. Smith, R. O. Awais, S. K. Luthra and E. O. Aboagye, Nucl. Med. Biol., 2012, 39, 1000–1005 CrossRef CAS PubMed.
- C. M. Waldmann, S. Hermann, A. Faust, B. Riemann, O. Schober, M. Schäfers, G. Haufe and K. Kopka, Bioorg. Med. Chem., 2015, 23, 5734–5739 CrossRef CAS PubMed.
- S. D. Boss, T. Betzel, C. Müller, C. R. Fischer, S. Haller, J. Reber, V. Groehn, R. Schibli and S. M. Ametamey, Bioconjugate Chem., 2016, 27, 74–86 CrossRef CAS PubMed.
- E. Laurens, S. D. Yeoh, A. Rigopoulos, D. Cao, G. A. Cartwright, G. J. O’Keefe, H. J. Tochon-Danguy, J. M. White, A. M. Scott and U. Ackermann, Nucl. Med. Biol., 2012, 39, 871–882 CrossRef CAS PubMed.
- D. Schrigten, H.-J. Breyholz, S. Wagner, S. Hermann, O. Schober, M. Schäfers, G. Haufe and K. Kopka, J. Med. Chem., 2012, 55, 223–232 CrossRef CAS PubMed.
- G. Smith, R. Sala, L. Carroll, K. Behan, M. Glaser, E. Robins, Q. D. Nguyen and E. O. Aboagye, Nucl. Med. Biol., 2012, 39, 652–665 CrossRef CAS PubMed.
- L. Jia, D. Jiang, P. Hu, X. Li, H. Shi, D. Cheng and L. Zhang, Nucl. Med. Biol., 2014, 41, 495–500 CrossRef CAS PubMed.
- V. Hugenberg, S. Hermann, F. Galla, M. Schäfers, B. Wünsch, H. C. Kolb, K. Szardenings, A. Lebedev, J. C. Walsh, V. P. Mocharla, U. B. Gangadharmath, K. Kopka and S. Wagner, Nucl. Med. Biol., 2016, 43, 424–437 CrossRef CAS PubMed.
- A. Udemba, G. Smith, Q.-D. Nguyen, M. Kaliszczak, L. Carroll, R. Fortt, M. J. Fuchter and E. O. Aboagye, Org. Biomol. Chem., 2015, 13, 5418–5423 CAS.
- T. Läppchen, R. P. M. Dings, R. Rossin, J. F. Simon, T. J. Visser, M. Bakker, P. Walhe, T. van Mourik, K. Donato, J. R. van Beijnum, A. W. Griffioen, J. Lub, M. S. Robillard, K. H. Mayo and H. Grüll, Eur. J. Med. Chem., 2015, 89, 279–295 CrossRef PubMed.
- A. Haslop, A. Gee, C. Plisson and N. Long, J. Labelled Compd. Radiopharm., 2013, 56, 313–316 CrossRef CAS PubMed.
- A. Haslop, L. Wells, A. Gee, C. Plisson and N. Long, Mol. Pharmaceutics, 2014, 11, 3818–3822 CrossRef CAS PubMed.
- S. Maschauer, K. Michel, P. Tripal, K. Büther, T. Kuwert, O. Schober, K. Kopka, B. Riemann and O. Prante, Am. J. Nucl. Med. Mol. Imaging, 2013, 3, 425–436 CAS.
- S. Maschauer and O. Prante, Carbohydr. Res., 2009, 344, 753–761 CrossRef CAS PubMed.
- C. R. Fischer, C. Müller, J. Reber, A. Müller, S. D. Krämer, S. M. Ametamey and R. Schibli, Bioconjugate Chem., 2012, 23, 805–813 CrossRef CAS PubMed.
- S. Maschauer, C. Greff, J. Einsiedel, J. Ott, P. Tripal, H. Hübner, P. Gmeiner and O. Prante, Bioorg. Med. Chem., 2015, 23, 4026–4033 CrossRef CAS PubMed.
- F. Pisaneschi, R. L. Slade, L. Iddon, G. P. C. George, Q.-D. Nguyen, A. C. Spivey and E. O. Aboagye, J. Labelled Compd. Radiopharm., 2014, 57, 92–96 CrossRef CAS PubMed.
- C. R. Fischer, V. Groehn, J. Reber, R. Schibli, S. M. Ametamey and C. Müller, Mol. Imaging Biol., 2013, 15, 649–654 CrossRef PubMed.
- C. Lang, S. Maschauer, H. Hu, P. Gmeiner and O. Prante, J. Med. Chem., 2013, 56, 9361–9365 CrossRef CAS PubMed.
- S. Maschauer, J. Einsiedel, H. Hübner, P. Gmeiner and O. Prante, J. Med. Chem., 2016, 59, 6480–6492 CrossRef CAS PubMed.
- C.-M. Yook, S. J. Lee, S. J. Oh, H.-J. J. Ha and J. J. Lee, J. Labelled Compd. Radiopharm., 2015, 58, 317–326 CrossRef CAS PubMed.
- V. Hugenberg, B. Riemann, S. Hermann, O. Schober, M. Schäfers, K. Szardenings, A. Lebedev, U. Gangadharmath, H. Kolb, J. Walsh, W. Zhang, K. Kopka and S. Wagner, J. Med. Chem., 2013, 56, 6858–6870 CrossRef CAS PubMed.
- H. Schieferstein, T. Betzel, C. R. Fischer and T. L. Ross, EJNMMI Res., 2013, 3, 68 CrossRef PubMed.
- L. Mirfeizi, A. A. Rybczynska, A. van Waarde, L. Campbell-Verduyn, B. L. Feringa, R. A. J. O. Dierckx and P. H. Elsinga, Nucl. Med. Biol., 2014, 41, 203–209 CrossRef CAS PubMed.
- G. M. Entract, F. Bryden, J. Domarkas, H. Savoie, L. Allott, S. J. Archibald, C. Cawthorne and R. W. Boyle, Mol. Pharmaceutics, 2015, 12, 4414–4423 CrossRef CAS PubMed.
- M. Pretze and C. Mamat, J. Fluorine Chem., 2013, 150, 25–35 CrossRef CAS.
- H. Schieferstein and T. L. Ross, Eur. J. Org. Chem., 2014, 3546–3550 CrossRef CAS.
- T. J. Tewson, Nucl. Med. Biol., 1997, 24, 755–760 CrossRef CAS PubMed.
- V. J. Majo, N. R. Simpson, J. Prabhakaran, J. J. Mann and J. S. D. Kumar, J. Labelled Compd. Radiopharm., 2014, 57, 705–709 CrossRef CAS PubMed.
- Z. Zhang, J. Lau, H.-T. Kuo, C. Zhang, N. Hundal-Jabal, N. Colpo, F. Bénard and K.-S. Lin, Bioorg. Med. Chem. Lett., 2016, 26, 584–588 CrossRef CAS PubMed.
- Y.-C. Huang, Y.-C. Chang, C.-N. Yeh and C.-S. Yu, Molecules, 2016, 21, 387 CrossRef PubMed.
- O. Sadovski, J. W. Hicks, J. Parkes, R. Raymond, J. Nobrega, S. Houle, M. Cipriano, C. J. Fowler, N. Vasdev and A. A. Wilson, Bioorg. Med. Chem., 2013, 21, 4351–4357 CrossRef CAS PubMed.
- T. M. Shoup, A. A. Bonab, A. A. Wilson and N. Vasdev, Mol. Imaging Biol., 2015, 17, 257–263 CrossRef CAS PubMed.
- W. C. Silvers, H. Cai, O. K. Öz and X. Sun, Bioorg. Med. Chem. Lett., 2016, 26, 924–927 CrossRef CAS PubMed.
- M. B. Skaddan, L. Zhang, D. S. Johnson, A. Zhu, K. R. Zasadny, R. V. Coelho, K. Kuszpit, G. Currier, K.-H. Fan, E. M. Beck, L. Chen, S. E. Drozda, G. Balan, M. Niphakis, B. F. Cravatt, K. Ahn, T. Bocan and A. Villalobos, Nucl. Med. Biol., 2012, 39, 1058–1067 Search PubMed.
- M. Glaser, E. Årstad, A. Gaeta, J. Nairne, W. Trigg and E. G. Robins, J. Labelled Compd. Radiopharm., 2012, 55, 326–331 CrossRef CAS.
- B. H. Rotstein, H.-Y. Wey, T. M. Shoup, A. A. Wilson, S. H. Liang, J. M. Hooker and N. Vasdev, Mol. Pharmaceutics, 2014, 11, 3832–3838 CrossRef CAS PubMed.
- E. Sawatzky, E. Al-Momani, R. Kobayashi, T. Higuchi, S. Samnick and M. Decker, ChemMedChem, 2016, 11, 1540–1550 CrossRef CAS PubMed.
- R. R. Flavell, C. Truillet, M. K. Regan, T. Ganguly, J. E. Blecha, J. Kurhanewicz, H. F. VanBrocklin, K. R. Keshari, C. J. Chang, M. J. Evans and D. M. Wilson, Bioconjugate Chem., 2016, 27, 170–178 CrossRef CAS PubMed.
- A. Baranwal, H. H. Patel and J. Mukherjee, J. Labelled Compd. Radiopharm., 2014, 57, 86–91 CrossRef CAS PubMed.
- A. Baranwal and J. Mukherjee, Bioorg. Med. Chem. Lett., 2015, 25, 2902–2906 CrossRef CAS PubMed.
- I. Al Jammaz, B. Al-Otaibi, S. Amer, N. Al-Hokbany and S. Okarvi, Nucl. Med. Biol., 2012, 39, 864–870 CrossRef CAS PubMed.
- M. Simpson, L. Trembleau, R. W. Cheyne and T. A. D. Smith, Appl. Radiat. Isot., 2011, 69, 418–422 CrossRef CAS PubMed.
- I. AlJammaz, B. Al-Otaibi, H. AlHindas and S. M. Okarvi, Nucl. Med. Biol., 2015, 42, 804–808 CrossRef CAS PubMed.
- M. Onega, J. Domarkas, H. Deng, L. F. Schweiger, T. A. D. Smith, A. E. Welch, C. Plisson, A. D. Gee and D. O’Hagan, Chem. Commun., 2010, 46, 139–141 RSC.
- S. Dall’Angelo, N. Bandaranayaka, A. D. Windhorst, D. J. Vugts, D. van der Born, M. Onega, L. F. Schweiger, M. Zanda and D. O’Hagan, Nucl. Med. Biol., 2013, 40, 464–470 CrossRef PubMed.
- X.-G. Li, S. Dall’Angelo, L. F. Schweiger, M. Zanda and D. O’Hagan, Chem. Commun., 2012, 48, 5247–5249 RSC.
- O. Keinänen, X.-G. Li, N. K. Chenna, D. Lumen, J. Ott, C. F. M. Molthoff, M. Sarparanta, K. Helariutta, T. Vuorinen, A. D. Windhorst and A. J. Airaksinen, ACS Med. Chem. Lett., 2016, 7, 62–66 CrossRef PubMed.
- M. Onega, J. Domarkas, H. Deng, L. F. Schweiger, T. A. D. Smith, A. E. Welch, C. Plisson, A. D. Gee and D. O’Hagan, Chem. Commun., 2010, 46, 139–141 RSC.
- M. M. Alauddin, P. S. Conti and J. D. Fissekis, J. Labelled Compd. Radiopharm., 2002, 45, 583–590 CrossRef CAS.
- Z. Li, H. Cai and P. S. Conti, Nucl. Med. Biol., 2011, 38, 201–206 CrossRef CAS PubMed.
- H. Anderson, N. Pillarsetty, M. Cantorias and J. S. Lewis, Nucl. Med. Biol., 2010, 37, 439–442 CrossRef CAS PubMed.
- B. S. Moon, N. H. Jo, K. C. Lee, M. I. El-Gamal, G. Il An, S. H. Hong, T. H. Choi, W. K. Choi, J.-H. Park, J.-H. Cho, G. J. Cheon and C.-H. Oh, Bull. Korean Chem. Soc., 2010, 31, 3309–3312 CrossRef CAS.
- H. Zhang, M. V. Cantorias, N. Pillarsetty, E. M. Burnazi, S. Cai and J. S. Lewis, Nucl. Med. Biol., 2012, 39, 1182–1188 CrossRef CAS PubMed.
- H. Cai, Z. Li and P. S. Conti, Nucl. Med. Biol., 2011, 38, 659–666 CrossRef CAS PubMed.
- K. Chen, Z. Li and P. S. Conti, Nucl. Med. Biol., 2012, 39, 1019–1025 CrossRef CAS PubMed.
- V. Carroll, B. W. Michel, J. Blecha, H. Vanbrocklin, K. Keshari, D. Wilson and C. J. Chang, J. Am. Chem. Soc., 2014, 136, 14742–14745 CrossRef CAS PubMed.
- I. Lee, J. Yang, J. H. Lee and Y. S. Choe, Bioorg. Med. Chem. Lett., 2011, 21, 5765–5769 CrossRef CAS PubMed.
- C. Besanceney-Webler, H. Jiang, T. Zheng, L. Feng, D. Soriano Del Amo, W. Wang, L. M. Klivansky, F. L. Marlow, Y. Liu and P. Wu, Angew. Chem., Int. Ed., 2011, 50, 8051–8056 CrossRef CAS PubMed.
- M. Pretze, F. Wuest, T. Peppel, M. Köckerling and C. Mamat, Tetrahedron Lett., 2010, 51, 6410–6414 CrossRef CAS.
- K.-Z. Hu, H. Wang, T. Huang, G. Tang, X. Liang, S. He and X. Tang, Nucl. Med. Biol., 2013, 40, 926–932 CrossRef CAS PubMed.
- H. Liu, S. Liu, Z. Miao, H. Jiang, Z. Deng, X. Hong and Z. Cheng, Mol. Pharmaceutics, 2013, 10, 3384–3391 CrossRef CAS PubMed.
- S. Gao, G. Tang, S. Zhu, K. Hu, S. Yao, C. Tang, C. Yang, Y. Wang, J. Li, X. Pan, J. Guo, Q. Wang, R. Gao, W. Zhang, J. Wang, J. Huang and L. Zang, J. Radioanal. Nucl. Chem., 2016, 309, 1257–1264 CrossRef CAS.
- Z. Li, H. Cai, M. Hassink, M. L. Blackman, R. C. D. Brown, P. S. Conti and J. M. Fox, Chem. Commun., 2010, 46, 8043–8045 RSC.
- E. J. Keliher, T. Reiner, A. Turetsky, S. A. Hilderbrand and R. Weissleder, ChemMedChem, 2011, 6, 424–427 CrossRef CAS PubMed.
- T. Reiner, E. J. Keliher, S. Earley, B. Marinelli and R. Weissleder, Angew. Chem., Int. Ed., 2011, 50, 1922–1925 CrossRef CAS PubMed.
- P. Carberry, B. P. Lieberman, K. Ploessl, S. R. Choi, D. N. Haase and H. F. Kung, Bioconjugate Chem., 2011, 22, 642–653 CrossRef CAS PubMed.
- H.-L. Huang, C.-N. Yeh, K.-W. Chang, J.-T. Chen, K.-J. Lin, L.-W. Chiang, K.-C. Jeng, W.-T. Wang, K.-H. Lim, C. G. Chen, K.-I. Lin, Y.-C. Huang, W.-J. Lin, T.-C. Yen and C.-S. Yu, Bioorg. Med. Chem. Lett., 2012, 22, 3998–4003 CrossRef CAS PubMed.
- H.-L. Huang, Y.-C. Huang, W.-Y. Lee, C.-N. Yeh, K.-J. Lin and C.-S. Yu, PLoS One, 2014, 9, e104118 Search PubMed.
- R. Hoareau, L. Gobbi, S. Grall-Ulsemer and L. Martarello, J. Labelled Compd. Radiopharm., 2014, 57, 715–720 CrossRef CAS PubMed.
- Z. Li, L. Lang, Y. Ma and D. O. Kiesewetter, J. Labelled Compd. Radiopharm., 2008, 51, 23–27 CrossRef CAS.
- R. Löser, S. Fischer, A. Hiller, M. Köckerling, U. Funke, A. Maisonial, P. Brust and J. Steinbach, Beilstein J. Org. Chem., 2013, 9, 1002–1011 CrossRef PubMed.
- J. Pan, M. Pourghiasian, N. Hundal, J. Lau, F. Bénard, S. Dedhar and K.-S. Lin, Nucl. Med. Biol., 2013, 40, 850–857 CrossRef CAS PubMed.
- H. Doi, M. Goto and M. Suzuki, Bull. Chem. Soc. Jpn., 2012, 85, 1233–1238 CrossRef CAS.
- F. Kügler, J. Ermert and H. H. Coenen, J. Labelled Compd. Radiopharm., 2013, 56, 609–618 CrossRef PubMed.
- C. Lemaire, L. Libert, A. Plenevaux, J. Aerts, X. Franci and A. Luxen, J. Fluorine Chem., 2012, 138, 48–55 CrossRef CAS.
- C. Lemaire, P. Damhaut, A. Plenevaux, R. Cantineau, L. Christiaens and M. Guillaume, Appl. Radiat. Isot., 1992, 43, 485–494 CrossRef CAS.
- H. Suzuki, N. Yazawa, Y. Yoshida, O. Furusawa and Y. Kimura, Bull. Chem. Soc. Jpn., 1990, 63, 2010–2017 CrossRef CAS.
- D. R. Hwang, C. S. Dence, Z. A. McKinnon, C. J. Mathias and M. J. Welch, Nucl. Med. Biol., 1991, 18, 247–252 CAS.
- A. A. Wilson, R. F. Dannals, H. T. Ravert and H. N. Wagner, J. Labelled Compd. Radiopharm., 1990, 28, 1189–1199 CrossRef CAS.
- F. Kügler, W. Sihver, J. Ermert, H. Hübner, P. Gmeiner, O. Prante and H. H. Coenen, J. Med. Chem., 2011, 54, 8343–8352 CrossRef PubMed.
- K. Dahl, M. Schou and C. Halldin, J. Labelled Compd. Radiopharm., 2012, 55, 455–459 CrossRef CAS.
- S. R. Taylor, M. P. Roberts, N. A. Wyatt, T. Q. Pham, D. Stark, T. Bourdier, P. Roselt, A. Katsifis and I. Greguric, Aust. J. Chem., 2013, 66, 491–499 CrossRef CAS.
- G.-C. Li, R. Zhang, L.-J. Li and K.-J. Jiang, J. Radioanal. Nucl. Chem., 2013, 295, 385–391 CrossRef CAS.
- V. Shalgunov, J.-P. van Wieringen, H. Janssen, P. M. Fransen, R. A. J. O. Dierckx, M. Michel, J. Booij and P. H. Elsinga, J. Nucl. Med., 2015, 56, 133–139 CrossRef CAS PubMed.
- G. I. Warnock, J. Aerts, M. A. Bahri, F. Bretin, C. Lemaire, F. Giacomelli, F. Mievis, N. Mestdagh, T. Buchanan, A. Valade, J. Mercier, M. Wood, M. Gillard, A. Seret, A. Luxen, E. Salmon and A. Plenevaux, J. Nucl. Med., 2014, 55, 1336–1341 CrossRef CAS PubMed.
- Z. Li, X. Zhang, X. Zhang, M. Cui, J. Lu, X. Pan and X. Zhang, J. Med. Chem., 2016, 59, 10577–10585 CrossRef CAS PubMed.
- I. N. Petersen, J. Villadsen, H. D. Hansen, A. A. Jensen, S. Lehel, N. Gillings, M. M. Herth, G. M. Knudsen and J. L. Kristensen, Bioorg. Med. Chem., 2016, 24, 5353–5356 CrossRef CAS PubMed.
- M. G. Strebl, C. Wang, F. A. Schroeder, M. S. Placzek, H.-Y. Wey, G. C. Van de Bittner, R. Neelamegam and J. M. Hooker, ACS Chem. Neurosci., 2016, 7, 528–533 CrossRef CAS PubMed.
- X.-G. Li, M. Haaparanta and O. Solin, J. Fluorine Chem., 2012, 143, 49–56 CrossRef CAS.
- R. J. Abdel-Jalil, M. Aqarbeh, D. Löffler, B. Shen, S. A. Orabi, W. Voelter and H.-J. Machulla, J. Radioanal. Nucl. Chem., 2010, 283, 239–243 CrossRef CAS.
- C. R. Conard and M. A. Dolliver, Org. Synth., 1932, 12, 22 CrossRef.
- Z. Li, M. Cui, J. Zhang, J. Dai, X. Zhang, P. Chen, H. Jia and B. Liu, Eur. J. Med. Chem., 2014, 84, 628–638 CrossRef CAS PubMed.
- X. Cui, X. Zhang, C. Peng, J. Dai, B. Liu and M. Cui, RSC Adv., 2016, 6, 44646–44654 RSC.
- L. Li, M. N. Hopkinson, R. L. Yona, R. Bejot, A. D. Gee and V. Gouverneur, Chem. Sci., 2011, 2, 123–131 RSC.
- J. Way, V. Bouvet and F. Wuest, Curr. Org. Chem., 2013, 17, 2138–2152 CrossRef CAS.
- F. Kügler, J. Ermert, P. Kaufholz and H. H. Coenen, Molecules, 2015, 20, 470–486 CrossRef PubMed.
- J. D. Way, M. Wang, I. Hamann, M. Wuest and F. Wuest, Nucl. Med. Biol., 2014, 41, 660–669 CrossRef CAS PubMed.
- J. D. Way and F. Wuest, J. Labelled Compd. Radiopharm., 2014, 57, 104–109 CrossRef CAS PubMed.
- Y. Yagi, H. Kimura, K. Arimitsu, M. Ono, K. Maeda, H. Kusuhara, T. Kajimoto, Y. Sugiyama and H. Saji, Org. Biomol. Chem., 2015, 13, 1113–1121 CAS.
- G. Yuan, G. B. Jones, N. Vasdev and S. H. Liang, Bioorg. Med. Chem. Lett., 2016, 26, 4857–4860 CrossRef CAS PubMed.
- M. Glaser, E. Årstad, A. Gaeta, J. Nairne, W. Trig and E. G. Robins, J. Labelled Compd. Radiopharm., 2012, 55, 326–331 CrossRef CAS.
- H. M. Betts and E. G. Robins, J. Labelled Compd. Radiopharm., 2014, 57, 215–218 CrossRef CAS PubMed.
- R. Iwata, C. Pascali, A. Bogni, G. Horvath, Z. Kovács, K. Yanai and T. Ido, Appl. Radiat. Isot., 2000, 52, 87–92 CrossRef CAS PubMed.
- P. A. Schubiger, L. Lehmann and M. Friebe, PET Chemistry – The Driving Force in Molecular Imaging, 2007 Search PubMed.
- C. Lemaire, M. Guillaume, R. Cantineau, A. Plenevaux and L. Christiaens, Appl. Radiat. Isot., 1991, 42, 629–635 CrossRef CAS.
- M. Namavari, A. Bishop, N. Satyamurthy, G. Bida and J. R. Barrio, Int. J. Radiat. Appl. Instrum., Part A, 1992, 43, 989–996 CrossRef CAS.
- S. Forsback, O. Eskola, M. Haaparanta, J. Bergman and O. Solin, Radiochim. Acta, 2008, 96, 845–848 CrossRef CAS.
- F. Dolle, S. Demphel, F. Hinnen, D. Fournier, F. Vaufrey and C. Crouzel, J. Labelled Compd. Radiopharm., 1997, 41, 105–114 CrossRef.
- C. H. K. Kao, W. L. Hsu, H. L. Xie, M. C. Lin, W. C. Lan and H. Y. Chao, Ann. Nucl. Med., 2011, 25, 309–316 CrossRef PubMed.
- B. B. Azad, R. Chirakal and G. J. Schrobilgen, J. Labelled Compd. Radiopharm., 2007, 50, 1236–1242 CrossRef CAS.
- C. Lemaire, S. Gillet, S. Guillouet, A. Plenevaux, J. Aerts and A. Luxen, Eur. J. Org. Chem., 2004, 2899–2904 CrossRef CAS.
- C. Lemaire, A. Plenevaux, R. Cantineau, L. Christiaens, M. Guillaume and D. Comar, Appl. Radiat. Isot., 1993, 44, 737–744 CrossRef CAS.
- C. Lemaire, P. Damhaut, A. Plenevaux and D. Comar, J. Nucl. Med., 1994, 35, 1996–2002 CAS.
- D. Yin, L. Zhang, G. Tang, X. Tang and Y. Wang, J. Radioanal. Nucl. Chem., 2003, 257, 179–185 CrossRef CAS.
- R. N. Krasikova, V. V. Zaitsev, S. M. Ametamey, O. F. Kuznetsova, O. S. Fedorova, I. K. Mosevich, Y. N. Belokon, Š. Vyskočil, S. V. Shatik, M. Nader and P. A. Schubiger, Nucl. Med. Biol., 2004, 31, 597–603 CrossRef CAS PubMed.
- R. N. Krasikova, O. S. Fedorova, I. K. Mosevich, O. F. Kuznetsova, M. Korsakov, S. M. Ametamey and P. A. Schubiger, J. Labelled Compd. Radiopharm., 1999, 42, S102–S104 Search PubMed.
- B. Shen, W. Ehrlichmann, M. Uebele, H. J. Machulla and G. Reischl, Appl. Radiat. Isot., 2009, 67, 1650–1653 CrossRef CAS PubMed.
- L. C. Libert, X. Franci, A. R. Plenevaux, T. Ooi, K. Maruoka, A. J. Luxen and C. F. Lemaire, J. Nucl. Med., 2013, 54, 1154–1161 CrossRef CAS PubMed.
- C. Lemaire, L. Libert, X. Franci, J.-L. Genon, S. Kuci, F. Giacomelli and A. Luxen, J. Labelled Compd. Radiopharm., 2015, 58, 281–290 CrossRef CAS PubMed.
- M. Pretze, D. Franck, F. Kunkel, E. Foßhag, C. Wängler and B. Wängler, Nucl. Med. Biol., 2017, 45, 35–42 CrossRef CAS PubMed.
- F. Basuli, H. Wu, C. Li, Z.-D. Shi, A. Sulima and G. L. Griffiths, J. Labelled Compd. Radiopharm., 2011, 54, 633–636 CrossRef CAS.
- H. T. Ravert, D. P. Holt and R. F. Dannals, J. Labelled Compd. Radiopharm., 2014, 57, 695–698 CrossRef CAS PubMed.
- V. Bernard-Gauthier and R. Schirrmacher, Bioorg. Med. Chem. Lett., 2014, 24, 4784–4790 CrossRef CAS PubMed.
- C.-Y. Shiue, M. Watanabe, A. P. Wolf, J. S. Fowler and P. Salvadori, J. Labelled Compd. Radiopharm., 1984, 21, 533–547 CrossRef CAS.
- S. B. Höfling, S. Maschauer, H. Hübner, P. Gmeiner, H.-J. Wester, O. Prante and M. R. Heinrich, Bioorg. Med. Chem. Lett., 2010, 20, 6933–6937 CrossRef PubMed.
- N. Vasdev, P. N. Dorff, J. P. O’Neil, F. T. Chin, S. Hanrahan and H. F. VanBrocklin, Bioorg. Med. Chem., 2011, 19, 2959–2965 CrossRef CAS PubMed.
- J. A. Hendricks, E. J. Keliher, B. Marinelli, T. Reiner, R. Weissleder and R. Mazitschek, J. Med. Chem., 2011, 54, 5576–5582 CrossRef CAS PubMed.
- X. Huang, R. J. Gillies and H. Tian, J. Labelled Compd. Radiopharm., 2015, 58, 156–162 CrossRef CAS PubMed.
- T. Läppchen, M. L. H. Vlaming, E. Custers, J. Lub, C. F. Sio, J. DeGroot and O. C. Steinbach, Appl. Radiat. Isot., 2012, 70, 205–209 CrossRef PubMed.
- P. Slobbe, A. D. Windhorst, M. Stigter-van Walsum, R. C. Schuit, E. F. Smit, H. G. Niessen, F. Solca, G. Stehle, G. A. M. S. van Dongen and A. J. Poot, Nucl. Med. Biol., 2014, 41, 749–757 CrossRef CAS PubMed.
- W. Li, W. Thompson, T. Fisher, J. S. Wai, D. Hazuda, H. D. Burns and T. G. Hamill, J. Labelled Compd. Radiopharm., 2010, 53, 517–520 CAS.
- O. Tietz, S. K. Sharma, J. Kaur, J. Way, A. Marshall, M. Wuest and F. Wuest, Org. Biomol. Chem., 2013, 11, 8052–8064 CAS.
- N. Turkman, A. Shavrin, V. Paolillo, H. H. Yeh, L. Flores, S. Soghomonian, B. Rabinovich, A. Volgin, J. Gelovani and M. Alauddin, Nucl. Med. Biol., 2012, 39, 593–600 CrossRef CAS PubMed.
- J. Way and F. Wuest, Nucl. Med. Biol., 2013, 40, 430–436 CrossRef CAS PubMed.
- H. Zhang, R. Huang, N. Pillarsetty, D. L. J. Thorek, G. Vaidyanathan, I. Serganova, R. G. Blasberg and J. S. Lewis, Eur. J. Nucl. Med. Mol. Imaging, 2014, 41, 322–332 CrossRef CAS PubMed.
- J. Kaur, O. Tietz, A. Bhardwaj, A. Marshall, J. Way, M. Wuest and F. Wuest, ChemMedChem, 2015, 10, 1635–1640 CrossRef CAS PubMed.
- P. K. Garg, S. Garg and M. R. Zalutsky, Nucl. Med. Biol., 1994, 21, 97–103 CrossRef CAS PubMed.
- I. Koslowsky, J. Mercer and F. Wuest, Org. Biomol. Chem., 2010, 8, 4730–4735 CAS.
- C. R. Berry, P. K. Garg, M. R. Zalutsky, R. E. Coleman and T. R. DeGrado, J. Nucl. Med., 1996, 37, 2011–2016 CAS.
- C. R. Berry, T. R. DeGrado, F. Nutter, P. K. Garg, E. B. Breischwerdt, K. Spaulding, K. D. Concannon, M. R. Zalutsky and E. Coleman, Vet. Radiol. Ultrasound, 2002, 43, 183–186 CrossRef PubMed.
- I. Al Jammaz, B. Al-Otaibi, S. Okarvi and J. Amartey, J. Labelled Compd. Radiopharm., 2006, 49, 125–137 CrossRef CAS.
- I. Al Jammaz, B. Al-Otaibi, S. Amer and S. M. Okarvi, Nucl. Med. Biol., 2011, 38, 1019–1028 CrossRef CAS PubMed.
- T. L. Ross, M. Honer, C. Müller, V. Groehn, R. Schibli and S. M. Ametamey, J. Nucl. Med., 2010, 51, 1756–1762 CrossRef CAS PubMed.
- T. Betzel, C. Müller, V. Groehn, A. Müller, C. R. Fischer, S. D. Krämer, R. Schibli and S. M. Ametamey, Bioconjugate Chem., 2013, 24, 205–214 CrossRef CAS PubMed.
- K. S. Jang, Y. W. Jung, P. S. Sherman, C. A. Quesada, G. Gu and D. M. Raffel, Bioorg. Med. Chem. Lett., 2013, 23, 1612–1616 CrossRef CAS PubMed.
- K. S. Jang, Y. Jung, G. Gu, R. A. Koeppe, P. S. Sherman, C. A. Quesada and D. M. Raffel, J. Med. Chem., 2013, 56, 7312–7323 CrossRef CAS PubMed.
- C. Drerup, J. Ermert and H. H. Coenen, Molecules, 2016, 21, 1160 CrossRef PubMed.
- A. Maisonial, B. Kuhnast, J. Papon, R. Boisgard, M. Bayle, A. Vidal, P. Auzeloux, L. Rbah, M. Bonnet-Duquennoy, E. Miot-Noirault, M.-J. Galmier, M. Borel, S. Askienazy, F. Dollé, B. Tavitian, J.-C. Madelmont, N. Moins and J.-M. Chezal, J. Med. Chem., 2011, 54, 2745–2766 CrossRef CAS PubMed.
- U. Ackermann, D. Sigmund, S. D. Yeoh, A. Rigopoulos, G. O’Keefe, G. Cartwright, J. White, A. M. Scott and H. J. Tochon-Danguy, J. Labelled Compd. Radiopharm., 2011, 54, 788–794 CrossRef CAS.
- B. Carney, G. Carlucci, B. Salinas, V. Di Gialleonardo, S. Kossatz, A. Vansteene, V. A. Longo, A. Bolaender, G. Chiosis, K. R. Keshari, W. A. Weber and T. Reiner, Mol. Imaging Biol., 2016, 18, 386–392 CrossRef CAS PubMed.
- J. Marik and J. L. Sutcliffe, Appl. Radiat. Isot., 2007, 65, 199–203 CrossRef CAS PubMed.
- S. Richter and F. Wuest, Molecules, 2014, 19, 20536–20556 CrossRef PubMed.
- T. Kudo, M. Ueda, H. Konishi, H. Kawashima, Y. Kuge, T. Mukai, A. Miyano, S. Tanaka, S. Kizaka-Kondoh, M. Hiraoka and H. Saji, Mol. Imaging Biol., 2011, 13, 1003–1010 CrossRef PubMed.
- V. Awasthi, G. Pathuri, H. B. Agashe and H. Gali, J. Nucl. Med., 2011, 52, 147–153 CrossRef CAS PubMed.
- X. Yang, R. C. Mease, M. Pullambhatla, A. Lisok, Y. Chen, C. A. Foss, Y. Wang, H. Shallal, H. Edelman, A. T. Hoye, G. Attardo, S. Nimmagadda and M. G. Pomper, J. Med. Chem., 2016, 59, 206–218 CrossRef CAS PubMed.
- A. Hoehne, D. Behera, W. H. Parsons, M. L. James, B. Shen, P. Borgohain, D. Bodapati, A. Prabhakar, S. S. Gambhir, D. C. Yeomans, S. Biswal, F. T. Chin and J. Du Bois, J. Am. Chem. Soc., 2013, 135, 18012–18015 CrossRef CAS PubMed.
- J. Ides, D. Thomae, L. Wyffels, C. Vangestel, J. Messagie, J. Joossens, F. Lardon, P. Van der Veken, K. Augustyns, S. Stroobants and S. Staelens, Nucl. Med. Biol., 2014, 41, 477–487 CrossRef CAS PubMed.
- N. Matusiak, R. Castelli, A. W. Tuin, H. S. Overkleeft, R. Wisastra, F. J. Dekker, L. M. Prély, R. P. M. Bischoff, A. van Waarde, R. A. J. O. Dierckx and P. H. Elsinga, Bioorg. Med. Chem., 2015, 23, 192–202 CrossRef CAS PubMed.
- P. Ghosh, K. C. Li and D. Y. Lee, Appl. Radiat. Isot., 2011, 69, 609–613 CrossRef CAS PubMed.
- H. Xu, Z. Wang, Y. Wang, S. Hu and N. Liu, PLoS One, 2013, 8, e57897 CAS.
- M. Glaser, E. Årstad, S. K. Luthra and E. G. Robins, J. Labelled Compd. Radiopharm., 2009, 52, 327–330 CrossRef CAS.
- T. Ganguly, S. Dannoon, M. R. Hopkins, S. Murphy, H. Cahaya, J. E. Blecha, S. Jivan, C. R. Drake, C. Barinka, E. F. Jones, H. F. Vanbrocklin and C. E. Berkman, Nucl. Med. Biol., 2015, 42, 780–787 CrossRef CAS PubMed.
- H.-W. Kao, C.-L. Chen, W.-Y. Chang, J.-T. Chen, W.-J. Lin, R.-S. Liu and H.-E. Wang, Bioorg. Med. Chem., 2013, 21, 912–921 CrossRef CAS PubMed.
- C. Neto, C. Fernandes, M. C. Oliveira, L. Gano, F. Mendes, T. Kniess and I. Santos, Nucl. Med. Biol., 2012, 39, 247–260 CrossRef CAS PubMed.
- F. Svensson, T. Kniess, R. Bergmann, J. Pietzsch and F. Wuest, J. Labelled Compd. Radiopharm., 2011, 54, 769–774 CrossRef CAS.
- S. Dannoon, T. Ganguly, H. Cahaya, J. J. Geruntho, M. S. Galliher, S. K. Beyer, C. J. Choy, M. R. Hopkins, M. Regan, J. E. Blecha, L. Skultetyova, C. R. Drake, S. Jivan, C. Barinka, E. F. Jones, C. E. Berkman and H. F. VanBrocklin, J. Med. Chem., 2016, 59, 5684–5694 CrossRef CAS PubMed.
- N. Harada, H. Kimura, S. Onoe, H. Watanabe, D. Matsuoka, K. Arimitsu, M. Ono and H. Saji, J. Nucl. Med., 2016, 57, 1978–1984 CrossRef PubMed.
- D. E. Olberg, J. M. Arukwe, D. Grace, O. K. Hjelstuen, M. Solbakken, G. M. Kindberg and A. Cuthberson, J. Med. Chem., 2010, 53, 1732–1740 CrossRef CAS PubMed.
- Y. Chen, M. Pullambhatla, C. A. Foss, Y. Byun, S. Nimmagadda, S. Senthamizhchelvan, G. Sgouros, R. C. Mease and M. G. Pomper, Clin. Cancer Res., 2011, 17, 7645–7653 CrossRef CAS PubMed.
- N. Malik, H. J. Machulla, C. Solbach, G. Winter, S. N. Reske and B. Zlatopolskiy, Appl. Radiat. Isot., 2011, 69, 1014–1018 CrossRef CAS PubMed.
- B. D. Zlatopolskiy, R. Kandler, F. M. Mottaghy and B. Neumaier, Appl. Radiat. Isot., 2012, 70, 184–192 CrossRef CAS PubMed.
- B. D. Zlatopolskiy, P. Krapf, R. Richarz, H. Frauendorf, F. M. Mottaghy and B. Neumaier, Chem. – Eur. J., 2014, 20, 4697–4703 CrossRef CAS PubMed.
- B. D. Zlatopolskiy, R. Kandler, D. Kobus, F. M. Mottaghy and B. Neumaier, Chem. Commun., 2012, 48, 7134–7136 RSC.
- B. Kuhnast, F. Hinnen, B. Tavitian and F. Dollé, J. Labelled Compd. Radiopharm., 2008, 51, 336–342 CrossRef CAS.
- N. Arksey, T. Hadizad, B. Ismail, M. Hachem, A. C. Valdivia, R. S. Beanlands, R. A. DeKemp and J. N. DaSilva, Bioorg. Med. Chem., 2014, 22, 3931–3937 CrossRef CAS PubMed.
- M. P. Roberts, T. Q. Pham, J. Doan, C. D. Jiang, T. W. Hambley, I. Greguric and B. H. Fraser, J. Labelled Compd. Radiopharm., 2015, 58, 473–478 CrossRef CAS PubMed.
- P. Krapf, R. Richarz, E. A. Urusova, B. Neumaier and B. D. Zlatopolskiy, Eur. J. Org. Chem., 2016, 430–433 CrossRef CAS.
- K. Hashizume, N. Hashimoto and Y. Miyake, J. Org. Chem., 1995, 60, 6680–6681 CrossRef CAS.
- J.-H. Chun and V. W. Pike, Eur. J. Org. Chem., 2012, 4541–4547 CrossRef CAS PubMed.
- L. Wang, O. Jacobson, D. Avdic, B. H. Rotstein, I. D. Weiss, L. Collier, X. Chen, N. Vasdev and S. H. Liang, Angew. Chem., Int. Ed., 2015, 54, 12777–12781 CrossRef CAS PubMed.
- S. K. Fehler, S. Maschauer, S. B. Höfling, A. L. Bartuschat, N. Tschammer, H. Hübner, P. Gmeiner, O. Prante and M. R. Heinrich, Chem. – Eur. J., 2014, 20, 370–375 CrossRef CAS PubMed.
- L. Barré, L. Barbier and M. Lasne, J. Labelled Compd. Radiopharm., 1993, 32, 101–102 Search PubMed.
- I. Ekaeva, L. Barre, M.-C. Lasne and F. Gourand, Appl. Radiat. Isot., 1995, 46, 777–782 CrossRef CAS.
- T. Ludwig, J. Ermert and H. H. Coenen, Nucl. Med. Biol., 2002, 29, 255–262 CrossRef CAS PubMed.
- U. Mühlhausen, J. Ermert and H. H. Coenen, J. Labelled Compd. Radiopharm., 2009, 52, 13–22 CrossRef.
- T. Stoll, J. Ermert, S. Oya, H. F. Kung and H. H. Coenen, J. Labelled Compd. Radiopharm., 2004, 47, 443–455 CrossRef CAS.
- A. Helfer, J. Castillo Meleán, J. Ermert, A. Infantino and H. H. Coenen, Appl. Radiat. Isot., 2013, 82, 264–267 CrossRef CAS PubMed.
- T. L. Ross, J. Ermert and H. H. Coenen, Molecules, 2011, 16, 7621–7626 CrossRef CAS PubMed.
- T. Tominaga, H. Ito, Y. Ishikawa, R. Iwata, K. Ishiwata and S. Furumoto, J. Labelled Compd. Radiopharm., 2016, 59, 117–123 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2017 |