
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Teichoic acids: synthesis and applications
Daan
van der Es
,
Wouter F. J.
Hogendorf
,
Herman S.
Overkleeft
,
Gijsbert A.
van der Marel
and
Jeroen D. C.
Codée
*
Leiden Institute of Chemistry, Leiden University, PO Box 9502, 2300 RA Leiden, The Netherlands. E-mail: jcodee@chem.leidenuniv.nl
First published on 19th December 2016
Abstract
This review describes the developments in the synthesis of teichoic acids (TA) – glycosylated poly(alditolphosphates) – and the application of these fragments in immunological studies. These structurally diverse biopolymers are omnipresent constituents of the Gram-positive bacterial cell wall where they fulfill a variety of vital functions. They have been and continue to be attractive synthetic targets because of their challenging structures and the fact that their microheterogeneity precludes their isolation in single and pure enough form from natural sources. Progress in glycosylation chemistry and the development of effective phosphorylation chemistry has driven TA synthesis over the years, and highly complex and large TA structures can now reliably be targeted. Starting from the first TA synthesis in 1981, this review highlights the progress made in the field over the years. The synthesized TA fragments have been used to unravel their role in immunology and it is described how focused libraries of TAs have been used to discover the active principles of the TA polymers that interact with the innate immune system. Recently, synthetic TA fragments have also found applications as well-defined synthetic antigens for the generation of novel vaccine modalities to combat Gram-positive bacterial infections. It is foreseen that synthetic TA fragments will be valuable tools in the future to unravel the mode of action of these biomolecules at the molecular level. They will be instrumental in discovering and characterizing their designated biological binding partners, be it pattern recognition receptors or carbohydrate binding lectins or biomachinery enzymes. This review thus serves to showcase the potential of organic synthesis for (chemical) biology and immunology.
 Daan van der Es | Daan van der Es recently completed his PhD thesis entitled “Synthesis of Phosphodiester-Containing Bacterial Cell Wall Components: Teichoic Acids, Capsular Polysaccharides and Phosphatidyl Glycerol Analogues” under the guidance of Gijs van der Marel and Jeroen Codée. He currently is an assistant professor in the Medicinal Chemistry department at the Leiden Academic Center for Drug Research where he is involved in the design and synthesis of novel ligands and functionalized probes for G-protein coupled receptors. |
 Wouter F. J. Hogendorf | Wouter Hogendorf completed his PhD thesis entitled “Synthetic Methods to Glycerol Teichoic Acids” under the supervision of Gijs van der Marel and Jeroen Codée. He has developed automated solid phase and fluorous phase synthesis strategies for the assembly of teichoic acids. Additionally, he developed the first synthetic teichoic acid based vaccine against E. faecalis. After a post-doctoral position with Michael Bols and Christian Pedersen, working on the synthesis of C. difficile lipoteichoic acids, he worked as a post-doctoral researcher with John Nielsen. He is currently a researcher at Novo Nordisk working on the design and synthesis of new drugs. |
 Herman S. Overkleeft | Herman Overkleeft is Professor in Bioorganic Chemistry at Leiden University. He is the recipient of several Grants and Awards, including an ERC Advanced Grant, the Golden Medal from the Royal Dutch Chemical Society, a Wilhelm Friedrich Bessel Forschungspreis from the Humboldt Foundation and the Jeremy Knowles Award from the Royal Chemical Society. He is co-author on over 400 publications ranging a wide number of topics from organic synthesis, chemical biology, glycobiology oncology to immunology. His current research interests include the design, synthesis and application in medicinal chemistry and chemical biology research of inhibitors for glycosidases, glycosyl transferases and proteases. |
 Gijsbert A. van der Marel | Gijs van der Marel is Professor in Synthetic Organic Chemistry of the Leiden Institute of Chemistry at Leiden University. Throughout his career he has been interested in the development of synthetic methodology to assemble biopolymers: oligonucleotides, peptides and oligosaccharides. He has co-authored over 800 publications and he has supervised more than 80 PhD students. His current research interests include the development of fully synthetic vaccines, the synthesis of complex oligosaccharides and glycopeptides and glycolipids and the development of teichoic acid chemistry. |
 Jeroen D. C. Codée | Jeroen Codée completed his PhD in 2004 under the guidance of Jacques van Boom and Stan van Boeckel, investigating novel glycosylation methodology directed at the synthesis of glycosaminoglycans. After a two-year post-doctoral position in the group of Peter Seeberger at the Eidgenössische Technische Hochschule (ETH) Zürich, he returned to Leiden University where he now is associate Professor at the Leiden Institute of Chemistry. His group focusses on unraveling the glycosylation reaction mechanism, the assembly of complex bacterial glycans and the development of automated solid phase procedures for the synthesis of oligosaccharides and teichoic acids. |
Introduction
Teichoic acids (TAs) are structurally diverse anionic glycopolymers and prime constituents of the Gram-positive bacterial cell wall (teichoic is derived from the ancient Greek stem τειχ- meaning wall-related). These phosphodiester-based biomolecules include the membrane anchored lipoteichoic acid (LTA) and the peptidoglycan-bound wall teichoic acid (WTA). They perform crucial functions in the cell envelope (see Fig. 1) as they provide control over the rigidity and porosity of the cell wall and influence the bacterium's morphology.1–4 They are engaged in cell-division, cation homeostasis and protection against antimicrobial peptides and antibiotics. LTAs are involved in biofilm formation5 and binding of bacteriophages.6 Recently, WTAs have been shown to mask bacterial epitopes (including LTA-based epitopes) for binding of antibodies.7 Gram-positive bacteria lacking TAs can only be cultured under very specific circumstances, indicating the vital function of these structurally diverse biopolymers.8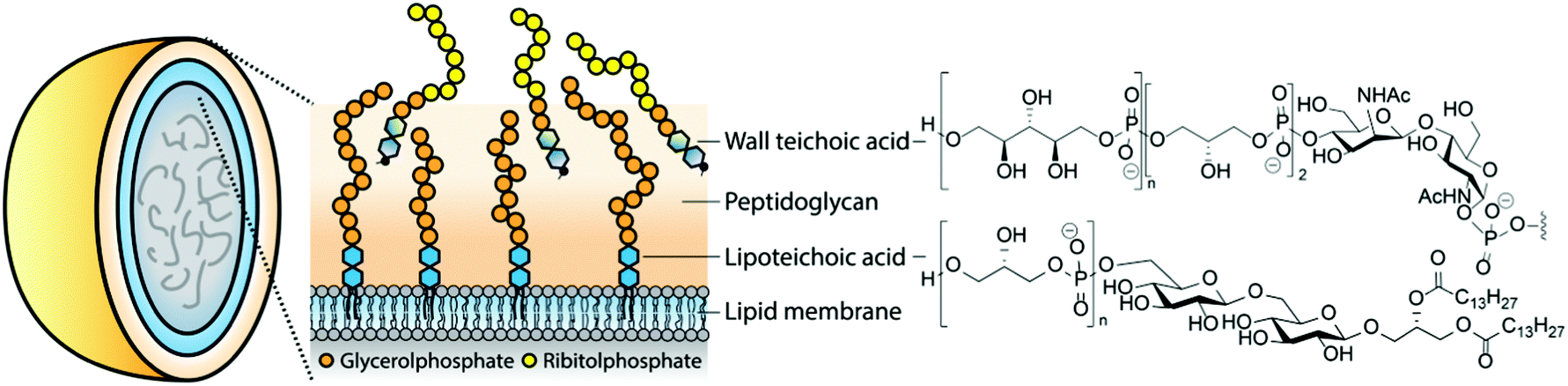 | ||
| Fig. 1 Examples of teichoic acids in the Gram-positive bacterial cell wall. | ||
As TAs are abundant in the Gram-positive bacterial cell-wall, they represent obvious targets for the immune system. It has been speculated that LTA is the Gram-positive equivalent to the Gram-negative lipopolysaccharide (LPS), a very powerful innate immune system stimulating agent responsible for the induction of septic shock during infections with Gram-negative bacteria. However, there has been much debate on the role of TAs in the immune response. Initial studies were hampered by difficulties encountered in TA isolation and purification. Both partially degraded and impure preparations9–11 were used providing a cloudy image of the immunological effect of these components. Improved purification protocols for LTA (mild butanol extraction as opposed to the previously employed hot phenol extraction methods) have delivered pure LTA preparations that have been shown to be capable of inducing a bona fide innate immune response.12 This review provides an overview of the synthetic chemistry that has been devised to generate well-defined TA-fragments. In doing so, it illustrates the development in organic synthesis approaches and strategies to provide both structurally complex and structurally diverse TAs. Applications of the synthesized TAs in the elucidation of their involvement in innate immune activation, and their role as antigens in novel vaccine modalities is described. The reported studies clearly show that structural variation in the TA molecules has a profound effect on their interaction with the immune system.
TA structures
The two main classes of TAs, LTAs and WTAs, can each be divided in sub-categories. Fischer13–15 introduced the first classification of LTAs and he defined four distinct LTA classes (Fig. 2A). Type I encompasses linear glycerol phosphate repeats, that can be decorated with alanine or carbohydrate residues. Type II and III LTA's consist of galactosylated glycerol phosphate repeats where the carbohydrate residue is part of the polymer chain. The repeating unit of type II LTA is a digalactosyl glycerol phosphate and type III is built up from monogalactosyl glycerol phosphates. Notably, both are encountered in only one bacterial species.16 The LTA from Streptococcus pneumonia has been designated as type IV LTA and with the detection of more diverse LTA structures, other classes have been introduced.4 For example, the LTA isolated from Clostridium difficile,17 was designated as type V LTA. This TA features a backbone that is not connected through the polyol moiety, but instead features a phosphodiester linkage between two C-6 hydroxyl functionalities.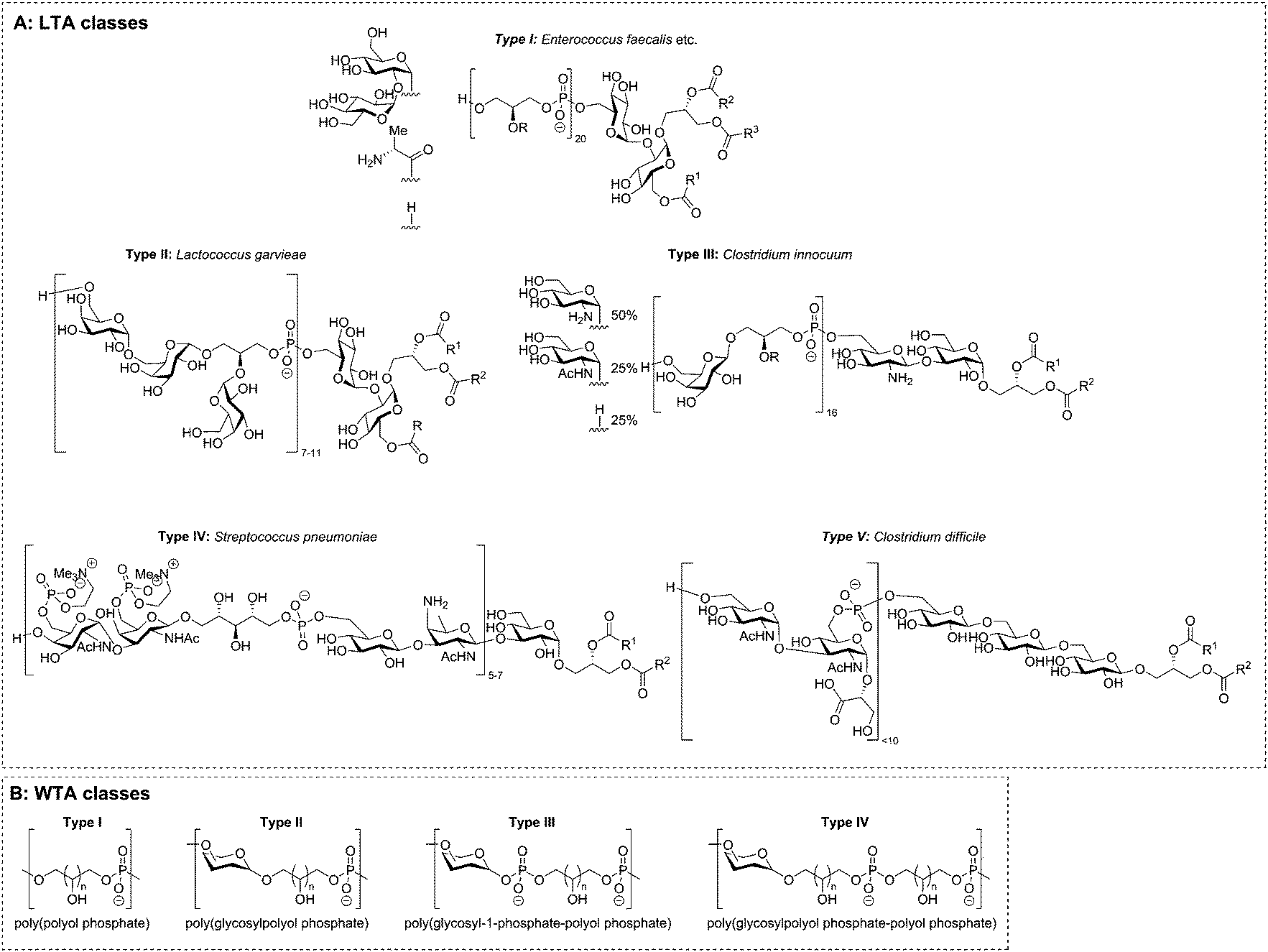 | ||
| Fig. 2 Teichoic acid classes as proposed by Fischer (A) and Stackebrandt (B). | ||
The WTAs can also be divided in different classes. Based on their work on the genus Nocardiopsis, Stackebrandt et al.18,19 suggested four classes for the structure and linkage of the repeating units (Fig. 2B). Similar to Fischer's LTA class I, WTA type I includes all polyol phosphates (glycerol phosphate, ribitol phosphate, etc.) linked from polyol to polyol via a phosphodiester linkage. In the backbone of type II WTAs a carbohydrate residue is incorporated, with the polyol connected to the anomeric center of carbohydrate residue through an acetal linkage. Type III and IV are copolymers of glycosyl-1-phosphates and type I repeating units (type III) or of type I and type II repeating units (type IV). The isolation of even more complicated copolymers will eventually necessitate the definition of more types, but no such TAs have hitherto been isolated. Besides these different types of TAs, bacteria can also produce exo-polysaccharides, some of which feature alditol residues and phosphodiesters, making classification of these biopolymers an even more difficult task.
Biosynthesis of TAs
The biosynthesis of LTA19,20 and WTA21 differs significantly, taking place in different cell locations and using different building blocks. Type I LTAs are generally synthesized on the glycolipid anchor, on the outside of the cell membrane, by transferring the glycerol phosphate building block from phosphatidylglycerol (PG) onto the nascent TA chain.22 In S. aureus, the synthesis is initiated by the glycosyltransferase YpfP that generates the glycolipid anchor (Fig. 3A). This anchor is synthesized on the inside of the lipid membrane and flipped to the outside by LtaA. The construction of the poly-glycerol phosphate chain is executed by LTA synthase (LtaS),23 a membrane bound polymerase featuring five transmembrane domains and an extracellular catalytic domain. LtaS transfers the glycerol phosphate from phosphatidyl glycerol (PG, Fig. 3B) by action of an active-site threonine residue that first covalently captures the glycerol phosphate moiety of PG thereby releasing diacylglycerol. Next, the glycerol phosphate moiety is transferred to the growing TA chain. D-Alanylation of LTA fragments is carried out by an ensemble of enzymes, DltA, B, C and D.19 This membrane bound multi-enzyme system first binds alanine from the cytoplasmic space and transfers it to the outside of the cell. There the load is delivered to the actual transferase, which attaches the alanine residue to the membrane anchored LTA. It is postulated that glycosylation of teichoic acids proceeds in a similar manner, but the enzymes involved have yet to be characterized.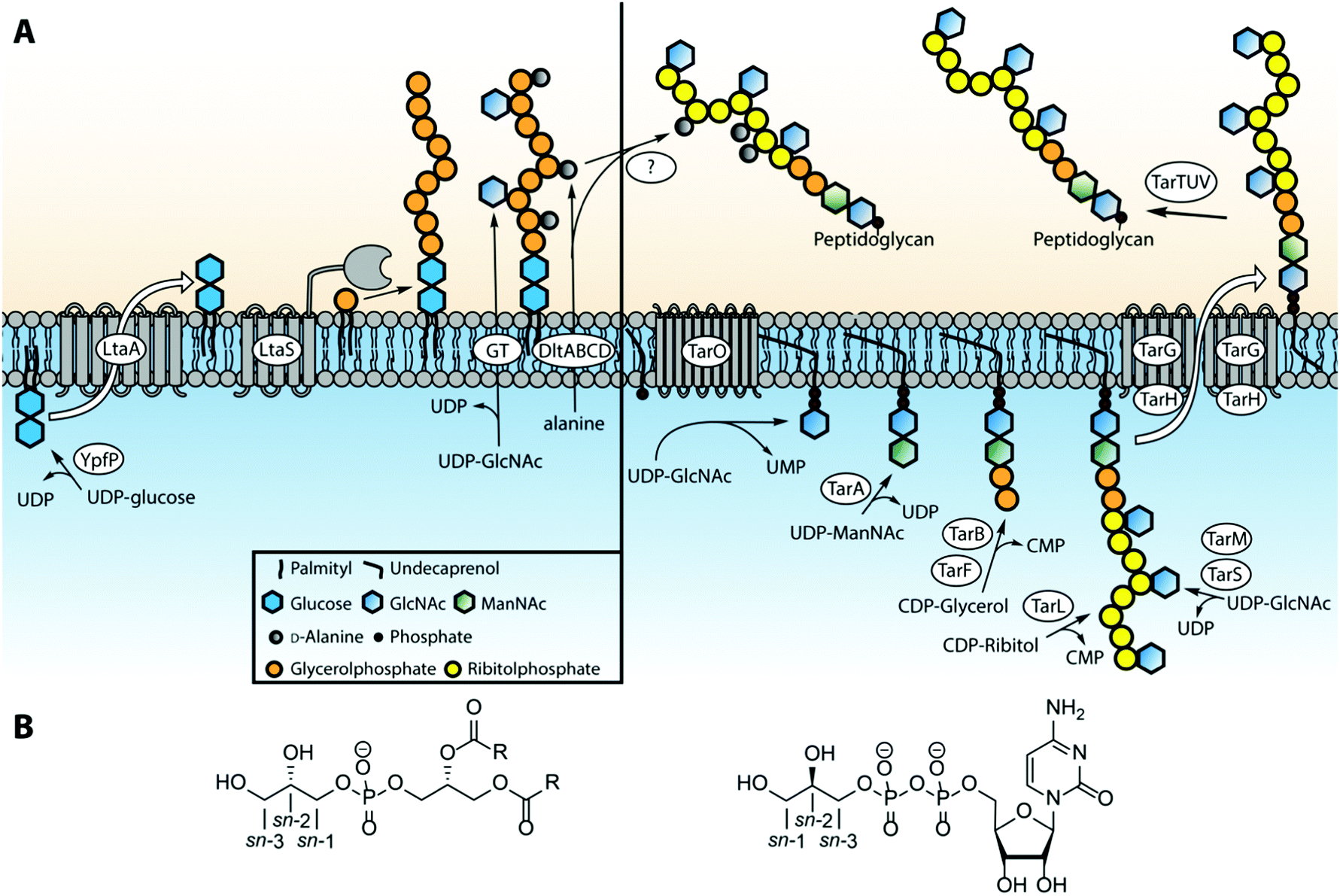 | ||
| Fig. 3 (A) Biosynthetic pathways of LTA and WTA in Staphylococcus aureus. (B) Structures of phosphatidylglycerol (containing sn-glycerol-1-phosphate) and CDP-glycerol (containing sn-glycerol-3-phosphate). | ||
Type IV LTA biosynthesis24 differs significantly from type I LTA synthesis. The proposed type IV LTA biosynthesis pathway involves cytoplasmic assembly of the complex repeating unit on an undecaprenyl carrier. After phosphocholination of the monomeric repeating unit the building blocks are oligomerized on the cytoplasmic side of the cell membrane. Finally the complete dolichol-TA structure is transported over the membrane and the oligomer is transferred onto the lipid anchor to finish the LTA synthesis. Interestingly, the oligomer can also be transferred to the peptidoglycan to yield S. pneumonia WTA. The enzymes involved in the biosynthesis of types II, III and V LTA are currently unknown, but it is likely that the mechanism is more similar to WTA than type I LTA biosynthesis.
Most work on WTA biosynthesis has concentrated on type I WTAs, in particular B. subtilis poly-glycerol or ribitol phosphate WTA and S. aureus poly(ribitol phosphate).21 The common linkage unit that connects the poly(alditol phosphate) chain to the peptidoglycan is a β-N-acetyl-mannosamine-(1,4)-α-N-acetyl-glucosamine-1-phosphate disaccharide that is linked through a phosphodiester linkage to a muramic acid moiety in the peptidoglycan.2 The synthesis of this linker is performed on an undecaprenyl phosphate carrier by the consecutive action of TarO and TarA (or TagO and TagA for glycerol teichoic acids). Next, a glycerol phosphate residue is transferred to the linker disaccharide by TarB (or TagB) using cytidine diphosphate glycerol (CDP-glycerol) as the glycerol phosphate donor (see Fig. 3B). This sets the stage for the assembly of the poly(glycerol phosphate) by TagF or the poly(ribitol phosphate) by TarK and L in B. subtilis. Of note, the stereochemistry of the glycerol phosphate that is transferred during WTA synthesis (sn-glycerol-3-phosphate, for sn-numbering see Fig. 3B) differs from the stereochemistry of the glycerol phosphate building blocks in LTA synthesis (sn-glycerol-1-phosphate). For the assembly of poly(ribitol phosphate) WTA chain in S. aureus first another glycerol phosphate moiety is attached by TarF, after which TarL generates the ribitol phosphate polymer using CDP-ribitol as a substrate. Finally the poly(glycerol phosphate) and poly(ribitol phosphate) WTA chain are decorated with carbohydrate residues by various enzymes (TarM and TarS in S. aureus25). The complete polymer is subsequently transported through the membrane by a membrane-bound hetero tetrameric complex consisting of TarG and TarH and transferred from the undecaprenyl support to the peptidoglycan. The mechanism for this last step is not clear at present, but it is thought that the set of enzymes TarTUV is involved. Interestingly, the transfer of D-alanine residues is executed at this final stage of WTA synthesis. This mechanism is unclear as well but it has been proposed that the DltABCD system involved in LTA-alanylation is responsible and that D-alanine may be transferred from LTA to WTA.19
Chemical synthesis of TAs
The microheterogeneity and polymeric nature of TAs make it impossible to isolate fragments of these biopolymers having a single substitution pattern and well-defined length. Organic synthesis can generate well-defined fragments with predetermined substituents.26 Non-natural and more stable analogues can be generated to probe the effect of substituents at the molecular level and synthetically accessed material is devoid of biological impurities that can thwart (immunological) evaluation of the material. Finally, organic synthesis can provide TA fragments equipped with appropriate conjugation handles that can be exploited in the generation of semi-synthetic vaccine modalities and analytic tools, such as TA-microarrays.Synthetic interest in TAs started in the early 1980's and the first chemical synthesis of a teichoic acid fragment was reported by Van Boeckel et al. in 1981, who described the assembly of a Bacillus atrophaeus (formerly known as B. subtilis var. niger) trimer repeat.27 As depicted in Scheme 1, they coupled phosphodiester 2 and primary alcohol 1 using 1-(2,4,6-triisopropylbenzenesulfonyl)-3-nitro-1H-1,2,4-triazole (TPSNT) as condensing agent to construct the phosphotriester linkage. The phosphate on the primary alcohol was masked as an aniline phosphoramidate. The resulting dimer was treated with isopentyl nitrite to remove the aniline and liberate the phosphodiester of 3, that could be elongated in a second coupling cycle to give protected timer 5. Deprotection of the oligomer was achieved by first unmasking the tribromoethyl groups, using zinc, followed by removal of the ortho-chlorophenyl group. Removal of the acetyl esters, followed by cleavage of the THP-acetal under acidic conditions yielded deprotected 6, in 73% yield28 from 5.
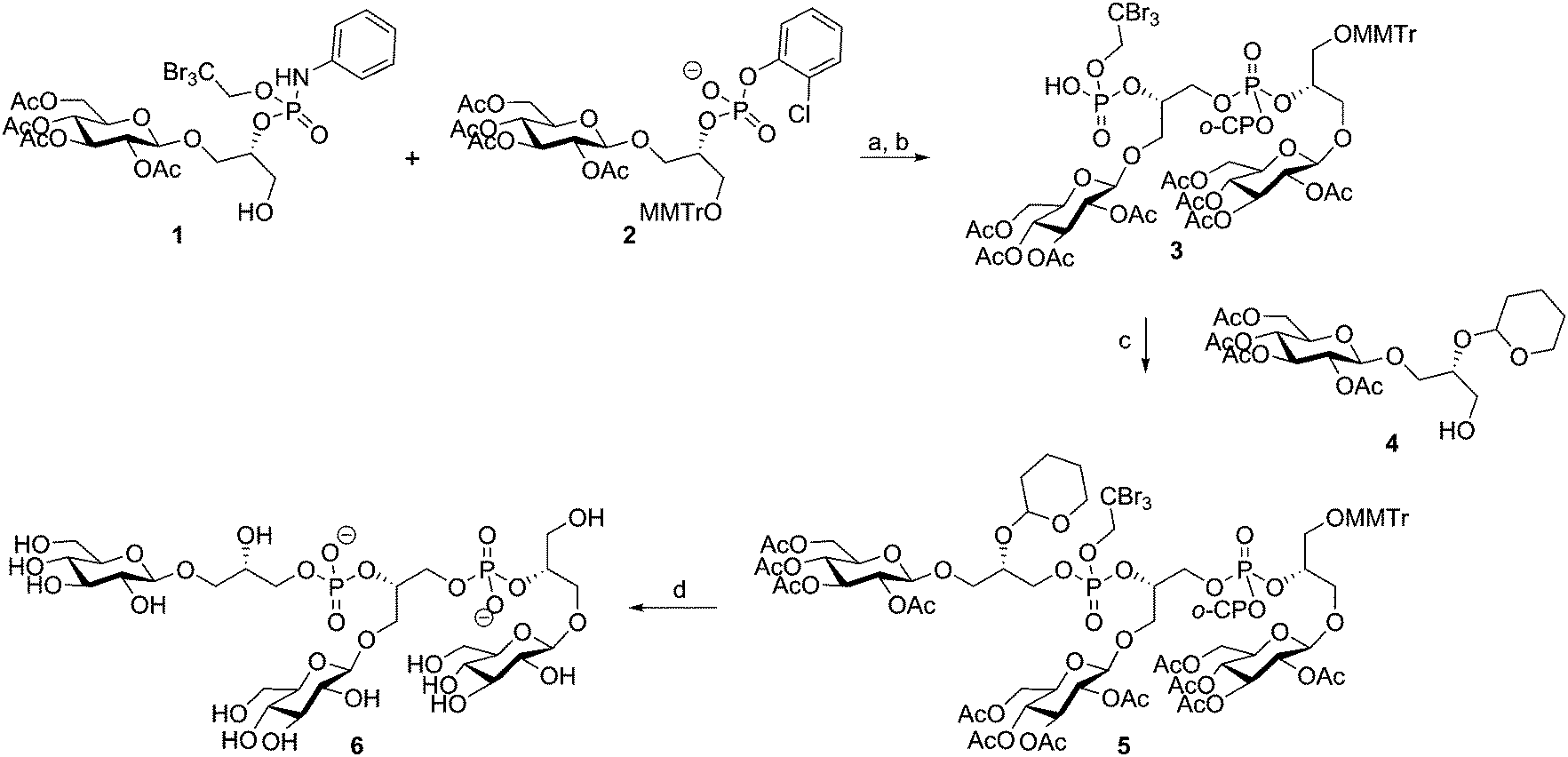 | ||
| Scheme 1 van Boeckel's Bacillus atrophaeus TA-synthesis. Reagents and conditions: (a) TPSNT, pyridine, 56%; (b) isopentyl nitrite, pyridine/AcOH, 80%; (c) TPSNT, pyridine, 79%; (d) (i) Zn, pTSOH, pyridine, 88%, (ii) tetramethylguanidinium-4-nitrobenzaldoximate, THF, (iii) aq. NH3, 87% over 2 steps, (iv) 0.1 M HCl, dioxane/H2O, 96%. | ||
Continuing their efforts towards the assembly of well-defined TA fragments, the Van Boom group synthesized a fragment of S. aureus LTA, composed of three glycerol phosphate residues coupled to a gentiobiose based glycolipid anchor.29 Van Boom and co-workers were also the first to report on automated solid phase synthesis of a TA-fragment. Using state-of-the-art phosphoramidite chemistry as introduced by Beaucage,30 Westerduin et al. assembled a pentameric Bacillus licheniformis WTA-fragment as depicted in Scheme 2.31 Using an automated DNA/RNA synthesizer, the fragment was assembled employing coupling cycles that entailed: (i) removal of the DMT group (2% trichloroacetic acid in DCM); (ii) phosphoramidite coupling using building block 8 under the agency of tetrazole in MeCN; (iii) capping of the unreacted alcohols (Ac2O, 2,6-lutidine and N-methylimidazole); (iv) oxidation of the formed phosphite triesters using I2 in 2,6-lutidine and 1,4-dioxane. Three repetitions of this coupling cycle delivered the resin-bound target compound. Upon ammonia treatment of the resin, the product was cleaved from the resin and the base labile protecting groups (the acetyl esters and the cyanoethyl phosphotriesters) removed. The target compound was purified by size exclusion chromatography to provide, after global deprotection by hydrogenolysis, pentamer 12 in 29% from immobilized 10. At the time 50 equivalents of phosphoramidite 8 were required to push the reaction to completion but the synthesis was accomplished in a time in which the chemistry and automated synthesis methodology were only just conceived. Recent syntheses (vide infra) require a significantly smaller amount of building block, but build on exactly the same chemistry. Surprisingly, it wasn't until the recent syntheses reported by Hogendorf et al. that more TA fragments were synthesized using this effective synthesis technique.
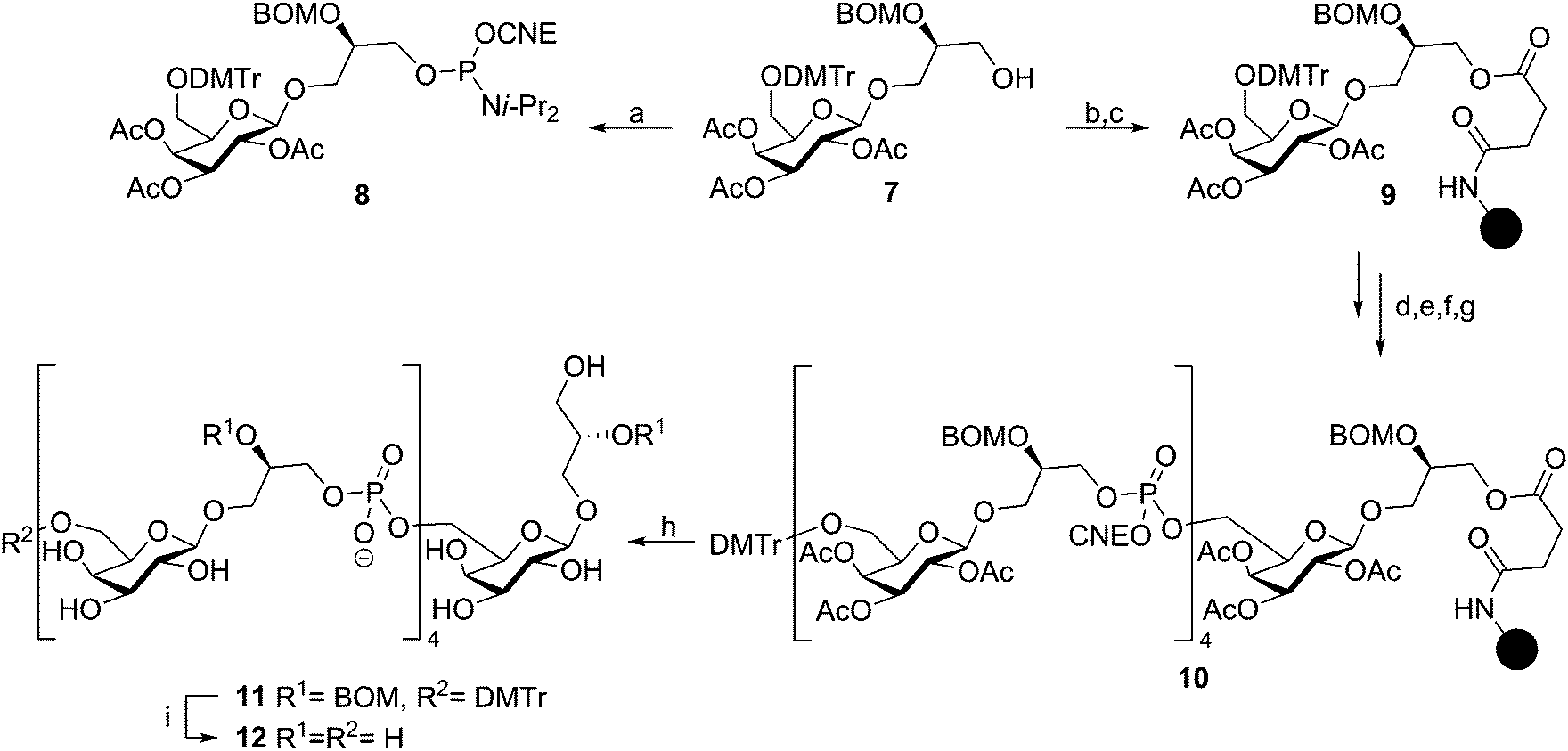 | ||
| Scheme 2 First automated solid phase synthesis of an TA-fragment by Westerduin et al. Reagents and conditions: (a) 2-cyanoethyl N,N-diisopropylchlorophosphoramidite, DIPEA, DCM, 98%; (b) succinic anhydride, pyridine, DMAP, 45 °C, 90%; (c) (i) DIC, HOBT, MeCN, (ii) N-acetylimidazole, DCM; (d) 2% TCA, DCM; (e) 8, tetrazole, MeCN; (f) Ac2O, 2,6-lutidine, N-methylimidazole; (g) I2, 2,6-lutidine, dioxane; (h) NH3, H2O; (i) Pd/C, H2. | ||
In 1992 the Kusumoto group published the synthesis of the proposed structure of Streptococcus pyogenes LTA.32 Using phosphoramidite chemistry they synthesized tetramer 18 using a block coupling strategy (Scheme 3). To this end, dimeric phosphotriester 15 was generated from glycerol nucleophile 13 and phosphoramidite 14. Selective TBS removal followed by phosphoramidite introduction then yielded 16, where selective PMB removal from 15 yielded alcohol 17. These two diglycerol phosphates were condensed to give a tetramer repeat, of which the primary alcohol was liberated by TBS removal and subsequently transformed into a phosphoramidite moiety to give tetramer 18. Coupling of this fragment to kojibiose glycolipid 19 yielded protected LTA fragment 20, which was deprotected in a single hydrogenation event to yield 21. Two years later, the same glycerol phosphate oligomer was coupled to the glycolipid anchor from E. hirae, bearing an additional lipid tail.33 Both fragments were evaluated for their innate immune-stimulating potential but no activity was established.34
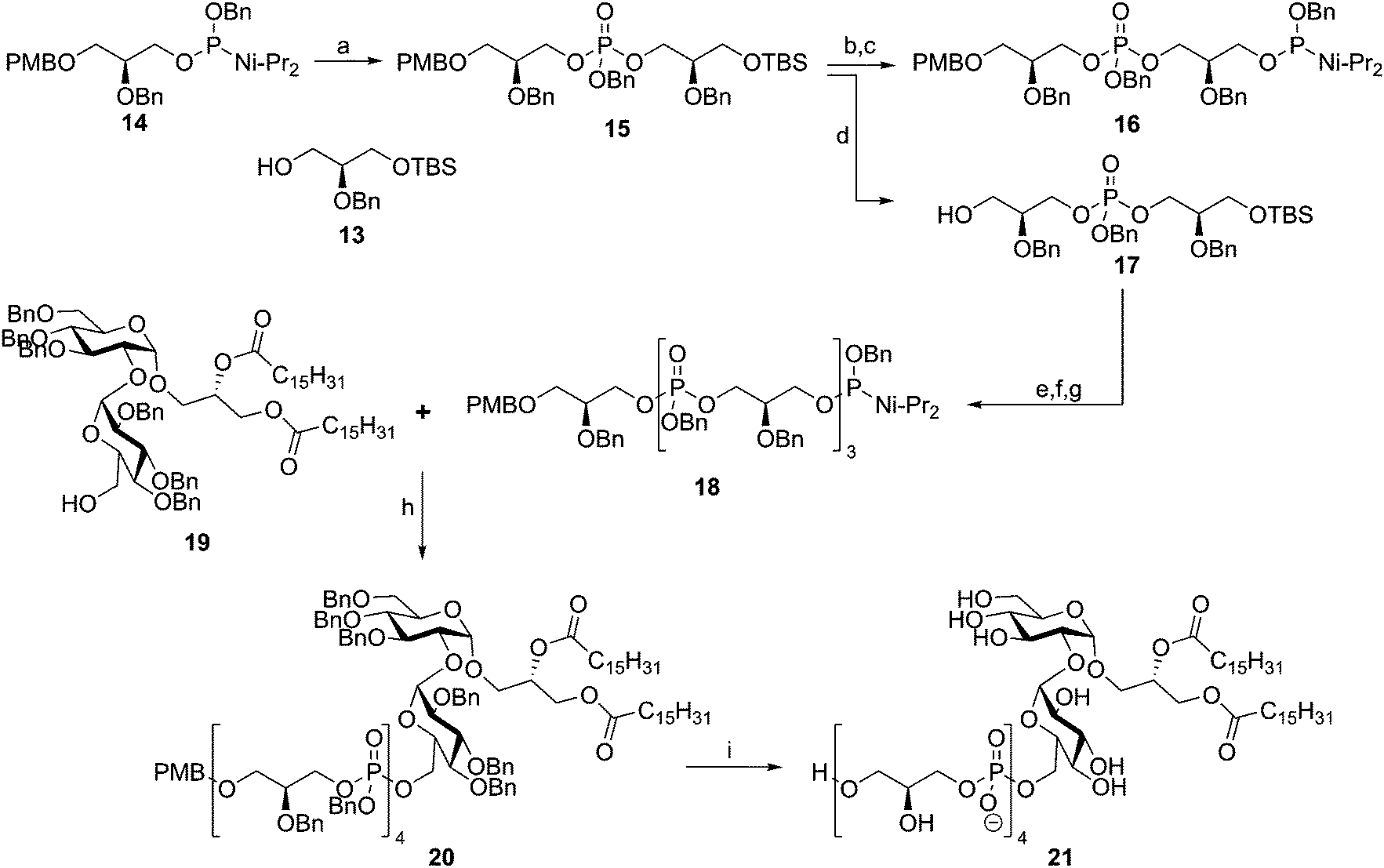 | ||
| Scheme 3 Fukase's Streptococcus pyogenes LTA synthesis. Reagents and conditions: (a) (i) 13, tetrazole, DCM, MeCN, (ii) m-CPBA, 89%; (b) TBAF, THF, 99%; (c) bis(N,N-diisopropylamino)-O-benzyl-phosphoramidite, tetrazole, DCE, MeCN, 84%; (d) DDQ, H2O, DCM, 82%; (e) (i) 16, tetrazole, DCM, MeCN, (ii) m-CPBA; (f) TBAF, THF, 88%; (g) bis(N,N-diisopropylamino)-O-benzyl-phosphoramidite, tetrazole, DCE, MeCN; (h) (i) tetrazole, DCM, MeCN, (ii) m-CPBA; (i) Pd(0), H2, THF, MeOH, 48%. | ||
In 2003, after almost a decade of silence in TA-synthesis, Schmidt and co-workers reported on the synthesis of a Staphylococcus aureus LTA fragment.35 Notably, this group succeeded in the generation of fragments having different substituents on the glycerol phosphate backbone, including the labile alanine esters (Scheme 4). The disaccharide glycerolipid anchor was made from gentiobiose and was equipped with a phosphoramidite to yield 26. Glycerol phosphoramidites 23 and 24 were synthesized from commercial solketal and either equipped with an N-acetylglucosamine or a PMB group on the 2-OH. A TBDPS-ether was selected to serve as temporary protecting group for the primary alcohol to be elongated during the synthesis. Starting from di-O-benzyl glycerol 22 six coupling and desilylation steps yielded hexamer glycerol phosphate 25. The coupling between hexamer glycerol phosphate 25 and lipid anchor phosphoramidite 26 proceeded uneventfully and subsequent PMB cleavage set the stage for the introduction of the alanine residues. Both D- and L-alanine residues were incorporated to investigate how the stereochemistry at the αC-atom affects the activity of the TA fragment. The alanine residues were introduced using Cbz-protected alanine in conjunction with a PyBOP, NMI condensation cocktail. Global deprotection of the LTA fragment was achieved using hydrogenolysis over Pearlman's catalyst (Pd(OH)2) in a mixture of DCM/MeOH/H2O to solubilize the amphiphilic molecule. Target compounds 28 and 29, featuring four D-alanine or four L-alanine esters were obtained in 22% and 17%, respectively. Immunological evaluation of the compounds,36–38 alongside isolated LTA (using the optimized mild n-butanol extraction procedure) and TAs lacking the lipid anchor as well as the lipid anchor alone revealed that the synthetic material with the natural D-alanine substituents was capable of inducing pro-inflammatory cytokines in human whole blood to a similar extent as the isolated material. In a human whole blood cytokine response assay the synthetic material and the isolated LTA stimulated the production of IL-1β, IL-6, IL-8, IL-10 and TNF-α to a similar extent.37 The L-alanine bearing fragment was 100 times less active than the LTA fragment featuring the naturally configured D-alanine residues. Deacylated, isolated LTA (without lipid anchor or alanine residues) displayed no immunostimulatory activity38 and was incapable of blocking the activity of native LTA, indicating that it had lost its affinity for its cellular binding partner. The lipid anchor alone showed weak immunostimulatory activity. Combined, these results show that for optimal innate immune stimulatory activity LTA fragments should bear both the lipid anchor and a number of D-alanine residues.
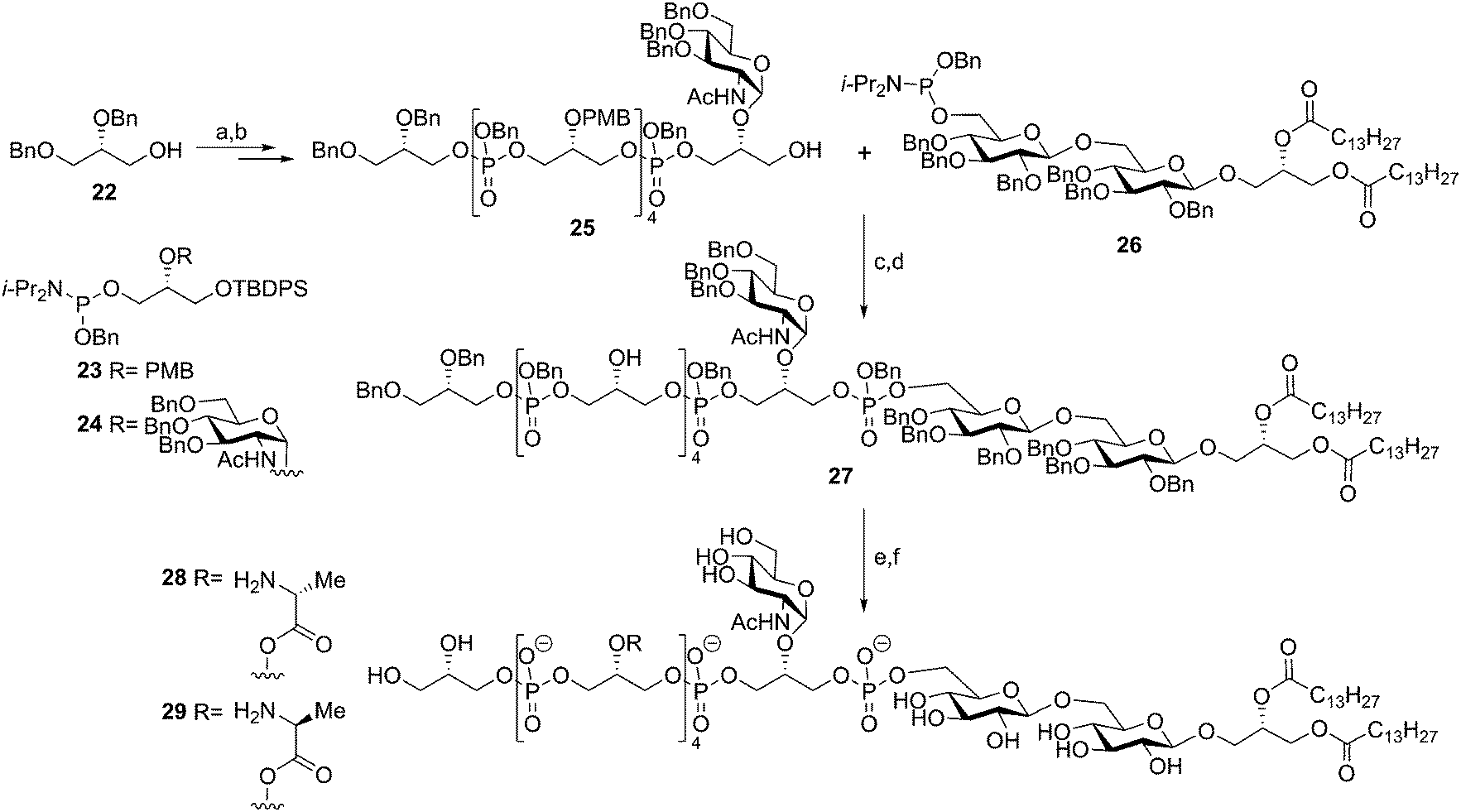 | ||
| Scheme 4 Schmidt's Staphylococcus aeureus LTA synthesis. Reagents and conditions: (a) (i) 23 or 24, tetrazole, DCM, (ii) tBuO2H; (b) TBAF; (c) (i) tetrazole, DCM, (ii) tBuO2H, 75%; (d) CAN, MeCN/toluene/H2O, 67%; (e) PyBOP, N-methylimidazole, Z-protected alanine, DCM; (f) Pd(OH)2-C, H2, DCM/MeOH/H2O, 28: 33% over 2 steps, 29: 25% over 2 steps. | ||
Subsequently, the Schmidt laboratory explored, through the chemical synthesis of a variety of LTA fragments, the importance of all structural components for immunostimulatory activity. A convergent approach was devised that made it possible to prepare several constructs by minor alterations of the synthetic route. In 2005, the syntheses of fragments 30 and 31 were described (see Fig. 4).39 Whereas LTA 30 only lacked the gentiobiosyl moiety, derivative 31 features more radical changes: both gentiobiosyl and GlcNAc moieties were omitted and the D-alanine esters were replaced by the more stable amide analogues on the C-2 glycerol residues of opposed stereochemistry. The effects of these modifications on the immunostimulatory properties were minimal, showing that the gentiobiosyl and GlcNAc were not necessary for cytokine (IL-8, TNF-α) production in human whole blood. The presence of the alanylated glycerol phosphate backbone proved more important. This finding was confirmed by the synthesis and evaluation of TA derivatives 32a–e. These compounds consist of the complete gentiobiosyl diacyl glycerol moiety connected to an oligoglycerol phosphate backbone ranging in length between two (32a) and six residues (32e) and containing between one and five alanine modifications (Fig. 4).40,41 Biological evaluation showed that a minimum of two alanylated glycerol phosphates in the construct is required for significant activity (10-fold increase of cytokine induction compared to one alanylated residue). The gentiobiosyl diacyl glycerol anchor (structure not shown) alone did not induce any immune response. The role of TLR2 in the induction of pro-inflammatory cytokines was investigated using peritoneal macrophages from wild type and TLR2 knock out mice. Stimulation of these cells by isolated LTA and LTAs 31 and 32e led to the release of TNF-α and IL-8 by the wild type cells, but not by the TLR –/– macrophages. Stimulation assays with TLR2 transfected HEK 293 cells corroborated the TLR2 dependent activation by the LTAs. The group of Schmidt also reported the synthesis of a construct that contained all the natural substitutions and a second glycolipid moiety (structure 33, Fig. 4).42 It was shown in a human whole blood cytokine release assay that the titers of released IL-8 and TNF-α were a factor 10 higher compared to the monoglycolipid 28 (see Scheme 4). It has been speculated that the altered presentation of the crucial recognition elements, such as the D-alanyl esters, by this bis-amphiphilic construct is responsible for the increase in activity.
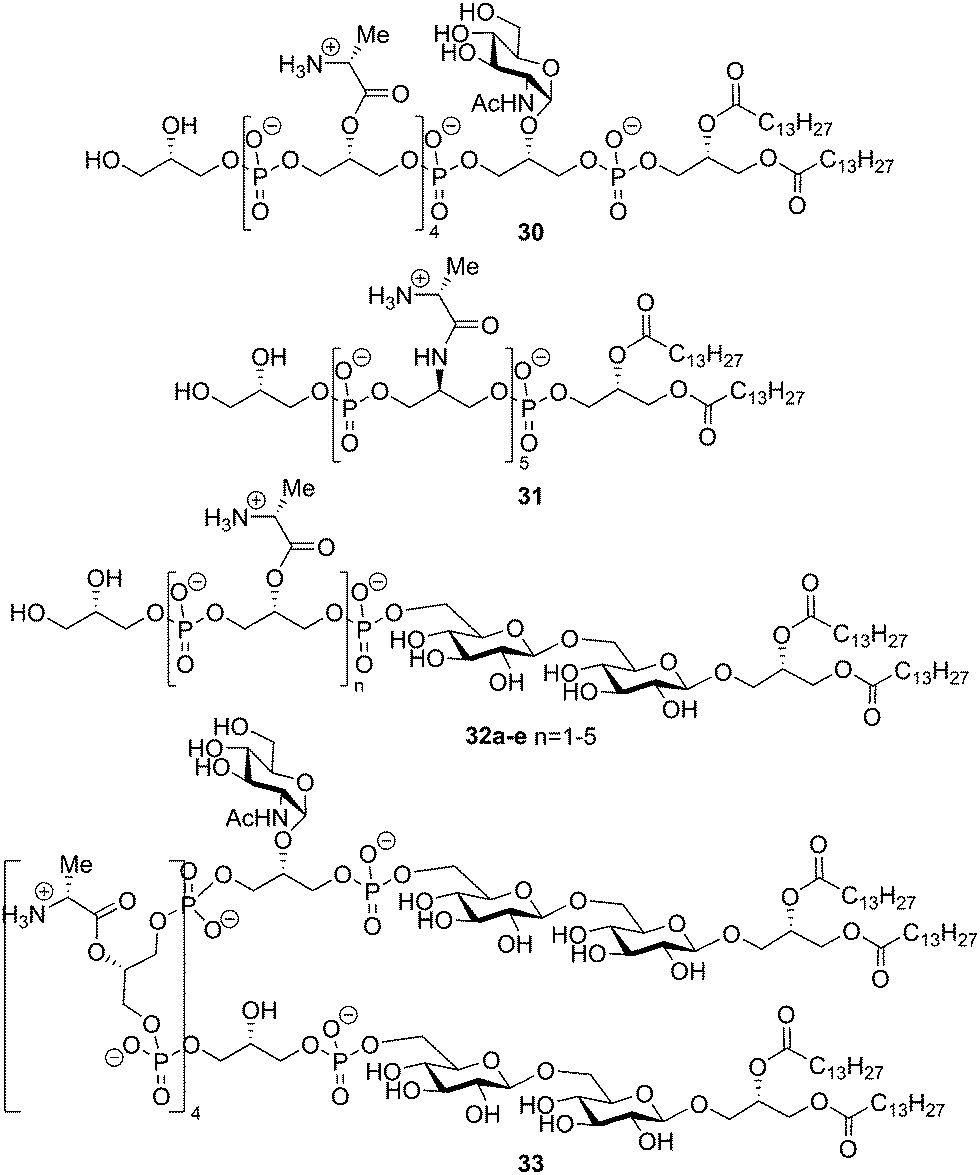 | ||
| Fig. 4 Schmidt's synthetic LTA derivatives based on Staphylococcus aureus. | ||
The type I LTA from Streptococcus sp DSM 8747, a mutant which is genetically related to Streptococcus pneumonia, consists of a galactofuranoside diacyl glycerol glycolipid with the oligoglycerol phosphate backbone connected to the C-6 of the galactofuranose residue. The backbone is substituted for about 30% at the glycerol C-2 position with D-alanine esters.43 In 2010, Schmidt and coworkers reported on the synthesis of a fragment of this LTA.44 The β-galactofuranoside diacylglycerol phosphoramidite 40 was constructed via the imidate glycosylation procedure using anchimeric assistance of a temporary 2′-benzoyl ester, followed by several consecutive (protecting group) manipulations (see Scheme 5). Block coupling of the previously reported PMB-protected pentamer en route to 25 and glycolipid phosphoramidite 40 using tetrazole followed by oxidation yielded the fully protected intermediate in 90%. At this stage the PMB ethers were selectively cleaved (85% yield) and the resulting tetraol was decorated with alanyl moieties using Cbz-D-alanine and PyBOP (74% yield). Global deprotection of the fully protected 43 (containing four alanine esters) followed by purification resulted in the target LTA fragment 44 provided with roughly two alanine esters. It is unclear why the alanine esters in this particular molecule were prone to hydrolysis, as this phenomenon was not observed in similar syntheses reported earlier by the group of Schmidt.35,39,40,42 Unfortunately, no immunological evaluation of the synthesized LTA molecule has been reported to date.
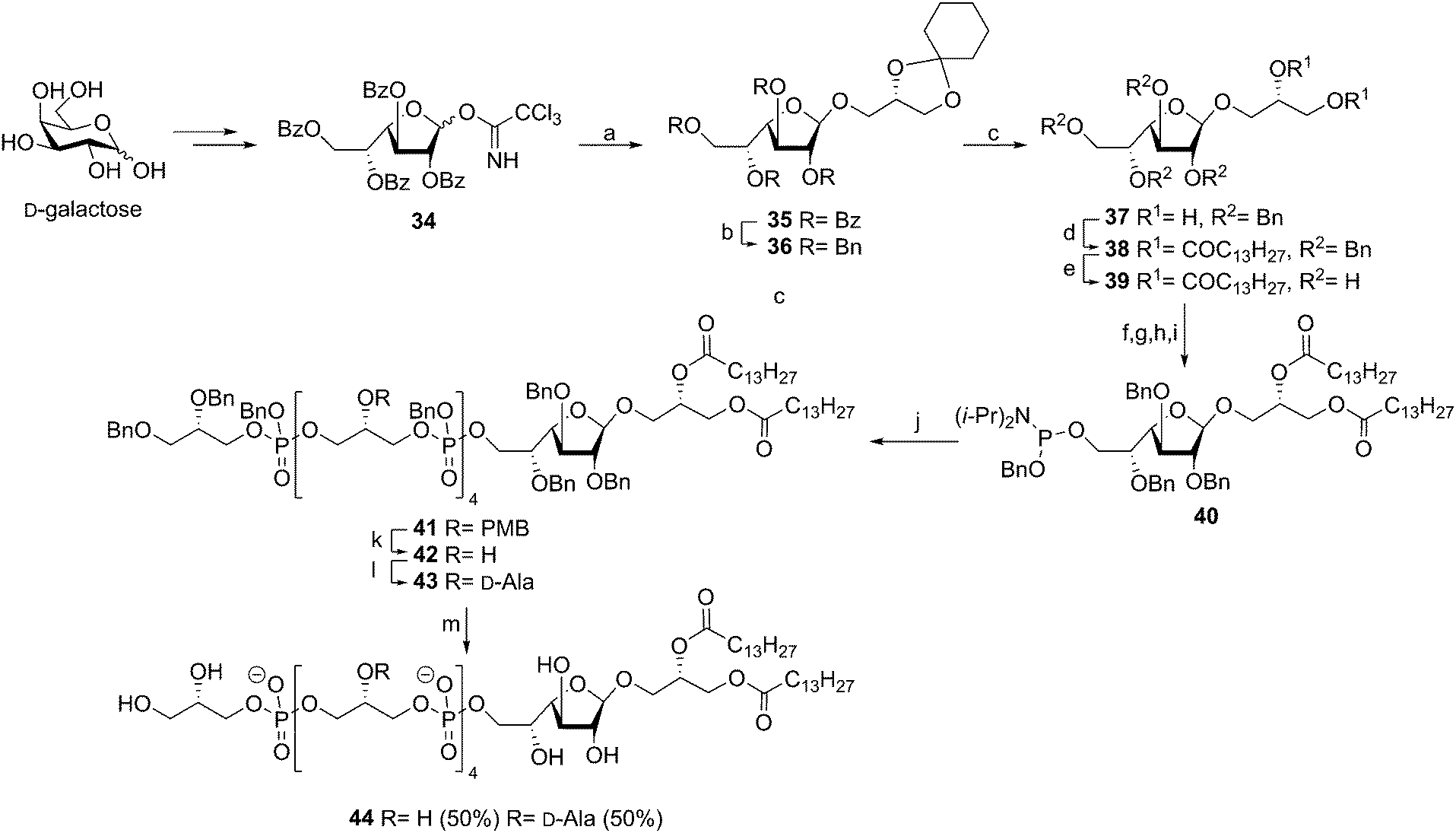 | ||
Scheme 5 Schmidt's Streptococcus species DSM 8747 LTA synthesis. Reagents and conditions: (a) TMSOTf, DCM, 89%; (b) NaOMe, MeOH; then BnBr, NaH, DMF, 96%; (c) p-TsOH, DCM/MeCN, ethylene glycol, 76%; (d) DCM, DMAP, DCC, quant; (e) Pd/C, H2, TFA, EtOAc/MeOH, 90%; (f) TBDMSCl, DMAP, Et3N, DCM, 63%; (g) BnBr, NaH, DMF, 90%; (h) TBAF, DCM, 76%; (i) BnOP(Ni-Pr2)2, diisopropanylammonium tetrazolide, 81%; (j) GTA pentamer, tetrazole, DCM, then t-BuOOH, 90%; (k) CAN, MeCN, toluene, H2O, 85%; (l) N-methylimidazole, Cbz-D-alanine triethylammonium salt, PyBOP, DCM, 74%; (m) Pd(OH)2, H2, DCM/MeOH/H2O (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:2), 28%. :3:2), 28%. | ||
The most impressive synthetic achievement in the TA arena is undoubtedly the successful assembly of the complex Streptococcus pneumoniae type IV LTA.45 This synthesis is summarized in Scheme 6 and is based on the block coupling of ribitol tetrasaccharide 54 and trisaccharide lipid anchor 55. The tetrasaccharide unit 54 was synthesized by a series of glycosylations, starting with the α-selective coupling of 46 and 47, followed by the acetonitrile mediated β-selective coupling of the deacetylated disaccharide to tetrabenzyl donor 45. Another acetonitrile mediated coupling between 48 and 49 yielded the ribitol coupled galactosazide, which could be coupled to the synthesized trisaccharide. Desilylation followed by introduction of the phosphocholine residues furnished the ribitol tetrasaccharide 54. Trisaccharide lipid anchor phosphoramidite 55 was synthesized by sequentially coupling 52, 51, and 50 to glycerol residue 53. The crucial coupling between 54 and 55 yielded fragment 56 in 68% yield, showcasing the effectiveness of phosphoramidite chemistry. Compound 56 was deprotected by hydrogenolysis over Pd(OH)2 in THF/H2O to provide target compound 57 after hydrophobic interaction chromatography (HIC). Unfortunately, no yield was reported for the final deprotection step. The trisaccharide lipid anchor and the tetrasaccharide ribitol were also deprotected to give fragments 59 and 58 for immunological evaluation. Preliminary immunological evaluation indicated that the compound, as well as the trisaccharide lipid anchor lacking the tetrasaccharide ribitol phosphate part, are capable of stimulating the production of IL-8 in a human whole blood assay and an assay using human mononuclear cells (MNCs). Using TLR2 and TLR4/MD2/CD14 transfected HEK 293 cells the intermediacy for these PRRs in immunestimulation was ruled out. Binding studies of the synthetic fragments with L-ficolin, and its fibrinogen recognition domain (FBG) indicated that compounds 57 and 58 could be recognized by this soluble lectin from the complement system.46 The interaction of the synthetic LTA and the ficolin receptor triggers the activation of the lectin complement pathway of the innate immune system. The binding of the FBG domain with the TA fragments was further investigated in competitive binding studies in which fragment 57 competed with monosaccharides (mannose, glucose, N-acetyl galactosamine) and phosphocholine for FBG binding. These studies indicated that the GalNAc and phosphocholine residues are important structural features for interaction of the S. pneumonia LTA with L-ficolin.
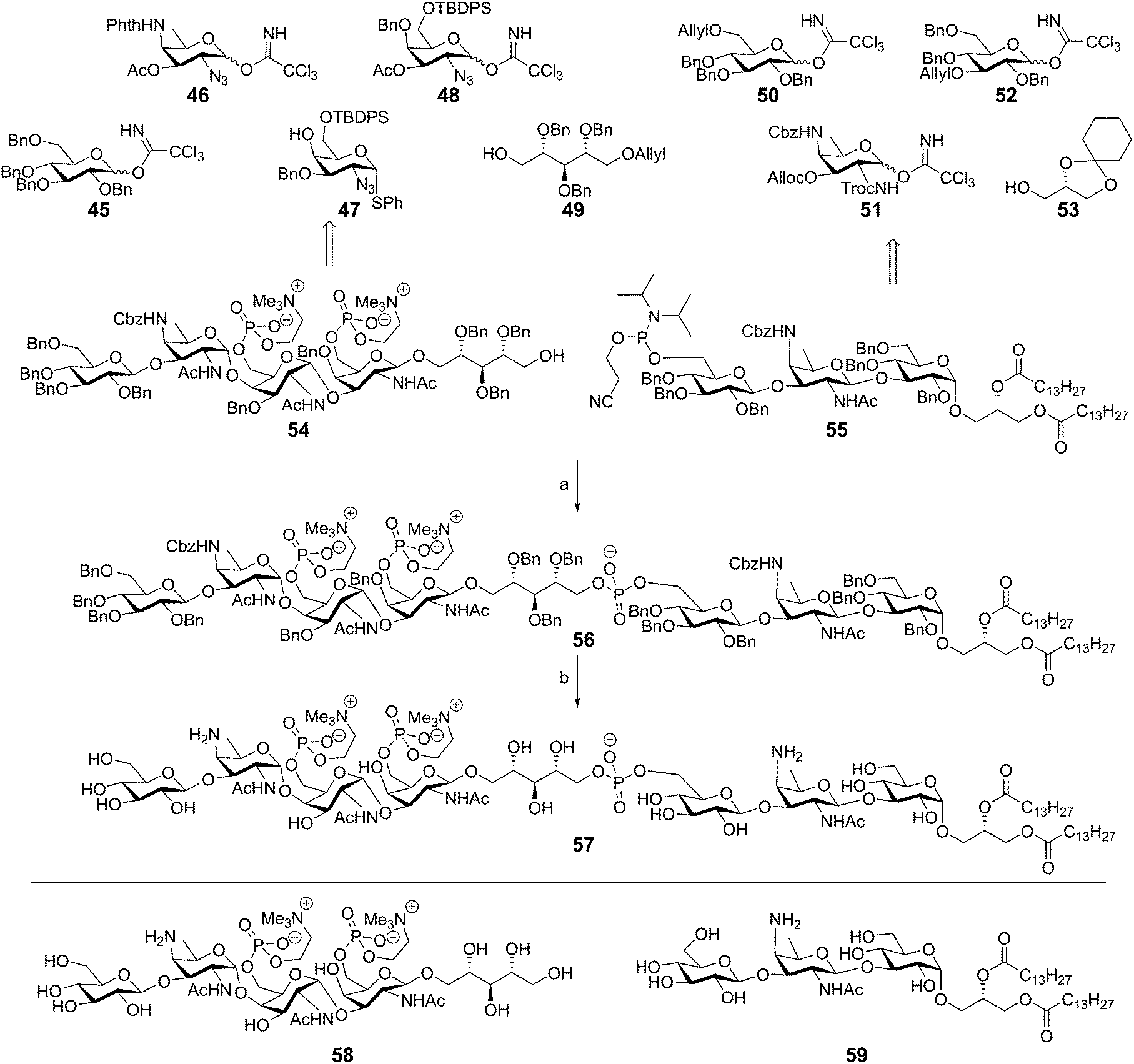 | ||
| Scheme 6 Pedersen's Streptococcus pneumoniae LTA synthesis. Reagents and conditions: (a) (i) tetrazole, MeCN (ii) tBuOOH, Me2NH, EtOH, 68%; (b) Pd(OH)2/C, H2, THF, H2O. | ||
Recently, two reports have described the assembly of Clostridium difficile TA fragments. This LTA has an “uncommon” structure as the diglucosylphosphate repeating units are not connected through the anomeric center of the reducing end glucosyl moiety, but through the C-6–OH of this residue. The anomeric center of this glucosyl moiety is decorated with a glycerate moiety. Deviating from the classical DNA chemistry, that employs the acid labile dimethoxytrityl group for temporary protection, the group of Seeberger reported an enol ether based assembly strategy as shown in Scheme 7.47 The key building block 68 was synthesized starting with two sequential glycosylations on glycerate 60 using trichloroimidates 63 and 61, giving diglycosyl-glycerate 66. The azides on this disaccharide were converted into acetamides and the levulinoyl group was removed to provide the primary alcohol. Subsequent allyl isomerization yielded key enol ether 67, which was transformed into phosphoramidite building block 68. The assembly sequence of dimer repeat 70 started with attachment of the spacer to alcohol 67. To this end phosphoramidite 62 was activated with 5-(ethylthio)tetrazole (ETT) and the intermediate phosphite was oxidized with I2 in water. These conditions also cleaved the enol ether to yield spacer equipped monomer 69 in 98% yield. Subsequent elongation using phosphoramidite 68 proceeded in 78% yield and furnished protected dimeric fragment 70. Deprotection of monomer 69 and dimer 70 furnished the C. difficile TA fragments that were analyzed for their ability to bind antibodies in serum, taken from C. difficile-infected patients using a microarray platform. Two out of three sera contained IgG-antibodies against dimer 71 indicating that these LTA fragments could be used as antigens in future vaccine modalities. To generate a conjugate vaccine, TA fragment 72 was coupled to CRM197, a commonly used (licensed) carrier protein, through an adipic acid linker.48 This vaccine modality (73) was combined with two different adjuvants, alum and complete Freund's adjuvant. It was shown that the stand-alone conjugate was highly immunogenic, indicating that the conjugate was endowed with intrinsic adjuvant activity. However, the conjugate was not able to stimulate bone marrow dendritic cells in vitro. The conjugate adsorbed onto alum provided the most robust IgG response of the three vaccine formulations and this construct was used in a mouse model. It was shown to offer protection against C. difficile as revealed by diminished C. difficile fecal levels in vaccinated mice with respect to no-treated or CRM-treated mice.
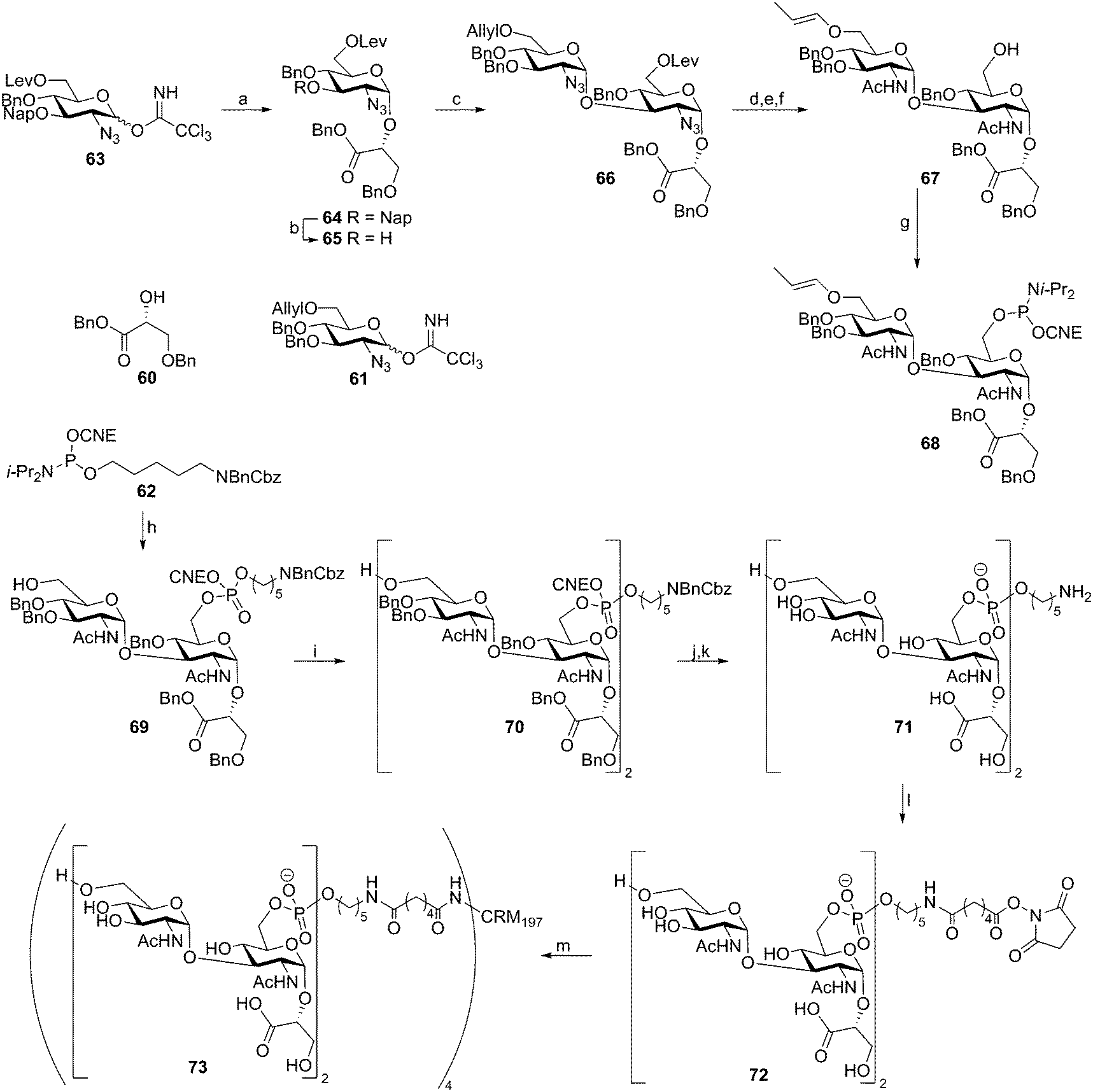 | ||
| Scheme 7 Seeberger's Clostridium difficile TA synthesis. Reagents and conditions: (a) 60, TMSOTf, DCM, Et2O, −20 °C to −10 °C, 81% (α:β = 9:1); (b) DDQ, DCM, phosphate buffer, pH = 7.2, 0 °C to rt, 80%; (c) 61, TMSOTf, DCM, Et2O, −20 °C to −10 °C, 69% (α:β = 8:1); (d) AcSH, pyridine, 67%; (e) Ir(COD)(Ph2MeP)2, THF; (f) N2H4 hydrate, AcOH, pyridine, DCM, 78% over 2 steps; (g) 2-cyanoethyl bis(N,N-diisopropylamino)phosphoramidite, tetrazole, diisopropylamine, DCM, MeCN, 82%; (h) (i) 67 5-(ethylthio)tetrazole, MeCN, (ii) I2, H2O, THF, 98%; (i) (i) 68, 5-(ethylthio)tetrazole, MeCN, (ii) I2, H2O, THF, 78%; (j) NEt3; (k) H2 (4 bar), Pd/C, H2O, AcOH; (l) di-N-succinimidyl adipate, NEt3, DMSO; (m) CRM197, 100 mM sodium phosphate, pH 7.4. | ||
Hogendorf et al. reported the synthesis of a set of C. difficile LTA fragments, differing in the number of repeating units (1–5) and containing the C. difficile trisaccharide glycolipid anchor (Scheme 8).49 This synthesis was based on the use of the ‘classic’ DMTr-temporary protecting group. The key DMTr-phophoramidite building block 80 was assembled using glucosazide synthons 74 and 75. First, benzylglycerate 60 was glycosylated in a stereoselective manner with donor 75 to give 76. Desilylation and ensuing elongation with building block 74 then gave diglycosyl-glycerate 78. The azides in this molecule were transformed into acetamide groups and the primary TBDPS group was changed into a DMTr-ether. Fmoc removal and phosphitylation gave central building block 80. Anchor 81 was coupled to this phosphoramidite, and subsequent oxidation and DMTr cleavage yielded lipid-decorated monomer 82 in 62% yield over 3 steps. Repetition of this sequence using phosphoramidite 80 yielded the di-, tri-, tetra- and pentamers (83, 84, 85 and 86 respectively). Deprotection followed by hydrophobic interaction chromatography gave the five C. difficile LTA fragments (87–91), which were tested on their ability to stimulate the production of pro-inflammatory cytokines by human mononuclear cells and in a whole blood assay. However, no significant immunostimulatory effect was observed.
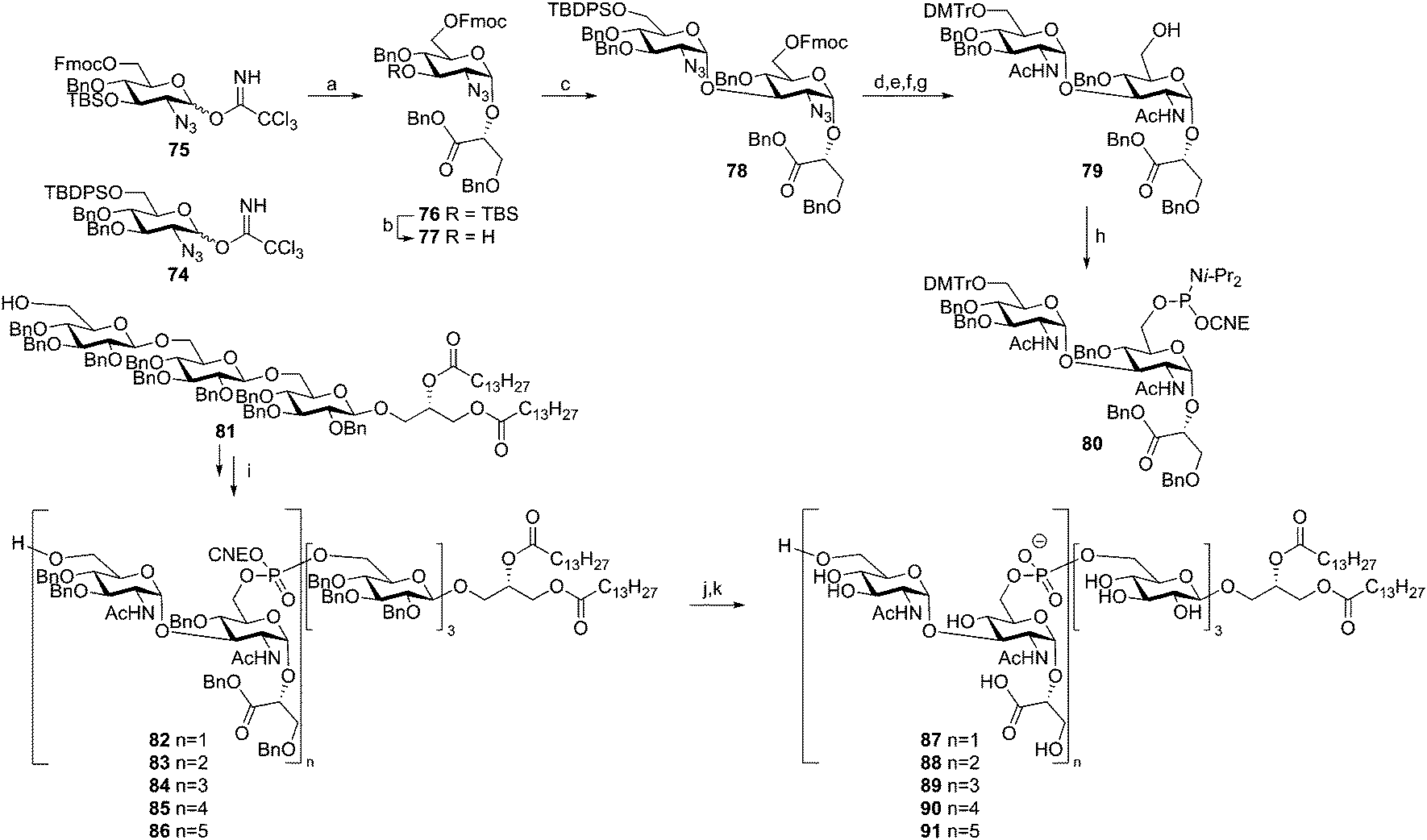 | ||
| Scheme 8 Hogendorf's Clostridium difficile LTA synthesis. Reagents and conditions: (a) 60, TMSOTf, Et2O, −35 °C to 0 °C, 88%; (b) HF·pyridine, THF, 98%; (c) 74, TMSOTf, Et2O, −35 °C to 0 °C, 60% (α:β = 15:1); (d) AcSH, pyridine, 68%; (e) HF·pyridine, THF, 95%; (f) DMTr-Cl, pyridine, 91%; (g) DBU, DCM, 94%; (h) 2-cyanoethyl bis(N,N-diisopropylamino)phosphoramidite, tetrazole, diisopropylamine, DCM, 84%; (i) (i) 80, DCI, MeCN, (ii) I2, H2O, pyridine, THF, (iii) DCA, TES-H, DCM, 82: 62%, 83: 74%, 84: 66%, 85: 56%, 86: 42%; (j) DBU, DCM, 76–97%; (k) Pd black, H2, THF, H2O, AcOH. | ||
Enterococcus faecalis and E. faecium are normally harmless commensal bacteria inhabiting the gastrointestinal tract, but they are also the causative agents of sepsis, endocarditis, wound infections and urinary tract infections.50 They are amongst the most prevalent nosocomial infectious agents and especially multi-drug resistant enterococci pose a great problem. The LTA of E. faecalis and feacium belongs to the class of type I LTAs. The poly(glycerol phosphate) chain of these bacteria can be decorated with either D-alanine residues or carbohydrate appendages. E. faecalis LTA can carry kojibiosyl groups51 and E. faecium has α-D-glucopyranosyl substituents.52 Our group developed several synthetic strategies to assemble a variety of these enterococcal LTA fragments. Initially solution phase chemistry was explored to generate a kojibiosyl functionalized glycerol phosphate hexamer as depicted in Scheme 9. The kojibiosyl synthon was synthesized using benzylidene protected glucose donors to allow for stereoselective construction of the α-glycosidic linkages. Using kojibiosyl glycerol phosphoramidite 100 and glycerol phosphoramidite 102, a hexamer was synthesized starting from dibenzylglycerol 101. Aminohexanol phosphoramidite 103 was coupled to the decorated hexamer to yield fully protected 104, which could be deprotected to yield spacer functionalized E. faecalis TA hexamer 105.
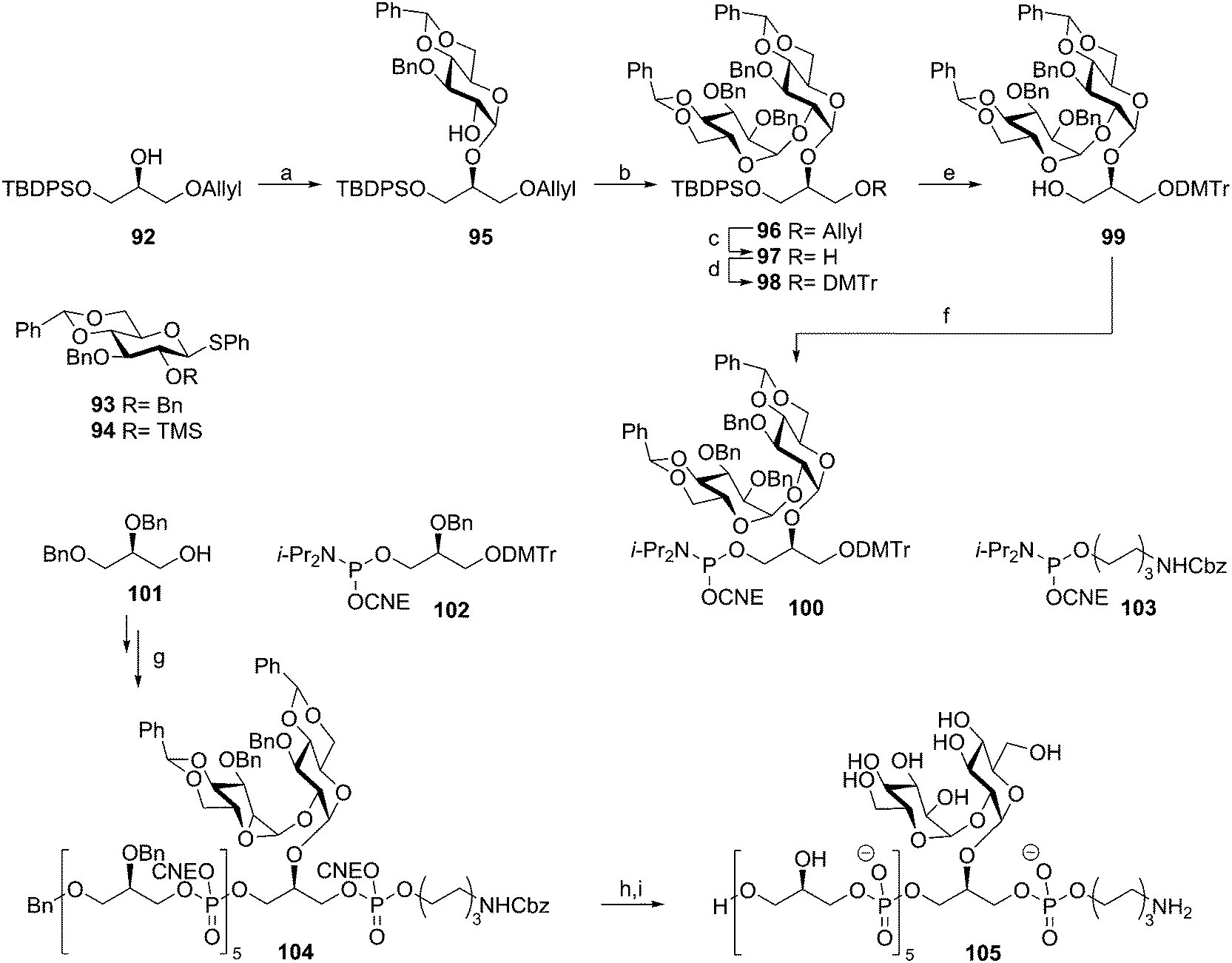 | ||
| Scheme 9 Hogendorf's Enterococcus faecalis TA synthesis. Reagents and conditions: (a) (i) 94, Tf2O, Ph2SO, TTBP, DCM, (α:β = 6:1), (ii) Na2CO3, MeOH, 46%; (b) 93, Tf2O, Ph2SO, TTBP, DCM, 82%; (c) (i) Ir(COD)(Ph2MeP)2, THF, (ii) I2, aq. NaHCO3, 67%; (d) DMTr-Cl, Et3N, DCM, 98%; (e) TBAF, THF, 91%; (f) 2-cyanoethyl N,N-diisopropylchlorophosphoramidite, DIPEA, DCM, 72%; (g) (i) 102, 100, or 103, DCI, MeCN, (ii) I2, THF, H2O, pyridine, (iii) DCA, TES-H, DCM or pyridinium p-toluenesulphonate, MeOH, DCM, 15% over 17 steps; (h) NH3, H2O, dioxane, 100%; (i) Pd black, H2, H2O, dioxane, 76%. | ||
To streamline the assembly of TA fragments, a light-fluorous phase synthesis strategy was developed. In this approach a C8F17 fluorous support was used that allowed for the rapid purification of the growing oligomers using fluorous solid phase extraction (F-SPE). Building on the fluorous-propylsulfonylethoxycarbonyl (FPse) group, introduced by Ali et al.,53 a set of LTA-oligomers was assembled54 (Scheme 10A). Using glycerol phosphoramidite 102 in twelve consecutive coupling-oxidation-DMTr cleavage reaction sequences, dodecamer 112 was effectively generated in 25% overall yield. The intermediate pentamer 111 could also be coupled to phosphoramidites 108, 109 or 110 and spacer phosphoramidite 103 to yield fluorous supported 113, 114, and 115 respectively. Subsequent deprotection yielded glucose, N-acetyl glucosamine and glucosamine decorated hexamers 116, 117 and 118. To further explore the scope of this method, the more complex TA from Spirilliplanes yamanashiensis was targeted (Scheme 10B). Building block 119 was synthesized and coupled to fluorous support 107 to end up, after six elongation cycles with large hexamer 120 (4.7 kDa), which could be effectively purified using the C8F17 tail and F-SPE. Final deprotection yielded TA-fragment 121.
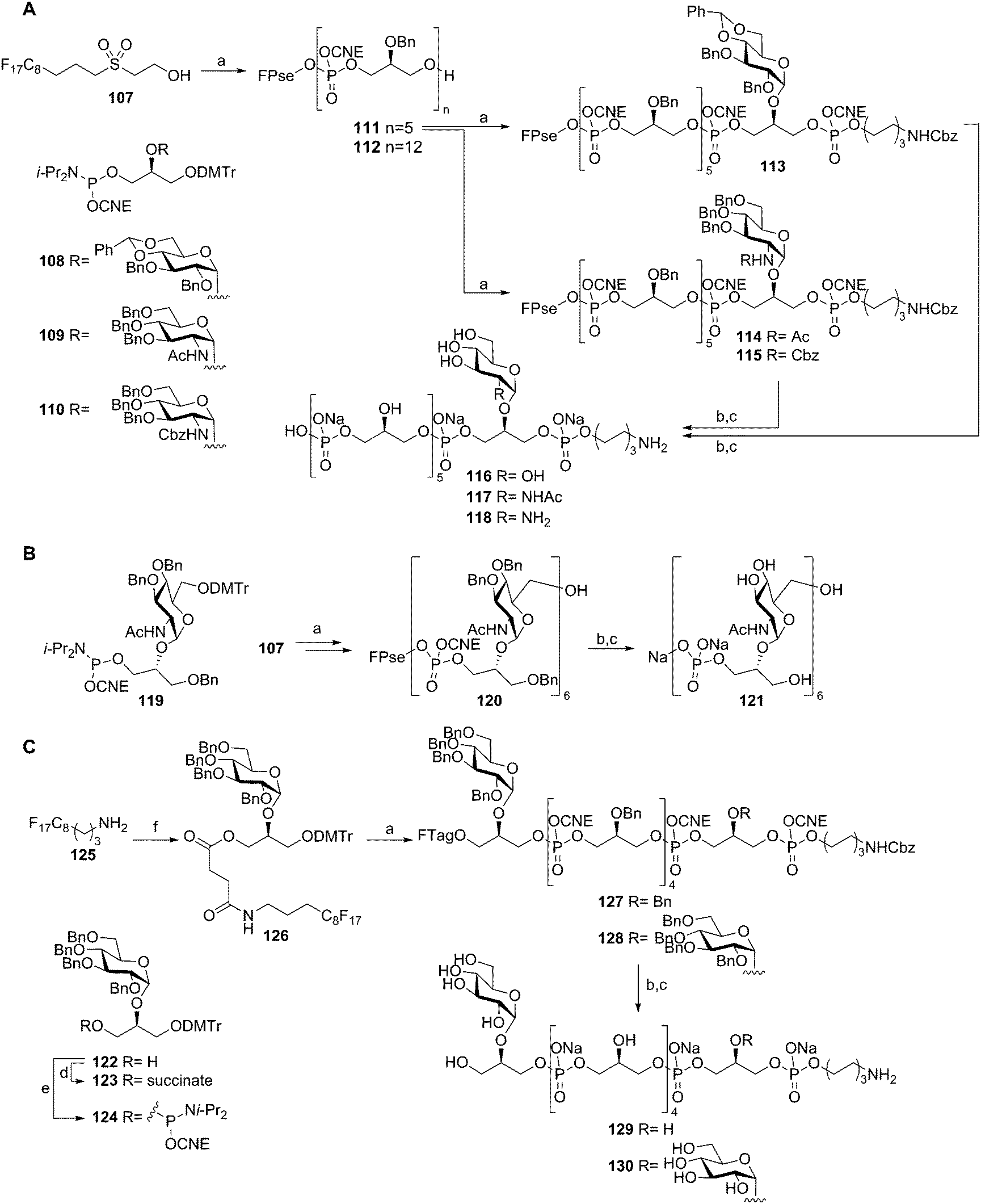 | ||
| Scheme 10 TA-synthesis using a Pse-based fluorous linker (A), Spirilliplanes yamanashiensis-TA synthesis (B), TA-synthesis using a succinate based fluorous linker (C). Reagents and conditions: (a) (i) phosphoramidite, DCI, MeCN, (ii) I2, H2O, pyridine, THF, (iii) DCA, TES-H, DCM, towards 113: pyridinium p-toluene sulphonate, MeOH, (iv) F-SPE, 71–99% over 4 steps; (b) conc. NH4OH, 40 °C; (c) Pd black, H2, H2O, AcOH, 76–98% over 2 steps; (d) succinic anhydride, Et3N, DCM, 96%; (e) 2-cyanoethyl N,N-diisopropylchlorophosphoramidite, Et3N, DCM, 74%; (f) (i) 123, BOP, DIPEA, DMF, DCM, (ii) DCA, TES-H, DCM, (iii) F-SPE, 90% over 3 steps. | ||
The fluorous-propylsulfonylethoxycarbonyl (FPse) linker system delivers TA fragments with a terminal phosphate monoester. Because immunological evaluation of these LTA fragments showed that the terminal phosphate group was detrimental for the recognition of the fragments by anti-LTA antibodies, another fluorous linker system was developed that delivers TA fragments with a terminal alcohol group55 (Scheme 10C). To this end fluorous amine 125 was condensed with succinate equipped building block 123 giving fluorous synthon 126. Applying the above described F-SPE-strategy, using perbenzylated building block 124, hexamers 127 and 128 were synthesized. Deprotection using concentrated ammonia cleaved the fluorous succinate ester, releasing the hexamers with a terminal alcohol. Hydrogenolysis then furnished hexamers 129 and 130.
Almost 25 years after the first automated solid phase TA synthesis, this methodology was revisited for the generation of a small set of TA fragments.56 Employing classical automated DNA synthesis, the aforementioned building blocks 102, 124 and 103 were used to generate the set of TAs depicted in Scheme 11. To this end, aminopropyl functionalized controlled pore glass (CPG) was loaded with succinate functionalized glycerol building block 132 or 123. The DMTr protecting group was cleaved using 3% DCA in toluene and the released alcohols were coupled with phosphoramidite 102 using 5-BTT as activator to yield the phosphite intermediates, which were oxidized to the corresponding phosphotriesters using I2 in pyridine. The remaining unreacted alcohols were capped using acetic anhydride and 2,6-lutidine. After repetitive coupling-oxidation-capping-deprotection cycles spacer phosphoramidite 103 was coupled. The products were cleaved from the resin under basic conditions and purified by HPLC. Finally, hydrogenolysis yielded a series of glycerol phosphate oligomers equipped with an aminohexanol spacer, varying in length from 6 to 20 glycerol phosphate repeating units and incorporating glycoside residues at different positions on the backbone. The set of TAs was evaluated for their ability to bind to opsonophagocytic antibodies, raised against E. faecalis LTA, in an opsonophagocytic killing inhibition assay (OPIA). It was observed that longer fragments showed increased inhibition of the antibodies and especially the glycosylated fragments 141 and 129 were found to be potent inhibitors. Of note, the kojibiosyl functionalized TA fragment 105 proved to be a relatively weak inhibitor for opsonophagocytic killing. Along with the most potent inhibitor, hexamer 129, 105 was used to generate a conjugate vaccine by coupling to BSA as a carrier protein57 (Scheme 12). To this end TA-fragments 105 and 129 were equipped with a maleimide handle and linked to thiol-functionalized BSA to deliver conjugates with approximately 20 TA fragments per BSA carrier. Immunization of rabbits with the BSA conjugate of 129 provided serum containing opsonophagocytic antibodies able to mediate killing of clinical E. faecalis strains. Notably, also E. faecium and S. aureus strains were killed by the serum. In a passive immunization experiment the serum was shown to offer protection in a Rat endocarditis model. The fragments have not yet been evaluated for their innate immune system stimulating activity.
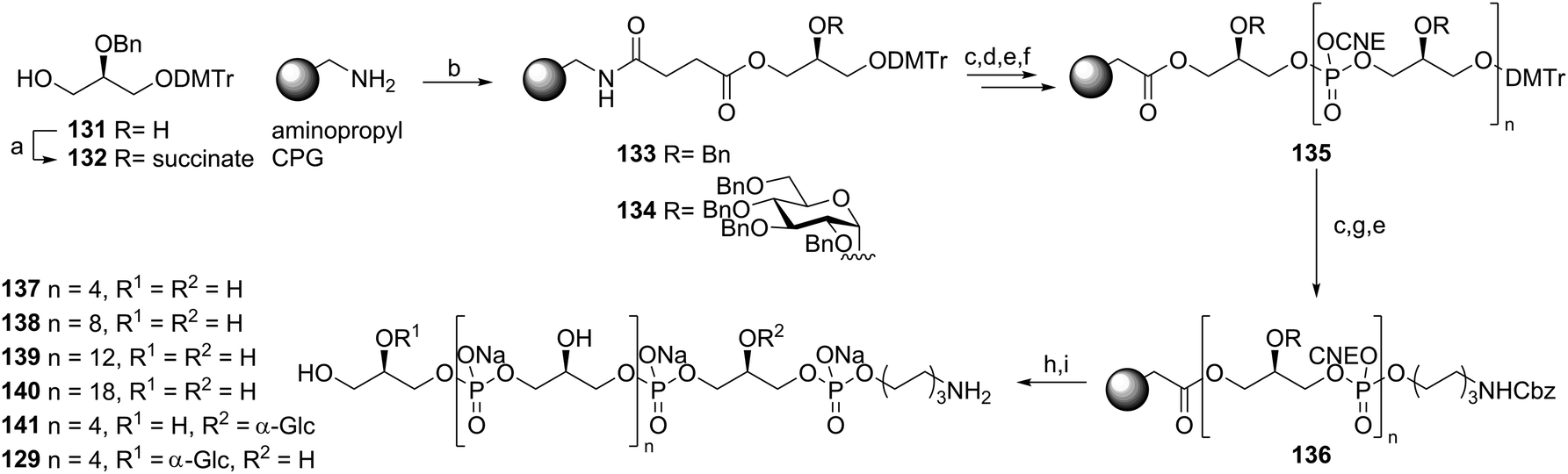 | ||
| Scheme 11 Automated solid phase TA synthesis. Reagents and conditions: (a) succinic anhydride, Et3N, DCM, 99%; (b) 132 or 123, DIC, MeCN; (c) 3% DCA, toluene; (d) 102 or 124, 5-BTT, MeCN; (e) I2, pyridine, H2O, MeCN; (f) Ac2O, N-methylimidazole, 2,6-lutidine, MeCN; (g) 103, 5-BTT, MeCN; (h) conc. NH4OH; (i) Pd black, H2, H2O, dioxane, AcOH. | ||
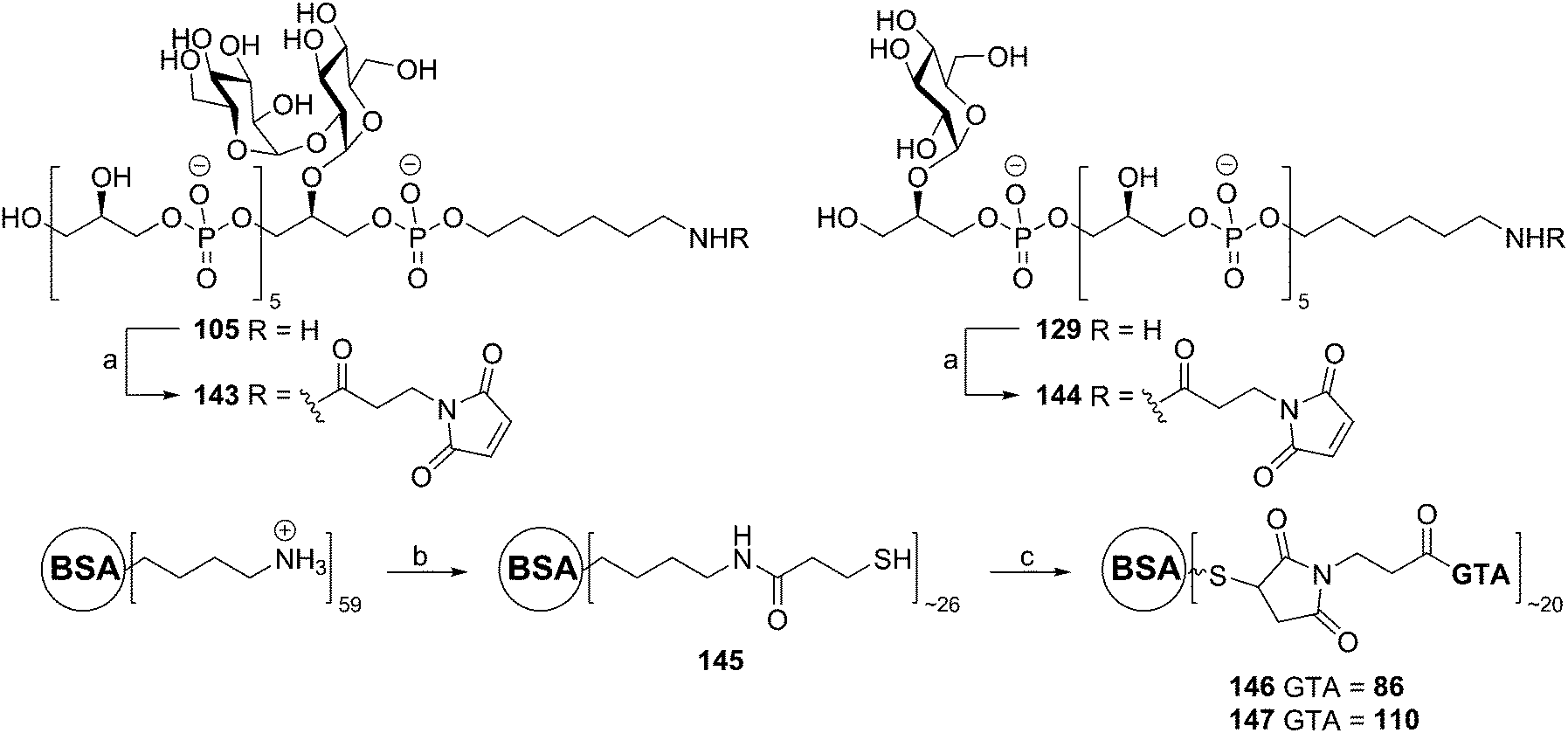 | ||
| Scheme 12 Generation of a glycerol phosphate conjugate vaccine. Reagents and conditions: (a) N-succinimidyl-3-maleimidopropionate, aq. NaHCO3, 143: 86%, 144: 67%; (b) (i) N-succinimidyl-3-thio-propionatehomodisulfide, PBS (pH = 8.0), DMF, (ii) dithiothreitol; (c) 143 or 144, PBS (pH = 7.2). | ||
Snapper and co-workers also reported an approach to generate an anti-staphylococcal vaccine based on synthetic poly(glycerol phosphate).58 They applied a synthetic strategy that involves benzoyl protected glycerol phosphoramidite 153 (Scheme 13). This building block was obtained by installing a levulinoyl protecting group on solketal, removing the isopropylidene group and protecting the primary alcohol with a DMTr ether. Installation of a benzoyl group and removal of the temporary Lev group with hydrazine then yielded alcohol 152, which was equipped with a phosphoramidite functionality. This synthon was used in an automated solid phase synthesis to generate a decamer glycerol phosphate. Deprotection of the oligomer and simultaneous cleavage from the solid support was achieved under basic conditions. The product was purified by dialysis and conjugated to tetanus toxoid using the 4-formylbenzaldehyde/6-hydrazinonicotinamide conjugation method. Little experimental details and no analytical data were provided to assess the quality of the generated material. Given the propensity of acyl groups to migrate from the secondary alcohol to the primary one in glycerol substrates and the minimal purification procedure, the identity of decamer 154 may be questionable. The immunological evaluation of the conjugate revealed that the vaccine cannot trigger an innate immune response, as exposure of peritoneal cells to the conjugate did not lead to significant IL-6 production. To provide an effective vaccine, the conjugate was adsorbed onto alum and combined with the CpG-ODN adjuvant. This vaccine formulation was capable of eliciting an immune response, as judge from a significant boost in anti-poly(glycerol phosphate) IgG levels. The serum raised with the vaccine was further characterized in an opsonophagocytic killing assay and shown to be capable of mediating S. aureus killing in vitro. Finally, the vaccine was evaluated in a mouse bacteremia model, where it was shown to offer protection against infection by S. aureus.
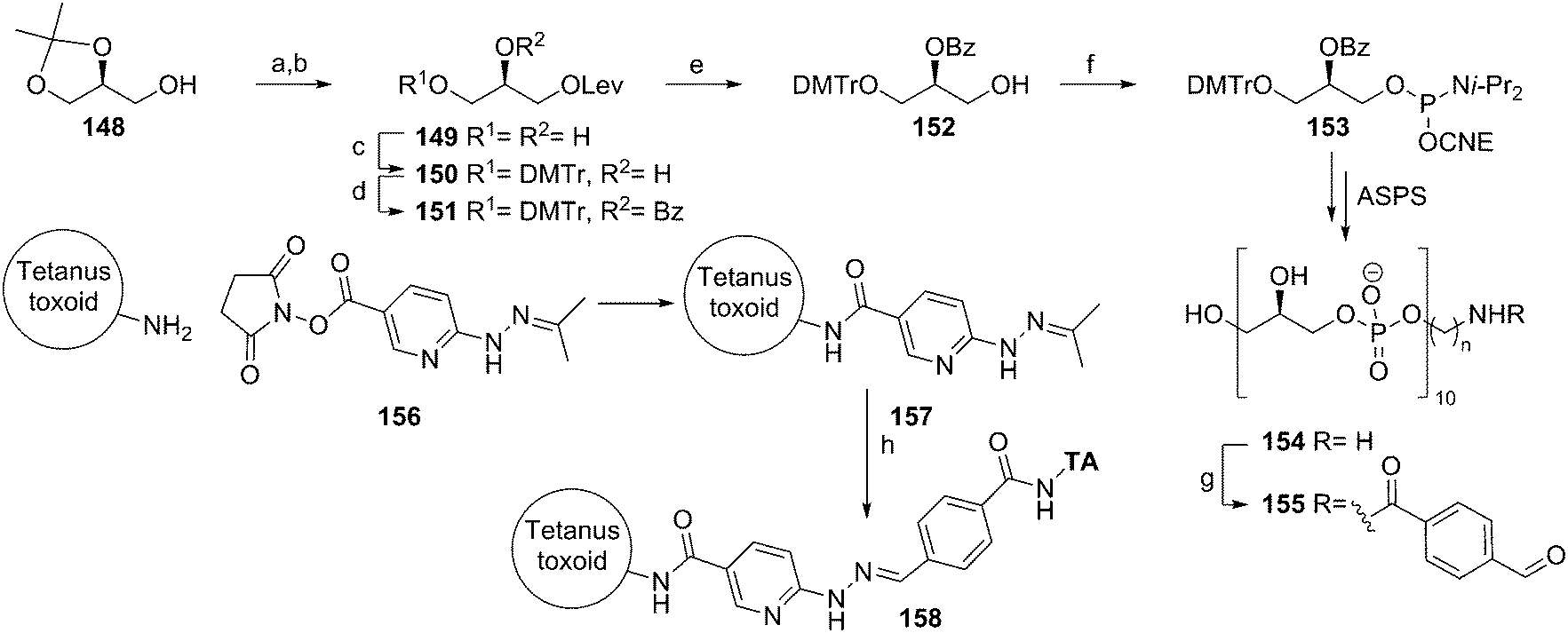 | ||
| Scheme 13 Snapper's anti-staphylococcal vaccine synthesis. Reagents and conditions: (a) levulinic acid; (b) 75% AcOH, H2O; (c) DMTr-Cl; (d) benzoylchloride; (e) N2H4·H2O, AcOH, pyridine; (f) 2-cyanoethyl N,N-diisopropylchlorophosphoramidite; (g) 4FB-OSu; (h) PBS, aniline. | ||
In comparison to the amount of LTA syntheses reported to date, relatively little effort has been devoted to the synthesis of WTA fragments. Pozsgay and co-workers described the assembly of oligo(ribitol phosphates) to allow for their immunological evaluation59 (Scheme 14). Using 2,3,4-benzyl protected, DMTr-equipped ribitol-1-phosphoramidite 159, the automated solid phase synthesis of these oligomers was attempted. They reported however that an “intractable” mixture was obtained upon cleavage of the products from the resin. Therefore this method was excluded from further investigations and a solution phase approach was undertaken. In these syntheses, mild aqueous acetic acid mediated cleavage conditions were applied to remove the DMTr group, as it was thought that the acidic conditions could jeopardize the phosphotriester bonds. Building on CBz-protected aminoethanol spacer 160, up to 12 ribitol phosphate repeating units were installed. The 8-mer and 12-mer were deprotected and conjugated to BSA, yielding the two oligo(ribitol-1-phosphate) conjugates. So far no immunological evaluation of these compounds has been reported.
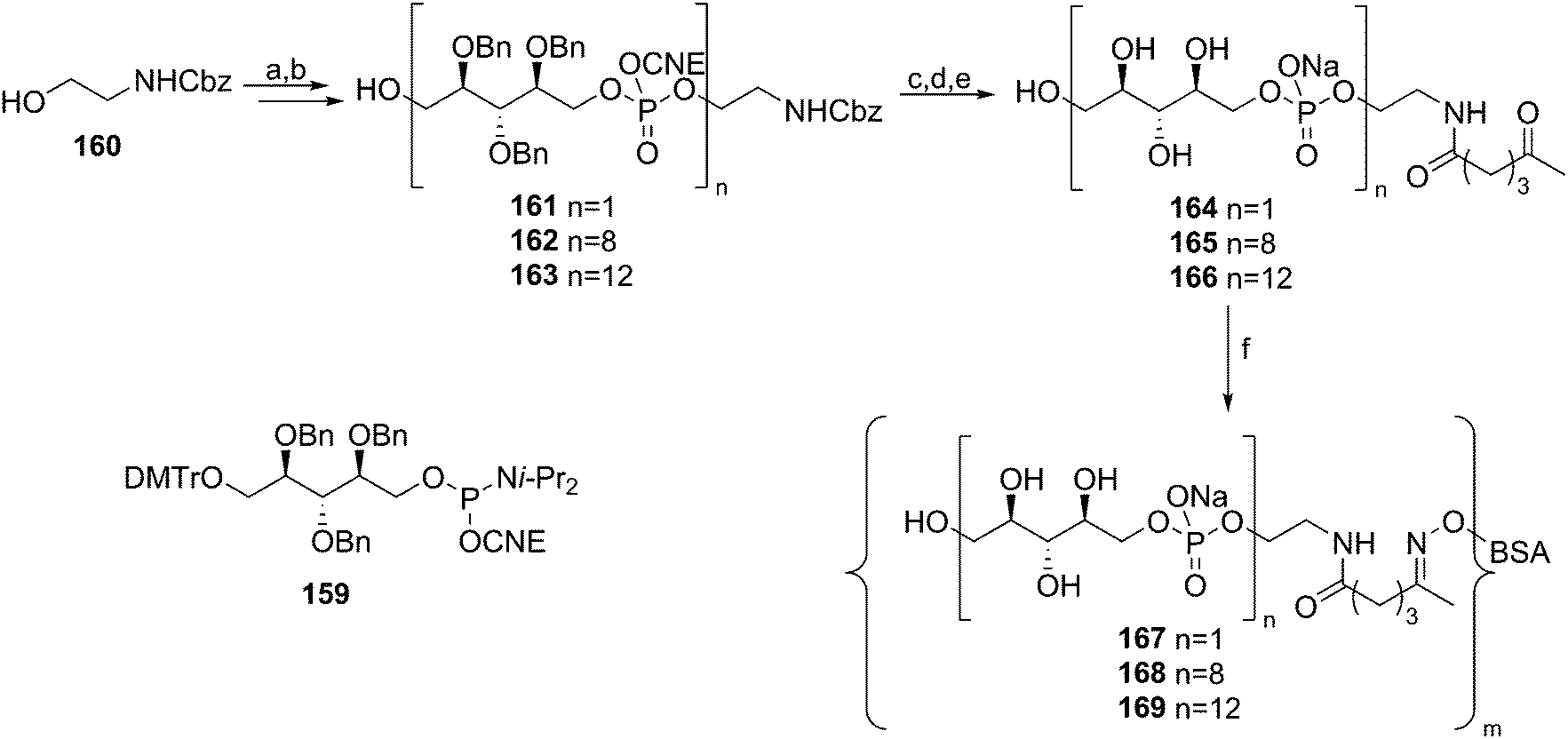 | ||
| Scheme 14 Pozsgay's ribitolphosphate–BSA conjugate synthesis. Reagents and conditions: (a) (i) 159, tetrazole, MeCN, (ii) I2, pyridine, THF, H2O; (b) AcOH, H2O, DCM; (c) conc. NH4OH, MeOH, 50 °C; (d) Pd/C, H2, t-BuOH, H2O; (e) 5-ketohexanoic anhydride, Et3N, MeOH, H2O; (f) aminooxy-BSA, PBS (pH = 7.4), EDTA, glycerol. | ||
Recently we described the synthesis of various WTA structures of E. faecium.60 The E. faecium TA is built up from -6-(GalNAc-α(1–3)-GalNAc-β(1–2)-GroP)- repeating units and because the stereochemistry of the glycerol moiety had not been established, fragments with both sn-1 and sn-3 glycerol phosphates were assembled. Key digalactosyl glycerol phosphoramidites 181 and 182, which were used for the assembly of mono- di- and trimers 183–188 were assembled as depicted in Scheme 15. For the construction of both the β-galactosamine and α-galactosamine linkage it was decided to use galactosazide synthons, allowing for a more straightforward protecting group strategy. Building blocks 171 and 172 were synthesized in several steps from protected galactal 170 and they were subsequently reacted in a stereoselective fashion, using triflic acid in DCM, providing digalactosazide 173 as a single diastereoisomer. After hydrolysis and introduction of a trifluoroimidate leaving group, the acetonitrile-mediated glycosylation with either stereoisomer of the semi-protected glycerol acceptor (175 and 176) yielded the targeted glycerol disaccharides 177 and 178 in high yield and excellent stereochemistry. Several protecting group manipulations were executed to yield alcohols 179 and 180, which could be phosphitylated to provide phosphoramidites 181 and 182. Assembly of the oligomeric fragments commenced with the condensation, oxidation and detritylation of spacer phosphoramidite 103 and either 179 or 180, yielding spacer equipped monomers 183 and 184. Repetition of the aforementioned coupling cycle on 183 and 184 using phosphoramidites 181 and 182 provided di- and trimers 185–188. Deprotection of all synthesized fragments using palladium catalyzed hydrogenolysis gave the deprotected fragments (189–194), the NMR spectra of which were compared with the NMR spectrum obtained using a sample of isolated WTA. This provided evidence for the absolute stereochemistry of the E. faecium WTA glycerol phosphate indicating it to be sn-glycerol-3-phosphate. This conclusion is corroborated by the fact that the biosynthesis of WTAs generally uses CDP-sn-3 glycerol synthons (vide supra). The immunological evaluation of the fragments is currently ongoing.
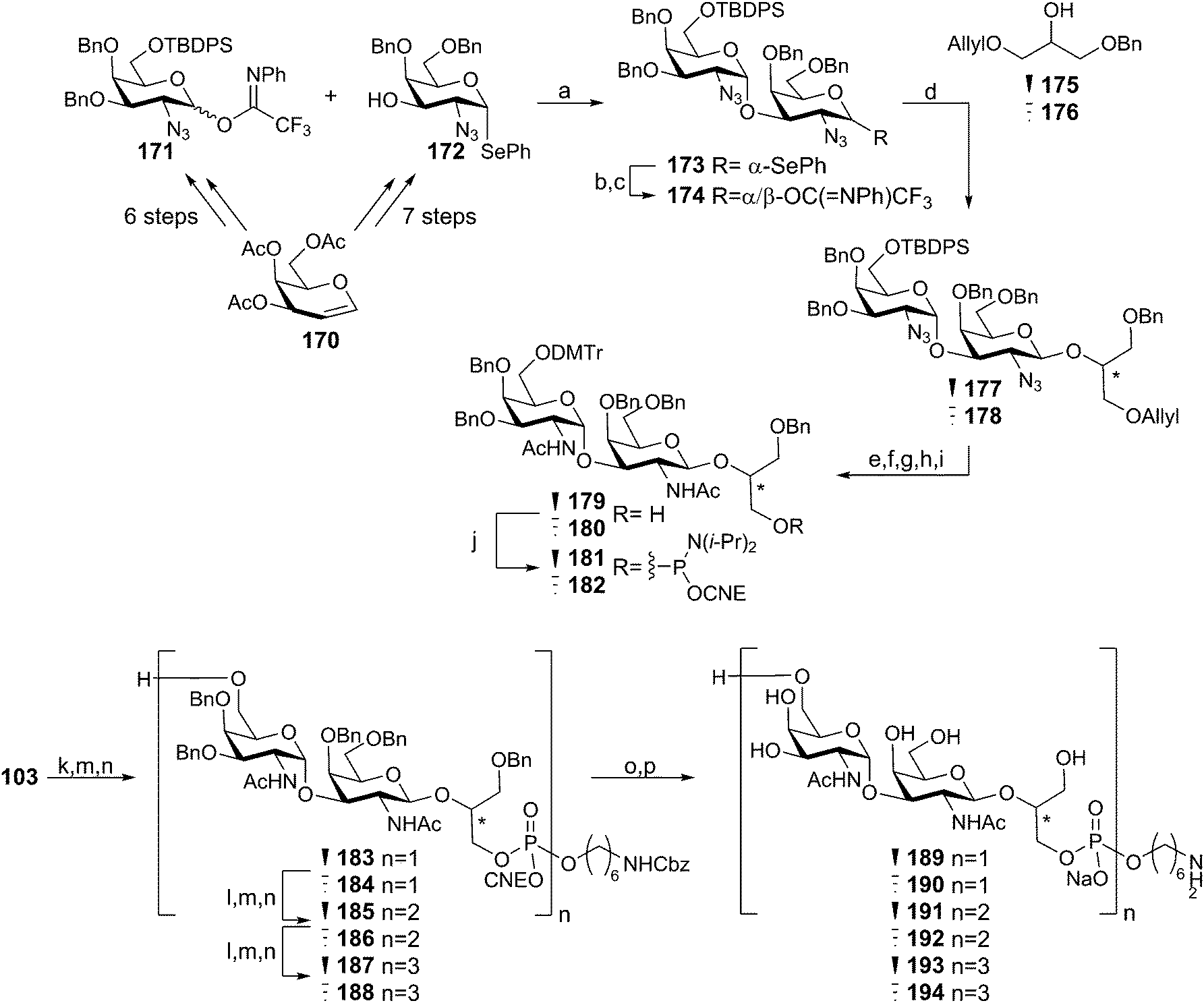 | ||
Scheme 15
Enterococcus faecium WTA synthesis. Reagents and conditions: (a) TfOH, DCM, 0 °C, 58%; (b) NIS, THF/H2O; (c) CF3(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NPh)Cl, K2CO3, acetone, quant over 2 steps; (d) 175 or 176, TfOH, MeCN/EtCN/DCM, −40 °C, 177: 90% 178: 80%; (e) PMe3, dioxane/H2O; (f) Ac2O, NEt3, DCM; (g) TBAF, THF; (h) DMTrCl, NEt3, DCM; (i) (1) Ir(COD)(Ph2MeP)2, THF; (2) NaHCO3, I2, H2O/THF, 179: 28% over 5 steps, 180: 46% over 5 steps; (j) di-isopropylethylamine, N,N-di-isopropylamino-2-cyanoethyl-chlorophosphite, DCM, 181: 85% 182: 62%; (k) DCI, 179 or 180, MeCN; (l) DCI, 181 or 182, MeCN; (m) CSO, MeCN; (n) 3% TCA, DCM, 183: 72%, 184: 80%, 185: 62%, 186: 64%, 187: 69%, 188: 67%; (o) NH3(conc), dioxane; (p) Pd(0), H2, AcOH, H2O, 189: 73%, 190: 57%, 191: 44%, 192: 45%, 193: 49%, 194: 56%. NPh)Cl, K2CO3, acetone, quant over 2 steps; (d) 175 or 176, TfOH, MeCN/EtCN/DCM, −40 °C, 177: 90% 178: 80%; (e) PMe3, dioxane/H2O; (f) Ac2O, NEt3, DCM; (g) TBAF, THF; (h) DMTrCl, NEt3, DCM; (i) (1) Ir(COD)(Ph2MeP)2, THF; (2) NaHCO3, I2, H2O/THF, 179: 28% over 5 steps, 180: 46% over 5 steps; (j) di-isopropylethylamine, N,N-di-isopropylamino-2-cyanoethyl-chlorophosphite, DCM, 181: 85% 182: 62%; (k) DCI, 179 or 180, MeCN; (l) DCI, 181 or 182, MeCN; (m) CSO, MeCN; (n) 3% TCA, DCM, 183: 72%, 184: 80%, 185: 62%, 186: 64%, 187: 69%, 188: 67%; (o) NH3(conc), dioxane; (p) Pd(0), H2, AcOH, H2O, 189: 73%, 190: 57%, 191: 44%, 192: 45%, 193: 49%, 194: 56%. | ||
Conclusion
Because of their biological relevance, the fact that they often cannot be isolated in sufficient purity from natural sources and their challenging structures, teichoic acids have been attractive targets for organic synthesis for over 35 years now. Developments in synthetic carbohydrate chemistry and nucleotide chemistry have led to increasingly sophisticated TA syntheses. The advent of phosphoramidite chemistry, a major step forward in the construction of phosphotriester linkages, has enabled the assembly of highly complex TAs as it allows the union of large and complex TA fragments in a very reliable and efficient manner. Further progress in synthetic chemistry, including the development of reliable and stereoselective glycosylation chemistry, and automated solid-phase methods will allow more efficient TA syntheses and the generation of TA libraries.Synthetic TAs have been instrumental in unraveling their mode of action in immunology. By the hand of synthetic Staphylococcus aureus LTA molecules the active principle of this biopolymer has been elucidated and the involvement of TLR2 in the activation of the immune system by this TA confirmed. For other TAs however the involvement of this PRR in immune activation remains to be established. Given the wide structural variety of TAs is, different biological binding partners are likely involved. Carbohydrate binding lectins of the complement system have already been implied to be key interaction partners and the generation of libraries of TAs will allow for the establishment of detailed structure–activity relationships in the near future. Synthetic TAs of different bacteria have been probed for their use as synthetic antigens in novel vaccine formulations. Initial results with model vaccines against E. faecalis, S. aureus and C. difficile are promising and it is not unlikely that TAs of other Gram-positives will be used for similar purposes. These synthetic antigens will also prove to be valuable tools in unraveling the molecular details behind their mode of action, in terms of antigen uptake, processing and presentation.
The availability of well-defined and functionalized TA fragments will also open up avenues to explore the biosynthesis of these important cell wall components providing leads for the development of novel antibiotic regimes that interfere with the assembly of these vital bacterial biopolymers.
Acknowledgements
The Netherlands Organisation for Scientific Research (NWO) is acknowledged for financial support.References
- F. C. Neuhaus and J. Baddiley, Microbiol. Mol. Biol. Rev., 2003, 67, 686–723 CrossRef CAS PubMed.
- C. Weidenmaier and A. Peschel, Nat. Rev. Microbiol., 2008, 6, 276–287 CrossRef CAS PubMed.
- S. Brown, J. P. Santa Maria and S. Walker, Annu. Rev. Microbiol., 2013, 67, 313–336 CrossRef CAS PubMed.
- M. G. Percy and A. Gründling, Annu. Rev. Microbiol., 2014, 68, 81–100 CrossRef CAS PubMed.
- F. Fabretti, C. Theilacker, L. Baldassarri, Z. Kaczynski, A. Kropec, O. Holst and J. Huebner, Infect. Immun., 2006, 74, 4164–4171 CrossRef CAS PubMed.
- M. P. Chapot-Chartier, Front. Microbiol., 2014, 5, 1–10 Search PubMed.
- S. Gautam, T. Kim, E. Lester, D. Deep and D. A. Spiegel, ACS Chem. Biol., 2016, 11, 25–30 CrossRef CAS PubMed.
- J. P. Santa Maria, A. Sadaka, S. H. Moussa, S. Brown, Y. J. Zhang, E. J. Rubin, M. S. Gilmore and S. Walker, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12510–12515 CrossRef CAS PubMed.
- S. Morath, A. Geyer, I. Spreitzer, C. Hermann and T. Hartung, Infect. Immun., 2002, 70, 938–944 CrossRef CAS PubMed.
- M. Hashimoto, K. Tawaratsumida, H. Kariya, A. Kiyohara, Y. Suda, F. Krikae, T. Kirikae and F. Gotz, J. Immunol., 2006, 177, 3162–3169 CrossRef CAS.
- M. Hashimoto, K. Tawaratsumida, H. Kariya, K. Aoyama, T. Tamura and Y. Suda, Int. Immunol., 2006, 18, 355–362 CrossRef CAS PubMed.
- S. Morath, S. von Aulock and T. Hartung, J. Endotoxin Res., 2005, 11, 348–356 CrossRef CAS PubMed.
- W. Fischer, Advances in Microbial Physiology, 1988, vol. 29, pp. 233–302 Search PubMed.
- W. Fischer, Glycolipids, Phosphoglycolipids, and Sulfoglycolipids, Springer, US, Boston, MA, 1990, pp. 123–234 Search PubMed.
- W. Fischer, New Comprehensive Biochemistry, 1994, vol. 27, pp. 199–215 Search PubMed.
- H. U. Koch and W. Fischer, Biochemistry, 1978, 17, 5275–5281 CrossRef CAS PubMed.
- C. W. Reid, E. Vinogradov, J. Li, H. C. Jarrell, S. M. Logan and J. R. Brisson, Carbohydr. Res., 2012, 354, 65–73 CrossRef CAS PubMed.
- I. B. Naumova, A. S. Shashkov, E. M. Tul'skaya, G. M. Streshinskaya, Y. I. Kozlova, N. V. Potekhina, L. I. Evtushenko and E. Stackebrandt, FEMS Microbiol. Rev., 2001, 25, 269–284 CrossRef CAS PubMed.
- O. Schneewind and D. Missiakas, J. Bacteriol., 2014, 196, 1133–1142 CrossRef PubMed.
- N. T. Reichmann and A. Gründling, FEMS Microbiol. Lett., 2011, 319, 97–105 CrossRef CAS PubMed.
- J. G. Swoboda, J. Campbell, T. C. Meredith and S. Walker, ChemBioChem, 2010, 11, 35–45 CrossRef CAS PubMed.
- M. C. Ganfield and R. A. Pieringer, J. Biol. Chem., 1980, 255, 5164–5169 CAS.
- A. Grundling and O. Schneewind, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8478–8483.
- D. Denapaite, R. Brückner, R. Hakenbeck and W. Vollmer, Microb. Drug Resist., 2012, 18, 344–358 CrossRef CAS PubMed.
- V. Winstel, G. Xia and A. Peschel, Int. J. Med. Microbiol., 2014, 304, 215–221 CrossRef CAS PubMed.
- C. M. Pedersen and R. R. Schmidt, Microbial Glycobiology, Elsevier, 1st edn, 2010, pp. 455–476 Search PubMed.
- C. A. A. van Boeckel, G. M. Visser, J. P. G. Hermans and J. H. van Boom, Tetrahedron Lett., 1981, 22, 4743–4746 CrossRef CAS.
- C. A. A. van Boeckel, G. M. Visser, J. P. G. Hermans and J. H. van Boom, Recl. Trav. Chim. Pays-Bas, 1983, 102, 526–537 CrossRef CAS.
- J. J. Oltvoort, M. Kloosterman, C. A. A. van Boeckel and J. H. van Boom, Carbohydr. Res., 1984, 130, 147–163 CrossRef CAS.
- S. L. Beaucage and M. H. Caruthers, Tetrahedron Lett., 1981, 22, 1859–1862 CrossRef CAS.
- P. Westerduin, G. H. Veeneman, Y. Pennings, G. A. van der Marel and J. H. van Boom, Tetrahedron Lett., 1987, 28, 1557–1560 CrossRef CAS.
- K. Fukase, T. Matsumoto, N. Ito, T. Yoshimura, S. Kotani and S. Kusumoto, Bull. Chem. Soc. Jpn., 1992, 65, 2643–2654 CrossRef CAS.
- K. Fukase, T. Yoshimura, S. Kotani and S. Kusumoto, Bull. Chem. Soc. Jpn., 1994, 67, 473–482 CrossRef CAS.
- H. Takada, Y. Kawabata, R. Arakaki, S. Kusumoto, K. Fukase, Y. Suda, T. Yoshimura, S. Kokeguchi, K. Kato and T. Komuro, Infect. Immun., 1995, 63, 57–65 CAS.
- A. Stadelmaier, S. Morath, T. Hartung and R. R. Schmidt, Angew. Chem., Int. Ed., 2003, 42, 916–920 CrossRef CAS PubMed.
- R. R. Schmidt, C. M. Pedersen, Y. Qiao and U. Zähringer, Org. Biomol. Chem., 2011, 9, 2040 CAS.
- S. Deininger, A. Stadelmaier, S. von Aulock, S. Morath, R. R. Schmidt and T. Hartung, J. Immunol., 2003, 170, 4134–4138 CrossRef CAS.
- S. Morath, A. Stadelmaier, A. Geyer, R. R. Schmidt and T. Hartung, J. Exp. Med., 2002, 195, 1635–1640 CrossRef CAS PubMed.
- I. Figueroa-Perez, A. Stadelmaier, S. Morath, T. Hartung and R. R. Schmidt, Tetrahedron: Asymmetry, 2005, 16, 493–506 CrossRef CAS.
- I. Figueroa-Perez, A. Stadelmaier, S. Deininger, S. von Aulock, T. Hartung and R. R. Schmidt, Carbohydr. Res., 2006, 341, 2901–2911 CrossRef CAS PubMed.
- S. Deininger, I. Figueroa-Perez, S. Sigel, A. Stadelmaier, R. R. Schmidt, T. Hartung and S. Von Aulock, Clin. Vaccine Immunol., 2007, 14, 1629–1633 CrossRef CAS PubMed.
- A. Stadelmaier, I. Figueroa-Perez, S. Deininger, S. von Aulock, T. Hartung and R. R. Schmidt, Bioorg. Med. Chem., 2006, 14, 6239–6254 CrossRef CAS PubMed.
- P. Roethlisberger, N. Iida-Tanaka, K. Hollemeyer, E. Heinzle, I. Ishizuka and W. Fischer, Eur. J. Biochem., 2000, 267, 5520–5530 CrossRef CAS PubMed.
- Y. Qiao, B. Lindner, U. Zähringer, P. Truog and R. R. Schmidt, Bioorg. Med. Chem., 2010, 18, 3696–3702 CrossRef CAS PubMed.
- C. M. Pedersen, I. Figueroa-Perez, B. Lindner, A. J. Ulmer, U. Zähringer and R. R. Schmidt, Angew. Chem., Int. Ed., 2010, 49, 2585–2590 CrossRef CAS PubMed.
- C. M. Pedersen, I. Figueroa-Perez, J. Boruwa, B. Lindner, A. J. Ulmer, U. Zähringer and R. R. Schmidt, Chem. – Eur. J., 2010, 16, 12627–12641 CrossRef CAS PubMed.
- C. E. Martin, F. Broecker, S. Eller, M. A. Oberli, C. Anish, C. L. Pereira and P. H. Seeberger, Chem. Commun., 2013, 49, 7159 RSC.
- F. Broecker, C. E. Martin, E. Wegner, J. Mattner, J. Y. Baek, C. L. Pereira, C. Anish and P. H. Seeberger, Cell Chem. Biol., 2016, 23, 1014–1022 CrossRef CAS PubMed.
- W. F. J. Hogendorf, N. Gisch, D. Schwudke, H. Heine, M. Bols and C. M. Pedersen, Chem. – Eur. J., 2014, 20, 13511–13516 CrossRef CAS PubMed.
- R. J. Willems and W. van Schaik, Future Microbiol., 2009, 4, 1125–1135 CrossRef PubMed.
- C. Theilacker, Z. Kaczynski, A. Kropec, F. Fabretti, T. Sange, O. Holst and J. Huebner, Infect. Immun., 2006, 74, 5703–5712 CrossRef CAS PubMed.
- S. Kodali, E. Vinogradov, F. Lin, N. Khoury, L. Hao, V. Pavliak, C. H. Jones, D. Laverde, J. Huebner, K. U. Jansen, A. S. Anderson and R. G. K. Donald, J. Biol. Chem., 2015, 290, 19512–19526 CrossRef CAS PubMed.
- A. Ali, R. J. B. H. N. van den Berg, H. S. Overkleeft, D. V. Filippov, G. A. van der Marel and J. D. C. Codée, Tetrahedron Lett., 2009, 50, 2185–2188 CrossRef CAS.
- W. F. J. Hogendorf, L. N. Lameijer, T. J. M. Beenakker, H. S. Overkleeft, D. V. Filippov, J. D. C. Codée and G. A. Van der Marel, Org. Lett., 2012, 14, 848–851 CrossRef CAS PubMed.
- W. F. J. Hogendorf, A. Kropec, D. V. Filippov, H. S. Overkleeft, J. Huebner, G. A. van der Marel and J. D. C. Codée, Carbohydr. Res., 2012, 356, 142–151 CrossRef CAS PubMed.
- W. F. J. Hogendorf, N. Meeuwenoord, H. S. Overkleeft, D. V. Filippov, D. Laverde, A. Kropec, J. Huebner, G. A. Van der Marel and J. D. C. Codée, Chem. Commun., 2011, 47, 8961–8963 RSC.
- D. Laverde, D. Wobser, F. Romero-Saavedra, W. Hogendorf, G. van der Marel, M. Berthold, A. Kropec, J. Codee and J. Huebner, PLoS One, 2014, 9, e110953 Search PubMed.
- Q. Chen, J. Dintaman, A. Lees, G. Sen, D. Schwartz, M. E. Shirtliff, S. Park, J. C. Lee, J. J. Mond and C. M. Snapper, Infect. Immun., 2013, 81, 2554–2561 CrossRef CAS PubMed.
- A. Fekete, P. Hoogerhout, G. Zomer, J. Kubler-Kielb, R. Schneerson, J. B. Robbins and V. Pozsgay, Carbohydr. Res., 2006, 341, 2037–2048 CrossRef CAS PubMed.
- D. van der Es, N. A. Groenia, D. Laverde, H. S. Overkleeft, J. Huebner, G. A. van der Marel and J. D. C. Codée, Bioorg. Med. Chem., 2016, 24, 3893–3907 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |