
A bonding evolution analysis for the thermal Claisen rearrangement: an experimental and theoretical exercise for testing the electron density flow
Patricio
González-Navarrete
 *,
Juan
Andrés
* and
V. Sixte
Safont
*,
Juan
Andrés
* and
V. Sixte
Safont
Departamento de Química Física y Analítica, Universitat Jaume I, 12071, Castelló de la Plana, Spain. E-mail: andres@qfa.uji.es; pgonzale@uji.es
First published on 30th November 2017
Abstract
A comprehensive theoretical investigation of the thermal Claisen rearrangement of allyl vinyl ether (AVE) to allylacetaldehyde has been carried out. We present the use of the electron localization function (ELF) to monitor the bonding evolution aspects in the course of this thermal rearrangement and the results are compared with a photo-impulsive process where instantaneous vibration frequencies are monitored [I. Iwakura, Phys. Chem. Chem. Phys., 2011, 12, 5546–5555]. Our results reveal an asynchronous electron density rearrangement inasmuch that the breaking of the C3–O bond and the formation of C1–C5 do not take place simultaneously. We also demonstrate how the bonding evolution brings about the natural appearance of the curly arrows representing the electronic flow in molecular rearrangements. This holds the key to gaining an unprecedented insight into the mapping of the electron density flow while the bonds change throughout the reaction progress.
1. Introduction
During the progress of a given chemical reaction, molecular structural changes from reactants to products take place via transition structures and/or possible intermediates. The results of fundamental research studies – both theoretical and experimental – have allowed the localization and determination of those stationary points on potential energy surfaces (PESs) to fully clarify the mechanism behind chemical reactions. Nevertheless, a further step is based on the idea that it is reasonable to think that an adequate representation of these chemical events should be given by a physical observable defined in coordinate space. The electron density, ρ(r), in contrast to the electronic wave-function, is a physical observable and therefore represents a well-defined property for analysis. ρ(r) is an experimentally accessible scalar field and a local function defined within the exact many-body theory. A deeper analysis in chemical reactivity can thus be achieved in order to identify the electron density flow as a function of reaction progress, while chemical events such as breaking/forming bonds and/or rearrangements of pairs of electrons are monitored. In this sense, previous theoretical studies have been reported and a connection between electron density ρ(r) distribution and chemical reactivity is found.1–13 In addition, developments in ultrafast electron and X-ray diffraction have led to experiments where molecular dynamics can be followed on the time scale of a chemical reaction.14–16 Examples include the seminal work of Zewail on femtosecond dynamics17 or those based on X-ray diffraction,18,19 electron diffraction,20 or laser-induced recollision.21–24 In particular, an ultrafast spectroscopy system with a visible ultrashort-pulse laser developed by Kobayashi et al.25–27 makes it possible to obtain time-dependent frequency shifts of relevant molecular vibrational modes throughout the reaction.28,29 This development is considered an innovative window to analyze the reaction mechanism of complex chemical rearrangements and also allows for a clear visualization of ultrafast structural changes in molecules during bond breaking/forming chemical events. Similarly, the remarkable properties of modern ultrashort X-ray and electron pulses seem to offer a very feasible alternative in the domain of ultrafast electronic processes of molecular systems. In particular, the degenerated Cope rearrangement of semibullvalene has been used as an example to illustrate the X-ray imaging of chemically active valence electrons during a pericyclic reaction.30 This approach allows the extraction of the changes in ρ(r) throughout the reaction progress, which are directly related to bond-making and bond-breaking processes, namely, the chemical valence electron density from the overall X-ray scattering pattern – which itself is dominated by the core electrons. This makes it possible to image the flow of valence electrons in space and time, thus demonstrating the asynchronous nature of bond-breaking and forming processes in pericyclic reactions.The motivation behind this study essentially arises from the work concerning the photo-impulsive Claisen rearrangement of allyl vinyl ether (AVE) reported by Iwakura and co-workers.31,32 The photo-impulsive reaction is induced with Raman processes, where only a fraction of molecular vibration modes are excited to high-level vibrational excited states with a few-optical-cycle visible pulse, which ensures that the reaction is triggered coherently. Although the thermal reaction may not be completely ruled out, the photo-impulsive process in the ground state, which is neither a photo- nor a thermal-reaction, follows the same reaction pathway as that of the symmetry-allowed thermal rearrangement in the ground state. The frontier orbitals of the photo-impulsive reaction in the electronic ground state can therefore be thought to be the same as those of the thermal reaction.28 Taking this into account, we present an alternative representation of the reaction mechanism for the thermal Claisen rearrangement of AVE in its respective ground state within the framework of the bonding evolution theory (BET),33 which combines the joint use of the electronic localization function (ELF)34,35 and Thom's catastrophe theory (CT)36 allowing a direct comparison with photo-impulsive reaction systems.
2. Computational details
The optimization and characterization of all stationary points on the PES as well as the calculation of the intrinsic reaction coordinate (IRC)37,38 pathway have been performed using Gaussian 09.39 The B3LYP40,41 electron density functionals, together with the 6-311+G(d,p)42 basis set, have been used for all atoms; as in previous studies the use of this methodology has been successfully tested.6,7,10,11 For each point obtained on the IRC pathway, the topological analysis of the ELF was performed using the TopMod package,43 considering a cubical grid with a step-size smaller than 0.05 Bohr. The topological partition of the ELF gradient field yields basins of attractors that can be thought of as corresponding to atomic cores, bonds, and lone pairs. In molecules, two types of basins are found: (i) core basins surrounding nuclei and labeled C(A) (where A is the atomic symbol of the element), and (ii) valence basins that are characterized by the number of core basins with which they share a boundary. This number is called the synaptic order.44 Hence, there are monosynaptic, disynaptic, trisynaptic basins, and so on. Monosynaptic basins, labeled as V(A), correspond to the lone pairs of the Lewis model, and polysynaptic basins correspond to the shared pairs of the Lewis model. In particular, disynaptic basins, labeled as V(A,X), correspond to two-center bonds, trisynaptic basins, labeled as V(A,X,Y), to three-center bonds, and so on. The valence shell of a molecule is the union of its valence basins. As hydrogen nuclei are located within the valence shell, they are counted as a formal core in the synaptic order, since hydrogen atoms have a valence shell; they are therefore called protonated disynaptic. Additionally, the basin population obtained by integration of the electronic density defines the number of electrons shared in a bond or in lone pairs. Taking this into account, the electronic density flow can be evaluated, thus indicating the changes in the structure of the system, that is, the connectivity among atoms along the reaction coordinate. It also serves as a basis for a better understanding of such processes by undertaking a meaningful assessment of the physical origins of potential energy barriers. Thus, the reaction is represented as a sequence of ELF topological domains called structural stability domains (SSDs). Accordingly, by using the molecular structure defined through the ELF topology, the reaction mechanism can be rationalized in terms of chemical events (i.e., bond forming or breaking processes, creation and annihilation of electron pairs) that are directly related to the corresponding SSD. For the sake of brevity, details concerning the theoretical aspects of the ELF, CT, and BET analyses as applied to single-step intermolecular processes are available elsewhere,5 while its applicability to more complex processes including multi-step and/or intermolecular reactions has also been previously shown by us.6,7,10,113. Results and discussion
The single step process of the thermal Claisen rearrangement of AVE to allylacetaldehyde proceeds via a six-membered transition state (TS) with an activation energy barrier of 26.2 kcal mol−1, while the reaction is calculated to be exothermic by 18.6 kcal mol−1, see Scheme 1. In AVE, 23 ELF basins have been localized: 6 core basins, 8 hydrogenated, 7 disynaptic basins (accounting for C–C and C–O bonds), and 2 monosynaptic basins (V1(O) and V2(O) accounting for the lone pairs of the oxygen atom, although for the sake of clarity the contribution of these two basins will be considered as the union of both, V1U2(O)). The localization and characterization of the ELF-basins for AVE and allylacetaldehyde as well as their redistribution in the course of the SSDs are depicted in Scheme 2, while the reaction energy profile with their corresponding SSDs are reported in Fig. 1. Note that the presence of two disynaptic basins in AVE between C1–C2 and C4–C5 accounts for the double nature of C1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2 and C4C5 bonds. The analysis of the ELF topology thus reveals eight different SSDs, which can be viewed as a sequence of chemical events. Each SSD is characterized by a particular number, and nature, of ELF-basins. As can be seen in Fig. 1, SSD-I extends from an IRC value of −10.7 to −1.36 amu1/2 Bohr, and, as explained above, it consists of 23 ELF-basins. This ELF-basin distribution changes at SSD-II in which 22 ELF basins are found: 6 core basins, 8 hydrogenated, 6 (instead of seven) disynaptic and 2 monosynaptic basins, see Scheme 2. Along SSD-II, which extends from an IRC value of −1.33 to −1.07 amu1/2 Bohr, the disynaptic basin V(C3,O) has been annihilated and therefore is not present, see Scheme 2. In this way, by analyzing the changes in the number and/or the nature of the ELF basins along the IRC reaction pathway, we detect seven changes in the ELF-topological description, thus giving rise to the eight SSDs found. The observed changes are explained in more detail, and are related to chemical events, in what follows.
C2 and C4C5 bonds. The analysis of the ELF topology thus reveals eight different SSDs, which can be viewed as a sequence of chemical events. Each SSD is characterized by a particular number, and nature, of ELF-basins. As can be seen in Fig. 1, SSD-I extends from an IRC value of −10.7 to −1.36 amu1/2 Bohr, and, as explained above, it consists of 23 ELF-basins. This ELF-basin distribution changes at SSD-II in which 22 ELF basins are found: 6 core basins, 8 hydrogenated, 6 (instead of seven) disynaptic and 2 monosynaptic basins, see Scheme 2. Along SSD-II, which extends from an IRC value of −1.33 to −1.07 amu1/2 Bohr, the disynaptic basin V(C3,O) has been annihilated and therefore is not present, see Scheme 2. In this way, by analyzing the changes in the number and/or the nature of the ELF basins along the IRC reaction pathway, we detect seven changes in the ELF-topological description, thus giving rise to the eight SSDs found. The observed changes are explained in more detail, and are related to chemical events, in what follows.
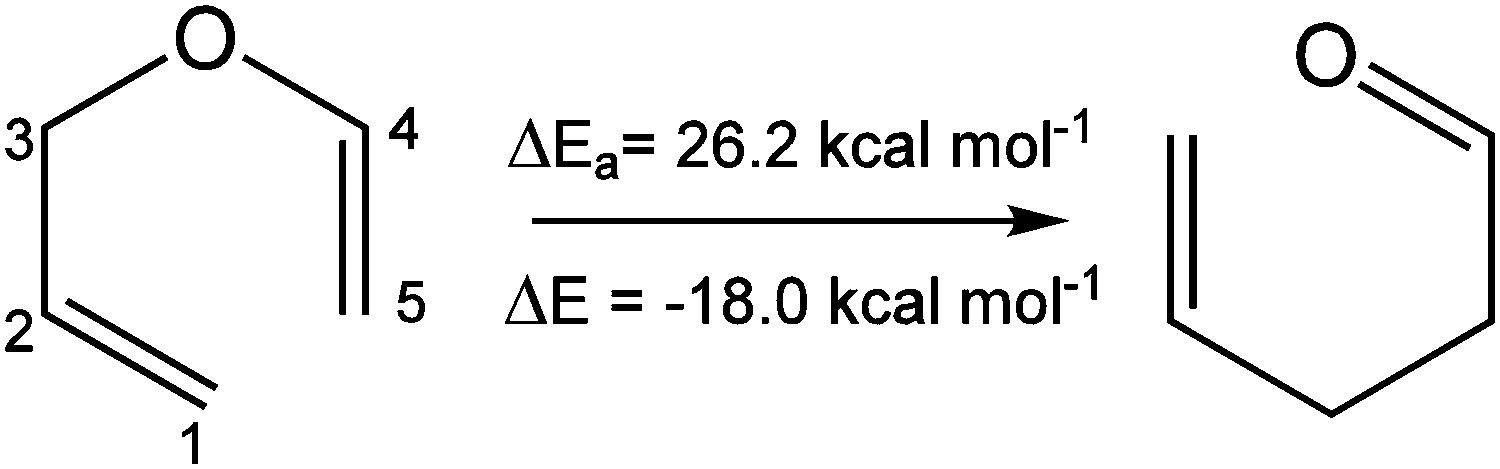 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 Distribution of the ELF-basins from AVE to Allylacetaldehyde in every ELF-SSD. Color code: black for core basins, gray for disynaptic basin, and circumferences for monosynaptic basins. For the sake of clarity, protonated disynaptic basins are omitted. | ||
 | ||
| Fig. 1 IRC reaction pathway with their corresponding structural stability domains (SSDs) for the thermal rearrangement of AVE to allylacetaldehyde. Below the graph, a schematic representation of the reaction mechanism for each SSD from the ELF analysis (full lines and ellipses represent disynaptic and monosynaptic basins, respectively). Dotted lines indicate a large basin population. For the sake of clarity, protonated disynaptic basins and monosynaptic Vi=1,2(O) basins are omitted. | ||
In addition, for each point obtained on the IRC pathway, the basin populations of some specific attractors have been evaluated with the aim of following their respective evolutions in the course of the reaction (see Fig. 2, Table 1 and Scheme 2). The sequence of the ELF-SSDs summarizes the whole process as follows: (1) breaking of the C3–O bond; (2) reduction of the double bonds C1C2 and C4C5 to single ones; (3) formation of the single bond C1–C5; and (4) formation of the double bonds C2C3 and OC4. This response from the system accounts for an asynchronous redistribution of the electronic flow in the course of this concerted reaction mechanism, where C3–O bond-breaking and C1–C5 formation processes, as well as the reduction of double bonds to single ones (and vice versa), do not occur simultaneously. A detailed ELF analysis shows that the first ELF-topological change connecting SSD-I and SSD-II is associated with the breaking process of the C3–O bond, since the annihilation of the disynaptic basin V(C3,O) is observed. An apparent reduction in the population of the disynaptic basin V(C3,O) accounts for the weakening of the bond C3–O in the course of SSD-I, while an increment in the population of the monosynaptic basins V1U2(O) is also observed, thus indicating that part of the electron density from the C3–O bond is transferred to the valence shell of the oxygen atom. SSD-II is predicted to be rather short and, therefore, no drastic changes in the basin populations are observed. The second ELF-topological change connecting SSD-II and SSD-III accounts for the reduction of the double C4C5 bond to the single C4–C5 bond; the disynaptic basins Vi=1,2(C4,C5) merge into a single disynaptic basin (C4,C5). The same holds true for the next ELF-topological change connecting SSD-III and SSD-IV, where the disynaptic basins Vi=1,2(C1,C2) merge into a single one V(C1,C2). Note that the TS is localized in the course of SSD-IV. Interestingly, neither topological signature nor breaking/forming processes confirm that the formation of the C1–C5 bond does not occur in the TS. In addition, significant changes in the basin populations of some basins are observed, in particular, an important increase in the basin population of the disynaptic basins V(O,C4) and V(C2,C3), whereas the disynaptic basins V(C1,C2) and V(C4,C5) reduce their respective populations considerably. The electronic flow in the course of SSD-IV prepares the system for the imminent formation of C1–C5. Thus, the subsequent ELF-topological changes result in the creation of the non-bonding monosynaptic basin V(C5), and afterwards V(C1). It is worth noting that the appearance of these two monosynaptic basins in the course of the reaction does not really necessarily represent a diradical character. Bear in mind that the whole process has been considered on a uniform closed shell singlet spin state, and therefore, it lacks multireference character.
 | ||
| Fig. 2 Basin populations along the IRC pathway for the thermal rearrangement of AVE to allylacetaldehyde. | ||
| SSD-I | SSD-II | SSD-III | SSD-IV | SSD-V | SSD-VI | SSD-VII | SSD-VIII | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rxa | AVE | −10.7b | −1.36c | −1.33b | −1.07c | −1.04b | −0.98c | −0.95b | 0.0(TS) | 0.95c | 0.98b | 1.04c | 1.07b | 1.83c | 1.86b | 2.20c | 2.23b | 11.5c | Allylacetaldehyde |
| a The reaction coordinate (Rx) is in amu1/2 Bohr. b Reaction coordinates at the initial point of the SSD. c Reaction coordinate at the last point of the SSD. | |||||||||||||||||||
| V1(C1,C2) | 1.71 | 1.71 | 1.55 | 1.55 | 1.50 | 1.50 | 1.51 | 3.18 | 3.00 | 2.76 | 2.76 | 2.75 | 2.56 | 2.37 | 2.36 | 2.30 | 2.29 | 2.01 | 2.00 |
| V2(C1,C2) | 1.71 | 1.71 | 1.69 | 1.69 | 1.70 | 1.70 | 1.68 | — | — | — | — | — | — | — | — | — | — | — | — |
| V1(C2,C3) | 2.06 | 2.06 | 2.29 | 2.30 | 2.36 | 2.36 | 2.38 | 2.38 | 2.69 | 2.99 | 2.99 | 3.01 | 3.01 | 3.13 | 3.14 | 3.19 | 1.62 | 1.72 | 1.72 |
| V2(C2,C3) | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 1.57 | 1.71 | 1.72 |
| V(C3,O) | 1.31 | 1.22 | 0.45 | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
| V(O,C4) | 1.50 | 1.46 | 1.57 | 1.58 | 1.61 | 1.62 | 1.63 | 1.63 | 1.78 | 1.97 | 1.98 | 2.00 | 2.01 | 2.16 | 2.16 | 2.21 | 2.22 | 2.41 | 2.41 |
| V1U2(O) | 4.62 | 4.74 | 5.30 | 5.73 | 5.68 | 5.51 | 5.58 | 5.53 | 5.45 | 5.37 | 5.37 | 5.37 | 5.37 | 5.37 | 5.32 | 5.28 | 5.28 | 5.14 | 4.65 |
| V1(C4,C5) | 1.79 | 1.81 | 1.76 | 1.76 | 1.75 | 3.34 | 3.33 | 3.33 | 3.21 | 3.07 | 2.76 | 2.74 | 2.72 | 2.64 | 2.47 | 2.40 | 2.39 | 2.07 | 2.06 |
| V2(C4,C5) | 1.79 | 1.46 | 1.62 | 1.62 | 1.60 | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
| V(C1) | — | — | — | — | — | — | — | — | — | — | — | 0.19 | 0.33 | — | — | — | — | — | |
| V(C5) | — | — | — | — | — | — | — | — | — | 0.31 | 0.32 | 0.33 | 0.51 | — | — | — | — | — | |
| V(C1,C5) | — | — | — | — | — | — | — | — | — | — | — | — | — | 0.85 | 0.97 | 0.98 | 1.75 | 1.81 | |
Following the creation of these two non-bonding basins, a new topological change is observed connecting SSD-VI and SSD-VII; the monosynaptic basins V(C5) and V(C1) merge into the single disynaptic basin V(C1,C5), accounting for the formation of the C1–C5 bond for the very first time. As mentioned above, the bond breaking/forming processes for the C3–O/C1–C5 bonds, respectively, do not take place simultaneously, thus demonstrating the asynchronicity of the process despite the fact that the reaction proceeds via a concerted mechanism. Finally, due to a regular increment in the population of the disynaptic basin V(C2,C3), the last topological change connecting SSD-VII and SSD-VIII is observed, accounting for the transformation of the single C2–C3 bond into double C2C3. In terms of ELF analysis, the disynaptic basin V(C4,C5) splits into two disynaptic basins Vi=1,2(C4,C5). In particular, note that for the double bond OC4 in the allylacetaldehyde only a single disynaptic basin is observed despite its double nature and, therefore, an ELF topological change accounting for the double nature is not expected. Nevertheless, a significant increment in the population of the disynaptic basin V(O,C4) can be observed in the course of the reaction, thereby indicating a change in the bond order between C4 and O atoms. According to the above findings, the reaction mechanism can be illustrated as depicted in Scheme 3, where the evolution of the basin populations in each SSD accounts for the curly arrows which stand for electron density flow.
 | ||
| Scheme 3 Schematic representation of the curly arrows in every ELF-SSDs localized in the course of the thermal Claisen rearrangement of AVE to allylacetaldehyde. For the sake of clarity, protonated disynaptic basins and monosynaptic Vi=1,2(O) basins are omitted. | ||
The results obtained from the photo-impulsive reaction stimulated by a Raman process using a 5 fs pulse for the Claisen rearrangement of AVE31,32 reveal that the C3–O bond is weakened or broken in the first step of the process, since the νsC3–O–C4 of ether (890 cm−1) disappears at about 800 fs. This process is directly related to the first topological change connecting SSD-I and SSD-II, since the annihilation of the disynaptic basin V(C3,O4) reflects the breaking process of the C3–O bond as the first event in the reaction. In addition, the νsCC of the vinyl and allyl groups appears at around 1650 cm−1 just after photo-irradiation; nevertheless from 500 to 800 fs the νsCC splits into two red- and blue-shifted bands, indicating that the electrons transfer from the allyl group to the vinyl group. This behavior can also be perceived in the evolution of the basin populations: after the annihilation of the disynaptic basin V(C3,O), reflecting the breaking process of the C3–O bond, a plausible increment in the population of the monosynaptic basins V1U2(O) and the disynaptic basin V(O,C4) is observed, indicating a charge transfer from the allyl moiety to the vinyl group. Likewise, while the basin populations of the disynaptic basins V1U2(C1,C2) and V1U2(C4,C5) are slightly different at a very early stage of the reaction, this difference in their corresponding populations is more pronounced in the course of SSD-IV (see Table 1 and Fig. 2), thereby corroborating a higher concentration of electron density in the vinyl moiety. Then, the electron density flows from the vinyl group to the allyl group to form the C1–C5 bond. This is indicated by the appearance of νsC–C–C at 1000 cm−1, confirming the presence of the C1–C5 bond. According to our results, under the framework of the ELF-topology the presence, first, of the monosynaptic basin V(C5), then V(C1), and subsequently V(C1,C5) also indicates the direction of the flow of the electron density from the vinyl to the allyl moiety as well as the formation of the C1–C5 bond. While the photo-impulsive process first shows the weakening of the C3–O bond, giving rise to the formation of a bis-allyl-like intermediate, the simultaneous C3–O bond-breaking and C1–C5 bond-formation via an aromatic six-membered structure subsequently takes place. Similarly, the evolution of the ELF basins supports the sequence of events, also demonstrating the presence of a bis-allyl-like species in the course of the reaction, although the breaking/forming processes are predicted in an asynchronous way.
4. Conclusions
In summary, a complete bonding evolution analysis provides a significant guide to elucidate chemical reaction mechanisms. Thus, valuable information is obtained to facilitate the visualization and conceptualization of chemical reactions in terms of chemical events such as the breaking/forming of chemical bonds or the transformation of formally double to simple bonds. This fact allows a Lewis-type representation of curly arrows associated with electron density flow. The topological changes take place by means of consecutive steps, in which the C3–O breaking bond process precedes the process of formation of the C1–C5 bond. This result is in agreement with the experimental data reported by visible ultrashort pulse laser that allows time-dependent frequency shifts of relevant molecular vibrational modes to be obtained as the reaction progresses. The present methodology is based on physical laws and quantum theory grounds, and it can be considered an appropriate tool to tackle chemical reactivity with a wide range of possible applications. This analysis can be used for the study of different organic and inorganic chemical reactions, thus changing the way in which we think about reaction mechanisms.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are grateful to Generalitat Valenciana for PrometeoII/2014/022 and ACOMP/2015/1202, to Universitat Jaume I for project UJI-B2016-25, and to Ministerio de Economía y Competitividad (Spain) for project CTQ2015-65207-P. The authors also thank the Servei d'Informàtica, Universitat Jaume I, for generous allocation of computer time.References
- J. Andrés, S. Berski, L. R. Domingo and P. González-Navarrete, J. Comput. Chem., 2012, 33, 748–756 CrossRef PubMed.
- P. González-Navarrete, J. Andrés and S. Berski, J. Phys. Chem. Lett., 2012, 3, 2500–2505 CrossRef PubMed.
- P. González-Navarrete, L. R. Domingo, J. Andrés, S. Berski and B. Silvi, J. Comput. Chem., 2012, 33, 2400–2411 CrossRef PubMed.
- V. Polo and J. Andres, J. Comput. Chem., 2005, 26, 1427–1437 CrossRef CAS PubMed.
- V. Polo, P. Gonzalez-Navarrete, B. Silvi and J. Andres, Theor. Chem. Acc., 2008, 120, 341–349 CrossRef CAS.
- M. Oliva, V. S. Safont, P. González-Navarrete and J. Andrés, Theor. Chem. Acc., 2017, 136, 51 CrossRef.
- J. Andres, L. Gracia, P. Gonzalez-Navarrete and V. S. Safont, Comput. Theor. Chem., 2015, 1053, 17–30 CrossRef CAS.
- J. Andres, S. Berski, J. Contreras-Garcia and P. Gonzalez-Navarrete, J. Phys. Chem. A, 2014, 118, 1663–1672 CrossRef CAS PubMed.
- J. Andres, P. Gonzalez-Navarrete and V. S. Safont, Int. J. Quantum Chem., 2014, 114, 1239–1252 CrossRef CAS.
- P. Gonzalez-Navarrete, F. R. Sensato, J. Andres and E. Longo, J. Phys. Chem. A, 2014, 118, 6092–6103 CrossRef CAS PubMed.
- V. S. Safont, P. Gonzalez-Navarrete, M. Oliva and J. Andres, Phys. Chem. Chem. Phys., 2015, 17, 32358–32374 RSC.
- J. Andres, S. Berski and B. Silvi, Chem. Commun., 2016, 52, 8183–8195 RSC.
- S. Berski and P. Durlak, New J. Chem., 2016, 40, 8717–8726 RSC.
- A. H. Zewail, Science, 2010, 328, 187–193 CrossRef CAS PubMed.
- H. N. Chapman, Nature, 2010, 467, 409–410 CrossRef CAS PubMed.
- P. Wernet, Phys. Chem. Chem. Phys., 2011, 13, 16941–16954 RSC.
- A. H. Zewail, J. Phys. Chem. A, 2000, 104, 5660–5694 CrossRef CAS.
- B. Perman, V. Srajer, Z. Ren, T. Y. Teng, C. Pradervand, T. Ursby, D. Bourgeois, F. Schotte, M. Wulff, R. Kort, K. Hellingwerf and K. Moffat, Science, 1998, 279, 1946–1950 CrossRef CAS PubMed.
- R. Neutze, R. Wouts, D. van der Spoel, E. Weckert and J. Hajdu, Nature, 2000, 406, 752–757 CrossRef CAS PubMed.
- H. Ihee, V. A. Lobastov, U. M. Gomez, B. M. Goodson, R. Srinivasan, C. Y. Ruan and A. H. Zewail, Science, 2001, 291, 458–462 CrossRef CAS PubMed.
- J. Itatani, J. Levesque, D. Zeidler, H. Niikura, H. Pepin, J. C. Kieffer, P. B. Corkum and D. M. Villeneuve, Nature, 2004, 432, 867–871 CrossRef CAS PubMed.
- H. J. Woerner, J. B. Bertrand, D. V. Kartashov, P. B. Corkum and D. M. Villeneuve, Nature, 2010, 466, 604–607 CrossRef CAS PubMed.
- Y. Arasaki, K. Takatsuka, K. Wang and V. McKoy, J. Chem. Phys., 2010, 132 Search PubMed.
- P. Hockett, C. Z. Bisgaard, O. J. Clarkin and A. Stolow, Nat. Phys., 2011, 7, 612–615 CrossRef CAS.
- T. Kobayashi, T. Saito and H. Ohtani, Nature, 2001, 414, 531–534 CrossRef CAS PubMed.
- A. Baltuska, T. Fuji and T. Kobayashi, Opt. Lett., 2002, 27, 306–308 CrossRef PubMed.
- T. Kobayashi, A. Shirakawa and T. Fuji, IEEE J. Sel. Top. Quantum Electron., 2001, 7, 525–538 CrossRef CAS.
- I. Iwakura, A. Yabushita, J. Liu, K. Okamura and T. Kobayashi, Phys. Chem. Chem. Phys., 2012, 14, 9696–9701 RSC.
- I. Iwakura, A. Yabushita and T. Kobayashi, Chem. Phys. Lett., 2011, 501, 567–571 CrossRef CAS.
- T. Bredtmann, M. Ivanov and G. Dixit, Nat. Commun., 2014, 5, 5589 CrossRef CAS PubMed.
- I. Iwakura, A. Yabushita and T. Kobayashi, Chem. Lett., 2010, 39, 374–375 CrossRef CAS.
- I. Iwakura, Phys. Chem. Chem. Phys., 2011, 13, 5546–5555 RSC.
- X. Krokidis, S. Noury and B. Silvi, J. Phys. Chem. A, 1997, 101, 7277–7282 CrossRef CAS.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- B. Silvi and A. Savin, Nature, 1994, 371, 683–686 CrossRef CAS.
- R. Thom, Structural stability and morphogenesis; an outline of a general theory of models, W. A. Benjamin, Reading, Mass., 1st English edn, 1975 Search PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- S. Noury, X. Krokidis, F. Fuster and B. Silvi, Comput. Chem., 1999, 23, 597–604 CrossRef CAS.
- B. Silvi, J. Mol. Struct., 2002, 614, 3–10 CrossRef CAS.
| This journal is © the Owner Societies 2018 |